

Abstract
Serine/arginine splicing factor 10 (SRSF10) is a member of the family of mammalian splicing regulators known as SR proteins. Like several of its SR siblings, the SRSF10 protein is composed of an RNA binding domain (RRM) and of arginine and serine-rich auxiliary domains (RS) that guide interactions with other proteins. The phosphorylation status of SRSF10 is of paramount importance for its activity and is subjected to changes during mitosis, heat-shock, and DNA damage. SRSF10 overexpression has functional consequences in a growing list of cancers. By controlling the alternative splicing of specific transcripts, SRSF10 has also been implicated in glucose, fat, and cholesterol metabolism, in the development of the embryonic heart, and in neurological processes. SRSF10 is also important for the proper expression and processing of HIV-1 and other viral transcripts. We discuss how SRSF10 could become a potentially appealing therapeutic target to combat cancer and viral infections.
Keywords: RNA binding proteins, SR proteins, alternative splicing, splicing, stress
INTRODUCTION
The vast majority of human genes use alternative pre-mRNA splicing (AS) to generate multiple protein variants often with distinct functions. Modulation of AS can also lead to RNA degradation by multiple mechanisms. In some cases, AS will produce mRNA variants carrying premature stop codons (PTCs) that will elicit degradation by nonsense-mediated RNA decay (NMD), thereby preventing protein production (Lykke-Andersen and Jensen 2015). Splicing of cryptic introns or repression of splicing, leading to intron retention, may also result in RNA decay by the nuclear RNA exosome (Kilchert et al. 2016). The contributions of AS extend to all cellular processes, and important biological roles of AS have been documented from cell fate determination to brain development (Gabut et al. 2011; Han et al. 2013; Venables et al. 2013; Su et al. 2018; Yamazaki et al. 2018). In addition, alterations in AS can contribute to a variety of diseases including cancer (Shkreta et al. 2013; Chabot and Shkreta 2016). Recent work also suggests that the intestinal microbiome of individuals with autism spectrum disorders may produce molecules that alter AS in the brain (Sharon et al. 2019).
Continued progress on our understanding of AS has been achieved by identifying the RNA binding proteins (RBPs) that modulate splice site selection by recruiting components of the generic splicing machinery (the spliceosome) or by preventing spliceosome assembly (Fu and Ares 2014; Van Nostrand et al. 2020). Among such RBPs, SR proteins often interact with enhancer elements found in alternative exons to antagonize the activity of nearby silencer elements whose activities are often mediated by hnRNP proteins such as A1, K, and PTBP1 (Martinez-Contreras et al. 2007; Michelle et al. 2011). These respective positive and negative functions for SR and hnRNP proteins are often reversed when they bind to introns. While RBPs appear to coexist in complexes (Damianov et al. 2016; Gueroussov et al. 2017), the rules of assembly and how their interactions coordinate splicing control are poorly understood. Likewise, how these interactions are modulated by stress and environmental cues is only beginning to be investigated.
Another layer of splicing control is provided by coupling splicing decisions with transcription. Histone marks and chromatin remodeling factors impact splice site selection by altering the speed of transcription and the recruitment of splicing regulators (Naftelberg et al. 2015). This link between transcription and splicing is complex because RBPs can function in transcription (Moumen et al. 2005; Ji et al. 2013), transcription factors can directly control splicing decisions (Han et al. 2017), and the availability of splicing factors can influence the dynamic release of a nascent transcript from its gene (Hochberg-Laufer et al. 2019).
Although SR proteins and SR-like proteins were the first group of splicing regulators identified in Drosophila and then mammals (Baker 1989; Ge and Manley 1990; Krainer et al. 1990), they have also been implicated in nuclear-export of mature mRNAs, mRNA decay, transcription, translation, and genome stability (Howard and Sanford 2015; Jeong 2017). In contrast to other SR proteins, SRSF10 was first identified as a factor with inhibitory splicing activity, and later shown to also be capable of positive contributions. Our recent appreciation of what SRSF10 can achieve is now revealing an even greater level of regulatory complexity (in addition to the fact that SRSF10 probably holds the title for the splicing regulator with the largest number of aliases—i.e., SRrp40, SRp38, FUSIP, NSSR1, TASR2, and SFRS13A).
