

Abstract

SET domain-containing protein 2 (SETD2), a histone methyltransferase, has been identified as a target of interest in certain hematological malignancies, including multiple myeloma. This account details the discovery of EPZ-719, a novel and potent SETD2 inhibitor with a high selectivity over other histone methyltransferases. A screening campaign of the Epizyme proprietary histone methyltransferase-biased library identified potential leads based on a 2-amidoindole core. Structure-based drug design (SBDD) and drug metabolism/pharmacokinetics (DMPK) optimization resulted in EPZ-719, an attractive tool compound for the interrogation of SETD2 biology that enables in vivo target validation studies.
Keywords: Histone methyltransferase, SETD2
The lysine N-methyltransferase SETD2,1 is the only known enzyme capable of the trimethylation of histone H3 lysine 36 (H3K36)2 and has been reported to play a role in transcriptional elongation3,4 and alternative splicing.5,6 Although SETD2 has been shown to have a role as a tumor suppressor,7−10 our target identification efforts identified it as a potential target of interest for the treatment of some cancers.11 Inhibitors of SETD2 have been described in the literature;12 however, none are suitable for in vivo studies. To further validate this target, we sought a suitable small molecule inhibitor that could be used to thoroughly interrogate SETD2 biology both in vitro and in vivo. Herein, we detail the efforts that led to the identification of a selective SETD2 inhibitor with these desirable properties.
The modulation of epigenetic targets with small molecules has been part of our strategy to understand the biology of various histone methyltransferases.13 During the course of these efforts, we developed a histone methyltransferase-biased library enriched with chemotypes that possessed desirable physiochemical properties, which could be elaborated into tool compounds. We screened our in-house library against SETD2 in a radiometric assay that monitored the methylation of a histone peptide and identified two hits of interest, compounds 1 and 2 (Figure 1).
Figure 1.

Initial screening hits and results of the initial optimization.
Hits 1 and 2 were confirmed in the SETD2 biochemical assay with IC50 values of 166 and 22.1 μM, respectively. Further workup in our hit validation funnel demonstrated that 1 and 2 were true hits. Both 1 and 2 show reversible inhibition of SETD2, with an uncompetitive mechanism with respect to the substrate S-adenosylmethionine (SAM) and a noncompetitive mechanism with respect to the peptide substrate. Further, compound 2 displayed well-behaved binding in a surface plasmon resonsance (SPR) binding assay with a KD value of 40 μM, and we obtained a ligand-bound X-ray cocrystal structure at 2.30 Å between compound 2, SAM, and SETD2 (Figure 2).
Figure 2.

X-ray structure of HTS hit 2. Compound 2 is represented as sticks, and SAM is represented as lines. (A) Surface representation of SETD2 showing the location of compound 2 at right relative to the SAM binding site. (B) Binding mode of compound 2 showing key interactions with SETD2 and the proximity to the SAM binding site.
The structure of the ligand-bound complex shows that 2 occupies a portion of the enzyme’s peptide binding site, with the 7-methylindole buried deep within the lysine channel where histone 3 lysine 36 would normally be positioned for methylation by SAM. In this binding pose, the indole aromatic rings make a face-to-face π-stacking interaction with Tyr1666. The indole N–H and the indolecarboxamide carbonyl interact with the backbone carbonyl and N–H from Phe1606, respectively, via H-bonding, and the cyclohexane ring of the ligand makes predominantly nonpolar contacts with the protein. The β-alanine terminus was poorly resolved, presumably due to its high degree of flexibility and mobility within the crystal lattice. Although the noncompetitive mechanism of 2 with respect to the peptide substrate may seem at odds with the observed binding location in the X-ray structure, this type of relationship was observed previously14−16 and may occur for a variety of reasons, such as exosite binding. Additional follow-up efforts led to the identification of compounds 3, with a biochemical IC50 value of 2.08 μM, and 4, with a biochemical IC50 value of 4.24 μM, as potential starting points for further elaboration.
