

Abstract

This work surveys a variety of diamino-heterocycles as an isosteric replacement for the piperazine substructure of our previously disclosed piperarinyl-tetrahydroisoquinoline containing CXCR4 antagonists. A late-stage Buchwald coupling route was developed for rapid access to final compounds from commercial building blocks. Among 13 analogs in this study, compound 31 embodying an aza-piperazine linkage was found to have the best overall profile with potent CXCR4 inhibitory activity and favorable in vitro absorption, distribution, metabolism, and excretion (ADME) properties. An analysis of the calculated physiochemical parameters (ROF, cLogD) and the experimental ADME attributes of the analogs lead to the selection of 31 for pharmacokinetic studies in mice. Compared with the clinical compound AMD11070, compound 31 has no CYP450 3A4 or 2D6 inhibition, higher metabolic stability and PAMPA permeability, greatly improved physiochemical parameters, and superior oral bioavailability (%F = 24). A binding rationale for 31 within CXCR4 was elucidated from docking and molecular simulation studies.
Keywords: GPCR, chemokine receptor, CXCR4 antagonist, tetrahydroisoquinoline, ADME
The C-X-C chemokine receptor type 4 (CXCR4) is a seven-transmembrane G-protein-coupled receptor (GPCR) and falls within the Class A rhodopsin-like GPCR family.1,2 The sole endogenous ligand of CXCR4, the chemokine CXCL12 (stromal cell-derived factor 1 (SDF-1)), plays a vital role in homing hematopoietic stem cells (HSCs) to the bone marrow and in HSC quiescence.3,4 When CXCL12 binds to CXCR4, it activates different signaling pathways that lead to biological responses such as chemotaxis, cell survival and proliferation, intracellular calcium flux, and gene transcription.5 CXCR4 receptor levels of expression are low in healthy tissues, but they were found to be a prognostic marker in many different cancers such as breast, lung, leukemia, ovarian, prostate, and colorectal cancers.6 Organs and tissues that express high levels of CXCL12, such as the liver, lung, bone marrow, and lymph nodes, attract the migration of CXCR4-expressing cancer cells.7
Previous research on CXCR4 antagonists has resulted in several types of small-molecule-based drugs such as AMD3100 (1), AMD11070 (2), GSK812397 (3), and IT1t (4) (Figure 1).8 Of these, the first U.S. Food and Drug Administration (FDA) approved CXCR4 antagonist drug was Plerixafor (1, AMD3100), which was initially developed as an anti-HIV agent functioning as a potent antagonist, but drug toxicity prevented it from being administered daily.9 Clinical investigations of Plerixafor led to its FDA-approval for HSC mobilization in non-Hodgkin’s lymphoma and multiple myeloma patients needing autologous transplants.10 The poor oral bioavailability displayed by Plerixafor led to the discovery of AMD11070 (2) as the first orally active CXCR4 antagonist, which is currently in clinical trials.11 Efforts to increase the potency by modifying the structural features of AMD11070 led to the novel hybrid piperazine (3, GSK812397), whereas the isothiourea (4, IT1t) series was discovered through high-throughput screening efforts.12,13 Later efforts to replace the benzimidazole ring of AMD11070 were successful as interest in oral CXCR4 antagonists grew. Our laboratory discovered that a tetrahydroisoquinoline (THIQ) ring was a suitable replacement for the benzimidazole core, and our parent compound TIQ-15 (5) had good drug-like properties.14 Follow-up medicinal chemistry produced a second-generation series of 2-piperazinyl-TIQ antagonists (6) with very good overall properties. To expand the structural variability, we are describing herein a series of piperazine ring replacements (7) focusing on diamine heterocycles and other isosteric moieties using a late-stage Buchwald–Hartwig coupling synthetic approach.
Figure 1.

Notable CXCR4 antagonists and Medchem strategy.
The target compounds (Schemes 1 and 2, Table 1) were selected based on the criteria of having a diamino group that was in a 1,2, 1,3, or 1,4 carbon spacing, including a mono- or bicyclic ring system, and improving drug-like properties versus the butyl amine compounds AMD11070 (2) and TIQ-15 (3). Of particular interest were cases that provided isosteric and homologous piperazine ring replacements such as the bicyclic octahydropyrrolo-pyrrole ring (28g–28j), which has been shown to be a scaffold in other GPCR efforts. This would allow us to compare directly to our previous effort involving compounds containing substituted piperazines (6).15,16 Our first synthetic approach began with the previously described racemic intermediate 2-bromo-tetrahydroisoquinoline-methylester 8, which was directly subjected to Buchwald coupling conditions with the isomeric Boc-protected 4-amino-piperidines (Scheme 1).15 The resulting 2-amino-tetrahydroisoquinolines (9a/9b) were reduced to the carboxaldehydes (10a/10b) and reacted with chiral methylamino-tetrahydroquinoline 11. In the case of the isomer where the piperidine nitrogen was attached to the phenyl ring (12b), only the R,S-isomer could be cleanly isolated, whereas in the case of the isomeric N-Boc-piperidine, the isomers (12a, 13) were separated. All three materials were subjected to standard Boc deprotection conditions to yield the final compounds (14–16).