While the more archetypical SR proteins SRSF1 and SRSF2 make several critical contributions to a variety of biological processes including, respectively, oncogenesis and organogenesis (Xiao et al. 2007b; Das and Krainer 2014), the atypical SRSF10 is emerging as a similarly talented SR protein whose various contributions will be reviewed here. First, we review structural features of SRSF10 and describe the growing list of its splicing variants. We then summarize some of the studies that have led to our current understanding of how SRSF10 modulates splice site selection. Finally, we describe the many contributions of SRSF10 to cellular pathways, organ function and viral replication. We conclude our review by pointing out areas that need further research and highlighting features that position SRSF10 as a potential target for clinical applications.
STRUCTURE OF SRSF10
Transcript variants and protein isoforms
The human SRSF10 pre-messenger RNA transcript produces two main mRNAs resulting from splicing to the 3′ splice site (ss) of exon 7a or 7d to yield, respectively, SRSF10-2 FL (262 aa) and the smaller SRSF10-1 (183 aa) protein (Fig. 1A). Most functional work published on SRSF10 concerns SRSF10-2 FL, as confirmed by immunoblot analysis. However, mRNA expression studies are often an aggregate of multiple variants. The other splice variants include SRSF10-9, which is produced by joining exon 7a to the 3′ss of 7d, skipping the 7b and 7c portions, to yield a shortened RS2 domain (Fig. 1A). The relative mRNA expression of SRSF10-2 and SRSF10-1 differ in human tissues, with SRSF10-2 being abundant in brain tissues, and SRSF10-1 being more predominant in tissues like the adrenal gland and liver (Frederiksen et al. 2021). The production of proteins from mRNA variants SRSF10-4 and SRSF10-8 (exons 6 to 7c splicing) remains to be confirmed. The alternative splicing of a NAGNAG type 3′ss in E6 leads to the absence of a serine in the RS1 domain in the SRSF10-3, SRSF10-5, and SRSF10-8 proteins (Fig. 1A). The SRSF10 pre-mRNA also contains two competing 5′ss in exon 2 (E2) (Meinke et al. 2020); the upstream one is recognized by the minor spliceosome which splices it to E4 to produce the full-length protein coding mRNA, whereas the downstream 5′ss used for E2a is spliced to exon E3 by the major spliceosome. Full-length SRSF10 autoregulates its own production by promoting E2a/E3 splicing (Meinke et al. 2020). The E2a|E3-containing variant SRSF10-7 carries a premature termination codon (PTC) located less than 50 nt upstream of the E2a/E3 junction that may generate a shorter protein with only the RRM domain (Fig. 1A,B). While this PTC may trigger NMD on transcripts that have also undergone exons 3/4 splicing, transcripts made by use of the alternative polyadenylation site in exon 3 (Fig. 1B) appear less affected (Neumann et al. 2020). Notably, relative to 34°C, E2a/E3 splicing decreases at 38°C in both mouse hepatocytes and human HEK293 cells, possibly indicating decreased phosphorylation of SRSF10 by the temperature-dependent CLK kinases (Haltenhof et al. 2020). This striking temperature-driven alternative splicing control of mRNA decay has been observed for many RBPs and constitutes an important layer of rhythmic gene expression (Neumann et al. 2020).
FIGURE 1.