To assess the importance of substitution at the 7-position of the indole, we utilized a small collection of compounds (Table 1) to assess the impact of this structural motif. We found that compounds containing the 7-methylindole displayed significant activity in the biochemical assay. Moreover, the X-ray structure of 2 showed that the indole was positioned deep within the lysine channel and was highly engaged with the protein, which was further validated by analogs that explored the structure–activity relationship (SAR) of the indole unit based on compound 3 (Table 1). In combination with this methyl, some substitution with fluorine was tolerated, as shown by 8 and 9, with substitution at the 4-position leading to significant improvements in the activity. The indole unit was generally tolerated better as compared to other heterocycles, which is consistent with the observed crystal structure. We therefore focused our synthetic efforts on 7-methylindole- and 4-fluoro-7-methylindole-containing analogs.
Table 1. Effect of Indole Substitution and the Core Change.


Standard deviations for the active compounds are less than 13% of the IC50 values.
Despite improvements in the biochemical potency for derivatives of compound 3 and our efforts to improve the physical and DMPK properties of the molecules, progressing this subseries remained difficult. We therefore refocused our optimization efforts on compound 4. Our work in this new series can be seen in Table 2. For compounds with an adequate biochemical potency, we demonstrated the activity in a cellular context using an in-cell Western (ICW) cell biochemical assay in A549 cells by monitoring the H3K36 trimethyl mark. We began by surveying the impact of the meta-substitution on the arene, as it had a notably improved activity over that of an exemplary analog with an ortho-substitution. The morpholine analog 17 had a submicromolar activity and, when combined with the C4 fluoride substitution on the indole core, the biochemical potency for 18 increased sixfold. Further investigation of the amine meta-substituent led to 20, which had a fivefold increase in its biochemical potency over that of 18. Next, we focused on the addition of a second meta-substituent, which was observed to further improve the biochemical potency in some cases, as seen in compounds 21–23.
Table 2. Effect of R1, R2, R3, and R4 Substitutions of 4.


Standard deviations range from 3% to 63% of the IC50 values.
Standard deviations range form 7% to 67% of the IC50 values.
The translation of the biochemical activity into a cellular assay continued to prove challenging at this stage. We noted that many of the more biochemically potent compounds contained amines that would be expected to be quite basic and that compounds 22 and 23 in general displayed a large shift between the biochemical and cell biochemical assays. Several factors might explain such a shift, including the impact of basicity on the partition between cellular compartments17 and the effect of serum binding in the assay medium. To probe this hypothesis, we prepared nonbasic pyrrolidine analogs with an amide substituent. This effort furnished 24 and 25 with overall lower shifts between the biochemical and cellular assays, with 25 displaying a low-nanomolar potency in the cell biochemical assay.
During these studies, we were able to obtain an X-ray structure for 20 of the ligand-bound complex, as shown in Figure 3. The positioning of the indole ring is comparable to that in the structure for compound 2, with similar π-stacking and hydrogen bonding interactions. The central aromatic ring of 20 makes π-stacking contacts with Tyr1604, and the N-aminopiperidine substituent points out toward the solvent. The steric disposition of the N-aminopiperidine in this structure was consistent with the observation that meta-substituents on the central aryl ring had higher cellular activities.
Figure 3.

X-ray structure of 20 overlaid with 2. Compound 20 is shown as green sticks, and compound 2 is shown as a ball and stick representation. The protein structures in the 20 and 2 costructures are shown in gray and blue, respectively.
While 25 provided many key features that were desirable for an in vivo tool compound, there were a few additional areas to optimize. Analogs in Table 2 displayed a high lipophilicity and aromatic character; as a result, low aqueous solubilities were observed for several analogs, and low fractions absorbed were observed for those analogs we studied in vivo. The pharmacokinetic (PK) behavior of compound 25 in mice is shown in Table 3 as an example of this. While compound 25 shows a moderate clearance, we did not pursue in vivo studies, in part due to the low fraction absorbed (Fa).
