

Abstract
Accumulation of microtubule-associated tau protein is thought to cause neuron loss in a group of neurodegenerative diseases called tauopathies. In diseased brains, tau molecules adopt pathological structures that propagate into insoluble forms with disease-specific patterns. Several types of posttranslational modifications in tau are known to modulate its aggregation propensity in vitro, but their influence on tau accumulation and toxicity at the whole-organism level has not been fully elucidated. Herein, we utilized a series of transgenic Drosophila models to compare systematically the toxicity induced by five tau constructs with mutations or deletions associated with aggregation, including substitutions at seven disease-associated phosphorylation sites (S7A and S7E), deletions of PHF6 and PHF6* sequences (ΔPHF6 and ΔPHF6*), and substitutions of cysteine residues in the microtubule binding repeats (C291/322A). We found that substitutions and deletions resulted in different patterns of neurodegeneration and accumulation, with C291/322A having a dramatic effect on both tau accumulation and neurodegeneration. These cysteines formed disulfide bonds in mouse primary cultured neurons and in the fly retina, and stabilized tau proteins. Additionally, they contributed to tau accumulation under oxidative stress. We also found that each of these cysteine residues contributes to the microtubule polymerization rate and microtubule levels at equilibrium, but none of them affected tau binding to polymerized microtubules. Since tau proteins expressed in the Drosophila retina are mostly present in the early stages of tau filaments self-assembly, our results suggest that disulfide bond formation by these cysteine residues could be attractive therapeutic targets.
Introduction
Abnormal accumulation of the microtubule-binding protein tau is associated with a group of neurodegenerative diseases called tauopathies (1). Tau folds into paired helical filaments that are deposited in neurofibrillary tangles, which is a pathological feature of tauopathies (2). Tau is an intrinsically disordered protein, and its structures are regulated by posttranslational modifications and interactions with other proteins and structures such as microtubules (3). Once tau proteins acquire pathological, aggregation-prone structures, they act as ‘seeds’ to convert other tau proteins into disease-specific aggregates, and they spread to other cells (4). Since the accumulation of tau underlies neuron loss in diseased brains (5), targeting the early steps of the generation of abnormal tau species may be an efficient strategy for suppressing neuronal loss.
Six isoforms of tau resulting from alternative splicing are expressed in the adult human brain. Each isoform contains a microtubule-binding region composed of either three or four repeats (R1–R4) in the C-terminal half, a flanking basic proline-rich region, and zero to two (0N–2N) insertions in the N-terminal domain (2,6,7). In the microtubule-binding region, two hexanucleotide segments, the 275VQIINK280 sequence in R2 (PHF6*) and 306VQIVYK311 in R3 (PHF6), are critical for tau assembly. While both are reported to mediate an inter-molecular interaction for tau self-assembly to form a β-sheet-like structure, PHF6 is believed to play an initiating role (8,9). N-terminal projection domains mediate tau dimerization and oligomerization (10). Tau undergoes extensive posttranslational modifications, which also affects tau assembly. Tau is excessively phosphorylated in diseased brains and recognized by Alzheimer’s disease (ad) diagnostic antibodies such as AT8 (S199, S202, T205), AT100 (T212 and S214) and PHF1 (S396 and S404). Substitution of these Ser and Thr residues with glutamic acid to mimic phosphorylation generates a pathological conformation that is characteristic of tau in ad (11,12). Tau has two cysteine residues (Cys291 in R2 and Cys322 in R3) that can interact with another tau molecule or other proteins via thiol-disulfide exchange (13,14). These cysteine residues contribute to the formation of dimers and granular oligomers (15), one of the toxic intermediate structures of tau (16). Cysteine sulfenic acid (Cys-SOH) is a mediator of redox signaling, and oxidative stress is known to contribute to disease pathogenesis (17). Modification of these regions and residues affects the kinetics and final structures of tau in vitro (13,14). However, the behavior and toxicity of these tau species are not fully elucidated at the whole-organism level.
In the present study, we established a series of transgenic flies carrying 2N4R tau with known mutations or deletions that alter aggregation propensity. We found that these mutant tau strains induced neurodegeneration, and they were both quantitatively and qualitatively different from wild-type (WT) strains. Expression of tau in which cysteine residues were mutated to alanine showed dramatically decreased neurodegeneration. These cysteine residues form disulfide bonds to stabilize tau in the fly retina and mouse primary cultured neurons, and they contribute to tau accumulation caused by oxidative stress. These cysteine residues affect tau functions by promoting microtubule polymerization without affecting its interaction with formed microtubules, suggesting a novel mechanism that regulates tau abnormalities.
Results
Substitution of cysteine residues and deletion of PHF6 sequences dramatically suppresses 4R tau toxicity in Drosophila
To explore the relationship between tau aggregation status and toxicity, we established a series of transgenic flies carrying WT 2N4R tau (4Rtauwt) or 2N4R tau with deletions or amino acid substitutions that are known to affect tau aggregation, such as ΔPHF6, ΔPHF6*, S7A, S7E and C291/322A (8,9,11,12,15) (Fig. 1A). These constructs were placed under the control of a Gal4-responsive upstream activation sequence (UAS), and integrated into the genome via a site-directed insertion system to ensure equivalent expression levels (18).
Figure 1 .
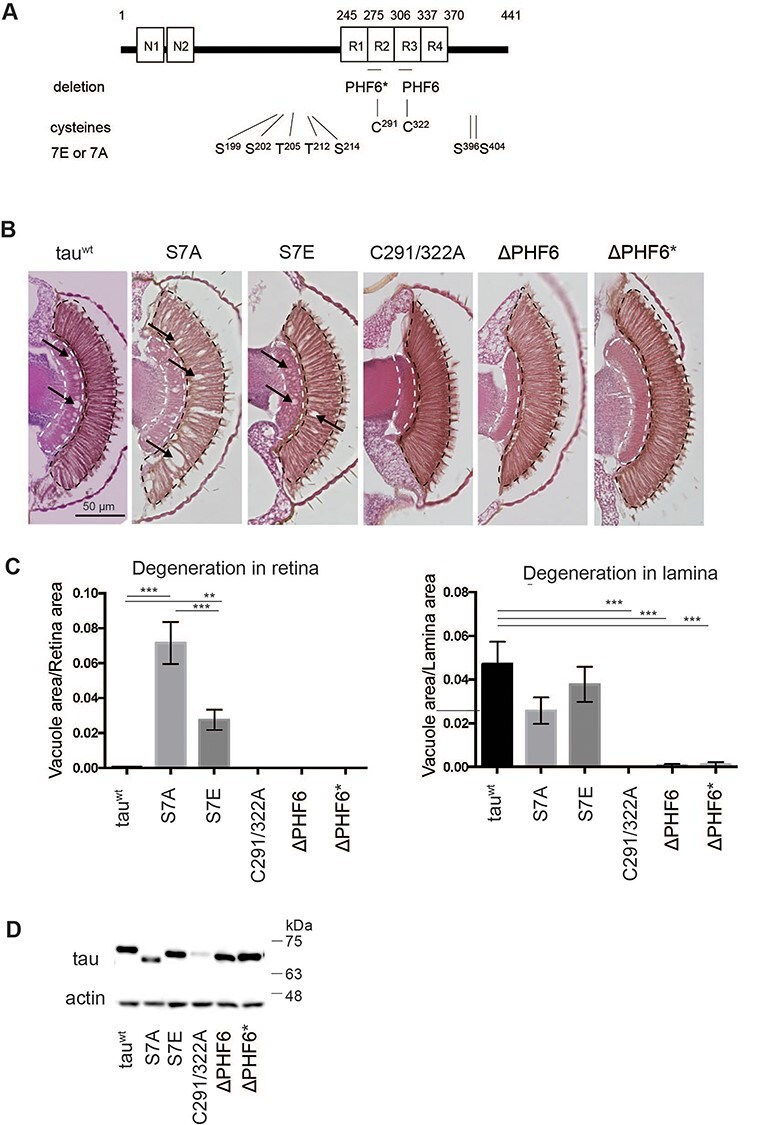
Tau proteins with different aggregation propensities induce different patterns of neurodegeneration in the fly retina. (A) Schematic representation of tau mutations investigated in this study. (B) Retinal sections expressing tau mutants. Transgenic flies carrying 4Rtauwt, 4RtauS7A, 4RtauS7E, 4RtauΔPHF6, 4RtauΔPHF6* and 4RtauC291/322A under the control of the UAS promoter were established by site-directed insertion at the attP2 site. Tau expression was driven by the pan-retinal driver gmr-GAL4. Flies were at 10 days old after eclosion. (C) Area of vacuoles in the retina and lamina (means ± SE, n = 5 or 6). (D) Tau protein levels analyzed by western blotting of head extracts with anti-tau antibody (T46). Actin was used as a loading control.