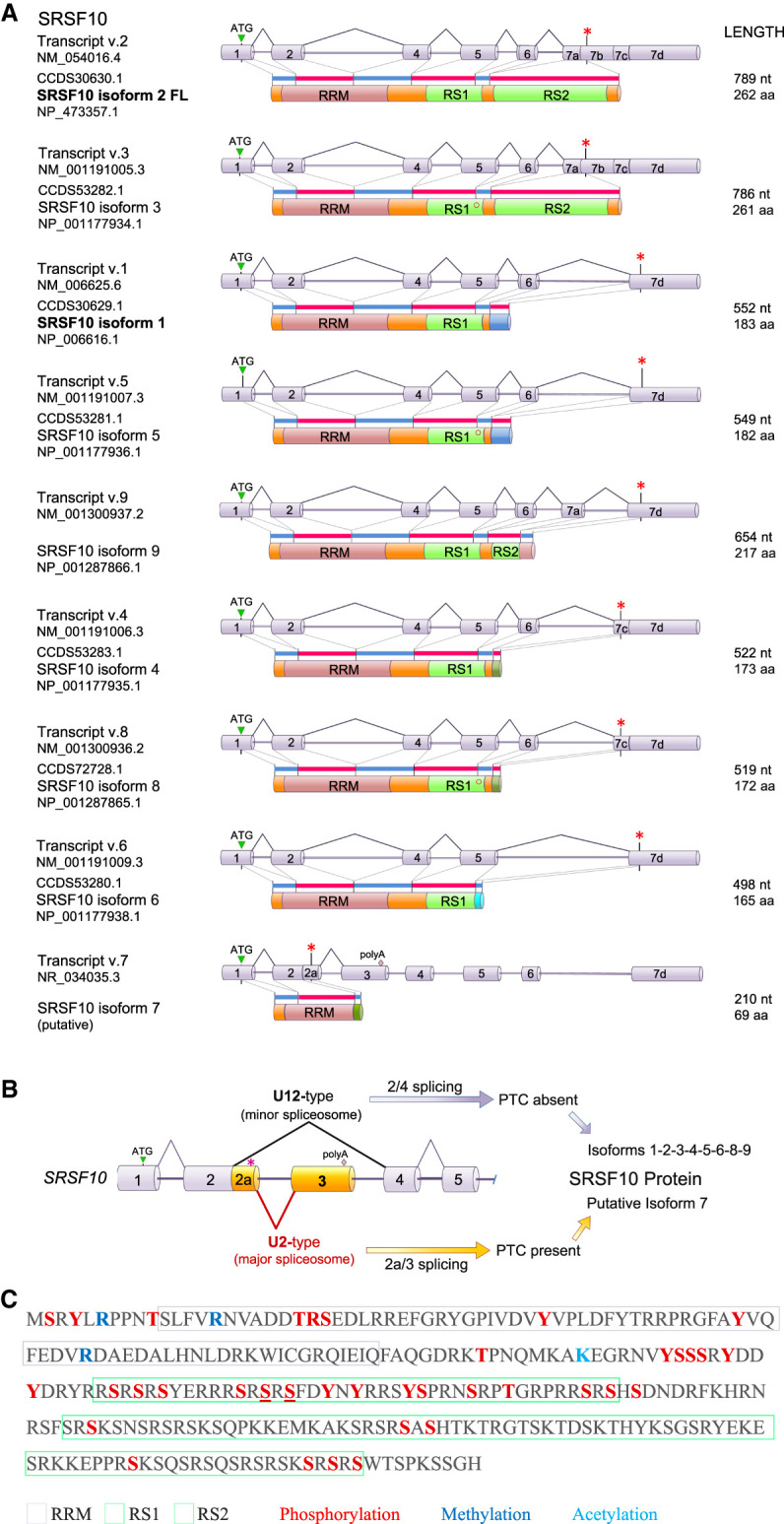
Structure of the human SRSF10 gene, transcripts and SRSF10 proteins. (A) The structure of SRSF10 mRNA variants produced by alternative splicing and corresponding protein isoform structures are shown. The length in nucleotides (nt) of the mRNA and the coding portion in amino acids (aa) are indicated, as well as their RefSeq accession numbers. Stop codons are indicated by asterisks (*). Exon 6 can be alternatively spliced to produce two forms; the shorter one lacking a serine in the RS1 domain (o). (B) Structure of the poison-exon region of SRSF10. The premature termination codon (PTC shown as *) is in the exon 2a region. (C) Known post-translational modifications on SRSF10 based on data compiled in https://www.phosphosite.org/proteinAction?id=6181&showAllSites=true.
Protein domains and post-translational modifications
The main longer SRSF10-2 protein is perfectly conserved in mammals and is made up of three modules (Fig. 1C). The first module is an amino-terminal RNA recognition motif (RRM) that is responsible for binding directly to sequence motifs such as AGAGAV (V = A or G or C) (Ray et al. 2009, 2013) or AAAGACAAA (Shin and Manley 2002). Two adjacent RS domains (RS1 and RS2) form the central and carboxy-terminal modules. The RS1 domain is relatively small (∼60 residues) and bears the greatest similarity to typical RS domains. The RS2 domain (∼90 residues) only contains four RS tetrapeptides (Fig. 1C). While sequence binding elements are usually found at or near regulated exons, other non-GA-rich motifs, such as AC-rich motifs are overrepresented around SRSF10-regulated exons (Wu et al. 2018). This may indicate that SRSF10 cooperates with other RNA-binding proteins to regulate alternative exon splicing. A cooperative mode would be consistent with the emerging mechanism of action for other splicing regulators that function as part of multi-RBP assemblies (Damianov et al. 2016; Gueroussov et al. 2017).