Scheme 1. Synthesis of Compounds 14–16.

Reagents. i. Pd2(dba)3, (±)-BINAP, Cs2CO3, tert-butyl piperidin-4-ylcarbamate or tert-butyl 4-aminopiperidine-1-carboxylate, PhMe, 100 °C or μwave 140 °C; ii. DIBAL-H, −78 °C, PhMe ; iii. 11, NaBHOAc3, DCM, rt; iv. TFA, DCM, rt.
Scheme 2. Synthesis of Compounds 24, 29–37.
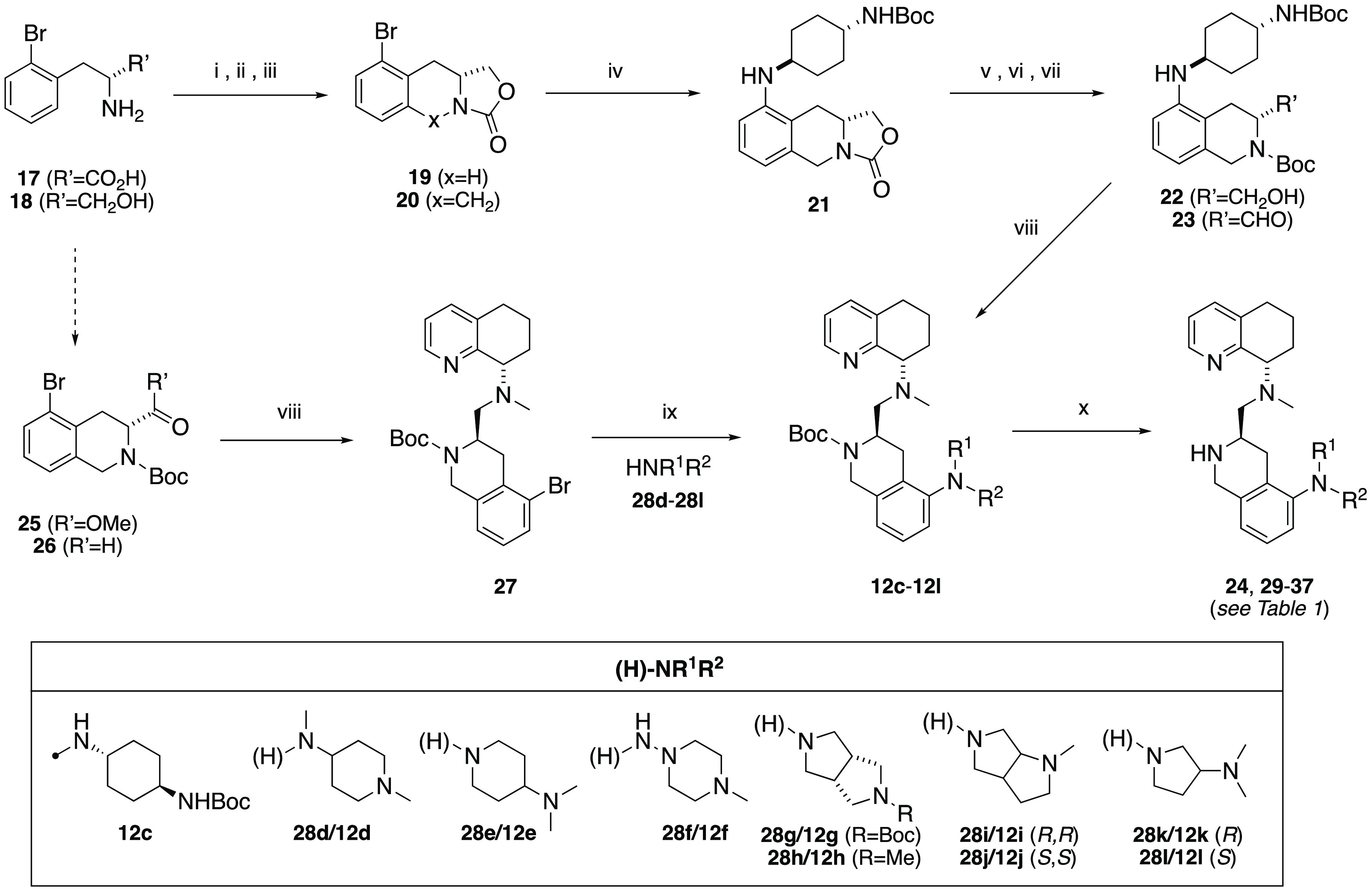
Reagents. i. NaBH4; ii. (EtO)2CO, KOH; iii. (CH2O)3, BF3–OEt2; iv. Pd2(dba)3, (±)-BINAP, trans-1,4-BocNHC6H10NH2, Cs2CO3, PhMe, 100 °C; v. KOH, MeOH; vi. Boc2O, THF; vii. DMSO, Pyr-SO3, NEt3, DCM; viii. 11, NaBHOAc3, DCM, rt ; ix. Pd2(dba)3, (±)-BINAP, 28d–28l, Cs2CO3, PhMe, 100 °C or μwave 140 °C; x. TFA, DCM, rt.
Table 1. Biological and ADME Assay Data for 2-Aminoheterocyclic Tetrahydroisoquinoline CXCR4 Antagonists.