Table 3. Detailed Profile Assessments for 25 and 32.
| compound | 25 | 32 |
|---|---|---|
| hepatocyte CLint, human/mouse (mL/min/kg) | 64/239 | 51/721 |
| Caco-2 PappA-B (10–6 cm/s), efflux ratio | 3.2, 5.6 | |
| PSA (Å2) | 68 | 87 |
| cLogD | 3.2 | 0.32 |
| CYP 450 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4 inhibition, IC50 (μM)a | >50, >50, 16, 36, >50, > 50, >50, >50 | |
| hERG IC50 (μM) | 4.5 | |
| PK parameters (mouse) | ||
| CL (mL/min/kg) | 24.2 | 53.3 |
| Vss (L/kg) | 1.04 | 8.13 |
| terminal t1/2 (h) | 0.7 | 3.13 |
| estimated Fa | <0.01 (50 mpk) | 0.7 (30 mpk) |
| time over ICW IC50 (h) | 0 (50 mpk) | ∼6 (30 mpk), ∼12 (100 mpk) |
Values are estimated IC50 values based on the percent (%) inhibition at a single concentration (50 μM).
To address this, we investigated the impact of alternatives to the compounds’ central aromatic core using a saturated system similar to those of our original HTS hits 1 and 2.18,19 The resulting compounds (Table 4) displayed activities that were highly stereoselective, favoring the (1R,3S)-isomer. The activity generally tracked with those found in the corresponding aromatic series of compounds. Thus, merging those lessons from our SAR studies as outlined above, the optimal activity was found in aminopyrrolidine compounds such as 30 and 32. This scaffold displayed much more favorable physical chemical, and ADME properties when compared to those of the analogous compounds with an aryl core. Additionally, the ADME properties measured in vitro generally translated well into in vivo PK. We chose to focus on compound 32 as it showed high potencies in both the biochemical and cell biochemical assays. We determined the Ki value for 32 to be 3.3 nM with an uncompetitive mechanism versus SAM and a noncompetitive mechanism versus the peptide (Supporting Information Figure 1). Although it displayed a relatively high intrinsic clearance in vitro in mouse hepatocytes, we reasoned that its decreased aromatic character might provide some benefits in oral absorption. The results for mouse PK studies for compound 32 are shown in Table 3. We found that compound 32 had a much higher oral absorption in mice as compared to that of compound 25. Although 32 exhibited a relatively high in vivo clearance, the fraction absorbed at 10 mpk was estimated to be 0.6, indicating that 32 was well absorbed in a conventional formulation (0.5% CMC/0.1% Tween80), which can be attributed to its improved solubility and dissolution. Gratifyingly, oral exposure increased greater than dose-proportional as doses increased up to 100 mpk, thus providing sufficient exposure relative to the ICW IC50 for the use of compound 32 as a SETD2-inhibitory tool compound. Furthermore, 32 displayed a >8000-fold selectivity in a panel of 14 other histone methyltransferases, showing a remarkable level of selectivity over these closely related and potentially confounding targets. It showed minimal activity in a panel of 47 common off-target receptors and enzymes, showing only modest micromolar activity against three GPCRs. Finally, the compound showed no activity greater than 30% inhibition at 10 μM in a panel of 45 kinases (see the Supporting Information)
Table 4. Structure–Activity Relationship of the 1,3-cis-Diaminocyclohexyl Substitution.
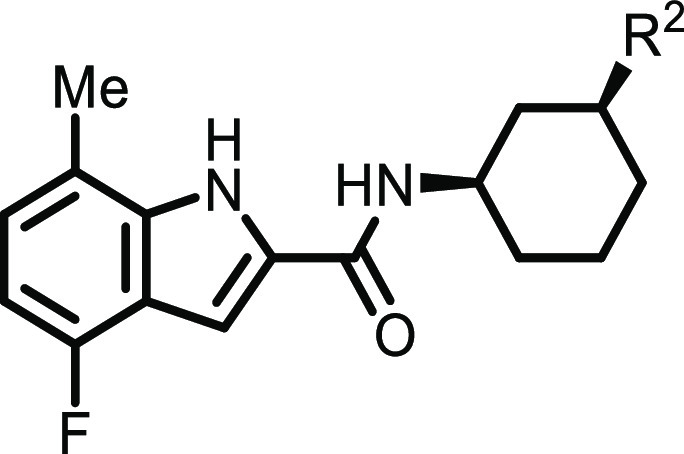

Standard deviations range from 3% to 48% of the IC50 values.
