

Abstract
Rationale: Although it is clear that cystic fibrosis (CF) airway disease begins at a very young age, the early and subsequent steps in disease pathogenesis and the relative contribution of infection, mucus, and inflammation are not well understood.
Objectives: As one approach to assessing the early contribution of infection, we tested the hypothesis that early and continuous antibiotics would decrease the airway bacterial burden. We believed that, if they do, this might reveal aspects of the disease that are more or less sensitive to decreasing infection.
Methods: Three groups of pigs were studied from birth until ∼3 weeks of age: 1) wild-type, 2) CF, and 3) CF pigs treated continuously with broad-spectrum antibiotics from birth until study completion. Disease was assessed with chest computed tomography, histopathology, microbiology, and BAL.
Measurements and Main Results: Disease was present by 3 weeks of age in CF pigs. Continuous antibiotics from birth improved chest computed tomography imaging abnormalities and airway mucus accumulation but not airway inflammation in the CF pig model. However, reducing bacterial infection did not improve two disease features already present at birth in CF pigs: air trapping and submucosal gland duct plugging. In the CF sinuses, antibiotics did not prevent the development of infection or disease or the number of bacteria but did alter the bacterial species.
Conclusions: These findings suggest that CF airway disease begins immediately after birth and that early and continuous antibiotics impact some, but not all, aspects of CF lung disease development.
Keywords: lung physiology, respiratory mucosa, multidetector computed tomography, antibacterial agents, mucus
At a Glance Commentary
Scientific Knowledge on the Subject
How early cystic fibrosis (CF) lung disease begins and the role of bacteria in early pathogenesis is unclear.
What This Study Adds to the Field
Using a CF pig model, we found that CF airway disease begins immediately after birth. Both bacteria-dependent and bacteria-independent disease mechanisms were identified. Thus, early antibiotic treatment—or other therapeutic interventions—may significantly alter the course of disease.
Lung disease and progressive respiratory failure are the primary causes of morbidity and mortality in people with cystic fibrosis (CF). Although airway infection, mucus obstruction, and inflammation are all important in the pathogenesis of CF airway disease (1–5), how these features interact and progress with time is less clear.
Bacterial infection of the airways is a universal feature of CF. It is present very early in CF and can be detected even before the onset of overt symptoms in infants with CF (6–8). Antibiotics directed at airway infection have dramatically increased life expectancy and improved quality of life for people with CF (9–11), but how they impact pathogenesis and disease markers is less clear. Airway mucus accumulation likely also occurs early in CF. For example, in AREST-CF (Australian Respiratory Early Surveillance Team for Cystic Fibrosis) studies, air trapping was identified in up to two-thirds of infants with CF, often in the absence of culturable bacteria in the BAL (7, 12). In addition, children with CF had higher BAL fluid mucin levels than control subjects without CF (13). Finally, airway inflammation is also a key feature of early CF lung disease (14–20). For example, neutrophil elastase was detectable in approximately 25% of BAL fluid samples from infants with CF at 1 year of age (15). Moreover, neutrophil elastase in BAL fluid was associated with the development of bronchiectasis in children with CF and worse lung function (21–23). These findings suggest that inflammation is a key mediator of early structural CF lung disease.
We previously established that, on the day of birth, the airways of CF pigs lack mucus accumulation and are not inflamed (24, 25). However, they already have host defense defects, including an abnormally acidic airway surface liquid pH that impairs both bacterial killing by antimicrobial factors and mucociliary transport (25–32). Within months of birth, their intrapulmonary airways and sinuses spontaneously develop classic CF airway disease (25, 33). Thus, although it is clear that CF airway and sinus disease begins at a very young age, or maybe even before birth, the early and subsequent steps in disease pathogenesis and the relative contribution of infection, mucus accumulation, and inflammation are not well understood. As one approach to assessing the contribution of infection to early disease, we tested the hypothesis that early and continuous antibiotics would decrease the airway bacterial burden. We believed that, if it does, it might reveal aspects of the disease that are more or less sensitive to decreasing infection. Here, we show that disease is present by 3 weeks of age in CF pigs and that continuous antibiotics from birth affects some—but not all—aspects of airway disease in the CF pig model.
Methods
Additional methods are provided in the online supplement.
Study Approval
All animal studies were approved by the University of Iowa Animal Care and Use Committees.
Study Design
CF pigs expressing porcine CFTR complementary DNA via the intestinal fatty acid binding protein promoter (CFTR−/−;TgFABP>pCFTR) were developed previously (33). These pigs do not exhibit the meconium ileus phenotype that is lethal without surgical correction and do not express CFTR in their airways. A total of 21 CF pigs were used in this study, and 7 received continuous antibiotic therapy. Twelve non-CF pigs were used as control animals. The CF pig cohort receiving continuous antibiotic therapy was treated with a combination of ceftiofur hydrochloride (4 mg/kg intramuscularly once daily, from birth to 10 d) (Zoetis, Kalamazoo, MI), ciprofloxacin oral suspension (15 mg/kg orally twice daily, from ∼10 d until study completion) (Bayer), cephalexin oral suspension (30 mg/kg orally twice daily, from ∼10 d until study completion) (Teva), and trimethoprim-sulfamethoxazole oral suspension (30 mg/kg orally twice daily, from ∼10 d until study completion) (Teva). The majority of CF pigs were killed at predefined time points although some were killed slightly earlier, as advised by the University of Iowa veterinarian staff, owing to worsening respiratory status. We refer to the pigs in this study as “3-week-old” pigs. Average study age ± SEM was 21 ± 2 days old for non-CF pigs, 16 ± 1 for CF pigs, and 21 ± 1 for CF pigs treated continuously with broad-spectrum antibiotics from birth until study completion (CF+ABT). We previously published multidetector computed tomography (CT) airway and lung volume data from some of the pigs reported in the current study (34).