Expression of human tau in the eyes driven by the pan-retinal driver gmr-GAL4 causes a rough-eye phenotype due to apoptosis during development in the retina (19,20) and progressive neurodegeneration in the lamina, the first synaptic neuropil of the optic lobe containing photoreceptor axons (21). Herein, fly eyes expressing 4Rtauwt, 4RtauS7A and 4RtauS7E exhibited neurodegeneration in the retina and lamina (Fig. 1B), while the patterns of neurodegeneration in the eyes were different. Introduction of the S7A mutation suppressed degeneration in the neuropil and enhanced vacuole formation in the cell body region, while the introduction of the S7E mutation enhanced neurodegeneration in both areas with more severe degeneration in the neuropil than in the cell body region. Meanwhile, 4RtauΔPHF6, 4RtauΔPHF6* and 4RtauC291/322A generated much less neurodegeneration in both areas than 4Rtauwt (Fig. 1C).
Western blotting revealed that tau protein levels were similar among all strains except 4RtauC291/322A, in which levels were much lower (Fig. 1D, detailed analyses below). These results suggest that differences in neurodegeneration among tau strains 4Rtauwt, 4RtauΔPHF6, 4RtauΔPHF6*, 4RtauS7A and 4RtauS7E are not simply due to the accumulation of tau proteins: rather, they reflect qualitative differences.
C291 and C322 contribute to tau stability and toxicity in the fly retina
We focused on tauC291/322A because it had the most prominent effect on neurodegeneration. Western blotting analyses revealed that tauC291/322A protein levels were significantly lower than those of tauwt (Fig. 2A). To dissect the roles of C291 and C322, we established transgenic fly lines carrying 2N4R tau with each of the cysteine residues substituted with alanine (i.e. 4RtauC291A and 4RtauC322A). Lines 4RtauC291A and 4RtauC322A also showed lower protein levels, albeit not as low as 4RtauC291/322A (Fig. 2A). At the mRNA level, there was no significant difference in expression among these tau transgenes (Fig. 2B).
Figure 2 .
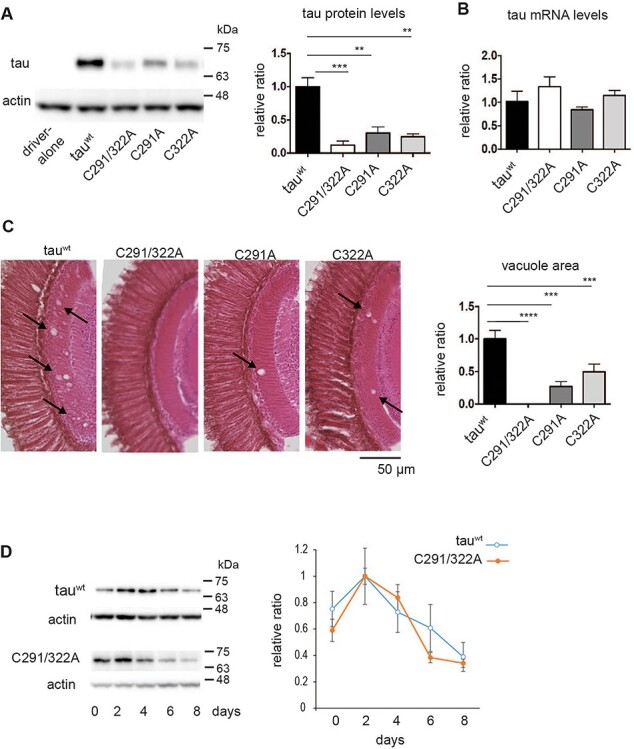
Cysteine residues stabilize tau proteins in the fly retina. (A) Tau protein levels analyzed by western blotting of head extracts with anti-tau antibody (T46). Actin was used as a loading control (means ± SE, n = 4, *P < 0.05, Student’s t-test). (B) mRNA levels analyzed by RT-qPCR. No significant differences were detected (mean ± SD, n = 3, one-way ANOVA followed by Tukey’s HSD multiple comparisons test). Flies were 2 days old after eclosion. (C) Substitution of cysteine residues to alanine mitigates tau toxicity in a Drosophila model. Neurodegeneration in the lamina caused by tau was observed as an increase in the vacuole area (indicated by arrows). Quantitation of the vacuole area is shown on the right (mean ± SE, n.s., n = 3, ****P < 0.0001, ***P < 0.001, **P < 0.01, one-way ANOVA followed by Tukey’s HSD multiple comparisons test). Flies were 10 days old. (D) Turnover of tauC291/322A is faster than that of tauwt in the fly retina. Tau expression was driven by elav-GeneSwitch. Newly eclosed flies were fed RU486-containing food for 2 days, then transferred to food without RU486 (day 0). Flies were collected every 2 days, and head homogenates were subjected to western blotting with anti-tau antibody (T46). TauC291/322A levels were significantly lower at day 6 or at day 8 compared to day 2, while tauwt levels were not (P < 0.001 for tauC291/322A, one-way ANOVA with post hoc Tukey HSD). Mean ± SE, n = 3.
To rule out the possibility that differences in protein levels among these tau species were associated with the site at which these transgenes were integrated, we established another set of transgenic flies carrying 4Rtauwt and 4RtauC291/322A at an integration site on a different chromosome (attP40). These flies also displayed lower protein levels of 4RtauC291/322A than 4Rtauwt, further indicating that the insertion was not responsible for the reduction in tau expression levels (Supplementary Material, Fig. S1).
We analyzed neurodegeneration in fly retina tissue expressing 4Rtauwt, 4RtauC291/322A, 4RtauC291A and 4RtauC322A. We found that severity of neurodegeneration was correlated with protein levels; compared with 4Rtauwt, 4RtauC291/322A, 4RtauC291A, and 4RtauC322A caused less neurodegeneration, and substitution of both C291 and C322 had a more prominent effect than either substitution of C291 or C322 (Fig. 2C).
Since these mutations decreased tau protein levels, we investigated whether these cysteine residues stabilize tau proteins. To analyze the turnover of tau proteins, we expressed tau transiently in neurons and observed how tau protein levels decayed. Expression of 4Rtauwt and 4RtauC291/322A driven by elav-GeneSwitch was induced in flies by feeding RU486 for 2 days, then turned off by transferring flies to food without RU486. 4Rtauwt levels were decreased to 50% of maximum levels on day 8, while 4RtauC291/322A levels were 50% lower on day 6 (Fig. 2D). These results indicate that Cys291 and Cys322 regulate tau stability in adult fly eyes.
Cysteine residues are critical for tau protein stability in mouse primary cultured neurons
We next investigated whether these cysteine residues also stabilize tau in mammalian neurons. Mouse primary neurons were separately transfected with the same amount of plasmid carrying 4Rtauwt or 4RtauC291/322A. Western blotting analyses revealed that substitution of these cysteine residues to alanine decreased tau levels dramatically. Real-time quantitative reverse transcription (qRT-PCR) analysis indicated that cells transfected with 4RtauC291/322A expressed higher tau mRNA levels than 4Rtauwt, suggesting that lower tau protein levels were not caused by lower transcription, but were instead associated with post-transcriptional events (Fig. 3B).
Figure 3 .
