

Abstract
Deposition of human serum amyloid A (SAA) amyloids in blood vessels, causing inflammation, thrombosis, and eventually organ damage, is commonly seen as a consequence of certain cancers and inflammatory diseases and may also be a risk after SARS-COV-2 infections. Several attempts have been made to develop peptide-based drugs that inhibit or at least slow down SAA amyloidosis. We use extensive all-atom molecular dynamic simulations to compare three of these drug candidates for their ability to destabilize SAA fibrils and to propose for the best candidate, the N-terminal sequence SAA1-5, a mechanism for inhibition. As the lifetime of peptide drugs can be increased by replacing l-amino acids with their mirror d-amino acids, we have also studied corresponding d-peptides. We find that DRI-SAA1-5, formed of d-amino acids with the sequence of the peptide reversed, has similar inhibitory properties compared to the original l-peptide and therefore may be a promising candidate for drugs targeting SAA amyloidosis.
Keywords: Molecular dynamics simulations, serum amyloid A
Various cancer and inflammatory diseases can promote overexpression of serum amyloid A1,2 to 1000 times higher concentrations than seen usually.3 The resulting higher chance for misfolding and aggregation leads often to serum amyloid A (SAA) amyloidosis as a secondary disease bringing on widespread damage to tissues and organs, with kidney failure a common complication.4 Similar SAA concentrations have also been reported after SARS-COV-2 infections,5−7 suggesting an increased risk for SAA amyloidosis. It has been speculated that SAA amyloids are also implicated in the multisystem inflammatory syndrome in adults (MIS-A) which has a similar spectrum of symptoms as SAA amyloidosis.8 Hence, there is an interest in finding drug candidates that can inhibit or at least slow down SAA amyloid formation.
Several attempts have been made to develop such drugs based on synthesis of small peptides. These peptides are chosen from the sequence of amyloidogenic proteins for their complementarity to the regions on the SAA protein that are responsible for self-association.9−13 This peptide-based approach has been successfully applied to combat aggregation of amyloid-β (Aβ) peptides (connected to Alzheimer’s disease) and α-synuclein (implied in Parkinson’s disease), where so-designed inhibitors are able to prevent fibril formation and to disaggregate preformed fibrils, both in cell culture models and in animal models.9−15 Earlier biophysical studies have confirmed that the N-terminal sequence RSFFS of the SAA protein (to which we refer in the following as R5S) and the Aβ peptide segment Aβ17–20 (LVFF), called by us L4F, can significantly suppress the SAA aggregation process.10 However, that study considered as drug target only the short SAA1–12 fragment,16,17 which has a different aggregation propensity than larger segments.12 For this reason, we choose in the present study assemblies of larger SAA fragments as drug targets but select again these two peptides as possible drug candidates. In addition, we add as a third inhibitor candidate the peptide FVFLM of the protein SERPINA1, found in the urine and placenta18 of pregnant women suffering from preeclampsia. The choice of this peptide, called by us F5M, is motivated by an in silico study of Kouza et al.19 who showed that FVFLM strongly binds with central hydrophobic domain Aβ16–20 (KLVFF), thereby disrupting the Aβ aggregation pathway. We hypothesize that a similar mechanism is likely for inhibition of SAA aggregation. All three inhibitor candidates are listed in Figure 1 together with their names used in this study.
Figure 1.
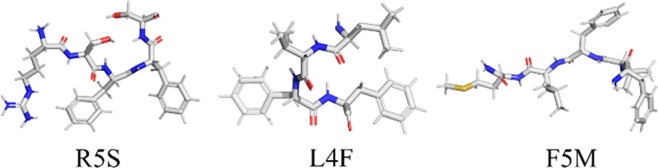
Inhibitor candidates evaluated in this study. Carbons are colored in gray, nitrogen in blue, oxygen in red, and sulfur in yellow.
SAA fibrils are not formed by the full-sized protein but by shorter fragments derived after enzymatic cleavage. The most common fragment is SAA1–76, but the only experimentally resolved human SAA fibril structure is for the segment SAA2–55, deposited in the Protein Data Bank under identifier 6MST.20 Hence, in the present paper we compare through unbiased all-atom molecular dynamics (MD) simulations the ability of the three inhibitor candidates to reduce the stability of this fibril structure, a necessary condition for lowering the risk for amyloidosis. As regular MD of a few hundred nanoseconds were sufficient for our purpose, we did not utilize a more advanced enhanced sampling technique relying on artificial dynamics; albeit, enhanced sampling methods such as replica exchange molecular dynamics (REMD) have been employed in the past successfully to study the Aβ fibril–drug interaction at the molecular level.21
Our results suggest that the fragment SAA1–5 (RSFFS, to which we refer as R5S) has a larger effect on fibril stability than L4F (the LVFF segment Aβ17–20) and F5M, the segment FVFLM of SEREPINA1. Since short half-life time and the resulting need for frequent drug administration are common complications in peptide-based therapies that would also hamper the use of R5S as a potential drug, we have also studied versions of R5S where the l-amino acids are replaced by d-amino acids. This is because peptides made of d-amino acids are resistant to proteolytic digestion, resulting in longer half-life times. Hence, following our earlier work22 we have studied both a variant where the sequence is unchanged (D-R5S) and one where also the sequence is reversed (DRI-R5S). The later variant is chosen because such d-retro-inverso peptides resemble the parent peptide and have often similar biological activity.23−25
All systems investigated by us are listed in Table S1 of the Supporting Information. Their preparation and the protocols and techniques used in the setup and analysis of our simulations are described in the Materials and Method section, which can be also found in the Supporting Information. As amyloid formation decreases the entropy of the SAA chains, overcoming this loss of entropy requires an energetic bias toward the fibril structure. Peptide inhibitors work by reducing the energetic stabilization of the fibril structure upon binding to the fibril. Hence, for evaluating the peptide inhibitor candidates we have to ensure first that in our simulations the fibril stability is not already artificially decreased by other interfering factors such as, for instance, the chosen ionic strength. Another potential systematic error could result from the missing first residue in the experimentally determined SAA2–55 fibril model. This residue, an arginine, is at neutral pH protonated and, therefore, able to form salt bridges with negatively charged residues. These salt bridges are missing in simulations of our model which as a consequence may underestimate the stability of the fibril. In order to exclude such systematic errors, we first compare simulations of the fibril with and without the N-terminal arginine and without added salt (except for counterions) and at 150 mM NaCL (the physiological intracellular salt concentration). The time evolution of the root-mean-square deviation (RMSD) to the respective start configuration is shown in Figure 2. As the RMSD is calculated over all the backbone atoms and all chains in the respective fibril model, we call it a global RMSD.