Disease Assessment
At study completion, the presence of CF was assessed using multiple modalities, including using CT scanning of the sinuses and lungs (at airway pressures of 0 and 25 cm H2O), BAL, bacterial cultures, histopathological analysis/scoring, and airway mucus quantification. Using the CT image datasets, both air trapping and lung parenchymal heterogeneity measurements were performed.
Statistics
The descriptive statistics are provided as mean ± SEM, median and interquartile range, or stacked bar plots. Tests were conducted at the α = 0.05 level, and all statistical analyses were performed using SAS 9.4 (SAS Institute Inc.) or Prism 8 (GraphPad). Complete statistical analysis details are included in the online data supplement.
Results
Three-Week-Old CF Pigs Have More Lung Bacteria Than Non-CF Pigs
To assess bacteria in the lung and to avoid confounding variables associated with BAL sampling, we sterilely obtained sections of lung and quantitatively determined the numbers of bacteria. We previously found that, within hours of birth, newborn CF pigs have more bacteria present in their lungs than do non-CF pigs (25) (Figure 1A). By 3 weeks of age, the numbers of bacteria had increased in the lungs of both non-CF pigs (newborn: 0.84 ± 0.29 vs. 3-week-old: 2.39 ± 0.57 log10 CFU/ml, P < 0.05) and CF pigs (newborn: 2.21 ± 0.47 vs. 3-week-old: 5.03 ± 0.61 log10 CFU/ml, P < 0.05). CF pigs had more bacteria in their lungs than non-CF pigs (Figure 1A).
Figure 1.

Continuous antibiotics decrease lung bacterial abundance in 3-week-old cystic fibrosis (CF) pigs. (A) Lung bacteria counts in newborn non-CF and CF pigs (previously published data from Stoltz and colleagues [2010]) (25) and in 3-week-old non-CF pigs, CF pigs, and CF pigs receiving antibiotics from birth (CF+ABT). (B) Number of individual bacterial species in amounts greater than 102 CFU/g present in newborn and 3-week-old pig lungs. (C) Percentage of newborn CF pigs (n = 12), 3-week-old CF pigs (n = 13), and CF+ABT pigs (n = 6) with recovered lung bacteria greater than 102 CFU/g from various genera. Each symbol represents data from an individual animal. Horizontal line denotes mean ± SEM. Mann-Whitney test or Kruskal-Wallis test followed by Dunn’s multiple comparison test, *P < 0.05.
No dominant bacterial species emerged by 3 weeks of age in the CF lung (Figures 1B and 1C). In previously published data, Staphylococcus and Enterococcus were the most commonly isolated bacterial species from newborn CF pig lungs (Figure 1C) (25). However, Streptococcus species were the most common isolates from the lungs of 3-week-old CF pigs (Figure 1C). Pseudomonas aeruginosa was present in the lungs of one 3-week-old CF pig. These results suggest that CF pigs have host defense defects against multiple different organisms rather than favoring one or a few specific bacterial organisms, consistent with results obtained in newborn pigs (25).
Chronic Antibiotic Treatment Reduces Lung Bacteria in 3-Week-Old CF Pigs
To determine whether continuous antibiotic treatment would reduce the lung bacterial burden in CF pigs, we treated another group of CF pigs with multiple antibiotics with broad antibacterial coverage at high doses starting at birth (CF+ABT pigs). From birth to 7–10 days of age, antibiotic-treated pigs received ceftiofur (a third-generation cephalosporin administered parenterally), which was followed by oral cephalexin, ciprofloxacin, and trimethoprim-sulfamethoxazole until study completion. This chronic, broad-spectrum antibiotic treatment regimen reduced bacterial counts in the lungs of 3-week-old CF pigs to levels found in non-CF pigs (CF+ABT: 1.60 ± 0.91 vs. non-CF: 2.39 ± 0.57 log10 CFU/ml, P > 0.05) (Figures 1A and 1B).
Prevention of Airway Infection Reduces Some but Not All Features of CF Lung Disease
We investigated whether CF pigs had evidence of airway disease by 3 weeks of age and hypothesized that reducing the lung bacterial load would impact early disease development. At birth, histological scoring showed no airway inflammation and minimal airway mucus in non-CF and CF pigs (Figures 2A and 2B and Figure E1 in the online supplement). At 3 weeks, non-CF-pig airways still had minimal airway inflammation and mucus (Figures 2A, 2B, and E1). However, airway inflammation had increased in 3-week-old CF pigs compared with non-CF pigs (Figures 2A–2C and E1). Antibiotics did not decrease the inflammation (Figures 2A and E1). Airway mucus was increased in 3-week-old CF pigs compared with non-CF pigs (Figures 2B–2D and E1). MUC5AC and MUC5B contributed to this mucus phenotype (Figure E2). Antibiotics decreased mucus accumulation to an intermediate phenotype between non-CF pigs and CF pigs (Figures 2B and E1), such that there was a nonstatistical difference when compared with non-CF pigs (P = 0.16) or with CF pigs (P = 0.90).
Figure 2.

Cystic fibrosis (CF) pigs develop lung disease by 3 weeks of age with airway neutrophilic inflammation and mucus accumulation. (A) Airway neutrophil inflammation scoring in newborn and 3-week-old pig lungs. (B) Airway mucus scoring in newborn and 3-week-old pig lungs. For both the inflammation and the mucus scoring scale, a higher number indicates more severe disease, with “3” being the worst. Shown is the percentage of pigs with a score of 0, 1, 2, or 3 within a group. For newborns, n = 3–4/group. For 3-week-old pigs, n = 12 for non-CF pigs, n = 14 for CF pigs, and n = 7 for CF+ABT pigs. Individual data points are shown in Figure E1. (C) Image of neutrophils (asterisk) in a CF pig airway. Scale bar = 50 μm. (D) Airways with regions of luminal mucus in bronchi and bronchioles (arrowheads) in a 3-week-old CF pig. Scale bar = 750 μm. Mann-Whitney test or Kruskal-Wallis test followed by Dunn’s multiple comparison test, *P < 0.05. Br = bronchi; CF+ABT = CF pigs treated continuously with broad-spectrum antibiotics from birth until study completion.
