

Abstract
Rationale: Cigarette smoke (CS) inhalation triggers oxidative stress and inflammation, leading to accelerated lung aging, apoptosis, and emphysema, as well as systemic pathologies. Metformin is beneficial for protecting against aging-related diseases.
Objectives: We sought to investigate whether metformin may ameliorate CS-induced pathologies of emphysematous chronic obstructive pulmonary disease (COPD).
Methods: Mice were exposed chronically to CS and fed metformin-enriched chow for the second half of exposure. Lung, kidney, and muscle pathologies, lung proteostasis, endoplasmic reticulum (ER) stress, mitochondrial function, and mediators of metformin effects in vivo and/or in vitro were studied. We evaluated the association of metformin use with indices of emphysema progression over 5 years of follow-up among the COPDGene (Genetic Epidemiology of COPD) study participants. The association of metformin use with the percentage of emphysema and adjusted lung density was estimated by using a linear mixed model.
Measurements and Main Results: Metformin protected against CS-induced pulmonary inflammation and airspace enlargement; small airway remodeling, glomerular shrinkage, oxidative stress, apoptosis, telomere damage, aging, dysmetabolism in vivo and in vitro; and ER stress. The AMPK (AMP-activated protein kinase) pathway was central to metformin’s protective action. Within COPDGene, participants receiving metformin compared with those not receiving it had a slower progression of emphysema (−0.92%; 95% confidence interval [CI], −1.7% to −0.14%; P = 0.02) and a slower adjusted lung density decrease (2.2 g/L; 95% CI, 0.43 to 4.0 g/L; P = 0.01).
Conclusions: Metformin protected against CS-induced lung, renal, and muscle injury; mitochondrial dysfunction; and unfolded protein responses and ER stress in mice. In humans, metformin use was associated with lesser emphysema progression over time. Our results provide a rationale for clinical trials testing the efficacy of metformin in limiting emphysema progression and its systemic consequences.
Keywords: aging, comorbidities, metformin, cigarette smoke, chronic obstructive pulmonary disease
At a Glance Commentary
Scientific Knowledge on the Subject
The current treatments for pulmonary emphysema have a limited ability to halt the lung tissue destruction or the multiple and disabling systemic pathologies associated with chronic obstructive pulmonary disease (COPD), which include accelerated aging, apoptosis, inflammation, and oxidative stress in the lung and other organs. Metformin is a widely used drug that is associated with beneficial effects in diseases characterized by accelerated aging and metabolic stress and is associated with an increased lifespan in mice.
What This Study Adds to the Field
Metformin has protective effects against cigarette smoke–induced pulmonary and systemic pathologies in a murine model of COPD. In particular, metformin protects the mice against cigarette smoke–induced accelerated aging, inflammation, oxidative stress, telomere damage, mitochondrial dysfunction, and unfolded protein responses associated with endoplasmic reticulum stress. These protective effects are linked to activation of 5′ AMPK (AMP-activated protein kinase) and GDF-15 (growth differentiation factor 15), which are major bioenergetic sensors and metabolic regulators. Importantly, in humans, metformin use is associated with protection against emphysema progression over time, as measured by computed tomography in the COPDGene (Genetic Epidemiology of COPD) cohort. Thus, we provide a scientific rationale to stimulate the potential application of metformin in trials aimed at limiting emphysema progression.
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality worldwide (1). Most patients with COPD develop radiologically defined pulmonary emphysema (2), and in a significant proportion of them, this is the dominant feature of the disease. The best-studied cause of emphysema is the chronic inhalation of cigarette smoke (CS) (1), which, in addition to injuring the lung, is associated with a range of systemic effects, including loss of tissue of mesenchymal origin, best manifested by cachexia with loss of muscle mass and function (3), kidney disfunction, and comorbidities that independently increase the risk of death (4).
The deleterious effects of CS are associated with increases in inflammation (5), alveolar epithelial-cell apoptosis (6), accelerated aging, and oxidative stress (6) in the lungs as well as systemically (7). CS also induces accumulation of nonfunctional and potentially cytotoxic, misfolded proteins (unfolded protein responses [UPRs]) and endoplasmic reticulum (ER) stress that contribute to lung cell apoptosis and inflammation (8). These findings, together with mitochondrial dysfunction and disruption of mitophagy with incomplete degradation of damaged mitochondria (9), suggest that accelerated aging may be a potential mechanism contributing to emphysema development. Consistent with this, we have shown that patients with COPD develop comorbidities typically seen in much older patients, such as osteoporosis, atherosclerosis, skin cancer, cataracts, and dementia, 10 years earlier than patients without a COPD diagnosis (10). Importantly, current treatment for emphysema is based on the use of inhaled medications that improve respiratory symptoms and lung function but have limited if any effect on lung tissue regeneration and the systemic effects of the disease (11).
Metformin is prescribed to treat diabetes mellitus type 2. Apart from increasing insulin sensitivity, metformin usage has been linked with a reduced risk of cancer and cardiovascular diseases (12), suggesting a beneficial role in other diseases associated with aging. Indeed, metformin-dependent activation of 5′ AMPK (AMP-activated protein kinase) lengthens the lifespan and attenuates the deleterious effects of aging in preclinical models (13). A benefit of metformin has also been observed in conditions associated with inflammatory organ injury and fibrosis via regulation of cellular redox homeostasis (14).
To test the hypothesis that metformin could ameliorate the pulmonary and systemic lesions caused by CS, we compared emphysema-like lung disease, alveolar-cell apoptosis, oxidative stress, and aging in the lung, kidney, and quadriceps muscle in air-exposed mice versus CS-exposed mice that received metformin-enriched versus metformin-free chow for the second half of the CS exposures. To explore the impact of metformin in human disease, we also measured its acute effect on mitochondrially dependent functions and the AMPK pathway in CS-exposed human bronchial epithelial cells (HBECs). In humans, we explored whether the use of metformin was associated with slower emphysema progression among participants in the COPDGene (Genetic Epidemiology of COPD) study with spirometry-confirmed COPD.
Methods
Animal Studies
A full description of the methods is available in the online supplement. All procedures performed on mice were approved by the Brigham and Women’s Hospital Institutional Animal Care and Use Committee.
In summary, C57BL/6, wild-type mice were exposed to air or CS by using a Teague TE-10z device for 2 hours daily on 6 days a week for 24 weeks, with the total particulate matter delivered ranging from 150 and 200 mg/m3 (15, 16). Sex-matched mice were fed either custom-made, fat-enriched, 1% metformin–enriched or metformin-free chow every day during the last 12 weeks of the exposures (see Figure E1 in the online supplement).
We quantified emphysema and small airway remodeling (SAR) by using morphometry on Gill-stained and Masson’s trichrome–stained, formalin-fixed, paraffin-embedded tissue (15, 16); respiratory mechanics (15); and cell counts in BAL-fluid samples (16). Kidney sections were stained with hematoxylin and eosin to determine the glomerular diameter and glomerular shrinkage (17). Quadriceps sections were stained for reduced nicotinamide adenine dinucleotide to quantify type I and type II muscle fiber proportions (18). Lung, kidney, and quadriceps sections were assayed for caspase-3 (a marker of intrinsic apoptosis [16]), 4-hydroxy-2-nonenal (a marker of lipid peroxidation [16]), thiobarbituric acid–reactive substances, and H2AX (γ-H2A.X variant histone), the levels of which increase with oxidative stress and aging; p21 and p16 (senescence-associated cyclin-dependent kinase inhibitors); sirtuin-1 (an antiaging enzyme that reduces telomeric attrition [19]); and DNA fragmentation (determined via TUNEL [terminal deoxynucleotidyl transferase dUTP nick end labeling]). The level of each molecule was quantified as described in the online supplement.
Telomere length and levels of mTERT (murine telomerase reverse transcriptase) (20) were measured by using a quantitative PCR method in the lung, kidney, and quadriceps (21). To evaluate the underlying molecular mechanisms of metformin-associated protection of the lung against CS-induced pathologies, we performed quantitative proteomics on lung protein lysates from both air- and CS-treated metformin-enriched chow–fed and metformin-free chow–fed groups. Proteomic data were obtained by using high-performance liquid chromatography– electrospray ionization–tandem mass spectrometry in a positive-ion mode on a Thermo Fisher Scientific Orbitrap Fusion Lumos tribrid mass spectrometer fitted with an EASY-Spray source. Progenesis QI proteomics software (Waters) was used to perform ion intensity–based, label-free quantification (22). Unbiased gene-annotation enrichment analysis of the proteins in the CS group that were significantly affected by metformin treatment was performed by using the high-throughput functional annotation tool of the Database for Annotation, Visualization, and Integrated Discovery (https://david.ncifcrf.gov/).