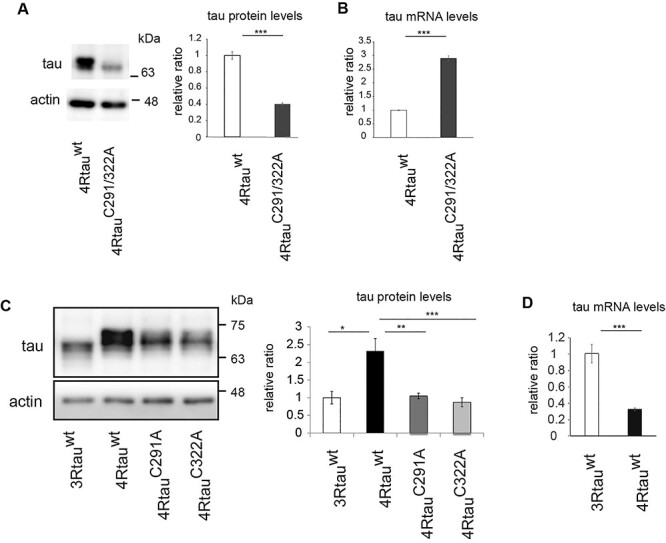
Cysteine residues positively regulate tau protein levels in mouse primary cortical neurons. (A) 4RTauwt and 4RtauC291/322A were introduced into mouse primary cortical neurons. After 4 days, neurons were harvested and subjected to western blotting with anti-human tau antibody (HT7). Actin was used as a loading control. Representative images (left) and quantitation (right) are shown (mean ± SE, n = 3, ***P < 0.005, Student’s t-test). (B) mRNA levels analyzed by RT-qPCR (mean ± SD, n = 3, ***P < 0.005, Student’s t-test). (C) 3Rtauwt, 4Rtauwt, 4RtauC291A and 4RtauC322A were introduced into mouse primary cortical neurons and subjected to western blotting with anti-human tau antibody (HT7). Representative images (left) and quantitation (right) are shown. Actin was used as a loading control; mean ± SD, n = 3, ***P < 0.005, one-way ANOVA followed by Tukey’s HSD multiple comparisons test. (D) mRNA levels of 3Rtauwt and 4Rtauwt were analyzed by qRT-qPCR; mean ± SD, n = 3, ***P < 0.005, Student’s t-test.
We subsequently explored the effects of single substitution of these Cys residues on tau levels. Alanine substitution at Cys291 (4RtauC291A) and at Cys322 (4RtauC322A) both caused a reduction in tau protein levels, indicating that each of these sites is important for tau stability in mammalian neurons (Fig. 3C). Tau Cys322 is only found in the 4R isoform of tau, and we found that levels of 2N3R tau (3Rtauwt) expressed in mouse primary neurons were lower than those of 4Rtauwt (Fig. 3C). Additionally, 3Rtauwt mRNA levels were higher than those of 4Rtauwt (Fig. 3D). Comparison among the levels of 3Rtauwt, 4RtauC291 and 4RtauC322A showed that 3Rtauwt levels were similar to those of 4RtauC291A and 4RtauC322A (Fig. 3D). These results suggest that the two cysteine residues in 4R tau contribute to the difference in stability between 3R and 4R tau.
Tau forms disulfide bonds in cultured neurons and in the Drosophila brain
Residues C291 and C322 in tau are reported to form homophilic or heterophilic disulfide bonds in vitro (13,14,22,23). We, therefore, investigated whether C291 and/or C322 are involved in disulfide bond formation in tau proteins in cultured neurons. Mouse primary neurons were transfected with vectors encoding tauwt, 4RtauC291/322A, 4RtauC291A and 4RtauC322A. Cells were lysed in buffer containing iodoacetic acid (IAA) that irreversibly modifies free thiols in proteins. The IAA-modified tau proteins were then immunoprecipitated from cell lysates and treated with dithiothreitol (DTT) to disrupt any pre-existing disulfide bonds in tau before IAA treatment. The newly exposed free thiol groups could then be relabeled with biotinylated iodoacetamide (BIAM) (24). We found that 4Rtauwt was labeled with BIAM, indicating that C291 and Cys322 were involved in disulfide bonds before IAA treatment (Fig. 4A, arrowhead). BIAM labeling was also observed with tauC291A and tauC322A, indicating that substitution of either Cys291 or Cys322 was not sufficient to block disulfide bond formations (Fig. 4A). By contrast, BIAM labeling was not observed with tauC291/322A (Fig. 4A). These results suggest that both of these residues are involved in the formation of disulfide bonds in cultured neurons.
Figure 4 .
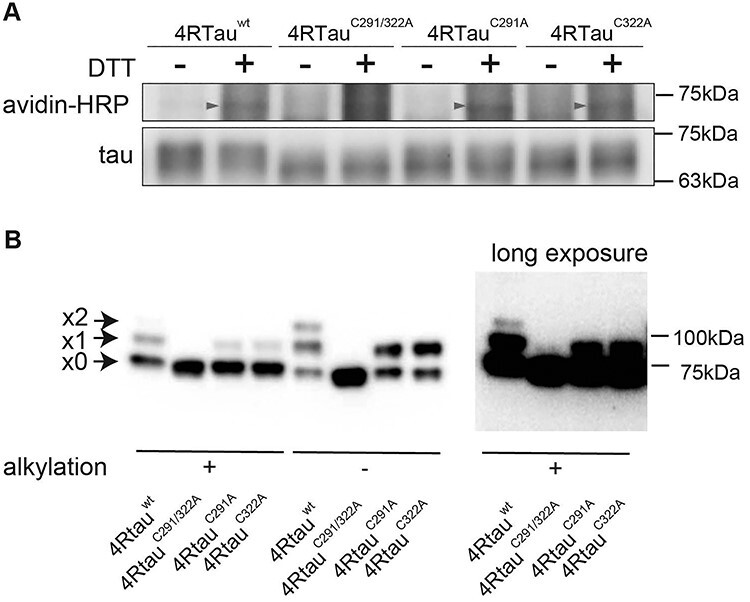
Tau forms disulfide bonds in mouse primary cultured neurons and the fly retina. (A) 4Rtauwt, 4RtauC291/322A, 4RtauC291A and 4RtauC322A were expressed in mouse primary cortical neurons and subjected to BIAM analyses. Biotinylation was observed with tauwt, tauC291A and tauC322A, but not with tauC291/322A (avidin-HRP, arrowheads). The 2N4R isoform of tau was used. (B) Tau forms disulfide bonds via C291 or C322 in the fly retina. Fly head lysates were first treated with an alkylating reagent to block free thiols (alkylation [+]). Lysates were then treated with DTT to reduce disulfide bonds and incubated with m-PEG to label the reduced cysteine residues. Tau forming disulfide bonds were detected by PEG-maleimide labeling in fly heads expressing 4Rtauwt, 4RtauC291A or 4RtauC322A, but not those expressing 4RtauC291/322A. Samples were also prepared without alkylation, which exposes all cysteines for tagging if there is no oxidation (alkylation [−]). Arrowheads from the top indicate two, one or zero cysteine residues forming disulfide bonds. The top two bands were not observed with 4RtauC291/322A even after long exposure (right, long exposure).
We also analyzed the formation of disulfide bonds in tau expressed in the fly retina using maleimide-polyethylene glycol (m-PEG) labeling, another method to detect disulfide bonds. Fly head lysates were treated with an alkylating reagent to block free thiols (alkylation [+]). Lysates were then treated with DTT to reduce disulfide bonds and incubated with m-PEG to label the reduced cysteine residues. Samples were subjected to SDS-PAGE and blotted with anti-tau antibody to detect m-PEG-tagged tau through mobility shift in immunoblots (25,26). To evaluate the efficiency of m-PEG tagging and oxidation of samples during the experiments, samples were also prepared without alkylation, which should expose all cysteines for tagging if there is no oxidation during the experiment (alkylation [−]). If there was no oxidation during the experiment and all the cysteine residues were labeled, the x2 band alone with 4Rtauwt and the x1 band alone with tauC291A and tauC322A would be observed.
m-PEG tagging of 4Rtauwt proteins resulted in three bands, x0, x1 and x2 (Fig. 4B). The most rapidly migrating band (x0) was the major band in alkylation [+], and the two slower migrating bands (x1) and (x2) were more intense in alkylation [−]. Furthermore, m-PEG-tagged tauC291/322A yielded only the most rapidly migrating band, x0 (Fig. 4B), while tauC291A and tauC322A yielded the two fast-migrating bands x0 and x1 (Fig. 4B). Thus, the bands x0, x1 and x2 presumably corresponded to tau with zero, one and two disulfide bonds, respectively. The x1 and x2 bands of 4Rtauwt indicate that one or two cysteine residues in 4Rtauwt were involved in disulfide bonds in fly eyes (Fig. 4B). Together, these results suggest that Cys291 and Cys322 form multiple patterns of reversible disulfide bonds in mature neurons in vivo.