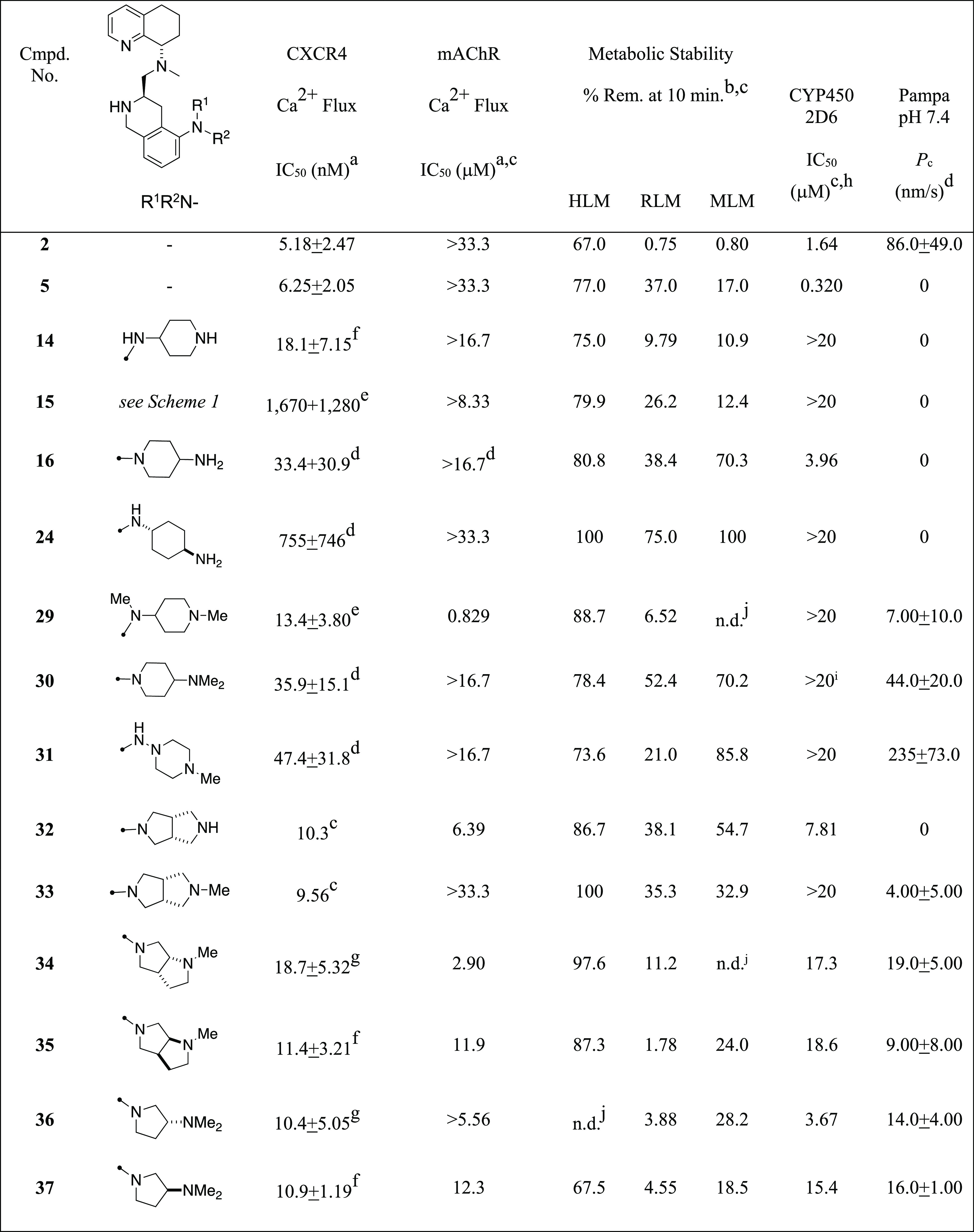
Concentration of compound inhibiting the Ca2+ flux (release) by 50%, reported as a single experiment (n = 1) or the mean of several experiments (n > 1).
Metabolic stability was determined as the percentage of test compound remaining after incubation for 10 min at 37 °C in liver microsome preparations (CYP450 and other NADP-dependent enzymes).
n = 1.
Reported error represents the standard deviation of multiple experiments (n = 2).
Reported error represents the standard deviation of multiple experiments (n = 3).
Reported error represents the standard deviation of multiple experiments (n = 4).
Reported error represents the standard deviation of multiple experiments (n = 5).
Lower limit (<20 μM) in the CYP450 2D6 assay is shown; all compounds had >20 μM activity against CYP450 3A4.
CYP 2C9 IC50 = 6.67 μM.
Value not determined (n.d.).