In light of the findings from the unbiased proteomic analyses, indicating a protective effect of metformin against the UPR and ER stress, we used RT-PCR analysis to assess the levels of the three main mediators of the UPRs, namely ATF-6 (activating transcription factor 6), PERK (protein kinase RNA–like ER kinase), and IRE1-α (serine/threonine–protein kinase/endoRNase inositol–requiring enzyme 1 α) and its downstream effector molecule XBP-1 (X-box binding protein 1) in the lungs from the four murine experimental groups, as well as the levels of the energy-sensor molecule GDF-15 (growth differentiation factor 15) (23), which is known to be a key regulator of UPRs (see Table E1 in the online supplement for the primers used). In addition, we assessed two crucial molecules in ER stress responses: EDEMs (ER-associated degradation–enhancing α-mannosidase–like proteins) (24), which function as sensors of UPR responses, and BIP (binding immunoglobulin protein; also known as GRP-78), an XBP-1–induced ER chaperone with antiapoptotic properties due to its ability to control the activation of transmembrane ER stress sensors (IRE1, PERK, and ATF6) (25).
The levels of P-AMPK (phospho-AMPK)/AMPK and P-ACC (phospho– acetyl-CoA carboxylase)/ACC (acetyl-CoA carboxylase) were evaluated by using Western blotting and immunofluorescence. The levels of Camkk2 (calcium/calmodulin-dependent protein kinase kinase 2, which activates AMPK by phosphorylating its α subunit [26]), Ulk1 (which mediates AMPK-induced autophagy in response to dynamic changes in cellular nutrients and energy levels [27]), and other mediators of the AMPK pathway (see the online supplement) were assessed by using quantitative PCR in murine lung lysates.
In Vitro Studies
The mitochondrial oxygen consumption rate (OCR) was measured in HBEC cultures in the presence of increasing concentrations of CS extract without or without metformin (Seahorse Bioscience XF96 Extracellular Flux Analyzer). Total and phosphorylated levels of AMPK, ACC, and mTOR (mammalian target of rapamycin),which is known to be inhibited by metformin (28), were also measured in HBEC lysates by using sandwich ELISAs.
Metformin and Emphysema in the COPDGene Study
The COPDGene study is a multicenter observational study intended to identify genetic factors associated with COPD incidence and progression, which enrolled non-Hispanic White or Black individuals, aged 45–80 years, with at least a 10–pack-year smoking history. Participants underwent whole-lung volumetric computed tomography (CT) at study entry and at Year 5, permitting longitudinal assessment of lung CT characteristics. Full details are reported elsewhere (29). All participants with COPD (defined by a post-bronchodilator FEV1/FVC ratio of <0.70) and usable CT scan information at baseline and follow-up were included in this analysis. We excluded participants whose smoking status changed between visits, as this has been shown to change CT imaging characteristics independent of emphysema (30). Participant-reported medication inventories were obtained at baseline and at the 5-year follow-up. Metformin use was defined as self-reported metformin use at baseline. The outcomes of interest were the percentage of emphysema, defined as the percentage of lung voxels with attenuation less than −950 Hounsfield units, and the adjusted lung density (ALD), defined as the average attenuation at the 15th percentile of lung voxels corrected for total lung volume (31). An individual with worsening emphysema would be expected to have an increasing percentage of emphysema and a decreasing ALD.
Statistics
For the murine and in vitro studies, data were analyzed by using one-way ANOVA, Holm-Sidak, or Dunn methods for multiple group comparisons, which were followed by pairwise testing by using SigmaPlot (Systat Software Inc.). Data not normally distributed are presented as the median and interquartile range, and pairwise testing was performed by using the Mann-Whitney U test. Normally distributed data are presented as the mean and SEM, and pairwise testing was performed by using the Student’s t test.
To estimate the association of metformin use and changes in emphysema in COPDGene, we constructed linear mixed models with random intercepts by CT scanner model and study site by using an unstructured covariance matrix to account for repeated measures. The change in the percentage of emphysema and ALD attributable to metformin use was represented by a treatment-by-time interaction. We adjusted for covariates associated with emphysema progression and other potential confounders: age, sex, race, time-varying body mass index, use of ACE (angiotensin-converting enzyme) or angiotensin II receptor blockers (32), comorbidity count (33), and time-varying CT scanner model. Because smoking is anticipated to confound the metformin–emphysema association, we included a dual adjustment for the smoking status and time-varying pack-years of smoking. To investigate whether associations were attributable to metformin specifically or were relevant to other medications targeting glycemic control, we performed sensitivity analyses introducing concurrent sulfonylurea or insulin use (and their interaction with time) into the model. An additional sensitivity analysis was performed comparing participants who reported metformin use at both baseline and the 5-year follow-up with those who did not report metformin use at either visit. A two-sided P value of <0.05 was considered to indicate statistical significance.
Results
Animal Studies
Metformin-enriched chow–fed mice exposed to CS for 6 months had a greater alveolar internal surface area (Figures 1A and 1B) and a reduced mean linear intercept (Figure E2A) compared with CS-exposed, metformin-free chow–fed mice, even when adjusting for increasing extents of lung deflation (Figure E2B). Metformin protected against CS-induced extracellular matrix deposition around the small airways (Figures 1C and 1D). Metformin did not have a significant effect on the lung function of air- or CS-exposed, metformin-enriched chow–fed mice compared with air- or CS-exposed, metformin-free chow–fed mice when looking at both the pressure–volume loops (Figure E2C and E2D) and the quasistatic lung compliance (Figure E2E).
Figure 1.
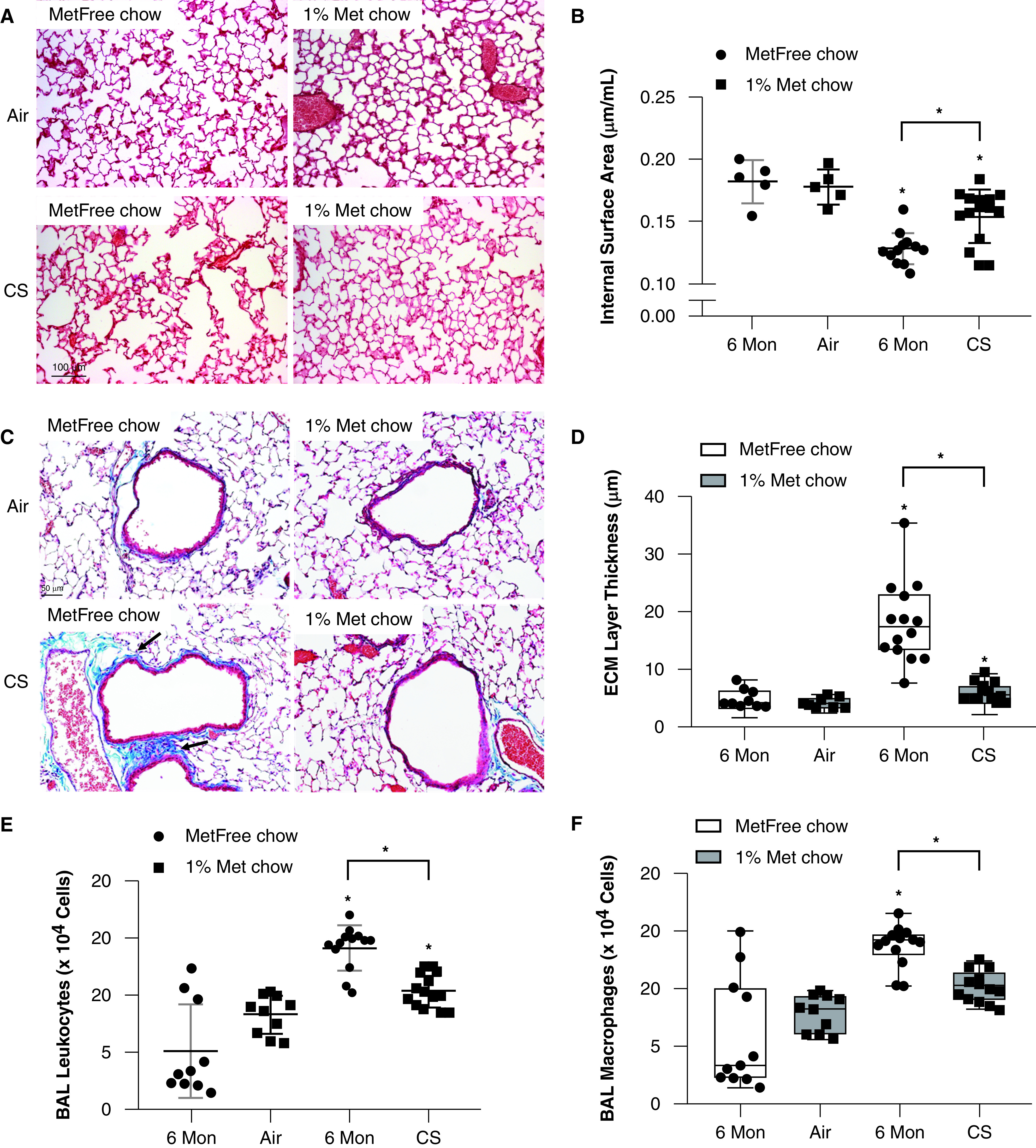
Metformin ameliorates the development of airspace enlargement, small airway remodeling, and inflammation in the lungs of mice exposed to cigarette smoke (CS). Wild-type mice were exposed to air or CS for 24 weeks and fed metformin-free (MetFree) or metformin-enriched (Met) chow during the last 12 weeks of exposure. (A) The lung parenchyma of Met chow–fed, CS-exposed mice showed milder airspace enlargement than that of the MetFree chow–fed, CS-exposed mice. Scale bars: CS, 50 μm; air, 100 μm. (B) Metformin treatment was protective against CS-induced decreases in the internal surface area quantified in formalin-fixed, paraffin-embedded Gill-stained lung sections and adjusted for the total lung volume (5 air-exposed mice/group and 13–15 CS-exposed mice/group). N = 5–15 mice/group. (C) Representative images of extracellular matrix (ECM) deposition around small airways (in bright blue color; see arrows) in Masson’s Trichrome–stained lung sections. Scale bar, 50 μm. (D) The thickness of the ECM protein layer deposited around small airways (300–799 μm in diameter) was significantly lower in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. (E and F) The numbers of total leukocytes and macrophages quantified in BAL samples were lower in MetFree chow–fed, CS-exposed mice than in Met chow–fed, CS-exposed mice. *P < 0.05 for each time point indicated. Mon = months.