Figure 2.
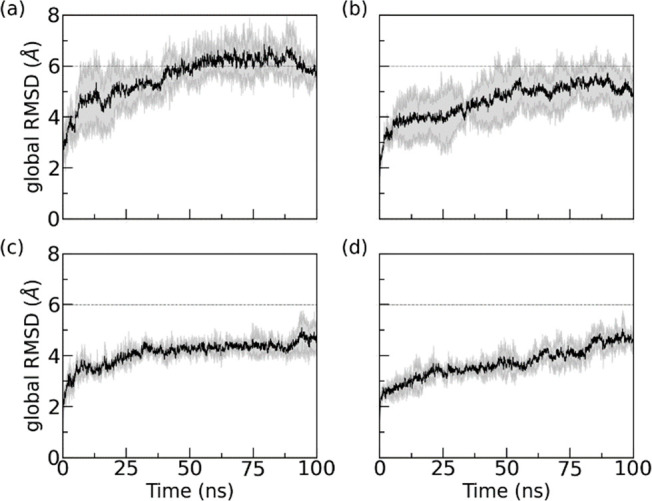
Time evolution of global RMSD of control simulations. RMSD values are calculated in reference to the experimentally resolved structure (PDB ID: 6MST) considering only backbone atoms in all chains of the respective fibril model. The data are averaged over three trajectories, with the shaded region indicating the standard deviation of the average. Systems displayed are those with arginine at 0 mM (a), with arginine at 150 mM NaCl (b), without arginine at 0 mM (c), and without arginine at 150 mM (d). In order to allow for a more easy comparison of the four systems, we have drawn dashed lines at 6 Å to guide the eyes.
Comparing the four simulations, we observe first that the RMSD is lower in simulations of the fibril with physiological salt concentration than it is in the simulations of the corresponding systems without salt. Hence, omitting salt artificially decreases fibril stability. While this effect is small in simulations where the first residue is missing, raising from (4.4 ± 0.4) Å in 0 mM to (4.7 ± 0.4) Å in 150 mM, it is more pronounced in simulations where the arginine is added. Here, the final RMSD value at 100 ns is (5.8 ± 0.4) Å in 0 mM NaCl and (5.0 ± 0.8) Å in 150 mM NaCl.
Similarly, we find in accordance with previous experimental results20 that addition of the N-terminal arginine reduces the stability of the fibril even when comparing the two models at the same salt concentrations. The presence of arginine breaks stabilizing contacts at the cross-stacking interface and N-terminal hydrophobic cluster (see Figure S1 in the Supporting Information), with the effect lower when the electrostatic interactions are screened by the Na+ and Cl– ions. We remark that by repeating the above four simulations now adding the R5S inhibitor, we find the same trend as for the control simulations (in the absence of any inhibitor); see Supporting Information Figure S2. Hence, for our evaluation and the comparison of the inhibitors, relying on longer trajectories, we use the experimental fibril model without added N-terminal arginine and choose a salt concentration of 150 mM.
With the optimal simulation conditions determined, we then compare for all three inhibitor candidates the stability of the SAA fibril in the presence of the inhibitor with the stability of the control. Our results are shown in Figure 3, where we show in the top row the global RMSD to the start configuration and in the middle row the chain RMSD with respect to the start configuration. In Figure 2 is the global RMSD calculated for the backbone atoms in all chains of the fibril model, whereas the chain RMSD is the average over chain-wise calculated RMSD values. Hence, the global RMSD measures the structural deviation of the entire fibril while the chain RMSD measures the structural distortion of each protein chain within the fibril. On the other hand, the residue-wise root-mean-square-fluctuation (RMSF) shown in the bottom row describes the flexibility of a certain residue in the chains that make up the fibril.
Figure 3.
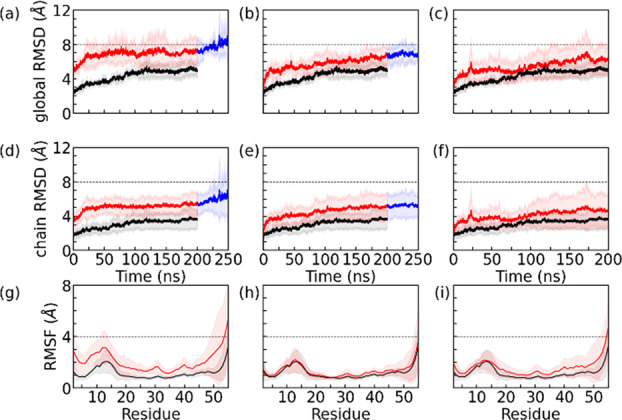
Time evolution of the global and chain RMSD measured in simulations of the fibril in the absence (in black) and presence of three inhibitors (in red). Residue-wise RMSF values are calculated over the final 100 ns of each trajectory. Data in the presence of R5S, L4F, and F5M inhibitors are shown in the first, second, and third columns, respectively. The simulations with R5S and L4F inhibitors present were later extended up to 250 ns. This additional timespan is drawn in blue. Chain RMSD and RMSF values are averaged over all six chains in the SAA fibril and all three trajectories. The shaded region represents the standard deviation. The RMSD and RMSF values are calculated in reference to the experimentally resolved structure considering all backbone atoms in residues 2–55. For better visualization of subtle differences, we have drawn the dashed lines at suitable global/chain RMSD and RMSF to guide the eyes.
