

SUMMARY
CD8 T cell responses against different tumor neoantigens occur simultaneously, yet little is known about the interplay between responses and its impact on T cell function and tumor control. In mouse lung adenocarcinoma, we found that immunodominance is established in tumors, wherein CD8 T cell expansion is predominantly driven by the antigen that most stably binds MHC. T cells responding to subdominant antigens were enriched for a TCF1+ progenitor phenotype correlated with response to immune checkpoint blockade (ICB) therapy. However, the subdominant T cell response did not preferentially benefit from ICB due to a dysfunctional subset of TCF1+ cells marked by CCR6 and Tc17 differentiation. Analysis of human samples and sequencing datasets revealed that CCR6+ TCF1+ cells exist across human cancers and are not correlated with ICB response. Vaccination eliminated CCR6+ TCF1+ cells and dramatically improved the subdominant response, highlighting a strategy to optimally engage concurrent neoantigen responses against tumors.
Keywords: TCF1, neoantigen, immunodominance, CCR6, Tc17, vaccine, lung cancer
Graphical Abstract
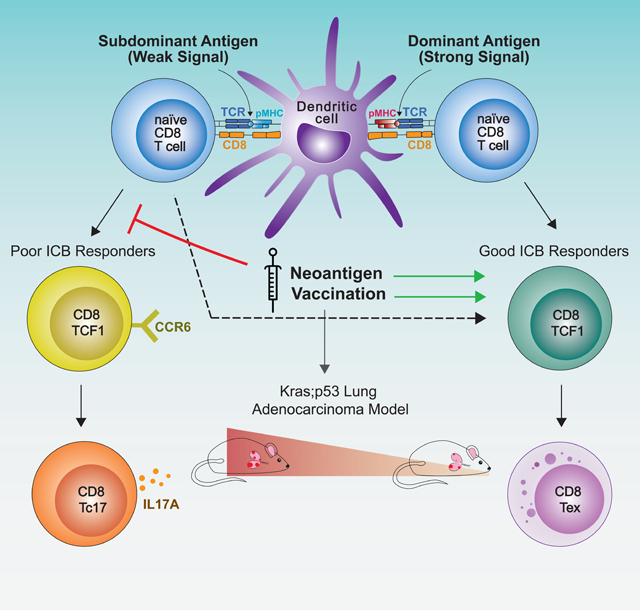
IN BRIEF
Analysis of antigen dominance hierarchy and dynamics in a lung cancer model highlights the opportunity of optimizing T cell responses against subdominant neoantigens for better checkpoint blockade response through vaccination that adjusts T cell progenitor subpopulations
INTRODUCTION
Accumulating evidence indicates that peptides derived from mutated proteins presented on major histocompatibility (MHC) molecules, termed neoantigens, drive productive T cell responses to tumors (Schumacher et al., 2019). Neoantigen-specific CD8 T cells expand in response to immune checkpoint blockade (ICB) therapies (e.g. anti-PD1/anti-PDL1) and patient responses correlate with high neoantigen and/or mutational burden (Keenan et al., 2019). Additionally, adoptive T cell therapies targeting neoantigens can promote meaningful tumor regression (Tran et al., 2017; Zacharakis et al., 2018). These observations provide strong rationale for development of immunotherapies directly targeting neoantigens and, in particular, have fueled recent large-scale efforts to develop neoantigen-targeted cancer vaccines (Hollingsworth and Jansen, 2019).
To identify therapeutically actionable neoantigens for immunotherapies, computational pipelines have been developed to predict immunogenic epitopes from exome sequencing of patient tumors (Peters et al., 2020; Wells et al., 2020). While these algorithms identify tens to hundreds of potential neoantigens, the vast majority of these epitopes do not elicit a detectable T cell response. Studies profiling T cell reactivity against predicted neoantigens have generally identified only a few, and sometimes zero, bona fide T cell-reactive epitopes across most cancer types (Linette et al., 2019; McGranahan et al., 2016; Scheper et al., 2019; Simoni et al., 2018). If this low frequency of T cell reactivity reflects the number of neoantigens capable of eliciting an anti-tumor response, this could pose a significant challenge in the selection of relevant neoantigens for immunotherapies.
Poor T cell reactivity against computationally predicted neoantigens may be in part attributable to shortcomings of prediction algorithms or detection limits of T cell assays. There are also plausible biological explanations, including selection against tumor cells expressing immunogenic neoantigens, resulting in loss of T cell-reactive antigens over time (i.e. immunoediting). Immunoediting has been observed in mouse models (DuPage et al., 2012; Matsushita et al., 2012; Milo et al., 2018) and in conjunction with clinical response to ICB and adoptive T cell therapies (Anagnostou et al., 2017; Verdegaal et al., 2016). While it is difficult to quantify the extent of immunoediting during tumor progression in humans (Rosenthal et al., 2019), evidence in mice suggests it may depend on the tumor type or tissue context (DuPage et al., 2011; DuPage et al., 2012; Matsushita et al., 2012). In addition, antigen immunodominance may be established during anti-tumor immune responses (Schreiber et al., 2002). As observed in acute viral infection, one or two epitopes may saturate the T cell response and suppress responses to additional antigens (Yewdell, 2006). Supporting this idea, healthy donor T cells can respond to tumor antigens that did not elicit a response from endogenous tumor infiltrating T cells, suggesting that responses against some antigens are depleted or suppressed during tumor progression (Stronen et al., 2016). Further, vaccines against melanoma neoantigens generate de novo responses, highlighting a degree of unrealized neoantigen immunogenicity in tumors (Carreno et al., 2015; Ott et al., 2017; Sahin et al., 2017). Thus, T cell responses against some neoantigens might outcompete others and narrow the T cell response to fewer epitopes.
To study the dynamics of anti-tumor T cell responses, our group previously engineered a Kras/p53-driven (KP) autochthonous mouse model of lung adenocarcinoma to express tumor-specific neoantigens (DuPage et al., 2011; DuPage et al., 2009). Tumors are initiated from single transformed cells in situ and develop over approximately five months, allowing ample time for tumor-immune crosstalk and longitudinal sampling of the T cell response at different tumor stages. Two model CD8 T cell neoantigens are expressed in tumor cells as a fusion to Luciferase: SIINFEKL (SIIN) from chicken ovalbumin and a synthetic peptide, SIYRYYGL (SIY) (termed LucOS). In this KP LucOS model, the CD8 T cell response initially slows tumor progression, but exhausts over time. Importantly, neoantigen and MHC expression is sustained in advanced tumors, indicating mechanisms other than immunoediting hinder the T cell response.
In the LucOS model, SIIN and SIY are concurrently expressed, providing an experimental model to study the interaction of distinct neoantigen-specific CD8 T cell compartments in the context of an anti-tumor response. Here, we find an antigen dominance hierarchy is established between SIIN and SIY in lung tumors that limits expansion of the subdominant CD8 T cell response and represses differentiation from a progenitor to an exhausted cell state. Progenitor CD8 T cells are correlated with response to ICB in mice and humans (Philip et al., 2021), yet their biology remains poorly understood. We uncover a previously undescribed subpopulation of progenitor cells that can arise from antigen subdominance and correlates with impaired functionality and poor response to ICB. Therapeutic vaccination eliminates this subpopulation and greatly expands the subdominant response, highlighting vaccination as a strategy to optimally engage simultaneous CD8 T cell responses against multiple neoantigens in tumors.
RESULTS
Longitudinal Analysis Uncovers Heterogeneity Between CD8 T Cell Responses to Different Neoantigens in Lung Adenocarcinoma
To investigate how CD8 T cell responses to tumor neoantigens may interact, we longitudinally characterized SIIN- versus SIY-specific CD8 T cells in tumors initiated by intratracheal instillation of lentivirus containing LucOS and Cre recombinase in KrasLSL-G12D/+;p53fl/fl mice (Figure 1A; Dupage et al., 2011). At the peak of the response (5 weeks), we observed that SIIN drove substantially greater CD8 T cell expansion compared to SIY in the tumor-bearing lung (Figures 1B, 1C, S1A and S1B). The SIIN response contracted sharply thereafter, whereas the SIY response persisted better over time, resulting in similar numbers of SIIN- and SIY-specific cells by 12 weeks. Both populations increasingly displayed hallmarks of dysfunction over time, exhibiting progressive decline in proliferation (Ki67) and upregulation of multiple inhibitory receptors (PD1, LAG3, TIM3; Figure 1D, 1E, S1C and S1D). Notably, inhibitory receptor expression was lower on SIY-specific cells at 5 and 8 weeks compared to SIIN-specific cells, suggesting SIY-specific cells exhibit slower kinetics of dysfunction. A greater proportion of SIY-specific cells also expressed a marker of long-lived T cells, IL-7R, which may contribute to their persistence (Figure 1F). 30% of SIIN-specific cells produced IFNɣ compared to 15% of SIY-specific cells at 5 weeks, indicating that SIIN-specific cells have greater effector function early in the response (Figure 1G). However, few cells were IFNɣ+ TNFɑ+, or “polyfunctional”, a phenotype associated with better tumor control (Spranger et al., 2014). Additionally, only a small proportion of cells expressed the cytotoxicity-associated molecule Granzyme B (GZMB; SIIN 7% versus SIY 4%; Figure 1H). These data are consistent with both SIIN- and SIY-specific CD8 T cells having poor anti-tumor activity overall; however, SIIN-specific cells have greater effector function compared to SIY-specific cells.
Figure 1. Longitudinal Analysis Uncovers Heterogeneity Between CD8 T Cell Responses to Different Neoantigens in Lung Adenocarcinoma.
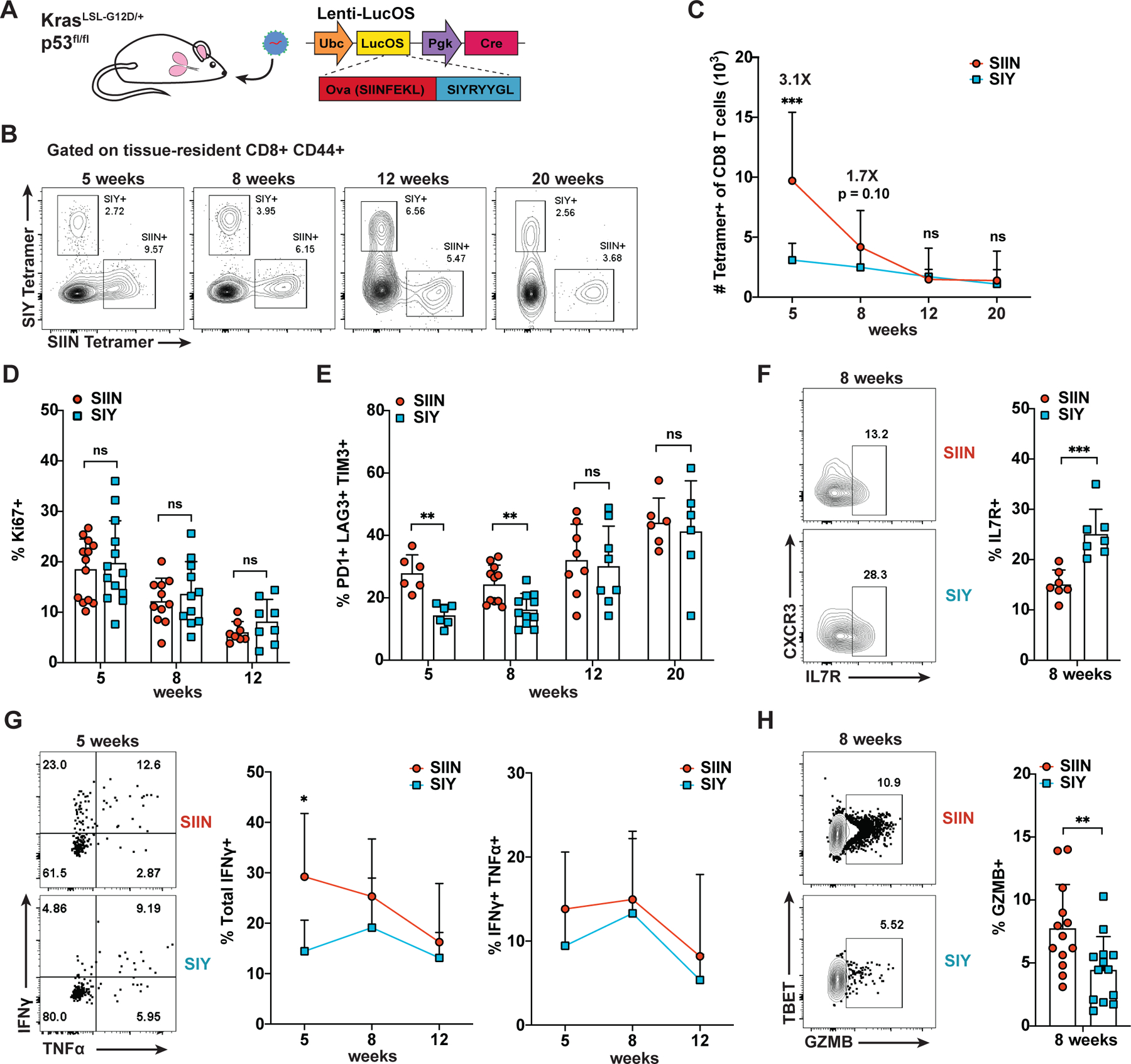
(A) KP LucOS genetically engineered mouse model of lung adenocarcinoma expressing tumor-specific neoantigens SIINFEKL (SIIN) and SIYRYYGL (SIY).
(B-C) Percentage (B) and absolute number (C) of SIIN- and SIY-specific CD8+ CD44+ T cells in tumor-bearing lung tissue by H-2Kb peptide-MHC tetramer staining and flow cytometry.
(D-H) Percentage of SIIN- and SIY-specific CD8 T cells expressing Ki67 (D), PD1, LAG3 and TIM3 (E), IL7R (F), IFNɣ and TNFα (G) and GZMB (H) by flow cytometry.
(B-H) are representative of ≥ 3 independent experiments per time point, n ≥ 5 mice per group. Results here and in the following figures are expressed as mean + SD. Statistics were calculated by two-tailed Student’s t test: ns = not significant, * p < 0.05, ** p < 0.01, *** p < 0.001.
Single-Cell RNA-Sequencing Reveals Enrichment of a TCF1+ Progenitor Cell Phenotype Amongst SIY-Specific CD8 T Cells
To further investigate phenotypic differences between SIIN- and SIY-specific CD8 T cells, we performed single-cell RNA-sequencing (scRNA-seq) with paired T cell receptor (TCR) sequencing at 5 weeks post-tumor initiation. Comparison of transcriptional profiles by uniform manifold approximation and projection (UMAP) and unsupervised clustering revealed similar distribution of SIIN- and SIY-specific cells; however, some clusters were significantly enriched for cells directed against one antigen versus the other (Figures 2A–2C; Table S3). To explore this transcriptional heterogeneity, we classified single cells using “ProjecTILs” T cell atlases (Andreatta et al., 2021) derived from mouse lymphocytic choriomeningitis virus (LCMV) infection and B16 melanoma and MC38 colorectal carcinoma tumor infiltrating lymphocytes (TILS; Figures 2D and S2A; Table S1). Consistent with Figure 1, SIIN-specific cells were enriched for “effector” and “exhausted” T cell signatures, while SIY-specific cells were enriched for signatures of less differentiated “naïve” and “memory precursor” states. Further, cluster 7, which was almost exclusively composed of SIY-specific cells, displayed hallmarks of suppressed activation, including poor clonal expansion and high expression of AY036118.1 (ETS-related transcription factor 1, ERF1), a transcriptional repressor of c-myc and cell proliferation (Figures S2C, S2E and S4B; Verykokakis et al., 2007). Intriguingly, SIY-specific cells were also enriched for signatures of “progenitor” cells, a population marked by stem-like ability to replenish more cytotoxic, but terminally-exhausted cells (Figure 2D and Table S3; Utzschneider et al., 2016). Recent studies have shown ICB acts to promote differentiation of progenitor cells, rather than to reverse dysfunction of terminally-exhausted cells (Kurtulus et al., 2019; Miller et al., 2019; Sade-Feldman et al., 2019; Siddiqui et al., 2019); hence, progenitor cells appear to be the therapeutically relevant target of ICB therapy. Progenitor cells are commonly defined by expression of the transcription factor T cell factor 1 (TCF1) and the absence of terminal exhaustion markers, such as Havcr2 (TIM3; Miller et al., 2019). SIY-specific cells were preferentially assigned to clusters 4 and 8 (C4 and C8) that were enriched for progenitor gene signatures and expressed Tcf7 (encoding TCF1) and other progenitor markers, including Il7r (Figures 2C–2E and S2C; Tables S1 and S3). Conversely, SIIN-specific cells were preferentially assigned to clusters C2 and C3, which expressed markers of cytotoxicity and exhaustion, including Gzmb and Havcr2.
Figure 2. Single-Cell RNA-Sequencing Reveals Enrichment of a TCF1+ Progenitor Cell Phenotype Amongst SIY-Specific CD8 T Cells.
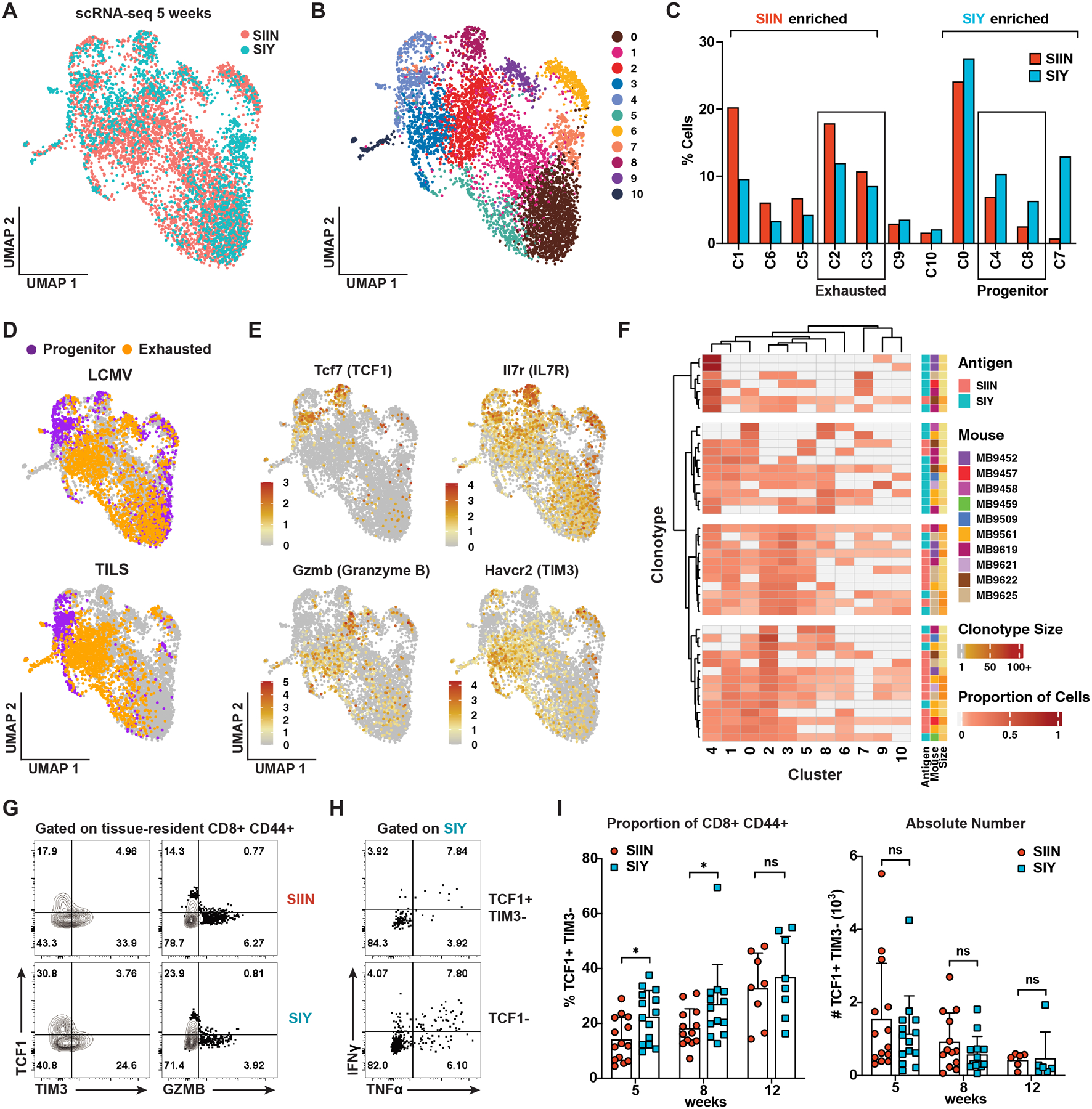
(A) UMAP embedding of scRNA-seq 5’ gene expression data comparing SIIN- and SIY-specific CD8 T cells at 5 weeks post-tumor initiation. n = 10 mice, 4,023 SIIN and 1,861 SIY cells.
(B) Unsupervised clustering of the scRNA-seq data in (A).
(C) Assignment of SIIN- versus SIY-specific cells to the clusters in (B). Brackets indicate significant enrichment (p < 0.05) for SIIN- or SIY-specific cells.
(D) Classification of individual cells from (A) using the ProjecTILs pipeline (Andreatta et al., 2021). SIY-specific cells are enriched for a progenitor phenotype (TIL p = 0.002; LCMV p = 2.51E-15); SIIN-specific cells are enriched for an exhausted phenotype (TIL p = 4.01E-17; LCMV p = 1.52E-10).
(E) Log-normalized expression of genes associated with progenitor (Tcf7 and Il7r) or exhausted Gzmb and Havcr2) cell states.
(F) Heatmap depicting proportions of TCR clonotypes (rows) assigned to each cluster (columns), for clonotypes ≥ 5 cells with ≥ 1 cell assigned to TCF1+ progenitor clusters C4 or C8.
(G-H) Percentage of SIIN- versus SIY-specific cells expressing TCF1, TIM3 and GZMB (G) and the proportion of SIY-specific TCF1+ TIM3− and TCF1− cells expressing IFNɣ and TNFα (H) at 5 weeks by flow cytometry.
(I) Percentage and absolute number of SIIN- and SIY-specific TCF1+ TIM3− cells over time by flow cytometry.
(G-I) are representative of 3 independent experiments, n ≥ 5 mice per group.
The proximity of clusters C4 and C8 to C2 and C3 on the UMAP suggests transcriptional similarity between these populations, consistent with the previously proposed lineage relationship between progenitor and exhausted cells (Figure 2B; Siddiqui et al., 2019). We further examined this relationship by analyzing the distribution of TCR clonotypes (≥ 5 cells) with at least one cell assigned to progenitor clusters C4 or C8 (Figure 2F). Unsupervised hierarchical clustering largely segregated SIIN and SIY clonotypes, revealing antigen-specific distribution patterns across clusters of cell states (Figure 2F). SIIN clonotypes were distributed across most clusters, but were enriched for cells assigned to exhausted cluster C2 (Figures 2F and S4C). This supports a lineage relationship between progenitor and exhausted cell states and suggests SIIN clonotypes containing progenitor cells are well-progressed on the path to exhaustion. In contrast, both progenitor cell-containing and total SIY clonotypes were biased in cell distribution to progenitor clusters C4 and C8 (Figures 2F, S4A and S4C), suggesting SIY-specific cells are skewed towards a less differentiated state. Further, while SIIN and SIY clonotype numbers were comparable (≥ 2 cells: SIIN 153, SIY 149), we observed that SIY clonotypes were smaller in size, indicating that clonal expansion may be repressed amongst the SIY response (Figures S4B).
Flow cytometric analysis confirmed that SIY-specific cells were enriched for a progenitor cell phenotype, referred to hereafter as “TCF1+ progenitor” and defined as CD8ɑ+ CD44+ TCF1+ TIM3− cells (Figure 2G). Consistent with previous reports (Miller et al., 2019; Siddiqui et al., 2019), the majority of these cells expressed the inhibitory receptor PD1 (Figure S2F). Few expressed GZMB, indicating they are generally not cytotoxic, but they were more proliferative than TCF1− cells and produced IFNɣ and TNFɑ (Figure 2G, 2H and S2G). SIY-specific cells were enriched for TCF1+ progenitor cells compared to SIIN-specific cells at 5 and 8 weeks; however, their proportion became similar by 12 weeks (Figure 2I). The percentage of TCF1+ cells increased over time, but the absolute number decreased, mirroring the overall contraction of the CD8 T cell response (Figure 2I). Altogether, these data indicate that SIY-specific cells are enriched for a less differentiated, TCF1+ progenitor state early in the response to KP LucOS lung tumors, while SIIN-specific cells differentiate more rapidly to an exhausted cell state.
