

Abstract
Obesity is the result of numerous, interacting behavioral, physiological, and biochemical factors. One increasingly important factor is the generation of additional fat cells, or adipocytes, in response to excess feeding and/or large increases in body fat composition. The generation of new adipocytes is controlled by several “adipocyte-specific” transcription factors that regulate preadipocyte proliferation and adipogenesis. Generally these adipocyte-specific factors are expressed only following the induction of adipogenesis. The transcription factor(s) that are involved in initiating adipocyte differentiation have not been identified. Here we demonstrate that the transcription factor, CREB, is constitutively expressed in preadipocytes and throughout the differentiation process and that CREB is stimulated by conventional differentiation-inducing agents such as insulin, dexamethasone, and dibutyryl cAMP. Stably transfected 3T3-L1 preadipocytes were generated in which we could induce the expression of either a constitutively active CREB (VP16-CREB) or a dominant-negative CREB (KCREB). Inducible expression of VP16-CREB alone was sufficient to initiate adipogenesis as determined by triacylglycerol storage, cell morphology, and the expression of two adipocyte marker genes, peroxisome proliferator activated receptor gamma 2, and fatty acid binding protein. Alternatively, KCREB alone blocked adipogenesis in cells treated with conventional differentiation-inducing agents. These data indicate that activation of CREB was necessary and sufficient to induce adipogenesis. Finally, CREB was shown to bind to putative CRE sequences in the promoters of several adipocyte-specific genes. These data firmly establish CREB as a primary regulator of adipogenesis and suggest that CREB may play similar roles in other cells and tissues.
Excess body fat, or obesity, is a major health concern in the United States and other developed nations. It has been estimated that 26% of Americans are overweight (2), with 5 to 14% of men and 7 to 24% of women considered obese depending on the definition employed (2, 5, 6, 12, 22, 45, 57). Similar or even higher estimates for the prevalence of obesity have been reported in other countries (42). Obesity contributes to an increased rate of mortality (20) by virtue of its role in the development of cardiovascular disease, diabetes, pulmonary dysfunction, and gallstones (5, 10, 12).
Weight gain and obesity occur when energy intake by an individual exceeds the rate of energy expenditure (23). Energy intake and expenditure are in turn determined by multiple, interacting factors ranging from dietary composition and feeding and exercise habits to physiologic factors and biochemical pathways that modulate lipid and overall energy metabolism (58). At the cellular level obesity was originally considered a hypertrophic disease resulting from an increase in fat cell size or volume (30). However, several studies have demonstrated a hyperplastic component to obesity. For example, sequential biopsies in children indicate that fat cell numbers increase when body fat reaches 25% of total weight (26, 35). Similarly, obese adults have increased numbers of fat cells (30), and preadipocytes from obese subjects proliferate more rapidly in culture than cells from lean individuals (30, 51). New fat cells could arise from a preexisting population of undifferentiated progenitor cells or through the dedifferentiation of adipocytes to preadipocytes which then proliferate and redifferentiate into mature adipocytes. In either case, the generation of new fat cells demonstrates the crucial role of adipocyte proliferation and differentiation in the development of obesity.
The isolation and characterization of cell lines that progress from undifferentiated progenitor cells to mature adipocytes following appropriate stimulation has made it possible to identify factors that participate in adipocyte development (40). Among these factors, the nuclear hormone receptor, peroxisome proliferator activated receptor gamma 2 (PPARγ2), members of the CCAAT-enhancer binding protein (CEBP) family of transcription factors, and adipocyte determination-differentiation factor 1 (ADD1-SREBP) appear to play paramount roles in adipocyte differentiation (40, 58). Ectopic expression of PPARγ2 has been shown to drive the differentiation of preadipocytes to mature adipocytes in the presence of PPAR ligands (64), and PPARγ2 has been shown to bind to the promoters of several adipocyte-specific genes as a heterodimer with the cis-retinoic acid receptor alpha (RXRα) (62, 63). CEBPβ, which is expressed early in the adipocyte differentiation program, has likewise been shown to promote the differentiation of fibroblasts to adipocytes (75) and increase the expression of PPARγ2 (68). CEBPα is expressed relatively late in adipogenesis and appears to accelerate or potentiate the differentiation process as well as stimulate the expression of certain adipocyte-specific genes (40). While expressed late in adipocyte development, overexpression of CEBPα in fibroblasts will induce their differentiation to mature fat cells like PPARγ2 and CEBPβ (24). Expression of ADD1-SREBP1 alone is not sufficient to induce adipogenesis, but this factor has been shown to stimulate the expression of two genes involved with lipid metabolism: fatty acid synthetase and lipoprotein lipase (33). In addition, ADD1-SREBP1 appears to increase the percentage of cells that undergo adipocyte differentiation under conditions that favor adipogenesis. In addition to these factors, c-Jun, c-Fos, and c-Myc are also induced early in adipogenesis during the period of clonal expansion (40). Interestingly, all the aforementioned factors are undetectable or expressed at very low levels in preadipocytes, and their expression increases only after the induction of adipogenesis. This suggests that the expression of these factors and induction of adipogenesis is under the control of an unidentified factor(s) present in undifferentiated preadipocytes.
We have hypothesized that the transcription factor cyclic AMP (cAMP) response element binding protein (CREB) may play a crucial role in initiating adipogenesis. This hypothesis is based on our previous reports showing that both CREB phosphorylation and transcriptional activity were stimulated by agents that induce adipocyte differentiation such as dibutyryl cAMP (Bt2cAMP) acting through the cAMP-dependent protein kinase A (PKA), and insulin via an ERK1/ERK2 kinase cascade (34) and decreased nuclear protein phosphatase 2A (PP2A) activity (49, 50). A number of groups have demonstrated similar increases in CREB phosphorylation and activity in response to other growth factors, including nerve growth factor and fibroblast growth factor, via ERK1/ERK2 and p38 mitogen-activated protein (MAP) kinase pathways, respectively (27, 60). Other growth factor-regulated protein kinases such as protein kinase c (PKC) (69), certain Ca2+–calmodulin-dependent protein kinases (55), and p70 S6 kinase have also been shown to phosphorylate CREB. CREB is also regulated by several viral proteins, some of which alter cellular growth and differentiation. For example, we have demonstrated that the small tumor antigen (small-t) of simian virus 40 (SV40) enhances CREB phosphorylation and activity by inhibiting nuclear PP2A activity (67). Other studies have shown that human T lymphotropic virus type 1 Tax protein and the X protein of hepatitis B virus alter CREB DNA binding activity (3, 8, 25, 41), whereas adenovirus E1A proteins regulate the binding of CREB to the transcriptional coactivators, CREB binding protein and P300 (7, 37, 39). Finally, other members of the CREB-activating transcription factor (ATF) family of transcription factors that bind the same cis-acting promoter sequences as CREB are targets for various growth factor signaling systems and viral transforming proteins (1, 14, 29, 38, 41, 60). Together, these data strongly implicate CREB as a potential regulator of cell proliferation and differentiation.
In this report we demonstrate that CREB is constitutively expressed in 3T3-L1 fibroblasts prior to the induction of adipogenesis or the appearance of “adipocyte-specific” markers. Moreover, both CREB phosphorylation and transcriptional activity were induced in either preadipocytes or mature adipocytes following treatment with differentiation-inducing cocktail containing insulin, Bt2cAMP, and dexamethasone. To directly demonstrate that CREB induces adipogenesis, we generated stably transfected 3T3-L1 preadipocytes cells lines in which we could induce the expression of either constitutively active (VP16-CREB) or dominant-negative (KCREB) CREB proteins. With these cell lines we found that expression of constitutively active CREB alone was sufficient to induce adipocyte differentiation (based on cell morphology, triacylglycerol storage, and the appearance of adipocyte markers), whereas dominant-negative CREB effectively blocked adipogenesis in cells treated with conventional differentiation-inducing agents. How does CREB stimulate adipocyte development? Here we show that CREB binds to putative cAMP response elements (CREs) in the promoters of several adipocyte-specific gene promoters and that CREB regulates transcription from the promoters of the adipocyte-specific genes for phosphoenolpyruvate carboxykinase (PEPCK), fatty acid binding protein (FABP [aP2/422]), and fatty acid synthetase (FAS).
MATERIALS AND METHODS
Materials.
