

Abstract

The ring strain present in azetidines can lead to undesired stability issues. Herein, we described a series of N-substituted azetidines which undergo an acid-mediated intramolecular ring-opening decomposition via nucleophilic attack of a pendant amide group. Studies were conducted to understand the decomposition mechanism enabling the design of stable analogues.
Keywords: azetidine, decomposition, ring-opening, pKa, 15N NMR
Azetidines are commonly utilized in medicinal chemistry design and can be found in numerous marketed drugs and clinical candidates (Figure 1). The small ring provides a rigid scaffold that enables productive protein–ligand interactions, and the lower MW relative to larger heterocycles can translate to improved ligand efficiency (LE). The presence of the azetidine ring nitrogen enables a range of synthetic options as well as the potential to incorporate a basic nitrogen in target molecules. Numerous examples of improved potency and/or pharmacokinetic performance for azetidines vs related heterocycles have been reported in the literature.1−4 However, the small ring size of azetidines introduces strain and the potential for decomposition pathways that may not be observed with larger ring systems.5 The strain also introduces the potential for metabolic ring-opening via reaction with glutathione.6 Metabolic ring opening has also been reported for structurally similar oxetanes.7 Herein, we describe a chemical stability issue observed for a specific aryl-azetidine chemotype as a caution to medicinal chemists who may be working in a similar design space.
Figure 1.

Examples of azetidine-containing drugs and clinical candidates.
During a medicinal chemistry effort for an undisclosed program, N-linked heteroaryl azetidines were demonstrated to provide overall superior drug profiles relative to closely related analogues with larger ring azetidine replacements. A significant portion of the analogue campaign focused on variation of the azetidine N-substituent. Based on observations from synthesis and purification efforts, it became apparent that closely related analogues exhibited significant differences regarding chemical stability at acidic pH, which was confirmed by measuring aqueous T1/2 values by NMR at pH 1.8. For example, while the 3-pyridyl analogue 1 provided a relatively short T1/2 of 3.8 h, the closely related 2- and 4-pyridyl analogues 2 and 3 were stable to these conditions (Table 1). While the instability observed with 1 would not impede small scale synthesis or evaluation in standard biology and ADME assays, it could result in downstream complications for drug manufacturing and formulation efforts leading to unwanted delays to project progression. Therefore, early in the program, a significant effort was focused on understanding and addressing this stability issue.
Table 1. Summary of Stability Data.

Aqueous T1/2 at pH 1.8 determined by 1H NMR. The solution concentrations studied were not sufficient to break the buffer capacity of the system.
Azetidine nitrogen 15N shift.
QM calculated pKa values were determined with Jaguar, version 7.6, Schrodinger, LLC, New York, NY, 2009.
Azetidine nitrogen pKa.
<5% degradation observed at 24 h.
∼50% degradation was observed by the first time point (3–5 min).
To better understand the scope of the problem, additional aqueous stability data were generated for a variety of analogues (Table 1). The N-phenyl (4), 4-methoxy-phenyl (5), and 4-cyano-phenyl (6) analogues were the least stable in this analysis with the 4-cyano compound 6 showing a very short T1/2 of <10 min. All three N-pyridyl analogues (1–3) provided better stability than the N-phenyl analogues (4–6) with the 2- and 4-pyridyl examples showing excellent stability.
Expansion of the strained azetidine ring in 1 to a pyrrolidine provided a stable analogue (7), and increasing the length of the alkyl chain spanning the amide and azetidine groups from one methylene (1) to two (8) or three (9) led to progressively longer T1/2 values, suggesting the potential for an intramolecular decomposition pathway involving azetidine ring-opening via attack by the pendant amide group (Figure 2).
Figure 2.

