

Abstract
Purpose of review:
The giant protein titin forms the “elastic” filament of the sarcomere, essential for the mechanical compliance of the heart muscle. Titin serves a biological spring, and therefore structural modifications of titin affect function of the myocardium and are associated with heart failure and cardiomyopathy.
Recent findings:
In this review, we discuss the current understanding of titin’s biophysical properties and how modifications contribute to cardiac function and heart failure. In addition, we review the most recent data on the clinical impact and phenotype heterogeneity of TTN truncating variants, including diseases involving striated muscles, and prospects for future therapies.
Summary:
Because of the giant structure of the titin protein and the complexity of its function, titin’s role in health and disease is not yet completely understood. Future research efforts need to focus on novel therapeutic approaches able to modulate titin transcriptional and post-translational modification.
Keywords: dilated cardiomyopathy, titin, diastolic dysfunction, sarcomere, genetic variants, heart failure, genetic testing
Introduction
The heart’s primary functions are to both collect blood from the body as well as generate contractile force and deliver blood to the lungs and systemic organ tissues. The relaxation phase is termed diastole while the contractile phase is terms systole. Both of these functions must be perfectly coordinated such that the input of blood to the heart equals the output of blood the rest of the body. Dysfunction of either systole or diastole will cause an upstream accumulation of fluid in either systemic tissue or the lungs leading to congestive heart failure (CHF). CHF is a complex syndrome that affects 6.2 million people in the United States and 23 million people worldwide.1, 2 While the clinical presentation of CHF is homogenous with development of pulmonary edema, systemic edema, and progressive dyspnea; the underlying causes and findings of CHF are heterogenous. CHF can be classified by physiologic presentation with abnormalities either in contractile function of the heart termed systolic dysfunction, or abnormalities in relaxation of the heart termed diastolic dysfunction. Systolic dysfunction can be further classified based on the anatomic appearance of the dysfunctional ventricle which can show cavitary enlargement of the ventricle with a thin wall, or a reduction in the size of the ventricular cavity and a thick wall, termed dilated cardiomyopathy (DCM) or hypertrophic cardiomyopathy (HCM) respectively.
CHF can also be defined by the cause or etiology that leads to cardiac dysfunction. The most common cause of heart failure in the world is ischemic, where obstruction or occlusion of the coronary arteries leads to decreased myocardial perfusion causing ischemia and eventually infarction of cardiac tissue. Nonischemic cardiomyopathy (NICM) is defined as a loss of function of one or both ventricles in absence of significant coronary artery disease.3 NICM can be secondary to a number of conditions that compromise the structure and contractile function of the heart tissue including infiltrative conditions, metabolic diseases, myocarditis, medications, and toxins. However, in up to 50% of NICM patients, the etiology is idiopathic and can often be ascribed to genetic causes.
In nearly all causes of heart failure, there are changes and dysfunction of the cardiac sarcomere, which is the fundamental intracellular structure that generates contractile force. An essential component of the sarcomere, is the protein titin which is the largest macromolecule in the human body and is composed of 27,000–33,000 amino acids.4 Titin is heavily expressed in striated muscle tissue including cardiac myocytes. This filamentous protein is greater than 1 micrometer in length and spans half the length on an entire sarcomere connecting the Z-disk (via N-terminal domain) to the M-line (via C-terminal domain)5 (Figure 1). Titin is encoded by the gene TTN, which contain 363 exons6. Modifications of TTN at the genetic, transcriptional, and post-translational levels are associated with the development of all forms of heart failure. The importance of TTN is further highlighted by the observation that gene mutations that result in early termination or truncations (TTNtv) are the most common cause of genetic forms of DCM and account for 20–25% of cases7.
Figure 1. The sarcomere structure and titin.
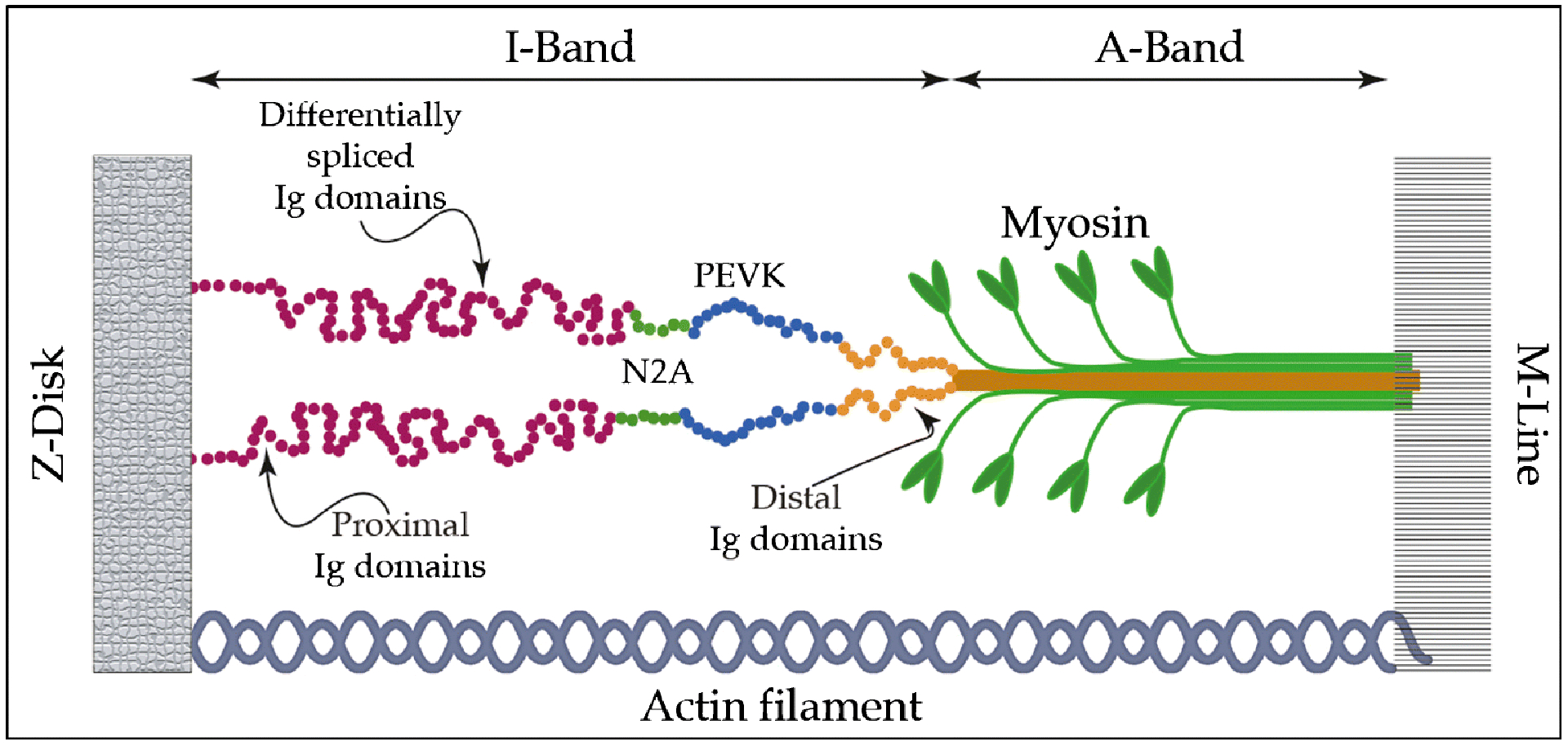
The basic elements of the sarcomere include myosin, which forms the thick filament, actin forming the thin filament, and a third elastic filament constituted by titin. Titin is a giant protein of 27,000 to 33,000 amino acids with a molecular weight of 3M Dalton, which spans from the M-band to the Z-disk. The I-band region of the molecule, including a series of immunoglobulin-like (Ig) domains, functions as a molecular spring. In adult cardiac muscle, this includes N2BA isoform with expanded N2A element shown in green. (Ig) domains make up a proximal (red) and distal (orange) tandem (Ig) segments. In blue, the PEVK sequence.