Cell culture media and supplies were from Gibco-BRL (Beverly, Mass.), Gemini Bioproducts (Gaithersburg, Md.), and Specialty Media, Inc. (Lavallette, N.J.). 3T3-L1 fibroblasts were provided by Ted Ciraldi (LaJolla, Calif.). Luciferase assay reagents were obtained from Analytical Luminescence Laboratory (San Diego, Calif.) and chloramphenicol acetyltransferase (CAT) enzyme-linked immunoassay assay kits were from Boehringer Mannheim (Indianapolis, Ind.). A plasmid containing an enhancerless thymidine kinase (TK) promoter linked to four copies of the Gal4 regulatory sequence driving expression of a luciferase reporter gene (pGal4TK-LUC) was provided by James Hoeffler (Invitrogen, Carlsbad, Calif.). An expression vector (pRSV-KCREB) for the dominant-negative CREB inhibitor protein, KCREB, was provided by Richard Goodman (Oregon Health Sciences University, Portland). CREB- and P-CREB-specific antibodies were purchased from New England Biolabs (Beverly, Mass.). Antibodies to PPARγ2, RXRα, CEBPα and -β, and VP16 were purchased from Santa Cruz Biotechnology (Santa Cruz, Calif.). Cell Titer 96 AQ reagents were from Promega Corp. (Madison, Wis.), and the Ecdysone Inducible Expression System (pIND, pVgRXR vectors, zeocin, and muristerone), and total RNA isolation reagents were from Invitrogen. A biotinylated, 60-base oligonucleotide complementary to the mouse FABP (or aP2/422; bases 1 to 60 of the open reading frame, 5′-GTAATCATCGAAGTTTTCACTGGAGACAAGCTTCCAGGTTCCCACAAAGGCATCACACAT-3′), and 20 bp double-stranded oligonucleotides for gel retardation assays (see Fig. 8) were purchased from Gene Link (Thornwood, N.Y.). All other reagents were of molecular biology grade or better and were purchased from Sigma Chemical Co. (St. Louis, Mo.).
FIG. 8.

CREB binds putative CRE sequences in the promoters of several adipocyte-specific genes. (A) The promoter regions of several adipocyte-specific genes were visually inspected for the presence of putative CRE sequences. Potential CREs present in these promoters are indicated by the box-enclosed regions which surround the nucleotides homologous to those in the consensus CRE sequences shown at the top of the figure. (B) Next, 20-bp double-stranded oligonucleotides, end labeled with [γ-32P]ATP and polynucleotide kinase, were incubated with purified, recombinant CREB protein as described in Materials and Methods. The reactions were separated on nondenaturing, 6% polyacrylamide gels and exposed to Kodak X-ARomat film. The figure shows a representative autoradiogram of the free (bottom) and CREB-bound complexes in comparison to reactions performed with a nonspecific (NS) oligonucleotide lacking a CRE sequence. (C) Next, 5 μg of nuclear extract protein prepared from 3T3-L1 fibroblasts was incubated with the indicated, labeled oligonucleotides either in the absence (−) or presence (+) of CREB-specific antibody. The reactions were separated on polyacrylamide gels as described above and exposed to film. The figure shows a representative autoradiogram of unbound and protein-bound oligonucleotides. (D) A total of 5 μg of nuclear extract protein prepared from 3T3-L1 untreated (−) fibroblasts or cells treated with muristerone to induce KCREB expression (+) was incubated with the indicated, labeled oligonucleotides. The reactions were separated on polyacrylamide gels as described above and exposed to film. The figure shows a representative autoradiogram of unbound and protein-bound oligonucleotides.
Cell lines and transfection procedures.
3T3-L1 fibroblasts were passaged in low-glucose Dulbecco modified Eagle medium (DMEM) plus 10% fetal calf serum (FCS)–1 mM l-glutamine. 3T3-L1 fibroblasts were differentiated into adipocytes after they reached confluency by the addition of differentiation medium (high-glucose DMEM containing 10% FCS, 1 mM l-glutamine, 300 μM isobutylmethylxanthine or Bt2cAMP, 1 μM dexamethasone, and 1 μg of insulin per ml). After 2 days, the 3T3-L1 cells were transferred to adipocyte growth medium (high-glucose DMEM plus 10% FCS, 1 mM l-glutamine, and 1 μg of insulin per ml) and refed every 2 days. Differentiation of fibroblasts to mature adipocytes was confirmed by Oil Red O staining of lipid vesicles.
Plates of 3T3-L1 fibroblasts and adipocytes were grown to 70 to 80% confluency and transfected with the indicated plasmids with Superfect Reagent (Qiagen, Valencia, Calif.) according to the manufacturer's recommendations. Cells stably transfected with the plasmid pVgRXR were selected in conventional medium containing 500 μg of Zeocin per ml, and cells stably transfected with pIND-VP16-CREB, pIND-KCREB (or pIND-VP16-KCREB and pIND-LacZ) plasmids were selected in medium containing 500 μg of Geneticin per ml. Large, rapidly growing, well-separated colonies were isolated 10 to 12 days after selection was begun with either antibiotic. Isolated clones were passaged in low-glucose DMEM containing 10% FCS, 1 mM l-glutamine, and 500 μg (each) of Zeocin and Geneticin per ml. VP16-CREB or KCREB expression was induced through the addition of 10 μM muristerone to the growth medium as indicated in the figure legends. The effect of VP16-CREB and KCREB expression on 3T3-L1 proliferation was assessed by measuring the cell number with the Cell-Titer 96 Aq reagent system (Promega Corp., Madison, Wis.). Cells were treated with various reagents at the concentrations and times specified in the figure legends.
Differentiation of 3T3-L1 preadipocytes to mature adipocytes was followed by observing the accumulation of triacylglycerol in Oil Red O staining vesicles and by the appearance of adipocyte markers, FABP (aP2/422) and PPARγ2. Differentiation assays were performed on cells growing on eight-chamber microscope slides. The cells were treated with the indicated agents in high-glucose medium. Ten days following the initiation of differentiation, the cells were stained with Oil Red O as previously described earlier (34) and counterstained with hematoxylin to visualize cell morphology. Cells were observed by bright-field microscopy, and representative fields were photographed with Kodak 200 film. Alternately, cells growing on multiwell slides were lysed directly in Laemmli sodium dodecyl sulfate (SDS) gel loading buffer, and the lysates were subjected to Western blot analysis for PPARγ2 expression. Total RNA was isolated from cells grown in duplicate wells by using the Total RNA Isolation Kit from Invitrogen and subjected to Northern blot analysis with a probe to FABP.
Luciferase assays were performed on a Monolight 2010 luminometer by using the Enhanced Luciferase Assay kit (Analytical Luminescence Laboratory, San Diego, Calif.) according to the supplier's directions. Transfection efficiencies were normalized by cotransfecting the cells with a plasmid containing a chimeric SV40 promoter–β-galactosidase gene, and β-galactosidase levels were measured as previously described. All experiments were repeated at least three times, and consistent results were obtained in all cases.
Lipid accumulation was quantitated by isopropanol extraction of Oil Red O from stained cells and optical density determinations at 518 nm as previously described (28).
Ecdysone-inducible VP16-CREB and KCREB expression system.
The edison-inducible expression system was employed to prepare stably transfected 3T3-L1 cells in which we could induce the expression of VP16-CREB and KCREB. The open reading frame for KCREB was isolated from the plasmid, pRSV-KCREB, as an HindIII-EcoRI fragment. This fragment was subjected to PCR with a 5′ primer that introduced a new HindIII site and a consensus Kozak translation initiation sequence (GCCACC) immediately upstream of the first methionine codon. The resulting PCR product was purified by electrophoresis on a 1% agarose gel and ligated into the HindIII and EcoRI sites of the plasmid, pIND. The open reading frame for VP16 (amino acids 412 to 490) was excised from the plasmid, pVP16 (Arthur Gutierrez-Hartman, University of Colorado Health Sciences Center, Denver) as a HindIII-BamHI fragment. This fragment was also subjected to PCR to introduce a Kozak sequence immediately upstream of the translation start site. This fragment was directly ligated to a BglII-EcoRI fragment containing the DNA binding domain (amino acids 217 to 327) of CREB-327 excised from the plasmid pRSET-CREB (James Hoeffler, Invitrogen). This chimeric VP16-CREB gene was ligated into the HindIII and EcoRI sites of pIND. As a control, we generated a chimeric gene composed of the VP16 transactivation domain linked to the (non)DNA-binding domain of KCREB. The resulting plasmids were confirmed by restriction enzyme mapping and sequencing.
Western and northern blot analysis.
Lysates and total RNA from 3T3-L1 fibroblasts and adipocytes treated as described in the figure legends was prepared as described above. After we corrected for protein concentrations, the lysates prepared in Laemmli SDS loading buffer were resolved on 10% polyacrylamide-SDS gels and transferred to nitrocellulose. The nitrocellulose blots were blocked with phosphate-buffered saline containing 5% dry milk and 0.1% Tween 20 and then treated with antibodies that recognize phosphorylated CREB (P-CREB), total CREB, CEBPα and -β, RXRα, PPARγ2, or VP16. The blots were washed and subsequently treated with goat anti-rabbit immunoglobulin G-conjugated to alkaline phosphatase (for CREB, P-CREB, CEBPα and -β, RXRα, and PPARγ2 antibodies) or anti-goat–alkaline phosphatase conjugate (VP16 antibody). After the blots were washed, specific immune complexes were visualized with 5-bromo-4-chloro-3-indolylphosphate (BCIP) and nitroblue tetrazolium.
FABP expression was measured by Northern blot analysis of total RNA isolated from 3T3-L1 cells as described above. Approximately 5 μg of total RNA from each sample was separated by electrophoresis on denaturing (formaldehyde) 1% agarose gels run at 5 V/cm until the bromophenol blue tracking dye had migrated half the length of the gel. The gels were soaked in several changes of distilled water overnight at 4°C, stained with ethidium bromide, and briefly examined in UV light to ensure RNA integrity and equivalent RNA amounts in each lane. The gels were destained in several changes of 5× sodium chloride-sodium citrate (SSC) buffer. RNA was transferred onto nitrocellulose membranes. The resulting blots were heated to 80°C under vacuum for 2 h. The membranes were blocked for 30 min at 70°C, and then a biotin-labeled FABP-specific oligonucleotide probe (250 fg/ml) was added for an additional 30 min. Blots were washed in three changes of 0.1× SSC containing 1% SDS at 70°C for 15 min in each wash. Specific hybridization complexes were then visualized with BCIP and nitroblue tetrazolium.