Mechanism of decomposition.
Replacement of the dimethyl amide in 1 with an azetidine amide (10) led to enhanced stability, while the related pyrrolidine and piperidine amides (11 and 12) showed T1/2 values roughly similar to that of the acyclic reference amide 1. These findings further support the above-mentioned decomposition hypothesis as the amide in 10 would be expected to be a poorer nucleophile than the acyclic amide 1 because delocalization of the nitrogen lone pair in 10 would be disfavored due to increased azetidine ring strain. Such ring strain would be relatively minor for the pyrrolidine or piperidine analogues 11 and 12, which is supported by the similar half-lives for these analogues relative to 1. The greater stability of the morpholine analogue 13 relative to piperidine 12 also supports the proposed decomposition mechanism as the electron-withdrawing morpholine ring oxygen would reduce the nucleophilicity of the morpholine amide relative to the piperidine amide.
The proposed decomposition mechanism is supported by HRMS data generated with compound 1 (Figure 3). Incubation of 1 at acidic pH catalyzes a rearrangement followed by loss of NHMe2 to provide the lactone intermediate 14 which is a major component at early time points. Further rearrangement of lactone 14 provides lactam 15, which is the major component at the later time points. The structural assignment of 15 was confirmed by NMR. A related intramolecular ring-opening of an azetidine with a pendant BOC group has been reported.8
Figure 3.
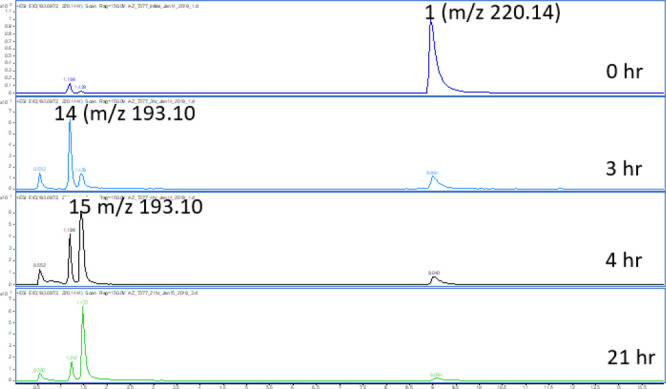
HRMS (pH 1.0) time course results showing the conversion of 1 → 14 → 15.
Having confirmed the degradation mechanism, our attention then focused on identifying the features of the N-aryl/heteroaryl group which impart optimal stability with the goal of enabling prospective design of analogues devoid of this issue. To better understand the impact of pH on stability, analogue 5 was evaluated at three pH values, confirming that more rapid decomposition occurs at low pH (pH = 1.8, T1/2 = 0.5 h; pH = 2.7, T1/2 = 1.2 h; pH = 7.0, stable). Given that the rate of decomposition is sensitive to pH and likely involves protonation of the azetidine nitrogen as a precursor to ring opening, the pKa of the azetidine nitrogen is a key determinant of stability, and both calculated and measured pKa values are provided in Table 1. For the pyridyl analogues, calculated values are provided for both the pyridine (cpKa1) and azetidine (cpKa2) nitrogens. NMR was utilized to determine measured values as this method enables definitive structural assignment of pKa. For the N-phenyl analogue 4, the azetidine nitrogen pKa was determined to be 4.3, and the calculated value is 2.9. For analogue 1, the pyridyl nitrogen pKa was determined to be 6.3, similar to the calculated value of 5.5, and the calculated pKa of the azetidine nitrogen is −1.1, which is too low to be confirmed by the NMR method. The enhanced stability of 1 vs 4 is likely due to the very low pKa of the azetidine nitrogen in 1, leading to a lack of azetidine N-protonation at pH 1.8. The enhanced stability observed with the conjugated pyridines 2 and 3 likely results from delocalization of the azetidine nitrogen lone pairs leading to the enhanced basicity of the pyridine nitrogens and reduced basicity of the azetidine nitrogens of 2 and 3 relative to 1. As was the case for 1, azetidine nitrogen pKa values could not be measured for 2 and 3, but the calculated values clearly show the impact of conjugation on pKa and the observed enhancement of aqueous stability at pH = 1.8. The conjugation differences for 1–3 are also reflected in the 15N NMR spectra as the azetidine nitrogen peak shifts for 2 and 3 are considerably downfield of 1, illustrating the greater sp2 character for the conjugated examples (Table 1). While the conjugation theory appears to explain the observed results for the pyridyl analogues 1–3, it is curious to note the extremely poor stability of conjugated 4-cyano-phenyl analogue 6. The 15N shift of the azetidine nitrogen in 6 is in the same range as that observed for the stable pyridyl analogues 2 and 3, confirming the expected