To quantify airway mucinous changes in differently sized airways (Figure E3), we stained lung histological sections with periodic acid Schiff after diastase treatment (dPAS) and measured 1) the percentage dPAS staining of the submucosal gland and surface airway epithelia area and 2) the amount of dPAS-positive material (mucus) in the airway lumens. The percentage of tracheal submucosal gland epithelial area that was dPAS positive was similar between 3-week-old non-CF pigs, CF pigs, and CF+ABT pigs (Figure E4A). In all groups, dPAS staining of the surface epithelium was greatest in the trachea and decreased along the length of the airway tree, with the bronchioles having the least amount of surface epithelial dPAS staining. There was no difference between the three groups (Figure E4B). Both gland-containing and non–gland-containing CF airways were variably affected by mucus accumulation (Figures 3A–3F). Greater luminal dPAS-positive material tended to be present in the small bronchi of CF pigs, and antibiotic treatment significantly decreased this response (Figure 3G), suggesting a relationship between bacterial infection and mucus accumulation.
Figure 3.

Antibiotics decrease mucus accumulation in the airways of 3-week-old cystic fibrosis (CF) pigs. (A–F) Airways from 3-week-old CF pigs. (A) Areas of atelectasis (asterisk) and air trapping (arrow) surround a small collapsed airway (arrowhead). (B) Small collapsed bronchus with stringy adherent mucus that emanated from (C) mucus cells in submucosal glands (arrows) and mucosal recesses (arrowheads). In more distal, non–gland-containing airways, (D) mucus sometimes filled the lumens of bronchioles (arrowhead) and (D–F) was sometimes observed along the airway wall. dPAS stain was used. Scale bars are as follows: (A) 1,000 μm, (B) 200 μm, (C) 50 μm, (D) 200 μm, (E) 200 μm, and (F) 100 μm. (G) Percentage of airway lumen area with dPAS-positive staining in regions from trachea to bronchioles of 3-week-old non-CF, CF, and CF+ABT pigs. Data points represent average measurements from individual animals. The horizontal line denotes the median value with the interquartile range. The total number of airways studied across all animals for each airway level are as follows: trachea, 90; large bronchi, 49; medium bronchi, 102; small bronchi, 215; and bronchioles, 164. A Kruskal-Wallis test was followed by Dunn’s multiple comparison test, *P < 0.05. CF+ABT = CF pigs treated continuously with broad-spectrum antibiotics from birth until study completion; dPAS = diastase-treated tissues with PAS stain; PAS = periodic acid Schiff.
On chest CT imaging, 3-week-old CF pigs had evidence of lung disease, including parenchymal abnormalities (Figure 4A). These changes were heterogeneous, consistent with prior studies reporting substantial heterogeneity of CF lung abnormalities (35–39). As a test of heterogeneity in the lung fields, we measured the Hounsfield unit (HU) interquartile range, which we termed the “CT lung heterogeneity” score (Figure E5). Regions with healthy lung are less heterogenous in appearance, whereas regions with greater lung disease have greater HU variation, i.e., an increased HU interquartile range (Figure E5). In newborn pigs, the CT lung heterogeneity score revealed no genotype-dependent differences (Figures 4A and 4B). However, by 3 weeks of age, CF pigs had an increased heterogeneity score compared with non-CF pigs in all six lung regions (Figures 4A–4C and E5). Continuous antibiotic treatment decreased the heterogeneity score to non-CF levels in all lung regions except for the right cranial region (Figure 4C). These findings suggested that bacterial abundance influenced lung disease. To further evaluate that suggestion, we asked whether the heterogeneity score was related to the bacterial abundance in the same lung region. We found a significant positive correlation between the number of bacteria in a lung region and the respective CT lung heterogeneity score (r = 0.79, P = 0.0001) (Figure 4D). Thus, by 3 weeks of age, CT imaging revealed evidence of lung disease in CF pigs, and early and continuous antibiotic treatment prevented the changes.