In this review, we will highlight the structural components of titin and how they contribute to the essential biophysical properties of cardiac function, we will review how transcriptional and post-translational modifications of titin are associated with heart failure, we will discuss mechanisms by which TTNtv lead to DCM, and we will conclude with therapeutic modalities for treatment of CHF by modification of titin.
Biophysical properties of titin spring elements
The “spring protein” titin can be considered as a series of elastic elements with a modular structure: these modules are connected in series. TTN is organized into four major domains. The N-terminal domain anchors titin to the Z-disk of the sarcomere, while the C-terminal domain anchors titin to the M-band. The remaining two domains impart important biophysical characteristics to titin. The I-band is responsible for the elastic behavior of titin and accountable for the passive elasticity of cardiac muscle. The A-band, an inextensible domain responsible for the organization of the sarcomere. The A-band is rigidly attached to the myosin filament with no or minimal possibility for elongation8–11.
The elastic, I-band does not represent a uniform molecular spring, but instead consists of four structurally distinct segments: two tandem immunoglobulin-like (Ig) domains and two additional spring elements. The two (Ig) domain regions have different mechanical properties: the distal region consisting of (Ig) domains arranged in tandem, and a proximal region consisting of differentially spliced (Ig) domains (Figure 1). In addition to the (Ig) domains, spring-like properties of titin are imparted by the N2A element consisting of several (Ig) domains interspersed with unique sequences, and the PEVK element, considered to be a naturally disordered protein region, consisting for around 70% of proline (P), glutamate (E), valine (V), and lysine (K) residues.
These four spring elements react in different ways under a stretching force, suggesting that these regions have different mechanical properties.12, 13 The N2A and PEVK segments are flexible random-coil like sequences, with dissimilar persistence length (a measure of molecule flexibility).14,15, 16 On the contrary, both proximal and distal (Ig) domains, have a β-sheet (BS) structure with the same persistence length. BS structures show high strength, offering resistance when loaded, approximately ten times higher than the maximum force in an alpha helix. From a biomechanical point of view, single molecule atomic force microscopy (AFM) studies show that β-sheets resemble a “worm like chain” where increased force leads to discreet unfolding of components of the BS (Figure 2). Interestingly, even though the two (Ig) domains have the same persistence length, they show different unfolding as well as extensibility behavior, making the proximal less stable than the distal one. When force is applied, the distal (Ig) domains stretch through an unfolding intermediate state. This unfolding intermediate state provides increased mechanical resistance.14, 17 The mechanical unfolding of the proximal domain does not involve an unfolding intermediate state and therefore is an all-or-none event. Unfolding forces for these (Ig) domains are different: 127 (± 18) pN: for the proximal, and 210 (± 32) pN for the distal domain. In addition to the different mechanical stability, the two (Ig) domains also show a difference in contour length increment during domain unfolding (contour length is the molecule length at maximum physically possible extension). The measured contour length increment for the distal one is ΔLc = (27,9±0.79) nm while for the proximal is ΔLc = (30±0.62) nm.
Figure 2. Structural components of titin unfold at different extension forces, imparting a wide range of extensibility.