Focusing first on the top two rows in Figure 3, we see that global and chain RMSD are correlated. Both RMSD values are growing for all three inhibitors faster and to higher values than for the control, with the effect most pronounced for Inh1. For instance, the global RMSD values averaged over the final 100 ns and the three trajectories are for the control (0.49 ± 0.02) nm, for R5S (0.74 ± 0.05) nm, for L4F (0.66 ± 0.03) nm, and for F5M (0.60 ± 0.04) nm. The correlation between global and chain RMSD is especially obvious for R5S inhibitor where the sudden rise in global RMSD at 200 ns is mirrored by one in chain RMSD. The correlation between the two quantities suggests that the dissolution of the fibril goes in hand with a change in chain structure. In order to quantify the structural change of the SAA chains, we have also calculated the residue-wise backbone RMSF along the trajectories and show this quantity in the bottom row of Figure 3. Here we find that the large structural deviations, seen by visual inspection in the β-arch formed by residues 2–22 and in the C-terminal cavity formed by residues 45–55, is correlated with high flexibility of the residues in these two regions. Note that the RMSF distribution of each residue is for each of the inhibitor simulations broader than for the control (the fibril without inhibitor candidate added). Hence, the inhibitor disturbs the chain conformations which in turn leads to dissolution of the fibril geometry.
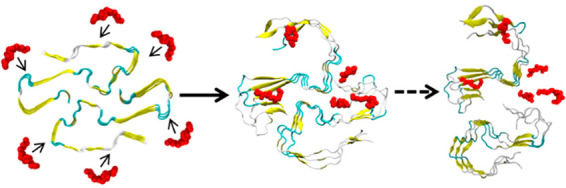
Note that we simulated initially the control and the inhibitor-containing system for 200 ns. As the differences between R5S and L4F inhibitors were small and, unlike the control or F5M inhibitor, the RMSD in both systems appeared to continue growing, we extended simulations for R5S and L4F inhibitors by 50 ns, with the extension drawn in a different color. With this extension, it appears from Figure 3 and visual inspection of the snapshots in Figure 4 that the structural deviations are largest in the presence of R5S inhibitor.
Figure 4.
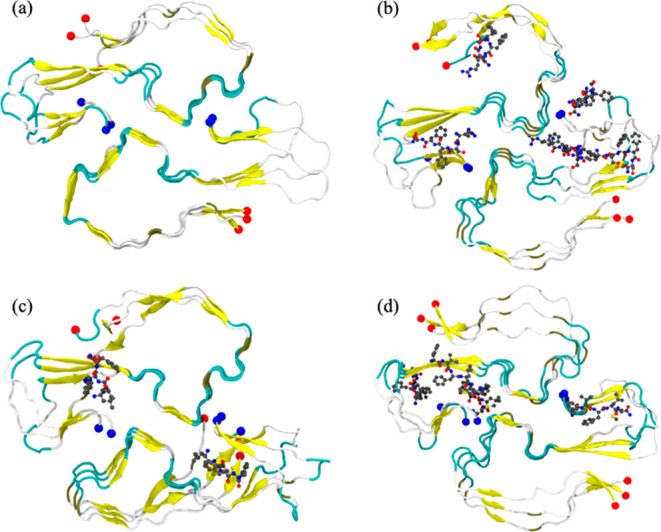
Representative final configuration extracted from a control simulation of the SAA2–55 fibril is shown in (a). Corresponding final snapshots extracted from simulations in the presence of R5S, L4F, and F5M inhibitors are shown in (b), (c), and (d), respectively. N- and C-terminal residues are marked by blue and red spheres, respectively.
We have quantified this observation by measuring for both the control and inhibitor simulations the solvent-accessible surface area (SASA), the number of stacking contacts (Nstacking), the number of stacking hydrogen bonds (NHB-stacking), the number of packing contacts (Npacking), the number of packing hydrogen bonds (NHB-packing), and the interstrand packing distance (Dpacking). We list in Table 1 the mean and standard deviation of these quantities, as calculated over the last 100 ns of each system for three independent 200 ns-long trajectories. The first of these quantities, the solvent-accessible surface area (SASA) of the hydrophobic residues, is a measure for the solvent exposure of the hydrophobic residues in the SAA fibril. It is measured in the VMD package by running a spherical probe of 1.4 Å radius over the fibrillar surface. In the case of R5S inhibitor, the hydrophobic SASA increases by about ∼26% over the control, suggesting the inhibitor causes conformational changes in which the tightly packed hydrophobic core becomes hydrated. This is not the case for the other two inhibitors, where the SASA values increase by only about 11%.
Table 1. Solvent-Accessible Surface Area (SASA), Number of Stacking Contacts (Nstacking), and Stacking Hydrogen Bonds (NHB-stacking); Number of Packing Contacts (Npacking) and Packing Hydrogen Bonds (NHB-packing); and Interstrand Packing Distance (Dpacking)a.
| System | Control | R5S | L4F | F5M |
|---|---|---|---|---|
| SASA (Å2) | 6298 ± 388 | 7931 ± 639 | 6962 ± 46 | 6966 ± 399 |
| %ΔSASA | - | 26 | 11 | 11 |
| Nstacking | 16100 ± 376 | 14643 ± 509 | 15667 ± 412 | 15118 ± 861 |
| %ΔNstacking | - | –9 | –3 | –6 |
| Npacking | 2265 ± 216 | 1149 ± 492 | 1782 ± 170 | 1648 ± 302 |
| %ΔNpacking | - | –49 | –21 | –27 |
| NHB-stacking | 56 ± 7 | 53 ± 6 | 53 ± 7 | 50 ± 8 |
| %ΔNHB-stacking | - | –5 | –5 | –10 |
| NHB-packing | 7 ± 2 | 4 ± 2 | 6 ± 2 | 5 ± 2 |
| %ΔNHB-packing | - | –41 | –14 | –34 |
| Dpacking (Å) | 9.5 ± 0.3 | 12.2 ± 5.0 | 9.6 ± 0.3 | 9.7 ± 0.4 |
| %ΔDpacking | - | 29 | 2 | 2 |
Averages are calculated over the three independent trajectories considering only the final 100 ns of each 200 ns long trajectory, with the corresponding standard deviations also shown. The change between simulations in the presence and absence of the inhibitors is listed as percent changes.