Metformin was also protective against CS-induced increases in BAL-fluid total leukocyte (Figure 1E), macrophage (Figures 1F and E3A), and neutrophil (Figures E3B and E3C) counts. Lymphocyte counts were not affected by CS exposure (Figures E3D and E3E).
In addition, metformin ameliorated the CS-induced pulmonary oxidative stress levels (Figures 2A, 2B, and 2D) and protected against CS-induced alveolar epithelial-cell apoptosis (Figures 2C–2F) and accelerated aging (Figures 3A–3E). In the lung, metformin protected against CS-induced decreases in mTERT mRNA levels and increases in γ-H2AX (phosphorylated form of H2AX), which is associated with the induction of DNA double-strand breaks as a result of oxidative stress and aging (Figures 3F–3H) (34).
Figure 2.

Metformin protects the lung against cigarette smoke (CS)-induced apoptosis and oxidative stress. Wild-type mice were exposed to air or CS for 24 weeks and were fed metformin-free (MetFree) or metformin-enriched (Met) chow during the last 12 weeks of exposure. (A and B) The levels of MDA and 4-hydroxy-2-nonenal (4-HNE) were lower in CS-exposed, Met chow–fed mice than in CS-exposed, MetFree chow–fed mice, respectively. (C) The number of cells positive for the apoptosis marker caspase-3 normalized for squared micrometers of lung tissue was lower in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. (D) Representative double-immunofluorescence pictures of pulmonary alveoli positive for 4-HNE (green arrows), caspase-3 (red arrows), or both (yellow arrows). N = 5–15 mice/group. Scale bar, 50 μm. (E and F) The number of alveolar cells with DNA fragmentation (see arrows) as determined by using a TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay and normalized for cells/μm2 of lung tissue was higher in MetFree chow–fed, CS-exposed mice than in Met chow–fed, CS-exposed mice. Scale bar, 100 μm. *P < 0.05 versus the air group that received the same chow or versus the group indicated. MDA = malondialdehyde; Mon = months.
Figure 3.

Metformin protects the lung against cigarette smoke (CS)-induced aging and telomere damage. Wild-type mice were exposed to air or CS for 24 weeks and were fed metformin-free (MetFree) or metformin-enriched (Met) chow during the last 12 weeks of exposure. (A and B) The numbers of alveolar cells/μm2 of alveolar tissue expressing the aging markers p16 and p21 were lower in CS-exposed, Met chow–fed mice than in CS-exposed, MetFree chow–fed mice, respectively. (C) Representative double-immunofluorescence pictures showing a lower number of cells positive for the aging markers p16 (green arrows), p21 (red arrows), or both (yellow arrows) in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. Scale bar, 50 μm. (D) The number of alveolar cells/μm2 of alveolar tissue expressing the prosurvival marker sirtuin-1 was higher in CS-exposed, Met chow–fed mice than in CS-exposed, MetFree chow–fed mice. (E) Representative immunofluorescence pictures showing a higher number of cells positive for sirtuin-1 (arrows) in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. Scale bar, 50 μm. (F) The mRNA mTERT (murine telomerase reverse transcriptase) levels quantified by using RT-PCR analysis were higher in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. (G) The number of alveolar cells/μm2 of alveolar tissue expressing γ-H2AX (the phosphorylated form of H2AX [γ-H2A.X variant histone]) was higher in CS-exposed, MetFree chow–fed mice than in Met chow–fed mice. (H) Representative immunofluorescence pictures showing a higher number of cells positive for γ-H2AX (arrows) in MetFree chow–fed, CS-exposed mice than in Met chow–fed, CS-exposed mice. Scale bar, 50 μm. *P < 0.05 versus the air group that received the same chow or versus the group indicated. Mon = months.
CS exposure in mice induces both glomerular and tubular kidney damage (17). Metformin was protective against CS-induced glomerular shrinkage (Figure 4A and 4B), oxidative stress (Figures 4C, 4D, and 4F), apoptosis (Figures 4E–4H), and decreases in the level of sirtuin-1 (Figures 4I and 4L) in both glomerular and tubular renal cells.
Figure 4.
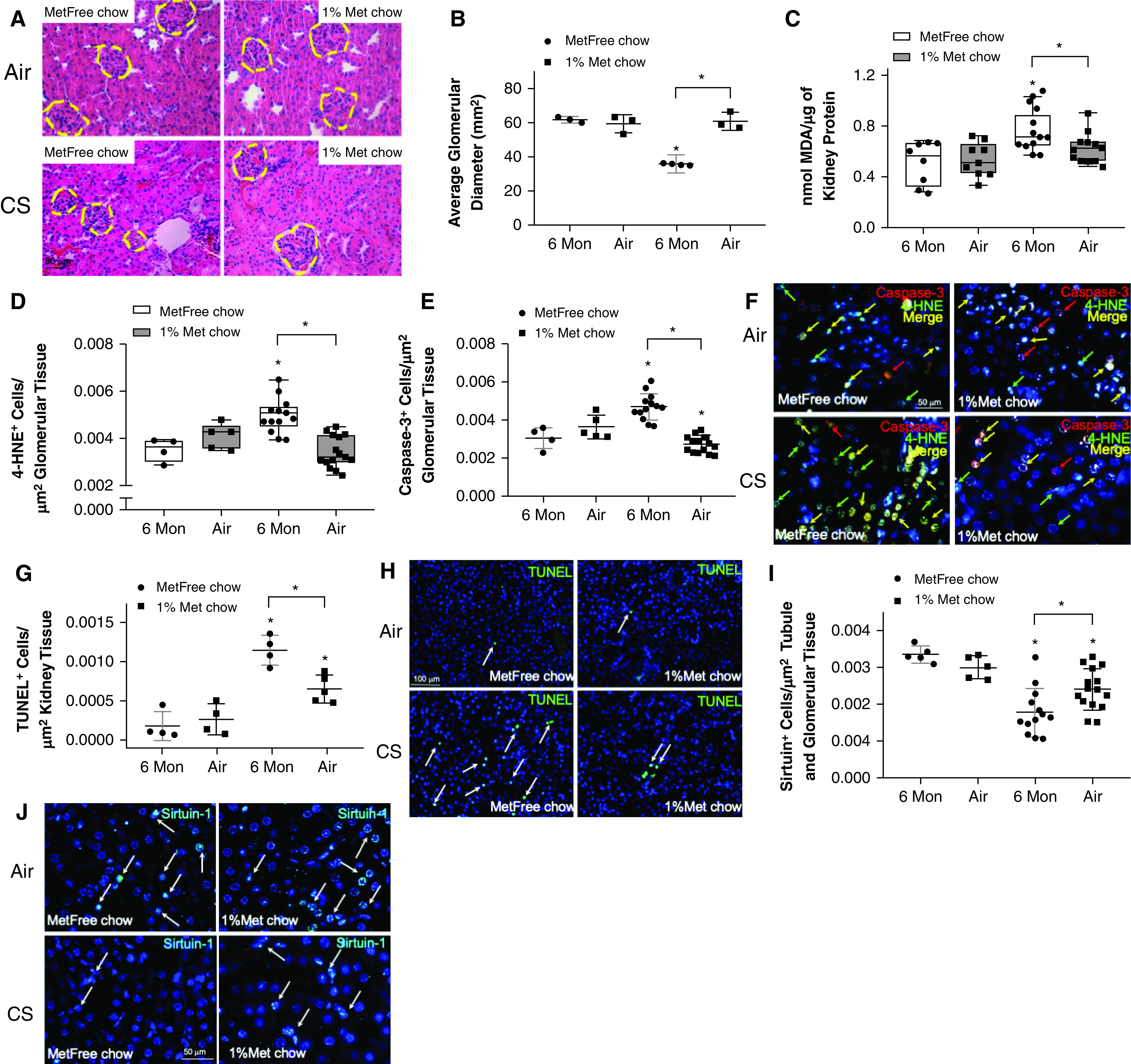
Metformin protects against cigarette smoke (CS)-induced glomerular shrinkage by reducing oxidative stress, apoptosis, and aging in the kidneys. Wild-type mice were exposed to air or CS for 24 weeks and were fed metformin-enriched (Met) or metformin-free (MetFree) chow during the last 12 weeks of exposure. (A) Representative images of glomeruli in hematoxylin and eosin–stained kidney sections. Met chow–fed mice were protected against CS-induced glomerular shrinkage compared with MetFree chow–fed, CS-exposed mice. Scale bar, 50 μm. (B) The glomerular diameters were quantified in 3–4 mice/group. (C–E) The levels of MDA and the numbers of alveolar cells expressing 4-hydroxy-2-nonenal (4-HNE) and caspase-3 normalized for cells/μm2 of renal tissue were lower in CS-exposed, Met chow–fed mice than in CS-exposed, MetFree chow–fed mice, respectively. (F) Representative double-immunofluorescence pictures showing a lower number of cells positive for caspase-3 (red arrows), 4-HNE (green arrows), or both (yellow arrows) in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. Scale bar, 50 μm. (G and H) The number of cells with DNA fragmentation (see arrows) as determined by using a TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay and normalized for cells/μm2 of renal tissue was higher in MetFree chow–fed, CS-exposed mice than in Met chow–fed, CS-exposed mice. Scale bar, 100 μm. (I) The number of cells/μm2 of renal tissue expressing sirtuin-1 was higher in CS-exposed, MetFree chow–fed mice than in CS-exposed, Met chow–fed mice. (J) Representative immunofluorescence pictures showing a higher number of cells positive for sirtuin-1 (arrows) in MetFree chow–fed, CS-exposed mice than in Met chow–fed, CS-exposed mice. Scale bar, 50 μm. *P < 0.05 versus the air group that received the same chow or versus the group indicated. MDA = malondialdehyde; Mon = months.