Gel retardation assays.
Gel retardation assays were performed in reactions containing 1 μg of nonspecific, competitor DNA as previously described (34).
RESULTS
Insulin or Bt2cAMP alone can induce adipocyte differentiation in 3T3-L1 cells.
Maximal differentiation of 3T3-L1 preadipocytes to mature adipocytes generally requires the addition of a mixture of insulin or insulin-like growth factor-1, a glucocorticoid, and a cAMP mimetic (40). However, Green and Kehinde (28) demonstrated that 3T3-L1 cells would undergo adipogenesis and accumulate triacylglycerol when treated with insulin alone, and Yarwood et al. (74) recently demonstrated that cAMP agonists potentiate growth factor-induced adipogenesis. We assessed the ability of these agents to induce adipose differentiation by treating 3T3-L1 fibroblasts with increasing concentrations of either insulin or Bt2cAMP for 48 h and then measured triacylglycerol levels after an additional 8 days in culture. We found that 3T3-L1 cells accumulated triacylglycerol when treated with insulin or Bt2cAMP alone, in a manner dependent on the concentration of each agent used (Fig. 1). Maximal triacylglycerol levels were noted with 3 mM Bt2cAMP or 10 mg of insulin per ml. No lipid accumulation was observed at Bt2cAMP concentrations below 3 μM or with insulin concentrations of <3 μg/ml. The maximal levels of lipid stored in the cells treated with Bt2cAMP or insulin alone were just slightly less than lipid levels measured in L1 fibroblasts treated with a mixture of 0.3 mM Bt2cAMP and 10 μg of insulin per ml (typical concentrations used in differentiation experiments). L1 cells treated with a mixture of 10 μg of insulin per ml, 1 μM dexamethasone, and 0.3 mM Bt2cAMP accumulated only 30% more triacylglycerol than cells treated with optimal doses of Bt2cAMP or insulin alone. Thus, high levels of insulin or cAMP mimetics, both of which we have shown stimulate CREB in 3T3-L1 cells (34), are able to induce adipogenesis in the absence of other differentiation-inducing agents.
FIG. 1.

Insulin or Bt2cAMP alone are sufficient to induce adipogenesis in 3T3-L1 cells. 3T3-L1 fibroblasts were passaged as described in Materials and Methods. The cells were treated with the indicated final concentrations of either Bt2cAMP (○) or insulin (□) for 48 h in high-glucose medium. The cells were then refed every 2 days for 10 days with high-glucose medium containing 10 μg of insulin per ml. On day 10 cells were fixed and stained with Oil Red O. Oil Red O was extracted from the cells with 200 μl of isopropanol and measured at 518 nm. Levels of Oil Red O staining were corrected for nonspecific binding levels of stain to untreated cells. For comparison, levels of Oil Red O staining in cells treated with either 10 μg of insulin per ml and 0.3 mM Bt2cAMP (dotted line) or else 10 μg of insulin per ml, 0.3 mM Bt2cAMP, and 1 μM dexamethasone (solid line) are also shown.
CREB is constitutively expressed prior to and during adipogenesis and regulated by differentiation-inducing agents.
The first clue that CREB might be involved in adipocyte differentiation was observed in assays in which total CREB (unphosphorylated plus phosphorylated) protein and Ser133 phosphorylated CREB (phospho-CREB or P-CREB) were measured in NIH 3T3-L1 cells (Fig. 2). CREB was present in 3T3-L1 fibroblasts prior to the induction of adipogenesis and throughout the differentiation process at relatively stable levels (Fig. 2A and B, total CREB panels). This is in sharp contrast to other adipocyte-specific transcription factors such as CEBPα and -β and RXRα, which are undetectable in untreated preadipocytes (Fig. 2B, day 0). CEBPβ first became detectable in our experiments approximately 30 min following treatment with inducing agents and then continues to increase for at least 48 h (Fig. 2A and B), while RXRα and CEBPα do not appear until days 2 and 8, respectively (Fig. 2B). The expression of these factors corresponds to previous reports describing the appearance of these and other adipocyte-specific factors only after the induction of adipogenesis (40, 44, 58).
FIG. 2.

CREB is expressed before and during adipogenesis, and differentiation-inducing agents stimulate CREB phoshorylation and transcriptional activity. (A) 3T3-L1 preadipocytes were grown to confluency as described in Materials and Methods. The cells were refed with complete growth medium containing 1 μg of insulin per ml, 1 μM dexamethasone, and 0.3 mM Bt2cAMP for the times indicated above each lane. Approximately 25 μg of cell lysate protein from each sample was separated on 10% acrylamide–SDS gels and transferred to nitrocellulose membranes. Duplicate membranes were subjected to Western analysis by using antibodies specific for Ser133 phosphorylated CREB (Phospho-CREB), total CREB, or CEBPβ protein as indicated. (B) Preadipocytes were grown to confluency and then refed with medium containing insulin, dexamethasone, and Bt2cAMP for 48 h. The cells were then refed every 2 days with medium containing 1 μg of insulin per ml. Cell lysates were prepared on the days indicated above each lane, and 25 μg of lysate protein from each sample was separated on 10% polyacrylamide–SDS gels and transferred to nitrocellulose membranes. Individual membranes were probed with antibodies specific for phospho-CREB, total CREB, CEBPα and -β, and RXRα as indicated. (C) 3T3-L1 fibroblasts and adipocytes were grown as described in Materials and Methods. Cells were transfected with the plasmid, pGal4TK-Luc, alone or with the plasmid pRSV-Gal4-CREB by using Superfect transfection reagent. The plasmids are described in the main text. At 24 h after transfection, the cells were treated with 0.5 mM Bt2cAMP alone or with a mixture of 1 μg of insulin per ml, 1 μM dexamethasone, and 0.5 mM Bt2cAMP for 4 h. The control cells received no treatment. Luciferase levels were measured in cell lysates as an index of transcription from the Gal4-TK promoter. Levels of transcription are shown relative to levels measured in untreated control cells transfected with pGal4TK-Luc alone.
Not only was CREB present in 3T3-L1 cells before and during adipogenesis, but phosphorylation of CREB was rapidly stimulated in cells treated with a differentiation-inducing mixture containing insulin, Bt2cAMP, and dexamethasone (Fig. 2A). Phospho-CREB levels increased approximately 20-fold within 10 min of treatment, remained elevated for another 20 min, and then began to decline slowly. Interestingly, variations in CREB phosphorylation were also noted during the 10-day differentiation process (Fig. 2B). High levels of phospho-CREB were detected in cell lysates prepared on days 2, 6, and 10, whereas lower but significant levels of phospho-CREB were detected in lysates from days 4 and 8. These results appear to reflect increases in CREB phosphorylation due to refeeding of the cells with serum-containing medium. Our ability to stimulate CREB phosphorylation with differentiation-inducing agents clearly points to a role for CREB in adipogenesis. Other CRE binding factors, including ATF-1, ATF-2, and CREM, were not detected on Western blots of preadipocyte or adipocyte cell lysates (data not shown), suggesting that such proteins do not participate in adipogenesis.
We next assessed the ability of differentiation-inducing agents to regulate CREB transcriptional activity. For these experiments, 3T3-L1 preadipocytes or mature adipocytes were transfected with a plasmid from which a chimeric protein composed of the CREB transactivation domain (amino acids 1 to 261 of CREB-327) linked to the Gal4 DNA-binding domain (amino acids 1 to 174) was expressed. The transcriptional activity of this chimeric protein was measured by cotransfecting the cells with a plasmid containing a Gal4-responsive promoter linked to a luciferase reporter gene (pGal4TK-Luc). As shown in Fig. 2C, transcription from the Gal4-responsive promoter was unaffected by any treatment in the absence of Gal4-CREB protein. However, in either preadipocytes or mature adipocytes expressing Gal4-CREB, the differentiation-inducing mixture of insulin, Bt2cAMP, and dexamethasone or the use of Bt2cAMP alone stimulated transcription from the Gal4-responsive promoter by 10- to 12-fold. These results are consistent with our previous data showing that insulin and cAMP mimetics alone stimulate CREB-mediated transcription via phosphorylation of CREB serine 133 (34). The ability of adipogenesis-inducing agents to stimulate both CREB phosphorylation and transcriptional activity, and the constitutive expression of CREB prior to and during differentiation led us to hypothesize that CREB might play a role in initiating and maintaining the adipocyte differentiation program.
Generation of stably transfected 3T3-L1 fibroblasts that inducibly express constitutively active and dominant-negative forms of CREB.