The second and third synthetic sequences investigated began with 2-bromo-d-phenyl alanine (17, Scheme 2). We had previously described a route where we utilized this starting material to construct several chiral intermediates.15 In this account, we further explored the versatility of this synthon. Reduction to the amino alcohol (18) and formation of the oxazolidinone (19) enabled a facile Pictet–Spangler to tricyclic intermediate 20. The next target selected was based on butyl amine side-chain replacements we had previously discovered, where the trans-diamino cyclohexane group was found to have the best overall properties.17 Buchwald coupling with N-Boc-1,4-trans-diamino-cyclohexane provided compound 21. Hydrolysis of the oxazolidinone, Boc protection (to 22), and Parikh–Doering oxidation provided aldehyde 23. Reductive amination with tetrahydroquinoline (THQ) 11 provided 12c and, after Boc deprotection, the final compound 24. Although this route was scalable, it provided to be quite lengthy. The third and most versatile route we employed utilized the previously described carboxaldehye 26. This compound was accessible via DIBAL-H reduction of the methyl ester 25. The THQ top piece 11 was attached via reductive amination. The 2-bromo advanced intermediate 27 could then be altered via a late-stage Buchwald coupling reaction with a variety of amines (28d–28l) under standard conditions in toluene in good yields (40–60%). The resulting 2-aminoderivatives (12d–12l) were Boc-deprotected to provide the final desired compounds 29–37.
Next, we subjected the compounds (14–16, 24, 29–37) to a battery of assays to evaluate their potential as CXCR4 antagonist-based drug leads. The tests included both CXCR4 and muscarinic acetylcholine receptor (mAChR) calcium flux measurements, metabolic stability in liver microsomes, inhibition of CYP450 enzymes 3A4 and 2D6, and PAMPA membrane permeability.15 The results for these compounds showed the following trends (Table 1). First, the on-target CXCR4 calcium flux assay indicates that all but two of the substitutions produced potent antagonist responses (IC50 values <50 nM), with 9 of the 13 compounds having values below 20 nM. These compounds had only slightly higher CXCR4 calcium flux potency than both 2 and 5 (two- to three-fold), and all of them had fewer free nitrogen–hydrogen bonds that could improve the permeability by the reduction of H-bond donor count. Only the S,S-syn isomer 15 and the trans-1,4-diamino-cylcohexane 24 showed high calcium flux potency (IC50 > 200 nM). These two activity outliers show that there is a maximum tolerable spacing between the diamine nitrogen atoms and also confirm our previous discovery of the R-stereochemical preference on the THIQ ring.14 An equally distinguishing test result was regarding mAChR activity, where half of the compounds showed modest to weak activity in the 0.8 to 12 μM range, whereas the others had no measurable results below 20 μM. mAChR responses are involved in neuromuscular and autonomic nervous system control, and potential side effects include reduced smooth muscle contraction and gland secretion, which are therefore undesirable in our series.16 Surprisingly, four of the five compounds (32, 34, 35, 37) with mAChR activity had a pyrrolidine linked to the aromatic ring system. However, two of these (35, 37) were just above the 10 μM IC50 mark with therapeutic ratios of 1000 versus calcium flux and could still be considered viable for lead progression.
The other three assays utilized profiled the absorption, distribution, metabolism, and excretion (ADME) properties of our compounds. Many of these compounds had good stability in human liver microsomes (HLMs, >70% remaining at 10 min), and this did not seem to be a limitation of this series. Whereas some of the compounds had good stability in one or two of these species, the compounds with good metabolic stability (16, 30, 31) in all three species could translate to higher plasma levels, increasing the success potential in rodent and human models of cancer. Second, the CYP450 3A4 and 2D6 enzyme assays produced results where only one of the compounds showed inhibition against the 3A4 isozyme; however, because significant 2D6 inhibition was observed in the parent compound (5), this property was of particular significance to us. Six of the compounds (16, 32, 34–37) showed modest to weak inhibition of 2D6 (IC50 values in the 4–19 μM range), but this was 10 to 50 times higher than that for TIQ-15 (5). The other seven (14, 15, 24, 29–31, 33) showed no 2D6 inhibition (IC50 values >20 μM). The third and final characteristic we measured was the permeability or the ability to be passively absorbed through the intestinal wall utilizing a PAMPA permeability assay.18 Five of the compounds showed no permeability (14–16, 24, 32), and six showed low to modest permeability (29, 33–37), indicating the likelihood of a significant amount of protonated material (>99%) compared with free base in the equilibrium; however, two compounds showed significant permeability potential (30, 31), with only the N-aza-piperazine 31 exceeding the 100 nm/s threshold.