Standard deviations range from 0.5% to 57% of the IC50 values.
Again, we confirmed the binding of 32 by cocrystallization, as shown in Figure 4. Like earlier analogs, the position of the indole was conserved, and the same contacts that were observed previously were seen here as well. The cyclohexane ring appears to make nonpolar contacts and principally serves to orient the pyrrolidine substituent in an appropriate direction toward the solvent. While the conformation of the ligand is well-defined by the electron density, the amino acid side chains of the protein loop proximal to the pyrrolidine sulfonamide (Lys1673 and Glu1674) are poorly defined, presumably reflecting the flexible nature of this region. As a result, no specific interactions between this portion of the ligand and the protein, either directly or via bound waters, were observed.
Figure 4.

X-ray structure of 32 overlaid with 20. Compound 32 is shown as green sticks, and compound 20 is shown as a ball and stick representation. The protein structures in the 32 and 20 costructures are shown in gray and blue, respectively.
Compounds 32 and 33 were studied for their effects on the proliferation of two multiple myeloma cell lines, KMS34 and KMS11, and the data can be seen in Table 5. The potent antiproliferative activity of compound 32 coupled with its encouraging PK behavior in mice suggested that it might prove suitable for study in appropriate xenograft models.20
Table 5. Anti-Proliferation Data of Relevant Cell Lines for 32 and 33 in 14 Day Long-Term Proliferation (LTP) Assays.
| compound | KMS34 LTP day 14 IC50 (μM) | KMS11 LTP day 14 IC50 (μM) |
|---|---|---|
| 32 | 0.025 | 0.211 |
| 33 | 0.038 | 0.476 |
Compound 32 and its related analogs disclosed in Table 4 can be prepared as outlined in Scheme 1 (analogs from Tables 1 and 2, see the Supporting Information). The synthesis begins with the condensation of hydrazine 34 with ethyl pyruvate, followed by a cyclic [3,3]-sigmatropic rearrangement and subsequent ammonium elimination in the presence of p-TsOH via a Fishcher indole cyclization protocol. Basic saponification afforded the carboxylic acid 36 in a high yield. The installation of the amine partner was accomplished by a HATU-mediated coupling with (1R,3R)-3-aminocyclohexan-1-ol, followed by a mild oxidation of the alcohol to the cyclohexanone 37. Then, condensation of the secondary amine partner with 37 and a subsequent in situ reduction give a mixture of diastereomers (2:1 cis/trans). Finally, isolation by prep-HPLC chromatography completes the synthesis for EPZ-719 (32).
Scheme 1. Synthesis of EPZ-719.

Reagents and conditions are as follows: (a) ethyl pyruvate, H2SO4, EtOH, 88%; (b) p-TsOH, toluene, 100 °C, 23%; (c) NaOH, THF, MeOH, water, rt, 81%; (d) (1R,3R)-3-aminocyclohexan-1-ol, HATU, diisopropylethylamine, DMF, rt, 76%; (e) PCC, EtOAc, rt, 48%; (f) (S)-N-methyl-N-(pyrrolidin-3-yl)methanesulfonamide, NaBH3CN, MeOH, rt, 52%; (g) prep-HPLC.
In summary, we began a campaign to identify an appropriate compound that could be used to interrogate SETD2 biology in vivo. A screen of our focused library directed toward epigenetic targets provided hit compounds 1 and 2. 1 and 2 were modified in a sequential medicinal chemistry optimization process to lead to compound 32, which met our goals for potency, selectivity, and ADME behavior suitable for in vitro and in vivo investigations of SETD2 biology. Structural insights gained from crystallographic studies served to elucidate the binding behavior of the inhibitors but proved limited in optimizing solvent-facing portions of the molecules due to the flexibility of the protein. Close attention to the physical chemical properties of the inhibitors, in particular basicity, lipophilicity, and aromatic character, led to compounds with attractive cellular activities and in vivo exposures. Additional results of in vivo studies on SETD2 biology and tumor growth inhibition in xenograft models are in preparation for a future manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00272.