To directly assess the participation of CREB in adipogenesis, we generated stably transfected 3T3-L1 cell lines in which we could induce the expression of constitutively active or dominant-negative forms of CREB with the insect hormone homolog, muristerone (ecdysone-inducible expression system; Invitrogen). This system allowed us to directly modulate CREB transcriptional activity without relying on pharmacological agents that might regulate other signaling pathways and transcription factors. Constitutively active CREB consisted of the transactivation domain of the viral VP16 protein (amino acids 412 to 490) linked to the CREB DNA-binding domain (amino acids 217 to 327). KCREB, a protein which binds to endogenous CREB and prevents its binding to CRE sequences in gene promoters (65), was employed as the dominant-negative CREB. As a control, we also prepared a chimeric gene composed of the VP16 transactivation domain linked to the KCREB (non)DNA-binding domain. We believed that this protein would also act as a dominant-negative CREB because of the effect of the KCREB DNA-binding region, even though it contained the VP16 transactivation domain. VP16-KCREB would therefore serve as a control for nonspecific or indirect effects, such as squelching due to the transactivation portions of the proteins. The open reading frames for these genes were ligated into the vector, pIND, and transfected into 3T3-L1 fibroblasts previously transfected with the plasmid, pVgRXR, and selected in zeocin. After selection for pIND-carrying cells in Geneticin, two clones expressing VP16-CREB (2-4 and 9-7), two clones expressing KCREB (2-1 and 2-10), and two clones expressing VP16-KCREB (2-6 and 9-3) in response to muristerone were isolated and characterized.
The muristerone-induced appearance of VP16-CREB protein in clones 2-4 and 9-7 was examined by Western blot analysis by using antibodies specific for VP16 (Fig. 3). VP16-CREB levels increased slowly over the first 4 h after muristerone addition in both cell lines but rose rapidly to maximal levels in 20 to 24 h. Thereafter, protein levels decrease slowly even in the continued presence of muristerone. Removal of muristerone from the cells slightly increased the rate of VP16-CREB disappearance. The kinetics of KCREB induction were monitored by Western blot analysis with antibodies to total CREB (level corrected for endogenous CREB content in untreated cells) and differed significantly from VP16-CREB expression. KCREB levels increased much more rapidly than VP16-CREB in both clones and continued to rise throughout the course of the assay. Although removal of muristerone at 20 h decreased the rate of KCREB expression, KCREB levels continued to increase.
FIG. 3.

Time course of VP16-CREB or KCREB expression in stably transfected 3T3-L1 cells after muristerone induction. 3T3-L1 cells stably transfected with the plasmid, pVgRXR, and either pIND-VP16-CREB or pIND-KCREB were generated as described in Materials and Methods. Individual clones inducibly expressing VP16-CREB, designated 2-4 and 9-7, and clones inducibly expressing KCREB, designated 2-1 and 2-10, were isolated. The expression of VP16-CREB or KCREB was monitored in these clonal cell lines versus time after treatment with muristerone at a final concentration of 10 μM. At 20 h, duplicate wells of cells were refed with medium lacking muristerone (levels indicated by dashed lines) for comparison to cells in medium with muristerone (solid lines). Levels of VP16-CREB and KCREB were measured by separating 25 μg of protein from lysates prepared at the times shown on 10% acrylamide–SDS gels. Proteins were transferred to nitrocellulose membranes subsequently probed with antibodies to VP16 (for VP16-CREB) or CREB (for KCREB). Since the CREB antibody detected both KCREB and endogenous CREB proteins, levels of KCREB expression were corrected for endogenous CREB levels measured in untreated cells (not shown). The optical densities of the bands on the blots was determined by using Scan Analysis Software. A representative blot for each protein is displayed to the right of the graphs.
To test the ability of KCREB and VP16-CREB expression to influence gene transcription, a plasmid containing a truncated, CRE-containing portion of the phosphoenolpyruvate carboxykinase (PEPCK) gene promoter linked to a luciferase reporter gene (−109 pPC-Luc) was transfected into each of the clones. In KCREB clones 2-1 and 2-10, transcription from this promoter was efficiently stimulated 2.5- to 3.5-fold by treatment of the cells with Bt2cAMP (Fig. 4A). Prior overnight treatment of the cells with muristerone alone had no effect on PEPCK promoter-driven transcription but efficiently inhibited Bt2cAMP-stimulated transcription. The ability of KCREB to block Bt2cAMP-stimulated transcription appeared to be due to its inability to bind CRE sequences and block binding of endogenous CREB to CRE elements (Fig. 4B), a result consistent with its reported function (65). Alternately, muristerone treatment of VP16-CREB clones 2-4 and 9-7 stimulated luciferase production from the PEPCK promoter fragment by 3.5- to 4-fold compared to the levels measured in untreated cells. These data confirmed our hypothesis that VP16-CREB would stimulate transcription from CREB-regulated promoters in the absence of signals directed toward activating endogenous CREB. VP16-KCREB blocked Bt2cAMP-stimulated transcription and CRE DNA-binding activity (data not shown).
FIG. 4.

Effect of VP16-CREB or KCREB on CRE-dependent gene transcription and 3T3-L1 proliferation. (A) Stably transfected 3T3-L1 clonal cell lines inducibly expressing VP16-CREB (clones 2-4 and 9-7) or KCREB (clones 2-1 and 2-10) were transfected with the plasmid, −109 pPC-Luc, which contains a CREB-responsive portion of PEPCK gene promoter linked to a luciferase reporter gene by using Superfect reagent. The cells were cotransfected with the plasmid pSV-βGal. The following day the cells were treated with 0.3 mM Bt2cAMP, 10 μM muristerone, or both agents together as indicated. After 4 h cell lysates were prepared, and the luciferase activity was measured as an index of transcriptional activity. Levels are shown relative to the levels of luciferase activity in untreated, control cells (No Add'n). (B) 3T3-L1 cells stably transfected with pIND-KCREB (clone 2-10) were stimulated with the indicated final concentrations of muristerone for 20 h. Nuclear extracts were prepared, and electrophoretic mobility shift assays were performed with a 32P-labeled oligonucleotide containing the consensus CRE sequence (TGACGTCA). Reactions were separated on nondenaturing 6% polyacrylamide gels and exposed to film. The figure shows a representative autoradiograph. (C) Stably transfected 3T3-L1 clonal cell lines inducibly expressing VP16-CREB (clones 2-4 and 9-7) or KCREB (clones 2-1 and 2-10) were transferred to duplicate wells of 96-well plates (5,000 cells/well). After 24 h, one well was treated with 10 μg of muristerone (squares, solid line), and the remaining well was left untreated (circles, dotted line). Cell numbers were determined in each well with the Cell Titer 96 AQ reagent system at 24, 48, 72, and 96 h after plating of the cells. Values are shown relative to levels measured in wells containing untreated, control cells at the 24-h time point for each cell line and are the averages of three assays.
Two possible mechanisms by which CREB could potentiate adipose differentiation would be by increasing cell proliferation related to clonal expansion or by inhibiting cell growth as a prelude to terminal differentiation. To examine these possibilities, changes in the rate of cell growth in control versus muristerone-treated VP16-CREB- and KCREB-expressing clones was measured. As shown in Fig. 4C, no significant differences in the rate of cell proliferation were noted over a period of 72 h after muristerone treatment between control and treated VP16-CREB- or KCREB-expressing cells.
CREB activation is necessary and sufficient to induce adipogenesis.
Based on our data showing that CREB phosphorylation and transcriptional activity were stimulated by agents that induce adipose differentiation, we hypothesized that the expression of VP16-CREB would initiate or potentiate adipogenesis, whereas KCREB (and the VP16-KCREB control), would inhibit differentiation in preadipocytes treated with differentiation-inducing agents. This hypothesis was first tested in experiments in which triacylglycerol storage was monitored as an index of adipose differentiation by Oil Red O staining (Fig. 5). Each of the VP16-CREB and KCREB cell lines, as well as control cells (stably transfected with the pIND-LacZ expression vector) showed no signs of triacylglycerol accumulation if propagated in the absence of differentiation-inducing agents. Likewise, all cell lines exhibited significant triacylglycerol accumulation and large, rounded morphology 10 days following exposure to a differentiation-inducing mixture of insulin, Bt2cAMP, and dexamethasone. Thus, each of the cell lines exhibited normal differentiation characteristics. No triacylglycerol accumulation was observed in control cells treated with muristerone alone, but triacylglycerol vesicles were readily apparent in control cells exposed to both muristerone and the conventional differentiation-inducing mixture. These data indicated that muristerone alone had no significant impact on cell phenotype.
FIG. 5.

VP16-CREB stimulates and KCREB inhibits adipogenesis in 3T3-L1 cells as determined by triacylglycerol storage. 3T3-L1 preadipocyte cell lines inducibly expressing VP16-CREB (clones 2-4 and 9-7) or KCREB (clones 2-1 and 2-10) or control cells (stably transfected with the plasmids, pVgRXR and pIND-LacZ) were grown to confluence as described in Materials and Methods. The cells were treated with the reagents indicated above each column of photographs. Cells treated with differentiation mixture received 10 μg of insulin per ml, 1 μM dexamethasone, and 0.3 mM Bt2cAMP for 48 h and then were refed every 2 days with conventional medium containing 10 μg of insulin per ml. Muristerone was added to medium at a final concentration of 10 μM for the entire 10-day differentiation period. After 10 days in culture, the cells were stained with Oil Red O to visualize triacylglycerol vesicles and then counterstained with hematoxylin. The photographs show cells on day 10 of each treatment.