Metformin protected against CS-induced reductions of the sirtuin-1 level in quadriceps myocytes (Figures 5A and 5B), whereas CS-exposed, metformin-enriched chow–fed mice had longer telomeres in their quadriceps (Figure 5C) and lower levels of γ-H2AX than CS-exposed, metformin-free chow–fed mice (Figures 5D and 5E). Importantly, metformin partially protected against CS-induced loss of type I peripheral muscle fibers (Figures 5F and 5G) (18). The number of caspase-3–positive quadriceps cells was lower in CS-exposed, metformin-enriched chow–fed mice than in CS-exposed, metformin-free chow–fed mice (Figure E4A). However, no difference in 4-hydroxy-2-nonenal–positive quadriceps cells was found between these two groups (Figure E4B).
Figure 5.

Metformin protects against cigarette smoke (CS)-induced skeletal muscle aging and waste by preserving quadriceps telomere length and type I fibers. (A) The number of cells/μm2 of peripheral muscle tissue expressing the prosurvival marker sirtuin-1 was higher in CS-exposed, metformin-enriched (Met) chow–fed mice than in metformin-free (MetFree) chow–fed mice. (B) Representative immunofluorescence pictures showing a higher number of cells positive for sirtuin-1 (arrows) in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. Scale bar, 50 μm. (C) The quadriceps telomere length was higher in Met chow–fed, CS-exposed mice than in MetFree chow–fed, CS-exposed mice. (D) The number of cells/μm2 of peripheral muscle tissue expressing γ-H2AX (the phosphorylated form of H2AX [γ-H2A.X variant histone]) was higher in CS-exposed, MetFree chow–fed mice than in CS-exposed, Met chow–fed mice. (E) Representative immunofluorescence pictures showing a higher number of cells positive for γ-H2AX (arrows) in MetFree chow–fed, CS-exposed mice than in Met chow–fed, CS-exposed mice. Scale bar, 50 μm. (F) CS-exposed, Met chow–fed mice had a greater number of reduced nicotinamide adenine dinucleotide (NADH) muscle fibers (arrows in G) than CS-exposed, MetFree chow–fed mice. (G) The NADH staining of type I and type II muscle fibers in formalin-fixed, paraffin-embedded skeletal muscle sections. The type I fibers stain with dark blue, and the type II fibers stain with light blue. Met chow–fed mice were protected against CS-induced loss of type I fibers compared with MetFree chow–fed, CS-exposed mice. Scale bar, 50 μm. *P < 0.05 versus the air group that received the same chow or versus the group indicated. Mon = months.
A volcano plot comparison of the proteomic analyses of lungs from metformin-enriched chow–fed and metformin-free chow–fed mice (Figure 6A) showed significant differences in the mean expression of several proteins as a result of metformin treatment, highlighting a protective effect of metformin against CS-induced autophagy and mitochondrial dysfunction in the lung. Unbiased gene-annotation enrichment analysis of the proteins in the CS-exposed group that were significantly affected by metformin treatment confirmed that within the Gene Ontology class biological processes as well as the Kyoto Encyclopedia of Genes and Genomes pathways, several pathways associated with metformin in CS-exposed mice were related to mitochondrial function, findings that were distinct from those associated with the other pairwise comparisons (Figure 6B, blue font; Figure 6C; and Figure E5). The second pathway that was most strongly associated with the use of metformin in CS-exposed mice was related to ER function and the UPR (see Figure 6B, orange font, and Figure E5).
Figure 6.

Proteomic analyses of proteins associated with metformin-induced protection of the lung against cigarette smoke (CS)-induced mitochondrial damage and endoplasmic reticulum (ER) stress. (A) A volcano plot shows the log2 fold change plotted against log10-adjusted P values for CS-exposed versus CS-exposed and metformin-treated (CSM) samples for all significantly affected proteins, with mitochondria-associated proteins being highlighted in blue and ER proteins being shown in orange. The horizontal line represents the cutoff for a P value of <0.05, and the two vertical lines represent the cutoff values of 1.5-fold change in either the positive or negative direction. (B) Scatter plot of the GO biological processes category showing enrichment findings for the significantly affected proteins of the different pairwise comparisons. CS is used to indicate CS exposure alone. (C) Scatter plot of the mitochondrial proteins significantly affected by metformin treatment. The vertical dashed green line represents the cutoff P value of <0.05. n = 4 per group. (D) Scatter plot of the mitochondrial proteins associated with COPD pathogenesis and CS-induced lung damage with proteins significantly affected by metformin treatment highlighted. The vertical dashed green line represents the P cutoff value of < 0.05. n = 4 per group. The entire list of proteins associated with CS exposure and metformin treatment is available in the online supplement. A = air-exposed, metformin-free chow–fed mice; AM = air-exposed, metformin-enriched chow–fed mice; COPD = chronic obstructive pulmonary disease; GO = Gene Ontology.
Furthermore, unlike CS-exposed and metformin-free chow–fed mice, CS-exposed and metformin-enriched chow–fed mice had a predominant expression pattern of mitochondrial proteins known to be associated with COPD and the AMPK pathway, autophagy, pulmonary inflammation, and oxidative stress (Figure 6D) that was more similar to that of the air-exposed groups. See Tables E2–E4 for the complete list and function of proteins found to be affected by metformin treatment.
A protective action of metformin against CS is likely mediated by AMPK. CS increased P-AMPK (Figure 7A and E6A), which was accompanied by increased expression of Camkk2, which is known to phosphorylate and activate AMPK (Figure 7B), and the downstream molecule Ulk1, which is implicated in autophagy (Figure 7C). Interestingly, we observed that CS-exposed, metformin-enriched chow–fed mice had decreased AMPK phosphorylation, and Camkk2 and Ulk1, as compared with the control group (Figures 7A–7C). These findings suggest that metformin normalizes the AMPK phosphorylation and related autophagy signaling pathways. It is also possible that CS alone can increase expression of Ulk1 and Camkk2, independently of AMPK activation (35, 36). Indeed, additional experiments revealed that phosphorylation of ACC, as a downstream target of AMPK and regulator of fatty acid oxidation, was increased in the lungs from air-exposed, metformin-enriched chow–fed, air-exposed mice compared with metformin-free chow–fed, air-exposed mice but was not significantly different between metformin-free chow–fed, CS-exposed mice and metformin-enriched chow–fed, CS-exposed mice (Figures E6B–E6D).