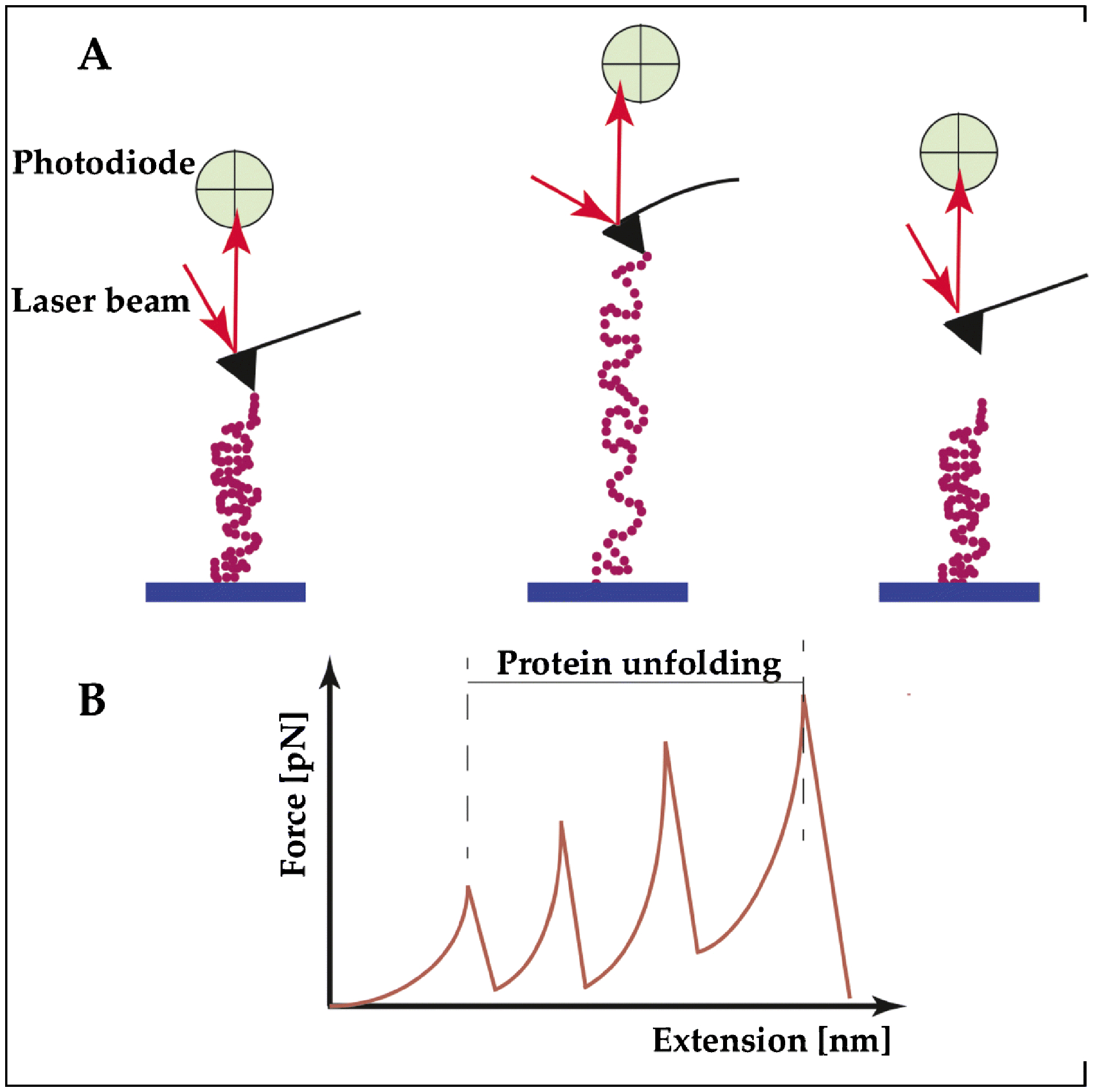
(A) Example of a single-molecule force spectroscopy using the atomic force microscope (AFM) to study the contribution of the individual building blocks of titin under stretching. Left: The TTN segment to be tested is deposited on a substrate, and then attached at one end at the tip of an elastic AFM cantilever. Middle: The peptide is pulled up by a piezoelectric controller. When the proteins start to unfold, the AFM cantilever is deflected and its deflection is measured by a laser beam. This deflection is transformed in a force curve. Right: The TTN peptide detaches when it reaches the maximum stretch. (B) Titin proteins have an intrinsic elasticity that causes them to remain rigid at small length scales yet display significant flexibility over longer lengths. One of the theories that describes this behavior and the progressive unfolding of the different segments of TTN is the “worm-like chain” model of semiflexible polymers, as shown in panel B.
In the absence of external force, the (Ig) domains are in their natural, folded state and the linker regions between the domains adopt a bent configuration. When the sarcomeres are “passively” stretched, e.g., by the antagonist muscle or diastolic pressure, the (Ig) domains and the PEVK region extend serially. When low forces are applied to stretch the sarcomere, the linker regions connecting the (Ig) domains are unbent even though the (Ig) domains remain partially folded:18, 19 These mechanisms allows initial extensibility at low force.18, 20 When the stretch force increases, the PEVK and (Ig) domains begin to extend and provide ample extensibility.13, 19–21 Upon release, the unfolded (Ig) domains perform a folding contraction under force, which generates work (Figure 2).
The biophysical characteristics of the I-band contribute to the physiology of global cardiac function. The elastic nature of the I-band and the wide range of extensibility depending on force applied contributes to length dependent activation described in the Frank-Starling curve. The Frank-Starling curve depicts an increase in the contractile force of cardiac myocytes with increased ventricular filling during diastole. When there is more cardiac filling and increased left ventricular end diastolic pressure, unfolding of the titin spring domains provides increased restorative force that contributes to increased force of contraction.22, 23
Transcriptional modifications of TTN modify extensibility and biophysical properties
The elastic components of titin impart important functional properties to the sarcomere and mechanics of cardiomyocyte function. One way that the spring function of titin can be modified or tuned is through transcriptional modifications that increase or decrease passive tension and elasticity. TTN contains 363 exons and demonstrates extensive transcriptional modification via alternative splicing of these exons. There are different splicing events that transcribe multiple isoforms of TTN, particularly in the I-band containing multiple (Ig) domains.24 These isoforms vary significantly in length and compliance. The 3 most well described TTN isoforms are FCT, N2B, and N2BA. FCT is the longest of the 3 isoforms and is primary expressed in fetal tissue with a shift towards predominately transcribing N2B and N2BA in the post-natal period. N2B is a shorter, stiffer isoform containing a tandem N2B and short PEVK segment, while N2BA is a longer more compliant molecule that has a longer PEVK segment and increased number of (Ig) domains.24, 25 Although both the N2B and N2BA isoforms are co-expressed in adult cardiomyocytes, the ratio of the two isoforms determines the relative stiffness of global cardiac tissue. Transcriptional upregulation of the more compliant isoform, N2BA, has been described in various cardiovascular disease states associated with decreased systolic function and ventricular dilation.26 Furthermore, echocardiographic correlation has demonstrated that patients with higher expression of N2BA have increased end diastolic volume: pressure ratio which correlates with decreased ventricular stiffness.27 The mechanisms and regulation by which different TTN isoforms are selected and expressed is incompletely understood, however one important factor is RNA binding motif protein 20 (RBM20). RBM20 is a protein that is incorporated into the spliceosome, which splices out introns and selects exons for incorporation into mature RNA. Humans and model animals with RBM20 mutations have very high relative expression of the N2BA isoform. Furthermore, RBM20 mutants develop severe DCM, which demonstrates that when there is high cardiac expression of a longer titin isoform that contains more springy (Ig) elements, there is decreased ventricular passive tension and development of ventricular dilation.28, 29
Post-translational modifications further alter titin’s mechanical properties
In addition to the molecular secondary and tertiary structure of the spring elements that are modified by isoform selection, post-translational modifications also contribute to the mechanical properties of titin. Phosphorylation of the N2B element show an increase in the persistence length of the titin as measured by AFM single-molecule force spectroscopy. This indicates that the bending rigidity of the titin is increased by phosphorylation of the N2B region.30 In addition, phosphorylation of the PEVK element by several kinases including by protein kinase C alpha (PKCα) has been shown to increase titin stiffness and change cardiac tension.23, 31–33 Furthermore, Lanzicher et al. have recently shown that the compliance of the N2A element depends on the local effects on binding of signaling molecules (CARP) and that this contributes to strain- and phosphorylation- dependent mechano-signaling.34 Signaling mechanisms that lead to phosphorylation and de-phosphorylation of titin are complex, however identification and modification of these pathways may be important in understanding heart failure and identifying potential therapies. As one possible example, patients with diastolic dysfunction attributed to diabetes have increased activation of PKCα and phosphorylation of the PEVK making titin stiffer and likely contributing the decreased relaxation of global ventricular tissue.35
Truncation effects on CA2+ sensitivity, length dependent activation, and phosphorylation