However, in both of the VP16-CREB-expressing cell lines treatment with muristerone alone was sufficient to induce triacylglycerol accumulation and rounded cell morphology. These data indicated that VP16-CREB expression, stimulated by muristerone, was capable of initiating adipogenesis. Alternately, both KCREB-expressing cell lines failed to exhibit signs of differentiation when treated with muristerone prior to and during their exposure to the conventional differentiation-inducing mixture. Likewise, cells inducibly expressing VP16-KCREB failed to differentiate when treated with inducing agents (data not shown). The ability to block adipogenesis by inhibiting endogenous CREB activity indicated that CREB is required to induce normal adipose differentiation.
These data were confirmed by measuring the expression of the adipocyte-specific markers PPARγ2 and FABP (aP2/422) in cell lysates prepared on days 0 and 10 of treatment (Fig. 6). As expected, no expression of these factors was noted in untreated control or in VP16-CREB- or KCREB-expressing cell lines. However, when treated with the differentiation-inducing cocktail, PPARγ2 and FABP expression were observed in day 10 samples from all cell lines. As observed for triacylglycerol storage, muristerone had no effect on the anticipated expression of PPARγ2 and FABP in uninduced or induced control cells. No PPARγ2 or FABP were present on day 0 in either VP16-CREB cell line, but they were easily detected in day 10 lysates from muristerone-treated cells in the absence of other differentiation-stimulating agents. On the other hand, expression of KCREB (or VP16-KCREB [data not shown]) before and during the application of the conventional differentiation-inducing mixture completely blocked the appearance of both PPARγ2 and FABP in the day 10 samples. Once again, these data support the hypothesis that the activation of CREB activation is sufficient and necessary to initiate the adipocyte differentiation program.
FIG. 6.

VP16 CREB stimulates and KCREB inhibits adipogenesis in 3T3-L1 cells as determined by expression of adipocyte-specific genes. Control and VP16-CREB- and KCREB-expressing cell lines were grown and treated as described in the legend to Fig. 5. On days 0 and 10 of the experiment, whole-cell lysates and total RNA was prepared from duplicate wells of cells. Approximately 25 μg of protein in the cell lysates was separated on 10% polyacrylamide–SDS gels and transferred to nitrocellulose blots. The blots were probed with a polyclonal antibody to PPARγ2, and the specific PPARγ2 band is indicated by an asterisk in the top row of blots. Likewise, 10 μg of total RNA was separated on 1% denaturing agarose gels and transferred to nitrocellulose blots. The blots were probed with an alkaline phosphatase-conjugated single-stranded oligonucleotide to FABP (aP2/422).
A number of factors appeared to influence the ability of VP16-CREB and KCREB to regulate adipogenesis. For example, the muristerone-induced expression of VP16-CREB alone is sufficient to induce adipogenesis and lipid accumulation. However, when VP16-CREB-expressing (muristerone-induced) cells are also treated with insulin, triacylglycerol accumulation is enhanced compared to cells treated with muristerone alone (Fig. 7). Further increases in lipid accumulation were observed in VP16-CREB-expressing cells treated with insulin and dexamethasone. Although insulin and dexamethasone appeared to potentiate lipid storage in these cells, these agents did not alter the percentage of cells undergoing adipogenesis. Whether the differences in lipid accumulation reflect overall changes in differentiation-related processes or simply increases in glucose uptake and/or triacylglycerol synthesis and storage has not been determined.
FIG. 7.
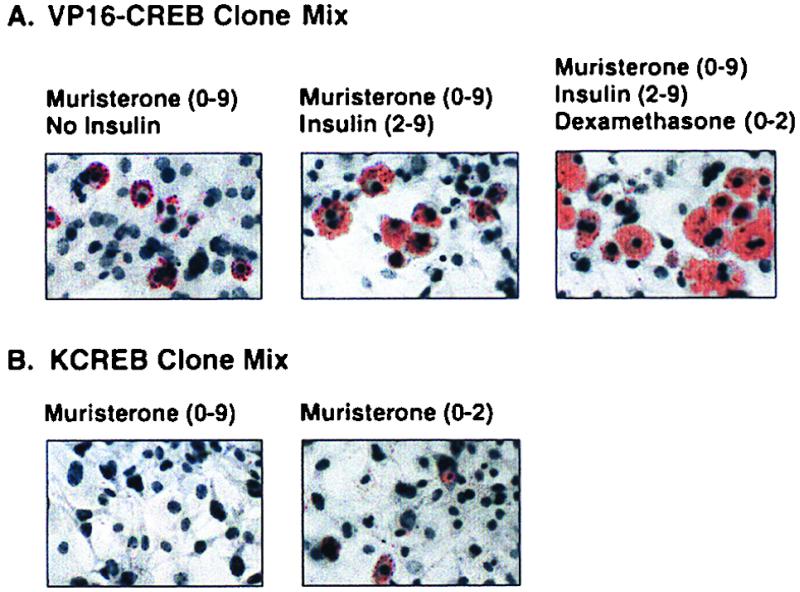
Parameters of adipogenesis in VP16-CREB- or KCREB-expressing 3T3-L1 cells. Mixtures of clonal cell lines expressing VP16-CREB (clone 2-4 plus clone 9-7) or KCREB (clone 2-1 plus clone 2-10) were passaged as described in Materials and Methods as indicated. Cultures of these mixed cell populations were treated with the agents listed above each photograph. The numbers in parentheses indicates the days on which the agents were included in the growth medium for the cells. After 9 days in culture, the cells were fixed and stained with Oil Red O and counterstained with hematoxylin.
In similar experiments we noted that the “complete” inhibition of adipogenesis required the constitutive expression of KCREB. As shown in Fig. 7, when KCREB expression was induced with muristerone for only the first 48 h of the experiment approximately 5 to 10% of the cells exhibited low levels of triacylglycerol storage and a rounded morphology. In contrast, no cells exhibited lipid accumulation when treated with muristerone to induce KCREB expression for the entire 9- or 10-day differentiation period.
CREB regulates adipocyte-specific genes.
How does CREB modulate adipogenesis? Most likely this process occurs through the activation of genes that drive clonal expansion and/or the adipogenic cascade and/or the expression of adipocyte phenotype markers. As an initial evaluation of this hypothesis, we tested the ability of CREB to bind to known DNA binding and/or regulatory sequences from several adipocyte-specific gene promoters, including PEPCK, FABP, FAS, PPARγ2, stearoyl-coenzyme A desaturase (SCD), and CEBPα, -β, and -δ. These sites were selected based on their ability to mediate transcriptional regulation in response to cAMP mimetics and/or insulin, their participation in gene expression during adipogenesis (13, 15, 43, 44, 47, 73), and their significant homology to the consensus CRE sequence (Fig. 8A). We first assessed the ability of purified, recombinant CREB to bind double-stranded oligonucleotide probes of these sequences in gel retardation assays. Recombinant CREB was able to bind to the probes of sequences for the promoters of the PEPCK, FABP, FAS, SCD, and CEBPβ and -δ genes but not of the PPARγ2 or CEBPα genes (Fig. 8B). Likewise, endogenous CREB present in 3T3-L1 fibroblast nuclear extracts was shown to bind some of these promoter sequences in “supershift” gel retardation assays. Reactions containing antibody which recognizes total CREB exhibited an additional “supershifted” band that was absent in reactions lacking the CREB antibody with oligonucleotides to putative CRE sequences in the PEPCK, FABP, FAS, and CEBPβ promoters (Fig. 8C). No supershifted complex was observed in reactions performed with a nonspecific probe, although a factor(s) present in the nuclear extracts was able to bind this sequence. The DNA-binding activity observed in nuclear extracts appeared to be due primarily to CREB since very little binding was observed in reactions performed with nuclear extracts from cells expressing KCREB (Fig. 8D), which specifically blocks CREB DNA binding activity. These data provide preliminary evidence that CREB may participate in adipogenesis by binding to regulatory elements in the promoters of certain adipocyte-specific genes in a coordinated fashion with other regulatory factors.
The ability of CREB to regulate transcription from three adipocyte-specific gene promoters is demonstrated in Fig. 9. In these experiments, 3T3-L1 preadipocytes were transfected with plasmids containing the “full-length” promoters of the PEPCK, FABP, and FAS genes linked to a luciferase reporter gene. Transcription from all of these promoters could be stimulated by treating the cells with the conventional differentiation mixture. Cotransfection of the cells with a VP16-CREB expression vector also stimulated transcription from each of the promoters. Alternately, cotransfection of the cells with a KCREB expression vector consistently decreased basal transcription levels slightly from all three promoters and completely blocked the induction of luciferase production from them by the differentiation mixture. Thus, CREB not only binds to putative CRE sequences in these gene promoters but appears to directly modulate transcription from them.
FIG. 9.

CREB regulates transcription from adipocyte-specific gene promoters. Control or VP16-CREB- or KCREB-expressing 3T3-L1 fibroblasts were transfected with plasmids containing the full-length promoters of the PEPCK, FABP, or FAS genes linked to luciferase. The cells were cotransfected with the internal control plasmid, pRSV-βGal. The following day the cells were treated with muristerone to induce either VP16-CREB or KCREB expression as indicated and/or with the conventional differentiation mixture of 10 μg of insulin per ml, 1 μM dexamethasone, and 3 mM Bt2cAMP for 4 h. Cell lysates were then prepared, and luciferase activity was measured as an index of transcriptional activity. Levels of transcription are shown relative to levels measured in untreated cells for each promoter tested and were then corrected for transfection efficiency.