Figure 7.
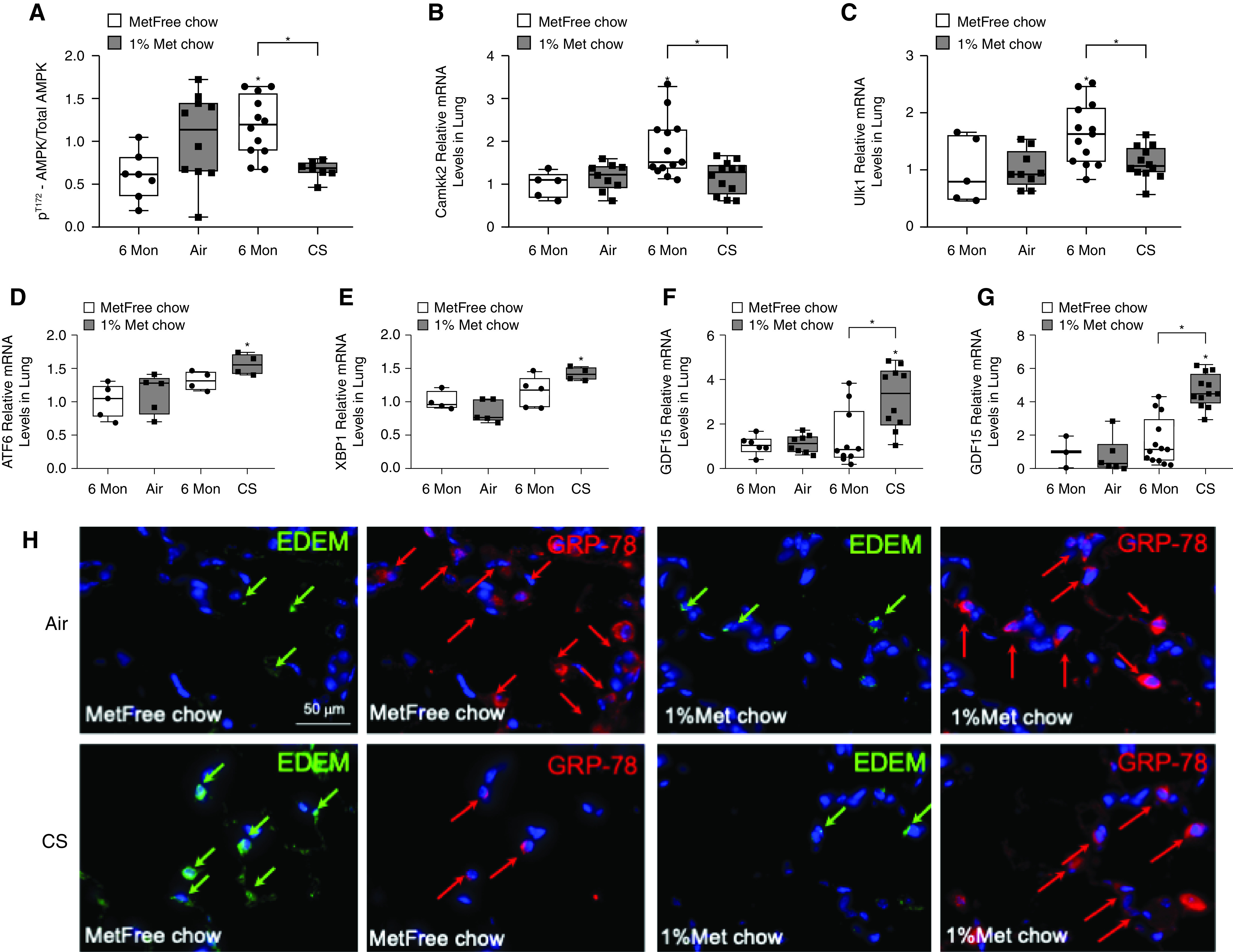
Metformin effects in cigarette smoke (CS)-exposed mice are associated with changes in components of the AMPK (AMP-activated protein kinase) pathway and in key stress sensors regulating unfolded protein responses (UPRs) and endoplasmic reticulum (ER) stress. (A) Western blot analyses of the lung homogenates were performed, and the levels of phosphoT172-AMPK/total AMPK were quantified and found to be decreased in CS-exposed, metformin-enriched (Met) chow–fed mice compared with CS-exposed, metformin-free (MetFree) chow–fed mice. Pulmonary (B) Camkk-2 (calcium/calmodulin-dependent protein kinase kinase 2) and (C) Ulk-1 levels were quantified by using RT-PCR analysis in the four experimental groups of mice and were found to be decreased in CS-exposed, Met chow–fed mice compared with CS-exposed, MetFree chow–fed mice. (D and E) The mRNA levels of the UPR sensors ATF-6 (activating transcription factor 6) and XBP-1 (X-box binding protein 1) were higher in CS-exposed, Met chow–fed mice than in CS-exposed, MetFree chow–fed mice and (F and G) were associated with higher gene expression levels of GDF15 (growth differentiation factor 15), a rescue cytokine that is expressed during cellular stress, in both the lungs and the kidneys, respectively. (H) Double-immunofluorescence staining for ER stress sensors, EDEMs (ER-associated degradation–enhancing α-mannosidase–like proteins) (green arrows) and BIP (binding immunoglobulin protein, also known as GRP-78) (red arrows) showing a decrease in EDEMs and an increase in GRP-78 in CS-exposed, Met chow–fed mice compared with CS-exposed, MetFree chow–fed mice. Scale bar, 50 μm. *P < 0.05 versus the air group that received the same chow or versus the group indicated. Mon = months.
On the basis of the new findings from the unbiased proteomic analyses, we assessed the effect of metformin on key regulatory molecules of UPRs and ER stress. In particular, we assessed the levels of the three main mediators of UPRs, namely ATF-6, PERK, and IRE1-α and its downstream effector molecule XBP-1 (37), in the lungs and observed a significative upregulation of the ATF-6 arm and XBP-1 in CS-exposed, metformin-enriched chow–fed mice compared with CS-exposed, metformin-free chow–fed mice (Figures 7D and 7E). These metformin-induced changes were associated with increases in another key metabolic stress–induced sensor, GDF-15, which is a rescue cytokine that is expressed during cellular stress (38) and is known to augment, upon stressful stimuli, the expression of ATF-6 and XBP-1 as transcriptional stress markers (39) (Figures 7F and 7G). Furthermore, lower levels of EDEMs and higher levels of GRP-78 were observed in the lungs of CS-exposed, metformin-enriched chow–fed mice compared with CS-exposed, metformin-free chow–fed mice (Figure 7H), confirming a protective action of metformin against UPR stress responses and ER stress. No changes in PERK and IRE1-α (Figures E7A and E7B, respectively) were found across treatment groups.
We monitored the weight of mice and observed that both air-exposed metformin-free chow–fed mice and CS-exposed metformin-free chow–fed mice substantially increased their weight during the 6-month exposures compared with their initial weight. As expected, CS-exposed, metformin-free chow–fed mice gained less weight than air-exposed, metformin-free chow–fed mice. In contrast, both air-exposed, metformin-enriched chow–fed mice and CS-exposed metformin-enriched chow–fed mice maintained their weight throughout the exposures (Figure E8).
In Vitro HBECs
Subsequent experiments tested the effects of metformin and CS in lung epithelial cells in vitro. Simultaneous treatment with metformin protected the HBECs from CS-induced decreases in the uncoupled OCR (FCCP-OCR) in a concentration-dependent fashion (Figure E9A). The FCCP-OCR is the maximal rate of the electron transport chain and is a sensitive marker of mitochondrial dysfunction (40). In CS-treated cells compared with untreated control cells, CS mediated a decline in the FCCP-OCR, which was associated with decreased AMPK phosphorylation. Notably, inclusion of metformin diminished such adverse effects caused by CS extract (Figure E9A and E9B) by preventing the decrease in the FCCP-OCR in CS-treated cells, but inclusion of metformin did not prevent the decrease in AMPK phosphorylation (Figure E9C). Additional experiments confirmed that metformin diminished the phosphorylation (activation) of mTOR in CS-treated HBECs (Figure E9D). These results indicate that metformin may prevent bioenergetic dysfunction in CS-treated HBECs.