An analysis of the computational physiochemical properties of these compounds provided more insight (Figure 2). First, according to the determined Lipinski parameters, all of the compounds meet the criteria of the rule of five.19 In all, six parameters were analyzed (Table S1) and are shown as a radar plot versus the accepted ranges for each (Figure 2A, defined by the green background).20 In general, the mean values were well within the range of the accepted parameters (black dotted line). Because the two first-generation CXCR4 antagonists (2,5) also lie within the acceptable range for most of these parameters (except rotatable bonds), a more in-depth analysis might reveal significant differences. Whereas the 2-amino substituents were chosen based on their structural aspects, the differences in the CXCR4 activity for the majority of the products were also within 10-fold of each other. These differences were not manifested in ligand efficiencies, as the heavy-atom counts were almost identical (Table S2); however, upon determining cLogP values, we observed modest differences in the lipophilic efficiency (LipE, Table S2), showing that most compounds (except 15 and 24) displayed a value in the range of 3.8 to 5.5.21 In comparison with the known butyl amine first-generation antagonists (2, 5), the LipE values for these compounds were also closely matched. The main difference was seen in the cLogDpH7.4 values generated (Table S2, Figure 2B). Here the butyl amine benchmark compounds (2,5) produced negative values (−0.04, −0.95), whereas a wider range from negative to positive was produced for the compounds in this study. Of note are compounds 14 and 31, which lie on the extreme of cLogDpH7.4 values (−1.45 vs 1.13), whereas the remainder of the compounds fall within this range. Another surprising comparison was the low PAMPA permeability of compound 29, which might be explained by the higher basicity of the N-methylpiperidine versus the N-methyl aza piperazine of 31 (calculated pKa values of 9.14 vs 7.26, SI). The golden triangle analysis (Figure 2B) was performed by interchanging cLogD to provide further insight.22 Only one compound (31) fell within the defined limits of molecular weight (MW) and LogD defined by the triangle, for which inclusion has been shown to correlate to better oral bioavailability. Experimental support of the golden triangle observation is provided by a graphical comparison of microsomal and permeability data. Here we plotted both human (HLM) and mouse (MLM) liver microsomal stability versus PAMPA permeability, which has been shown to correlate to human oral absorption (Figure 2C).20,23 In this diagram, one compound (31) clearly fits within the >20% FPO portion (upper right quadrant) of this plot and is superior to all of the other compounds in both HLM and MLM results.
Figure 2.
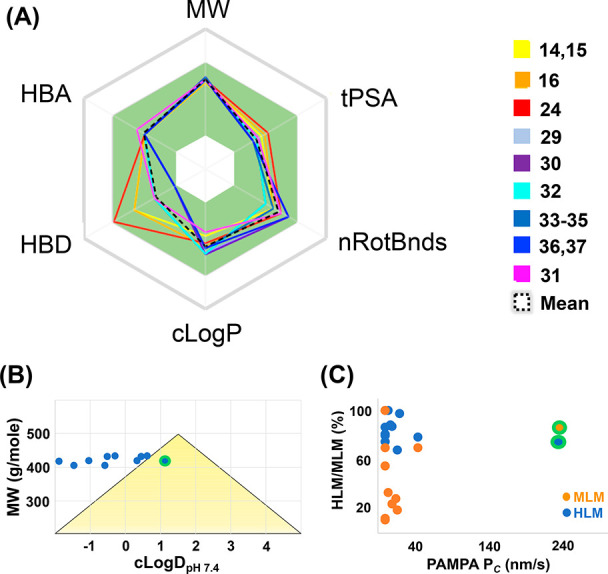
Graphical representation of physiochemical and ADME parameters for compounds in Table 1. Numbers are provided in Tables S1 and S3. (A) Radar plot of the Lipinski ROF+1. Compounds are color-coded. The mean value is designated as the black dotted line; acceptable ROF limits are within the green area. (B) Golden triangle plot. (C) Metabolic stability versus permeability. The location of 31 is highlighted by a green halo.
To attain a better understanding of how our compounds act upon on the CXCR4 receptor on the molecular level, we undertook a computer modeling study. Previous computational and mutagenesis studies with 5 and similar molecules have identified interactions with Asp97 and Glu288 to be crucial.25 With the former study in mind, using the Schrodinger Maestro suite, we prepared compound 31 as the dication conformer, and it was docked into the optimized CXCR4:1T1t crystal structure (PDB: 3ODU).24 The grids generated included water molecules. In this case, induced-fit docking afforded two plausible binding poses (Figure 3A,B). In the crystal structure, IT1t (4) formed two salt bridges between residues Asp97 and E288 of the CXCR4 receptor. Both docked poses of 31 also formed two salt bridges with these same residues. Within pose 1 (Figure 3A), the piperazine nitrogen forms an electrostatic interaction with Asp97, and the THIQ nitrogen atom of 31 forms a salt bridge with Glu288. Secondary interactions are formed with Trp94 (π-stacking), His113 (π-stacking), and the aromatic portion of the THIQ ring. While in binding pose 2, similar to our previous poses of THIQ molecules, the π-stacking network with the aromatic portion of the THIQ ring remained intact, but the salt bridges were formed with the alternative basic centers of the molecule (Figure 3B).15,25 On the basis of our previous observations, each binding pose was plausible due to the similarity between the salt bridge residues and that of IT1t. Thus further computational analysis of the binding poses 1 and 2 of compound 31 was completed using binding pose metadynamics (BPMD) and molecular dynamics (MD) simulations (Figure 3C, SI).26,27 In this case, pose 1 of compound 31 possessed the least instability (blue line, PoseScore = 1.774) and maintained most of its interactions, whereas pose 2 seemed less stable (green line, PersScore: 0.864) in the receptor throughout the BPMD sampling protocol (Figure 3C). Also, a 100 ns MD simulation of compound 31 denoted that pose 1 was highly stable with a root-mean-squared deviation (rmsd) of <2 Å throughout the entirety of the simulation. Furthermore, pose 1 of compound 31 maintained strong interactions with Trp94, Asp97, His113, and Glu288 (Figure S1). Our results also indicated that the pyridine nitrogen of the top ring, within pose 1, developed a water-mediated interaction with Glu288 and Tyr255 during the simulation. Alternatively, pose 2 of compound 31 possessed a PoseScore of >2 and had an average rmsd of >2 Å within the MD simulation (Figure 3C, Supporting Information (SI)). Furthermore, pose 2 of compound 31 lost its hydrophobic and electrostatic interactions with Trp94 (π-stacking), His113 (π-stacking), and Glu288 (Figure S2). Further evidence is provided by docking compound 31 into the 3ODU grid without water molecules, where the highest ranked pose is in the same orientation and forms similar interactions with the receptor (Figure S3) as pose 1. Altogether, the data suggested that pose 1 and pose 2 of compound 31 were both plausible binding poses, but our computational modeling strongly favors binding pose 1.