DISCUSSION
The data presented here demonstrate that CREB activation is necessary and sufficient to initiate the adipocyte differentiation program. This conclusion is based on the constitutive expression of CREB in 3T3-L1 fibroblasts prior to the induction of adipogenesis and throughout the differentiation process. Furthermore, both CREB phosphorylation and transcriptional activity are rapidly induced in 3T3-L1 fibroblasts by conventional differentiation-inducing agents, and CREB appears to bind to and stimulate transcription from the promoters of several adipocyte-specific genes. Most importantly, we have directly demonstrated that CREB stimulates adipogenesis through our ability to induce adipocyte differentiation with constitutively active VP16-CREB and to completely block the efficacy of normal differentiation-inducing agents with dominant-negative KCREB. Obviously, there are caveats to these conclusions based on studies with VP16-CREB and KCREB. The properties of VP16-CREB may differ significantly form those of wild-type CREB, and KCREB may alter the function of factors other than CREB. However, the strength of our conclusion is founded on complementary results generated with positive and negative forms of CREB that elicit opposing responses. In addition, the ability of the chimeric VP16-KCREB protein to block adipogenesis indicates that our data are not due to indirect or nonspecific effects such as transcriptional squelching. Our conclusion is further supported by the ability of VP16-CREB and KCREB to regulate transcription from well-defined, CRE-containing, adipocyte-specific gene promoters.
The induction of adipogenesis by VP16-CREB alone indicates that CREB activation is sufficient to induce this process, whereas the ability of KCREB to block adipogenesis indicates that CREB activation is a necessary step in adipocyte development. These conclusions are significant because factors previously identified as participants in adipogenesis are only expressed in significant levels after initiation of the differentiation program. Our results suggest that CREB is a primary inducer of adipogenesis and, therefore, a potential target for intercellular signaling mechanisms that recruit the development of new fat cells in hyperplastic obesity. Further, CREB and the signaling systems that impinge on CREB may prove to be targets for therapeutic agents to treat or prevent obesity. Interestingly, preliminary experiments in our laboratory indicate that constitutive overexpression of KCREB in mature adipocytes leads to their dedifferentiation with loss of triacylglycerol vesicles, even in the presence of insulin (data not shown). Unger and colleagues (76) have recently reported a similar reversal of adipocyte phenotype in normal rats after overexpression of leptin. These studies support the contention that adipocyte development and function can be regulated at various levels, thus opening the door to novel strategies designed to address obesity and related disorders such as insulin resistance.
Our data further confirm the concept that CREB and other ATF-cAMP response element modulator (CREM)-inducible cAMP early repressor (ICER) family members play important roles in multiple cellular activities, most notably proliferation and differentiation. Initial clues to CREB's participation in these activities came from studies showing that several growth factors and other extracellular stimuli activate CREB. We demonstrated that insulin stimulates CREB phosphorylation in 3T3-L1 fibroblasts and adipocytes and HepG2 cells through an ERK1/ERK2 signaling system (34) and a decrease in nuclear PP2A activity (49, 50). Greenberg, and colleagues have reported a similar signaling cascade to CREB for nerve growth factor in neuronal cells (27, 71, 72). Likewise, fibroblast growth factor (60) and insulin-like growth factor 1 (46) also stimulates CREB phosphorylation and activity in neuronal cells, but this process appears to be mediated by p38 MAP kinase rather than ERK1/ERK2. CREB and related proteins have also been implicated in the G1-S transition of the cell cycle in studies showing that cyclin A gene transcription is stimulated by cAMP agonists via CRE sequences in the cyclin A gene promoter (19).
In addition to this circumstantial evidence promoting a role for CREB and related factors in cell growth and differentiation, several groups have recently reported direct evidence supporting this hypothesis. For example, Shimomura et al. (56) have reported that a dominant-negative ATF-1 protein blocks cAMP-induced neurite outgrowth in PC12 cells. Likewise, ectopic expression of a dominant-negative CREB protein in pituitary somatotrophic cells leads to somatotroph hypoplasia and dwarfism in transgenic mice (59). Targeted expression of a dominant-negative CREB in cardiac myocytes has been shown to produce idiopathic-dilated cardiomyopathy with exaggerated heterogeneity in the myocyte phenotype (21). Surface antigen receptor activation of B lymphocyte proliferation appears to involve enhanced CREB phosphorylation in response to elevated PKA and PKC activity and downregulation of PP2A (4, 69, 70), and the expression of dominant-negative CREB in T lymphocytes blocks their proliferation after activation (9). CREB null transgenic mice exhibit perinatal mortality, reduced corpus callosum and anterior commissures in the brain, decreased thymic cellularity, and impaired T lymphocyte development (52). cAMP signaling to CREM and ICER via PKA has been shown to play a role in hepatocyte proliferation (53, 54), and CREB phosphorylation directly inhibits hepatic stellate cell proliferation (31). Similarly, cAMP-induced ICER IIγ expression blocks the proliferation of either mouse pituitary tumor cells or human choriocarcinoma cells at the G2-M boundary (48). Lamas et al (36) have reported that the CREB inhibitor, ICER, modulates pituitary corticotroph proliferation. In other studies, the tissue-specific extinguisher locus (TSE-1) identified by Fournier and colleagues (11, 32, 61), which presumably blocks PKA signaling to CREB and other factors, accounts for loss of hepatocyte phenotype markers in hepatoma-fibroblast hybrids. The data presented here extend the multifunctional role of CREB by demonstrating for the first time that activation of this factor is necessary and sufficient to induce a differentiation program by using constitutively active and dominant-negative forms of CREB.
One concern raised by these studies regards the paradoxical role of cAMP signaling in both adipogenesis and lipolysis. Our data are consistent with previous reports demonstrating a key role for cAMP in potentiating adipogenesis (40, 74). However, other laboratories have shown that β3-adrenergic stimulation of cAMP-PKA signalling increases lipolysis (16, 18, 66), and targeted knockout of the RIIβ subunit of PKA leads to decreased obesity in mice (17). These contradictory processes may be reconciled based on different roles for cAMP-PKA signaling between undifferentiated fibroblasts compared to mature adipocytes. Similarly, differentiation of fibroblasts to adipocytes is induced by the transient application of high levels of cAMP mimetics, whereas β3-adrenergic stimulation or RIIβ subunit knockout probably represents protracted increases in cAMP-PKA signaling. Thus, differences in experimental models may account for the seemingly contradictory role of cAMP in adipogenesis and lipolysis. Moreover, it should be remembered that cAMP and PKA regulate numerous intracellular systems and not just CREB and that, more importantly, CREB function can be regulated by a variety of growth factors and not just increases in cAMP. Together, these concepts support a model in which multiple signals may impinge upon CREB to induce adipogenesis in fibroblasts, whereas lipolysis is the result of cAMP-PKA signaling to increase the activity of lipolytic pathways in mature adipocytes. Obviously, this is an area which will require significant investigation to unravel the underlying factors, their roles, and their interactions.
Another question not fully addressed by these studies concerns the target(s) which CREB modulates in order to induce adipogenesis. Our preliminary data indicate that CREB can bind to putative CREs in the promoters of several adipocyte-specific genes. Most the sequences we examined (with the exception of the CEBPδ sequence) have been shown by other groups to interact with nuclear factors and to participate in gene expression in response to cAMP and/or insulin (13, 15, 43, 44, 47, 73). Furthermore, the genes encoding PEPCK, FABP, and CEBPβ have been shown to be acutely regulated by cAMP or insulin, and the PEPCK and CEBPβ sequences we tested have been shown to confer cAMP and CREB responsiveness on these genes. Certainly, our data are insufficient to permit us to conclude that CREB directly regulates the genes we selected. However, the results provide tantalizing evidence that CREB may regulate certain adipocyte-specific genes, which would support a role for CREB in adipogenesis. We have initiated experiments to directly asses CREB's role in regulating a group of candidate genes, as well as identify other “CREB-regulated, adipocyte-specific” genes via gene microarray analysis.
The binding of CREB to an oligonucleotide probe corresponding to a sequence in the CEBPβ promoter was particularly interesting. As noted before, CEBPβ is expressed very early in adipogenesis and will induce the differentiation of fibroblasts to adipocytes when expressed ectopically (75). Our data suggest that one mechanims by which CREB may induce adipocyte differentiation is through an ability to stimulate CEBPβ expression, which may be sufficient to induce the entire adipogenic cascade. If true, it should be possible to block CREB-induced adipogenesis by inhibiting CEBPβ expression or activity. Figure 2B shows that CREB undergoes cyclical increases and decreases in phosphorylation (and presumably in transcriptional activity) during adipogenesis. These results imply that CREB may be crucial at other steps in adipocyte differentiation—from an initial stimulation of CEBPβ expression to the late expression of genes encoding PEPCK, FABP, and FAS.