Figure 4.
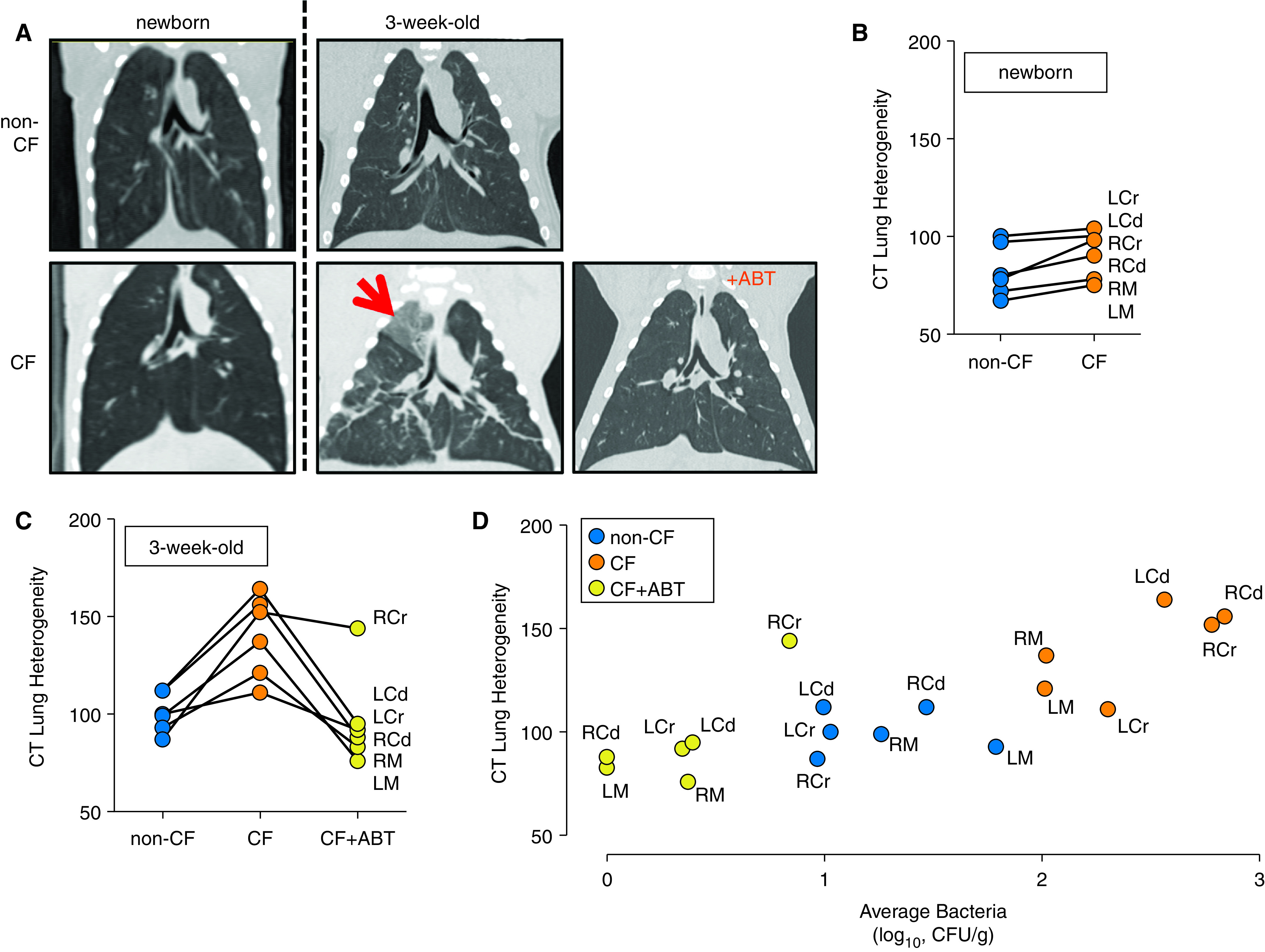
Antibiotics improve lung disease on computed tomography (CT) imaging. (A) Coronal CT images of newborn and 3-week-old non–cystic fibrosis (CF) pig lungs, CF pigs lungs, and lungs of CF pigs receiving antibiotics from birth (CF+ABT) at an airway pressure of 25 cm H2O. The arrow denotes lung parenchyma with increased opacification. (B) CT lung heterogeneity score in newborn non-CF (n = 8) and CF (n = 8) pigs described by the Hounsfield unit IQR of anatomically determined lung zones. (C) CT lung heterogeneity score in 3-week-old non-CF pigs (n = 9), CF pigs (n = 9), and CF+ABT pigs (n = 7). For B and C, data points represent average Hounsfield unit IQR in a lung zone for all animals of a genotype. (D) Plotted are the CT lung heterogeneity score and average bacteria present within specified regions. Data points represent the average IQR and bacterial count in a specific lung zone for all animals of a genotype/treatment group. Zones include cranial, middle, and caudal regions of left and right lungs (B–D). Cd = caudal region; Cr = cranial region; IQR = interquartile range; L = left lung; M = middle region; R = right lung.
We hypothesized that two aspects of CF already present in newborn CF pigs would not be affected by bacterial eradication. First, in a previous study, we found that CF pigs exhibited airflow obstruction and air trapping when they were less than 24 hours old (40). These abnormalities could not be attributed to mucus accumulation or inflammation because the newborn pigs lacked those changes, suggesting that developmental airway abnormalities were responsible (24, 25, 40). The increased air trapping in newborn CF versus non-CF lungs persisted in 3-week-old pigs (Figures 5A, 5B, and E6). Antibiotic treatment did not statistically reduce air trapping, in agreement with an abnormality caused by congenital airway abnormalities (40). Second, at birth mucus strands fail to detach from CF submucosal gland duct openings, in the absence of infection or inflammation (28). In newborn pigs, submucosal gland duct plugging was more frequently observed in CF than in non-CF airways (Figures 5C, 5D, and E7). This was also the case in 3-week-old pigs. Continuous antibiotic treatment had no significant effect on duct plugging (Figures 5D and E7).
Figure 5.

Antibiotics have no effect on air trapping or submucosal gland duct plugging in cystic fibrosis (CF) pigs. (A) Air trapping in 3-week-old non-CF pig lungs, CF pig lungs, and lungs of CF pigs receiving antibiotics from birth (CF+ABT). Shown are representative multi–detector row computed tomography images obtained at an airway pressure of 0 cm H2O. Air trapping is identified by arrows. (B) Quantitative assessment of air trapping. Shown is the percentage of pigs with a score of 0, 1, 2, 3, 4, 5, or 6 within a group. A higher number indicates greater air trapping, with “6” being the worst. Both left and right lungs were divided into six zones (three zones per lung): the cranial, middle, and caudal. Each zone was scored for the presence of air trapping in a binary manner (0 = no air trapping; 1 = air trapping). Scores range from 0 to 6 for each animal. For 3-week-old pigs, n = 11 for non-CF pigs, n = 10 for CF pigs, and n = 6 for CF+ABT pigs. Individual data points are shown in Figure E6. (C) In affected 3-week-old CF pigs, tracheal submucosal glands and ducts were dilated and filled with thick mucinous tethers (arrow). Scale bar = 50 μm. (D) Quantification of tracheal submucosal gland duct plugging in newborn and 3-week-old pigs. Shown is the percentage of pigs with a score of 0, 1, 2, 3, or 4 within a group. A higher number indicates more duct plugging, with “4” being the worst. For newborns, n = 3–4/group. For 3-week-old pigs, n = 9 for non-CF pigs, n = 14 for CF pigs, and n = 7 for CF+ABT pigs. Individual data points are shown in Figure E7. A Mann-Whitney test or Kruskal-Wallis test was followed by Dunn’s multiple comparison test, *P < 0.05. SMG = submucosal gland.