Figure 3.

Computational induced-fit docking studies with compound 31 in the CXCR4:IT1t grid (3ODU).24 (A) Pose 1. (B) Pose 2. (C) Metadynamic study results showing the rmsd change over time for poses 1 and 2.
Next, two selectivity assessments were performed on compound 31. First, a seven-target chemokine selectivity panel (CXCR1,2,3; CCR4,5,6,7) showed no inhibition of ligand-induced calcium flux by the compound at concentrations up to 10 μM. Next, a hERG K+ channel assessment was performed using an automated patch clamp functional assay showing weak inhibition (IC50 = 6.8 μM). Given these and previous outcomes, compound 31 was then investigated in pharmacokinetic (PK) experiments in mice along with AMD11070 (2). To evaluate the oral administration potential, we dosed mice in two routes with drug levels estimated to provide accurate data (3 mg/kg IV; 30 mg/kg PO). The plasma levels of the drug compounds were measured using LC-MS/MS methods centered on the parent molecular ion. The PK parameters were calculated and are provided for each compound in Table 2. Overall, compound 31 showed superior performance to AMD11070 in every parameter by a significant amount. The maximum concentrations (Cmax) show a 20-fold difference favoring 31 (791 vs 41 ng/mL). When normalizing for molecular weight, the maximum drug levels for 2 and 31 adequately cover (117 vs 1881 nM) the CXCR4 calcium flux potencies (23- versus 40-fold); however, at the 8 h time mark, there is a significant difference where the drug plasma coverage is barely adequate for AMD11070 (19 nM; four-fold), whereas the levels for 31 are significantly higher (706 nM; 15-fold). This would indicate a less frequent dosing requirement for 31 compared with 2. Furthermore, this observation is supported by the clearance rates and half lives for the two compounds. Compound 2 has a higher clearance rate (7.23 versus 1.83 L/h/kg) and a slightly shorter half life than 31 (3.1 versus 3.5 h). Overall, a more than 10 times higher amount of compound 31 was absorbed (%F = 24 versus 2.4) with 40 times higher overall drug levels (AUC of 4001 versus 101) and similar high volumes of distribution (9.35 versus 32.2) indicating the superior exposure of compound 31 compared with 2. Whereas the CXCR4 calcium flux potency is 10 times higher for 31, the higher drug levels overcome the therapeutic exposure requirement, especially at the 8 h time mark. Although this result with 31 might indicate that compounds 30 and 33 might also produce favorable results in PK studies, time and resources combined to limit these investigations.
Table 2. Mouse PK Parameters for Compounds 2 and 31.
| compound |
||
|---|---|---|
| 2 | 31 | |
| Cmax (ng/mL) | 41.0 | 791 |
| C8h (ng/mL)b | 6.73 | 297 |
| AUC0–8h (h*ng/mL)b | 100.6 | 4001c |
| clearance rate (L/h/kg) | 7.23 | 1.83 |
| Vd (L/kg) | 32.2 | 9.35 |
| T1/2 (h) | 3.1 | 3.5c |
| FPO (%)a,b | 2.4 | 24c |
Doses: 3 mg/kg IV and 30 mg/kg PO.
Measurements conducted up to 8 h.
Values for 31 exceed the 8 h time point and are underestimated.
In summary, a late-stage alteration strategy using Buchwald coupling methodology allowed the quick evaluation of a variety of 2-amino substituents designed to emulate and vary the piperazinyl group. The efforts identified unfavorable substitutions and orientations where there is a limit to the type of diamino fragment. Many examples failed to provide the combined properties of low off-target effects (mAChR, CYP450 2D6), high metabolic stability, and suitable permeability; however, four compounds (14, 30, 31, 33) provided good properties in four areas with good CXCR4 potency, no mAChR activity, no CYP450 2D6 inhibition, and higher metabolic stability. Compound 31 provided the best permeability and overall properties compared with these other compounds and 11070 (2). The mouse oral PK study showed that the calculated and experimental parameters (Figure 2B,C; Tables S1 and S2) correlated to the superior performance of compound 31 in most parameters (%F, Cmax, AUC, CL) compared with AMD11070 (Table 2). Therefore, compound 31 would make a good next-generation CXCR4 antagonist for further study, especially in mouse models of cancer immune responses and cancer metastasis. Progress in these areas will be the subject of future reports.