Enrichment of TCF1+ Progenitor Cells in the SIY Response is Driven by an Antigen Dominance Hierarchy
Having observed that SIIN drove a larger CD8 T cell response than SIY, we hypothesized that competition between the T cell responses might underlie enrichment of TCF1+ progenitor cells amongst the SIY response. To test this, we engineered lentiviruses to express SIIN-only or SIY-only as fusions to luciferase (LucSIIN and LucSIY; Figure 3A). Strikingly, in LucSIY mice, SIY-specific cells expanded similarly to SIIN-specific cells in LucOS and LucSIIN mice (Figures 3A, 3B and S3A). Furthermore, enrichment for TCF1+ progenitor cells in the SIY response was no longer observed (Figures 3C and S3B). A higher proportion of SIY-specific cells in LucSIY mice expressed markers of effector memory and exhausted cells (CX3CR1, GZMB, PD1, LAG3, TIM3; Figures S3C–S3F). Thus, the SIY response in LucSIY mice more closely resembled the response against SIIN in LucOS and LucSIIN mice. These data demonstrate that an antigen dominance hierarchy can form in tumors and this significantly influences the phenotype of cells responding to the subdominant antigen. Competition with SIIN promotes a TCF1+ progenitor phenotype amongst SIY-specific cells and hinders differentiation to effector and exhausted cell states.
Figure 3. Enrichment of TCF1+ Progenitor Cells in the SIY Response is Driven by an Antigen Dominance Hierarchy.
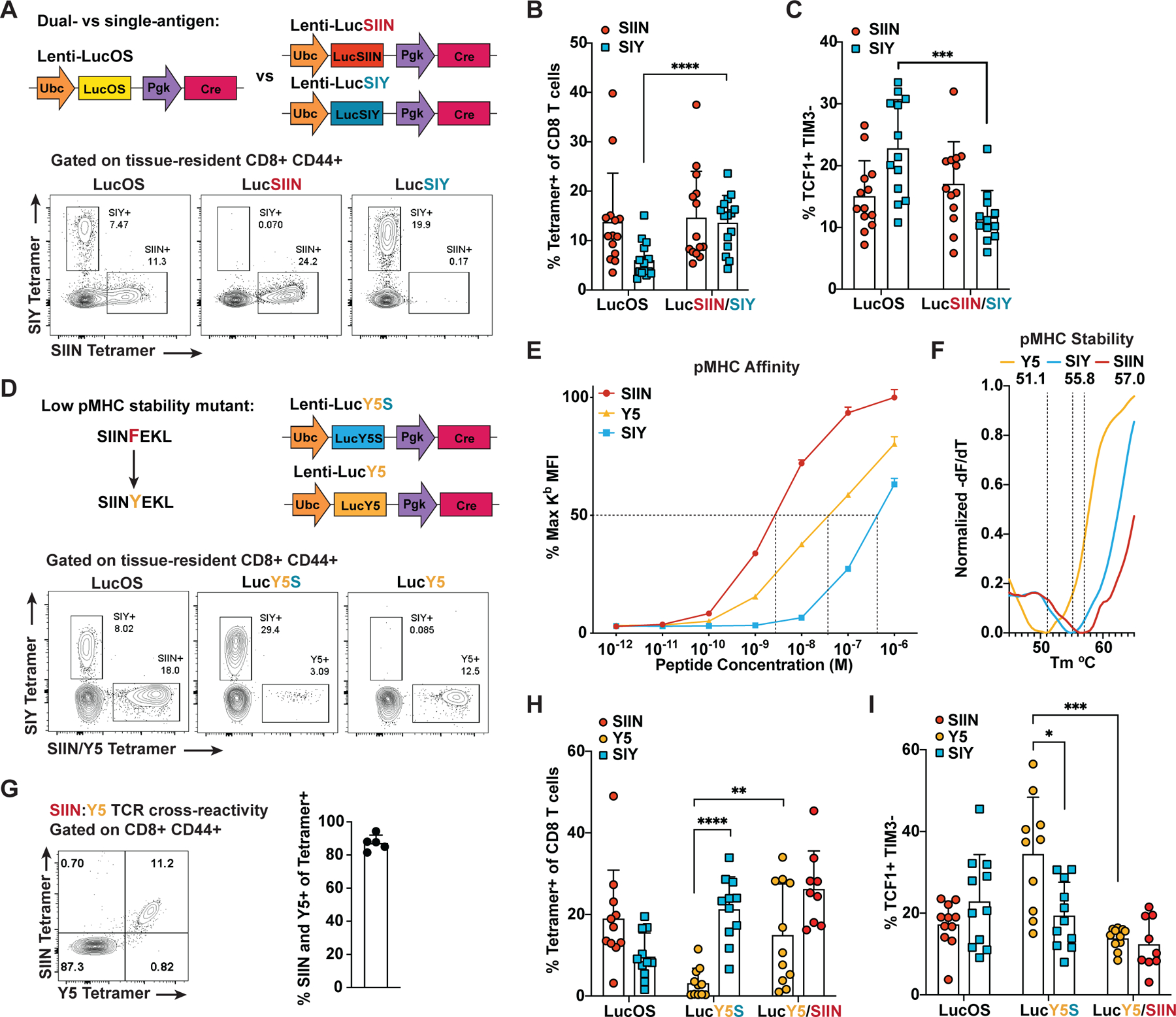
(A) Lentiviral constructs expressing SIIN and SIY (LucOS), SIIN-only (LucSIIN) and SIY-only (LucSIY) and representative flow cytometry plots of antigen-specific cells in the lung 5–6 weeks post-tumor initiation.
(B and C) Percentage of SIIN- and SIY-specific cells of CD8 T cells (B) and the proportion that are TCF1+ TIM3− (C) in LucOS, LucSIIN and LucSIY mice at 5–6 weeks by flow cytometry.
(D) Lentiviral constructs expressing SIINYEKL (Y5) in combination with SIY (LucY5S) or alone (LucY5) and representative flow cytometry plots of antigen-specific cells in the lung 5–6 weeks post-tumor initiation.
(E) pMHC affinity for each antigen assessed by flow cytometric quantification of H-2Kb stabilization on TAP-deficient RMA-S cells. Dotted lines = 50% binding concentration, which approximates the affinity (Kd) of each peptide for H-2Kb.
(F) pMHC complex stability (Koff) for each antigen measured by Differential Scanning Fluorimetry of pMHC thermal melting. Dotted lines = melting temperature (Tm) of each pMHC complex. Displayed Tm values are averaged from 3 experiments.
(G) TCR cross-reactivity measured by SIIN and Y5 tetramer co-staining of CD8 T cells from spleens of SIIN-vaccinated mice. Percentage of tetramer+ cells stained by both tetramers is graphed.
(H-I) Percentage of Y5-, SIIN- and SIY-specific cells of CD8 T cells (H) and the proportion that are TCF1+ TIM3− (I) at 5–6 weeks in LucOS, LucY5S, LucY5 and LucSIIN mice.
(A-F) and (H-I) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group. (E and G) are representative of 2 independent experiments.
In acute viral infection, several factors underlie antigen immunodominance, including: relative stability of peptide-MHC (pMHC) complexes, number of pMHC complexes on the surface of antigen-presenting cells, and functional avidity of the T cell repertoire for each antigen (Yewdell, 2006). SIINFEKL is known to bind H-2Kb MHC molecules with greater affinity and stability than SIYRYYGL (Eisen et al., 2012); thus, we hypothesized that differential MHC binding might contribute to the observed dominance hierarchy. To test this, we created lentiviruses expressing SIINYEKL (Y5), a point mutant of SIIN that binds H-2Kb molecules with reduced stability (Koff) and affinity (Kd) (Figure 3D–3F and S3G; Howarth et al., 2004). The Y5 mutation conserves key TCR contact residues and is largely cross reactive with the SIIN T cell precursor pool (Figure 3G and S3H; Bentzen et al., 2018). By comparing SIIN and Y5, we can decouple the contribution of pMHC binding and TCR repertoire/avidity to antigen dominance. Remarkably, when Y5 and SIY were expressed together (LucY5S), we found the antigen dominance hierarchy was reversed compared to LucOS, favoring SIY as the immunodominant antigen (Figures 3D and 3H). SIY-specific T cells expanded substantially more than Y5-specific cells and the Y5 response was enriched for TCF1+ progenitor cells (Figures 3H and 3I). When Y5 was expressed alone (LucY5), expansion of Y5-specific cells rebounded and enrichment for TCF1+ progenitor cells was no longer observed. These results demonstrate that differential pMHC binding can establish an antigen dominance hierarchy in cancer, and subdominant responses, irrespective of the antigen, are enriched for a TCF1+ progenitor cell phenotype. Y5 bound H-2Kb with higher affinity, but lower stability than SIY (Figure 3E and 3F), suggesting pMHC stability plays a larger role than affinity in establishing Y5 subdominance.
To evaluate the dynamics between antigens with similar MHC binding properties, we expressed two neoantigens derived from point mutations in murine methylcholanthrene (MCA)-induced sarcoma, mALG8 and mLAMA4 (Figure S3K; Gubin et al., 2014). Compared to mLAMA4, mALG8 binds H-2Kb with similar, but slightly lower affinity and stability (Figures S3I and S3J). Expression of mALG8 or mLAMA4 alone (LucA and LucL, respectively) in tumors resulted in responses of equivalent magnitude (Figure S3L). However, when co-expressed (LucAL), neither response became dominant in terms of T cell expansion, but both decreased in magnitude compared to the single-antigen setting (Figure S3L). The proportion of TCF1+ progenitor cells was also increased for both responses (Figure S3M), trending towards greater enrichment for the mALG8 response, the slightly weaker MHC binder. Thus, antigens with similar MHC binding properties are largely co-dominant, but their competition can drive enrichment for a TCF1+ progenitor phenotype, similar to subdominant antigens.
Subdominant Antigen-Specific T Cells Do Not Preferentially Benefit from anti-PD1/CTLA4 Therapy
Given TCF1+ progenitor cells play a key role in CD8 T cell responses to ICB therapy, we investigated whether SIY-specific cells in LucOS mice might preferentially benefit from ICB. In response to one week of anti-PD-1/CTLA-4 treatment, SIIN- and SIY-specific T cell expansion was greatest at 5 weeks, diminished at 8 weeks and absent by 12 weeks, mirroring the kinetics of T cell dysfunction and contraction of TCF1+ progenitor cells described above (Figures 4A and 4B). ICB treatment shifted cells away from a TCF1+ TIM3− phenotype and towards a TCF1− TIM3+ phenotype, consistent with increased differentiation to an exhausted state (Figure 4C). The absolute number of TCF1+ TIM3− cells increased, despite their decreased proportion, indicating that TCF1+ progenitor cells also expand with ICB (Figure 4D). ICB treatment also increased cell proliferation (Ki67) and cytotoxicity (GZMB; Figures 4E and 4F). Surprisingly, despite being enriched for TCF1+ progenitor cells, SIY-specific cells did not respond better to ICB compared to SIIN-specific cells. A somewhat greater shift in TCF1+ TIM3− to TCF− TIM3+ cells was observed, but did not translate into greater T cell expansion or increased effector functions (Figures 4B–4F).
Figure 4. Subdominant Antigen-Specific T Cells Do Not Preferentially Benefit from anti-PD1/CTLA4 Therapy.
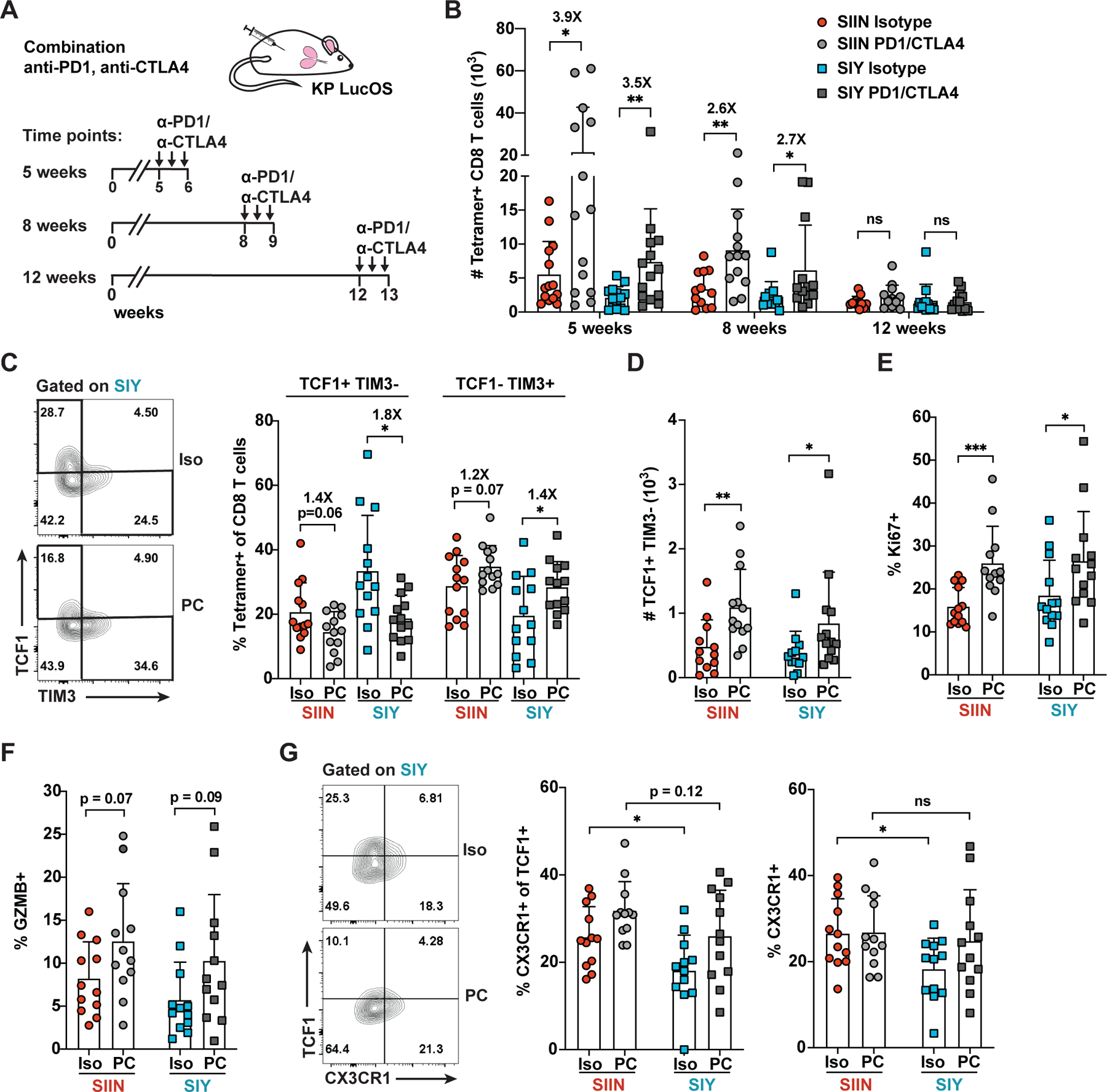
(A) Time course of anti-PD1/CTLA4 therapy initiated at 5, 8 or 12 weeks post-tumor initiation, comprised of 3 doses (arrows) of isotype control (Iso) or anti-PD1/CTLA4 (PC) antibodies in LucOS mice.
(B-G) Absolute number of SIIN- versus SIY-specific CD8 T cells (B), the proportion expressing TCF1+ TIM3− versus TCF1− TIM3+ (C), the absolute number of TCF1+ TIM3− cells (D), and the proportion expressing Ki67 (E), GZMB (F) and CX3CR1 (TCF1+ and total; G) assessed by flow cytometry in LucOS mice following treatment with Iso or PC starting at 8 weeks unless otherwise indicated. Representative of ≥ 3 independent experiments, n ≥ 5 mice per group.
Recent studies in chronic LCMV infection and cancer have identified the chemokine receptor CX3CR1 as a marker of cells recently differentiated from TCF1+ progenitor cells (Hudson et al., 2019; Zander et al., 2019). CX3CR1 expression correlates with cytotoxic function and depletion of CX3CR1+ cells results in a loss of disease control. CX3CR1 was expressed on a subset of SIIN- and SIY-specific TCF1+ cells and likely marks cells that are actively differentiating (Figure 4G). CX3CR1 expression was lower on SIY-specific TCF1+ cells and SIY-specific cells overall, suggesting SIY cells are repressed in their differentiation from a progenitor to an exhausted cell state (Figure 4G). ICB treatment was able to rescue much of this deficit, consistent with ICB promoting differentiation of progenitor cells (Figure 4G). Similarly, expression of SIY alone in LucSIY mice rescued CX3CR1+ cells to SIIN levels, which suggests that competition with the SIIN response represses SIY cell differentiation (Figure S3D). Notably, however, ICB was insufficient to fully rescue differentiation of SIY-specific cells, as the SIY response matched, but did not exceed the SIIN response, disproportionate with the enrichment of TCF1+ cells amongst the SIY response.
The Subdominant CD8 T Cell Response is Enriched for a CCR6+ TCF1+ Progenitor Cell Subset with a Tc17 Differentiation Trajectory
One possible explanation for a worse than expected SIY response to ICB is that SIY-specific TCF1+ progenitor cells are intrinsically less functional than their SIIN counterparts. To explore potential heterogeneity in progenitor cell phenotype or differentiation, we performed a separate analysis of progenitor clusters C4 and C8 and exhausted clusters C2 and C3 from the scRNA-seq data (Figure 5A). Both C4 and C8 were enriched for a number of genes previously associated with TCF1+ progenitor cells (e.g. Tcf7, Xcl1, Slamf6, Ccr7), but were localized distally on the UMAP and were characterized by unique gene signatures (Figure 5B; Table S3). C8 was marked by genes associated with memory T cells, including Klf2, S1pr1 and Il7r, while C4 expressed markers of T cell dysfunction, tolerance and anergy, and most notably, showed strong enrichment for a gene signature of Tc17 cells (Figure 5B, 5C, S5A and S5B; Tables S2 and S3).
Figure 5. The Subdominant CD8 T Cell Response is Enriched for a CCR6+ TCF1+ Progenitor Cell Subset with a Tc17 Differentiation Trajectory.
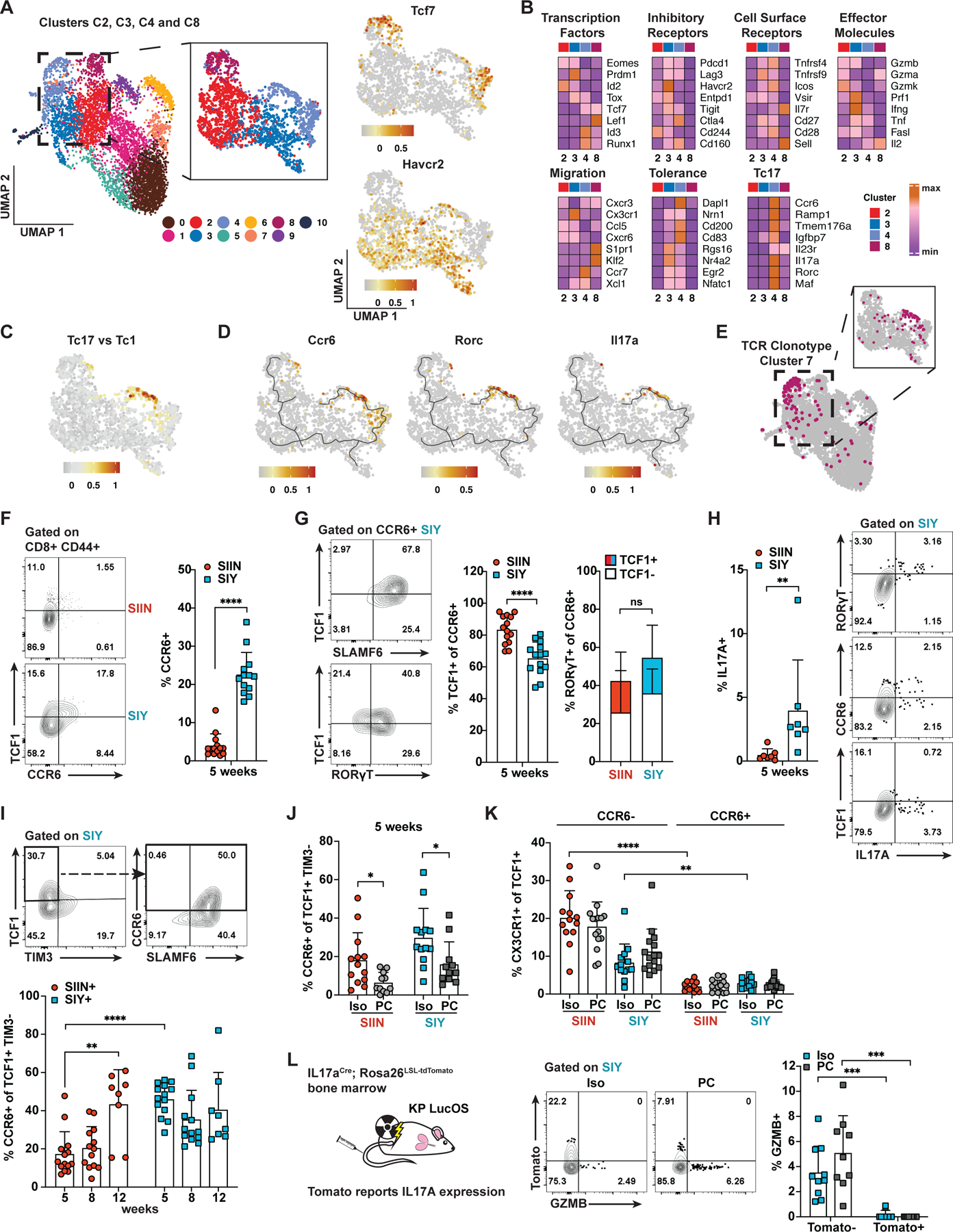
(A) UMAP embedding of clusters C2, C3, C4 and C8 from the scRNA-seq data in Figure 2B and log-normalized expression of Tcf7 and Havcr2.
(B) Expression (mean log(expression +1)) of genes associated with the indicated classifications across clusters C2, C3, C4 and C8.
(C) Scoring of cells from (A) for enrichment of a gene signature differentially upregulated in Tc17 versus Tc1 CD8 T cells in the skin of mice infected with S. epidermidis (Linehan et al., 2018; Table S2).
(D) Monocle3 lineage trajectories overlaid with UMAP visualizations of Ccr6, Rorc and Il17a expression.
(E) TCR clonotype cluster 7 (see Figures S4A and S4D) highlighted on the UMAP plots from (A).
(F-G) Percentage of SIIN- and SIY-specific cells expressing CCR6 (F) and the proportion of these cells expressing TCF1, SLAMF6 and RORɣT (G) at 5 weeks by flow cytometry. CCR6+ RORɣT+ cells are subsetted by TCF1 expression.
(H) Percentage of SIIN- and SIY-specific cells expressing IL17A at 5 weeks and flow cytometry plots showing co-expression of IL17A with CCR6, RORɣT and TCF1 on SIY-specific cells.
(I-J) Percentage of SIIN- and SIY-specific TCF1+ TIM3− cells expressing CCR6 over time and following one week of treatment with isotype (Iso) or anti-PD1/CTLA4 (PC) antibodies starting at 5 weeks.
(K) Percentage of SIIN- and SIY-specific TCF1+ cells that express CX3CR1 subsetted by CCR6 expression following Iso or PC treatment at 5 weeks.
(L) Irradiated KP mice were reconstituted with IL17aCre × Rosa26LSL-tdTomato bone marrow and LucOS tumors were initiated. Flow cytometric analysis of the percentage of SIY-specific cells expressing Tomato (marking current/prior IL17A expression) following one week of Iso or PC treatment starting at 5 weeks is shown. Data are compiled from 2 independent experiments. Data in (F-K) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group.