SRSF10 is phosphorylated at serines, threonines and tyrosines (Fig. 1C), but it is unclear how many phosphorylation events can occur on a single protein. Phosphorylation can however be extensive enough to noticeably change the gel migration of SRSF10 when compared to a dephosphorylated version (Shi and Manley 2007). The kinases that use SRSF10 as a substrate include the SRPK and CLK kinases as well as DYRK1A (Shi and Manley 2007; Ninomiya et al. 2011; Sohail et al. 2021). SRPK1 and CLK1 display a symbiotic relationship in phosphorylating SR proteins. Following phosphorylation of SRSF1 by CLK1, the release of SRSF1 requires SRPK1 (Aubol et al. 2016). Whether this coordination applies to SRSF10 remains unknown. The protein phosphatase PP1 is the only enzyme known to dephosphorylate SRSF10 (Shi and Manley 2007). Mass spectrometry analysis has also identified acetylated and methylated residues in SRSF10 (Fig. 1C) whose contributions to function remain unclear.
MECHANISM OF ACTION OF SRSF10: FROM NEGATIVE TO POSITIVE
SRSF10 as a splicing repressor
The first report of a role for SRSF10 in splicing came from in vitro splicing assays. The in vitro splicing of a single-intron containing pre-mRNA was repressed by baculovirus-produced SRSF10 (Cowper et al. 2001). SRSF10 also promoted distal 5′ss selection on a substrate that contained two competing 5′ss (Cowper et al. 2001). This result contrasts with the impact of typical SR proteins, such as SRSF1, which by stimulating U1 snRNP binding at both competing 5′ss would lead to a net increase in the use of the proximal 5′ss (Chabot 1996). The splicing inhibition caused by SRSF10 was likely due to an interaction between dephosphorylated SRSF10 (dSRSF10) and U1 snRNP, and possibly other snRNPs. This interaction would interfere with the stable binding of U1 snRNP to a 5′ss (Shin et al. 2004). A general reduction in the stability of U1 snRNP binding by dSRSF10 can therefore explain why SRSF10 can inhibit splicing of a single-intron pre-mRNA, and improves net distal 5′ss usage on a substrate carrying competing 5′ss (Fig. 2A). Because a typical SR protein like SRSF1 normally improves U1 snRNP binding (Eperon et al. 1993), and because SRSF10 has a dominant negative impact on the activity of SR proteins in in vitro splicing assays (Cowper et al. 2001), SRSF10 first became known as a protein that antagonizes the function of typical SR proteins.
FIGURE 2.
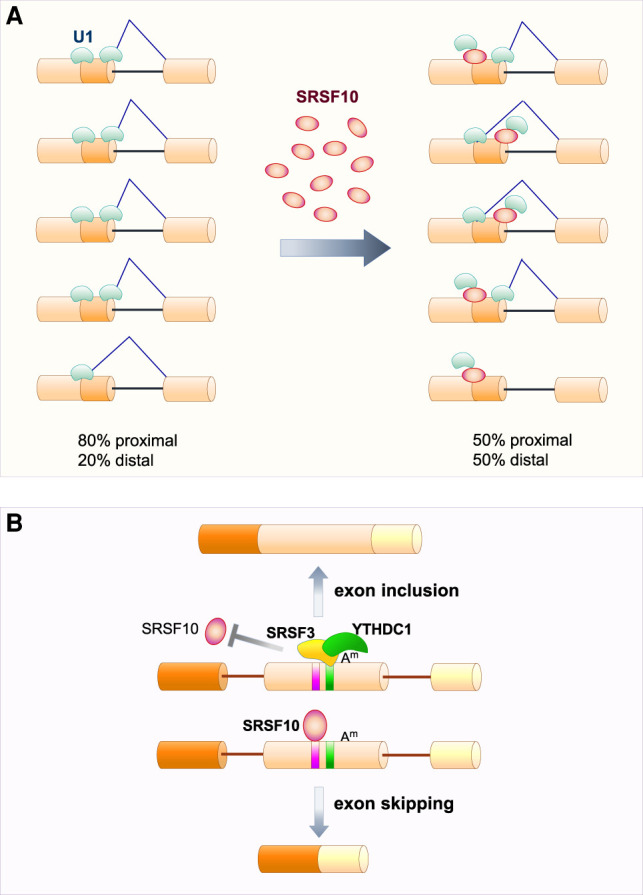
SRSF10 as a splicing repressor. (A) In a competitive environment, blocking splicing can shift splice site selection. When two 5′ splice sites are bound by U1 snRNP, the one that is most proximal to the 3′ splice site is used preferentially. SRSF10 binding can destabilize the interaction of U1 snRNP with 5′ splice sites to prevent splicing and therefore shift overall splicing to the distal 5′ splice site. (B) The exonic m6A (Am) is bound by the reader protein YTHDC1, that in turn recruits and stabilizes SRSF3 binding to promote exon inclusion by blocking SRSF10 binding which normally promotes exon skipping.