How does CREB regulate growth in certain cell lines and differentiation in others? One possible mechanism hinges on the availability or accessibility of proliferation-related genes in some cells and tissues versus the accessibility of differentiation-inducing genes and phenotype markers in other cell types. Applying this mechanism to adipogenesis suggests that only differentiation-inducing and/or adipocyte-specific genes rather than proliferation-inducing are accessible to CREB in preadipocytes. Another possible mechanisms focuses on the interactions of CREB with other transcription factors that, in concert, exert proliferative versus differentiation-inducing effects in a cell- or tissue-dependent manner. Interactions between CREB and other transcription factors have been described in several systems, but their role in adipogenesis remains unclear. A number of possible mechanisms may account for CREB's participation in both proliferation and differentiation pathways. It will be interesting to determine which mechanisms are actually functioning in these capacities and to define potential interactions between the mechanisms in the coordinate regulation of these processes.
ACKNOWLEDGMENTS
This research was supported by Public Health Service grants GM47117 and DK53969 (to D.J.K.) and DK0235, and Veterans Administration Merit and Career Development Awards (to J.E.B.R.).
We thank Richard Goodman (Vollum Institute, Oregon Health Science University, Portland) for reviewing the manuscript and providing suggestions for its improvement.
REFERENCES
- 1.Abdel-Hafiz H A-M, Heasley L E, Kyriakis J M, Avruch J, Kroll D J, Johnson G L, Hoeffler J P. Activating transcription factor-2 DNA-binding activity is stimulated by phosphoryation catalyzed by p42 and p54 microtubule-associated protein kinases. Mol Endocrinol. 1992;6:2079–2089. doi: 10.1210/mend.6.12.1337144. [DOI] [PubMed] [Google Scholar]
- 2.Abraham S, Johnson C L. Prevalence of severe obesity in adults in the United States. Am J Clin Nutr. 1980;33:364–369. doi: 10.1093/ajcn/33.2.364. [DOI] [PubMed] [Google Scholar]
- 3.Adya N, Zhao L-J, Huang W, Boros I, Giam C-Z. Expansion of CREBs DNA recognition specificity by Tax results from interaction with Ala-Ala-Arg at positions 282-284 near the conserved DNA-binding domain of CREB. Proc Natl Acad Sci USA. 1994;91:5642–5646. doi: 10.1073/pnas.91.12.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amato S F, Nakajima K, Hirano T, Chiles T C. Transcriptional regulation of the junB gene in B lymphocytes: role of protein kinase A and a membrane Ig-regulated protein phosphatase. J Immunol. 1997;159:4676–4685. [PubMed] [Google Scholar]
- 5.Anonymous. Build study, 1979. Washington, D.C.: Society of Actuaries and Association of Life Insurance Medical Directors of America; 1980. [Google Scholar]
- 6.Anonymous. National health and nutrition examination survey III, 1988–94: public use data on CD-ROM. Vol. 1996. 1998. [Computer file.] U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics, Washington, D.C. [Google Scholar]
- 7.Arany Z, Newsome D, Oldread E, Livingston D M, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 8.Barnabas S, Hai T, Andrisani O M. The hepatitis B virus X protein enhances the DNA binding potential and transcriptional efficacy of bZip transcription factors. J Biol Chem. 1997;272:20684–20690. doi: 10.1074/jbc.272.33.20684. [DOI] [PubMed] [Google Scholar]
- 9.Barton K, Muthusamy N, Chanyangam M, Fischer C, Clendenin C, Leiden J M. Defective thymocyte proliferation and IL-2 production in transgenic mice expressing a dominant negative CREB. Nature (London) 1996;379:81–85. doi: 10.1038/379081a0. [DOI] [PubMed] [Google Scholar]
- 10.Black D, James W P T, Besser G M. Obesity. A report of the Royal College of Physicians. J R Coll Phys (London) 1983;17:5–65. [PMC free article] [PubMed] [Google Scholar]
- 11.Boshart M, Weih F, Schmidt A, Fournier R E K, Schütz G. A cyclic AMP response element mediates repression of tyrosine aminotransferase gene transcription by the tissue-specific extinguisher locus Tse-1. Cell. 1990;61:905–916. doi: 10.1016/0092-8674(90)90201-o. [DOI] [PubMed] [Google Scholar]
- 12.Bray G A. Obesity in America: an overview. In: Bray G A, editor. Obesity in America. DHEW publication no. (NIH) 79-359. Vol. 1979. Washington, D.C.: Government Printing Office; 1979. pp. 1–19. [Google Scholar]
- 13.Casimir D A, Ntambi J M. cAMP activates the expression of stearoyl-CoA desaturase gene 1 during early preadipocyte differentiation. J Biol Chem. 1996;271:29847–29853. doi: 10.1074/jbc.271.47.29847. [DOI] [PubMed] [Google Scholar]
- 14.Chatton B, Bocco J L, Gaire M, Hauss C, Reimund B, Goetz J, Kedinger C. Transcriptional activation by the adenovirus larger E1a product is mediated by members of the cellular transcription factor ATF family which can directly associate with E1a. Mol Cell Biol. 1993;13:561–570. doi: 10.1128/mcb.13.1.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christy R J, Kaestner K H, Geiman D E, Lane M D. CCAAT/enhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci USA. 1991;88:2593–2597. doi: 10.1073/pnas.88.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins S, Daniel K W, Petro A E, Surwit R S. Strain-specific response to beta 3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. 1997;138:405–413. doi: 10.1210/endo.138.1.4829. [DOI] [PubMed] [Google Scholar]
- 17.Cummings D E, Branden E P, Planas J V, Motamed K, Idzerda R L, McKnight G S. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- 18.Danforth E J, Himms-Hagen J H. Obesity and diabetes and the beta 3-adrenergic receptor. Eur J Endocrinol. 1997;136:362–365. doi: 10.1530/eje.0.1360362. [DOI] [PubMed] [Google Scholar]
- 19.Desdouets C, Matesic G, Molina C A, Foulkes N S, Sassone-Corsi P, Brechot C, Sobczak-Thepot J. Cell cycle regulation of cyclin A gene expression by the cyclic AMP-responsive transcription factors CREB and CREM. Mol Cell Biol. 1995;15:3301–3309. doi: 10.1128/mcb.15.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drenick E J, Bale G S, Seltzer F. Excessive mortality and causes of death in morbidly obese men. JAMA. 1980;243:443–445. [PubMed] [Google Scholar]
- 21.Fentzke R C, Korcarz C E, Lang R M, Lin H, Leiden J M. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest. 1998;101:2415–2426. doi: 10.1172/JCI2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foreyt J P, Poston W S., II Obesity: a never-ending cycle? International J Fertility Women's Med. 1998;43:111–116. [PubMed] [Google Scholar]
- 23.Foster D W. Eating disorders: obesity, anorexia nervosa, and bulimia nervosa. In: Wilson J D, Foster D W, editors. William's textbook of endocrinology. W. B. Philadelphia, Pa: Saunders Co.; 1992. pp. 1335–1365. [Google Scholar]
- 24.Freytag S O, Paielli D L, Gilbert J D. Ectopic expression of the CCAAT/enhancer-binding protein a promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev. 1994;8:1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- 25.Giebler H A, Loring J E, van Orden K, Colgin M A, Garrus J E, Escudero K W, Brauweiler A, Nyborg J K. Anchoring of CREB binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol Cell Biol. 1997;17:5156–5164. doi: 10.1128/mcb.17.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ginsberg-Fellner F, Knittle J L. Weight reduction in young obese children. I. Effects of adipose tissue cellularity and metabolism. Pediatr Res. 1981;15:1381–1389. doi: 10.1203/00006450-198110000-00016. [DOI] [PubMed] [Google Scholar]
- 27.Ginty D D, Bonni A, Greenberg M E. Nerve growth factor activates a ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell. 1994;77:713–725. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 28.Green H, Kehinde O. An established preadipose cell line and its differentiation in culture II. Factors affecting the adipose conversion. Cell. 1975;5:19–27. doi: 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- 29.Gupta S, Campbell D, Derijard B, Davis R J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 30.Hirsch J, Batchelor B. Adipose tissue cellularity and human obesity. Clin Endocrinol Metab. 1976;5:299–311. doi: 10.1016/s0300-595x(76)80023-0. [DOI] [PubMed] [Google Scholar]
- 31.Houglum K, Lee K S, Chojkier M. Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. J Clin Investig. 1997;99:1322–1328. doi: 10.1172/JCI119291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones K W, Shapero M H, Chevrette M, Fournier R E K. Subtractive hybridization cloning of a tissue-specific extinguisher: TSE1 encodes a regulatory subunit of protein kinase A. Cell. 1991;66:861–872. doi: 10.1016/0092-8674(91)90433-y. [DOI] [PubMed] [Google Scholar]
- 33.Kim J B, Spiegelman B M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- 34.Klemm D J, Roesler W J, Boras T, Colton L A, Felder K, Reusch J E-B. Insulin stimulates cAMP-response element binding protein activity in HepG2 and 3T3-L1 cell lines. J Biol Chem. 1998;273:917–923. doi: 10.1074/jbc.273.2.917. [DOI] [PubMed] [Google Scholar]