Tc17 cells, marked by expression of the chemokine receptor CCR6, are an IL17A-producing CD8 T cell subset commonly associated with autoimmune inflammation (Srenathan et al., 2016). Ccr6 was highly expressed in progenitor cluster C4 and overlapped with Tcf7 expression; however, cells exhibiting other hallmarks of Tc17 cells, including Rorc (RORɣT) and Il17a expression, were predominantly found adjacent to Tcf7-expressing cells within C4 (Figure 5D). Monocle3 analysis of putative lineage trajectories (Cao et al., 2019; Trapnell et al., 2014) predicted a trajectory connecting Tcf7/Ccr6-expressing cells with the Tc17 population (Figure 5D). Further, unsupervised clustering of TCR clonotypes revealed a group of five clonotypes, clonotype cluster 7, that predominantly contained cells spanning both the Tcf7/Ccr6 and Rorc/Il17a regions of C4 (Figure 5E, S4A and S4D). Only a small number of TCR clonotypes expressed Il17a and might suggest a subset of TCRs promote Tc17 differentiation (Figure S4A). However, the majority of these clonotypes expressed Il17a in a small proportion of cells and were distributed across multiple clusters, indicating Tc17-associated clonotypes also give rise to other cell states.
Flow cytometry analyses confirmed the presence of CCR6+ SIIN and SIY-specific cells and revealed enrichment amongst the SIY response (Figure 5F). The majority of CCR6+ cells expressed TCF1 as well as SLAMF6, another marker used to define TCF1+ progenitor cells (Figure 5G and S5D; Miller et al., 2019). Most CCR6+ cells also expressed RORɣT and about half of these expressed TCF1 (Figure 5G). SIY-specific cells produced more IL17A compared to SIIN-specific cells, especially amongst the RORɣT+ and CCR6+ populations, indicating increased Tc17 differentiation of SIY-specific cells (Figure 5H). Comparing functionality of CCR6+ TCF1+ versus CCR6− TCF1+ cells, we found both populations proliferated similarly (Ki67), but GZMB expression was largely restricted to a small proportion of CCR6− TCF1+ cells (Figures S5E and S5F). CCR6+ TCF1+ cells had higher expression of tolerance/anergy markers (CD200, EGR2, CD83), inhibitory receptors (PD1, LAG3, TIGIT) and costimulatory receptors often upregulated on exhausted cells (ICOS, OX40) compared to CCR6− TCF1+ cells (Figure S5C). CCR6+ TCF1+ cells also expressed higher levels of TOX, a transcription factor closely tied with T cell dysfunction (Scott et al., 2019). Altogether, these results uncover previously undescribed heterogeneity amongst TCF1+ progenitor cells. High expression of markers of dysfunction/tolerance, low GZMB expression and Tc17 differentiation suggest CCR6+ TCF1+ cells represent an unconventional TCF1+ population with reduced functionality.
Remarkably, the CCR6+ subset made up nearly 40% of SIY-specific TCF1+ progenitor cells, accounting for much of the enrichment of TCF1+ progenitor cells amongst the SIY response at 5 and 8 weeks (Figures 5I and S5G). The proportion of CCR6+ TCF1+ progenitor cells increased over time for the SIIN response, correlating with the kinetics of dysfunction and loss of ICB response (Figure 5I and S5G). Thus, a higher proportion of CCR6+ TCF1+ versus CCR6− TCF1+ progenitor cells correlates with worse ICB response and suggests the CCR6+ subset has reduced capacity to respond to ICB. This may explain why SIY-specific cells do not respond better than SIIN-specific cells to ICB despite enrichment for TCF1+ progenitor cells. Consistent with this, the proportion of CCR6+ TCF1+ cells was reduced after one week of anti-PD1/CTLA4 ICB treatment (Figure 5J). Since total TCF1+ cell numbers increase with ICB response (Figure 4D), this indicates that ICB preferentially expands or recruits CCR6− TCF1+ cells. Additionally, little to no expression of CX3CR1 was observed on CCR6+ TCF1+ cells, while up to 20% of CCR6− TCF1+ cells expressed CX3CR1 (Figure 5K and S5H). This suggests CCR6+ TCF1+ cells are impaired in conventional differentiation to a cytotoxic state.
To assess the functionality of CCR6+ TCF1+ derived Tc17 cells, we utilized an IL17A reporter allele to permanently mark cells that have expressed IL17A with Tomato fluorescence (Figure 5L; Hirota et al., 2011). Consistent with the flow cytometry data, the majority of SIY-specific CCR6+ and RORɣT+ cells expressed Tomato (Figure S5I). Around 30% of TCF1+ cells also expressed Tomato and harbored most of the CCR6+ population (Figure S5I). Strikingly, Tomato+ cells expressed no GZMB, even following one week of anti-PD-1/CTLA4 ICB therapy (Figure 5L). This indicates cells that have expressed IL17A do not differentiate into cytotoxic cells. Further, the proportion of Tomato+ cells was reduced following ICB treatment and suggests ICB reduces differentiation down the Tc17 pathway (Figure S5J). Altogether, these data suggest that CCR6+ TCF1+ and Tc17 cells constitute functionally inferior cell states that contribute poorly to therapeutic response to ICB.
Therapeutic Vaccination Breaks Antigen Dominance and Eliminates the CCR6+ Subset of TCF1+ Progenitor Cells
CCR6 upregulation and Tc17 differentiation in autoimmune diseases is driven by reactivity to self-antigens (Srenathan et al., 2016). Hence, we hypothesized that suboptimal T cell priming conditions might induce this phenotype in tumors and sought to improve T cell priming conditions by therapeutic vaccination of LucOS mice with SIIN and SIY long peptides (Figure 6A). Vaccination vastly improved the SIIN- and SIY-specific CD8 T cell responses in the tumor-bearing lung, increasing T cell expansion, proliferation (Ki67) and cytotoxicity (GZMB) (Figure 6B–6D). Greater fold expansion was observed for the SIY versus SIIN response, suggesting SIY-specific cells preferentially benefit from vaccination. Spatial profiling by tissue-based cyclic immunofluorescence (t-CyCIF) showed increased infiltration of CD8 T cells into tumors post-vaccination, with greater proportions expressing Ki67 and GZMB (Figures 6E–6G, S6B and S6C). This correlated with decreased tumor size, particularly when immune cell infiltrate was excluded (Figure 6F and S6A). The effects of vaccination were antigen-specific, with no contribution of adjuvant alone (Figure S6D). Interestingly, little response was observed in the lung 7 days after the initial vaccine dose (Figures 6H and S6E). However, antigen-specific T cells were expanded in the blood, inguinal lymph nodes and spleen, especially for the SIY response (Figure 6I and S6F). These data indicate that vaccination acts predominantly by increasing priming and/or expanding activated cells in the periphery rather than boosting the existing response in the lung.
Figure 6. Therapeutic Vaccination Breaks Antigen Dominance and Eliminates the CCR6+ Subset of TCF1+ Progenitor Cells.
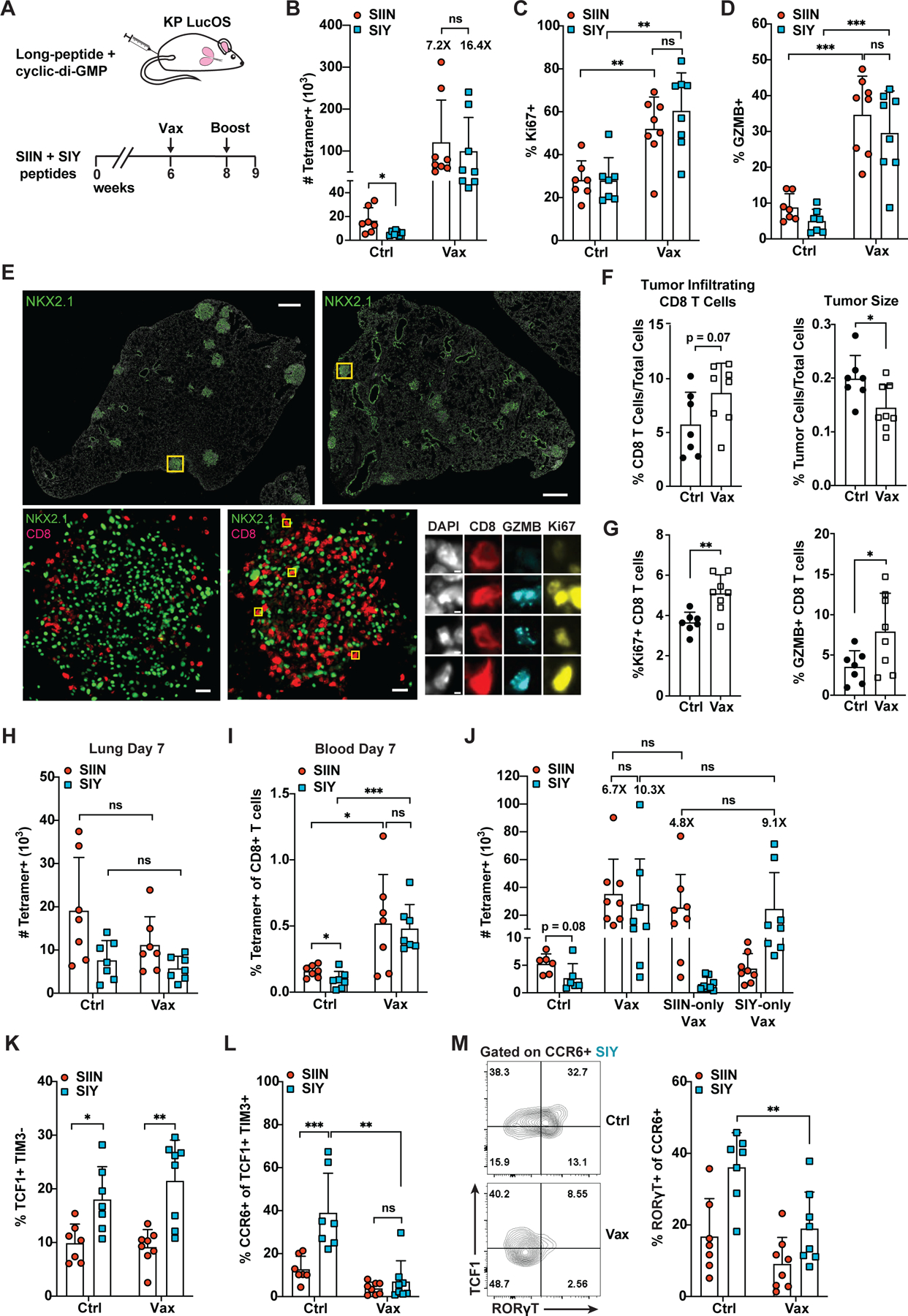
(A) SIIN and SIY long-peptide and cyclic di-GMP vaccine. Mice were vaccinated at 6 weeks (s.c. tail base), boosted at 8 weeks and analyzed 9 weeks post-tumor initiation.
(B-D) Absolute number of SIIN- and SIY-specific cells (B) and the percentage expressing Ki67 (C) and GZMB (D) in PBS control (Ctrl) versus vaccine (Vax) treated mice by flow cytometry.
(E-G) Tissue-based cyclic immunofluorescence (t-CyCIF) imaging of lung lobes of Ctrl versus Vax-treated mice (E) and quantification of CD8 T cell infiltration and tumor size (F) and the percentage of Ki67 and GZMB positive CD8 T cells in tumors (G). Results are averaged from all tumors from two lung lobes per mouse.
(H-I) Absolute number of SIIN- and SIY-specific cells in the lung (H) and the percentage in the blood (I) of Ctrl versus Vax treated mice 7 days after the initial vaccine dose.
(J) Absolute number of SIIN- and SIY-specific cells in mice treated with Ctrl, Vax, SIIN-only vaccine or SIY-only vaccine as in (A).
(K-L) Percentage of SIIN- and SIY-specific cells that are TCF1+ TIM3− (K) and the proportion of these cells that express CCR6 (L) in Ctrl versus Vax treated mice by flow cytometry.
(M) Proportion of CCR6+ SIIN- and SIY-specific cells expressing RORɣT in Ctrl versus Vax treated mice by flow cytometry.
(B-D) and (J-M) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group. (H-I) are compiled from 2 independent experiments.
Comparison of the SIIN/SIY pooled vaccine to SIIN-only and SIY-only vaccines demonstrated that the benefits of vaccination were restricted to the targeted antigen (Figure 6J and S6G). However, the SIY-only vaccine was able to increase the magnitude and functionality of the SIY response to a similar extent as the SIIN/SIY vaccine (Figure 6J and S6G). This indicates that targeting a subdominant antigen alone with vaccination can be sufficient to rescue the subdominant response to the level of the dominant response. The SIY response remained enriched for TCF1+ progenitor cells compared to the SIIN response after vaccination against SIIN/SIY or SIY-only (Figure 6K and data not shown); however, CCR6+ TCF1+ progenitor cells were all but eliminated (Figure 6L and S6G). Amongst the remaining CCR6+ cells, fewer expressed RORɣT, suggesting reduced propensity for Tc17 differentiation (Figure 6M). Altogether, these results provide strong evidence that CCR6 expression amongst TCF1+ cells and Tc17 differentiation is driven by suboptimal T cell priming. Moreover, they demonstrate that vaccination can “break” antigen dominance, improving the overall contribution and quality of the subdominant T cell response.
We further assessed whether vaccination could break the antigen dominance hierarchy between SIY and Y5 in LucY5S mice. Surprisingly, vaccination expanded only the dominant SIY response while subdominant Y5-specific cells remained low in number (Figure S6H). Still, Y5-specific cells benefitted functionally, with similar increases in GZMB and Ki67 expression and depletion of CCR6+ TCF1+ cells compared to SIY-specific cells (Figure S6H). Because Y5 is much less stable on MHC compared to SIY and SIIN (see Figure 3F), this suggests a minimum threshold of pMHC stability may be required to overcome antigen dominance in response to vaccination. Still, the functional enhancement of Y5-specific cells indicates that inclusion of unstable MHC binders in vaccines may still be beneficial overall.
CCR6+ TCF1+ Progenitor and Tc17 Cells are Found Across Human Cancers
To extend our findings to human cancer, we scored clusters from the mouse scRNA-seq dataset for enrichment of gene signatures (Table S4) derived from three published human CD8 T cell scRNA-seq datasets spanning lung, melanoma, colon, endometrial and renal cancers (Guo et al., 2018; Sade-Feldman et al., 2019; Wu et al., 2020). The mouse TCF1+ progenitor cluster, C8, was strongly enriched for gene signatures from clusters associated with progenitor and memory cell phenotypes, most notably two clusters described as containing TCF1+ progenitor cells in melanoma (Sade-Feldman CD8_4- and CD8_6-Memory/Effector) but also Guo CD8_C2-CD28 and C3-CX3CR1 and Wu 8.6.KLRB1 (Figures 7A and S7A). In contrast, mouse cluster C4, containing CCR6+ TCF1+ cells, showed weak to no enrichment for these signatures and was instead enriched for signatures of T cell exhaustion, including Sade-Feldman CD8_1-Exhaustion/CellCycle and Guo CD8_C6_LAYN (Figures 7A and S7A; Table S4). In a reciprocal analysis, we scored the tumor CD8 T cell subset of the Wu et al. pan-cancer dataset with gene signatures of mouse clusters C8 and C4 and found strong enrichment of C8 in the “progenitor region” of the UMAP, marked by TCF7 expression and cells assigned to the published progenitor-like cluster, 8.6.KLRB1 (Figures 7B and S7B). The C4 signature was weakly, but more broadly enriched across the UMAP, including in regions with HAVCR2 expression and cells assigned to published clusters with exhausted phenotypes, 8.3.Trm and 8.5.Mitosis (Figures 7B and S7B). These results suggest that both C8 and C4 cell populations resemble populations in human cancer; however, the C8 population is better aligned with progenitor cell states described in humans.
Figure 7. CCR6+ TCF1+ and Tc17 Cells are Found Across Human Cancers.
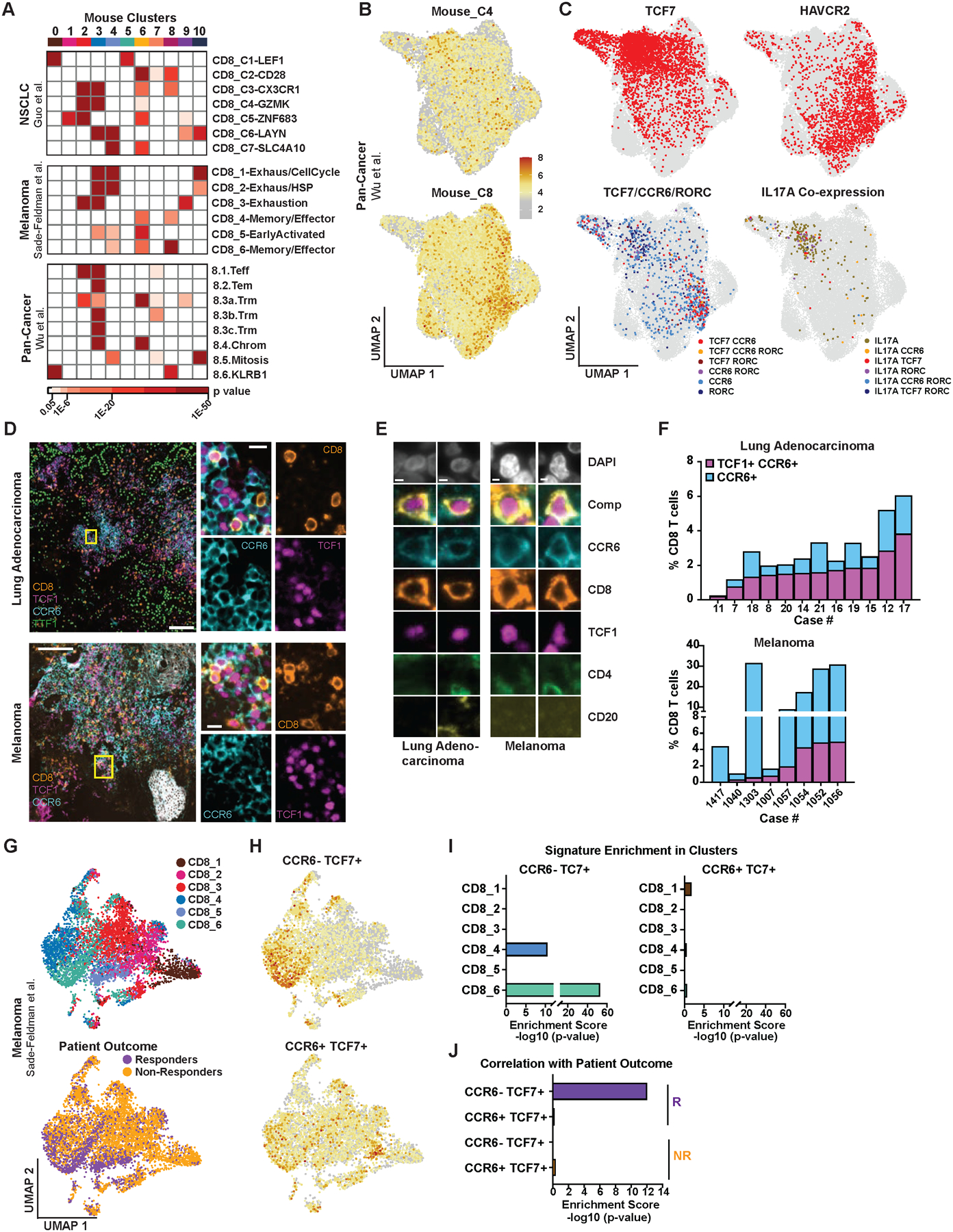
(A) Enrichment of CD8 gene signatures from three human cancer scRNA-seq datasets (Table S4) in the mouse scRNA-seq clusters from Figure 2B. Human clusters, right; mouse clusters, top. Colored boxes indicate significant enrichment, p ≤ 0.05.
(B-C) UMAP of tumor CD8 T cells from a human pan-cancer scRNA-seq dataset (Wu et al., 2020) showing enrichment of gene signatures derived from mouse progenitor clusters C4 and C8 (B) and expression of TCF7, HAVCR2, co-expression of TCF7, CCR6 and RORC and co-expression of IL17A with TCF7, CCR6 and/or RORC (C).
(D-F) t-CYCIF images of human lung adenocarcinoma and metastatic melanoma samples (D-E) and quantification of CD8 T cells expressing TCF1 and CCR6 (F).
(G) UMAP embedding of CD8 T cells from an anti-PD1 and/or anti-CTLA4 melanoma scRNA-seq dataset (Sade-Feldman et al., 2018) showing cell assignment to published clusters CD8_1 thru CD8_6 and responder versus non-responder patients.
(H) Scoring of cells in (G) for enrichment of gene signatures (Table S3) derived from mouse Ccr6− Tcf7+ and Ccr6+ Tcf7+ cells.
(I-J) Enrichment of the gene signatures from (H) in clusters CD8_1 thru CD8_6 (I) and responders versus non-responders (J).
In (C) and (I-J), mucosal invariant T (MAIT) cells that phenotypically resemble Tc17 cells were excluded from the analysis based on TCR sequence (Table S6; STAR methods).
In the reanalysis of the Wu et al. dataset, we further identified cells co-expressing TCF7 and CCR6, some of which also expressed RORC, indicating that CCR6+ TCF1+ cells are indeed found in human cancer (Figure 7C; Table S6). IL17A-expressing cells, some co-expressing TCF7, CCR6 and/or RORC, were also found (Figure 7C; Table S6). While CCR6+ TCF7+ cells were predominantly localized to the progenitor region of the UMAP, IL17A-expressing cells were distally localized in the region expressing HAVCR2 and exhibiting a more differentiated phenotype. We identified similar cell populations with analogous localization patterns in four additional scRNA-seq datasets from individual cancer types, including lung cancer, melanoma and basal cell carcinoma (Figure S7C; Guo et al., 2018, Sade-Feldman et al., 2019, Tirosh et al., 2016, Yost et al., 2019). We further validated the presence of CCR6+ TCF1+ cells in human tumor tissue by t-CyCIF imaging and found these cells made up 2% of CD8 T cells on average across early-stage lung adenocarcinoma and metastatic melanoma samples (Figures 7D and S7D; Tables S5 and S7). Our observations in mouse lung adenocarcinoma indicate that while only a small percentage of TCF1+ cells are antigen-experienced, the majority of CCR6+ cells are contained within the activated, tumor antigen-specific response (Figure S7G). Hence, despite comprising a small proportion of total CD8 T cells, CCR6+ TCF1+ cells likely constitute a larger fraction of tumor-reactive CD8 T cells compared to CCR6− TCF1+ cells in humans.
Finally, we directly investigated a correlation between CCR6+ TCF1+ cells and response to ICB by reanalyzing the CD8 T cell subset of the Sade-Feldman et al. melanoma dataset from patients treated with ICB (Sade-Feldman et al., 2019). Consistent with published observations, cells derived from responder versus non-responder patients segregated spatially on the UMAP, with responder cells colocalizing with TCF7 expression and progenitor-associated clusters CD8_4 thru CD8_6 (Figure 7G and S7C). Scoring the dataset with signatures derived from CCR6+ TCF7+ versus CCR6− TCF7+ cells from the mouse scRNA-seq data revealed significant enrichment of the CCR6− TCF7+ signature in clusters CD8_4 and CD8_6 and cells from responder patients (Figures 7H–7J). In contrast, the CCR6+ TCF7+ signature was enriched in exhaustion-associated cluster CD8_1, and no significant enrichment was observed in responder cells. These data indicate that CCR6− TCF1+, and not CCR6+ TCF1+ cells, are positively correlated with patient response to ICB. This is consistent with our findings in mouse lung adenocarcinoma and suggests CCR6+ TCF1+ cells contribute poorly to ICB response in human patients.
DISCUSSION
Neoantigens can drive potent anti-tumor T cell responses, yet T cell reactivity against predicted neoantigens is starkly limited across most cancers (Schumacher et al., 2019). Our study demonstrates that antigen dominance hierarchies in tumors may contribute to this narrowly focused CD8 T cell response. In KP mouse lung adenocarcinomas expressing pairs of immunogenic neoantigens, we found that CD8 T cell responses against different neoantigens compete and can result in the establishment of an immunodominant antigen. This profoundly impacted the response against the subdominant antigen, suppressing T cell expansion, differentiation and effector function. These findings demonstrate that CD8 T cell responses against concurrently expressed tumor neoantigens are interdependent, and their competition can limit the overall diversity and effectiveness of the anti-tumor response.
Multiple factors may contribute to the establishment of antigen dominance hierarchies in tumors (Schreiber et al., 2002; Yewdell, 2006). Here, using the Y5 mutant of SIIN that poorly binds MHC, we demonstrated that pMHC binding plays a central role. It has previously been shown that only tumor peptides with high affinity for MHC are efficiently cross-presented (Engels et al., 2013); hence, weak pMHC binding may limit opportunities for good interactions with antigen presenting cells (APCs). The subdominant SIY and Y5 responses in KP LucOS and LucY5S mice, respectively, were rescued when these antigens were expressed alone, despite pMHC binding being unchanged. Thus, it may be that competition between CD8 T cell responses for interactions with APCs amplifies a deficit in MHC binding and results in formation of antigen dominance hierarchies.