Finally, we also assessed lung disease with BAL. In 3-week-old pigs, BAL total cell counts, percentage neutrophils, cytokine levels (TNF-α [tumor necrosis factor-α], IL-6, IL-8, IL-10, and IL-13), bacterial counts, and bacterial load quantified by 16S quantitative PCR were similar between non-CF, CF, and CF+ABT pigs (Figures E8A–E8D and E9). Less than half of the bacterial species identified in lung tissue samples were also recovered in BAL samples of the same animal (Figure E8E). There was no relationship observed between tissue and BAL samples when comparing the number of bacteria recovered (Figure E8F) or the degree of BAL neutrophils/inflammation (Figure E8G). Bacteria were more likely to be recovered from tissue than from BAL samples, and a similar trend was present for inflammation. These findings suggest that, in very early CF (weeks after birth), BAL did not provide a sensitive method for detecting the presence of bacteria or inflammation.
Antibiotic Treatment Did Not Prevent Development of Sinus Disease in CF Pigs
Nearly 100% of people with CF develop chronic rhinosinusitis (41). Our previous work showed that CF pigs often develop mucopurulent sinus disease (42). Here, we found that, by 3 weeks of age, approximately 50% of CF pigs (6/13) had evidence of sinus disease at necropsy (defined as the presence of mucopurulent material in the ethmoid sinuses or nasal turbinates). Sinusitis was characterized by sinus opacification, mucus accumulation, neutrophil-rich inflammation, and bacteria (Figures 6A–6C). Non-CF pigs did not develop sinus disease. Similar to the lower airways, both MUC5AC and MUC5B mucins contributed to mucus accumulation in CF pigs (Figure E10). As in intrapulmonary airways, multiple different bacterial species were recovered from the CF sinuses (Figures 6D, 7, and E11).
Figure 6.

Continuous antibiotics fail to eradicate bacteria and prevent sinus disease in 3-week-old cystic fibrosis (CF) pigs. (A) Computed tomography imaging of ethmoid sinuses in 3-week-old non-CF pigs, CF pigs, and CF pigs receiving antibiotics from birth (CF+ABT). Arrows denote opacification of sinus space suggestive of mucus accumulation. (B) Three-week-old CF pigs had sinus disease with abundant luminal neutrophilic inflammation (asterisk), clumps of bacteria (arrowhead), and mucus accumulation (arrows) (scale bars: left panel = 25 μm and right panel = 1 mm). (C) Quantification of sinus bacteria in 3-week-old non-CF, CF, and CF+ABT pigs. (D) Number of individual bacterial species isolated from sinus samples greater than 104 CFU/sample in non-CF, CF, and CF+ABT pigs. Data points represent individual animals. Horizontal line denotes mean ± SEM. A Kruskal-Wallis test was followed by Dunn’s multiple comparison test, *P < 0.05.
Continuous antibiotic treatment from birth did not prevent sinusitis in CF pigs; the majority of CF+ABT pigs (5/7) had evidence of sinusitis at necropsy. In contrast with the response in intrapulmonary airways, antibiotics did not decrease bacterial infection in the sinuses (Figures 6Cb 6D, 7A, and 7B). However, the abundance of specific bacterial species was altered. For example, greater amounts of Streptococcus species were isolated in CF sinuses (Figure 7C), whereas Enterococcus species (Figure 7D) and P. aeruginosa (Figure 7E) were cultured in greater amounts from the CF+ABT sinuses. There was no difference in Staphylococcus species recovery between CF and CF+ABT groups (Figure 7F). Comparing the percentage of CF and CF+ABT pigs with a particular bacterial species, independent of abundance, revealed that Escherichia coli, Klebsiella, Pseudomonas, and Enterococcus were found at a higher frequency in the antibiotic-treated animals (Figure E12). These findings suggest that this antibiotic treatment regimen starting at birth does not impact the development of sinus infection or disease in CF pigs but might select for certain bacteria.
Figure 7.

A nonspecific host defense defect is present in 3-week-old cystic fibrosis (CF) pig sinuses. Quantification of bacterial species in sinuses of non-CF, CF, and CF+ABT pigs at 3 weeks of age identified as (A) gram-positive bacteria, (B) gram-negative bacteria, (C) Streptococcus species, (D) Enterococcus species, (E) Pseudomonas aeruginosa, and (F) Staphylococcus species. Data points represent individual animals. Horizontal line denotes mean ± SEM. A Kruskal-Wallis test was followed by Dunn’s multiple comparison test, *P < 0.05. CF+ABT = CF pigs treated continuously with broad-spectrum antibiotics from birth until study completion.
Discussion
Lung Disease Develops within Weeks of Birth in CF Pigs
Early studies of children dying of CF within the first year of life revealed pulmonary infection and inflammation (43). More recent studies in young children with CF also reported that airway disease begins very early in life, including within months of birth (44, 45). However, ethical and technical concerns limit studies in humans at very early time points. Therefore, we turned to CF pigs and found that, within 3 weeks after birth, they had developed lung disease.
Bacterial Infection Contributes to Some but Not All Aspects of CF Airway Disease
Antibiotics prevent development of some lung abnormalities
A prominent feature of CF lung disease is its heterogeneous distribution throughout the lung. We measured this heterogeneity with CT scans and found greater heterogeneity in 3-week-old CF pigs than in non-CF pigs. Importantly, antibiotic treatment prevented the development of these abnormalities. Moreover, this radiographic improvement correlated with bacterial abundance. Consistent with the radiographic changes, dPAS(+) material filled a greater percentage of the lumen of small bronchi in 3-week-old CF pigs, and antibiotic treatment prevented this development. These findings indicate that bacteria-dependent mechanisms contribute to the development of CF lung disease. Support for these findings come from studies of CF ferrets, for which antibiotic treatment is required for survival beyond the neonatal stage (46, 47). After that, continuous antibiotic treatment increased survival from ∼105 days to ∼1,143 days, an approximate 10-fold improvement in survival with antibiotic treatment, compared with antibiotic treatment for symptoms (48). And of course, with the advent of antibiotics and more aggressive treatment of infectious pulmonary exacerbations, the survival and quality of life of people with CF have dramatically improved.