Synthetic procedures for all compounds; biochemical, cellular, ADME, and pharmacokinetic assay protocols; and crystallography protocols(PDF)
Author Present Address
† Accent Therapeutics, 65 Hayden Avenue, Lexington, Massachusetts 02421, United States
Author Present Address
‡ Blueprint Medicines, 45 Sidney Street, Cambridge, Massachusetts 02139, United States.
Author Present Address
◆ 28-7 Therapeutics, 490 Arsenal Way, Suite 100B, Watertown, Massachusetts 02472, United States.
Author Present Address
∥ Munchhof LLC, 335 Frost Lane, Corvallis, Montana 59828, United States.
Author Present Address
± Remix Therapeutics, One Kendall Square, Building 200, Cambridge, Massachusetts 02139, United States.
Author Present Address
∇ Foghorn Therapeutics, 500 Technology Square, Suite 700, Cambridge, Massachusetts 02139, United States.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare the following competing financial interest(s): All authors are/were employees of Epizyme, Inc. Work was funded by Epizyme.
Notes
The coordinates of SETD2 in complex with 2, 20, and 32 have been deposited under PDB IDs 7LZB, 7LZD, and 7LZF, respectively.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters special issue “Epigenetics 2022”.
Supplementary Material
References
- Sun X. J.; Wei J.; Wu X. Y.; Hu M.; Wang L.; Wang H. H.; Zhang Q. H.; Chen S. J.; Huang Q. H.; Chen Z. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem. 2005, 280 (42), 35261–71. 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- Edmunds J. W.; Mahadevan L. C.; Clayton A. L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27 (2), 406–20. 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F.; Bugler B.; Schmidt C. K.; Guillou E.; Caron P.; Briois S.; Iacovoni J. S.; Daburon V.; Miller K. M.; Jackson S. P.; Legube G. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21 (4), 366–74. 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S.; Vitor A. C.; Sridhara S. C.; Martins F. B.; Raposo A. C.; Desterro J. M.; Ferreira J.; de Almeida S. F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife 2014, 3, e02482 10.7554/eLife.02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida S. F.; Grosso A. R.; Koch F.; Fenouil R.; Carvalho S.; Andrade J.; Levezinho H.; Gut M.; Eick D.; Gut I.; Andrau J. C.; Ferrier P.; Carmo-Fonseca M. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat. Struct. Mol. Biol. 2011, 18 (9), 977–83. 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- Luco R. F.; Pan Q.; Tominaga K.; Blencowe B. J.; Pereira-Smith O. M.; Misteli T. Regulation of alternative splicing by histone modifications. Science 2010, 327 (5968), 996–1000. 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh G. L.; Furge K.; Greenman C.; Chen L.; Bignell G.; Butler A.; Davies H.; Edkins S.; Hardy C.; Latimer C.; Teague J.; Andrews J.; Barthorpe S.; Beare D.; Buck G.; Campbell P. J.; Forbes S.; Jia M.; Jones D.; Knott H.; Kok C. Y.; Lau K. W.; Leroy C.; Lin M. L.; McBride D. J.; Maddison M.; Maguire S.; McLay K.; Menzies A.; Mironenko T.; Mulderrig L.; Mudie L.; O’Meara S.; Pleasance E.; Rajasingham A.; Shepherd R.; Smith R.; Stebbings L.; Stephens P.; Tang G.; Tarpey P. S.; Turrell K.; Dykema K. J.; Khoo S. K.; Petillo D.; Wondergem B.; Anema J.; Kahnoski R. J.; Teh B. T.; Stratton M. R.; Futreal P. A. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463 (7279), 360–3. 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duns G.; van den Berg E.; van Duivenbode I.; Osinga J.; Hollema H.; Hofstra R. M.; Kok K. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010, 70 (11), 4287–91. 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- Fontebasso A. M.; Schwartzentruber J.; Khuong-Quang D. A.; Liu X. Y.; Sturm D.; Korshunov A.; Jones D. T.; Witt H.; Kool M.; Albrecht S.; Fleming A.; Hadjadj D.; Busche S.; Lepage P.; Montpetit A.; Staffa A.; Gerges N.; Zakrzewska M.; Zakrzewski K.; Liberski P. P.; Hauser P.; Garami M.; Klekner A.; Bognar L.; Zadeh G.; Faury D.; Pfister S. M.; Jabado N.; Majewski J. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125 (5), 659–69. 10.1007/s00401-013-1095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi A. A.; Ostrovnaya I.; Reva B.; Schultz N.; Chen Y.-B.; Gonen M.; Liu H.; Takeda S.; Voss M. H.; Tickoo S. K.; Reuter V. E.; Russo P.; Cheng E. H.; Sander C.; Motzer R. J.; Hsieh J. J. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin. Cancer Res. 2013, 19 (12), 3259–3267. 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian A. R.; Thomenius M.; Totman J. A.. Methods of treating cancer by inhibiting SETD2. WO 2019/036466 A1, 2019.