Subdominant T cell responses in our model were associated with enrichment of TCF1+ progenitor cells that drive CD8 T cell responses to ICB therapy in mice and humans (Philip et al., 2021). However, we found that the subdominant SIY response in KP LucOS mice did not preferentially benefit from ICB therapy, possibly due to predominance of a dysfunctional subset of TCF1+ cells marked by CCR6 expression. Another recent study from our lab corroborates there being heterogeneity amongst TCF1+ cells and identifies a SLAMF6+ subset, that contains CCR6+ TCF1+ cells, as a population recently arrived from the tumor draining lymph node (Schenkel et al., in press Immunity). Analysis of human scRNA-seq datasets spanning multiple cancer types and imaging of human lung adenocarcinoma and melanoma samples indicates this heterogeneity is reflected across human cancers (this study and Schenkel et al., in press Immunity). Furthermore, while CCR6− TCF1+ cells were correlated with melanoma patient response to ICB therapy, no correlation was observed for CCR6+ TCF1+ cells. Therefore, CCR6+ TCF1+ cells appear to contribute poorly to ICB response in both mice and humans.
In KP LucOS mice, we identified a lineage trajectory from CCR6+ TCF1+ cells to a Tc17 population. Tc17 cells have previously been reported in cancer, but their role remains unclear. While Tc17 cells have been reported to mediate tumor control in some transplant tumor settings, their production of IL17A may also contribute to tumor-promoting inflammation (Chang et al., 2014; Srenathan et al., 2016; Zhao et al., 2020). Here, we found that CD8 T cells marked by an IL17A reporter did not express GZMB, even in response to ICB therapy, suggesting Tc17 cells do not contribute to cytotoxic responses against tumors. Furthermore, our lab previously showed that treatment of KP lung adenocarcinoma with a neutralizing antibody to IL17A significantly reduces tumor burden by inhibiting recruitment of tumor-promoting neutrophils (Jin et al., 2019). Similarly, in humans, tumor infiltration by myeloid derived suppressor cells correlates with accumulation of IL17A-producing cells (Wu et al., 2014; Zhuang et al., 2012). Thus, in addition to contributing poorly to ICB response, CCR6+ TCF1+ cell differentiation to a Tc17 phenotype may also promote tumorigenesis through IL17A production.
Tc17 differentiation is predominantly observed in autoimmune inflammation and is driven by autoreactive TCR signals (Srenathan et al., 2016). Hence, we hypothesized that CCR6+ TCF1+ cells might be derived from suboptimal TCR stimulation during priming and differentiation. Consistent with this, vaccination of KP LucOS mice initially expanded cells in the blood and peripheral lymphoid organs, especially for the subdominant SIY response. TCF1+ cells were also significantly expanded in the lung following the vaccine boost; however, the CCR6+ subset was lost. Increased antigen presentation with vaccination likely alleviates the constraints of low affinity/stability pMHC binding, improving TCR stimulation and reducing differentiation of CCR6+ TCF1+ cells. An increasingly immunosuppressive tumor microenvironment may further contribute to poor TCR stimulation as tumors progress and could explain why SIIN-specific cells develop a CCR6+ TCF1+ phenotype over time.
Vaccination of KP LucOS mice dramatically improved the magnitude and functionality of tumor-reactive CD8 T cells, and the response to vaccination was correlated with a reduction in tumor burden. Likewise, early clinical studies have demonstrated the therapeutic potential of neoantigen vaccines in melanoma, where they improve T cell priming and tumor control (Hollingsworth and Jansen, 2019). Vaccination has shown a remarkable ability to elicit de novo T cell responses (Ott et al., 2017; Sahin et al., 2017) and, in combination with anti-PD1 therapy, also promotes epitope spreading to neoantigens not included in the vaccine (Ott et al., 2020). Therefore, vaccination against a range of predicted immunogenic neoantigens may be effective at priming new T cell responses even in cancers with few pre-treatment T cell-reactive epitopes. Our data suggest the presence of CCR6+ TCF1+ cells is indicative of subdominant and/or poorly primed T cell responses in tumors. Consequently, CCR6+ TCF1+ cells might mark tumors that would preferentially benefit from vaccination, and should be further explored as a biomarker of patient response to vaccine therapies.
pMHC affinity is used for identification of neoepitopes for cancer vaccines. High affinity neoepitopes are typically chosen (Ott et al., 2017) and may selectively target immunodominant T cell responses. Our results indicate subdominant T cell responses against some lower pMHC affinity neoantigens may experience greater benefit from vaccination and remain enriched for TCF1+ progenitor cells. Since TCF1+ cells repopulate the cytotoxic T cell pool and are ICB-responsive, engaging subdominant T cell responses through vaccination might result in more durable tumor control and better response to ICB. Overall, these findings provide rationale for evaluating the relative response to high versus low pMHC affinity/stability antigens in clinical trials of pooled neoantigen cancer vaccines, where subdominant antigens may contribute more to tumor control than previously realized.
Limitations of the Study
In this study, neoantigen pairs were expressed clonally in tumors and the expression level of each antigen was held constant. Human tumors are generally thought to be more heterogenous, containing variable numbers of neoantigens expressed at different levels and with varying clonality. These factors may influence hierarchical ordering of antigens and could be explored in future studies by modifying antigen number, expression level and clonal fraction.
In the mouse scRNA-seq analysis, we mapped a differentiation trajectory of CCR6+ TCF1+ cells to a Tc17 population that lacks cytotoxic capacity, even after ICB. However, as lineage tracing by adoptive transfer is technically intractable in the KP lung cancer model, we were unable to directly delineate differentiation of CCR6+ TCF1+ cells. Use of novel genetic alleles, such as a fate-mapping CCR6 reporter, or investigation in other models may shed further light on the relative functionality and plasticity of CCR6+ TCF1+ cells in response to ICB.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tyler Jacks (tjacks@mit.edu).
Materials Availability
Plasmids generated in this study have been deposited to Addgene: Lenti-LucOS (Cat. No. 22777), Lenti-LucSIIN (Cat. No. 174043), Lenti-LucSIY (Cat. No. 174044), Lenti-LucY5 (Cat. No. 174046), Lenti-LucY5S (Cat. No. 174045), Lenti-LucAL (Cat. No. 174049), Lenti-LucA (Cat. No. 174047), Lenti-LucL (Cat. No. 174048).
Data and Code Availability
The mouse scRNA-seq data has been deposited to GEO and is publicly available as of the date of publication. The accession number is listed in the key resources table. This paper additionally included analyses of publicly available human scRNA-seq datasets. The accession numbers for these datasets are listed in the key resources table.
All original code for the mouse and human scRNA-seq analyses and the t-CYCIF imaging analyses has been deposited on GitHub and is publicly available as of the date of publication. The DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivomAb PD1 | BioXCell | Clone 29F.1A12; Cat# BE0273; RRID: AB_2687796 |
| InVivomAb CTLA4 | BioXCell | Clone 9H10; Cat# BE0131; RRID: AB_10950184 |
| InVivomAb rat IgG2a | BioXCell | Clone 2A3; Cat# BE0089; RRID: AB_1107769 |
| InVivomAb polyclonal Syrian Hamster IgG | BioXCell | Cat# BE0087; RRID: AB_1107782 |
| FC: CD8ɑ | BD Biosciences | Clone 53-6.7; Cat# 563786, 612759; RRID: AB_2732919, AB_2870090 |
| FC: CCR6 (CD196) | BioLegend | Clone 29-2L17; Cat# 129822; RRID: AB_2687019 |
| FC: CX3CR1 | BioLegend | Clone SA011F11; Cat# 149031; RRID: AB_2565939 |
| FC: CXCR3 (CD183) | BD Biosciences | Clone CXCR3-173; Cat# 562937, 741032; RRID: AB_2687551, AB_2740650 |
| FC: CD44 | BD Biosciences; BioLegend | Clone IM7; Cat# 563736, 612799, 103057, 103059; RRID: AB_2738395, AB_2870126, AB_2564214, AB_2571953 |
| FC: CD45 | ThermoFisher Scientific; BD Biosciences | Clone 30-F11; Cat# 47-0451-80, 562420; RRID: AB_1548790, AB_11154401 |
| FC: CD83 | BioLegend | Clone Michel-19; Cat# 121506; RRID: AB_572009 |
| FC: CD200 | BD Biosciences | Clone OX-90; Cat# 565546; RRID: AB_2739288 |
| FC: EGR2 | ThermoFisher Scientific | Clone erongr2; Cat# 17-6691-80; RRID: AB_11150966 |
| FC: Granzyme B | BioLegend, BD Biosciences | Clone GB11; Cat# 515408, 560213; RRID: AB_2562196, AB_1645453 |
| FC: H-2Kb | ThermoFisher Scientific | Clone AF6-88.5.5.3; Cat# 17-5958-82; RRID: AB_1311280 |
| FC: ICOS (CD278) | ThermoFisher Scientific | Clone 7E.17G9; Cat# 46-9942-80; RRID: AB_2744727 |
| FC: IFN-γ | BioLegend | Clone XMG1.2; Cat# 505822; RRID: AB_961359 |
| FC: IL17A | ThermoFisher Scientific | Clone 17B7; Cat# 11-7177-81; RRID: AB_763581 |
| FC: IL7R (CD127) | ThermoFisher Scientific | Clone A7R34; Cat# 25-1271-82; RRID: AB_469649 |
| FC: Ki67 | BD Biosciences | Clone B56; Cat# 561277, 563756; RRID: AB_10611571, AB_2732007 |
| FC: LAG3 (CD233) | BD Biosciences, ThermoFisher Scientific; BioLegend | Clone C9B7W; Cat# 741820, 46-2231-82, 11-2231-80, 125219; RRID: AB_11151334, AB_2572483, AB_2566571 |
| FC: OX40 (CD134) | BioLegend | Clone OX-86; Cat# 119419; RRID: AB_2687175 |
| FC: PD1 (CD279) | BioLegend | Clone RMP1-30; Cat# 109110, 109121; RRID: AB_572017, AB_2687080 |
| FC: RORƔT | ThermoFisher Scientific | Clone B2D; Cat# 25-6981-82, 17-6981-82; RRID: AB_2784671, AB_2573254 |
| FC: SLAMF6 (LY-108) | BD Biosciences | Clone 13G3; Cat# 742272; RRID: AB_2871448 |
| FC: TCF1/TCF7 | Cell Signaling Technology | Clone C63D9; Cat# 6444, 6709, 9066; RRID: AB_2797627, AB_2797631, AB_2797696 |
| FC: TIGIT | BD Biosciences | Clone 1G9; Cat# 565474; RRID: AB_2739254 |
| FC: TIM3 | BioLegend | Clone RMT3-23; Cat# 119721, 119725; RRID: AB_2616907, AB_2716066 |
| FC: TNFα | BioLegend | Clone MP6-XT22; Cat# 506306; RRID: AB_315427 |
| FC: TOX | Miltenyi Biotec | Clone REA473; Cat# 130-118-335; RRID: AB_2751485 |
| FC: H-2Kb SIINFEKL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb SIYRYYGL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb SIINYEKL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb mALG8 monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb mLAMA4 monomer | NIH Tetramer Core Facility | Custom |
| Hashtag: TotalSeq-C anti-mouse | BioLegend | Clones M1/42, 30-F11; Cat# 155861, 155863, 155865, 155867, 155869, 155871, 155873, 155875, 155877, 155879; RRID: AB_2800693, AB_2800694, AB_2800695, AB_2800696, AB_2800697, AB_2819910, AB_2819911, AB_2819912, AB_2819913, AB_2819914 |
| tCycif: anti-mouse B220 (CD45R) | ThermoFisher Scientific | Clone RA3-6B2; Cat# 41-0452-80; RRID: AB_2573598 |
| tCycif: anti-mouse CD4 | ThermoFisher Scientific | Clone 4SM95; Cat# 41-9766-82; RRID: AB_2573637 |
| tCycif: anti-mouse CD8a | Cell Signaling Technology | Clone D4W2Z; Cat# 98941S; RRID: AB_2756376 |
| tCycif: anti-mouse FOXP3 | ThermoFisher Scientific | Clone FJK-16 s; Cat# 11-5773-82; RRID: AB_465243 |
| tCycif: anti-human αSMA | Abcam | Clone EPR5368; Cat# ab202509; RRID: AB_2868435 |
| tCycif: anti-human CD4 | R&D Systems | Cat# FAB8165G; RRID: AB_2728839 |
| tCycif: anti-human CD8α | ThermoFisher Scientific | Clone AMC908; Cat# 50-0008-80; RRID: AB_2574148 |
| tCycif: anti-human CD20 | ThermoFisher Scientific | Clone L26; Cat# 50-0202-80; RRID: AB_11151691 |
| tCycif: anti-human CD45 | BioLegend | Clone HI30; Cat# 304008; RRID: AB_314396 |
| tCycif: anti-human CCR6 | Abcam | Clone EPR22259; Cat# ab243852; RRID: AB_2860033 |
| tCycif: anti-human FOXP3 | eBioscience | Clone 236A/E7; Cat# 41-4777-80; RRID: AB_2573608 |
| tCycif: anti-human TCF1 | Cell Signaling Technology | Clone C63D9; Cat# 6444S; RRID: AB_2797627 |
| tCycif: anti-human TTF1 (also reacts with mouse) | Abcam | Clone EPR595(2); Cat# ab206726; RRID: AB_2857980 |
| Purification: Ly6G Rat Monoclonal | BioLegend | Clone 1A8; Cat# 127602; RRID: AB_1089180 |
| Purification: EpCam (CD326) Rat Monoclonal | BioLegend | Clone G8.8; Cat# 118202; RRID: AB_1089027 |
| Purification: F4/80 Rat Monoclonal | BioLegend | Clone BM8; Cat# 123102; RRID: AB_893506 |
| Biological samples | ||
| Human formalin fixed paraffin embedded tissue samples from lung adenocarcinoma cases | Partners Healthcare Institutional Review Board at Brigham Health, Boston, MA, USA | Excess tissue, discarded tissue protocol number 2018P001627 |
| Human formalin fixed paraffin embedded tissue samples from metastatic melanoma cases | Specialized Histopathology Services - Longwood Core Brigham and Womens Hospital, Boston, MA, USA | Project: 05-042 - T Jacks Lab slides |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase IV | Worthington Biochemical | Cat# LS004189 |
| DNase I | Sigma-Aldrich | Cat# 10104159001 |
| Monensin | BioLegend | Cat# 420701 |
| Golgi Plug | BD Biosciences | Cat# 555028 |
| Phorbol-12-myristate-13-acetate (PMA) | EMD Millipore | Cat# 524400 |
| Ionomycin Calcium Salt | Sigma | Cat# 13909 |
| Streptavidin, allophycocyanin conjugate | Invitrogen | Cat# S32362 |
| Streptavidin, R-phycoerythrin conjugate | Invitrogen | Cat# S21388 |
| Mirus TransIT LT1 | Mirus Bio | Cat# MIR 2300 |
| SYPRO Orange Protein Gel Stain | Thermo Fisher Scientific | Cat# S6651 |
| Cyclic-di-GMP | Invitrogen | Cat# tlrl-nacdg |
| SIINFEKL peptide | New England Peptide | Custom |
| SIYRYYGL peptide | New England Peptide | Custom |
| SIINYEKL peptide | New England Peptide | Custom |
| ITYTWTRL peptide (mALG8) | Genscript | Custom |
| VGFNFRTL peptide (mLAMA4) | Genscript | Custom |
| SMLVLLPDEVSGLEQLESIINYEKLTEWTS | New England Peptide | Custom |
| SMLVLLPDEVSGLEQLESIINFEKLTEWTS peptide | New England Peptide | Custom |
| GRCVGSEQLESIYRYYGLLLKERSEQKLIS peptide | New England Peptide | Custom |
| Empty peptide-receptive H-2Kb molecules | Saini et al., 2019 | Generated in-house |
| Zombie Fixable Viability Kit | BioLegend | Cat# 423102 |
| Ghost Dye | Tonbo Biosciences | Cat# 13-0865-T500 |
| Critical commercial assays | ||
| Intracellular Fixation & Permeabilization Buffer Set Kit | ThermoFisher Scientific | Cat# 88-8824-00 |
| Dynabead Sheep Anti-Rat IgG | ThermoFisher Scientific | Cat#11035 |
| Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit | 10X Genomics | Cat# 000167 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10X Genomics | Cat#1000071 |
| Easysep Dead Cell Removal (Annexin V) Kit | StemCell | Cat#17899 |
| Deposited data | ||
| Mouse KP LucOS SIIN versus SIY scRNA-seq | GEO | GEO: GSE164177 |
| Sade-Feldman et al. scRNA-seq dataset | Sade-Feldman et al., 2019 | GEO: GSE120575 |
| Wu et al. scRNA-seq dataset | (Wu et al., 2020) | GEO: GSE139555 |
| Guo et al. scRNA-seq dataset | Guo et al., 2018 | GEO: GSE99254 |
| Yost et al. scRNA-seq dataset | (Yost et al., 2019) | GEO: GSE123813 |
| Tirosh et al. scRNA-seq dataset | Tirosh et al., 2016 | GEO: GSE72056; Broad Data Use Oversight System: DUOS-000002 |
| Mouse and human single cell RNA-sequencing analysis code | This Paper | GitHub: https://doi.org/10.5281/zenodo.5110770 |
| t-CYCIF analysis code | This Paper | GitHub: https://doi.org/10.5281/zenodo.5104869 |
| Experimental models: Cell lines | ||
| GreenGo 3TZ for lentiviral titering | Sánchez-Rivera et al., 2014 | Available upon request |
| 293FS* viral packaging cell line | This paper | Available upon request |
| RMA-S TAP-deficient cell line | Schumacher et al., 1990 | Available upon request |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J | Jackson Laboratories | Stock No: 008179 |
| Mouse: B6.129P2-Trp53tm1Brn/J | Jackson Laboratories | Stock No: 008462 |
| Mouse: Il17atm1.1(icre)Stck/J | Jackson Laboratories | Stock No: 016879 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratories | Stock No: 007914 |
| Recombinant DNA | ||
| Plasmid: Lenti-LucOS | DuPage et al., 2011 | Addgene Cat# 22777 |
| Plasmid: Lenti-LucSIY | This Paper | Addgene Cat# 174044 |
| Plasmid: Lenti-LucSIIN | This Paper | Addgene Cat# 174043 |
| Plasmid: Lenti-LucY5S | This Paper | Addgene Cat# 174045 |
| Plasmid: Lenti-LucY5 | This Paper | Addgene Cat# 174046 |
| Plasmid: Lenti-LucA | This paper | Addgene Cat# 174047 |
| Plasmid: Lenti-LucL | This paper | Addgene Cat# 174048 |
| Plasmid: Lenti-LucAL | This paper | Addgene Cat# 174049 |
| Software and algorithms | ||
| FlowJo | FlowJo | Version 10.7.1 https://www.flowjo.com/solutions/flowjo/downloads |
| Aperio ImageScope | Leica Biosystems | Version 12 https://www.leicabiosystems.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| ImageJ BaSiC Plugin | Peng et al., 2017 | https://www.helmholtz-muenchen.de/icb/research/groups/marr-lab/software/basic/index.html |
| ASHLAR | The Python Package Index | https://pypi.org/project/ashlar/ |
| ilastik | Berg et al., 2019 | https://www.ilastik.org/download.html |
| MATLAB | MathWorks | version 2018a https://www.mathworks.com/products/matlab.html |
| R/Rstudio | Rstudio | version 1.2.5042 https://www.rstudio.com/ |
| Seurat | Butler et al., 2018 | version 4.0.0 https://cran.r-project.org/web/packages/Seurat/index.html |
| CellRanger | 10X Genomics | version 3.1.0 https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| ComplexHeatmap | CRAN Gu et al., 2016 | version 2.4.3 https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Tidyr | CRAN | version 1.1.2 https://tidyr.tidyverse.org, https://github.com/tidyverse/tidyr |
| Cowplot | CRAN | version 1.1.0 https://wilkelab.org/cowplot/ |
| GGplot2 | CRAN | version 3.2.2 https://ggplot2.tidyverse.org, https://github.com/tidyverse/ggplot2 |
| GridExtra | CRAN | version 2.3 https://cran.r-project.org/web/packages/gridExtra/index.html |
| Plyr | CRAN | version 1.8.6 https://cran.r-project.org/web/packages/plyr/index.html |
| Dplyr | CRAN | version 1.0.2 https://cran.r-project.org/web/packages/dplyr/index.html |
| Monocle3 | Trapnell et al., 2014 | version 0.2.3.0 https://cole-trapnell-lab.github.io/monocle3/ |
| Patchwork | CRAN | version 1.1.1 https://github.com/thomasp85/patchwork |
| Viridis | CRAN | version 0.5.1 https://github.com/sjmgarnier/viridis |
| Circlize | CRAN | Version: 0.4.11 https://github.com/jokergoo/circlize |
| Stringr | CRAN | Version: 1.4.0 https://stringr.tidyverse.org, https://github.com/tidyverse/stringr |
| Philentropy | CRAN | Version: 0.4.0 https://github.com/drostlab/philentropy |
| Dendextend | CRAN | Version: 1.14.0 https://cran.r-project.org/web/packages/dendextend/index.html |
| FactoEXTRA | CRAN | Version: 1.0.7 http://www.sthda.com/english/rpkgs/factoextra |
| ProjecTILs | Andreatta et al., 2021 | v.0.5.1 https://github.com/carmonalab/ProjecTILs |
| gplots | Warnes, 2015 | v.3.1.0 https://cran.r-project.org/web/packages/gplots/index.html |
| RColorBrewer | Neuwirth, 2014 | v.1.1-2 https://cran.r-project.org/web/packages/RColorBrewer/index.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
KrasLSL-G12D/+;p53fl/fl (KP) and IL17aCre;Rosa26LSL-tdTomato mice have been previously described (DuPage et al., 2011; Hirota et al., 2011; Madisen et al., 2010) and were on a C57BL/6 genetic background. Male and female KP mice were used in all experiments and experimental arms were gender and age-matched within 3 weeks. 8 week old, female IL17aCre;Rosa26LSL-tdTomato were used for the collection of bone marrow to reconstitute lethally irradiated KP mice. All studies were performed under an animal protocol approved by the Massachusetts Institute of Technology (MIT) Committee on Animal Care. Mice were assessed for morbidity according to guidelines set by the MIT Division of Comparative Medicine and were humanely sacrificed prior to natural expiration.
Lentiviral Tumor Induction
2.5 × 104 PFU of lentivirus containing Cre recombinase and model neoantigens were injected intratracheally into KP mice as previously described (Dupage et al., 2011, Dupage et al., 2009). Mice were at least 8 weeks old at the time of injection. Details of the lentiviral constructs and lentivirus production are included below. Mice were randomized post-infection for therapy trials.
Bone Marrow Chimeras
Bone marrow was harvested from the femur and tibia of 8 week-old female IL17aCre;Rosa26LSL-tdTomato mice into sterile RPMI 1640 media. Total bone marrow was pelleted by centrifugation at 1200 rpm for 5 minutes, resuspended in PBS and filtered through a 70 μm cell strainer. 1×107 cells in 200 ul PBS were injected retroorbitally into lethally irradiated KP mice (two doses of 550 rad, three hours apart). 6 weeks after bone marrow transfer, mice were injected intratracheally with Lenti-LucOS lentivirus as described above.
METHOD DETAILS
Lentiviral Constructs and Lentivirus Production
The LucOS lentiviral construct was previously described (Dupage et al., 2011). The LucSIIN and LucSIY vectors were generated by deleting the 24 nucleotides of SIINFEKL or SIYRYYGL coding sequence, respectively, from the LucOS vector. The LucY5S vector was generated by creating a point mutation at amino acid position 5 in SIINFEKL in the LucOS construct. To make the LucY5 vector, the 24 nucleotides of SIYRYYGL coding sequence were deleted from LucY5S. The LucA and LucL vectors were generated by fusing the mALG8 or mLAMA4 neoepitope sequence plus approximately 6 flanking amino acids to luciferase. These sequences were cloned in tandom to create LucAL, with care to avoid creation of additional potentially immunogenic epitopes.