- 35.Knittle J L, Timmers K, Ginsberg-Fellner F. The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell number and size. J Clin Investig. 1979;63:239–246. doi: 10.1172/JCI109295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamas M, Molina C, Foulkes N S, Jansen E, Sassone-Corsi P. Ectopic ICER expression in pituitary corticotroph AtT20 cells: effects on morphology, cell cycle, and hormonal production. Mol Endocrinol. 1997;11:1425–1434. doi: 10.1210/mend.11.10.9987. [DOI] [PubMed] [Google Scholar]
- 37.Lee J-S, Zhang X, Shi Y. Differential interactions of the CREB/ATF family of transcription factors with p300 and adenovirus E1A. J Biol Chem. 1996;271:17666–17674. [PubMed] [Google Scholar]
- 38.Liu F, Green M R. A specific member of the ATF transcription factor family can mediate transcription activation by the adenovirus E1a protein. Cell. 1990;61:1217–1224. doi: 10.1016/0092-8674(90)90686-9. [DOI] [PubMed] [Google Scholar]
- 39.Lundblad J R, Kwok R P S, Laurance M E, Harter M L, Goodman R H. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature. 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 40.MacDougald O A, Lane M D. Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem. 1995;64:345–373. doi: 10.1146/annurev.bi.64.070195.002021. [DOI] [PubMed] [Google Scholar]
- 41.Maguire H F, Hoeffler J P, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 42.McIntyre A M. Burden of illness review of obesity: are the true costs realised? J R Soc Health. 1998;118:76–84. doi: 10.1177/146642409811800207. [DOI] [PubMed] [Google Scholar]
- 43.Moustaïd N, Beyers R S, Sul H S. Identification of an insulin response element in the fatty acid synthase promoter. J Biol Chem. 1994;269:5629–5634. [PubMed] [Google Scholar]
- 44.Niehof M, Manns M P, Trautwein C. CREB controls LAP/C/EBPβ transcription. Mol Cell Biol. 1997;17:3600–3613. doi: 10.1128/mcb.17.7.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pi-Sunyer F-X, Laferrere B, Aronne L J, Bray G A. Obesity—a modern-day epidemic. J Clin Endocrinol Metab. 1999;84:3–7. doi: 10.1210/jcem.84.1.5392-1. [DOI] [PubMed] [Google Scholar]
- 46.Pugazhenthi S, Boras T, O'Connor D, Meintzer M K, Heidenreich K A, Reusch J E. Insulin-like growth factor 1-mediated activation of the transcription factor cAMP response element binding protein in PC12 cells. Involvement of p38 mitogen-activated protein kinase-mediated pathway. J Biol Chem. 1998;274:2829–2837. doi: 10.1074/jbc.274.5.2829. [DOI] [PubMed] [Google Scholar]
- 47.Quinn P G. Inhibition by insulin of protein kinase A-induced transcription of the phosphoenolpyruvate carboxykinase gene. J Biol Chem. 1994;269:14375–14378. [PubMed] [Google Scholar]
- 48.Razavi R, Ramos J C, Yehia G, Schlotter F, Molina C A. ICER-IIgamma is a tumor suppressor that mediates the antiproliferative activity of cAMP. Oncogene. 1998;17:3015–3019. doi: 10.1038/sj.onc.1202225. [DOI] [PubMed] [Google Scholar]
- 49.Reusch J E-B, Hsieh P, Bhuripanyo P, Carel K, Leitner J W, Olefsky J M, Draznin B. Insulin inhibits nuclear phosphatase activity: requirement for the C-terminal domain of the insulin receptor. Endocrinology. 1995;136:2464–2469. doi: 10.1210/endo.136.6.7750468. [DOI] [PubMed] [Google Scholar]
- 50.Reusch J E-B, Hsieh P, Klemm D, Hoeffler J, Draznin B. Insulin inhibits dephosphorylation of adenosine 3′,5′-monophosphate response element-binding protein/activating transcription factor-1: effect on nuclear phosphoserine phosphatase-2a. Endocrinology. 1994;135:2418–2422. doi: 10.1210/endo.135.6.7988426. [DOI] [PubMed] [Google Scholar]
- 51.Roncari D A K, Lau D C W, Kindler S. Exaggerated replication in culture of adipocyte precursors from massively obese persons. Metabolism. 1981;30:425–427. doi: 10.1016/0026-0495(81)90174-8. [DOI] [PubMed] [Google Scholar]
- 52.Rudolph D, Tafuri A, Gass P, Hammerling G J, Arnold B, Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci USA. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Servillo G, Della Fazia M A, Sassone-Corsi P. Transcription factor CREM coordinates the timing of hepatocyte proliferation in the regenerating liver. Genes Dev. 1997;12:3639–3643. doi: 10.1101/gad.12.23.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Servillo G, Panna L, Foulkes N S, Magni M V, Della Fazia M A, Sassone-Corsi P. Cyclic AMP signaling pathway and cellular proliferation: induction of CREM during liver regeneration. Oncogene. 1997;14:1601–1606. doi: 10.1038/sj.onc.1200996. [DOI] [PubMed] [Google Scholar]
- 55.Sheng M, Thompson M A, Greenberg M E. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 56.Shimomura A, Okamoto Y, Hirata Y, Kobayashi M, Kawakami K, Kiuchi K, Wakabayashi T, Hagiwara M. Dominant negative ATF1 blocks cycli AMP-induced neurite outgrowth in PC12D cells. J Neurochem. 1998;70:1029–1034. doi: 10.1046/j.1471-4159.1998.70031029.x. [DOI] [PubMed] [Google Scholar]
- 57.Slyper A H. Childhood obesity, adipose tissue distribution, and the pediatric practitioner. Pediatrics. 1998;102:e4. doi: 10.1542/peds.102.1.e4. [DOI] [PubMed] [Google Scholar]
- 58.Spiegelman B M, Flier J S. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 59.Struthers R S, Vale W W, Arias C, Sawchenko P E, Montminy M R. Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREB mutant. Nature. 1991;350:622–624. doi: 10.1038/350622a0. [DOI] [PubMed] [Google Scholar]
- 60.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb M J. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 61.Thayer M J, Lugo T G, Leach R J, Fournier R E K. Regulation of chimeric phosphoenolpyruvate carboxykinase genes by the trans-dominant locus TSE1. Mol Cell Biol. 1990;10:2660–2668. doi: 10.1128/mcb.10.6.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tontonoz P, Graves R A, Budavari A I, Erdjument-Bromage H, Lui M, Hu E, Tmepst P, Spiegelman B M. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPARγ and RXRα. Nucleic Acids Res. 1994;22:5628–5634. doi: 10.1093/nar/22.25.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tontonoz P, Hu E, Devine J, Beale E G, Spiegelman B M. PPARγ2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. 1995;15:351–357. doi: 10.1128/mcb.15.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tontonoz P, Hu E, Spiegelman B M. Stimulation of adipogenesis in fibroblastsby PPARγ2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 65.Walton K M, Rehfuss R P, Chrivia J C, Lochner J E, Goodman R H. A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol Endocrinol. 1992;6:647–655. doi: 10.1210/mend.6.4.1350057. [DOI] [PubMed] [Google Scholar]
- 66.Weyer C, Gautier J F, Danforth E J. Development of beta 3-adrenoreceptor agonists for the treatment of obesity and diabetes—an update. Diabetes Metab. 1999;25:11–21. [PubMed] [Google Scholar]
- 67.Wheat W H, Roesler W J, Klemm D J. Simian virus 40 small tumor antigen inhibits dephosphorylation of protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol. 1994;14:5881–5890. doi: 10.1128/mcb.14.9.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Z, Xie Y, Bucher N L R, Farmer S R. Conditional ectopic expression of C/EBP-β in NIH 3T3 cells induces PPARγ and stimulates adipogenesis. Genes Dev. 1995;9:2350–2356. doi: 10.1101/gad.9.19.2350. [DOI] [PubMed] [Google Scholar]
- 69.Xie H, Rothstein T L. Protein kinase C mediates activation of nuclear cAMP response element-binding protein (CREB) in B lymphocytes stimulated through surface Ig. J Immunol. 1995;154:1717–1723. [PubMed] [Google Scholar]
- 70.Xie H, Wang Z, Rothstein T L. Signaling pathways for antigen-mediated induction of transcription factor CREB in B lymphocytes. Cell Immunol. 1996;169:264–270. doi: 10.1006/cimm.1996.0117. [DOI] [PubMed] [Google Scholar]
- 71.Xing J, Ginty D D, Greenberg M E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 72.Xing J, Kornhauser J M, Xia Z, Thiele E A, Greenberg M E. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946–1955. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang V W, Christy R J, Cook J S, Kelly T J, Lane M D. Mechanism of regulation of the 422(aP2) gene by cAMP during preadipocyte differentiation. Proc Natl Acad Sci USA. 1989;86:3629–3633. doi: 10.1073/pnas.86.10.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yarwood S J, Kilgour E, Anderson N G. Cyclic AMP potentiates growth hormone-dependent differentiation of 3T3-F442A preadipocytes: possible involvement of the transcription factor CREB. Mol Cell Endocrinol. 1998;138:41–50. doi: 10.1016/s0303-7207(98)00049-5. [DOI] [PubMed] [Google Scholar]
- 75.Yeh W-C, Cao Z, Classon M, McKnight S L. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–181. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- 76.Zhou Y-T, Zhuo-Wei W, Higa M, Newgard C B, Unger R H. Reversing adipocyte differentiation: Implications for treatment of obesity. Proc Natl Acad Sci USA. 1999;96:2391–2395. doi: 10.1073/pnas.96.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]