During mitosis and in response to heat-shock (43°C–45°C), SRSF10 becomes dephosphorylated. In vitro studies using extracts and artificial pre-mRNAs indicated that dephosphorylation converts SRSF10 into a general splicing repressor (Shin and Manley 2002; Shin et al. 2004). In reverse, phosphorylation switches SRSF10 from a repressor to a splicing activator (Feng et al. 2008). Baculovirus-produced SRSF10 lacking the RS2 domain (and thus similar in structure to SRSF10-1) remains inhibitory, and inhibition is stimulated by dephosphorylation, indicating that the RS1 domain carries the signal for dephosphorylation-dependent inhibition (Shin et al. 2005). In contrast, a derivative lacking the RS1 domain displays weak repressor activity mostly on the second step of splicing, and this repression is not affected by dephosphorylation (Shin et al. 2005). Removing both RS domains is required to eliminate full repressor activity (Shin et al. 2005). The fact that the hybrid protein SRSF2-RRM/SRSF10-RS can function as a general splicing activator indicates that inhibition by the RS domain of SRSF10 is dictated by the identity of the RRM (Shin et al. 2005). Consistent with this view, the RRM of SRSF10 is unable to provide general splicing activation when it is covalently linked to a RS domain that is acting positively in a different context (Shin et al. 2005).
SRSF10 recruitment to an exon can also promote its skipping. In general, the genetic depletion of SRSF10 promotes a higher number of exon inclusion events than exon skipping events, especially in human cells (i.e., inclusion events occur two to four times more frequently than skipping events) (Xiao et al. 2016; Wu et al. 2018). It is unclear if modulating snRNP binding, as suggested by in vitro work, is responsible for these effects. A specific rescue of this negative impact occurs when target pre-mRNAs contain an alternative exon subjected to N6-methyladenosine (m6A) modification. Here, m6A recruits the reader protein YTHDC1 along with SRSF3 to prevent SRSF10 binding and exon skipping (Fig. 2B; Xiao et al. 2016).
SRSF10 as a splicing activator
Upon phosphorylation, SRSF10 can function as a positive splicing regulator (Feng et al. 2008). This activity does not need other SR proteins but a cofactor that remains to be identified is required to help transition spliceosome assembly from complex A to complexes B and C. Mechanistically, phosphorylated SRSF10 strengthens the ability of the U1 and U2 snRNPs to stably recognize splicing signals on the pre-mRNA. Thus, both the negative and positive activities of SRSF10 converge on snRNP binding; dephosphorylated SRSF10 promoting poor U1 binding, while the reverse occurs with phosphorylated SRSF10.
In contrast to the above characterizations, the dephosphorylation of SRSF10 has also been associated with a positive splicing function in Bcl-x pre-mRNA splicing (Shkreta et al. 2016). Through alternative 5′ss usage, Bcl-x produces two splicing variants: pro-apoptotic Bcl-xS and anti-apoptotic Bcl-xL (Shkreta et al. 2015). In normal growth conditions, SRSF10 only weakly activates the 5′ss of Bcl-xS. Overexpression of a HA-tagged SRSF10 or of a variant lacking the RS2 domain in 293 cells provides strong stimulation of the Bcl-xS 5′ss, while a variant lacking the RS1 domain is inactive (Shkreta et al. 2016). DNA damage induced by oxaliplatin promotes the dephosphorylation of serines in the RS1 domain of SRSF10, and strongly activates the 5′ss of Bcl-xS. This splicing change is associated with a stronger interaction of SRSF10 with hnRNP K (a repressor of the Bcl-xS 5′ss) (Revil et al. 2009) and a dissociation of both SRSF10 and hnRNP K from the Bcl-x pre-mRNA, allowing the positive regulatory hnRNP F and H proteins to bind and increase access of U1 snRNP to the Bcl-xS 5′ss (Fig. 3A; Garneau et al. 2005; Revil et al. 2009; Dominguez et al. 2010; Shkreta et al. 2016). DNA damage by oxaliplatin also increases the splicing modulatory activity of SRSF10 on RBBP8 and PCBP4 transcripts (Shkreta et al. 2016). The impact of oxaliplatin on the activity of SRSF10 can differ on other transcripts. For example, treating cells with oxaliplatin co-opts SRSF10 into controlling the alternative splicing of TNFRSF10B, SPTAN1, MLH3, CASP8, CHK2, DOM3Z, and BRCA1 (Shkreta et al. 2016; Cloutier et al. 2018). In other cases, such as CASP9, oxaliplatin converts SRSF10 from acting as a repressor into an activator (Cloutier et al. 2018).