VMD is also used to calculate the total number of stacking contacts (Nstacking), stacking hydrogen bonds (NHB-stacking), packing contacts (Npacking), and packing hydrogen bonds (NHB-packing). We define contacts by a distance cutoff of 4.5 Å and hydrogen bonds by the condition that the distance between the N and O atoms is less than 3.0 Å and the angle of N–H···O is between 160° and 180°. Table 1 shows that the presence of the inhibitors decreases the number of hydrogen bonds and hydrophobic contacts involved in packing of the two folds more than the ones stabilizing the stacking of chains. For example, in the presence of R5S, we observe a 49% loss in contact at the packing interface, but the stacking contacts decrease by only 9%. This loss of contacts at the packing interface is also reflected by an increase of about 29% in the interstrand packing distance Dpacking (shown also in Table 1) in the presence of R5S. The corresponding change is much smaller for the other two inhibitors where Dpacking increases by only about 2%. Here, Dpacking is defined by the distance between the center of mass of two chains at the interface where they are packed (residues 28–31).
Our above analysis shows that R5S, the N-terminal sequence SAA protein (RSFFS), is more effective than the other two inhibitors in destabilizing the SAA fibril. Hence, we focus in the following on this inhibitor, trying to understand the mechanism by which R5S destabilizes SAA fibrils. For this purpose, we have first tried to quantify the destabilization of the SAA fibril by R5S by calculating the binding free energy of the chains. The later quantity is estimated by the MMPBSA approach26 as thermodynamic integration or other exact methods would have been too costly in terms of computation time, and values are listed in Tables S2 and S3 in the Supporting Information. As in many other studies,27,28 we did not consider the entropic contribution in the binding free energy calculation. This is discussed in detail in the trajectory analysis section in the Supporting Information.
We find that the presence of R5S reduces the fibril stability by about 60 kcal/mol, from (−311 ± 80) kcal/mol in the control to (−254 ± 60) kcal/mol. The loss of stability is mostly from the change in the packing free energy, i.e., the difference in free energy of the fibril 2F3L and of two separated proto-fibrils 1F3L, which changes from (−148 ± 54) kcal/mol for the control to (−38 ± 38) kcal/mol in the presence of the inhibitor. The change in packing free energy of about 90 kcal/mol is due to a loss of interactions between residues of about 252 kcal/mol that is only partially compensated by a gain in solvation free energy of about −160 kcal/mol. A less pronounced difference is seen for the elongation free energy, i.e., the difference in free energy by adding one layer to the fibril (2F3L compared with 2F2L + 2F1L), which decreases by about 20 kcal/mol, from (−143 ± 54) kcal/mol in the control to (−125 ± 53) kcal/mol in the presence of R5S. The difference is again due to a reduction of interactions between residues, with the loss of about 56 kcal/mol only partially compensated by a gain in the solvation free energy of about −37 kcal/mol. Hence, our MMPBSA analysis indicates that the loss in fibril stability results from a loss of electrostatic and van der Waals interactions.
We have therefore compared in the next step the map of inter- and intraresidue distances measured either in the simulations where R5S inhibitor is present or in the control (where neither R5S nor one of the other two inhibitors is present). These maps are shown in Figure 5, with the coloring marking the average distance between two residues.
Figure 5.

Intrapeptide side-chain distance map measured during simulations of SAA fibrils in the presence (a) and absence of (b) of peptide inhibitor R5S. The corresponding interpeptide side-chain distance maps are also shown in (d) and (e), respectively. For a comparison, we also show (c) intrapeptide and (f) interpeptide side-chain distance maps of the cryo-EM SAA fibril model (PDB-ID: 6MST). Residue pairs whose average contact distance exceed 1.0 nm are excluded. Distance maps are calculated using the last 100 ns in all three independent trajectories.
Visual inspection of the cryo-EM structure (PDB-ID: 6MST) (resolved at 3.0 Å resolution) together with the intrachain and interchain distance map, derived from this structure and shown in Figure 5c and 5f, demonstrates that the N-terminal β-arch formed by residues 2–22 is stabilized by the densely packed hydrophobic core formed by residues F4, F6, F11, M17, W18, and A20 and by salt bridges between residue-pairs D12-R15 and D16-R19. Of importance are also contacts between the C-terminal end encompassing residues 47–55 and the region encompassing residues 19–26 that stabilizes the C-terminal cavity, namely multiple hydrophobic contacts involving the residues A21, M24, W53, V52, and A54 and the salt bridges between E26-R47. These contacts are also seen in the control simulations (see Figure 5b and e), indicating that these contacts are conserved in the absence of any inhibitor. On the other hand, in the presence of the peptide inhibitor R5S, these contacts are rarely found (see Figure 5a and d). Similarly, while only few contacts at the N-terminal cross-stacking interface (encompassing residues 11–17) are lost in the control simulations (Figure 5e), the stacking contacts are lost in the presence of R5S (Figure 5d), which may be one reason for its ability to destabilize the SAA fibrillar architecture as it causes a loss of van der Waals energy of about 56 kcal/mol (see Supporting Information Tables S2 and S3).
Corresponding figures for the other two inhibitors, L4F and F5M, are shown in Figure S3 of the Supporting Information. Similar as in the case of R5S are the hydrophobic cluster at the N-terminus perturbed in the presence of L4F, as are the C-terminal cavity stabilizing contacts between the C-terminus (residues 47–55) and the residues 19–26. However, for F5M the interstacking interface at the N-terminus is conserved over the course of the simulation and the disruptive effect of F5M is even lower: the distance maps in Figure S3 do not show any obvious loss of contacts.
We next have calculated the residue-wise binding probability of R5S inhibitor toward the SAA fibril to understand the relation between R5S binding and the loss of intrachain and interchain contacts. For this purpose, we define a binding site as the closest residue with at least one heavy atom within 4.5 Å from the R5S and do not consider peptide inhibitors that move away from the fibril. Data are averaged over the final 100 ns of all three trajectories and shown in Figure 6. From the binding probability map we see that R5S preferentially binds with aromatic residues (F3, F4, F6, F11, W18, and Y21), hydrophobic residues (L7, A10, A14, M17, and A20), and polar and charged residues (S2, S5, E9, and S22) at the N-terminus, disrupting the most amyloidogenic and hydrophobic segment of the protein. We also observe lower binding probability toward the packing interface through hydrophobic and polar interactions. As a result it increases the interstrand packing distance by about 29%. The resulting loss in contacts between polar and charged residues leads to a reduction of electrostatic energy between residues by about 219 kcal/mol and of the van der Waals energy by about 34 kcal/mol, that decreases the packing energy of the fibril in the presence of R5S (see Supporting Information Tables S2 and S3). Interestingly, we do not find any binding affinity toward the aromatic and hydrophobic residues at the C-terminal end (residues 53–55) and the GPGG motif (residues 48–51).