Metformin and Emphysema in the COPDGene Study
Out of 3,804 participants who provided baseline data, 1,687 (44%) attended the follow-up visit and formed our analytic population (Table 1). In comparison with those who only attended the baseline visit, included participants had less severe COPD, a lower intensity of smoking, and a higher body mass index; notably, 42% of those who only attended the baseline visit had died before Year 5 (Table E5). Within the analytic population, baseline metformin users were older, heavier, and had more comorbidities. However, they were less likely to be current smokers, had a lower percentage of emphysema, and had a higher ALD at baseline, but they had no difference in terms of COPD severity. At 5-year follow-up, emphysema progressed in both metformin users and nonusers (Table 2). However, compared with metformin nonusers, metformin users experienced a slower progression in the percentage of emphysema (adjusted difference-in-difference of −0.92%; 95% confidence interval [CI], −1.7% to −0.14%) and experienced an attenuation of the decrease in ALD (adjusted difference-in-difference of 2.2 g/L; 95% CI, 0.43 to 4.0 g/L) at 5 years (Table 2). The losses in the FEV1 in metformin nonusers and users were −2 (95% CI, −2.6 to −1.5) ml/yr and −0.66 (95% CI, −2.9 to 1.6) ml/yr, respectively, with a slope difference of 1.4 (95% CI, −0.9 to 3.7) ml/yr being shown. These results were also consistent in a sensitivity analysis of 1,540 participants who reported persistent metformin use or nonuse at both visits (Table 2). There was no association between sulfonylurea or insulin use and the change in the percentage of emphysema or the ALD, and point estimates for metformin use were not qualitatively different with concurrent adjustment for these antidiabetic agents (Table E6).
Table 1.
Baseline Characteristics of Included Participants from COPDGene (n = 1,687)
| Characteristic | Metformin Nonusers (n = 1,572) [n (%) or Mean ± SD] | Metformin Users (n = 115) [n (%) or Mean ± SD] | P Value |
|---|---|---|---|
| Age, yr | 63 ± 8.4 | 65.6 ± 7.1 | <0.01 |
| Sex, F | 740 (47) | 40 (35) | 0.01 |
| Black race | 310 (20) | 17 (15) | 0.2 |
| GOLD stage | 0.1 | ||
| GOLD 1 (FEV1 ⩾ 80%) | 371 (24) | 19 (17) | |
| GOLD 2 (50% ⩽ FEV1 < 80%) | 760 (48) | 61 (53) | |
| GOLD 3 (30% ⩽ FEV1 < 50%) | 355 (23) | 32 (28) | |
| GOLD 4 (FEV1 < 30%) | 86 (6) | 3 (3) | |
| Post-bronchodilator FEV1pp, % | 64 ± 21 | 62 ± 19 | 0.4 |
| Pack-years of smoking at baseline | 64 ± 9.0 | 63 ± 8 | 0.3 |
| Current smoker | 579 (37) | 28 (24) | <0.01 |
| Additional pack-years at follow-up | 3.6 ± 2.6 | 4.3 ± 2.6 | 0.1 |
| Body mass index, kg/m2 | 28 ± 5.6 | 31.8 ± 5.6 | <0.01 |
| Comorbidity count* | 2.3 ± 1.7 | 4.2 ± 1.6 | <0.01 |
| ACE/ARB use | 394 (25) | 78 (68) | <0.01 |
| Baseline CT characteristics | |||
| Percentage of emphysema | 10.2 ± 10.6 | 7.3 ± 8.2 | <0.01 |
| Adjusted lung density, g/L | 73.8 ± 25.1 | 79 ± 21.4 | 0.03 |
Definition of abbreviations: ACE/ARB = angiotensin-converting enzyme/angiotensin II receptor blocker; COPDGene = Genetic Epidemiology of Chronic Obstructive Pulmonary Disease; CT = computed tomography; GOLD = Global Initiative for Obstructive Lung Disease; FEV1pp = FEV1% predicted.
Count of coronary artery disease, congestive heart failure, diabetes, stroke, high blood pressure, and high cholesterol.
Table 2.
Association of Metformin Use with Markers of Emphysema Progression within COPDGene
| Outcome | Difference between Follow-up and Baseline |
Difference-in-Difference* | P Value | |
|---|---|---|---|---|
| Metformin Users [Units (95% CI)] | Metformin Nonusers [Units (95% CI)] | |||
| Overall cohort (n = 1,687) | ||||
| Percentage of emphysema | 1.4 (0.62 to 2.2) | 2.3 (2.1 to 2.6) | −0.92 (−1.7 to −0.14) | 0.02 |
| Adjusted lung density, g/L | −2.7 (−4.5 to −0.97) | −5.0 (−5.5 to −4.4) | 2.2 (0.43 to 4.0) | 0.01 |
| Persistent use (n = 1,540)† | ||||
| Percentage of emphysema | 0.87 (−0.06 to 1.8) | 2.4 (2.1 to 2.7) | −1.5 (−2.5 to −0.59) | <0.01 |
| Adjusted lung density, g/L | −2.9 (−4.6 to −0.38) | −5.1 (−5.7 to −4.5) | 2.6 (0.48 to 4.7) | 0.02 |
Definition of abbreviations: CI = confidence interval; CT = computed tomography; COPDGene = Genetic Epidemiology of Chronic Obstructive Pulmonary Disease.
Adjusted for age, sex, race, smoking status, use of an ACE (angiotensin-converting enzyme) or angiotensin II receptor blocker, comorbidity count, time-varying body mass index, CT scanner model, and pack-years of smoking.
Subset of participants who were consistent users or nonusers of metformin at baseline and follow-up.
Discussion
To our knowledge, this is the first comprehensive analysis of the effects of metformin in patients with COPD and in an experimental model of CS exposure. In a murine model, we confirmed that metformin administered to mice 3 months after beginning a 6-month exposure to CS effectively protects mice from progressive CS-induced pathologies. In addition to benefitting the lungs, metformin was also observed to provide benefits to the kidney and skeletal muscles. Our mechanistic studies revealed that metformin improved mitochondrial function linked to the activation of AMPK and GDF-15, which are among the major bioenergetic sensors and metabolic regulators in the lungs and other organs, leading to an amelioration of mitochondrial function, UPRs, and ER stress. The observation that participants enrolled in the COPDGene study who were receiving metformin had a slower progression of emphysema than those who were not receiving metformin supports the concept that the benefit of metformin may extend to humans.
Metformin Effects In Vivo in CS-exposed Mice
We found that metformin effectively diminishes the development of CS-induced pulmonary airspace enlargement, oxidative stress, alveolar epithelial cell accelerated aging, pulmonary inflammation, telomere damage, and SAR in mice. Metformin is an established activator of AMPK, although the precise mechanisms of its action remain to be determined. Once activated, AMPK is beneficial in protecting and restoring bioenergetic homeostasis under conditions of limited oxygen and nutrient bioavailability, such as during CS exposure, that increase the intracellular AMP/ATP ratio (41). This also includes bioenergetic imbalances that are characterized by an elevated intracellular AMP/ATP ratio (41), as occurs after exposure to CS. Recent studies have suggested that AMPK activation can reduce CS-induced lung inflammation, cellular senescence, oxidative stress, and emphysema (42). In experimental models with metformin treatment, AMPK activation has been linked to caloric restriction (43), resulting in reductions in both the accumulation of oxidative damage and the chronic inflammation that adversely affect the lifespan (13). These observations are consistent with the benefits of AMPK activation in acute lung injury models, with benefits including reductions in the airway inflammation, airway remodeling, and small airway extracellular matrix deposition that are known to be linked with aging.
Although metformin-dependent activation of AMPK is well documented, it is important to note that the impact of CS alone on AMPK activity is less consistent, particularly when comparing in vivo and in vitro models (42, 44). For example, CS alone has been shown to increase the expression of the transcriptional regulators of AMPK signaling, including Ulk1 and Camkk2, even if downstream AMPK activation is suppressed (35, 36). Of note, AMPK pathway activation is observed upon bioenergetic alterations and oxidative stress in the lungs of CS-treated mice (45). Hereby, we demonstrate for the first time that metformin effectively protects against CS-induced activation of AMPK and its downstream signaling, including Ulk1-related autophagy/mitophagy in vivo.