- Zheng W.; Ibanez G.; Wu H.; Blum G.; Zeng H.; Dong A.; Li F.; Hajian T.; Allali-Hassani A.; Amaya M. F.; Siarheyeva A.; Yu W.; Brown P. J.; Schapira M.; Vedadi M.; Min J.; Luo M. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 2012, 134 (43), 18004–14. 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. H.; Boriack-Sjodin P. A.; Smith S.; Thomenius M.; Rioux N.; Munchhof M.; Mills J. E.; Klaus C.; Totman J.; Riera T. V.; Raimondi A.; Jacques S. L.; West K.; Foley M.; Waters N. J.; Kuntz K. W.; Wigle T. J.; Scott M. P.; Copeland R. A.; Smith J. J.; Chesworth R. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7 (2), 134–8. 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blat Y. Non-competitive inhibition by active site binders. Chem. Biol. Drug Des. 2010, 75 (6), 535–40. 10.1111/j.1747-0285.2010.00972.x. [DOI] [PubMed] [Google Scholar]
- Drew A. E.; Moradei O.; Jacques S. L.; Rioux N.; Boriack-Sjodin A. P.; Allain C.; Scott M. P.; Jin L.; Raimondi A.; Handler J. L.; Ott H. M.; Kruger R. G.; McCabe M. T.; Sneeringer C.; Riera T.; Shapiro G.; Waters N. J.; Mitchell L. H.; Duncan K. W.; Moyer M. P.; Copeland R. A.; Smith J.; Chesworth R.; Ribich S. A. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci. Rep. 2017, 7 (1), 17993. 10.1038/s41598-017-18446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. H.; Drew A. E.; Ribich S. A.; Rioux N.; Swinger K. K.; Jacques S. L.; Lingaraj T.; Boriack-Sjodin P. A.; Waters N. J.; Wigle T. J.; Moradei O.; Jin L.; Riera T.; Porter-Scott M.; Moyer M. P.; Smith J. J.; Chesworth R.; Copeland R. A. Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med. Chem. Lett. 2015, 6 (6), 655–9. 10.1021/acsmedchemlett.5b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black W. C.; Percival M. D. The consequences of lysosomotropism on the design of selective cathepsin K inhibitors. ChemBioChem 2006, 7 (10), 1525–35. 10.1002/cbic.200600149. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Macdonald S. J.; Peace S.; Pickett S. D.; Luscombe C. N. Increasing small molecule drug developability in sub-optimal chemical space. MedChemComm 2013, 4 (4), 673–680. 10.1039/c3md00003f. [DOI] [Google Scholar]
- Ritchie T. J.; Macdonald S. J.; Young R. J.; Pickett S. D. The impact of aromatic ring count on compound developability: further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discovery Today 2011, 16 (3–4), 164–71. 10.1016/j.drudis.2010.11.014. [DOI] [PubMed] [Google Scholar]
- SETD2 target engagement for 32 and related compounds was confirmed via a direct correlation between the reduction of H3K36me3 levels and in vitro cellular antiproliferative effects. These data were presented by Totman et al. at EHA 2021 (HemaSphere 2021, 5:S2, 44) and will be detailed in a SETD2 biology-focused manuscript in preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.