Antibiotics did not prevent development of some lung abnormalities
Airway inflammation scores were increased in 3-week-old CF pigs, and despite continuous antibiotic treatment, they remained elevated. In addition, some measures of mucus accumulation persisted. The similarity of these findings in CF and CF+ABT pigs suggests that factors other than infection also contribute to the development of CF lung disease. Mucus with abnormal properties would seem to be the feature of the disease most likely to be responsible. That factor would be consistent with the plugging of CF submucosal glands. The impairment of mucociliary transport that begins at birth in CF pigs would also contribute (28). Although antibiotics markedly improved survival in CF ferrets, after 1–3 years, the antibiotic-treated animals exhibited lung inflammation and structural changes (48). However, whether these changes would have been worse in the absence of antibiotics is not known.
Some abnormalities present at birth persisted in antibiotic-treated CF pigs
The loss of CFTR-mediated Cl− and HCO3 − secretion generates submucosal gland mucus strands that have increased elasticity and tensile strength (31). Hence, at birth, submucosal gland ducts in CF pigs are frequently plugged with mucus (28, 49). The plugging of submucosal gland ducts was also observed in CF rats after they developed submucosal glands (50). Thus, it was not surprising that a histopathological analysis of 3-week-old CF pigs also showed duct plugging or that antibiotic treatment did not significantly decrease plugging. We previously reported that newborn pigs showed evidence of air trapping on chest CT (40). In 3-week-old CF pigs, we also detected air trapping. Consistent with the presence of this abnormality at birth, it was not reversed by antibiotic treatment. In total, these data highlight the complex defects that lead to CF lung disease.
Antibiotics Failed to Eradicate Bacteria in CF Sinuses
We considered factors that might account for the divergent responses in the sinuses and lungs. First, the selected antibiotics might have had poorer penetration and antimicrobial activity in the CF sinuses. The underlying sinus hypoplasia might have contributed to this as well (42). Second, although lungs and sinuses likely have similar host defenses and defects in CF, the lung might have additional protective measures that, combined with antibiotic therapy, could delay the onset of CF, such as cough clearance. Third, the antibiotic-induced shift in bacterial populations might have contributed—at least in part—to persistent sinus disease.
BAL Is Not a Sensitive Method for Detecting Very Early CF Lung Disease
Whereas we observed clear differences in bacterial numbers in tissue samples from non-CF pigs, CF pigs, and CF+ABT pigs, the trend was less pronounced in BAL samples. The discordance between bacterial species recovered in lung tissue and bacterial species recovered in BAL fluid suggests poor reliability of BAL sampling to adequately represent lung bacteria and/or reduced microbial detection due to dilution in the BAL. Similarly, CF BAL neutrophil counts were not elevated, even though airway neutrophilic inflammation was present on histological samples from CF lung tissue. These findings have important implications for studies in young children with CF and CF animal models. For example, earlier reports have suggested that airway inflammation and mucus accumulation precede airway infection (13, 45). Yet these studies based their assessment of bacterial infection on BAL methodology. On the basis of our findings, BAL results might not accurately reflect airway infection and pathology in very early disease.
Limitations of this study are as follows. 1) It is possible that the antibiotic regimen that we selected might not have affected all bacteria present in the CF pig lung. However, the antibiotics used have very broad activity, and they reduced lung bacterial counts to non-CF levels. 2) We were unable to perform culture-independent 16S sequencing on lung tissue samples because of the low bacterial biomass present (see the online supplement). 3) The role of viruses in early disease was not studied (16). 4) Because CF lung disease is heterogeneous, it is possible that we missed airway disease using tissue sampling. However, by also performing whole-lung volumetric chest CT scanning, we were able to assess lung disease in an unbiased manner. 5) Only one time point was studied. 6) Whether these findings are directly applicable to infants with CF is unknown owing to limited sample size and potential species differences. 7) The role of pathogen-associated molecular patterns was not investigated but could be an important focus of future studies.
This Study Raises Questions for the Future
First, the loss of CFTR impairs multiple respiratory host defenses (25–29, 31, 49, 51). Small molecule CFTR modulators improve CFTR function throughout the lung and thus are expected to improve multiple host defenses (16, 52, 53). Would correcting just one of the host defense defects limit disease progression? Would correcting CFTR in submucosal glands or surface epithelia be sufficient for benefit? For surface epithelia, would restoring host defenses to the cartilaginous airways be beneficial? What about to the distal airways? Second, would continuous antibiotic treatment impact the long-term development of CF airway disease? If so, at what cost?—increased antibiotic resistance, altered immune system development, disruption of the gastrointestinal tract microbiome, other adverse effects? Third, continuous antibiotics begun at birth had a beneficial effect in CF pigs. Would early antibiotic therapy provide additional benefit in the setting of highly effective CFTR modulator therapy started at birth?
In conclusion, results from this study show that airway disease develops shortly after birth in CF pigs and that early and continuous antibiotics impact some—but not all—aspects of CF lung disease development. These findings could have important implications for how we treat early CF airway disease, including the development of novel assays and therapeutic approaches.
Acknowledgments
Acknowledgment
The authors thank J. Bartlett, L. Boyken, S. Ernst, N. Gansemer, K. Heilmann, L. Powers, M. Rector, M. Stroik, P. Taft, and C. Wohlford-Lenane for their excellent assistance; the University of Iowa Office of Animal Resources; and Drs. S. Bodduluri, D. Diekema, A. Fischer, P. McCray, L. Ostedgaard, J. Reinhardt, M. Welsh, and J. Zabner for helpful discussions and feedback.
Footnotes
Supported, in part, by the NIH (HL135433, HL140261, HL007485, HL136813, OD018526, HL091842, HL051670, GM007337, HL007638, and TR002537), the Cystic Fibrosis Foundation (STOLTZ16XX0 and STOLTZ19R0), and the University of Iowa Physician Scientist Training Pathway.