Figure 6.

Residue-wise binding probability (normalized) of R5S inhibitor for the SAA fibril. Data are averaged over the final 100 ns of all three independent runs.
We remark that the binding affinities of L4F, shown in Figure S4 of the Supporting Information, are similar to R5S but bind more toward certain aromatic and hydrophobic residues (e.g F3, F4, and M24) and less toward polar and charges residues. The differences reflect that L4F is purely hydrophobic while there are two polar serine (S) and one positively charged arginine (R) residue in R5S inhibitor. Interestingly, while F5M (FVFLM) is also hydrophobic, we did not observe the same binding affinity to the hydrophobic and amyloidogenic N-terminus (see Figure S5 in the Supporting Information), as we observed for either R5S or L4F.
As discussed above, our simulations show that all three small peptides interact with the SAA fibril mostly by hydrophobic interactions and π–π stacking. In the case of R5S there is additional stabilization by long-range electrostatic interactions involving the polar (S) and charged (R) residues of R5S. These additional interactions lead to the strong binding of all six R5S inhibitors to the SAA fibril fragment (a hexamer). On the other hand, in the case of L4F and F5M there are fewer of the peptides bound to the fibril. For example, in one of the L4F inhibitor trajectories, we find after 250 ns only two peptides still binding to the fibril. Hence, our data demonstrate the need to consider polar and charged residues in the design of peptides inhibiting SAA amyloidosis as sole hydrophobic interactions are not sufficient for a strong binding.
Our above study established that the peptide inhibitor R5S, SAA1–5(RSFFS), is able to destabilize small fibril fragments such as the 2-fold three-layer 2F3L. While the hexamer is above the critical size for SAA fibril stability,29 a potential application as a drug would require that R5S inhibitor can also dissolve fully grown SAA fibrils. Hence, in order to test whether the destabilizing effect of R5S persists with increasing number of layers, we have also simulated the 2F4L octamer as the next larger SAA assembly. Comparing the time evolution of the global RMSD in Figure 7, measured in trajectories derived with either R5S inhibitor present or absent, we still observe the destabilizing effect of R5S seen in simulations of the smaller 2F3L hexamer. While the stability of the SAA fibril increases with size, and therefore the measured RMSD values are smaller and increasing more slowly, the qualitative behavior is the same as seen in the simulations of the smaller 2F3L fibril. While the 2F4L octamer is still a small system, we believe that this similarity to the 2F3L hexamer indicates that the destabilizing effect of R5S inhibitor will likely persist for the much larger systems one would observe in vito or in vivo.
Figure 7.
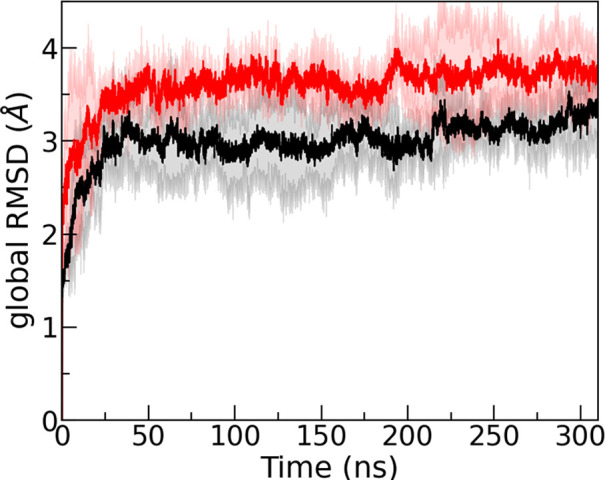
Time evolution of the global RMSD measured in simulations of the 2-fold-four-layer (2F4L) SAA fibril in the absence (in black) and presence of R5S inhibitor (in red). The shaded region represents the standard deviation. The RMSD values are calculated in reference to the experimentally resolved structure considering all backbone atoms.
In the preceding section we have shown that the peptide inhibitor R5S destabilizes SAA fibrils. This destabilization is a minimum condition for use of R5S as a potential drug candidate inhibiting SAA amyloid formation. However, R5S can only act as a drug if it survives long enough in the body to be able to dissolve SAA aggregates. However, peptide-based therapies are often hampered by short half-life times, a likely complication for the use of R5S. For this reason, we have also studied versions of R5S where the l-amino acids are replaced by d-amino acids, as such peptides are resistant to proteolytic digestion resulting in longer half-life times. Hence, following our earlier work,22 we have studied both a variant where the sequence is unchanged (D-R5S) and one where also the sequence is reversed (DRI-R5S). The later variant is chosen because such D-retro-inverso peptides resemble the parent peptide and often have similar biological activity.23−25
Our results are shown in Figure 8, where we draw the time evolution of the global RMSD of the SAA fibril relative to the initial cryo-EM structure. Shown are the results from simulations in the presence of DRI-R5S or D-R5S and the corresponding data obtained from simulations without any inhibitor or with L-R5S present. RMSD data over the course of three separate 200 ns simulations for each system indicate that the DRI-R5S is more effective to destabilize the fibril compared to D-R5S: the averaged RMSD values over the final 100 ns and three trajectories are (0.75 ± 0.04) nm and (0.62 ± 0.02) nm in the presence of DRI-R5S and D-R5S, respectively.
Figure 8.