TTNtv mutations may also affect the sensitivity of myofilaments to sarcomeric Ca2+changes. It is well known that phosphorylation of titin at different sites leads to alterations in passive stiffness. Phosphorylation of the N2B domain by protein kinase A (PKA) or cGMP dependent protein kinase (PKG) decrease titin stiffness. This differs from phosphorylation of PEVK region, which increases titin stiffness32. Vikhorev et al investigated the effects of TTNtv on the Ca2+ sensitivity, length dependent activation, and the modulation of these effects by in-vitro changes of phosphorylation. In this study, left ventricular tissue was isolated from explanted hearts of patients diagnosed with DCM carrying TTNtv and compared with healthy donor controls36. Results demonstrate that Ca2+ sensitivities of samples from TTNtv group are significantly higher compared with controls. Interestingly, donor controls that were found to have low phosphorylation of troponin I (TnI) also demonstrated increased Ca2+ sensitivity similar to tissue taken from TTNtv hearts. This suggests that increased calcium sensitivity in TTNtv may be related to TnI phosphorylation. In addition, in-vitro phosphorylation of TnI with PKA decreased Ca2+ sensitivity in all tissues, while de-phosphorylation of TnI with λ phosphatase significant increased Ca2+ sensitivity. Furthermore, the phosphorylation state of TnI was associated with length dependent activation, where stretched myofibrils from TTNtv tissue with low TnI phosphorylation, or de-phosporylation via λ phosphatase reduced length dependent activation. In addition, phosphorylation of TnI with PKA improved but did not restore length dependent activation.
TTNtv mutations cause late onset dilated cardiomyopathy
An estimated 50% of DCM are due to genetic causes that can either present as familial DCM or sporadic.37, 38 Truncation variants of the TTN gene (TTNtv) are the most frequent genetic cause of DCM accounting for up to 25% of DCM cases.37, 39, 40 These mutations are caused by single nucleotide polymorphisms leading to premature stop codons, splice site variants, and frameshift mutations. Unlike the biophysical changes to titin by transcriptional changes or post-translational changed described above, it has not been clearly elucidated how TTNtv lead to a DCM phenotype. There is however, mounting evidence that the functional consequences of these variants are attributed to haploinsufficiency and increased metabolic stress caused by abnormal TTN RNA transcripts that increase non-sense mRNA decay pathways and activation the mammalian target of rapamycin (mTOR) pathway3, 23. Activation of the mTOR pathway is observed in many causes of heart failure and my represent a final common pathway associated with heart failure.23 Interestingly, missense mutations of TTN do no demonstrate the same DCM phenotypic presentation at TTNtv. With improved sequencing capabilities, greater than 60,000 missense variants of the TTN have been identified, however the clinical significance of missense variations in TTN remains unknown.37
TTNtv demonstrate heterogenous penetrance
Despite the clear impact that TTNtv have on cardiac function, there is a wide spectrum of presentations and severity of DCM for patients with TTNtv.41 It is estimated that 2–3% of the general population live asymptomatically with TTNtvs, which may be attributed to age-related penetrance with an incidence of DCM in >95% in patients older than 40 years old.39 Furthermore, TTNtv appear to have a modifier gene effect where patients with TTNtv have increased risk of developing DCM when exposed to conditions of cardiac stress. This has been demonstrated in peripartum cardiomyopathy, and toxic cardiomyopathy, where patients with TTNtv have increased risk of developing DCM with similar exposures compared to patients without TTNtv.40, 42, 43 In a recent multicenter study of 537 TTNtv carriers, Akhtar at al. found that malignant ventricular arrhythmias are most commonly associated with severe LV systolic dysfunction, and that male sex and LV dysfunction were independent predictors of worse arrhythmic and heart failure outcomes.44
The location of the truncation mutation may correlate to disease pathogenicity. In prior studies, it is shown that the A-band of TTN was the most common location for pathogenic TTNtv in patients with DCM.39 This is likely because exons that transcribe sequences in the A-band are constituently expressed. Proximal exons, such as those in the I-band demonstrate more variability in their expression and incorporation into mature transcripts.45 Therefore if a truncation mutation is present in an exon that is not highly expressed, the clinical impact will be diminished.
Roberts et al. developed a score to predict pathogenicity of TTNtv called proportion spliced in (PSI). This is derived from RNA sequencing data in human LV tissue with end stage DCM (n=84).45 The PSI ranges from 0 to 1 and represents the ratio of TTN transcripts in which a given exon is spliced into expressed transcripts. An exon can be minimally expressed (PSI <0.15), variably expressed (PSI 0.15–0.9), or constitutively expressed (PSI >0.9). TTNtv expressed in non-constitutive exons had a lower risk of phenotypically apparent DCM. The investigators found that TTNtv located in constitutively expressed exons with PSI >0.9 were associated with a 93% probability of pathogenicity in patients with known DCM, providing a useful tool in clinical genetics of DCM.45
When comparing TTNtv of healthy individuals with patients with DCM, there is also a length dependent association with DCM that is apparent after controlling for PSI.45 The distance of TTNtv from the N terminus of the TTN protein has a significant linear correlation with specific indices of function on cardiac magnetic resonance imaging (MRI), including left ventricular ejection fraction (LVEF, P-0.0013), indexed LV stroke volume (LVSVi, P-0.0045), right ventricular ejection fraction (RVEF, P-0.017), and indexed RV stroke volume (RVSVi, P-0.049). This correlation was not seen with LV or RV end systolic or diastolic volumes.45
TTNtv are associated with severe muscular phenotypes
TTNtv have also been associated with various striated muscular disorders ranging from mild myopathy to debilitating muscular dystrophy with varying levels of cardiac involvement. Late onset tibial muscular dystrophy (TMD) affects the anterior compartment of the lower leg muscles and typically presents in the 4th decade of life.46 The FINmaj, named for its high prevalence in causing TMD in Finland, is caused by an insertion-deletion mutation in exon 364 of the TTN gene. Early adult onset TMD is characterized by the typical late onset mutation in combination with a frameshift mutation. These patients have evidence of fatty degeneration in their anterior muscle compartment on MRI imaging prior to age 20.46
Limb-girdle muscular dystrophy type 2J (LGMDT2J) is a more severe muscular dystrophy that has been associated with mutations in the TTN gene. It mainly affects proximal musculature such as the shoulders and hips and presents earlier in life in childhood. This disorder was first described in Finnish patients homozygous for the Finnish founder 11bp insertion-deletion mutation in TTN, called FINmaj.47
Early onset myopathy with fetal cardiomyopathy is associated with frameshift mutations in the titin exons mex1 and mex3 in families of Arab descent.48 It is characterized by early onset and progressive muscle weakness by the first year of life followed later by severe DCM and sudden cardiac death before adulthood. This is a rare disorder inherited in an autosomal recessive pattern and heterozygous carriers are usually asymptomatic.