Author Contributions: D.C.B., M.H.A.A., R.J.A., L.R.R., D.K.M., and D.A.S.: conceived of and designed study; D.C.B., M.H.A.A., R.J.A., A.A.P., L.R.R., D.P.C., and D.A.S.: performed experiments; D.C.B., M.H.A.A., R.J.A., A.A.P., L.R.R., M.I.A.P., P.T.E., C.W., T.J.G., D.B.H., D.K.M, and D.A.S.: analyzed data; D.C.B., M.H.A.A., R.J.A., A.A.P., L.R.R., D.P.C., M.I.A.P., T.J.G., D.B.H., E.A.H., D.K.M., and D.A.S.: interpreted results of experiments; D.C.B., R.J.A., D.K.M., and D.A.S.: prepared figures; D.C.B., M.H.A.A., and D.A.S.: drafted manuscript; D.C.B., M.H.A.A., R.J.A., A.A.P., L.R.R., D.P.C., M.I.A.P., P.T.E., C.W., T.J.G., D.B.H., E.A.H., D.K.M., and D.A.S.: edited, reviewed, and approved the final version of manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202102-0451OC on June 25, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chmiel JF, Davis PB. State of the art: why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir Res. 2003;4:8. doi: 10.1186/1465-9921-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363:2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wine JJ, Hansson GC, König P, Joo NS, Ermund A, Pieper M. Progress in understanding mucus abnormalities in cystic fibrosis airways. J Cyst Fibros. 2018;17:S35–S39. doi: 10.1016/j.jcf.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 5. Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372:415–417. doi: 10.1016/S0140-6736(08)61162-9. [DOI] [PubMed] [Google Scholar]
- 6. Grasemann H, Ratjen F. Early lung disease in cystic fibrosis. Lancet Respir Med. 2013;1:148–157. doi: 10.1016/S2213-2600(13)70026-2. [DOI] [PubMed] [Google Scholar]
- 7. Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF) Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180:146–152. doi: 10.1164/rccm.200901-0069OC. [DOI] [PubMed] [Google Scholar]
- 8. Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 9. Gibson RL, Emerson J, McNamara S, Burns JL, Rosenfeld M, Yunker A, et al. Cystic Fibrosis Therapeutics Development Network Study Group. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am J Respir Crit Care Med. 2003;167:841–849. doi: 10.1164/rccm.200208-855OC. [DOI] [PubMed] [Google Scholar]
- 10. Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, et al. Cystic Fibrosis Inhaled Tobramycin Study Group. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N Engl J Med. 1999;340:23–30. doi: 10.1056/NEJM199901073400104. [DOI] [PubMed] [Google Scholar]
- 11. Szaff M, Høiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand. 1983;72:651–657. doi: 10.1111/j.1651-2227.1983.tb09789.x. [DOI] [PubMed] [Google Scholar]
- 12. Hall GL, Logie KM, Parsons F, Schulzke SM, Nolan G, Murray C, et al. AREST CF. Air trapping on chest CT is associated with worse ventilation distribution in infants with cystic fibrosis diagnosed following newborn screening. PLoS One. 2011;6:e23932. doi: 10.1371/journal.pone.0023932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Esther CR, Jr, Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med. 2019;11:eaav3488. doi: 10.1126/scitranslmed.aav3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutièrrez JP, Hull J, et al. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156:1197–1204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- 15. Davies G, Thia LP, Stocks J, Bush A, Hoo AF, Wade A, et al. London Cystic Fibrosis Collaboration (LCFC) Minimal change in structural, functional and inflammatory markers of lung disease in newborn screened infants with cystic fibrosis at one year. J Cyst Fibros. 2020;19:896–901. doi: 10.1016/j.jcf.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 16. Deschamp AR, Hatch JE, Slaven JE, Gebregziabher N, Storch G, Hall GL, et al. Early respiratory viral infections in infants with cystic fibrosis. J Cyst Fibros. 2019;18:844–850. doi: 10.1016/j.jcf.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frayman KB, Wylie KM, Armstrong DS, Carzino R, Davis SD, Ferkol TW, et al. Differences in the lower airway microbiota of infants with and without cystic fibrosis. J Cyst Fibros. 2019;18:646–652. doi: 10.1016/j.jcf.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pittman JE, Wylie KM, Akers K, Storch GA, Hatch J, Quante J, et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis. Association of antibiotics, airway microbiome, and inflammation in infants with cystic fibrosis. Ann Am Thorac Soc. 2017;14:1548–1555. doi: 10.1513/AnnalsATS.201702-121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frayman KB, Armstrong DS, Carzino R, Ferkol TW, Grimwood K, Storch GA, et al. The lower airway microbiota in early cystic fibrosis lung disease: a longitudinal analysis. Thorax. 2017;72:1104–1112. doi: 10.1136/thoraxjnl-2016-209279. [DOI] [PubMed] [Google Scholar]
- 20. Rosenfeld M, Rayner O, Smyth AR. Prophylactic anti-staphylococcal antibiotics for cystic fibrosis. Cochrane Database Syst Rev. 2020;9:CD001912. doi: 10.1002/14651858.CD001912.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. AREST CF Investigators. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368:1963–1970. doi: 10.1056/NEJMoa1301725. [DOI] [PubMed] [Google Scholar]
- 22. Rosenow T, Mok LC, Turkovic L, Berry LJ, Sly PD, Ranganathan S, et al. The cumulative effect of inflammation and infection on structural lung disease in early cystic fibrosis. Eur Respir J. 2019;54:1801771. doi: 10.1183/13993003.01771-2018. [DOI] [PubMed] [Google Scholar]
- 23. Pillarisetti N, Williamson E, Linnane B, Skoric B, Robertson CF, Robinson P, et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am J Respir Crit Care Med. 2011;184:75–81. doi: 10.1164/rccm.201011-1892OC. [DOI] [PubMed] [Google Scholar]