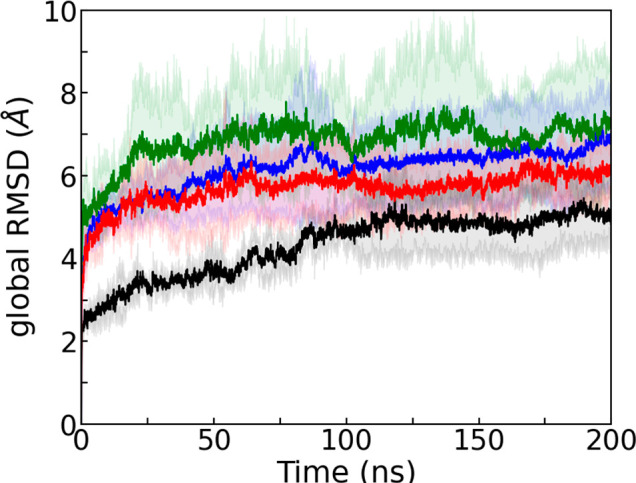
Time evolution of the global RMSD measured in simulations of the 2-fold-three-layer (2F3L) SAA fibril in the presence of DRI-R5S (blue) and D-R5S (red). For a comparison, we show also corresponding data obtained from simulations in the presence of L-R5S (green) and from the control simulation (black). The shaded region represents the standard deviation. The RMSD values are calculated in reference to the experimentally resolved structure considering all backbone atoms.
This observation is corroborated by the values of the hydrophobic SASA, Nstacking, NHB-stacking, Npacking, NHB-packing, and Dpacking shown in Table 2, which also lists the corresponding data for the control and the l-amino acid version of R5S. For example, the hydrophobic SASA increases by ∼21.6% in the presence of DRI-R5S, while the corresponding change in the presence of D-R5S is about 16%. While the percentage difference with respect to the control is for most of the quantities comparable for DRI-R5S with the corresponding changes for L-R5S (see Table 2), the fibril disruption is slightly smaller. This difference reflects that reversing the sequence and replacing l by d amino acids does not lead to exactly the same structure as seen in the l-parent. This has also been observed by us in earlier work for Aβ and amylin fibrils.22,30 However, while we do not find in the presence of either DRI-R5S or D-R5S the increase in interstrand packing distance that we see for L-R5S, taken together our data do suggest that DRI-R5S has a similar effectiveness to L-R5S in destabilizing SAA fibrils. The inter- and intrachain distance maps in the presence of DRI-R5S that we show in Figure S6 of the Supporting Information add further evidence for this conclusion. The distance map is similar to that of L-R5S, with the hydrophobic cluster, salt-bridges, and the N-terminal interstacking interfaces disrupted in the presence of DRI-R5S. The binding probability map in Figure S7 also shows similar binding models as L-R5S since DRI-R5S is again strongly binding to hydrophobic and aromatic residues in the N-terminal hydrophobic cluster or forming salt bridges with the charged residues E9, D12, D33, and K34.
Table 2. Mean Values of Solvent-Accessible Surface Area (SASA), Number of Stacking Contacts (Nstacking), Number of Packing Contacts (Npacking), and Interstrand Packing Distance (Dpacking)a.
| System | DRI-R5S | D-R5S | Control | L-R5S |
|---|---|---|---|---|
| SASA (Å2) | 7698 ± 488 | 7298 ± 329 | 6298 ± 391 | 7931 ± 639 |
| %ΔSASA | 22 | 16 | - | 26 |
| Nstacking | 14420 ± 498 | 15452 ± 679 | 16100 ± 376 | 14643 ± 509 |
| %ΔNstacking | –10 | –4 | - | –9 |
| Npacking | 1529 ± 124 | 1626 ± 252 | 2265 ± 216 | 1149 ± 492 |
| %ΔNpacking | –32 | –28 | - | –49 |
| Dpacking (Å) | 9.5 ± 0.3 | 9.6 ± 0.4 | 9.5 ± 0.3 | 12.2 ± 5.0 |
Averages are calculated over the three independent trajectories considering only the final 100 ns of each 200-ns-long trajectory. The corresponding standard deviations are listed in brackets. We also provide percent changes to describe the change of values calculated from simulations in the presence of an inhibitor, compared to the values calculated in the control simulations.
SAA amyloidosis is a common secondary disease connected with various cancers and inflammatory diseases,1,2 and it may also be a risk after SARS-COV-2 infections.8 In this paper we have studied three peptides that may serve as drug candidates for inhibiting or slowing down SAA amyloid formation. Using extensive all-atom molecular dynamics simulations, we compare the ability of the N-terminal sequence SAA1–5 (R5S), the Aβ peptide segment Aβ17–20 (L4F), and the peptide FVFLM (F5M) of the protein SERPINA to destabilize a small fragment of the experimentally resolved human SAA fibril (PDB-ID: 6MST). We find that the destabilizing effect is larger for the amphipathic R5S inhibitor than for the purely hydrophobic L4F and F5M and persists when increasing the fragment size. Going beyond confirming previous experimental results,10 our numerical investigation allows us to propose a mechanism for the inhibitor properties of R5S. We find that this peptide binds to the N-terminus, the most amyloidogenic and hydrophobic segment of the SAA fibril, mainly through short-range hydrophobic and π–π stacking interactions with aromatic and hydrophobic residues, but unlike for L4F and F5M inhibitors the binding interface is in addition stabilized via long-range interactions involving the polar and charged residues of R5S. The complex binding patterns of hydrophobic, polar, and ionic interactions disturb not only the densely packed hydrophobic core at the N-terminus but also the network of contacts that stabilize the SAA fibril geometry. From these observations we conclude that for the design of peptide inhibitors against SAA fibril formation, one should optimize not only the hydrophobic contacts of the peptide to the N-terminus but also salt bridge formation and polar contacts with the charged residues in this segment. In our case it is the presence of such contacts that set the R5S inhibitor apart from the other two peptides where the binding to the N-terminus is solely by hydrophobic contacts. Our proposed mechanism is consistent with the crucial role of the N-terminus (the first 11 residues) as starting point for the SAA fibril self-assembly.31 Hence, it is plausible that drug candidates such as our R5S inhibitor will have to target this domain, both to inhibit binding of chains in the early stages of the amyloidogenic pathway and to destabilize the aggregates.