Current guidelines for genetic testing
TTNtv-DCM is inherited in an autosomal dominant pattern although has variable penetrance with expression typically in the 5th to 6th decade of life.3 International societies provide guidance for genetic testing and call for an extensive family history with at least three generations, focusing on any incidence of cardiomyopathy or sudden cardiac death (SCD) at young age (<35 years).49–51 In patients with DCM, familial screening is indicated with physical exam, electrocardiogram, and echocardiogram for all first-degree relatives. In particular in patients with suspected familial DCM, genetic testing and counseling should be offered with the potential for genetic testing of relatives when a mutation is found.51 While there are no therapies currently available to reverse TTNtv-DCM, early diagnosis of DCM in patients with TTNtv may provide for earlier treatment of cardiomyopathy using standard evidence based medical therapies.
Targeted therapy for titin associated DCM
Identification of TTNtv DCM will increase as genetic testing becomes more integrated in clinical practice. This will not only affect prognosis of patients but also may pave the way to targeted therapies for patients as more is known about the transcriptional and post-translational regulation of titin as it relates to heart failure. There have been several proposed mechanisms for titin modifying therapy to treat CHF.23 For instance, there is increased activation of the mammalian target of rifampin (mTOR) associated with TTNtv. Rifampin, which inhibits mTOR, has been proposed as a potential targeted therapy for TTNtv DCM, however this has not been tested clinically.
Antisense oligonucleotide (AON) mediated exon skipping is a more direct gene therapy strategy for TTNtv that has been successfully applied to conditions such Duchenne’s muscular dystrophy, familial hypercholesterolemia, and spinal muscular atrophy.52–54 AON mediated exon skipping utilizes small single stranded oligonucleotides ranging from 18–30 base pairs to modify expression of mRNA, either by alternative splicing or recruiting RNAase.55 The overall goal is to “skip” the exon that contains the frameshift mutation and prevent early termination of the titin protein.
Other therapies such as T3 and insulin aim to upregulate either the shorter, stiffer N2B TTN isoform to increase passive tension and treat DCM.56 In contrast, medications such as digoxin upregulates the longer, more compliant N2BA isoform to decrease passive relaxation and potentially treat diastolic dysfunction.57 Similarly, metformin and Neuregulin-1 increase ERK2 mediated phosphorylation of the N2Bus element, decreasing passive tension and to treat diastolic dysfunction.23, 35 In addition to pharmacotherapies to modify titin in heart failure, dietary changes may also modify the biophysical properties of titin to improve cardiac function. This was suggested in mice who were fed a high amount of the polyamine, spermidine, and were shown to have longer lifespan, improved cardiac diastolic function, which was in part attributed to increased phosphorylation of the N2B segment of titin.58
Finally, among future perspectives in the therapy of titin cardiomyopathy, we should mention the use of human induced pluripotent stem cells-derived cardiomyocytes (iPSC-CMs) and the possibility to correct the mutant genomic sequence with “genome editing” by CRISPR/Cas 9 technology. These approaches are been actively investigated and, for selected diseases (in particular hematopoietic disorders) have entered in phase I clinical trials.59 However, in inherited heart muscle diseases, the genome editing of iPSC-CMs represent significant challenges, including editing efficiency in the heart, editing specificity (possible off target effects), and lack of ideal delivery systems (current based on AAV). Progress in genome editors and iPSC technology may overcome these challenges in the future.
Conclusion
Heart failure caused by both systolic and diastolic dysfunction remains a common disease process associated with high morbidity and mortality worldwide, therefore further understanding of the pathways and processes the contribute to its development are necessary to develop novel therapies or prevention strategies. A key molecule in the development of heart failure is the sarcomeric spring protein, titin, that imparts important biophysical properties to the sarcomere and global cardiac function. Modification of titin and its spring components via transcriptional and post translational changes are associated with the development of heart failure, and represent a potential therapeutic target. In addition, truncation mutations of TTN are an important cause of DCM, and likely are mediated by increased metabolic stress from non-sense mRNA decay leading to activation of the mTOR signaling pathway. Early identification of TTNtv in patients with DCM may prompt necessary treatment including maximum tolerated guideline directed medical therapy, primary or secondary implantable cardioverter defibrillator placement, cardiac resynchronization therapy, and even early consideration for heart transplantation. In addition, increased recognition will improve appropriate clinical and genetic screening of family members and potentially increase early identification of TTNtv carriers.