- 24. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest. 2016;126:879–891. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345:818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joo NS, Cho HJ, Khansaheb M, Wine JJ. Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J Clin Invest. 2010;120:3161–3166. doi: 10.1172/JCI43466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ermund A, Meiss LN, Dolan B, Bähr A, Klymiuk N, Hansson GC. The mucus bundles responsible for airway cleaning are retained in cystic fibrosis and by cholinergic stimulation. Eur Respir J. 2018;52:1800457. doi: 10.1183/13993003.00457-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xie Y, Lu L, Tang XX, Moninger TO, Huang TJ, Stoltz DA, et al. Acidic submucosal gland pH and elevated protein concentration produce abnormal cystic fibrosis mucus. Dev Cell. 2020;54:488–500.e5. doi: 10.1016/j.devcel.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luan X, Tam JS, Jagadeeshan S, Grishchenko N, Hassan N, Gioino P, et al. Airway submucosal glands from cystic fibrosis swine suffer from abnormal ion transport across the serous acini, collecting duct, and ciliated duct. Am J Physiol Lung Cell Mol Physiol. 2020;318:L931–L942. doi: 10.1152/ajplung.00219.2019. [DOI] [PubMed] [Google Scholar]
- 33. Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123:2685–2693. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adam RJ, Abou Alaiwa MH, Bouzek DC, Cook DP, Gansemer ND, Taft PJ, et al. Postnatal airway growth in cystic fibrosis piglets. J Appl Physiol (1985) 2017;123:526–533. doi: 10.1152/japplphysiol.00263.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosenow T, Oudraad MC, Murray CP, Turkovic L, Kuo W, de Bruijne M, et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) PRAGMA-CF: a quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. Am J Respir Crit Care Med. 2015;191:1158–1165. doi: 10.1164/rccm.201501-0061OC. [DOI] [PubMed] [Google Scholar]
- 36. Ramsey KA, Rosenow T, Turkovic L, Skoric B, Banton G, Adams AM, et al. AREST CF. Lung clearance index and structural lung disease on computed tomography in early cystic fibrosis. Am J Respir Crit Care Med. 2016;193:60–67. doi: 10.1164/rccm.201507-1409OC. [DOI] [PubMed] [Google Scholar]
- 37. Brody AS, Klein JS, Molina PL, Quan J, Bean JA, Wilmott RW. High-resolution computed tomography in young patients with cystic fibrosis: distribution of abnormalities and correlation with pulmonary function tests. J Pediatr. 2004;145:32–38. doi: 10.1016/j.jpeds.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 38. Mott LS, Park J, Gangell CL, de Klerk NH, Sly PD, Murray CP, et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis Study G. Distribution of early structural lung changes due to cystic fibrosis detected with chest computed tomography. J Pediatr. 2013;163:243–248. doi: 10.1016/j.jpeds.2012.12.042. [DOI] [PubMed] [Google Scholar]
- 39. Tomashefski JF, Jr, Bruce M, Goldberg HI, Dearborn DG. Regional distribution of macroscopic lung disease in cystic fibrosis. Am Rev Respir Dis. 1986;133:535–540. doi: 10.1164/arrd.1986.133.4.535. [DOI] [PubMed] [Google Scholar]
- 40. Adam RJ, Michalski AS, Bauer C, Abou Alaiwa MH, Gross TJ, Awadalla MS, et al. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med. 2013;188:1434–1441. doi: 10.1164/rccm.201307-1268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chaaban MR, Kejner A, Rowe SM, Woodworth BA. Cystic fibrosis chronic rhinosinusitis: a comprehensive review. Am J Rhinol Allergy. 2013;27:387–395. doi: 10.2500/ajra.2013.27.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang EH, Pezzulo AA, Meyerholz DK, Potash AE, Wallen TJ, Reznikov LR, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122:1898–1905. doi: 10.1002/lary.23392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersen D. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56:344–399. [Google Scholar]
- 44. Stick SM. The first 2 years of life: implications of recent findings. Curr Opin Pulm Med. 2009;15:615–620. doi: 10.1097/MCP.0b013e328331c8cd. [DOI] [PubMed] [Google Scholar]
- 45. Ranganathan SC, Hall GL, Sly PD, Stick SM, Douglas TA. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF) Early lung disease in infants and preschool children with cystic fibrosis: what have we learned and what should we do about it? Am J Respir Crit Care Med. 2017;195:1567–1575. doi: 10.1164/rccm.201606-1107CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TI, et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am J Respir Cell Mol Biol. 2014;50:502–512. doi: 10.1165/rcmb.2013-0261OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun X, Olivier AK, Yi Y, Pope CE, Hayden HS, Liang B, et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am J Pathol. 2014;184:1309–1322. doi: 10.1016/j.ajpath.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosen BH, Evans TIA, Moll SR, Gray JS, Liang B, Sun X, et al. Infection is not required for mucoinflammatory lung disease in CFTR-knockout ferrets. Am J Respir Crit Care Med. 2018;197:1308–1318. doi: 10.1164/rccm.201708-1616OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ostedgaard LS, Moninger TO, McMenimen JD, Sawin NM, Parker CP, Thornell IM, et al. Gel-forming mucins form distinct morphologic structures in airways. Proc Natl Acad Sci USA. 2017;114:6842–6847. doi: 10.1073/pnas.1703228114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Birket SE, Davis JM, Fernandez CM, Tuggle KL, Oden AM, Chu KK, et al. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight. 2018;3:e97199. doi: 10.1172/jci.insight.97199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Birket SE, Davis JM, Fernandez-Petty CM, Henderson AG, Oden AM, Tang L, et al. Ivacaftor reverses airway mucus abnormalities in a rat model harboring a humanized G551D-CFTR. Am J Respir Crit Care Med. 2020;202:1271–1282. doi: 10.1164/rccm.202002-0369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. 2017;195:1617–1628. doi: 10.1164/rccm.201609-1954OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Donaldson SH, Laube BL, Corcoran TE, Bhambhvani P, Zeman K, Ceppe A, et al. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight. 2018;3:e122695. doi: 10.1172/jci.insight.122695. [DOI] [PMC free article] [PubMed] [Google Scholar]