C291 and C322 contribute to tau accumulation under oxidative stress
Oxidative stress is believed to play a fundamental role in the pathogenesis of neurodegenerative diseases (27), as well as antioxidant defense systems that protect against tau toxicity, such as the expression of superoxide dismutase (SOD) (28,29). Since cysteine residues can form disulfide bonds in response to oxidative stress (30), we wondered whether C291 and C322 are involved in the effects of oxidative stress on tau metabolism. To test the effect of antioxidant, 4Rtauwt or 4RtauC291/322A was co-expressed with UAS-SOD1 (31,32). We found that co-expression of SOD1 reduced the abundance of 4Rtauwt protein by more than 40% (Fig. 5A). Co-expression of SOD1 also decreased the amount of 4RtauC291/322A, albeit to a lesser extent than tauwt (Fig. 5B). Levels of 4Rtauwt and 4RtauC291/322A mRNA were not diminished by SOD1 co-expression (Fig. 5C). The expression level of SOD1 co-expressed with 4Rtauwt and that with 4RtauC291/322A were similar (Fig. 5C). Co-expression of a control protein GFP did not affect 4Rtauwt levels (Supplementary Material, Fig. S2), indicating that the reduction in 4Rtauwt protein levels caused by SOD1 co-expression was not due to a nonspecific effect. These results suggest that these cysteine residues are involved in tau accumulation caused by oxidative stress.
Figure 5 .
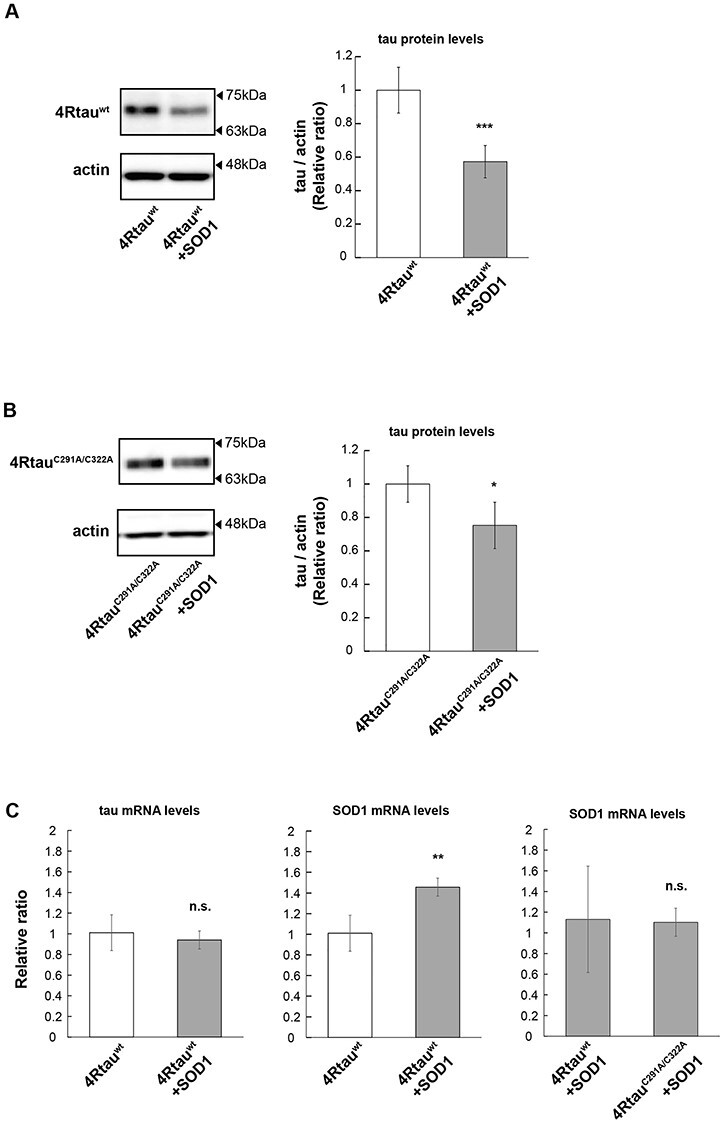
Overexpression of SOD1 reduces the levels of 4Rtauwt, and cysteine to alanine mutation suppresses this effect. (A and B) 4Rtauwt (A) or 4RtauC291/322A (B) was co-expressed with SOD1, and tau levels were analyzed by western blotting of head extracts with anti-tau antibody (T46). Actin was used as a loading control (mean ± SE, n = 3, *P < 0.05, Student’s t-test). (C) mRNA levels of tau or SOD1 were analyzed by RT-qPCR. (mean ± SD, n = 3, n.s. P > 0.05, **P < 0.01, one-way ANOVA followed by Tukey’s HSD multiple comparisons test). Flies were 2 days old after eclosion.
C291 and C322 in 4R tau play critical roles in microtubule polymerizing but not binding to polymerized microtubules in vitro
Since tau binding to microtubules has been reported to affect its metabolism (33,34), we hypothesized that changes in tau binding to microtubules underlies the effects of alanine substitution of C291 and C322. To analyze the roles of these cysteine residues in the interaction between tau and growing and polymerized microtubules, the microtubule polymerizing abilities of 4Rtauwt, 3Rtauwt and tau with cysteine substitutions to alanine were analyzed in vitro. We found that all were capable of promoting microtubule formation (Fig. 6A–E), and 4Rtauwt enhanced microtubule formation to a greater extent than 3Rtauwt (Fig. 6A). Regarding 4R tau, 4RtauC291A promoted microtubule formation at a similar rate to 4Rtauwt, but the amount of polymerized microtubules at equilibrium was lower. By contrast, with 4RtauC322A, both microtubule polymerization and the amount of microtubules at equilibrium were lower than observed for 4Rtauwt (Fig. 6B, D, and E). These results indicate that the C322 residue is involved in the functions of 4Rtau on microtubule polymerization and stabilization.
Figure 6 .