The proteomic data in our study (Figure 6) confirmed that metformin ameliorates CS-induced impairment of the lung bioenergetics and mitochondrial function and reduces the CS-induced proteostasis imbalance secondary to oxidative stress and inflammation. In particular, an unbiased gene-annotation enrichment proteomic analysis revealed that the pathways that were most associated with the protective effects of metformin use in CS-exposed mice were related to mitochondrial function (protein shuttles involved in substrate mobilization between the cytosol and mitochondria, increasing the substrate for mitochondrial metabolism) and UPRs and ER stress (ER–ribosome complex proteins). We also observed a significant increase in pulmonary and renal GDF-15 in CS-exposed mice receiving metformin compared with the irregular variation observed in the control animals (Figure 7). This suggests that critical levels of noxious stressors triggered by CS are necessary for the metformin to promote increases in levels of GDF-15. Although conflicting data exist regarding the role of GDF-15 (46), a recent study has notably shown that under stressful conditions, metformin elevates the circulating levels of GDF-15, therefore increasing glucose metabolism and decreasing the oxidative stress associated with aging (38). The increase in the GDF-15 levels was accompanied by an increase in the levels of the energy and stress sensors that regulate UPRs and ER stress (ATF-6, XBP-1, and GRP-78) and by a decrease in the ER stress marker EDEM. These data, together with an improved mitochondrial OCR in CS-exposed mice and HBECs, show that metformin improves cellular energy production by ameliorating mitochondrial metabolism and decreasing ER stress in CS-exposed lungs.
Chronic kidney disease is an important COPD comorbidity that is worsened by COPD exacerbations. Chronic CS exposure causes oxidative stress and apoptosis, an increased production of advanced glycosylation end products and RAGE (receptor for advanced glycosylation end products) in the renal epithelium, and glomeruli shrinkage, which causes microalbuminuria, in mice (17). We now show that metformin protects the kidneys from CS-induced oxidative stress, apoptosis, and aging in glomerular and tubular cells (Figure 4).
Functional impairment from sarcopenia, frequently present in patients with advanced emphysema, is associated with an accumulation of intramuscular CO2, resulting in AMPK activation and muscular hypometabolism (47). Our results show that metformin administration is not only associated with longer telomeres but is also associated with higher levels of sirtuin-1, which could contribute to the preservation of the critical mass of oxidative fibers that would otherwise be wasted in response to CS exposure. These results suggest that metformin could be helpful in patients with sarcopenia.
A stable weight is one indicator of metabolic health. Both air- and CS-exposed mice treated with metformin gained a small amount of weight for the entire 6 months, whereas metformin-free chow–fed mice increased their body weight by 50% over the same time frame, even though the animals had already reached their adult body weight.
Metformin Effects in HBECs In Vitro and Epidemiological Support for an Effect In Vivo
We show for the first time that metformin protects against CS-induced decreases in the mitochondrial FCCP-OCR in HBECs. Similar beneficial effects of metformin on the preservation of mitochondrial function have been shown in conditions associated with inflammatory lung injury and in fibrosis models of the lung and other organs (48). This is possible, as metformin-dependent activation of the AMPK signaling pathway promotes mitochondrial fission, biogenesis, and respiration and restores the mitochondrial life cycle in cultured cells and in vivo (48). Interestingly, although AMPK is a bioenergetic sensor and regulator of metabolism, we found that a CS-mediated decline in the OCR in HBECs is paralleled by lowered AMPK activation and increased mTOR activity in vitro; these signaling events are effectively prevented by the inclusion of metformin, likely through metformin preventing CS-induced mitochondrial proton leakage (Figure E9B). It is possible that despite a reduced OCR in CS-treated lung epithelial cells, AMPK activity is diminished via metabolic reprogramming (e.g., due to glycolytic flux). Another mechanism involved in the inhibition of AMPK is associated with inflammatory conditions that have been shown to reduce AMPK phosphorylation in vitro (49).
That the experimental observations we report could have translational applications to patients with emphysema is supported by the findings observed among participants with COPD within the COPDGene study, which indicate an association between metformin use and slower emphysema progression over 5 years of follow-up (Table 2). Furthermore, we have previously reported that metformin use is associated with improved respiratory function–related quality of life in the same cohort (50).
Limitations of the Study
We acknowledge that the animal experiments are not exactly representative of the therapeutic intervention that would guide its application in humans. However, the initiation of metformin 3 months after CS exposure best resembles the therapeutic approach to be considered in humans. In addition, our in vivo and in vitro findings suggest complex and yet understudied biological effects of metformin that can protect against the progression of CS-induced diseases; further studies are required to shed light on the mechanisms involved in such protection. In COPDGene, medication use was self-reported, and data on drug dosages and the duration of use were not recorded. Patients receiving metformin could have been using other confounding medications. However, as shown in Table 2, the difference in emphysema progression remained significant after adjusting for many potential confounders, including the use of ACE inhibitors. Our results may not be generalizable to participants with more severe COPD and a greater comorbidity burden, as these individuals were less likely to attend the follow-up visit, often because of mortality. Furthermore, the association between metformin use and emphysema progression may be confounded by healthy behaviors, as suggested by metformin users having a lower prevalence of smoking at baseline. This bias may be especially relevant to our sensitivity analysis among participants with persistent metformin use, although the results were qualitatively similar to those from the primary analysis. Overall, the longitudinal CT imaging in a well-characterized cohort of individuals with COPD provides a unique opportunity to examine the hypothesis that metformin ameliorates emphysema.
Conclusions
In this study, we show that in CS-exposed mice, the therapeutic administration of oral metformin results in the amelioration of airspace enlargement, pulmonary inflammation, SAR, peripheral muscle dysregulation, and kidney damage, with evidence of a slower aging process. The protective effects of metformin in the lung are associated with the amelioration of CS-induced mitochondrial metabolism impairment and ER stress (Figure 8). In human cells, metformin treatment improves the metabolic function disrupted by CS. The AMPK pathway is central to metformin’s protective action. Importantly, the use of metformin in selected patients in the COPDGene study is associated with a slower emphysema progression that is independent of confounding factors. For a disease with many comorbidities in which therapy is primarily limited to inhaled medications with limited systemic effects, the potential benefit provided by a relatively safe oral medicine seems compelling and worth pursuing in clinical trials.
Figure 8.

Potential protective effects of metformin against cigarette smoke (CS)-induced pathologies. In response to irritants from CS exposure, a series of events occurs in the lung. These events include the recruitment of inflammatory cells, cellular hypoxia, and the generation of excessive ROS, which lead to lung structural-cell apoptosis, accelerated lung aging, endoplasmic reticulum (ER) stress, and mitochondrial damage and impaired respiration. In turn, accelerated lung aging is associated with unfolded protein responses that lead to the accumulation of misfolded proteins in the cytoplasm that further increase inflammation and ER stress. All of these factors contribute to the development of emphysema. Metformin protects the lung against CS-induced pathologies in several ways: the green blocking sign indicates a facilitative effect of metformin, and the red blocking signs indicate an inhibitory effect from metformin. ROS = reactive oxygen species.
Acknowledgments
Acknowledgment
The authors thank Prof. Guy Brusselle for his intellectual contribution and valid suggestions during the entire study and thank Dr. Sara Cazorla for her help with the telomere assays.
Footnotes
Supported by the Flight Attendants’ Medical Research Institute (YFAC14004), the Parker B. Francis Foundation, the Asthma and Airway Disease Research Center Funds, and the NIH/NHLBI (HL149744 and HL132523) (F.P.). COPDGene (Genetic Epidemiology of Chronic Obstructive Pulmonary Disease [COPD]) is supported by the NHLBI (U01HL89897 and U01HL089856) and by the COPD Foundation through contributions made to an Industry Advisory Board comprising AstraZeneca, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion. T.D.W. is supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, and Center for Innovations in Quality, Effectiveness, and Safety (CIN 13-413). M.M. is supported by the National Institute of Environmental Health Sciences (P50ES018176), National Institute on Minority Health and Health Disparities (P50MD010431), and the U.S. Environmental Protection Agency (83615201 and 83615001). J.Z. is supported by the U.S. Department of Defense (W81XWH-17-1-0577) and the NIH (R01 HL139617-01).