Acknowledgments
We acknowledge the use of shared instrumentation provided by grants from the NSF (CHE1531620). This research was additionally funded, in part, by a Winship Invest$ Prostate Cancer Research Pilot Grant supported by the Dunwoody Golf Club Prostate Cancer Research Award, a philanthropic award provided by the Winship Cancer Institute of Emory University.
Glossary
Abbreviations
- CXCR1, CXCR2, CXCR3, CXCR4, CXC
chemokine receptor 1, 2, 3, 4
- CCR4, CCR5, CCR6, CCR7, CCR
chemokine receptor 4, 5, 6, 7
- mAChR
muscarinic acetylcholine receptor
- CXCL12
CXC chemokine ligand 12
- SDF-1
stromal cell-derived factor 1
- THIQ
tetrahydroisoquinoline
- THQ
tetrahydroquinoline
- GPCR
G-protein-coupled receptor
- CYP450
cytochrome P450
- HLM, RLM, MLM
human, rat, and mouse liver microsomes
- PAMPA
parallel artificial membrane permeability assay
- PK
pharmacokinetic
- hERG
human ether-a-go-go related gene
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00449.
Tables S1–S3. Figures S1–S3. Synthetic procedures for all compounds. Procedures for all assays and mouse PK studies. pKa calculations for 29 and 31. Additional computational docking studies on compounds 6 and 30 (PDF)
Author Contributions
The manuscript was written by L.J.W. and H.H.N. The synthetic medicinal chemistry work was devised and performed by H.H.N., Y.A.T., E.J., and L.J.W. Assays in Table 1 were performed by BMS authors. Modeling studies were done by N.S.A. and L.J.W. PK studies were overseen and performed by E.J.M., L.X., and Y.J. All authors have given approval to the final version of the manuscript.
D.C.L. is the principle investigator on a research grant from Bristol-Myers Squibb Research and Development to Emory University.
The authors declare the following competing financial interest(s): D.C.L., H.H.N., Y.A.T., V.M.T., R.J.W., E.J., E.J.M., M.B.K., and L.J.W. are coinventors on Emory-owned Intellectual Property that includes CXCR4 antagonists.
This paper was originally published ASAP on October 4, 2021. Due to a conversion error, there was a mistake in the units of the TOC graphic. The corrected version was reposted on October 14, 2021.
Supplementary Material
References
- Chen K.; Bao Z.; Tang P.; Gong W.; Yoshimura T.; Wang J. M. Chemokines in homeostasis and diseases. Cell. Mol. Immunol. 2018, 15, 324–334. 10.1038/cmi.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y.-R.; Kottmann A. H.; Kuroda M.; Taniuchi I.; Littman D. R. Function of the Chemokine Receptor CXCR4 in Haematopoiesis and in Cerebellar Development. Nature 1998, 393, 595–599. 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- Zou Y.-R.; Kottmann A. H.; Kuroda M.; Taniuchi I.; Littman D. R. The function of the chemokine receptor CXCR4 in hematopoiesis and in cellular development. Nature 1998, 393, 595–599. 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- Wright D. E.; Bowman E. P.; Wagers A. J.; Butcher E. C.; Weissman I. L. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J. Exp. Med. 2002, 195, 1145–1154. 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganju R. K.; Brubaker S. A.; Meyer J.; Dutt P.; Yang Y.; Qin S.; Newman W.; Groopman J. E. The alpha-chemokine, stromal cell-derived factor-1 alpha, binds to the transmembrane G-protein-coupled CXCR4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 1998, 273, 23169–23175. 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- Mishan M. A.; Ahmadiankia N.; Bahrami A. R. Bahrami AR. CXCR4 and CCR7: two eligible targets in targeted cancer therapy. Cell Biol. Int. 2016, 40, 955–967. 10.1002/cbin.10631. [DOI] [PubMed] [Google Scholar]
- Domanska U. M.; Kruizinga R. C.; Nagengast W. B.; Timmer-Bosscha H.; Huls G.; de Vries E. G.E.; Walenkamp A. M.E. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur. J. Cancer 2013, 49, 219–230. 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Choi W.-T.; Duggineni S.; Xu Y.; Huang Z.; An J. Drug discovery research targeting the CXC chemokine receptor 4 (CXCR4). J. Med. Chem. 2012, 55, 977–994. 10.1021/jm200568c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix C. W.; Collier A.; Lederman M.; Schols D.; Pollard R. B.; Brown S.; Jackson J. B.; Coombs R. W.; Glesby M. J.; Flexner C. W.; Bridger G. J.; Badel K.; MacFarland R. T.; Henson G. W.; Calandra G. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. JAIDS, J. Acquired Immune Defic. Syndr. 2004, 37, 1253–1262. 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Mozobil (Plerixafor, AMD3100), 10 years after its approval by the food and drug administration. Antiviral Chem. Chemother. 2019, 27, 1–8. 10.1177/2040206619829382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerlj R. T.; Bridger G. J.; Kaller A.; McEachern E. J.; Crawford J. B.; Zhou Y.; Atsma B.; Langille J.; Nan S.; Veale D.; Wilson T.; Harwig C.; Hatse S.; Princen K.; De Clercq E.; Schols D. Discovery of Novel Small Molecule Orally Bioavailable C-X-C Chemokine Receptor 4 Antagonists that are Potent Inhibitors of T-Tropic (X4) HIV-1 Replication. J. Med. Chem. 2010, 53, 3376–3388. 10.1021/jm100073m. [DOI] [PubMed] [Google Scholar]