Lentiviruses were produced in 293FS* cells by transfection with the lentiviral constructs above, psPAX2 and VSV-G packaging plasmids at a 4:3:1 ratio using Mirus TransIT LT1 (MirusBio). At 48- and 72-hours post-transfection, viral supernatant was collected, passed through 0.45 μm filters and concentrated by ultracentrifugation (25,000 rpm for 2 hours at 4°C). Concentrated lentivirus was resuspended in a 1:1 solution of Opti-MEM and HBSS and stored at −80°C. Viral titers were determined using the GreenGo 3TZ cell line as previously described (Sanchez-Rivera et al., 2014).
Anti-PD1/anti-CTLA4 Therapy
KP LucOS mice were treated for one week with InvivoMAb anti-PD1 (29F.1A12; BioXCell) and InvivoMAb anti-CTLA4 (9H10; BioXCell) monoclonal antibodies or Invivo isotype controls (Rag IgG2a, 2A3; Syrian Hamster, polyclonal; BioXCell). An initial dose of 200 μg each of anti-PD1 and anti-CTLA4 (or isotype control) was injected i.p. at day 0, followed by 200 μg anti-PD1 and 100 μg anti-CTLA (or isotype control) at day 3 and 6. All doses were delivered i.p. in 200 μl of PBS. Mice were analyzed one day following the last dose.
Therapeutic Vaccination
KP LucOS mice were vaccinated via subcutaneous tail-based injection with 30 amino acid peptides containing SIINFEKL, SIYRYYGL and/or SIINYEKL (10 nmol; New England Peptide) and cyclic di-GMP adjuvant (0.25 mg/ml; Invitrogen) 6 weeks post-tumor initiation. A booster injection was given 14 days later and mice were euthanized 9 weeks post-tumor initiation. All doses were delivered in two 50 μl boluses and control mice were injected with PBS. The long peptide sequences used were: SMLVLLPDEVSGLEQLESIINFEKLTEWTS, GRCVGSEQLESIYRYYGLLLKERSEQKLIS and SMLVLLPDEVSGLEQLESIINYEKLTEWTS (New England Peptide).
For assessment of SIIN-specific TCR cross-reactivity with Y5, wild-type mice were vaccinated against SIINFEKL as described above.
Tissue Collection and Flow Cytometry
To distinguish lung tissue-resident versus circulating immune cells, mice were injected retroorbitally with a fluorescently-conjugated anti-CD45 antibody (PE-CF594 or AlexaFluor780; 30-F11; BD Bioscience) 2–3 minutes prior to euthanasia (Anderson et al., 2014). Circulating CD8 T cells staining positive for the CD45 antibody were excluded from our analyses. Lung tissue was collected and minced by hand with spring scissors and incubated in 125 U/mL collagenase IV (Worthington Biochemical) and 40 U/mL DNase I (Sigma-Aldrich) for 30 minutes at 37°C. Following incubation, the tissue was dissociated using the m_lung_2.0.1 protocol on a gentleMACS Dissociator using gentleMACS C tubes (Miltenyi Biotec) and passed through a 70 μm cell strainer. Spleen and lymph nodes were dissociated through a 70 μm cell strainer into RPMI 1640 media containing 1% heat-inactivated fetal bovine serum. Cell suspensions were pelleted by centrifugation (1200 rpm for 5 minutes), resuspended in 1X RBC Lysis Buffer (eBioscience) and incubated on ice for 10 minutes to lyse red blood cells. Pellets were resuspended in PBS and single cell suspensions were transferred to a 96-well U-bottom plate. Cells were then stained with a fixable viability dye to exclude dead cells (20 minutes on ice; Zombie Dye; Invitrogen; Tonbo Ghost Dye; Tonbo Biosciences) and resuspended in FACS buffer (1% heat-inactivated FBS in PBS) and stained with the following surface antibodies for 15–30 minutes on ice: CD8ɑ (53-6.7), CCR6 (29-2L17), CX3CR1 (SA011F11), CXCR3 (CXCR3-173), CD44 (IM7), IL7R (A7R34), LAG3 (C9B7W), PD1 (RMP1-30), SLAMF6 (13G3), TIGIT (1G9), TIM3 (RMT3-23), OX40 (OX-86), ICOS (7E.17G9), CD200 (OX-90), CD83 (Michel-19) purchased from ThermoFisher Scientific, BD Biosciences or Biolegend (see Key Resources Table). Cells were simultaneously stained with H-2Kb peptide-MHC tetramers specific to SIINFEKL, SIYRYYGL, SIINYEKL, mALG8 or mLAMA4 (monomer, NIH Tetramer Core Facility; PE and APC streptavidin, Invitrogen). For intracellular staining, cells were fixed for 1 hour at room temperature using the eBioscience Fixation/Permeabilization Kit (ThermoFisher Scientific). Cells were then stained overnight at at 4°C with the following antibodies: TCF1/TCF7 (C63D9), RORɣT (B2D), TBET (eBio4B10), Granzyme B (GB11), Ki67 (B56), TOX (REA473), EGR2 (erongr2) purchased from Cell Signaling Technology, ThermoFisher Scientific, BD Biosciences, Biolegend or Miltenyi Biotec (see Key Resources Table). Samples were analyzed on a BD Biosciences LSR Fortessa or LSR II Flow Cytometry Analyzer.
For assaying cytokine production, T cells were enriched in lung samples by depletion of tumor cells and myeloid cells. Cells were incubated with 2 μg each of purified Ly-6G, EpCAM and F4/80 antibodies (Biolegend; 4°C for 20 minutes), then 125 μl of sheep anti-rat Dynabeads (Invitrogen; 4°C for 30 minutes with rotation). Note, to avoid depleting intravascularly labeled CD45+ cells, a mouse anti-mouse CD45.2 antibody (Brilliant Violet 510, Biolegend) was used for the intravascular stain. A Dynabeads magnet (Invitrogen) was applied to remove the beads and the T cell-containing supernatant was transferred to a clean tube, washed with 10 mL FACS buffer and pelleted by centrifugation (1200 rpm for 5 minutes). Pellets were resuspended in T-cell media (RPMI 1640 containing 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 1X MEM Non-essential amino acids, 2 mM L-glutamine, 0.275 mM beta-mercaptoethanol, and 50 U/mL penicillin-streptomycin) and transferred to a 96-well U-bottom plate. To assay IFNƔ and TNFɑ production, samples were pelleted by centrifugation (1200 rpm for 5 minutes) and were resuspended in T cell media plus 1X Monensin (Biolegend), 1X Golgi Plug (BD Bioscience) and SIINFEKL or SIYRYYGL peptide (167 nM; New England Peptide). For IL17A staining, cells were resuspended in T cell media containing PMA (2.5 ng/ml; EMD Millipore) and Ionomycin (1 μM; Sigma-Aldrich), 1X Monensin and 1X Golgi Plug. Samples were then incubated at 37°C for 4–5 hours. Ten percent of each sample was aliquoted and left unstimulated as a control. Cells were subjected to viability staining, surface staining and fixation as described above and stained with the following antibodies overnight at 4°C: IFN-ɣ (XMG1.2), TNFɑ (MP6-XT22) and IL17A (17B7) from ThermoFisher Scientific, BD Biosciences or Biolegend (see Key Resources Table).
scRNA-seq on Mouse CD8 T Cells
Lung single cell suspensions were prepared from 10 mice as described above. Dead cells were removed with an EasySep Dead Cell Removal Kit (Annexin V; StemCell). Cells were then stained with 1 μg TotalSeq-C hashtag antibodies 1–10 (M1/42, 30-F11; Biolegend) and surface antibodies against CD44 (IM7), CD8ɑ (53–6.7) and H-2Kb-SIINFEKL and H-2Kb-SIYRYYGL tetramers for 30 minutes on ice. FACS sorting was performed on a FACS Aria III (BD Biosciences) and CD44+ CD8α+ SIINFEKL+ and CD44+ CD8α+ SIYRYYGL+ T cells were sorted into separate tubes, counted and resuspended in PBS (no calcium or magnesium) with 0.04% BSA. Approximately 65,000 SIINFEKL+ cells and 14,000 SIYRYYGL+ cells were loaded across two channels of a Chromium single-cell 5’ chip (10X Genomics) according to manufacturer’s instructions. Single cells were partitioned into droplets with gel beads to form emulsions, after which cellular lysis and barcoded reverse transcription of mRNA was performed. Paired 5’ gene expression, hashtag barcode and V(D)J libraries were prepared using the following kits from 10X Genomics and protocols provided by the manufacturer: Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit v1.1; Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell. RNA expression libraries were sequenced individually using HiSeq X (Illumina). VDJ and hashtag barcode libraries were sequenced with HiSeq 2500 (Illumina).
Peptide Stabilization of H-2Kb on RMA-S Cells
TAP-deficient RMA-S cells were cultured in RPMI 1640 media (10% fetal bovine serum, 2 mM L-glutamine, 10 mM Hepes and 50 U/μl Penicillin-Streptomycin) at 26°C overnight (16 hours) to increase expression of empty MHC molecules on the cell surface. Cells were then plated at 100,000 cells per well in a 96-well U-bottom plate (150 μl per well). 10-fold serial dilutions of peptides (10 mM in DMSO stock) were generated in RPMI media at 4X final concentration in a 96-well U-bottom plate. 50 μl of peptide was added to the RMA-S cells, bringing the final volume to 200 μl. Cells were incubated with peptide for 2 hours at 26°C to allow for peptide binding followed by 1 hour at 37°C to degrade empty MHC molecules. Each condition was plated in triplicate. Cells were washed in FACS buffer (1% heat-inactivated FBS in PBS) and stained with an APC-conjugated H-2Kb (AF6-88.5.5.3; ThermoFisher Scientific) antibody for 15 minutes on ice. Plates were assayed on a BD Biosciences LSR II flow cytometry analyzer.
Differential Scanning Fluorimetry Assay for pMHC Thermal Melting
Empty peptide-receptive H-2Kb molecules (Saini et al., 2019) were diluted to 200 μg/ml in 50 mM HEPES pH 7.5 containing 1X SYPRO Orange dye. 18 μl of this solution was combined with 2 μl of 100 μM peptide (diluted in PBS) in triplicate in a 384 well quantitative PCR plate. The plate was incubated on ice for 10 minutes to allow for peptide-MHC interaction. The plate was then assayed on a Roche LightCycler 480 instrument. Fluorescence was acquired with excitation at 465 nM and emission at 580 nM (SYPRO Orange filter), Samples were equilibrated at 25 °C for 1 min and then heated with a gradient of 0.1 °C/s from 25 °C to 95 °C with 6 fluorescence readings for every 1 °C. Melting curves were normalized to the minimum and maximum fluorescence value and the inverse of the first derivative (−dF/dT) was then calculated. The minimum value of −dF/dT was taken at the Tm.
Histology and H&E Staining
Tumor-bearing lung lobes and a portion of the spleen were fixed in 4% paraformaldehyde overnight and embedded in paraffin. Hematoxylin and eosin stain (H&E stain) was performed with a standard method by the Hope Babette Tang Histology Facility at Koch Institute. Digitally scanned images of H&E slides were obtained with an Aperio ScanScope AT2 at 20X magnification and tumor area was annotated using Aperio ImageScope software.
t-CyCIF Staining and Imaging
The t-CyCIF experimental protocol was conducted as previously described (Du et al., 2019; Lin et al., 2018). In brief, the mouse and human lung adenocarcinoma and human melanoma FFPE slides were baked at 60°C for 30 min, dewaxed using Bond Dewax Solution (Leica Biosystems) at 72°C, and antigen retrieval was performed with Epitope Retrieval 1 Solution (Leica Biosystems) at 100°C for 20 minutes using the BOND RX Automated IHC/ISH Stainer (Leica Biosystems). All antibodies were diluted in Odyssey Intercept Buffer (plus Hoechst 33342 0.25 μg/mL; LI-COR Biosciences) and incubated overnight at 4°C in the dark. See the Key Resources Table for the complete list of antibodies. Slides were coverslipped using 20–50% glycerol solution (Sigma-Aldrich) in PBS. Images were taken using DAPI, FITC, Cy3, and Cy5 channels on the RareCyte CyteFinder Instrument (20x/0.75NA objective lens). After imaging, the fluorophores were inactivated with photobleaching solution (4.5% H2O2 and 20 mM NaOH in PBS) for 45 minutes under LED ligfFigurehts.
t-CyCIF Image Processing
The image processing of tissue cyclic immunofluorescence is organized in the following steps, each of which is described in detail below:
the software ASHLAR is used to stitch, register, and correct for image acquisition artifacts (using the BaSiC algorithm). The output of ASHLAR is a single pyramid ome.tiff file for each region imaged;
the ome.tiff file is re-cut into tiles (typically 5000×5000 pixels) containing only the highest resolution image for all channels. One random cropped image (250×250 pixels) per tile is outputted for segmentation training (using ImageJ/Fiji);
using the ilastik software the labelling of nuclear, cytoplasmic and background areas are trained on the cropped images. Based on the user training the Ilastik software outputs a 3-color RGB image with label probabilities;
the RBG probability images are thresholded and watershed in MATLAB to segment the nuclear area. The cytoplasmic measurements are derived by dilating the nuclear mask;
single-cell measurements are extracted for each channel (cell pixel median and mean for both nuclear and cytoplasmic area) as well as morphological measurements of area, solidity, and cell coordinates location.
BaSiC
The BaSiC ImageJ plugin tool was used to perform background and shading correction of the original images (Peng et al., 2017). The BaSiC algorithm calculates the flatfield, the change in effective illumination across an image, and the darkfield, which captures the camera offset and thermal noise. The dark field correction image is subtracted from the original image, and the result is divided by the flatfield image correction to obtain the final image.
ASHLAR
Alignment by Simultaneous Harmonization of Layer/Adjacency Registration (ASHLAR) is used to stitch together image tiles and register image tiles in subsequent layers to those in the first layer (Lin et al., 2018; Rashid et al., 2019). For the first image layer, neighboring image tiles are aligned to one another via a phase correlation algorithm that corrects for local state positioning error. A similar method is applied for subsequent layers to align tiles to their corresponding tile in the first layer. ASHLAR outputs an OME-TIFF file containing a multi-channel mosaic of the full image across all imaging cycles. Full codes available at: https://github.com/labsyspharm/ashlar.
Ilastik
Ilastik is a machine learning based bioimage analysis tool that is used to obtain nuclear and cytoplasmic segmentation masks from OME-TIFF files (Berg et al., 2019). For increased processing speed, randomly selected 250 × 250 pixel regions from the original OME-TIFF are used as training data. ilastik’s interactive user interface allows the user to provide training annotations on the cropped regions. Users are presented with a subset of the channels stacked images and label pixels as either nuclear area, cytoplasmic area, or background area. The annotations are used to train non-linear classifiers that are applied to the entire image to obtain probability masks describing the probabilities of each pixel belonging to the nuclear, cytoplasmic, or background area. A MATLAB (version 2018a) script uses these masks to construct binary masks for nuclear and cytoplasmic area.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-cell RNA Sequencing Analysis
Mouse CD8 scRNA-seq Data Processing, Cell Clustering, and Differential Expression Analysis
Cell Ranger, version 3.1.0 (Zheng et al., 2017), was used to process raw sequencing data for RNA expression, VDJ, and Cell Hashing libraries from mouse CD8 scRNA sequencing experiments. RNA expression data was aligned to the GRCm38/mm10 reference mouse transcriptome (version 3.0.0) and VDJ sequencing data was aligned to the prebuilt mouse (GRCm38/mm10) VDJ reference supplied by 10X Genomics (version 3.1.0; Zheng et al., 2017). Cell Ranger estimated 6,472 and 3,646 cells with 15,939 and 14,834 genes detected for RNA expression libraries of SIIN- and SIY- specific CD8 T-cells at a sequencing depth of 80.4% and 76.9%, respectively. VDJ libraries captured an estimated 4,713 and 2,705 cells with 3,857 and 2,196 of those cells containing productive V-J Spanning Pair for SIIN- and SIY- specific CD8 T-cell libraries, respectively. Cell Hashing libraries were estimated to capture 6,073 and 3,319 cells at a sequencing saturation of 10.3% and 13.2% for SIIN- and SIY- specific CD8 T Cell libraries, respectively. Cell Hashing data were demultiplexed with Seurat, version 4.0.0 (Butler et al., 2018; Stoeckius et al., 2018), using counts normalized by centered-log ratio (CLR) transformation at a positive quantile threshold of 0.98 to infer which mouse each cell was harvested from.
Seurat (version 4.0.0) was used to normalize, scale, select variable features, and perform differential gene expression analysis. To filter out dying cells with poor data quality and remove probable doublets, individual cells with <100 or >4000 detected genes, with >20,000 reads, or with >5% of reads aligned to mitochondrial genome in RNA expression libraries were removed from downstream analyses. Thereafter, any remaining cells, called as doublets, were also removed from downstream analyses. RNA expression data were normalized by total expression per cell and natural log transformed with a scale factor of 10,000. Expression for all genes in SIIN- and SIY- libraries were first centered by subtracting average expression of each gene and subsequently scaled by dividing gene expression levels by their standard deviations. SIIN- and SIY- libraries were then merged in Seurat (“merge.data = TRUE”). For each cell passing quality control thresholds, metadata assignments for V(D)J clonotypes and cell hashing (mouse of origin) were made using Python with Pandas and Numpy (Harris et al., 2020; McKinney, 2010).
The top 2,000 variable genes used for downstream dimensionality reductions were selected in Seurat using the vst method (Butler et al., 2018). Principal component analysis (PCA) was performed for the first 50 principal components (PCs). An estimation of PCs to include for downstream analyses was performed with the elbow method and JackStraw analysis. Based on this estimate, and in manual consideration of gene features of PCs, the first 30 PCs were selected for downstream analyses. A shared nearest neighbor graph (SNN, k = 20) was constructed with 30 PCs and used to perform Louvain clustering (default parameters, resolution)(Meo et al., 2011). The data was then visually projected in 2-dimensional space by using the Uniform Manifold Approximation and Projection (UMAP) algorithm (Butler et al., 2018) McInnes et al., 2018) in Seurat (default parameters). Differential gene expression was performed between all clusters using the default Wilcoxon Ranked Sum test (FindAllMarkers, min.pct = 0.25) (Table S3). The top 10 differentially expressed genes for each cluster are represented in a heatmap produced with ComplexHeatmap (cluster_columns = FALSE, cluster_rows = FALSE)(Figure S2D). Enrichment for SIIN or SIY was calculated by hypergeometric test (phyper, Stats R package; alpha = 0.05).
Gene expression scores were projected on UMAPs with Seurat using the FeaturePlot function (order = TRUE). UMAP embeddings that also depict gene expression scores were generated using the plot_cells function from Monocle3.
Subsetted C2, C3, C4, C8 Analysis and Trajectory Inference
A separate analysis of cells in the mouse CD8 scRNA-seq dataset assigned to C2, C3, C4 and C8 was performed with Monocle3, version 0.2.3.0 (Cao et al., 2019; Qiu et al., 2017; Trapnell et al., 2014). The raw RNA expression data for these cells were subsetted in Seurat and exported to Monocle3 for dimensionality reduction, clustering, and trajectory analyses. Preprocessing of raw expression data (normalization and PCA) was performed for 20 PCs, using all other default parameters. Further dimensionality reduction was performed with the UMAP algorithm (default parameters, except: umap.n_neighbors = 30L, umap.fast_sgd = FALSE, preprocess_method = “PCA”; Mclnnes et al., 2018). Leiden clustering (Tragg et al., 2018) was performed to group cells into clusters for trajectory analyses (resolution = 0.001) with otherwise default parameters. For trajectory analyses, a principal graph was learned and plotted in UMAP space (learn_graph, use_partition = FALSE, rann.k = 20). Monocle3 UMAP coordinates were exported to Seurat for gene expression and signature analysis. Differentially expressed genes were calculated in Seurat, as described above.
Gene lists reflecting CD8 T cell states and functionality were curated from a combination of the differentially expressed genes between Seurat clusters and literature review. Clusters C2, C3, C4 and C8 were scored for expression of these genes by the same methodology of Monocle3’s plot_genes_by_group function. Gene expression scores were then scaled for each cluster and visualized in heatmaps produced with ComplexHeatmap in R (cluster_columns = FALSE, cluster_rows = FALSE) (Figure 5B).
ProjecTILs T Cell Subtype Classification and Annotation
The mouse CD8 scRNA-seq data was aligned to the reference tumour-infiltrating lymphocytes (TIL) atlas and reference lymphocytic choriomeningitis virus (LCMV)-specific CD8 T cell atlas using the ProjecTILs R package (version 0.5.1; Andreatta et al., 2021). A normalized expression matrix (Seurat logNorm data) was provided as input and non-T cells were filtered using TILPRED-1.0 before alignment and batch-effect correction. Additional parameters for “make.projection” were kept as the default. Cell states were predicted for each cell using a nearest-neighbor algorithm (ProjecTILs “cellstate.predict”; Table S1) and overlaid onto the original mouse UMAP with TILPRED filtered cells set as “NA”. Cell states of interest were highlighted on UMAP plots using Seurat (Butler et al., 2018), with cells of interest plotted last. Cell state annotations were used “as-is” (without a confidence threshold). We further validated these assignments by applying a confidence score threshold of 0.5 for calling a cell state and confirmed that cell state clusters remain the same.
Enrichment of SIY and SIIN cells labeled as TIL (or LCMV) progenitor or exhausted states was evaluated using a hypergeometric test (phyper, Stats R package; alpha = 0.05). Cells annotated as progenitor or exhausted were tested similarly for enrichment in the mouse clusters.
Gene Signature Scoring and UMAP Projection
Scoring of published mouse gene signatures was performed with Seurat (version 4.0.0; Butler et al., 2018) using the AddModuleScore function, which calculates mean expression level for each signature subtracted by aggregated expression of control signatures for each cell. Signatures were filtered to exclude genes not detected in the query dataset. For projection of the TIL/LCMV progenitor exhausted and terminally exhausted signatures (Miller et al, 2019), deletional tolerance signature (Parish et al., 2009) and anergy signature (Safford et al., 2005), published gene signatures were filtered for genes captured in the mouse scRNA seq dataset (Table S2). Cells were then scored for each resultant signature using the AddModuleScore function in Seurat and highlighted on the Monocle3-based mouse UMAP of the mouse cluster C2, C3, C4 and C8 analysis.
For projection of the Tc17 versus Tc1 gene signatures from Linehan et al. (Linehan et al., 2018), raw RNA-seq sequence FASTQ files for Tc17 and Tc1 cells from the skin of Imiquimod-treated or S. epidermidis-infected mice were obtained from the Sequence Read Archive (SRA accession PRJNA419368; Imiquimod Tc17 runs: SRX3425166, SRX3425165, SRX3425145; Imiquimod Tc1 runs: SRX3425154, SRX3425153, SRX3425146; S. epidermidis Tc17 runs: SRX3425152, SRX3425151, SRX3425149; S. epidermidis Tc1 runs: SRX3425150, SRX3425148, SRX3425147). Sequences were aligned to the mouse genome (NCBI37/mm9 assembly) using Bowtie, v. 1.2.3 (Langmead et al., 2009), and feature counts were quantified using rsem, v. 1.3.1 (Li and Dewey, 2011), and UCSC mm9 genome annotation (genome.ucsc.edu). Pairwise gene expression differential analyses were conducted with the DESeq2 package, v. 1.16.1 (Love et al., 2014), in R (v. 3.6.0; r-project.org). The Tc17 vs Tc1 (S. epidermidis or Imiquimod; Table S2) signatures were scored using Seurat (AddModuleScore with default parameters) and highlighted on the Monocle3-based mouse UMAP of the mouse cluster C2, C3, C4 and C8 analysis.
CCR6+ TCF7+ and CCR6− TCF7+ signatures were derived from the mouse scRNA-seq data by identifying differentially expressed genes using Seurat (FindMarkers with default parameters) with a fold change > 2 and adjusted p-value < 0.05 (Table S3). Ccr6+ Tcf7+ marker genes were defined by comparing cells from cluster C4 expressing both Ccr6 and Tcf7 (log-normalized expression count > 0) to that of all other cells in the dataset except those in cluster C4. To define Ccr6− Tcf7+ marker genes, cells from cluster C8 expressing Tcf7 but not Ccr6 were compared to the expression of all other cells in the dataset except those in cluster C8. Mouse symbols were translated to human symbols using the Broad GSEA chip file “Mouse Gene Symbol Remapping to Human Orthologs MSigDB.v7.2.chip” (Liberzon et al., 2011; Subramanian et al., 2005), subsequently scored in the Sade-Feldman dataset and visualized with UMAPs using Seurat.