We finally have addressed a common problem in the use of peptides such as R5S as drug candidates: short half-life time and the resulting need for frequent administration that makes their use cumbersome. Following common protocols, we have therefore also studied versions of R5S where the l-amino-acids are replaced by their mirror d-amino acids, leading to peptides that are resistant to proteolytic digestion and have longer half-life times. Especially, we find that DRI-R5S, where not only l-amino acids are replaced by d-amino acids but also the sequence reversed, has similar inhibitory effects than the original L-R5S. The much longer lifetime may make DRI-R5S an attractive drug candidate targeting SAA amyloidosis, and we hope that our computational study will encourage soon experimental investigations of the inhibitory potency of DRI-R5S. While we are not aware of any human clinical trial of DRI-peptide inhibitors, we remark that similar tests as proposed for DRI-R5S have been done for the similar sized DRI-KLVFF (Aβ16–20), showing it to be stable against proteolysis and inhibiting Aβ1–42 fibril growth.32 The same group also attached a retro-inverted version of the HIV protein transduction domain “TAT”, to this peptide, allowing it now to cross the blood–brain barrier,33 and showed that its peripheral administration (at 100 nmol/kg) for 21 days into an Alzheimer’s disease mouse model significantly reduced the Aβ plaque count. Targeting SAA amyloidosis, DRI-R5S will not need to cross the blood–brain barrier, but we would expect that otherwise similar experimental protocols will apply.
Acknowledgments
The simulations in this work were done using the SCHOONER cluster of the University of Oklahoma, XSEDE resources allocated under grant MCB160005 (National Science Foundation), and TACC resources allocated under grant MCB20016 (National Science Foundation). We would like to thank Alan J. Ray for providing guidance and scripts for making figures.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00456.
Seven supporting figures (S1–S7), three supporting tables (S1–S3), and a description of systems preparation, simulation protocols, and analysis techniques (PDF)
Author Contributions
‡ A.K.J. and A.B.G have contributed equally and should both be considered first authors.
We acknowledge financial support from the National Institutes of Health under grant GM120634.
The authors declare no competing financial interest.
Supplementary Material
References
- Caughey B.; Baron G. S. Are Cheetahs on the Run from Prion-like Amyloidosis?. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (20), 7113–7114. 10.1073/pnas.0803438105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papendick R. E.; Munson L.; O’Brien T. D.; Johnson K. H. Systemic AA Amyloidosis in Captive Cheetahs (Acinonyx Jubatus). Vet. Pathol. 1997, 34 (6), 549–556. 10.1177/030098589703400602. [DOI] [PubMed] [Google Scholar]
- Gillmore J. D.; Lovat L. B.; Persey M. R.; Pepys M. B.; Hawkins P. N. Amyloid Load and Clinical Outcome in AA Amyloidosis in Relation to Circulating Concentration of Serum Amyloid A Protein. Lancet 2001, 358 (9275), 24–29. 10.1016/S0140-6736(00)05252-1. [DOI] [PubMed] [Google Scholar]
- Kofman T.; Grimbert P.; Canouï-Poitrine F.; Zuber J.; Garrigue V.; Mousson C.; Frimat L.; Kamar N.; Couvrat G.; Bouvier N.; Albano L.; Le Thuaut A.; Pillebout E.; Choukroun G.; Couzi L.; Peltier J.; Mariat C.; Delahousse M.; Buchler M.; Le Pogamp P.; Bridoux F.; Pouteil-Noble C.; Lang P.; Audard V. Renal Transplantation in Patients With AA Amyloidosis Nephropathy: Results From a French Multicenter Study. Am. J. Transplant. 2011, 11 (11), 2423–2431. 10.1111/j.1600-6143.2011.03620.x. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Yang J.-Z.; Bai W.-H.; Li Z.-Y.; Sun L.-F.; Yan J.-J.; Zhou C.-L.; Tang B.-P. Prognostic Value of Serum Amyloid A in Patients with COVID-19. Infection 2020, 48 (5), 715–722. 10.1007/s15010-020-01468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Xiang X.; Ren H.; Xu L.; Zhao L.; Chen X.; Long H.; Wang Q.; Wu Q. Serum Amyloid A Is a Biomarker of Severe Coronavirus Disease and Poor Prognosis. J. Infect. 2020, 80 (6), 646–655. 10.1016/j.jinf.2020.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanff T. C.; Mohareb A. M.; Giri J.; Cohen J. B.; Chirinos J. A. Thrombosis in COVID-19. Am. J. Hematol. 2020, 95 (12), 1578–1589. 10.1002/ajh.25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A. K.; Greenwood A. B.; Hansmann U. H. E. Presence of a SARS-CoV-2 Protein Enhances Amyloid Formation of Serum Amyloid A. J. Phys. Chem. B 2021, 125 (32), 9155–9167. 10.1021/acs.jpcb.1c04871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrocchi L. A.; Chen Y.; Waschuk S.; Wang F.; Cheung S.; Darabie A. A.; McLaurin J.; Fraser P. E. Design of Peptide-Based Inhibitors of Human Islet Amyloid Polypeptide Fibrillogenesis. J. Mol. Biol. 2002, 318 (3), 697–706. 10.1016/S0022-2836(02)00164-X. [DOI] [PubMed] [Google Scholar]
- Sosnowska M.; Skibiszewska S.; Kamińska E.; Wieczerzak E.; Jankowska E. Designing Peptidic Inhibitors of Serum Amyloid A Aggregation Process. Amino Acids 2016, 48 (4), 1069–1078. 10.1007/s00726-015-2167-y. [DOI] [PubMed] [Google Scholar]
- El-Agnaf O. M. A.; Paleologou K. E.; Greer B.; Abogrein A. M.; King J. E.; Salem S. A.; Fullwood N. J.; Benson F. E.; Hewitt R.; Ford K. J.; Martin F. L.; Harriott P.; Cookson M. R.; Allsop D. A Strategy for Designing Inhibitors of α -Synuclein Aggregation and Toxicity as a Novel Treatment for Parkinson’s Disease and Related Disorders. FASEB J. 2004, 18 (11), 1315–1317. 10.1096/fj.03-1346fje. [DOI] [PubMed] [Google Scholar]