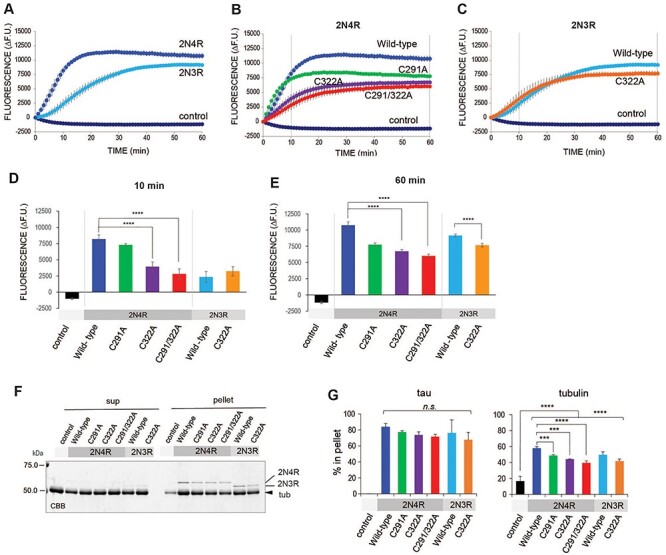
C291 or C322 contribute to tau functions in microtubule polymerization but not binding to formed microtubules. (A–C) Roles of cysteine residues of tau in tau functions related to microtubule polymerization in vitro. (A) Recombinant 2N4R and 2N3R tau promote polymerization of tubulins in vitro. Porcine tubulins were incubated with 2N4R or 2N3R wild-type tau. Microtubules monitored by fluorescence dye were increased in a time-dependent manner and reached equilibrium. Note that 2N4R tau stimulates microtubule formation faster than 2N3R tau. The total amount of polymerized microtubules was higher with 2N4R than 2N3R tau. Data represent the change in fluorescence units from 0 min (mean ± SEM, n = 4). (B and C) Tubulin polymerization activity of cysteine mutant tau 2N4R tau (B) and 2N3R tau (C) compared with WT tau. (B) 4RtauC291A increases microtubule formation at a similar rate to 4RtauWT, but decreases the amount of polymerized microtubules at equilibrium. By contrast, 4RtauC322A stimulates microtubule formation slower than 4RtauWT and increases the amount of microtubules. 4RtauC291A/C322A further affects tau functions, but differences from 4RtauC322A were not statistically significant. (C) Mutation of C322A in 2N3R tau slightly decreases the amount of polymerized microtubules at equilibrium. Data represent the change in fluorescence units from 0 min (means ± SEM, n = 4). (D, E) Quantification of polymerized microtubules in the growing phase (10 min, D) and at equilibrium (60 min, E; vertical lines in B and C). Data represent the change in fluorescence units from 0 min (means ± SEM, n = 4). Statistical significance was analyzed by one-way ANOVA followed by Tukey’s post hoc test. (F, G) Quantification of polymerized tubulins by SDS-PAGE. (F) Protein mixtures after a 60-min incubation were subjected to ultra-centrifugation to obtain the free (sup) and microtubule (pellet) fractions. Fractions were analyzed by SDS-PAGE followed by Coomassie brilliant blue staining. Bands corresponding to 2N4R, 2N3R and tubulin are indicated. (G) Quantitation of (F). Data represent the percentage of proteins fractionated in the pellet (means ± SEM, n = 3). Statistical significance was analyzed by one-way ANOVA followed by Tukey’s post hoc test.
In contrast to 4Rtau, 3Rtauwt and 3RtauC322A showed similar microtubule polymerization rates, with a slightly reduced amount of polymerized microtubules at equilibrium with 2N3RtauC322A (Fig. 6C, D, and E). These results indicate that the roles of C322 in microtubule stabilization in 4Rtau and 3Rtau are different.
The levels of microtubules formed with 4RtauC291/322A, 4RtauC291 and 4RtauC322 were lower than those formed with tauwt (Fig. 6F and G). The levels of polymerized microtubules at equilibrium are affected by microtubule growth and microtubule ‘catastrophe’ events. Tau proteins can bind to free tubulins to regulate the nucleation of microtubule bundles, microtubule elongation and stabilize polymerized microtubules to protect from microtubule ‘catastrophe’. To determine whether the reduced amount of polymerized microtubules at equilibrium with Cys-less tau was due to the reduced affinity of Cys-less tau for polymerized microtubules, we performed co-sedimentation analysis, which revealed that 4RtauC291/322A, 4RtauC291 and 4RtauC322 bound to polymerized microtubules at levels similar to that of 4Rtauwt (Fig. 6F and G). These results argue against the possibility that Cys-less tau species are less capable of binding to polymerized microtubules for their stabilization, and suggest that the effects of Cys-less tau species are associated with the elongation phase of microtubule polymerization. Taken together, these results suggest that cysteine residues in 4Rtau interact with free tubulins or newly formed microtubules, rather than with polymerized microtubules.
Discussion
Tau proteins can exist in various conformations and aggregation statuses, but their structure–toxicity relationships are not fully understood. Herein, we used a site-directed insertion system in Drosophila to compare the toxicity of a series of tau mutants in the same genetic background. We found that different types of neurodegeneration could be detected in this model. For example, tauS7A primarily affected the cell body region, while tauS7E primarily affected the neuropil (Fig. 1). These differences may reflect differences between tauS7A and tauS7E in intracellular localization; a previous report found that tauS7A localized in the cell body, while tauS7E is present in axons (35,36). These results suggest that different types of neurodegeneration can be induced by tau depending on its cellular distribution (35,36).
Among the tau mutants tested, deletion of PHF6 and deletion of PHF6* caused the most significant suppression of tau toxicity without affecting tau protein levels (Fig. 1). The recently reported cryo-EM structure of tau filaments demonstrated that the PHF6 segment is located in the region that forms an ordered core and serves as a platform for the incorporation of tau into the growing filament (37). Our results are consistent with those of a recent report showing that deletion of PHF6 suppresses tau toxicity in a Drosophila model (38) and supports the model that this ordered core is necessary for the seeded assembly of tau into pathological structures (37). In contrast, PHF6* is located in the region forming the fuzzy coat (37), and the role of this region in tau toxicity has not been fully understood. Our results revealed that expression of 4RtauΔPHF6*, similar to 4RtauΔPHF6, caused much less neurodegeneration compared to 4Rtauwt (Fig. 1), suggesting that PHF6* also play a critical role in tau toxicity.
In contrast to deletion of PHF6 or PHF6*, a reduction in neurodegeneration caused by cysteine substitutions was accompanied by lowered levels of tau proteins (Fig. 1). During the preparation of this manuscript, Prifti et al. also reported that alanine substitutions at C291 and C322 decrease tau accumulation and toxicity in a Drosophila model (39). Although deletion of PHF6 or PHF6* and cysteine substitutions are all reported to disrupt fibril formation in vitro (13,15,40), our results suggest that mechanisms by which these regions contribute to tau toxicity are different, and blocking cysteine residues decreases neurodegeneration via enhancement of tau degradation.
Disulfide bonds between pairs of cysteines determine protein structures and functions (41). Proteins with disulfide bonds are found primarily in relatively oxidizing environments in the cell such as the endoplasmic reticulum (ER) lumen, and most disulfide bonds are found in membrane proteins and secretory proteins formed in the ER lumen (41). However, in the microenvironment of the cytosol or in diseased states, oxidizing conditions can occur through elevated levels of reactive oxygen species (ROS) (30,42). Disulfide bond formation in tau mediates dimer formation to promote filament assembly in vitro (13), and administration of cysteine-capping reagents mitigates tau toxicity in a mouse model (15). Our current study confirmed disulfide bond formation in tau in vivo in mouse-cultured neurons as well as the fly retina (Figs 3 and 4). Our results also suggest that cysteine residues contribute to ROS-mediated accumulation of tau (Fig. 5), which may be one of the mechanisms by which oxidative stress contributes to disease pathogenesis.
Cysteine residues are involved in tau binding to microtubules stabilized by taxol (43–45), and they mediate thiol-disulfide exchange between αβ-tubulin or microtubules and tau (14). Thus, we hypothesized that the cysteine residues would affect tau toxicity by regulating microtubule binding. We were surprised to discover that the cysteine residues were required for efficient microtubule polymerization, but not for tau binding to polymerized microtubules (Fig. 6). Our results suggest novel models regarding the roles of cysteine residues of tau in microtubule formation; these cysteine residues may mediate interactions between tau and growing microtubules or recruit free tubulins to microtubule ends.
Tau is an intrinsically disordered protein, and pathological states can cause tau to assemble into oligomers and fibrils and eventually deposit in neurofibrillary tangles (46). Tau oligomerization is facilitated by intermolecular disulfide cross-linking between cysteine residues and by the PHF6 hexapeptide, which leads to the assembly of higher order assemblies (13). However, since tau filaments were not detected (19) and tau proteins were found mostly as detergent-soluble monomers in the fly model used here (Supplementary Material, Fig. S3), the effect of cysteine substitutions cannot be fully explained by suppression of formation of higher-order assemblies. Rather, the dramatic reduction observed with Cys-less tau may be associated with the stability of monomeric forms of tau. We showed that tau cysteine residues form disulfide bonds in vivo (Fig. 3), while the molecules that form disulfide bonds with tau proteins and stabilize them remain to be identified. While tubulins are strong candidates (Fig. 6 and (13,14)), the formation of an intramolecular disulfide bond by two cysteines may change tau protein structure and affect its stability. Substitution of either C291 or C322 had similar effects on 4R tau stability, supporting this conjecture (Fig. 3).