TCR Clonotype Analysis
Cells with productive TCR sequences in the mouse CD8 scRNA-seq dataset were assigned to clonotypes by Cell Ranger. While the vast majority (581 of 652) of clonotypes appear to originate exclusively from a single mouse, a minority (22 of 652) of clonotypes appear to have one or more cells that originate from multiple mice. These clonotypes were identified in Python with Pandas and excluded from downstream analyses. Most of the clonotypes detected by Cell Ranger contain less than 5 cells (548 of 652), which may reflect their underlying biological abundance (i.e. degree of clonal expansion). However, it is impossible to decouple this underlying biology from artifacts of technical dropout in these lowly-abundant clonotypes. As such, clonotypes containing less than 5 cells were removed in most analyses, resulting in a total of 103 clonotypes.
In order to understand how clonotypes were distributed across the Seurat clusters (C0–C10), a matrix was generated that quantifies the number of cells in each clonotype assigned to each Seurat cluster. To permit comparison of clonotypes of different sizes, this matrix was normalized to reflect the fraction of cells in each clonotype. To cluster clonotypes with similar distributions across Seurat clusters, Euclidean distance was first calculated between clonotypes in the normalized clonotype-cluster matrix. Visualizations of the clonotype-cluster matrix were made with ComplexHeatmap. Rows (clonotypes) in this heatmap were ordered by hierarchical clustering (method = “ward.D”) of default pairwise euclidean distances. Columns (clusters) were ordered by hierarchical clustering (method = “ward.D”) of pairwise Jaccard distance. Subsequently, complete linkage hierarchical clustering (hclust, R, method = “ward.D”) was performed on these distance calculations. A total of 12 clonotype clusters were generated by cutting the resulting dendrogram (h = 0.6, cutree, R).
Cells assigned to each clonotype were scored for expression of Gzmb, Havcr2, Cx3cr1, Tcf7, Ccr6, and Il17a by counting the number of cells with gene expression values > 0.5. To test enrichment of these genes, we performed a hypergeometric test (phyper, Stats R package; alpha = 0.05). To test enrichment of clonotypes specific for SIIN and SIY in C4+C8 and C2, we performed a 2-dimensional 2-sample KS test and visualized differences in proportion of cells with an empirical cumulative distribution plot. Additionally, to provide a metric for how equally distributed each clonotype is across clusters, we calculated the Gini index (Hurley, 2009) for each clonotype.
To visualize the location of cells from large and small clonotypes in UMAP space, we assigned each cell a value equivalent to the size of the clonotype assigned to each cell and plotted this feature in Seurat.
Enrichment of Human Signatures in Mouse Clusters
Marker genes for published CD8 T cell cluster signatures from Wu et al., Supplemental Table 4 (Wu et al., 2020), Guo et al., Supplemental Table 3 (Guo et al., 2018), and et al., Supplemental Table 4 (Sade-Feldman et al., 2019) were translated from human symbols to mouse symbols using the Mouse Genome Informatics (MGI) mouse/human orthology with phenotype annotations obtained on November 16, 2020 (Baldarelli et al., 2020) [http://www.informatics.jax.org/downloads/reports/HMD_HumanPhenotype.rpt]. For ambiguous mappings (i.e. human genes mapping to more than one mouse gene), we first prioritized the lowercase equivalent of the human gene, if available. For cases where lowercase equivalents were not an option, we checked to see if one of the possible translations was in the mouse dataset and if so, then we arbitrarily selected the first translation (but avoided symbols with the Gm* prefix if another option was available). For cases where neither the lowercase equivalent nor a match in the mouse dataset was found, the first translation was kept as a placeholder. In addition, genes without a mapping were dropped and not included in the final lists. Signature gene lists used for downstream analysis are provided in Table S4. Human signatures were scored in the mouse dataset using Seurat (AddModuleScore with default parameters) and visualized on the mouse Seurat-based UMAP (Figure S7A).
Heatmaps illustrating enrichment of human signatures in mouse clusters (Figure 7A) were generated in R using heatmap.2 and data was preprocessed using a custom script. For each human signature and mouse cluster, a Wilcoxon rank-sum test was performed to test for differential enrichment of a signature across clusters (i.e. a set of signature scores for a given cluster compared to signature scores across all other clusters) similar to DeTomaso et al. (DeTomaso et al., 2019). To reduce overall noise and highlight enrichment for clusters that included “sufficient” high-scoring cells, significance testing was conditioned according to distributional (i.e. quantile) cutoffs. Differential enrichment between sets of signature scores was only tested if the following conditions were met: the upper 2nd percentile of scores of cells within the cluster exceeded (i) the upper 2nd percentile of scores of cells outside the cluster, (ii) the upper 2nd percentile of scores of cells across the signature, and (iii) the upper 40th percentile of scores of cells across all signatures (i.e. a full matrix of signature scores per cell per cluster).
Analysis of External Human scRNA-seq Datasets
GSE139555 Wu et al.
Original cluster assignments for cells in the Wu et al. dataset (Wu et al., 2020) were retrieved from GEO (accession GSE139555; GSE139555_tcell_metadata.txt). Additionally, utility functions within the software library released in the GEO record (GSE139555_software.tar.gz) were used for extracting raw count tables with code updates to subset the dataset to contain cells assigned to CD8 T cell clusters in the original study (8.1.Teff, 8.2.Tem, 8.3a.Trm, 8.3b.Trm, 8.3c.Trm, 8.4.Chrom, 8.5.Mitosis, 8.6.KLRB1). Raw count tables for CD8 T cells per tumor sample, ignoring cells from normal adjacent tissue and peripheral blood samples across all patients, were then used in subsequent analyses. Counts per tumor sample were normalized using Seurat v3.2 SCTransform (Butler et al., 2018; Hafemeister and Satija, 2019), while regressing out percentage mitochondrial content per cell. Samples were integrated in a hierarchical fashion (per patient, then across patients) using the Seurat reference-based workflow (Stuart et al., 2019). Choice of reference sample did not affect result interpretation. PCA (Principal Component Analysis with top 30 components used for downstream analyses) dimensionality reduction and UMAP (Uniform Manifold Approximation and Projection) embeddings were generated using the integrated dataset.
For projection of mouse scRNA-seq cluster gene signatures onto the Wu analysis, the top 20 positive marker genes for each mouse cluster (C4 and C8, clustering resolution 0.7) were identified using Seurat (FindMarkers) with a fold change cutoff of 1.2 and adjusted p-value less than 0.05. Mouse symbols were translated to human symbols using the Broad GSEA chip file (MSigDB v7.2; https://data.broadinstitute.org/gsea-msigdb/msigdb/annotations_versioned/Mouse_Gene_Symbol_Remapping_Human_Orthologs_MSigDB.v7.2.chip)(Liberzon et al., 2011; Subramanian et al., 2005), subsequently scored in the Wu dataset and visualized on the Wu UMAP using Seurat. Signature gene lists used for downstream analysis (Table S3) reflect the collection of unique genes for which an ortholog has been mapped in human.
GSE120575 Sade-Feldman et al.
Log2-TPM counts were retrieved from GEO along with patient characteristics, including response to immune checkpoint blockade, and processed with Seurat v3.0.0 (Hafemeister and Satija, 2019). A Seurat object was created such that the counts slot and data slot were populated with TPM counts and log-TPM counts, respectively. Only cells assigned to published CD8 T cell clusters (Table S2; Sade-Feldman et al., 2019) were selected and used for downstream analyses. Highly variable genes were identified and the data was scaled (while regressing out percentage mitochondrial content per cell and the number of detected reads per cell) followed by dimensionality reduction with PCA and UMAP (using 24 principal components) using Seurat’s standard preprocessing procedure.
GSE99254 Guo et al.
Raw counts were retrieved from GEO (accession GSE99254). Cells from seven CD8 T cell clusters in the published analysis were retained for downstream analyses. This subset was processed using the SCT normalization flow of Seurat v3.0.0 (Hafemeister and Satija, 2019). Following dimensionality reduction using PCA, 12 principal components were used for downstream analyses including UMAP embeddings.
GSE72056 Tirosh et al.
Log2(TPM/10 + 1) counts were retrieved from GEO along with patient metadata and processed with Seurat v3.2 (Hafemeister and Satija, 2019). A Seurat object was created such that the counts slot and data slot were populated with TPM counts and log-TPM counts, respectively. The dataset was filtered to retain T cells based on metadata annotation and further filtered to retain only CD8 T cells by filtering on expression of CD8A (> 0) and CD4 (= 0). Only six patient samples with 50 or more CD8 T cells were selected and used for downstream analyses and integrated using Seurat’s reciprocal PCA (RPCA) integration flow. 30 principal components were used for downstream UMAP dimensionality reduction and data embedding.
GSE123813 Yost et al.
Raw counts were retrieved from GEO along with patient information and processed with Seurat v3.2 (Hafemeister and Satija, 2019). The dataset was filtered to retain basal cell carcinoma (BCC) T cells based on metadata annotation and further filtered to retain only CD8 T cells by filtering on expression of CD8A (> 0) and CD4 (= 0). A hierarchical integration flow based on SCT normalization was used, similar to the one described for Wu et al. above (GSE139555). Pre- and post-treatment samples per patient were integrated in the first round and cross-patient integration was performed using Seurat’s reference-based integration workflow. The code was largely based on that used for the GSE139555 analysis (described above). 30 principal components were used for downstream analyses.
For the analyses of GSE139555, GSE120575, GSE99254, GSE72056, GSE123813 above, the Seurat default “RNA” assay with log-normalized counts was used for feature plots, marker gene identification, and all expression-based analyses. For single gene and co-expression analyses, log-normalized expression count thresholds of 0.0 (raw count datasets GSE139555, GSE99254, GSE123813) and 0.5 (TPM datasets GSE72056, GSE120575) were used for considering a gene as expressed in a given cell.
Identification of mucosal associated invariant T (MAIT) cells
In the human scRNA-seq datasets we identified a population of mucosal associated invariant T (MAIT) cells that phenotypically resemble Tc17 cells (Toubal et al., 2019). MAIT cells express invariant TCRs that recognize antigen presented on the MHC-related molecule MR1. Cells with these TCRs were assigned to background (i.e. not highlighted) in the TCF7/CCR6/RORC/IL17A expression plots and excluded from statistical analyses of CCR6+/− TCF7+ signature enrichment in Figures 7C, 7I, 7J and S7C. Importantly, MAIT cells are also found in mice, but do not express CD8 and are not found amongst SIIN- or SIY-specific T cells that are MHC I Kb-restricted in KP LucOS lung tumors.
For Wu et al., clonotype assignments were retrieved from GEO record GSE139555. MAIT cells were identified based on their invariant TCR using the following logic: (alpha_v = “TRAV1-2”) AND one of ($alpha_j = “TRAJ33”) OR ($alpha_j = “TRAJ12”) OR ($alpha_j = “TRAJ20”) and assigned to the background set (not highlighted) in co-expression analysis (Toubal et al., 2019). For Sade-Feldman et al., clonotype assignments were requested and obtained from the authors (private communication). MAIT cells were identified using the logic described above. For Guo et al., cells identified as belonging to a MAIT cluster (CD8-C7-SLC4A10) in the published analysis (Guo et al., 2018) were tagged as MAIT cells. For Tirosh et al., we obtained raw scRNAseq FASTQ files through the Broad Institute Data Used Oversight System (duos.broadinstitute.org). This data was processed using MiXCR’s v3.0.13 “analyze shotgun” workflow (Bolotin et al., 2017) to catalog the T cell receptor repertoire. No cells satisfying the MAIT clonotype assignments described above were detected in this set. For Yost et al., clonotype assignments were requested and obtained from the authors (private communication). MAIT cells were identified using the logic described above.
Lists of MAIT cells per dataset are included in Table S6. Using a relaxed approach, cells without clonotype information were assumed to be non-MAIT. Overall, MAIT cells were not highlighted in co-expression plots and were dropped from CCR6+/− TCF7+ signature enrichment analyses.
Enrichment of CCR6+/− TCF7+ signatures in GSE120575 (Sade-Feldman et al.)
A hypergeometric test (phyper, Stats R package; alpha = 0.05) was used to test for enrichment of cells from published clusters in the set of cells with high CCR6+ TCF7+ or CCR6− TCF7+ signature scores (standardized signature score > 2). Similarly, cells annotated as responders/non-responders were tested for enrichment in the set of cells with high CCR6+/− TCF7+ signature scores (standardized signature score > 2). Response categories were determined by RESIST criteria: responders, complete or partial response; non-responders, stable or progressive disease (Sade-Feldman et al., 2019). MAIT cells were removed from the dataset prior to calculating proportions.
t-CyCIF Data Analysis Workflow
The data analysis is divided in a set of pre-processing steps in which data from different tissues is i) log2-transformed and aggregated together, ii) filtered for image analysis errors, and iii) normalized on a channel-by-channel basis across the entire data from a single experiment. All the steps are performed in MATLAB.
Data Aggregation
The image processing workflow outputs one ome.tiff image and one data file (.mat) for each tissue area imaged. The data matrices from each .mat file are concatenated into a single matrix for each metric measured (median/mean, nuclear/cytoplasmic) into a single structure (“AggrResults”). The morphological data (i.e., area, solidity, and centroid coordinates) is concatenated into a single structure (“MorpResults”), which also contains the indexing vector to keep track of the tissue of origin within the dataset.
Data Filtering
Single cells are filtered to identify and potentially exclude from subsequent analysis errors in segmentation and cells lost through the rounds of imaging. Two types of criteria are used to filter cells: morphological criteria based on cell object segmented area, which are applied to all the rounds for the cell object, and DAPI-based criteria which are applied to the DAPI measurement for each imaging round. The latter corrects for cell loss during cycling and computational misalignment, which are both round specific.
Morphological filtering criteria are: 1) nuclear area within a user-input range; 2) cytoplasmic area within a user-input range; 3) nuclear object solidity above a user-input threshold. DAPI-based criteria are: 1) nuclear DAPI measurement above a user-input threshold; 2) ratio between nuclear and cytoplasmic DAPI measurement above a user-input threshold. The filter information for the criteria is allocated to a logical (0–1) structure ‘Filter’, which is used to select the cells to analyze in the further analysis by indexing. The threshold selection is dataset dependent and is performed by data inspection. The values used in each dataset are available with the codes used for data analysis in the github repository https://github.com/santagatalab/2021_Burger_et_al_CyCIF_codes.
Data Normalization
Each channel distribution is normalized by probability density function (pdf) centering and rescaling. The aim is to center the distribution of the log2 fluorescent signal at 0 and rescale the width of the distribution to be able to compare across channels. The data is first log-transformed (base 2). The standard normalization is performed using a 2-component Gaussian mixture model, each model capturing the negative and the positive cell population. If the 2-component model fails to approximate the channel distribution, two other strategies are attempted: i) a 3-component model is used assuming the components with the two highest means are the negative and positive distribution (i.e., discarding the lowest component) or ii) the user selects a percentage ‘x’ of assumed positive cells and a single Gaussian distribution fit is performed on the remainder of the data to capture the negative distribution. The single Gaussian fit is then used as the lower component in a 2-component model to estimate the distribution of the positive population. The strategy chosen for each channel in each dataset is available in the github repository https://github.com/santagatalab/2021_Burger_et_al_CyCIF_codes. The “add_coeff” is defined as the intersection of the negative and positive distributions. The “mult_coeff” is defined as the difference between the mean of the negative and positive distributions. The full distribution is normalized by subtracting the add_coeff and dividing by the mult_coeff. The normalization is performed on the nuclear and cytoplasmic single-cell, single-channel distributions individually. The data preprocessing workflow is performed on all datasets. The individual analyses used in the paper are performed only in select datasets as follows.
Cell Type Calling
Cells from tissue-based experiments are classified into lineage compartments by cell type markers, by gating on the sign of the normalized values of cell type markers. Cells are scored by increasingly deeper lineage layers, starting with separating them into epithelial, immune and stromal compartments, and subdividing the immune population into lymphoid lineages. The cell types identified and the markers used to do so are included in Table S7. If a cell was called as more than one single cell-type, this is defined as a conflict. The conflicts are resolved by comparing the markers that triggered each of the cell type calls and assigning the cell type with the highest marker level. If the markers are within 10% of each other, the cell is deemed “not classifiable” for the specific layer.
Identification of CCR6+ TCF1+ CD8+ T Cells
The quantification of positive cells was performed on a single channel basis for CD8, CCR6 and TCF1. For CD8 and TCF1 signal the cells were called positive based on the normalization described above. However, in the lung cancer tissues imaged, CD20 positive B cells expressed high levels of CCR6, consistent with the current literature (Kadomoto et al., 2020). The variability of B cells between individual tissues and the spatial proximity between T cells and B cells can potentially lead to overestimation of CCR6 positivity. In order to avoid potential biases we modified the protocol in three ways: 1) the CCR6 protein was normalized at a single tissue basis to match the negative signal distribution, 2) when calculating the CCR6+ CD8+ cells (independent of TCF1 status) we discarded all cells closer than 32.5 μm (100 pixels), and 3) we imaged the CCR6 protein twice in the first and last round of the experiment.
Supplementary Material
Figure S1. Related to Figure 1. SIINFEKL- and SIYRYYGL-specific CD8 T cells in KP LucOS Lung Tumors Display Hallmarks of Dysfunction Over Time (A) Flow cytometry gating strategy for tumor SIIN- and SIY-specific CD8 T cells. A single cell suspension from tumor-bearing lung was gated on singlets and live (Zombie dye-negative) cells, then lymphocytes based on FSC/SSC. The lymphocyte gate was then analyzed for the proportion of tissue-resident CD8+ T cells based on the absence of staining by an intravascularly circulating anti-CD45 antibody (see STAR methods) and positive staining for CD8α. Tissue-resident CD8+ cells were further gated on positive CD44 staining prior to analysis of SIINFEKL (SIIN) and SIY (SIYRYYGL) peptide-MHC (H-2Kb) tetramer staining.
(B) Percentage of SIIN- versus SIY-specific cells of CD8 T cells assayed over time by flow cytometry.
(C) Representative flow cytometry plots of the percentage of SIIN- and SIY-specific cells expressing Ki67 over time.
(D) Representative flow cytometry plots comparing PD1, LAG3 and TIM3 expression between SIIN- and SIY-specific cells at 5 weeks. Naïve population (grey) represents CD44- tetramer- CD8α+ T cells. Geometric mean fluorescence intensity (gMFI) for PD1, LAG3 and TIM3 is plotted over time for SIIN- versus SIY-specific cells. All data points were compiled from a single experiment for gMFI comparison.
(B-D) are representative of ≥ 3 independent experiments per time point, n ≥ 5 mice per group.
Figure S2. Related to Figure 2. A Population of TCF1+ Progenitor Cells is Identified by scRNA-Sequencing of SIIN- and SIY-Specific CD8 T cells (A) Classification of individual cells from Figure 2A using mouse ProjecTILs atlases (Andreatta et al., 2021) from lymphocytic choriomeningitis virus (LCMV) infection and tumor infiltrating lymphocytes (TIL; B16 and MC38 transplant tumor models).
(B) Scoring of individual cells from Figure 2A for enrichment of progenitor versus terminally-exhausted gene signatures from chronic LCMV (clone 13) infection and B16 melanoma TIL mouse datasets (Miller et al. 2019). SIIN-specific cells are enriched for the terminally-exhausted signatures (tumor p = 2.90E-14; LCMV p = 2.24E-26), while SIY-specific cells are enriched for the tumor progenitor signature (tumor p = 0.004; LCMV p = 0.15). Statistics were calculated by hypergeometric test with a z-score cutoff of 0.5.
(C) Heatmap of the top 10 differentially expressed genes per cluster from Figure 2B. Select genes used to define the phenotypic labels assigned to each cluster are highlighted on the righthand side.
(D) Expression of the indicated progenitor-associated genes on cells from Figure 2A.
(E) Expression of AY036118.1 (ERF1), the top differentially expressed gene in cluster 7, and activation markers Cd69 and Cd44 on cells from Figure 2A.
(F-G) Flow cytometric analysis of PD1 (E) and Ki67 (F) expression by TCF1+ TIM3− (colored) versus TCF1− (black line) SIIN- and SIY-specific CD8 T cells at 5 weeks. The naïve population (grey) represents CD44- tetramer- CD8+ T cells. Representative of 3 independent experiments, n ≥ 5 mice per group.
Figure S3. Related to Figure 3. Expression of SIY Alone Results in Less Enrichment for TCF1+ Progenitor Cells and Greater Expression of Effector/Exhaustion Markers (A-B) Quantification of the absolute number of total (A) and TCF1+ TIM3− (B) SIIN- and SIY-specific CD8 T cells in LucOS versus LucSIIN and LucSIY mice at 5–6 weeks.
(C) Representative flow cytometry plots of PD1, LAG3 and TIM3 expression between SIIN- and SIY-specific cells in LucOS, LucSIIN and LucSIY mice at 5–6 weeks.
(D-F) Flow cytometric analysis of the proportion of SIIN- and SIY-specific cells expressing CX3CR1 (D), GZMB (E) and Ki67 (F) at 5–6 weeks.
(G) Melting curves associated with the Differential Scanning Fluorimetry (DSF) assay in Figure 3F. The inverse derivative (−dF/dT) was calculated from these curves to determine the pMHC melting temperature (Tm) as a measure of complex stability (Koff).
(H) Related to Figure 3G. Flow cytometric analysis of CD8 T cells from the spleens of SIIN-vaccinated wild-type mice co-stained with SIIN- and SIY-specific H-2Kb tetramers or Y5- and SIY-specific H-2Kb tetramers. The SIY-specific tetramer serves as a negative control for tetramer binding. Representative of 2 independent experiments, n ≥ 5 mice per experiment.
(I) Flow cytometric quantification of H-2Kb stabilization on the surface of TAP-deficient RMA-S cells across a peptide concentration gradient. Dotted lines = 50% binding concentration which approximates the affinity (Kd) of each peptide on H-2Kb. mLAMA4 and mALG8 are compared to SIIN, SIY and Y5, which are also shown in Figure 3E. Representative of 2 independent experiments.
(J) Differential Scanning Fluorimetry (DSF) assay for pMHC thermal melting as a measure of complex stability (Koff). The first derivative (−dF/dT) of the melting curve was calculated to determine the melting temperatures (Tm). Dotted lines = Tm (minimum −dF/dT value) of each pMHC complex in a representative experiment. mLAMA4 and mALG8 are compared to SIIN, SIY and Y5, which are also shown in Figure 3F. Displayed Tm values are averaged from 3 independent experiments.
(K) Diagram of lentiviral constructs expressing mALG8 and mLAMA4 (LucAL), mALG8-only (LucA) and mLAMA4-only (LucL). Representative flow cytometry plots showing the proportion of mALG8- versus mLAMA4-specific CD8 T cells in the lungs of KP tumor-bearing mice expressing each construct at 5–6 weeks.
(L-M) Percentage of mALG8- and mLAMA4-specific cells of CD8 T cells (L) and the proportion that are TCF1+ TIM3− (M) at 5–6 weeks in LucAL, LucA and LucL mice.
(A-D) and (K-M) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group.
Figure S4. Related to Figures 2 and 5. Analysis of TCR Clonotypes from SIIN versus SIY scRNA-seq Data Delineates Relationships Between CD8 T Cell States (A) Distribution of TCR clonotypes (≥ 5 cells) across all clusters. Proportion of cells scale: 1 = contains 100% of cells assigned to the clonotype, 0 = contains 0% of cells assigned to the clonotype. Hierarchical clustering was performed on the clonotypes (rows) and the clusters (columns). “Clonotype clusters”, antigen and mouse assignments, and the number of cells in each clonotype (size) are indicated on side-bars. A measure of the spread in distribution of each clonotype across clusters, or “sparsity” (Gini index; Hurley, 2009), is also included. Additional side-bars show the proportion of cells in each cluster expressing key phenotypic genes: Gzmb, Havcr2, Cx3cr1, Tcf7, Ccr6 and Il17a.
(B) UMAP depicting clonotype size for individual cells belonging to SIIN and SIY clonotypes. Scale bar indicates the number of cells in a clonotype. Cells that belong to very small clonotypes or do not have a clonotype assignment are shown in gray. The violin plot shows a comparison of the size of SIIN versus SIY clonotypes containing ≥ 2 cells.
(C) ECDF plots depicting the distribution of SIIN and SIY clonotypes to the indicated clusters. Enrichment is expressed as p-value by KS test.
(D) UMAP plots highlighting cells assigned to each of the clonotype clusters depicted on the clonotype cluster side-bar in (A). Clonotype cluster 7 is enriched for a Tc17 phenotype.