Acknowledgments:
Supported by the NIH R01HL69071, HL116906, HL147064, NCATS Colorado CTSA/UL1 TR002535 and UL1 TR001082 (LM), NIH 1K23HI067915, NIH 2UM1HG006542 and R01HL109209 (MRGT), NHLBI T32HL007822 (CT), MIUR PRIN - 20173ZW (OS) and in part by a Trans-Atlantic Network of Excellence grant from the Fondation Leducq 14-CVD03 (LM, MRGT, OS).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Virani SS; Alonso A; Benjamin EJ; Bittencourt MS; Callaway CW; Carson AP; Chamberlain AM; Chang AR; Cheng S; Delling FN; Djousse L; Elkind MSV; Ferguson JF; Fornage M; Khan SS; Kissela BM; Knutson KL; Kwan TW; Lackland DT; Lewis TT; Lichtman JH; Longenecker CT; Loop MS; Lutsey PL; Martin SS; Matsushita K; Moran AE; Mussolino ME; Perak AM; Rosamond WD; Roth GA; Sampson UKA; Satou GM; Schroeder EB; Shah SH; Shay CM; Spartano NL; Stokes A; Tirschwell DL; VanWagner LB; Tsao CW; American Heart Association Council on, E.; Prevention Statistics, C.; Stroke Statistics, S., Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141 (9), e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Zipes D; Braunwald E, Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed.; 2005. [Google Scholar]
- 3.Tharp CA; Haywood ME; Sbaizero O; Taylor MRG; Mestroni L, The Giant Protein Titin’s Role in Cardiomyopathy: Genetic, Transcriptional, and Post-translational Modifications of TTN and Their Contribution to Cardiac Disease. Front Physiol 2019, 10, 1436. [DOI] [PMC free article] [PubMed] [Google Scholar]; ••Review of the most recent advances in understanding biophysical properties of TTN in health and disease.
- 4.Maruyama K; Kimura S; Yoshidomi H; Sawada H; Kikuchi M, Molecular size and shape of beta-connectin, an elastic protein of striated muscle. J Biochem 1984, 95 (5), 1423–33. [DOI] [PubMed] [Google Scholar]
- 5.Tskhovrebova L; Trinick J, Roles of titin in the structure and elasticity of the sarcomere. J Biomed Biotechnol 2010, 2010, 612482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LeWinter MM; Wu Y; Labeit S; Granzier H, Cardiac titin: structure, functions and role in disease. Clin Chim Acta 2007, 375 (1–2), 1–9. [DOI] [PubMed] [Google Scholar]
- 7.LeWinter MM; Granzier HL, Titin is a major human disease gene. Circulation 2013, 127 (8), 938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trinick J, Titin as a scaffold and spring. Cytoskeleton. Curr Biol 1996, 6 (3), 258–60. [DOI] [PubMed] [Google Scholar]
- 9.Tskhovrebova L; Trinick J, Role of titin in vertebrate striated muscle. Philos Trans R Soc Lond B Biol Sci 2002, 357 (1418), 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Labeit S; Kolmerer B; Linke WA, The giant protein titin. Emerging roles in physiology and pathophysiology. Circ Res 1997, 80 (2), 290–4. [DOI] [PubMed] [Google Scholar]
- 11.Li H; Fernandez JM, Mechanical design of the first proximal Ig domain of human cardiac titin revealed by single molecule force spectroscopy. J Mol Biol 2003, 334 (1), 75–86. [DOI] [PubMed] [Google Scholar]
- 12.Linke WA; Rudy DE; Centner T; Gautel M; Witt C; Labeit S; Gregorio CC, I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol 1999, 146 (3), 631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trombitás K; Greaser M; Labeit S; Jin JP; Kellermayer M; Helmes M; Granzier H, Titin extensibility in situ: entropic elasticity of permanently folded and permanently unfolded molecular segments. J Cell Biol 1998, 140 (4), 853–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H; Linke WA; Oberhauser AF; Carrion-Vazquez M; Kerkvliet JG; Lu H; Marszalek PE; Fernandez JM, Reverse engineering of the giant muscle protein titin. Nature 2002, 418 (6901), 998–1002. [DOI] [PubMed] [Google Scholar]
- 15.Li H; Oberhauser AF; Redick SD; Carrion-Vazquez M; Erickson HP; Fernandez JM, Multiple conformations of PEVK proteins detected by single-molecule techniques. Proc Natl Acad Sci U S A 2001, 98 (19), 10682–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe K; Nair P; Labeit D; Kellermayer MS; Greaser M; Labeit S; Granzier H, Molecular mechanics of cardiac titin’s PEVK and N2B spring elements. J Biol Chem 2002, 277 (13), 11549–58. [DOI] [PubMed] [Google Scholar]
- 17.Marszalek PE; Lu H; Li H; Carrion-Vazquez M; Oberhauser AF; Schulten K; Fernandez JM, Mechanical unfolding intermediates in titin modules. Nature 1999, 402 (6757), 100–3. [DOI] [PubMed] [Google Scholar]
- 18.Linke WA; Ivemeyer M; Olivieri N; Kolmerer B; Rüegg JC; Labeit S, Towards a molecular understanding of the elasticity of titin. J Mol Biol 1996, 261 (1), 62–71. [DOI] [PubMed] [Google Scholar]
- 19.Freundt JK; Linke WA, Titin as a force-generating muscle protein under regulatory control. J Appl Physiol (1985) 2019, 126 (5), 1474–1482. [DOI] [PubMed] [Google Scholar]
- 20.Linke WA; Stockmeier MR; Ivemeyer M; Hosser H; Mundel P, Characterizing titin’s I-band Ig domain region as an entropic spring. J Cell Sci 1998, 111 (Pt 11), 1567–74. [DOI] [PubMed] [Google Scholar]
- 21.Linke WA; Ivemeyer M; Mundel P; Stockmeier MR; Kolmerer B, Nature of PEVK-titin elasticity in skeletal muscle. Proc Natl Acad Sci U S A 1998, 95 (14), 8052–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda N; Granzier HL, Titin/connectin-based modulation of the Frank-Starling mechanism of the heart. J Muscle Res Cell Motil 2005, 26 (6–8), 319–23. [DOI] [PubMed] [Google Scholar]
- 23.Tharp C; Mestroni L; Taylor M, Modifications of Titin Contribute to the Progression of Cardiomyopathy and Represent a Therapeutic Target for Treatment of Heart Failure. J Clin Med 2020, 9 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Updated review of the advances in therapeutic development in TTN cardiomyopathy.