In summary, our findings suggest that oxidative stress and disulfide bond formation in tau may be one of the triggering factors that lead to disease pathogenesis. Cysteine modifications may offer an effective strategy to block the generation and spreading of pathological tau species.
Materials and Methods
Chemicals and antibodies
Anti-human tau (HT7) antibody (Thermo), anti-tau (T46) (Thermo), anti-tau (TAU5) (Merk-Millipore) and anti-actin antibody (SIGMA) were purchased. Anti-tau antibody (TauC) was described previously (47). HRP streptavidin and RU486 were purchased from SIGMA.
Fly stocks and husbandry
Flies were maintained in standard cornmeal media at 25°C under light–dark cycles of 12:12 hours. To establish the transgenic fly line carrying the human 2N4R tau with or without mutations or deletions, cDNA was subcloned into pUASTattB, and injected to the oocytes carrying into P{CaryP}attP2. UAS-SOD1 (31) was obtained from Bloomington stock center (Stock #33605). For RU486 feeding, RU486 was mixed into the cornmeal media at the final concentration of 500 μm. Genotypes of the flies used in the experiments are described in Supplementary Material Table S1.
Plasmid construction for tau C291A, tau C322A and tauC291/322A
A fragment of cDNA coding human 2N4Rtau from nucleotide number 657–1105 for wild-type sequence, C291A mutation, C322A mutation and C291A/C322A double mutations were chemically synthesized by Eurofins Genomics. Fragments were digested with BstXI, and ligated into human 2N4R tau, which was also digested with BstXI. All tau genes (2N3R, 2N4R, C291A, C322A and C291/322A) were subcloned into pCAG vector for expression in mouse primary neurons or into pUASTattB for generation of transgenic fly lines.
SDS–PAGE and immunoblotting
SDS–PAGE for western blotting of tau and actin was performed using 10% (w/v) polyacrylamide gels. Proteins separated in the gel were transferred to a PVDF membrane (Merck Millipore) using a submerged blotting apparatus and then visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore). The chemiluminescent signal was detected by Fusion FX (Vilber) and intensity was quantified using ImageJ (NIH). Western blots were repeated a minimum of three times with different animals and representative blots are shown. Flies used for western blotting were 2-day-old after eclosion. To analyze the decay of tau protein levels after transient expression (Fig. 2), the same cohort of flies was divided into five groups of 15–20 flies, kept in RU486-containing food for 2 days, then transferred to regular cornmeal food. Heads were collected and homogenized for western blotting after 0, 2, 4, 6 or 8 days. Experiments were repeated three times with independent cohorts of flies.
Histological analysis
Neurodegeneration in the fly retina was analyzed as previously reported (48). Fly heads were fixed in Bouin’s fixative for 48 h at room temperature, incubated for 24 h in 50 mm Tris/150 mm NaCl, and embedded in paraffin. Serial sections (7 μm thickness) through the entire heads were stained with hematoxylin and eosin and examined by bright-field microscopy. Images of the sections that include the lamina were captured with Keyence microscope BZ-X700 (Keyence), and the vacuole area was measured using ImageJ (NIH). Heads from more than three flies (more than five hemispheres) were analyzed for each genotype.
qRT-PCR
qRT-PCR was carried out as previously reported (48). More than thirty flies for each genotype were collected and frozen. Heads were mechanically isolated, and total RNA was extracted using Isogen Reagent (NipponGene) according to the manufacturer’s protocol with an additional centrifugation step (11 000 × g for 5 min) to remove the cuticle membranes prior to the addition of chloroform. Total RNA was reverse transcribed using PrimeScript Master Mix (Takara Bio). qRT-PCR was performed using TOYOBO THUNDERBIRD SYBR qPCR Mix on a Thermal Cycler Dice Real-Time System (Takara Bio). The average threshold cycle value (CT) was calculated from at least three replicates per sample. Expression of genes of interest was standardized relative to rp49. Relative expression values were determined by the ΔΔCT method. Primers were designed using Primer-Blast (NIH). The following primers were used for RT-qPCR:
htau for 5′-CAAGACAGACCACGGGGCGG-3′.
htau rev 5′-CTGCTTGGCCAGGGAGGCAG-3′.
dSOD1 for 5′-CCCCACCAAGGTCAACATCA-3′.
dSOD1 rev 5′-TGACTTGCTCAGCTCGTGTC-3′.
rp49 for 5′-GCTAAGCTGTCGCACAAATG-3′.
rp49 rev 5′- GTTCGATCCGTAACCGATGT-3′.
Cell culture and transfection
Primary neurons were prepared from the mouse brain cortex at embryonic day 15 (E15) and plated on poly-l-lysine-coated dishes in DMEM and Ham’s F-12 (1:1) supplemented with 5% fetal bovine serum, 5% horse serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin at a density of 1.0 × 106 cells/ml. The medium was replaced to Neurobasal medium supplemented with 2% B-27 (Invitrogen), 0.5 mm l-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin after 4 h of plating. Equal amounts (8 μg) of plasmids encoding human tau were transfected into the suspension of neurons (8 × 106) before plating using Nucleofector with Mouse Neuron Nucleofector™ Kit (LONZA) according to the manufacturer’s protocol. All animal experiments were performed according to the guidelines for animal experimentation of Tokyo Metropolitan University. The study was approved by the Research Ethics and Safety Committee of Tokyo Metropolitan University (approval number, A30–1).
Biotinylation of cysteine residues in tau in primary neurons
Mouse primary neurons transfected with human tau were immediately frozen with liquid nitrogen and lysed on culture dishes with lysis buffer including 8 M urea and 30 mm iodoacetamide (IAA) and incubated for 15 min at 37°C. After dialysis to remove urea and IAA, lysates were incubated with biotinylated iodoacetamide (BIAM) to biotinylated cysteine residues in the absence or presence of dithiothreitol. Tau was immunoprecipitated from the cell lysates with monoclonal anti-tau antibody (TAU5) and protein G sepharose (GE healthcare). After separating with SDS-PAGE, biotinylation was detected with HRP streptavidin and tau was with anti-tau-C polyclonal antibody.
m-PEG labeling
Lysis buffer (100 mm Tris (pH 7.2), 1% SDS, 1 M N-ethylmaleimide, 0.5 M NaF, 0.5 M β-glycerol, 10 μm leupeptin and 0.1 mg/ml pefabloc (Sigma)) were bubbled with nitrogen gas to lessen the dissolved oxygen for 20 min then used to homogenate fly heads. Lysates were centrifuged to remove debris, and supernatants were incubated at 50°C for 25 min. Free N-ethylmaleimides were removed by desalting column (Zeba Spin Desalting Columns 7K (MWCO)). Samples were reduced by incubation with the addition of DTT at the final concentration of 50 mm for 20 min at 37°C and then applied to a desalting column. Samples were denatured by 0.5% SDS and incubated with 2 mm PEG-maleimide for 2 h at 37°C. Samples were mixed with Laemmli sample buffer (100 mm Tris (pH 6.8), 4% SDS), subjected to SDS-PAGE, and western blotting with anti-tau antibody.
Purification of recombinant proteins
Escherichia coli (E. coli) expression vectors, pRK172-2N4R and pRK172-2N3R human tau, were kind gifts from Dr M. Goedert. Mutation on specific cysteine to alanine residues was produced by site-directed mutagenesis by polymerase chain reaction. The positions of the amino acids were numbered according to the 441-amino acid isoform of human tau. Thus, C291 is localized in second repeat and C322 is localized in the third repeat of microtubule binding domain of tau. Recombinant tau was purified as described previously (49). Briefly, expression of recombinant human tau was induced in E. coli, BL21 (DE3) by 0.5 mm of isopropyl β-D-1-thiogalactopyranoside for 2 h. Tau expressing bacteria were harvested and lysed in homogenize buffer (50 mm PIPES, 1 mm EGTA and 1 mm DTT (pH 6.4)) with sonication. After centrifugation at 3000 × g at 4°C for 10 min, the soluble fractions were applied to phosphocellulose columns (P11, Whatman) and eluted by a stepwise gradient of 0.1–0.3 M NaCl. The soluble fractions were then precipitated by saturated ammonium sulfate and collected by ultracentrifugation at 100 000 × g at 2°C for 15 min. Resultant pellets were resuspended in a homogenized buffer containing 0.5 M NaCl and 2% 2-mercaptoethanol and heated at 100°C for 5 min. After the insoluble materials were removed by centrifugation at 15 000 × g for 15 min. The soluble heat stable fractions containing tau proteins were further fractionated using reverse-phase HPLC with Cosmosyl Protein-R column (Nacalai tesque). These fractions were dried and stored in −80°C until use. Purity and concentrations of proteins were evaluated by SDS-PAGE followed by Coomassie Brilliant Blue staining. We verified that purity of all recombinant proteins used here were better than 95% of total protein.