lone pair delocalization and sp2 character of the ring nitrogen. The key difference between 6 and the pyridines 1–3 is the lack of a basic aryl ring nitrogen in 6. Protonation of the more basic pyridyl nitrogens in 1–3 significantly reduces the pKa of the second protonation site on the azetidine ring nitrogen. Calculations suggest the azetidine pKa of 6 to be significantly higher than the conjugated examples 2 and 3, and the ability to generate a measured pKa for 6 provides further confirmation. While the measured pKa value of 0.5 would suggest a relatively low level of protonated species at pH 1.8 (∼5%), this is apparently sufficient to drive the rapid decomposition observed with 6.
Overall, the data suggested that azetidines linked to conjugated heteroaryls would provide enhanced chemical stability relative to other aryl substituents. With this information in hand, analogues were successfully designed which circumvented the stability issue, leading to the eventual selection of a clinical candidate for the program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00402.
Experimental details for the NMR determination of T1/2, the MS determination of the mechanism of degradation of 1, and synthetic method for the compounds (PDF)
The authors declare the following competing financial interest(s): All authors were employees and stockholders of Pfizer during the time this work was completed.
Supplementary Material
References
- Obach R. S.; LaChapelle E. A.; Brodney M. A.; Vanase-Frawley M.; Kauffman G. W.; Sawant-Basak A. Strategies Toward Optimization of the Metabolism of a Series of Serotonin-4 Partial Agonists: Investigation of Azetidines as Piperidine Isosteres. Xenobiotica 2016, 46, 1112–1121. 10.3109/00498254.2016.1152522. [DOI] [PubMed] [Google Scholar]
- Helal C. J.; Arnold E. P.; Boyden T. L.; Chang C.; Chappie T. A.; Fennell K. F.; Forman M. D.; Hajos M.; Harms J. F.; Hoffman W. E.; Humphrey J. M.; Kang Z.; Kleiman R. J.; Kormos B. L.; Lee C. W.; Lu J.; Maklad N.; McDowell L.; Mente S.; O’Connor R. E.; Pandit J.; Piotrowski M.; Schmidt A. W.; Schmidt C. J.; Ueno H.; Verhoest P. R.; Yang E. X. Application of Structure-Based Design and Parallel Chemistry to Identify a Potent, Selective, and Brain Penetrant Phosphodiesterase 2A Inhibitor. J. Med. Chem. 2017, 60, 5673–5698. 10.1021/acs.jmedchem.7b00397. [DOI] [PubMed] [Google Scholar]
- Tye H.; Mueller S. G.; Prestle J.; Scheuerer S.; Schindler M.; Nosse B.; Prevost N.; Brown C. J.; Heifetz A.; Moeller C.; Pedret-Dunn A.; Whittaker M. Novel 6,7,8,9-tetrahydro-5H-1,4,7,10a-tetraaza cyclohepta[f]indene analogues as potent and selective 5-HT2C agonists for the treatment of metabolic disorders. Bioorg. Med. Chem. Lett. 2011, 21, 34–37. 10.1016/j.bmcl.2010.11.089. [DOI] [PubMed] [Google Scholar]
- Driscoll J. P.; Sadlowski C. M.; Shah N. R.; Feula A. Metabolism and Bioactivation: It’s Time to Expect the Unexpected. J. Med. Chem. 2020, 63, 6303–6314. 10.1021/acs.jmedchem.0c00026. [DOI] [PubMed] [Google Scholar]
- Yu L. F.; Eaton J. B.; Fedolak A.; Zhang H. K.; Hanania T.; Brunner D.; Lukas R. J.; Kozikowski A. P. Discovery of Highly Potent and Selective α4β2-Nicotinic Acetylcholine Receptor (nAChR) Partial Agonists Containing an Isoxazolylpyridine Ether Scaffold that Demonstrate Antidepressant-like Activity. Part II. J. Med. Chem. 2012, 55, 9998–10009. 10.1021/jm301177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Q.; Gronberg G.; Bangur E. H.; Hayes M. A.; Castagnoli N.; Weidolf L. Metabolism of Strained Rings: Glutathione S-transferase-Catalyzed Formation of a Glutathione-Conjugated Spiro-azetidine without Prior Bioactivation. Drug Metab. Dispos. 2019, 47, 1247–1256. 10.1124/dmd.119.088658. [DOI] [PubMed] [Google Scholar]
- Toselli F.; Fredenwall M.; Svensson P.; Li X. Q.; Johansson A.; Weidolf L.; Hayes M. A. Hip To Be Square: Oxetanes as Design Elements To Alter Metabolic Pathways. J. Med. Chem. 2019, 62, 7383–7399. 10.1021/acs.jmedchem.9b00030. [DOI] [PubMed] [Google Scholar]
- Xiao J.; Wright S. W. Facile ring cleavage of basic azetidines. Tetrahedron Lett. 2013, 54, 2502–2505. 10.1016/j.tetlet.2013.03.012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.