- 24.LeWinter MM; Granzier H, Cardiac titin: a multifunctional giant. Circulation 2010, 121 (19), 2137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo W; Bharmal SJ; Esbona K; Greaser ML, Titin diversity--alternative splicing gone wild. J Biomed Biotechnol 2010, 2010, 753675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeWinter MM; Granzier HL, Cardiac titin and heart disease. J Cardiovasc Pharmacol 2014, 63 (3), 207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagueh SF; Shah G; Wu Y; Torre-Amione G; King NM; Lahmers S; Witt CC; Becker K; Labeit S; Granzier HL, Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110 (2), 155–62. [DOI] [PubMed] [Google Scholar]
- 28.Guo W; Schafer S; Greaser ML; Radke MH; Liss M; Govindarajan T; Maatz H; Schulz H; Li S; Parrish AM; Dauksaite V; Vakeel P; Klaassen S; Gerull B; Thierfelder L; Regitz-Zagrosek V; Hacker TA; Saupe KW; Dec GW; Ellinor PT; MacRae CA; Spallek B; Fischer R; Perrot A; Ozcelik C; Saar K; Hubner N; Gotthardt M, RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012, 18 (5), 766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brauch KM; Karst ML; Herron KJ; de Andrade M; Pellikka PA; Rodeheffer RJ; Michels VV; Olson TM, Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 2009, 54 (10), 930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkin J; Slater R; Del Favero G; Lanzicher T; Hidalgo C; Anderson B; Smith JE 3rd; Sbaizero O; Labeit S; Granzier H, Phosphorylating Titin’s Cardiac N2B Element by ERK2 or CaMKIIdelta Lowers the Single Molecule and Cardiac Muscle Force. Biophys J 2015, 109 (12), 2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rain S; Bos Dda S; Handoko ML; Westerhof N; Stienen G; Ottenheijm C; Goebel M; Dorfmuller P; Guignabert C; Humbert M; Bogaard HJ; Remedios CD; Saripalli C; Hidalgo CG; Granzier HL; Vonk-Noordegraaf A; van der Velden J; de Man FS, Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc 2014, 3 (3), e000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotter S; Gout L; Von Frieling-Salewsky M; Muller AE; Helling S; Marcus K; Dos Remedios C; Linke WA; Kruger M, Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res 2013, 99 (4), 648–56. [DOI] [PubMed] [Google Scholar]
- 33.Hamdani N; Herwig M; Linke WA, Tampering with springs: phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys Rev 2017, 9 (3), 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lanzicher T; Zhou T; Saripalli C; Keschrumrus V; Smith Iii JE; Mayans O; Sbaizero O; Granzier H, Single-Molecule Force Spectroscopy on the N2A Element of Titin: Effects of Phosphorylation and CARP. Front Physiol 2020, 11, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Recent study showing the complexity of TTN mechanics and the importance of phosphorylation and binding molecules in affecting TTN rigidity, relevant for future therapeutics developments.
- 35.Hopf AE; Andresen C; Kotter S; Isic M; Ulrich K; Sahin S; Bongardt S; Roll W; Drove F; Scheerer N; Vandekerckhove L; De Keulenaer GW; Hamdani N; Linke WA; Kruger M, Diabetes-Induced Cardiomyocyte Passive Stiffening Is Caused by Impaired Insulin-Dependent Titin Modification and Can Be Modulated by Neuregulin-1. Circ Res 2018, 123 (3), 342–355. [DOI] [PubMed] [Google Scholar]
- 36.Vikhorev Petr G, N. V. N, Yeung WaiChun, Li Amy, Lal Sean, dos Remedios Cristobal G, Blair Cheavar A, Guglin Maya, Campbell Kenneth S, Yacoub Magdi H, de Tombe Pieter, Marston Steven B, Titin-truncating mutations associated with dilated cardiomyopathy alter length-dependent activation and its modulation via phosphorylation. Cardiovascular Research 2020, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gigli M; Begay RL; Morea G; Graw SL; Sinagra G; Taylor MR; Granzier H; Mestroni L, A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front Cardiovasc Med 2016, 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNally EM; Mestroni L, Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res 2017, 121 (7), 731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Comprehensive review of the pathogenetic mechanisms, genetics and management of DCM.
- 39.Herman DS; Lam L; Taylor MR; Wang L; Teekakirikul P; Christodoulou D; Conner L; DePalma SR; McDonough B; Sparks E; Teodorescu DL; Cirino AL; Banner NR; Pennell DJ; Graw S; Merlo M; Di Lenarda A; Sinagra G; Bos JM; Ackerman MJ; Mitchell RN; Murry CE; Lakdawala NK; Ho CY; Barton PJ; Cook SA; Mestroni L; Seidman JG; Seidman CE, Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012, 366 (7), 619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ware JS; Li J; Mazaika E; Yasso CM; DeSouza T; Cappola TP; Tsai EJ; Hilfiker-Kleiner D; Kamiya CA; Mazzarotto F; Cook SA; Halder I; Prasad SK; Pisarcik J; Hanley-Yanez K; Alharethi R; Damp J; Hsich E; Elkayam U; Sheppard R; Kealey A; Alexis J; Ramani G; Safirstein J; Boehmer J; Pauly DF; Wittstein IS; Thohan V; Zucker MJ; Liu P; Gorcsan J 3rd; McNamara DM; Seidman CE; Seidman JG; Arany Z; Imac; Investigators, I., Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N Engl J Med 2016, 374 (3), 233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabish AM; Azzimato V; Alexiadis A; Buyandelger B; Knoll R, Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys Rev 2017, 9 (3), 207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ware JS; Amor-Salamanca A; Tayal U; Govind R; Serrano I; Salazar-Mendiguchia J; Garcia-Pinilla JM; Pascual-Figal DA; Nunez J; Guzzo-Merello G; Gonzalez-Vioque E; Bardaji A; Manito N; Lopez-Garrido MA; Padron-Barthe L; Edwards E; Whiffin N; Walsh R; Buchan RJ; Midwinter W; Wilk A; Prasad S; Pantazis A; Baski J; O’Regan DP; Alonso-Pulpon L; Cook SA; Lara-Pezzi E; Barton PJ; Garcia-Pavia P, Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J Am Coll Cardiol 2018, 71 (20), 2293–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Large population survey showing an association between genetic predisposition, in particular TTNtv, and risk of cardiac damage due to alcohol intake.
- 43.Garcia-Pavia P; Kim Y; Restrepo-Cordoba MA; Lunde IG; Wakimoto H; Smith AM; Toepfer CN; Getz K; Gorham J; Patel P; Ito K; Willcox JA; Arany Z; Li J; Owens AT; Govind R; Nunez B; Mazaika E; Bayes-Genis A; Walsh R; Finkelman B; Lupon J; Whiffin N; Serrano I; Midwinter W; Wilk A; Bardaji A; Ingold N; Buchan R; Tayal U; Pascual-Figal DA; de Marvao A; Ahmad M; Garcia-Pinilla JM; Pantazis A; Dominguez F; John Baksi A; O’Regan DP; Rosen SD; Prasad SK; Lara-Pezzi E; Provencio M; Lyon AR; Alonso-Pulpon L; Cook SA; DePalma SR; Barton PJR; Aplenc R; Seidman JG; Ky B; Ware JS; Seidman CE, Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140 (1), 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Large population survey suggesting that genetic factors, in particular TTN truncating variant, modify the risk of developing anthracycline cardiomyopathy.