In vitro tubulin polymerization assay
Effects of tau on tubulin polymerization were determined using fluorescence-based tubulin polymerization assay as described in manufactured instruction (Cytoskeleton Inc., Denver, CO). Briefly, purified wild-type and mutant tau (1 μm) were mixed with porcine tubulin (7.5 μm) in assembly buffer at 37°C. Polymerized tubulin was monitored by fluorescence (ex.360/em. 465) using Infinit F-200 Microplate Reader (TECAN, Männedorf/Switzerland) for 30 min at 1 min interval. After incubation, resultant solutions were subjected to centrifugation at 100 000g, for 15 min at 20°C. The supernatants (free tubulin fraction) and pellets (microtubule fraction) were subjected to SDS-PAGE to quantify the amount of tubulin constructed into the microtubule.
Statistics
Statistics were done with Microsoft Excel (Microsoft), GraphPad Prism (GraphPad) and R (R Foundation for Statistical Computing, Vienna, Austria.URL http://www.R-project.org/). Differences were assessed using the Student’s t-test or one-way ANOVA and Tukey’s honestly significant difference (HSD) post hoc test. P values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
The authors thank Dr Mel Feany, the Harvard Transgenic RNAi project (TRiP) (NIH/NIGMS R01-GM084947), the Bloomington Stock Center; and the NIG Drosophila stock center for fly stocks. We thank Dr Peter Davis for the AT8 and PHF1 antibodies. We thank Dr Michel Goedert for pRK172-2N4R and pRK172-2N3R human tau plasmids. We thank Drs. S.-I. Hisanaga, Wei Ran, and Yoshiyuki Soeda for critical comments.
Conflict of Interest statement. The authors declare that there is no conflict of interest.
Contributor Information
Taro Saito, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan; Department of Biological Sciences, Faculty of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Tomoki Chiku, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Mikiko Oka, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Satoko Wada-Kakuda, Department of Neuropathology, Faculty of Life and Medical Sciences, Doshisha University, Kyoto 6100394, Japan.
Mika Nobuhara, Department of Neuropathology, Faculty of Life and Medical Sciences, Doshisha University, Kyoto 6100394, Japan.
Toshiya Oba, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Kanako Shinno, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Saori Abe, Department of Biological Sciences, Faculty of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Akiko Asada, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan; Department of Biological Sciences, Faculty of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Akio Sumioka, Department of Life Science, Faculty of Science, Gakushuin University, Tokyo 1718588, Japan.
Akihiko Takashima, Department of Life Science, Faculty of Science, Gakushuin University, Tokyo 1718588, Japan.
Tomohiro Miyasaka, Department of Neuropathology, Faculty of Life and Medical Sciences, Doshisha University, Kyoto 6100394, Japan.
Kanae Ando, Department of Biological Sciences, Graduate School of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan; Department of Biological Sciences, Faculty of Science, Tokyo Metropolitan University, Tokyo 1920397, Japan.
Funding
Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (JSPS KAKENHI Grant number 17H05703 to K.A.); a research award from the Hoan-sha Foundation (to K.A.), the Takeda Science Foundation (to K.A.), a research award from the Japan Foundation for Aging and Health (to K.A), a Grant-in-Aid for Scientific Research on Challenging Research (Exploratory) (JSPS KAKENHI Grant number 19K21593 to K.A.); Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (JSPS KAKENHI Grant number 26117004 to T.M.).
References
- 1. Holtzman, D.M., Carrillo, M.C., Hendrix, J.A., Bain, L.J., Catafau, A.M., Gault, L.M., Goedert, M., Mandelkow, E., Mandelkow, E.M., Miller, D.S.et al. (2016) Tau: from research to clinical development. Alzheimers Dement., 12, 1033–1039. [DOI] [PubMed] [Google Scholar]
- 2. Gotz, J., Halliday, G. and Nisbet, R.M. (2019) Molecular pathogenesis of the Tauopathies. Annu. Rev. Pathol., 14, 239–261. [DOI] [PubMed] [Google Scholar]
- 3. Wesseling, H., Mair, W., Kumar, M., Schlaffner, C.N., Tang, S., Beerepoot, P., Fatou, B., Guise, A.J., Cheng, L., Takeda, S.et al. (2020) Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell, 183, 1699–1713.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goedert, M., Eisenberg, D.S. and Crowther, R.A. (2017) Propagation of tau aggregates and neurodegeneration. Annu. Rev. Neurosci., 40, 189–210. [DOI] [PubMed] [Google Scholar]
- 5. Ballatore, C., Lee, V.M. and Trojanowski, J.Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci., 8, 663–672. [DOI] [PubMed] [Google Scholar]
- 6. Gustke, N., Trinczek, B., Biernat, J., Mandelkow, E.M. and Mandelkow, E. (1994) Domains of tau protein and interactions with microtubules. Biochemistry, 33, 9511–9522. [DOI] [PubMed] [Google Scholar]
- 7. Chen, J., Kanai, Y., Cowan, N.J. and Hirokawa, N. (1992) Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature, 360, 674–677. [DOI] [PubMed] [Google Scholar]
- 8. von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E.M. and Mandelkow, E. (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA, 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. von Bergen, M., Barghorn, S., Li, L., Marx, A., Biernat, J., Mandelkow, E.M. and Mandelkow, E. (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem., 276, 48165–48174. [DOI] [PubMed] [Google Scholar]
- 10. Feinstein, H.E., Benbow, S.J., LaPointe, N.E., Patel, N., Ramachandran, S., Do, T.D., Gaylord, M.R., Huskey, N.E., Dressler, N., Korff, M.et al. (2016) Oligomerization of the microtubule-associated protein tau is mediated by its N-terminal sequences: implications for normal and pathological tau action. J. Neurochem., 137, 939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jeganathan, S., Hascher, A., Chinnathambi, S., Biernat, J., Mandelkow, E.M. and Mandelkow, E. (2008) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem., 283, 32066–32076. [DOI] [PubMed] [Google Scholar]
- 12. Bibow, S., Ozenne, V., Biernat, J., Blackledge, M., Mandelkow, E. and Zweckstetter, M. (2011) Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J. Am. Chem. Soc., 133, 15842–15845. [DOI] [PubMed] [Google Scholar]
- 13. Sahara, N., Maeda, S., Murayama, M., Suzuki, T., Dohmae, N., Yen, S.H. and Takashima, A. (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci., 25, 3020–3029. [DOI] [PubMed] [Google Scholar]
- 14. Martinho, M., Allegro, D., Huvent, I., Chabaud, C., Etienne, E., Kovacic, H., Guigliarelli, B., Peyrot, V., Landrieu, I., Belle, V. and Barbier, P. (2018) Two Tau binding sites on tubulin revealed by thiol-disulfide exchanges. Sci. Rep., 8, 13846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soeda, Y., Yoshikawa, M., Almeida, O.F., Sumioka, A., Maeda, S., Osada, H., Kondoh, Y., Saito, A., Miyasaka, T., Kimura, T.et al. (2015) Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun., 6, 10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sahara, N., Maeda, S. and Takashima, A. (2008) Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr. Alzheimer Res., 5, 591–598. [DOI] [PubMed] [Google Scholar]