Author Contributions: F.P. and B.C. conceived the project and designed the experiments. F.P., T.D.W., J.R.-Q., X.W., J.M., M.T., J.T., S.P., D.Z., K.H.C., E.C.-L., A.F., G.L.K., C.P.H., K.D., M.S., J.Z., and P.R.L. conducted experiments. F.P., T.D.W., J.R.-Q., X.W., K.H.C., C.A.O., A.F., G.L.K., N.N.H., K.D., J.G.L., J.Z., J.K., P.R.L., R.S., I.R., M.M., and B.C. contributed to data analysis. F.P., T.D.W., J.R.-Q., X.W., D.Z., K.H.C., A.F., G.L.K., C.P.H., N.N.H., K.D., M.S., Y.T., J.G.L., C.C., J.Z., J.K., P.R.L., I.R., M.M., and B.C. contributed to data interpretation. F.P., T.D.W., A.F., G.L.K., M.S., C.P.H., M.M., and B.C. wrote the manuscript. All co-authors reviewed and approved the final manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202012-4510OC on May 25, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Celli BR, Wedzicha JA. Update on clinical aspects of chronic obstructive pulmonary disease. N Engl J Med. 2019;381:1257–1266. doi: 10.1056/NEJMra1900500. [DOI] [PubMed] [Google Scholar]
- 2. Regan EA, Lynch DA, Curran-Everett D, Curtis JL, Austin JH, Grenier PA, et al. Genetic Epidemiology of COPD (COPDGene) Investigators. Clinical and radiologic disease in smokers with normal spirometry. JAMA Intern Med. 2015;175:1539–1549. doi: 10.1001/jamainternmed.2015.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Celli BR, Locantore N, Tal-Singer R, Riley J, Miller B, Vestbo J, et al. ECLIPSE Study Investigators. Emphysema and extrapulmonary tissue loss in COPD: a multi-organ loss of tissue phenotype. Eur Respir J. 2018;51:1702146. doi: 10.1183/13993003.02146-2017. [DOI] [PubMed] [Google Scholar]
- 4. Divo M, Cote C, de Torres JP, Casanova C, Marin JM, Pinto-Plata V, et al. BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186:155–161. doi: 10.1164/rccm.201201-0034OC. [DOI] [PubMed] [Google Scholar]
- 5. Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carnevali S, Petruzzelli S, Longoni B, Vanacore R, Barale R, Cipollini M, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284:L955–L963. doi: 10.1152/ajplung.00466.2001. [DOI] [PubMed] [Google Scholar]
- 7. Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33:1165–1185. doi: 10.1183/09031936.00128008. [DOI] [PubMed] [Google Scholar]
- 8. Kelsen SG. The unfolded protein response in chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13:S138–S145. doi: 10.1513/AnnalsATS.201506-320KV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. 2018;314:C73–C87. doi: 10.1152/ajpcell.00110.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Divo MJ, Celli BR, Poblador-Plou B, Calderón-Larrañaga A, de-Torres JP, Gimeno-Feliu LA, et al. EpiChron—BODE Collaborative Group. Chronic obstructive pulmonary disease (COPD) as a disease of early aging: evidence from the EpiChron cohort. PLoS One. 2018;13:e0193143. doi: 10.1371/journal.pone.0193143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195:557–582. doi: 10.1164/rccm.201701-0218PP. [DOI] [PubMed] [Google Scholar]
- 12. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009;182:8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laucho-Contreras ME, Taylor KL, Mahadeva R, Boukedes SS, Owen CA. Automated measurement of pulmonary emphysema and small airway remodeling in cigarette smoke-exposed mice. J Vis Exp. 2015:52236. doi: 10.3791/52236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Polverino F, Rojas-Quintero J, Wang X, Petersen H, Zhang L, Gai X, et al. A disintegrin and metalloproteinase domain-8: a novel protective proteinase in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;198:1254–1267. doi: 10.1164/rccm.201707-1331OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Polverino F, Laucho-Contreras ME, Petersen H, Bijol V, Sholl LM, Choi ME, et al. A pilot study linking endothelial injury in lungs and kidneys in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195:1464–1476. doi: 10.1164/rccm.201609-1765OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang D, Wang X, Li Y, Zhao L, Lu M, Yao X, et al. Thyroid hormone regulates muscle fiber type conversion via miR-133a1. J Cell Biol. 2014;207:753–766. doi: 10.1083/jcb.201406068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, et al. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci U S A. 2005;102:8222–8227. doi: 10.1073/pnas.0503095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Callicott RJ, Womack JE. Real-time PCR assay for measurement of mouse telomeres. Comp Med. 2006;56:17–22. [PubMed] [Google Scholar]
- 22. Parker SS, Krantz J, Kwak EA, Barker NK, Deer CG, Lee NY, et al. Insulin induces microtubule stabilization and regulates the microtubule plus-end tracking protein network in adipocytes. Mol Cell Proteomics. 2019;18:1363–1381. doi: 10.1074/mcp.RA119.001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinez CH, Freeman CM, Nelson JD, Murray S, Wang X, Budoff MJ, et al. COPDGene Investigators. GDF-15 plasma levels in chronic obstructive pulmonary disease are associated with subclinical coronary artery disease. Respir Res. 2017;18:42. doi: 10.1186/s12931-017-0521-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhattarai KR, Riaz TA, Kim HR, Chae HJ. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med. 2021;53:151–167. doi: 10.1038/s12276-021-00560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 26. Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, et al. Genetic Epidemiology of COPD (COPDGene) study design. COPD. 2010;7:32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ashraf H, Lo P, Shaker SB, de Bruijne M, Dirksen A, Tønnesen P, et al. Short-term effect of changes in smoking behaviour on emphysema quantification by CT. Thorax. 2011;66:55–60. doi: 10.1136/thx.2009.132688. [DOI] [PubMed] [Google Scholar]
- 31. Pompe E, Strand M, van Rikxoort EM, Hoffman EA, Barr RG, Charbonnier JP, et al. COPDGene Investigators. Five-year progression of emphysema and air trapping at CT in smokers with and those without chronic obstructive pulmonary disease: results from the COPDGene study. Radiology. 2020;295:218–226. doi: 10.1148/radiol.2020191429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parikh MA, Aaron CP, Hoffman EA, Schwartz JE, Madrigano J, Austin JHM, et al. The Multi-Ethnic Study of Atherosclerosis Lung Study. Angiotensin-converting inhibitors and angiotensin ii receptor blockers and longitudinal change in percent emphysema on computed tomography. Ann Am Thorac Soc. 2017;14:649–658. doi: 10.1513/AnnalsATS.201604-317OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Putcha N, Puhan MA, Drummond MB, Han MK, Regan EA, Hanania NA, et al. A simplified score to quantify comorbidity in COPD. PLoS One. 2014;9:e114438. doi: 10.1371/journal.pone.0114438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–1945. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pike LR, Singleton DC, Buffa F, Abramczyk O, Phadwal K, Li JL, et al. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J. 2013;449:389–400. doi: 10.1042/BJ20120972. [DOI] [PubMed] [Google Scholar]
- 36. Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287:31658–31665. doi: 10.1074/jbc.R112.356485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo FJ, Xiong Z, Lu X, Ye M, Han X, Jiang R. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell Signal. 2014;26:332–342. doi: 10.1016/j.cellsig.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 38. Coll AP, Chen M, Taskar P, Rimmington D, Patel S, Tadross JA, et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature. 2020;578:444–448. doi: 10.1038/s41586-019-1911-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gil CI, Ost M, Kasch J, Schumann S, Heider S, Klaus S. Role of GDF15 in active lifestyle induced metabolic adaptations and acute exercise response in mice. Sci Rep. 2019;9:20120. doi: 10.1038/s41598-019-56922-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12. doi: 10.1186/2049-3002-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 42. Lee JS, Park SJ, Cho YS, Huh JW, Oh YM, Lee SD. Role of AMP-activated protein kinase (AMPK) in smoking-induced lung inflammation and emphysema. Tuberc Respir Dis (Seoul) 2015;78:8–17. doi: 10.4046/trd.2015.78.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang CS, Li M, Ma T, Zong Y, Cui J, Feng JW, et al. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 2016;24:521–522. doi: 10.1016/j.cmet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 44. Morsch ALBC, Wisniewski E, Luciano TF, Comin VH, Silveira GB, Marques SO, et al. Cigarette smoke exposure induces ROS-mediated autophagy by regulating sestrin, AMPK, and mTOR level in mice. Redox Rep. 2019;24:27–33. doi: 10.1080/13510002.2019.1601448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Furlong HC, Stämpfli MR, Gannon AM, Foster WG. Cigarette smoke exposure triggers the autophagic cascade via activation of the AMPK pathway in mice. Biol Reprod. 2015;93:93. doi: 10.1095/biolreprod.115.132183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emmerson PJ, Duffin KL, Chintharlapalli S, Wu X. GDF15 and growth control. Front Physiol. 2018;9:1712. doi: 10.3389/fphys.2018.01712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Balnis J, Korponay TC, Jaitovich A. AMP-activated protein kinase (AMPK) at the crossroads between CO2 retention and skeletal muscle dysfunction in chronic obstructive pulmonary disease (COPD) Int J Mol Sci. 2020;21:955. doi: 10.3390/ijms21030955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. 2019;29:1511–1523. doi: 10.1016/j.celrep.2019.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park DW, Jiang S, Liu Y, Siegal GP, Inoki K, Abraham E, et al. GSK3β-dependent inhibition of AMPK potentiates activation of neutrophils and macrophages and enhances severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L735–L745. doi: 10.1152/ajplung.00165.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu TD, Fawzy A, Kinney GL, Bon J, Neupane M, Tejwani V, et al. Metformin use and respiratory outcomes in asthma-COPD overlap. Respir Res. 2021;22:70. doi: 10.1186/s12931-021-01658-3. [DOI] [PMC free article] [PubMed] [Google Scholar]