- Skibiszewska S.; Żaczek S.; Dybala-Defratyka A.; Jȩdrzejewska K.; Jankowska E. Influence of Short Peptides with Aromatic Amino Acid Residues on Aggregation Properties of Serum Amyloid A and Its Fragments. Arch. Biochem. Biophys. 2020, 681, 108264. 10.1016/j.abb.2020.108264. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Näslund J.; Lindqvist F.; Johansson J.; Karlström A. R.; Thyberg J.; Terenius L.; Nordstedt C. Arrest of -Amyloid Fibril Formation by a Pentapeptide Ligand (*). J. Biol. Chem. 1996, 271 (15), 8545–8548. 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- Soto C.; Sigurdsson E. M.; Morelli L.; Asok Kumar R.; Castaño E. M.; Frangione B. β-Sheet Breaker Peptides Inhibit Fibrillogenesis in a Rat Brain Model of Amyloidosis: Implications for Alzheimer’s Therapy. Nat. Med. 1998, 4 (7), 822–826. 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- Sato T.; Kienlen-Campard P.; Ahmed M.; Liu W.; Li H.; Elliott J. I.; Aimoto S.; Constantinescu S. N.; Octave J.-N.; Smith S. O. Inhibitors of Amyloid Toxicity Based on β-Sheet Packing of Aβ40 and Aβ42. Biochemistry 2006, 45 (17), 5503–5516. 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Xi W.; Hansmann U. H. E. Stability of the N-Terminal Helix and Its Role in Amyloid Formation of Serum Amyloid A. ACS Omega 2018, 3 (11), 16184–16190. 10.1021/acsomega.8b02377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egashira M.; Takase H.; Yamamoto I.; Tanaka M.; Saito H. Identification of Regions Responsible for Heparin-Induced Amyloidogenesis of Human Serum Amyloid A Using Its Fragment Peptides. Arch. Biochem. Biophys. 2011, 511 (1), 101–106. 10.1016/j.abb.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Buhimschi I. A.; Nayeri U. A.; Zhao G.; Shook L. L.; Pensalfini A.; Funai E. F.; Bernstein I. M.; Glabe C. G.; Buhimschi C. S. Protein Misfolding, Congophilia, Oligomerization, and Defective Amyloid Processing in Preeclampsia. Sci. Transl. Med. 2014, 6 (245), 245ra92–245ra92. 10.1126/scitranslmed.3008808. [DOI] [PubMed] [Google Scholar]
- Kouza M.; Banerji A.; Kolinski A.; Buhimschi I. A.; Kloczkowski A. Oligomerization of FVFLM Peptides and Their Ability to Inhibit Beta Amyloid Peptides Aggregation: Consideration as a Possible Model. Phys. Chem. Chem. Phys. 2017, 19 (4), 2990–2999. 10.1039/C6CP07145G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberta F.; Loerch S.; Rennegarbe M.; Schierhorn A.; Westermark P.; Westermark G. T.; Hazenberg B. P. C.; Grigorieff N.; Fändrich M.; Schmidt M. Cryo-EM Fibril Structures from Systemic AA Amyloidosis Reveal the Species Complementarity of Pathological Amyloids. Nat. Commun. 2019, 10 (1), 1104. 10.1038/s41467-019-09033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda T.; Kumar R.; Raman E. P.; Klimov D. K. Nonsteroidal Anti-Inflammatory Drug Naproxen Destabilizes Aβ Amyloid Fibrils: A Molecular Dynamics Investigation. J. Phys. Chem. B 2010, 114 (46), 15394–15402. 10.1021/jp107955v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P.; Nguyen N.; Hansmann U. H. E. D-Retro Inverso Amylin and the Stability of Amylin Fibrils. J. Chem. Theory Comput. 2020, 16 (8), 5358–5368. 10.1021/acs.jctc.0c00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsopoulos S.Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Woodhead Publishing, 2017. [Google Scholar]
- Feng Z.; Xu B. Inspiration from the Mirror: D-Amino Acid Containing Peptides in Biomedical Approaches. Biomol. Concepts 2016, 7 (3), 179–187. 10.1515/bmc-2015-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas N.; Gayen J.; Mahata M.; Su Y.; Mahata S. K.; O’Connor D. T. Novel Peptide Isomer Strategy for Stable Inhibition of Catecholamine Release: Application to Hypertension. Hypertension 2012, 60 (6), 1552–1559. 10.1161/HYPERTENSIONAHA.112.202127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R.; David L.; Gilson M. K. Accelerated Poisson–Boltzmann Calculations for Static and Dynamic Systems. J. Comput. Chem. 2002, 23 (13), 1244–1253. 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- Homeyer N.; Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2012, 31 (2), 114–122. 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- Jana A. K.; Tiwari M. K.; Vanka K.; Sengupta N. Unraveling Origins of the Heterogeneous Curvature Dependence of Polypeptide Interactions with Carbon Nanostructures. Phys. Chem. Chem. Phys. 2016, 18 (8), 5910–5924. 10.1039/C5CP04675K. [DOI] [PubMed] [Google Scholar]
- Wang W.; Hansmann U. H. E. Stability of Human Serum Amyloid A Fibrils. J. Phys. Chem. B 2020, 124 (47), 10708–10717. 10.1021/acs.jpcb.0c08280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi W.; Hansmann U. H. E. The Effect of Retro-Inverse D-Amino Acid Aβ-Peptides on Aβ-Fibril Formation. J. Chem. Phys. 2019, 150 (9), 95101. 10.1063/1.5082194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Khatua P.; Hansmann U. H. E. Cleavage, Downregulation, and Aggregation of Serum Amyloid A. J. Phys. Chem. B 2020, 124 (6), 1009–1019. 10.1021/acs.jpcb.9b10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M.; Moore S.; Mayes J.; Parkin E.; Beeg M.; Canovi M.; Gobbi M.; Mann D. M. A.; Allsop D. Development of a Proteolytically Stable Retro-Inverso Peptide Inhibitor of β-Amyloid Oligomerization as a Potential Novel Treatment for Alzheimer’s Disease. Biochemistry 2010, 49 (15), 3261–3272. 10.1021/bi100144m. [DOI] [PubMed] [Google Scholar]
- Parthsarathy V.; McClean P. L.; Hölscher C.; Taylor M.; Tinker C.; Jones G.; Kolosov O.; Salvati E.; Gregori M.; Masserini M.; Allsop D. A Novel Retro-Inverso Peptide Inhibitor Reduces Amyloid Deposition, Oxidation and Inflammation and Stimulates Neurogenesis in the APPswe/PS1ΔE9Mouse Model of Alzheimer’s Disease. PLoS One 2013, 8 (1), e54769 10.1371/journal.pone.0054769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.