- 17. Butterfield, D.A. and Boyd-Kimball, D. (2018) Oxidative stress, amyloid-beta peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer's disease. J. Alzheimers Dis., 62, 1345–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bateman, J.R., Lee, A.M. and Wu, C.T. (2006) Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics, 173, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wittmann, C.W., Wszolek, M.F., Shulman, J.M., Salvaterra, P.M., Lewis, J., Hutton, M. and Feany, M.B. (2001) Tauopathy in drosophila: neurodegeneration without neurofibrillary tangles. Science, 293, 711–714. [DOI] [PubMed] [Google Scholar]
- 20. Jackson, G.R., Wiedau-Pazos, M., Sang, T.K., Wagle, N., Brown, C.A., Massachi, S. and Geschwind, D.H. (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron, 34, 509–519. [DOI] [PubMed] [Google Scholar]
- 21. Iijima-Ando, K., Sekiya, M., Maruko-Otake, A., Ohtake, Y., Suzuki, E., Lu, B. and Iijima, K.M. (2012) Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer's disease-related tau phosphorylation via PAR-1. PLoS Genet., 8, e1002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenberg, K.J., Ross, J.L., Feinstein, H.E., Feinstein, S.C. and Israelachvili, J. (2008) Complementary dimerization of microtubule-associated tau protein: implications for microtubule bundling and tau-mediated pathogenesis. Proc. Natl. Acad. Sci. USA, 105, 7445–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhattacharya, K., Rank, K.B., Evans, D.B. and Sharma, S.K. (2001) Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem. Biophys. Res. Commun., 285, 20–26. [DOI] [PubMed] [Google Scholar]
- 24. Giannoni, E., Buricchi, F., Raugei, G., Ramponi, G. and Chiarugi, P. (2005) Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol., 25, 6391–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burgoyne, J.R., Oviosu, O. and Eaton, P. (2013) The PEG-switch assay: a fast semi-quantitative method to determine protein reversible cysteine oxidation. J. Pharmacol. Toxicol. Methods, 68, 297–301. [DOI] [PubMed] [Google Scholar]
- 26. Lee, Y.J. and Chang, G.D. (2019) Quantitative display of the redox status of proteins with maleimide-polyethylene glycol tagging. Electrophoresis, 40, 491–498. [DOI] [PubMed] [Google Scholar]
- 27. Zhu, X., Raina, A.K., Lee, H.G., Casadesus, G., Smith, M.A. and Perry, G. (2004) Oxidative stress signalling in Alzheimer's disease. Brain Res., 1000, 32–39. [DOI] [PubMed] [Google Scholar]
- 28. Dias-Santagata, D., Fulga, T.A., Duttaroy, A. and Feany, M.B. (2007) Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest., 117, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clausen, A., Xu, X., Bi, X. and Baudry, M. (2012) Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer's disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J. Alzheimers Dis., 30, 183–208. [DOI] [PubMed] [Google Scholar]
- 30. Cumming, R.C., Andon, N.L., Haynes, P.A., Park, M., Fischer, W.H. and Schubert, D. (2004) Protein disulfide bond formation in the cytoplasm during oxidative stress. J. Biol. Chem., 279, 21749–21758. [DOI] [PubMed] [Google Scholar]
- 31. Watson, M.R., Lagow, R.D., Xu, K., Zhang, B. and Bonini, N.M. (2008) A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem., 283, 24972–24981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tzou, F.Y., Su, T.Y., Lin, W.S., Kuo, H.C., Yu, Y.L., Yeh, Y.H., Liu, C.C., Kuo, C.H., Huang, S.Y. and Chan, C.C. (2021) Dihydroceramide desaturase regulates the compartmentalization of Rac1 for neuronal oxidative stress. Cell Rep., 35, 108972. [DOI] [PubMed] [Google Scholar]
- 33. Miyasaka, T., Sato, S., Tatebayashi, Y. and Takashima, A. (2010) Microtubule destruction induces tau liberation and its subsequent phosphorylation. FEBS Lett., 584, 3227–3232. [DOI] [PubMed] [Google Scholar]
- 34. Fujiwara, H., Watanabe, S., Iwata, M., Ueda, S., Nobuhara, M., Wada-Kakuda, S., Misonou, H. and Miyasaka, T. (2020) Inhibition of microtubule assembly competent tubulin synthesis leads to accumulation of phosphorylated tau in neuronal cell bodies. Biochem. Biophys. Res. Commun., 521, 779–785. [DOI] [PubMed] [Google Scholar]
- 35. Iwata, M., Watanabe, S., Yamane, A., Miyasaka, T. and Misonou, H. (2019) Regulatory mechanisms for the axonal localization of tau protein in neurons. Mol. Biol. Cell, 30, 2441–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kubo, A., Ueda, S., Yamane, A., Wada-Kakuda, S., Narita, M., Matsuyama, M., Nomori, A., Takashima, A., Kato, T., Onodera, O.et al. (2019) Ectopic expression induces abnormal Somatodendritic distribution of tau in the mouse brain. J. Neurosci., 39, 6781–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fitzpatrick, A.W.P., Falcon, B., He, S., Murzin, A.G., Murshudov, G., Garringer, H.J., Crowther, R.A., Ghetti, B., Goedert, M. and Scheres, S.H.W. (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Passarella, D. and Goedert, M. (2018) Beta-sheet assembly of tau and neurodegeneration in Drosophila melanogaster. Neurobiol. Aging, 72, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prifti, E., Tsakiri, E.N., Vourkou, E., Stamatakis, G., Samiotaki, M. and Papanikolopoulou, K. (2021) The two cysteines of tau protein are functionally distinct and contribute differentially to its pathogenicity in vivo. J. Neurosci., 41, 797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., Miyasaka, T., Murayama, S., Ikai, A. and Takashima, A. (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry, 46, 3856–3861. [DOI] [PubMed] [Google Scholar]
- 41. Sevier, C.S. and Kaiser, C.A. (2002) Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol., 3, 836–847. [DOI] [PubMed] [Google Scholar]
- 42. Diao, Y., Liu, W., Wong, C.C., Wang, X., Lee, K., Cheung, P.Y., Pan, L., Xu, T., Han, J., Yates, J.R., 3rd, Zhang, M. and Wu, Z. (2010) Oxidation-induced intramolecular disulfide bond inactivates mitogen-activated protein kinase kinase 6 by inhibiting ATP binding. Proc. Natl. Acad. Sci. USA, 107, 20974–20979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sillen, A., Barbier, P., Landrieu, I., Lefebvre, S., Wieruszeski, J.M., Leroy, A., Peyrot, V. and Lippens, G. (2007) NMR investigation of the interaction between the neuronal protein tau and the microtubules. Biochemistry, 46, 3055–3064. [DOI] [PubMed] [Google Scholar]
- 44. Kar, S., Fan, J., Smith, M.J., Goedert, M. and Amos, L.A. (2003) Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J., 22, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Melo, A.M., Coraor, J., Alpha-Cobb, G., Elbaum-Garfinkle, S., Nath, A. and Rhoades, E. (2016) A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. USA, 113, 14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dujardin, S., Commins, C., Lathuiliere, A., Beerepoot, P., Fernandes, A.R., Kamath, T.V., de Los Santos, M.B., Klickstein, N., Corjuc, D.L., Corjuc, B.T.et al. (2020) Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat. Med., 26, 1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ueno, H., Murayama, O., Maeda, S., Sahara, N., Park, J.M., Murayama, M., Sanda, A., Iwahashi, K., Matsuda, M. and Takashima, A. (2007) Novel conformation-sensitive antibodies specific to three- and four-repeat tau. Biochem. Biophys. Res. Commun., 358, 602–607. [DOI] [PubMed] [Google Scholar]
- 48. Saito, T., Oba, T., Shimizu, S., Asada, A., Iijima, K.M. and Ando, K. (2019) Cdk5 increases MARK4 activity and augments pathological tau accumulation and toxicity through tau phosphorylation at Ser262. Hum. Mol. Genet. in press, 28, 3062–3071. [DOI] [PubMed] [Google Scholar]
- 49. Xie, C., Soeda, Y., Shinzaki, Y., In, Y., Tomoo, K., Ihara, Y. and Miyasaka, T. (2015) Identification of key amino acids responsible for the distinct aggregation properties of microtubule-associated protein 2 and tau. J Neurochem, 135, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.