- Jenkinson S.; Thomson M.; McCoy D.; Edelstein M.; Danehower S.; Lawrence W.; Wheelan P.; Spaltenstein A.; Gudmundsson K. Blockade of X4-Tropic HIV-1 Cellular Entry by GSK812397, a Potent Noncompetitive CXCR4 Receptor Antagonist. Antimicrob. Agents Chemother. 2010, 54, 817–824. 10.1128/AAC.01293-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma G.; Streiff M. B.; Kovarik J.; Glickman F.; Wagner T.; Beerli C.; Zerwes H. G. Orally Bioavailable Isothioureas Block Function of the Chemokine Receptor CXCR4 in Vitro and in Vivo. J. Med. Chem. 2008, 51, 7915–7920. 10.1021/jm801065q. [DOI] [PubMed] [Google Scholar]
- Truax V. M.; Zhao H.; Katzman B. M.; Prosser A. R.; Alcaraz A. A.; Saindane M. T.; Howard R. B.; Culver D.; Arrendale R. F.; Gruddanti P. R.; Evers T. J.; Natchus M. G.; Snyder J. P.; Liotta D. C.; Wilson L. J. Discovery of Tetrahydroisoquinoline-Based CXCR4 Antagonists. ACS Med. Chem. Lett. 2013, 4, 1025–1030. 10.1021/ml400183q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. H.; Kim M. B.; Wilson R. J.; Butch C. J.; Kuo K. M.; Miller E. J.; Tahirovic Y. A.; Jecs E.; Truax V. M.; Wang T.; Sum C. S.; Cvijic M. E.; Schroeder G. M.; Wilson L. J.; Liotta D. C. Design, Synthesis, and Pharmacological Evaluation of Second-Generation Tetrahydroisoquinoline-Based CXCR4 Antagonists with Favorable ADME Properties. J. Med. Chem. 2018, 61, 7168–7188. 10.1021/acs.jmedchem.8b00450. [DOI] [PubMed] [Google Scholar]
- Bunnelle W. H.; Tietje K. R.; Frost J. M.; Peters D.; Ji J.; Li T.; Scanio M. J. C.; Shi L.; Anderson D. J.; Dyhring T.; Gronlien J. H.; Ween H.; Thorin-Hagene K.; Meyer M. D. Octahydropyrrolol[3,4-c]pyrrole: A Diamine Scaffold for Construction of Either α4β2 or α7-Selective Nicotinic Acetylcholine Receptor (nAChR) Ligands. Substitutions that Switch Subtype Selectivity. J. Med. Chem. 2009, 52, 4126–4141. 10.1021/jm900249k. [DOI] [PubMed] [Google Scholar]
- Jecs E.; Miller E. J.; Wilson R. J.; Nguyen H. H.; Tahirovic Y. A.; Katzman B. M.; Truax V. M.; Kim M. B.; Kuo K.; Wang T.; Sum C. S.; Cvijic M. E.; Schroeder G. M.; Wilson L. J.; Liotta D. C. ACS Med. Chem. Lett. 2018, 9, 89–93. 10.1021/acsmedchemlett.7b00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balimane P. V.; Han Y.-H.; Chong S. Current Industrial Practices of Accessing Permeability and P-Glycoprotein Interaction. AAPS J. 2006, 8, E1–E13. 10.1208/aapsj080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Ertl P.; Lewis R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discovery Today 2011, 16, 65–72. 10.1016/j.drudis.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. Setting Expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. 10.1016/j.bmcl.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Johnson T. W.; Dress K. R.; Edwards M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- Mandagere A. K.; Thompson T. N.; Hwang K.-K. Graphical Model for Estimating Oral Bioavailability of Drugs in Humans and Other Species from Their Caco-2 Permeability and in Vitro Liver Enzyme Metabolic Stability Rates. J. Med. Chem. 2002, 45, 304–311. 10.1021/jm010152k. [DOI] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y. T.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; Hamel D. J.; Kuhn P.; Handel T. M.; Cherezov V.; Stevens R. C. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman B. M.; Cox B. D.; Prosser A. R.; Alcaraz A. A.; Murat B.; Heroux M.; Tebben A.; Zhang Y.; Schroeder G. M.; Snyder J. P.; Wilson L. J.; Liotta D. C. Tetrahydroisoquinoline CXCR4 Antagonists Adopt a Hybrid Binding Mode within the Peptide Subpocket of the CXCR4 Receptor. ACS Med. Chem. Lett. 2019, 10, 67–73. 10.1021/acsmedchemlett.8b00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. J.; Tiwary P.; Borrelli K.; Feng S.; Miller E. B.; Abel R.; Friesner R. A.; Berne B. J. Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynmaics Simulations. J. Chem. Theory Comput. 2016, 12 (6), 2990–2998. 10.1021/acs.jctc.6b00201. [DOI] [PubMed] [Google Scholar]
- Fusani L.; Palmer D. S.; Somers D. O.; Wall I. D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60 (3), 1528–1539. 10.1021/acs.jcim.9b00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.