Figure S5. Related to Figure 5. scRNA-seq Analysis Reveals Phenotypic Heterogeneity Amongst TCF1+ Progenitor CD8 T Cells in KP LucOS Tumors (A) Expression of select genes from the heatmap in Figure 5B projected onto the UMAP from Figure 5A.
(B) Scoring of cells from Figure 5A for enrichment of a gene signature differentially upregulated in Tc17 versus Tc1 CD8 T cells in the skin of mice treated with Imiquimod (Linehan et al., 2018) and signatures of T cell deletional tolerance (Parish et al., 2009) and T cell anergy (Safford et al., 2005). See also Table S2.
(C) Flow cytometric analysis of CCR6+ versus CCR6− subsets of SIIN- and SIY-specific TCF1+ cells for expression of surface markers of dysfunction, tolerance and anergy at 5 weeks. Representative of ≥ 2 independent experiments per marker, n ≥ 5 mice per experiment.
(D) Percentage of SIIN- and SIY-specific CCR6+ cells expressing the TCF1+ progenitor cell-associated marker SLAMF6 at 5 weeks.
(E-F) Percentage of SIIN- and SIY-specific TCF1+ cells expressing Ki67 (E) and GZMB (F), separated into CCR6− versus CCR6+ subsets.
(G) Related to Figure 5I. Proportion of SIIN- and SIY-specific TCF1+ TIM3− cells that are CCR6+ versus CCR6− over time.
(H) Related to Figure 5K. Representative flow cytometric analysis of the proportion of SIIN- and SIY-specific TCF1+ cells that express CX3CR1 and CCR6 following one week of treatment with isotype control (Iso) or anti-PD1/CTLA4 (PC) antibodies starting at 5 weeks.
(I-J) Related to Figure 5L. Flow cytometric profiling of KP mice reconstituted with IL17aCre × Rosa26LSL-tdTomato bone marrow and infected with Lenti-LucOS lentiviruses. Cells that express or have previously expressed IL17A are marked by Tomato fluorescence. Representative flow cytometric analyses of CCR6, TCF1 and RORɣT expression on Tomato+ versus Tomato- SIY-specific CD8 T cells are shown (I). Graph depicts the proportion of Tomato+ SIY-specific cells following one week of treatment with isotype control (Iso) versus anti-PD1/CTLA4 (PC) antibodies at 5 weeks (J). Data are from 2 independent experiments.
(D-H) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group.
Figure S6. Related to Figure 6. Vaccination with SIIN and SIY Long Peptides Increases CD8 T Cell Proliferation and Cytotoxicity, Correlating with Decreased Tumor Burden (A) Representative t-CyCIF images of Nkx2.1 (epithelial/tumor cells), B220 (B cells), CD4 (T helper cells), FOXP3 (Treg cells) and CD8 (T cytotoxic cell) staining in lung tumors from control (Ctrl) versus SIIN/SIY vaccine (Vax) treated KP LucOS mice.
(B) Quantification of CD4+ T cells, Foxp3+ CD4+ Treg cells and CD20+ B cells in tumors from Ctrl versus Vax treated KP LucOS mice. Results represent averages of all individual tumor data from two lung lobes per mouse.
(C) Tumor size measurements based on cell counts within annotated tumor area and absolute annotated tumor area as determined by analysis of H&E stained slides and t-CyCIF cell counts.
(D) Absolute number of SIIN- and SIY-specific cells and the percentage expressing Ki67 and GZMB in mice treated with PBS versus cyclic-di-GMP adjuvant according to the vaccination regimen outlined in Figure 6A.
(E) Percentage of SIIN- and SIY-specific cells that are Ki67+, GZMB+ and TCF1+ TIM3− in Ctrl and Vax treated mice 7 days post-vaccination by flow cytometry. The proportion of TCF1+ TIM3− cells that express CCR6 is also shown. Data are compiled from 2 independent experiments.
(F) Flow cytometric analysis of the proportion of SIIN- and SIY-specific cells of CD8 T cells in the blood, spleen, inguinal lymph node (iLN) and mediastinal lymph node (mLN) in Ctrl and Vax treated mice 7 days post-vaccination. Fold change between Ctrl and Vax conditions in each compartment is graphed. Representative of 1–2 independent experiments.
(G) Flow cytometric analysis of the percentage of GZMB+ cells and the percentage of TCF1+ TIM3- cells expressing CCR6 amongst SIIN- and SIY-specific cells from mice treated with Ctrl, SIIN/SIY vaccine (Vax), SIIN-only vaccine or SIY-only vaccine, 3 weeks after the initial vaccination (9 weeks post-tumor initiation). Representative of 3 independent experiments, n ≥ 5 mice per group.
(H) Absolute number of Y5- and SIY-specific cells in mice treated with a Y5 and SIY long-peptide vaccine according to the regimen outlined in Figure 6A. The percentage of Y5- and SIY-specific cells expressing Ki67, GZMB, and TCF1+ TIM3- cells and the proportion of TCF1+ TIM3- cells expressing CCR6 is shown.
Figure S7. Related to Figure 7. Analysis of Tumor CD8 T Cells from Published Human scRNA-seq Datasets and Tissue Samples Highlights Human Relevance of Identified Mouse TCF1+ Progenitor Cell Subsets (A) Scoring of cells from the mouse SIIN versus SIY scRNA-seq data in Figure 2A for expression of gene signatures derived from the human scRNA-seq clusters described in Figure 7A (Guo et al., 2018; Sade-Feldman et al., 2019; Wu et al., 2020; see also Table S4). Signatures associated with progenitor and exhausted cell phenotypes are included.
(B) UMAP plots of tumor-derived CD8 T cells from the reanalysis of the Wu et al. scRNA-seq dataset in Figures 7B and 7C highlighting cells assigned to select clusters from the published Wu et al. analysis (Wu et al., 2020).
(C) Reanalysis of the CD8 T cell subset from four scRNA-seq datasets derived from multiple human cancer types: non-small cell lung adenocarcinoma (NSCLC), Guo et al.; melanoma, Tirosh et al. and Sade-Feldman et al.; basal cell carcinoma, Yost et al. (Guo et al., 2018; Sade-Feldman et al., 2019; Tirosh et al., 2016; Yost et al., 2019). Cells expressing TCF7, HAVCR2, CCR6, RORC and/or IL17A above a 0.0–0.5 log-normalized cutoff are highlighted on the respective UMAP plots. Mucosal invariant T (MAIT) cells that phenotypically resemble Tc17 cells (Toubal et al., 2019) were excluded from the analysis based on TCR sequence (Table S6; STAR methods).
(D) Representative single channel images for the tissue cyclic immunofluorescence (t-CyCIF) imaging analysis of human lung adenocarcinoma samples in Figure 7D.
(E-F) Quantification of CD8 T cells expressing TCF1 by t-CyCIF imaging in Figure 7D.
(G) Representative flow cytometry plots of the proportion of CCR6+ and TCF1+ TIM3− cells staining positive for SIIN- and SIY-specific H-2Kb tetramers. Representative of ≥ 3 independent experiments, n ≥ 5 mice per experiment.
Table S1. Related to Figures 2 and S2. ProjecTILs cell state assignments for the mouse scRNA-seq data and enrichment statistics for SIIN- and SIY-specific cells.
Table S2. Related to Figures 5, S2 and S5. Published mouse gene signatures projected onto the mouse scRNA-seq data.
Table S3. Related to Figures 2, 5 and 7. Top differentially expressed genes and gene signatures for the mouse scRNA-seq clusters and CCR6+/CCR6− TCF1+ cell subsets.
Table S4. Related to Figures 7 and S7. Published human gene signatures projected onto the mouse scRNA-seq data.
Table S5. Related to Figures 6 and 7. Cell type calling parameters for analysis of mouse and human t-CyCIF images.
Table S6. Related to Figures 7 and S7. Gene co-expression cell assignments for the tumor CD8 T cell analysis of human scRNA-seq datasets.
Table S7. Related to Figure 7. Clinical information for lung adenocarcinoma and melanoma samples profiled by t-CyCIF.
HIGHLIGHTS.
Peptide-MHC binding property underlies antigen dominance hierarchy in a lung cancer model
T cells responding to subdominant antigens are enriched in TCF1+ progenitors
CCR6+ TCF1+ progenitor cells contribute poorly to checkpoint blockade response
Vaccination can eliminate CCR6+ TCF1+ cells and improve the subdominant response
ACKNOWLEDGEMENTS
We would like to acknowledge R. Herbst, T. Delorey, D. Phillips, the Broad Genomics Platform and the Whitehead Institute Genomics Technology Core for assistance with scRNA-seq; C. Backlund for input on vaccination techniques; Koch Institute’s Robert A. Swanson Biotechnology Center for assistance with flow cytometry and histology; K. Yee, J. Texeira, K. Anderson, K. Mercer and M. Magendantz for administrative and logistical support; S. Spranger, M. Birnbaum, C. Jin and B. Eskiocak for their insightful intellectual input.
This work was supported by Howard Hughes Medical Institute (T.J.); Ludwig Center at Harvard (S.S.); NIH U54 CA225088 (S.S.); NIH T32HL007627(G.G.); NCI Cancer Center Support Grant P30-CA1405; Bridge Project, a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center (T.J. and S.S.); fellowship awards from Jane Coffin Childs Memorial Fund for Medical Research and Ludwig Center for Molecular Oncology at MIT (M.L.B.). T.J. is a David H. Koch Professor of Biology and Daniel K. Ludwig Scholar.
DECLARATION OF INTERESTS
T.J. is on the Board of Directors of Amgen and ThermoFisher Scientific, a co-founder of Dragonfly Therapeutics and T2 Biosystems, an SAB member of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics and President of Break Through Cancer. T.J. lab receives funding from the Johnson & Johnson Lung Cancer Initiative and The Lustgarten Foundation for Pancreatic Cancer Research, but this funding did not support this research. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until Aug. 31, 2020 was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. From Aug. 1, 2020, A.R. is an employee of Genentech (Roche Group). S.S. is a consultant for RareCyte, Inc. A.M.C. interned at Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
INCLUSION AND DIVERSITY STATEMENT
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
REFERENCES
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. (2017). Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 7, 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, et al. (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M, Corria-Osorio J, Muller S, Cubas R, Coukos G, and Carmona SJ (2021). Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun 12, 2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldarelli RM, Smith CM, Finger JH, Hayamizu TF, McCright IJ, Xu J, Shaw DR, Beal JS, Blodgett O, Campbell J, et al. (2020). The mouse Gene Expression Database (GXD): 2021 update. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, Church CD, Lyngaa R, Koelle DM, Becker JC, et al. (2018). T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nat Methods 16, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, Hemmers S, Putintseva EV, Obraztsova AS, Shugay M, et al. (2017). Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol 35, 908–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al. (2015). Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, Moghaddam SJ, and Dong C (2014). T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci U S A 111, 5664–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTomaso D, Jones MG, Subramaniam M, Ashuach T, Ye CJ, and Yosef N (2019). Functional interpretation of single cell similarity maps. Nat Commun 10, 4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Lin JR, Rashid R, Maliga Z, Wang S, Aster JC, Izar B, Sorger PK, and Santagata S (2019). Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging. Nat Protoc 14, 2900–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nature Protocols 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, and Jacks T (2012). Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen HN, Hou XH, Shen C, Wang K, Tanguturi VK, Smith C, Kozyrytska K, Nambiar L, McKinley CA, Chen J, et al. (2012). Promiscuous binding of extracellular peptides to cell surface class I MHC protein. Proc Natl Acad Sci U S A 109, 4580–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, Kranz DM, Meredith SC, Rowley DA, and Schreiber H (2013). Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 23, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Paloma J, Berger JO (2016). RobustGaSP: Robust Gaussian Stochastic Process Emulation, 2016. https://CRANR-projectorg/package=RobustGaSP. R package version 0.5.7. [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24, 978–985. [DOI] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, et al. (2020). Array programming with NumPy. Nature 585, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nature immunology 12, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth RE, and Jansen K (2019). Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth M, Williams A, Tolstrup AB, and Elliott T (2004). Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc Natl Acad Sci U S A 101, 11737–11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, et al. (2019). Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 51, 1043–1058 e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley N R. S (2009). Comparing Measures of Sparsity. IEEE Transactions on Information Theory 55, 4723–4741. [Google Scholar]
- Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, et al. (2019). Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell 176, 998–1013 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadomoto S, Izumi K, and Mizokami A (2020). The CCL20-CCR6 Axis in Cancer Progression. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan TE, Burke KP, and Van Allen EM (2019). Genomic correlates of response to immune checkpoint blockade. Nat Med 25, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50, 181–194 e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, and Sorger PK (2018). Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, et al. (2018). Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 172, 784–796 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette GP, Becker-Hapak M, Skidmore ZL, Baroja ML, Xu C, Hundal J, Spencer DH, Fu W, Cummins C, Robnett M, et al. (2019). Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc Natl Acad Sci U S A 116, 23662–23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. (2016). Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes Leland, Healy John, and Melville James (2018). Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426 (2018). [Google Scholar]
- McKinney W (2010). Data Structures for Statistical Computing in Python. PROC OF THE 9th PYTHON IN SCIENCE CONF (SCIPY 2010). [Google Scholar]
- Meo, Pasquale De, De Meo Pasquale, Ferrara Emilio, Fiumara Giacomo, and Provetti Alessandro (2011). Generalized Louvain Method for Community Detection in Large Networks. 2011 11th International Conference on Intelligent Systems Design and Applications. 10.1109/isda.2011.6121636. [DOI] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nature immunology 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo I, Bedora-Faure M, Garcia Z, Thibaut R, Perie L, Shakhar G, Deriano L, and Bousso P (2018). The immune system profoundly restricts intratumor genetic heterogeneity. Sci Immunol 3. [DOI] [PubMed] [Google Scholar]
- Neuwirth E (2014). RColorBrewer: ColorBrewer Palettes. R package version 1.1-2. [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ, et al. (2020). A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 183, 347–362 e324. [DOI] [PubMed] [Google Scholar]
- Parish IA, Rao S, Smyth GK, Juelich T, Denyer GS, Davey GM, Strasser A, and Heath WR (2009). The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood 113, 4575–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Thorn K, Schroeder T, Wang L, Theis FJ, Marr C, and Navab N (2017). A BaSiC tool for background and shading correction of optical microscopy images. Nat Commun 8, 14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Nielsen M, and Sette A (2020). T Cell Epitope Predictions. Annu Rev Immunol 38, 123–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, and Schietinger A (2021). CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P (2020). Embracing the dropouts in single-cell RNA-seq analysis. Nat Commun 11, 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid R, Gaglia G, Chen YA, Lin JR, Du Z, Maliga Z, Schapiro D, Yapp C, Muhlich J, Sokolov A, et al. (2019). Highly multiplexed immunofluorescence images and single-cell data of immune markers in tonsil and lung cancer. Sci Data 6, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, Lund T, Tanic M, Reading JL, Joshi K, et al. (2019). Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2019). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 176, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, et al. (2005). Egr-2 and Egr-3 are negative regulators of T cell activation. Nature immunology 6, 472–480. [DOI] [PubMed] [Google Scholar]
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226. [DOI] [PubMed] [Google Scholar]
- Saini SK, Tamhane T, Anjanappa R, Saikia A, Ramskov S, Donia M, Svane IM, Jakobsen SN, Garcia-Alai M, Zacharias M, et al. (2019). Empty peptide-receptive MHC class I molecules for efficient detection of antigen-specific T cells. Sci Immunol 4. [DOI] [PubMed] [Google Scholar]
- Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, et al. (2014). Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan SL, Gibbons G, Smith OC, Kim JY, Westcott P, Hwang WL, et al. (2021). Targeting of migratory cDC1 cells drives recruitment of TCF-1+ CD8+ T cells from tumor-draining lymph node to lung tumors. in press Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M, Dijkstra K, et al. (2019). Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med 25, 89–94. [DOI] [PubMed] [Google Scholar]
- Schreiber H, Wu TH, Nachman J, and Kast WM (2002). Immunodominance and tumor escape. Semin Cancer Biol 12, 25–31. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Heemels MT, Neefjes JJ, Kast WM, Melief CJ, and Ploegh HL (1990). Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell 62, 563–567. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Scheper W, and Kvistborg P (2019). Cancer Neoantigens. Annu Rev Immunol 37, 173–200. [DOI] [PubMed] [Google Scholar]
- Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110. [DOI] [PubMed] [Google Scholar]
- Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. [DOI] [PubMed] [Google Scholar]
- Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, and Gajewski TF (2014). Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srenathan U, Steel K, and Taams LS (2016). IL-17+ CD8+ T cells: Differentiation, phenotype and role in inflammatory disease. Immunol Lett 178, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J, et al. (2016). Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubal A, Nel I, Lotersztajn S, and Lehuen A (2019). Mucosal-associated invariant T cells and disease. Nat Rev Immunol 19, 643–657. [DOI] [PubMed] [Google Scholar]
- Traag VA, Waltman L and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Scientific Reports volume 9, Article number: 5233, 10.1038/s41598-019-41695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, and Rosenberg SA (2017). ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nature immunology 18, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. (2016). Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536, 91–95. [DOI] [PubMed] [Google Scholar]
- Verykokakis M, Papadaki C, Vorgia E, Le Gallic L, and Mavrothalassitis G (2007). The RAS-dependent ERF control of cell proliferation and differentiation is mediated by c-Myc repression. J Biol Chem 282, 30285–30294. [DOI] [PubMed] [Google Scholar]
- Warnes G (2015). gplots: Various R programming tools for plotting data. R Package Vers. [Google Scholar]
- Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. (2020). Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818–834 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang Z, Wang C, Zhang Z, Xia W, et al. (2014). gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40, 785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, et al. (2020). Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. [DOI] [PubMed] [Google Scholar]
- Yewdell JW (2006). Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 25, 533–543. [DOI] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W (2019). CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51, 1028–1042 e1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen X, Herjan T, and Li X (2020). The role of interleukin-17 in tumor development and progression. J Exp Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Peng LS, Zhao YL, Shi Y, Mao XH, Chen W, Pang KC, Liu XF, Liu T, Zhang JY, et al. (2012). CD8(+) T cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology 143, 951–962 e958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Figure 1. SIINFEKL- and SIYRYYGL-specific CD8 T cells in KP LucOS Lung Tumors Display Hallmarks of Dysfunction Over Time (A) Flow cytometry gating strategy for tumor SIIN- and SIY-specific CD8 T cells. A single cell suspension from tumor-bearing lung was gated on singlets and live (Zombie dye-negative) cells, then lymphocytes based on FSC/SSC. The lymphocyte gate was then analyzed for the proportion of tissue-resident CD8+ T cells based on the absence of staining by an intravascularly circulating anti-CD45 antibody (see STAR methods) and positive staining for CD8α. Tissue-resident CD8+ cells were further gated on positive CD44 staining prior to analysis of SIINFEKL (SIIN) and SIY (SIYRYYGL) peptide-MHC (H-2Kb) tetramer staining.
(B) Percentage of SIIN- versus SIY-specific cells of CD8 T cells assayed over time by flow cytometry.
(C) Representative flow cytometry plots of the percentage of SIIN- and SIY-specific cells expressing Ki67 over time.
(D) Representative flow cytometry plots comparing PD1, LAG3 and TIM3 expression between SIIN- and SIY-specific cells at 5 weeks. Naïve population (grey) represents CD44- tetramer- CD8α+ T cells. Geometric mean fluorescence intensity (gMFI) for PD1, LAG3 and TIM3 is plotted over time for SIIN- versus SIY-specific cells. All data points were compiled from a single experiment for gMFI comparison.
(B-D) are representative of ≥ 3 independent experiments per time point, n ≥ 5 mice per group.
Figure S2. Related to Figure 2. A Population of TCF1+ Progenitor Cells is Identified by scRNA-Sequencing of SIIN- and SIY-Specific CD8 T cells (A) Classification of individual cells from Figure 2A using mouse ProjecTILs atlases (Andreatta et al., 2021) from lymphocytic choriomeningitis virus (LCMV) infection and tumor infiltrating lymphocytes (TIL; B16 and MC38 transplant tumor models).
(B) Scoring of individual cells from Figure 2A for enrichment of progenitor versus terminally-exhausted gene signatures from chronic LCMV (clone 13) infection and B16 melanoma TIL mouse datasets (Miller et al. 2019). SIIN-specific cells are enriched for the terminally-exhausted signatures (tumor p = 2.90E-14; LCMV p = 2.24E-26), while SIY-specific cells are enriched for the tumor progenitor signature (tumor p = 0.004; LCMV p = 0.15). Statistics were calculated by hypergeometric test with a z-score cutoff of 0.5.
(C) Heatmap of the top 10 differentially expressed genes per cluster from Figure 2B. Select genes used to define the phenotypic labels assigned to each cluster are highlighted on the righthand side.
(D) Expression of the indicated progenitor-associated genes on cells from Figure 2A.
(E) Expression of AY036118.1 (ERF1), the top differentially expressed gene in cluster 7, and activation markers Cd69 and Cd44 on cells from Figure 2A.
(F-G) Flow cytometric analysis of PD1 (E) and Ki67 (F) expression by TCF1+ TIM3− (colored) versus TCF1− (black line) SIIN- and SIY-specific CD8 T cells at 5 weeks. The naïve population (grey) represents CD44- tetramer- CD8+ T cells. Representative of 3 independent experiments, n ≥ 5 mice per group.
Figure S3. Related to Figure 3. Expression of SIY Alone Results in Less Enrichment for TCF1+ Progenitor Cells and Greater Expression of Effector/Exhaustion Markers (A-B) Quantification of the absolute number of total (A) and TCF1+ TIM3− (B) SIIN- and SIY-specific CD8 T cells in LucOS versus LucSIIN and LucSIY mice at 5–6 weeks.
(C) Representative flow cytometry plots of PD1, LAG3 and TIM3 expression between SIIN- and SIY-specific cells in LucOS, LucSIIN and LucSIY mice at 5–6 weeks.
(D-F) Flow cytometric analysis of the proportion of SIIN- and SIY-specific cells expressing CX3CR1 (D), GZMB (E) and Ki67 (F) at 5–6 weeks.
(G) Melting curves associated with the Differential Scanning Fluorimetry (DSF) assay in Figure 3F. The inverse derivative (−dF/dT) was calculated from these curves to determine the pMHC melting temperature (Tm) as a measure of complex stability (Koff).
(H) Related to Figure 3G. Flow cytometric analysis of CD8 T cells from the spleens of SIIN-vaccinated wild-type mice co-stained with SIIN- and SIY-specific H-2Kb tetramers or Y5- and SIY-specific H-2Kb tetramers. The SIY-specific tetramer serves as a negative control for tetramer binding. Representative of 2 independent experiments, n ≥ 5 mice per experiment.
(I) Flow cytometric quantification of H-2Kb stabilization on the surface of TAP-deficient RMA-S cells across a peptide concentration gradient. Dotted lines = 50% binding concentration which approximates the affinity (Kd) of each peptide on H-2Kb. mLAMA4 and mALG8 are compared to SIIN, SIY and Y5, which are also shown in Figure 3E. Representative of 2 independent experiments.
(J) Differential Scanning Fluorimetry (DSF) assay for pMHC thermal melting as a measure of complex stability (Koff). The first derivative (−dF/dT) of the melting curve was calculated to determine the melting temperatures (Tm). Dotted lines = Tm (minimum −dF/dT value) of each pMHC complex in a representative experiment. mLAMA4 and mALG8 are compared to SIIN, SIY and Y5, which are also shown in Figure 3F. Displayed Tm values are averaged from 3 independent experiments.
(K) Diagram of lentiviral constructs expressing mALG8 and mLAMA4 (LucAL), mALG8-only (LucA) and mLAMA4-only (LucL). Representative flow cytometry plots showing the proportion of mALG8- versus mLAMA4-specific CD8 T cells in the lungs of KP tumor-bearing mice expressing each construct at 5–6 weeks.
(L-M) Percentage of mALG8- and mLAMA4-specific cells of CD8 T cells (L) and the proportion that are TCF1+ TIM3− (M) at 5–6 weeks in LucAL, LucA and LucL mice.
(A-D) and (K-M) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group.
Figure S4. Related to Figures 2 and 5. Analysis of TCR Clonotypes from SIIN versus SIY scRNA-seq Data Delineates Relationships Between CD8 T Cell States (A) Distribution of TCR clonotypes (≥ 5 cells) across all clusters. Proportion of cells scale: 1 = contains 100% of cells assigned to the clonotype, 0 = contains 0% of cells assigned to the clonotype. Hierarchical clustering was performed on the clonotypes (rows) and the clusters (columns). “Clonotype clusters”, antigen and mouse assignments, and the number of cells in each clonotype (size) are indicated on side-bars. A measure of the spread in distribution of each clonotype across clusters, or “sparsity” (Gini index; Hurley, 2009), is also included. Additional side-bars show the proportion of cells in each cluster expressing key phenotypic genes: Gzmb, Havcr2, Cx3cr1, Tcf7, Ccr6 and Il17a.