- 44.Akhtar MM; Lorenzini M; Cicerchia M; Ochoa JP; Hey TM; Sabater Molina M; Restrepo-Cordoba MA; Dal Ferro M; Stolfo D; Johnson R; Larranaga-Moreira JM; Robles-Mezcua A; Rodriguez-Palomares JF; Casas G; Pena-Pena ML; Lopes LR; Gallego-Delgado M; Franaszczyk M; Laucey G; Rangel-Sousa D; Basurte M; Palomino-Doza J; Villacorta E; Bilinska Z; Limeres Freire J; Garcia Pinilla JM; Barriales-Villa R; Fatkin D; Sinagra G; Garcia-Pavia P; Gimeno JR; Mogensen J; Monserrat L; Elliott PM, Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ Heart Fail 2020, 13 (10), e006832. [DOI] [PubMed] [Google Scholar]; •• Recent multicenter study looking at the prognostic factors of TTNtv DCM at the population level: the study showed that LV dysfunction and male gender may worsen the outcome of TTNtv carriers.
- 45.Roberts AM; Ware JS; Herman DS; Schafer S; Baksi J; Bick AG; Buchan RJ; Walsh R; John S; Wilkinson S; Mazzarotto F; Felkin LE; Gong S; MacArthur JA; Cunningham F; Flannick J; Gabriel SB; Altshuler DM; Macdonald PS; Heinig M; Keogh AM; Hayward CS; Banner NR; Pennell DJ; O’Regan DP; San TR; de Marvao A; Dawes TJ; Gulati A; Birks EJ; Yacoub MH; Radke M; Gotthardt M; Wilson JG; O’Donnell CJ; Prasad SK; Barton PJ; Fatkin D; Hubner N; Seidman JG; Seidman CE; Cook SA, Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015, 7 (270), 270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Savarese M; Sarparanta J; Vihola A; Udd B; Hackman P, Increasing Role of Titin Mutations in Neuromuscular Disorders. J Neuromuscul Dis 2016, 3 (3), 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng W; Chen H; Deng X; Yuan L; Yang Y; Song Z; Yang Z; Wu Y; Deng H, Identification of a Novel Mutation in the Titin Gene in a Chinese Family with Limb-Girdle Muscular Dystrophy 2J. Mol Neurobiol 2016, 53 (8), 5097–102. [DOI] [PubMed] [Google Scholar]
- 48.Hackman P; Savarese M; Carmignac V; Udd B; Salih MA, Salih Myopathy. In GeneReviews((R)), Adam MP; Ardinger HH; Pagon RA; Wallace SE; Bean LJH; Mirzaa G; Amemiya A, Eds. Seattle (WA), 1993. [PubMed] [Google Scholar]
- 49.Hershberger RE; Givertz MM; Ho CY; Judge DP; Kantor PF; McBride KL; Morales A; Taylor MRG; Vatta M; Ware SM, Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J Card Fail 2018, 24 (5), 281–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hershberger RE; Givertz MM; Ho CY; Judge DP; Kantor PF; McBride KL; Morales A; Taylor MRG; Vatta M; Ware SM; Practice AP; Guidelines C, Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2018, 20 (9), 899–909. [DOI] [PubMed] [Google Scholar]
- 51.Bozkurt B; Colvin M; Cook J; Cooper LT; Deswal A; Fonarow GC; Francis GS; Lenihan D; Lewis EF; McNamara DM; Pahl E; Vasan RS; Ramasubbu K; Rasmusson K; Towbin JA; Yancy C; American Heart Association Committee on Heart, F.; Transplantation of the Council on Clinical, C.; Council on Cardiovascular Disease in the, Y.; Council on, C.; Stroke, N.; Council on, E.; Prevention; Council on Quality of, C.; Outcomes, R., Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134 (23), e579–e646. [DOI] [PubMed] [Google Scholar]
- 52.Syed YY, Eteplirsen: First Global Approval. Drugs 2016, 76 (17), 1699–1704. [DOI] [PubMed] [Google Scholar]
- 53.Geary RS; Baker BF; Crooke ST, Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (kynamro((R))): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin Pharmacokinet 2015, 54 (2), 133–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood MJA; Talbot K; Bowerman M, Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum Mol Genet 2017, 26 (R2), R151–R159. [DOI] [PubMed] [Google Scholar]
- 55.Scoles DR; Minikel EV; Pulst SM, Antisense oligonucleotides: A primer. Neurol Genet 2019, 5 (2), e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu C; Yin Z; Ren J; McCormick RJ; Ford SP; Guo W, RBM20 is an essential factor for thyroid hormone-regulated titin isoform transition. J Mol Cell Biol 2015, 7 (1), 88–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liss M; Radke MH; Eckhard J; Neuenschwander M; Dauksaite V; von Kries JP; Gotthardt M, Drug discovery with an RBM20 dependent titin splice reporter identifies cardenolides as lead structures to improve cardiac filling. PLoS One 2018, 13 (6), e0198492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eisenberg T; Abdellatif M; Schroeder S; Primessnig U; Stekovic S; Pendl T; Harger A; Schipke J; Zimmermann A; Schmidt A; Tong M; Ruckenstuhl C; Dammbrueck C; Gross AS; Herbst V; Magnes C; Trausinger G; Narath S; Meinitzer A; Hu Z; Kirsch A; Eller K; Carmona-Gutierrez D; Buttner S; Pietrocola F; Knittelfelder O; Schrepfer E; Rockenfeller P; Simonini C; Rahn A; Horsch M; Moreth K; Beckers J; Fuchs H; Gailus-Durner V; Neff F; Janik D; Rathkolb B; Rozman J; de Angelis MH; Moustafa T; Haemmerle G; Mayr M; Willeit P; von Frieling-Salewsky M; Pieske B; Scorrano L; Pieber T; Pechlaner R; Willeit J; Sigrist SJ; Linke WA; Muhlfeld C; Sadoshima J; Dengjel J; Kiechl S; Kroemer G; Sedej S; Madeo F, Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016, 22 (12), 1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishiga M; Qi LS; Wu JC, Therapeutic genome editing in cardiovascular diseases. Adv Drug Deliv Rev 2021, 168, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]