(B) UMAP depicting clonotype size for individual cells belonging to SIIN and SIY clonotypes. Scale bar indicates the number of cells in a clonotype. Cells that belong to very small clonotypes or do not have a clonotype assignment are shown in gray. The violin plot shows a comparison of the size of SIIN versus SIY clonotypes containing ≥ 2 cells.
(C) ECDF plots depicting the distribution of SIIN and SIY clonotypes to the indicated clusters. Enrichment is expressed as p-value by KS test.
(D) UMAP plots highlighting cells assigned to each of the clonotype clusters depicted on the clonotype cluster side-bar in (A). Clonotype cluster 7 is enriched for a Tc17 phenotype.
Figure S5. Related to Figure 5. scRNA-seq Analysis Reveals Phenotypic Heterogeneity Amongst TCF1+ Progenitor CD8 T Cells in KP LucOS Tumors (A) Expression of select genes from the heatmap in Figure 5B projected onto the UMAP from Figure 5A.
(B) Scoring of cells from Figure 5A for enrichment of a gene signature differentially upregulated in Tc17 versus Tc1 CD8 T cells in the skin of mice treated with Imiquimod (Linehan et al., 2018) and signatures of T cell deletional tolerance (Parish et al., 2009) and T cell anergy (Safford et al., 2005). See also Table S2.
(C) Flow cytometric analysis of CCR6+ versus CCR6− subsets of SIIN- and SIY-specific TCF1+ cells for expression of surface markers of dysfunction, tolerance and anergy at 5 weeks. Representative of ≥ 2 independent experiments per marker, n ≥ 5 mice per experiment.
(D) Percentage of SIIN- and SIY-specific CCR6+ cells expressing the TCF1+ progenitor cell-associated marker SLAMF6 at 5 weeks.
(E-F) Percentage of SIIN- and SIY-specific TCF1+ cells expressing Ki67 (E) and GZMB (F), separated into CCR6− versus CCR6+ subsets.
(G) Related to Figure 5I. Proportion of SIIN- and SIY-specific TCF1+ TIM3− cells that are CCR6+ versus CCR6− over time.
(H) Related to Figure 5K. Representative flow cytometric analysis of the proportion of SIIN- and SIY-specific TCF1+ cells that express CX3CR1 and CCR6 following one week of treatment with isotype control (Iso) or anti-PD1/CTLA4 (PC) antibodies starting at 5 weeks.
(I-J) Related to Figure 5L. Flow cytometric profiling of KP mice reconstituted with IL17aCre × Rosa26LSL-tdTomato bone marrow and infected with Lenti-LucOS lentiviruses. Cells that express or have previously expressed IL17A are marked by Tomato fluorescence. Representative flow cytometric analyses of CCR6, TCF1 and RORɣT expression on Tomato+ versus Tomato- SIY-specific CD8 T cells are shown (I). Graph depicts the proportion of Tomato+ SIY-specific cells following one week of treatment with isotype control (Iso) versus anti-PD1/CTLA4 (PC) antibodies at 5 weeks (J). Data are from 2 independent experiments.
(D-H) are representative of ≥ 3 independent experiments, n ≥ 5 mice per group.
Figure S6. Related to Figure 6. Vaccination with SIIN and SIY Long Peptides Increases CD8 T Cell Proliferation and Cytotoxicity, Correlating with Decreased Tumor Burden (A) Representative t-CyCIF images of Nkx2.1 (epithelial/tumor cells), B220 (B cells), CD4 (T helper cells), FOXP3 (Treg cells) and CD8 (T cytotoxic cell) staining in lung tumors from control (Ctrl) versus SIIN/SIY vaccine (Vax) treated KP LucOS mice.
(B) Quantification of CD4+ T cells, Foxp3+ CD4+ Treg cells and CD20+ B cells in tumors from Ctrl versus Vax treated KP LucOS mice. Results represent averages of all individual tumor data from two lung lobes per mouse.
(C) Tumor size measurements based on cell counts within annotated tumor area and absolute annotated tumor area as determined by analysis of H&E stained slides and t-CyCIF cell counts.
(D) Absolute number of SIIN- and SIY-specific cells and the percentage expressing Ki67 and GZMB in mice treated with PBS versus cyclic-di-GMP adjuvant according to the vaccination regimen outlined in Figure 6A.
(E) Percentage of SIIN- and SIY-specific cells that are Ki67+, GZMB+ and TCF1+ TIM3− in Ctrl and Vax treated mice 7 days post-vaccination by flow cytometry. The proportion of TCF1+ TIM3− cells that express CCR6 is also shown. Data are compiled from 2 independent experiments.
(F) Flow cytometric analysis of the proportion of SIIN- and SIY-specific cells of CD8 T cells in the blood, spleen, inguinal lymph node (iLN) and mediastinal lymph node (mLN) in Ctrl and Vax treated mice 7 days post-vaccination. Fold change between Ctrl and Vax conditions in each compartment is graphed. Representative of 1–2 independent experiments.
(G) Flow cytometric analysis of the percentage of GZMB+ cells and the percentage of TCF1+ TIM3- cells expressing CCR6 amongst SIIN- and SIY-specific cells from mice treated with Ctrl, SIIN/SIY vaccine (Vax), SIIN-only vaccine or SIY-only vaccine, 3 weeks after the initial vaccination (9 weeks post-tumor initiation). Representative of 3 independent experiments, n ≥ 5 mice per group.
(H) Absolute number of Y5- and SIY-specific cells in mice treated with a Y5 and SIY long-peptide vaccine according to the regimen outlined in Figure 6A. The percentage of Y5- and SIY-specific cells expressing Ki67, GZMB, and TCF1+ TIM3- cells and the proportion of TCF1+ TIM3- cells expressing CCR6 is shown.
Figure S7. Related to Figure 7. Analysis of Tumor CD8 T Cells from Published Human scRNA-seq Datasets and Tissue Samples Highlights Human Relevance of Identified Mouse TCF1+ Progenitor Cell Subsets (A) Scoring of cells from the mouse SIIN versus SIY scRNA-seq data in Figure 2A for expression of gene signatures derived from the human scRNA-seq clusters described in Figure 7A (Guo et al., 2018; Sade-Feldman et al., 2019; Wu et al., 2020; see also Table S4). Signatures associated with progenitor and exhausted cell phenotypes are included.
(B) UMAP plots of tumor-derived CD8 T cells from the reanalysis of the Wu et al. scRNA-seq dataset in Figures 7B and 7C highlighting cells assigned to select clusters from the published Wu et al. analysis (Wu et al., 2020).
(C) Reanalysis of the CD8 T cell subset from four scRNA-seq datasets derived from multiple human cancer types: non-small cell lung adenocarcinoma (NSCLC), Guo et al.; melanoma, Tirosh et al. and Sade-Feldman et al.; basal cell carcinoma, Yost et al. (Guo et al., 2018; Sade-Feldman et al., 2019; Tirosh et al., 2016; Yost et al., 2019). Cells expressing TCF7, HAVCR2, CCR6, RORC and/or IL17A above a 0.0–0.5 log-normalized cutoff are highlighted on the respective UMAP plots. Mucosal invariant T (MAIT) cells that phenotypically resemble Tc17 cells (Toubal et al., 2019) were excluded from the analysis based on TCR sequence (Table S6; STAR methods).
(D) Representative single channel images for the tissue cyclic immunofluorescence (t-CyCIF) imaging analysis of human lung adenocarcinoma samples in Figure 7D.
(E-F) Quantification of CD8 T cells expressing TCF1 by t-CyCIF imaging in Figure 7D.
(G) Representative flow cytometry plots of the proportion of CCR6+ and TCF1+ TIM3− cells staining positive for SIIN- and SIY-specific H-2Kb tetramers. Representative of ≥ 3 independent experiments, n ≥ 5 mice per experiment.
Table S1. Related to Figures 2 and S2. ProjecTILs cell state assignments for the mouse scRNA-seq data and enrichment statistics for SIIN- and SIY-specific cells.
Table S2. Related to Figures 5, S2 and S5. Published mouse gene signatures projected onto the mouse scRNA-seq data.
Table S3. Related to Figures 2, 5 and 7. Top differentially expressed genes and gene signatures for the mouse scRNA-seq clusters and CCR6+/CCR6− TCF1+ cell subsets.
Table S4. Related to Figures 7 and S7. Published human gene signatures projected onto the mouse scRNA-seq data.
Table S5. Related to Figures 6 and 7. Cell type calling parameters for analysis of mouse and human t-CyCIF images.
Table S6. Related to Figures 7 and S7. Gene co-expression cell assignments for the tumor CD8 T cell analysis of human scRNA-seq datasets.
Table S7. Related to Figure 7. Clinical information for lung adenocarcinoma and melanoma samples profiled by t-CyCIF.
Data Availability Statement
The mouse scRNA-seq data has been deposited to GEO and is publicly available as of the date of publication. The accession number is listed in the key resources table. This paper additionally included analyses of publicly available human scRNA-seq datasets. The accession numbers for these datasets are listed in the key resources table.
All original code for the mouse and human scRNA-seq analyses and the t-CYCIF imaging analyses has been deposited on GitHub and is publicly available as of the date of publication. The DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivomAb PD1 | BioXCell | Clone 29F.1A12; Cat# BE0273; RRID: AB_2687796 |
| InVivomAb CTLA4 | BioXCell | Clone 9H10; Cat# BE0131; RRID: AB_10950184 |
| InVivomAb rat IgG2a | BioXCell | Clone 2A3; Cat# BE0089; RRID: AB_1107769 |
| InVivomAb polyclonal Syrian Hamster IgG | BioXCell | Cat# BE0087; RRID: AB_1107782 |
| FC: CD8ɑ | BD Biosciences | Clone 53-6.7; Cat# 563786, 612759; RRID: AB_2732919, AB_2870090 |
| FC: CCR6 (CD196) | BioLegend | Clone 29-2L17; Cat# 129822; RRID: AB_2687019 |
| FC: CX3CR1 | BioLegend | Clone SA011F11; Cat# 149031; RRID: AB_2565939 |
| FC: CXCR3 (CD183) | BD Biosciences | Clone CXCR3-173; Cat# 562937, 741032; RRID: AB_2687551, AB_2740650 |
| FC: CD44 | BD Biosciences; BioLegend | Clone IM7; Cat# 563736, 612799, 103057, 103059; RRID: AB_2738395, AB_2870126, AB_2564214, AB_2571953 |
| FC: CD45 | ThermoFisher Scientific; BD Biosciences | Clone 30-F11; Cat# 47-0451-80, 562420; RRID: AB_1548790, AB_11154401 |
| FC: CD83 | BioLegend | Clone Michel-19; Cat# 121506; RRID: AB_572009 |
| FC: CD200 | BD Biosciences | Clone OX-90; Cat# 565546; RRID: AB_2739288 |
| FC: EGR2 | ThermoFisher Scientific | Clone erongr2; Cat# 17-6691-80; RRID: AB_11150966 |
| FC: Granzyme B | BioLegend, BD Biosciences | Clone GB11; Cat# 515408, 560213; RRID: AB_2562196, AB_1645453 |
| FC: H-2Kb | ThermoFisher Scientific | Clone AF6-88.5.5.3; Cat# 17-5958-82; RRID: AB_1311280 |
| FC: ICOS (CD278) | ThermoFisher Scientific | Clone 7E.17G9; Cat# 46-9942-80; RRID: AB_2744727 |
| FC: IFN-γ | BioLegend | Clone XMG1.2; Cat# 505822; RRID: AB_961359 |
| FC: IL17A | ThermoFisher Scientific | Clone 17B7; Cat# 11-7177-81; RRID: AB_763581 |
| FC: IL7R (CD127) | ThermoFisher Scientific | Clone A7R34; Cat# 25-1271-82; RRID: AB_469649 |
| FC: Ki67 | BD Biosciences | Clone B56; Cat# 561277, 563756; RRID: AB_10611571, AB_2732007 |
| FC: LAG3 (CD233) | BD Biosciences, ThermoFisher Scientific; BioLegend | Clone C9B7W; Cat# 741820, 46-2231-82, 11-2231-80, 125219; RRID: AB_11151334, AB_2572483, AB_2566571 |
| FC: OX40 (CD134) | BioLegend | Clone OX-86; Cat# 119419; RRID: AB_2687175 |
| FC: PD1 (CD279) | BioLegend | Clone RMP1-30; Cat# 109110, 109121; RRID: AB_572017, AB_2687080 |
| FC: RORƔT | ThermoFisher Scientific | Clone B2D; Cat# 25-6981-82, 17-6981-82; RRID: AB_2784671, AB_2573254 |
| FC: SLAMF6 (LY-108) | BD Biosciences | Clone 13G3; Cat# 742272; RRID: AB_2871448 |
| FC: TCF1/TCF7 | Cell Signaling Technology | Clone C63D9; Cat# 6444, 6709, 9066; RRID: AB_2797627, AB_2797631, AB_2797696 |
| FC: TIGIT | BD Biosciences | Clone 1G9; Cat# 565474; RRID: AB_2739254 |
| FC: TIM3 | BioLegend | Clone RMT3-23; Cat# 119721, 119725; RRID: AB_2616907, AB_2716066 |
| FC: TNFα | BioLegend | Clone MP6-XT22; Cat# 506306; RRID: AB_315427 |
| FC: TOX | Miltenyi Biotec | Clone REA473; Cat# 130-118-335; RRID: AB_2751485 |
| FC: H-2Kb SIINFEKL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb SIYRYYGL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb SIINYEKL monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb mALG8 monomer | NIH Tetramer Core Facility | Custom |
| FC: H-2Kb mLAMA4 monomer | NIH Tetramer Core Facility | Custom |
| Hashtag: TotalSeq-C anti-mouse | BioLegend | Clones M1/42, 30-F11; Cat# 155861, 155863, 155865, 155867, 155869, 155871, 155873, 155875, 155877, 155879; RRID: AB_2800693, AB_2800694, AB_2800695, AB_2800696, AB_2800697, AB_2819910, AB_2819911, AB_2819912, AB_2819913, AB_2819914 |
| tCycif: anti-mouse B220 (CD45R) | ThermoFisher Scientific | Clone RA3-6B2; Cat# 41-0452-80; RRID: AB_2573598 |
| tCycif: anti-mouse CD4 | ThermoFisher Scientific | Clone 4SM95; Cat# 41-9766-82; RRID: AB_2573637 |
| tCycif: anti-mouse CD8a | Cell Signaling Technology | Clone D4W2Z; Cat# 98941S; RRID: AB_2756376 |
| tCycif: anti-mouse FOXP3 | ThermoFisher Scientific | Clone FJK-16 s; Cat# 11-5773-82; RRID: AB_465243 |
| tCycif: anti-human αSMA | Abcam | Clone EPR5368; Cat# ab202509; RRID: AB_2868435 |
| tCycif: anti-human CD4 | R&D Systems | Cat# FAB8165G; RRID: AB_2728839 |
| tCycif: anti-human CD8α | ThermoFisher Scientific | Clone AMC908; Cat# 50-0008-80; RRID: AB_2574148 |
| tCycif: anti-human CD20 | ThermoFisher Scientific | Clone L26; Cat# 50-0202-80; RRID: AB_11151691 |
| tCycif: anti-human CD45 | BioLegend | Clone HI30; Cat# 304008; RRID: AB_314396 |
| tCycif: anti-human CCR6 | Abcam | Clone EPR22259; Cat# ab243852; RRID: AB_2860033 |
| tCycif: anti-human FOXP3 | eBioscience | Clone 236A/E7; Cat# 41-4777-80; RRID: AB_2573608 |
| tCycif: anti-human TCF1 | Cell Signaling Technology | Clone C63D9; Cat# 6444S; RRID: AB_2797627 |
| tCycif: anti-human TTF1 (also reacts with mouse) | Abcam | Clone EPR595(2); Cat# ab206726; RRID: AB_2857980 |
| Purification: Ly6G Rat Monoclonal | BioLegend | Clone 1A8; Cat# 127602; RRID: AB_1089180 |
| Purification: EpCam (CD326) Rat Monoclonal | BioLegend | Clone G8.8; Cat# 118202; RRID: AB_1089027 |
| Purification: F4/80 Rat Monoclonal | BioLegend | Clone BM8; Cat# 123102; RRID: AB_893506 |
| Biological samples | ||
| Human formalin fixed paraffin embedded tissue samples from lung adenocarcinoma cases | Partners Healthcare Institutional Review Board at Brigham Health, Boston, MA, USA | Excess tissue, discarded tissue protocol number 2018P001627 |
| Human formalin fixed paraffin embedded tissue samples from metastatic melanoma cases | Specialized Histopathology Services - Longwood Core Brigham and Womens Hospital, Boston, MA, USA | Project: 05-042 - T Jacks Lab slides |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase IV | Worthington Biochemical | Cat# LS004189 |
| DNase I | Sigma-Aldrich | Cat# 10104159001 |
| Monensin | BioLegend | Cat# 420701 |
| Golgi Plug | BD Biosciences | Cat# 555028 |
| Phorbol-12-myristate-13-acetate (PMA) | EMD Millipore | Cat# 524400 |
| Ionomycin Calcium Salt | Sigma | Cat# 13909 |
| Streptavidin, allophycocyanin conjugate | Invitrogen | Cat# S32362 |
| Streptavidin, R-phycoerythrin conjugate | Invitrogen | Cat# S21388 |
| Mirus TransIT LT1 | Mirus Bio | Cat# MIR 2300 |
| SYPRO Orange Protein Gel Stain | Thermo Fisher Scientific | Cat# S6651 |
| Cyclic-di-GMP | Invitrogen | Cat# tlrl-nacdg |
| SIINFEKL peptide | New England Peptide | Custom |
| SIYRYYGL peptide | New England Peptide | Custom |
| SIINYEKL peptide | New England Peptide | Custom |
| ITYTWTRL peptide (mALG8) | Genscript | Custom |
| VGFNFRTL peptide (mLAMA4) | Genscript | Custom |
| SMLVLLPDEVSGLEQLESIINYEKLTEWTS | New England Peptide | Custom |
| SMLVLLPDEVSGLEQLESIINFEKLTEWTS peptide | New England Peptide | Custom |
| GRCVGSEQLESIYRYYGLLLKERSEQKLIS peptide | New England Peptide | Custom |
| Empty peptide-receptive H-2Kb molecules | Saini et al., 2019 | Generated in-house |
| Zombie Fixable Viability Kit | BioLegend | Cat# 423102 |
| Ghost Dye | Tonbo Biosciences | Cat# 13-0865-T500 |
| Critical commercial assays | ||
| Intracellular Fixation & Permeabilization Buffer Set Kit | ThermoFisher Scientific | Cat# 88-8824-00 |
| Dynabead Sheep Anti-Rat IgG | ThermoFisher Scientific | Cat#11035 |
| Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit | 10X Genomics | Cat# 000167 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10X Genomics | Cat#1000071 |
| Easysep Dead Cell Removal (Annexin V) Kit | StemCell | Cat#17899 |
| Deposited data | ||
| Mouse KP LucOS SIIN versus SIY scRNA-seq | GEO | GEO: GSE164177 |
| Sade-Feldman et al. scRNA-seq dataset | Sade-Feldman et al., 2019 | GEO: GSE120575 |
| Wu et al. scRNA-seq dataset | (Wu et al., 2020) | GEO: GSE139555 |
| Guo et al. scRNA-seq dataset | Guo et al., 2018 | GEO: GSE99254 |
| Yost et al. scRNA-seq dataset | (Yost et al., 2019) | GEO: GSE123813 |
| Tirosh et al. scRNA-seq dataset | Tirosh et al., 2016 | GEO: GSE72056; Broad Data Use Oversight System: DUOS-000002 |
| Mouse and human single cell RNA-sequencing analysis code | This Paper | GitHub: https://doi.org/10.5281/zenodo.5110770 |
| t-CYCIF analysis code | This Paper | GitHub: https://doi.org/10.5281/zenodo.5104869 |
| Experimental models: Cell lines | ||
| GreenGo 3TZ for lentiviral titering | Sánchez-Rivera et al., 2014 | Available upon request |
| 293FS* viral packaging cell line | This paper | Available upon request |
| RMA-S TAP-deficient cell line | Schumacher et al., 1990 | Available upon request |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J | Jackson Laboratories | Stock No: 008179 |
| Mouse: B6.129P2-Trp53tm1Brn/J | Jackson Laboratories | Stock No: 008462 |
| Mouse: Il17atm1.1(icre)Stck/J | Jackson Laboratories | Stock No: 016879 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratories | Stock No: 007914 |
| Recombinant DNA | ||
| Plasmid: Lenti-LucOS | DuPage et al., 2011 | Addgene Cat# 22777 |
| Plasmid: Lenti-LucSIY | This Paper | Addgene Cat# 174044 |
| Plasmid: Lenti-LucSIIN | This Paper | Addgene Cat# 174043 |
| Plasmid: Lenti-LucY5S | This Paper | Addgene Cat# 174045 |
| Plasmid: Lenti-LucY5 | This Paper | Addgene Cat# 174046 |
| Plasmid: Lenti-LucA | This paper | Addgene Cat# 174047 |
| Plasmid: Lenti-LucL | This paper | Addgene Cat# 174048 |
| Plasmid: Lenti-LucAL | This paper | Addgene Cat# 174049 |
| Software and algorithms | ||
| FlowJo | FlowJo | Version 10.7.1 https://www.flowjo.com/solutions/flowjo/downloads |
| Aperio ImageScope | Leica Biosystems | Version 12 https://www.leicabiosystems.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| ImageJ BaSiC Plugin | Peng et al., 2017 | https://www.helmholtz-muenchen.de/icb/research/groups/marr-lab/software/basic/index.html |
| ASHLAR | The Python Package Index | https://pypi.org/project/ashlar/ |
| ilastik | Berg et al., 2019 | https://www.ilastik.org/download.html |
| MATLAB | MathWorks | version 2018a https://www.mathworks.com/products/matlab.html |
| R/Rstudio | Rstudio | version 1.2.5042 https://www.rstudio.com/ |
| Seurat | Butler et al., 2018 | version 4.0.0 https://cran.r-project.org/web/packages/Seurat/index.html |
| CellRanger | 10X Genomics | version 3.1.0 https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| ComplexHeatmap | CRAN Gu et al., 2016 | version 2.4.3 https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Tidyr | CRAN | version 1.1.2 https://tidyr.tidyverse.org, https://github.com/tidyverse/tidyr |
| Cowplot | CRAN | version 1.1.0 https://wilkelab.org/cowplot/ |
| GGplot2 | CRAN | version 3.2.2 https://ggplot2.tidyverse.org, https://github.com/tidyverse/ggplot2 |
| GridExtra | CRAN | version 2.3 https://cran.r-project.org/web/packages/gridExtra/index.html |
| Plyr | CRAN | version 1.8.6 https://cran.r-project.org/web/packages/plyr/index.html |
| Dplyr | CRAN | version 1.0.2 https://cran.r-project.org/web/packages/dplyr/index.html |
| Monocle3 | Trapnell et al., 2014 | version 0.2.3.0 https://cole-trapnell-lab.github.io/monocle3/ |
| Patchwork | CRAN | version 1.1.1 https://github.com/thomasp85/patchwork |
| Viridis | CRAN | version 0.5.1 https://github.com/sjmgarnier/viridis |
| Circlize | CRAN | Version: 0.4.11 https://github.com/jokergoo/circlize |
| Stringr | CRAN | Version: 1.4.0 https://stringr.tidyverse.org, https://github.com/tidyverse/stringr |
| Philentropy | CRAN | Version: 0.4.0 https://github.com/drostlab/philentropy |
| Dendextend | CRAN | Version: 1.14.0 https://cran.r-project.org/web/packages/dendextend/index.html |
| FactoEXTRA | CRAN | Version: 1.0.7 http://www.sthda.com/english/rpkgs/factoextra |
| ProjecTILs | Andreatta et al., 2021 | v.0.5.1 https://github.com/carmonalab/ProjecTILs |
| gplots | Warnes, 2015 | v.3.1.0 https://cran.r-project.org/web/packages/gplots/index.html |
| RColorBrewer | Neuwirth, 2014 | v.1.1-2 https://cran.r-project.org/web/packages/RColorBrewer/index.html |