FIGURE 3.

SRSF10 as a splicing activator. (A) Control of Bcl-x splicing by SRSF10. Phosphorylated SRSF10 has minimal impact on Bcl-x splicing in normal growth conditions in 293 cells. Following DNA damage by oxaliplatin, the dephosphorylation of SRSF10 increases the interaction with the repressor hnRNP K and their dissociation from the Bcl-x pre-mRNA, allowing hnRNP F/H to disrupt G-quadruplexes and favor use of the 5′ splice site of Bcl-xS. (B) Global positive and negative effects on exon inclusion can be explained by SRSF10 binding and stimulating splice site usage either on the alternative (on the right) or a constitutive (on the left) exon.
A study performed in chicken cells indicates that the impact of SRSF10 on alternative splicing depends on the position of putative SRSF10 binding motifs on the target pre-mRNAs (Zhou et al. 2014b). In a manner reminiscent to authentic SR proteins, when SRSF10 binds to cassette exons, it tends to promote their inclusion, while its binding to the downstream constitutive exon is associated with exclusion of the upstream alternative exon. The insertion of SRSF10 consensus binding sequences within an SRSF10-repressed exon was sufficient to induce inclusion (Zhou et al. 2014b). These different outcomes can be explained by the positive effect that SRSF10 exerts on the splice site that is nearest to where SRSF10 putatively binds (Fig. 3B; Zhou et al. 2014b). In the SMN2 transcript, SRSF10 binds in the intron downstream from exon 7B to repress its inclusion (see below) (Frederiksen et al. 2021). Overall, while the phosphorylation status of SRSF10 appears to determine its function as a repressor or a stimulator of constitutive splicing, this distinction becomes more complex when dealing with alternatively spliced pre-mRNAs in which the binding position of SRSF10 relative to splice sites and other regulatory elements also makes a strong contribution.
SRSF10 as a master regulator of splicing factors
The importance of producing adequate amounts of splicing regulatory proteins such as SR proteins and hnRNP proteins is underscored by the presence of exonic ultraconserved elements in several of their cognate genes (Ni et al. 2007). This is also true for SRSF10. These ultraconserved elements are part of a program that controls alternative splicing to produce mRNAs that are subjected to degradation through NMD. In this regulatory pathway, a high level of a splicing activator protein will promote inclusion of an alternative poison exon carrying a stop codon in the cognate transcript, triggering NMD. Alternatively, a high level of a splicing repressor protein will repress the inclusion of a coding exon, creating a frameshift and the inclusion of PTC that will trigger NMD. Several cases of splicing factor autoregulation of splicing have been documented (Muller-McNicoll et al. 2019), as well as cases of splicing factors regulating the alternative splicing of other regulators (Sun et al. 2017; Desai et al. 2020). Notably, the expression of 14 SR proteins is coordinated through splicing of their poison exons during cell differentiation and tumorigenesis (Leclair et al. 2020). Two studies have uncovered a predominant role for SRSF10 in autoregulation and in cross-splicing regulation of other SR proteins (Leclair et al. 2020; Meinke et al. 2020). Specifically, in mice, blocking feedback regulation by preventing production of the inactive PTC-containing variant Srsf10-7 leads to an overexpression of Srsf10, and of all other SR proteins (Meinke et al. 2020). Correspondingly, overexpression of Srsf10 elicits the production of the PTC-containing Srsf10 variant (Meinke et al., 2020), while human SRSF10 provokes the skipping of poison exons in several SR protein transcripts (Leclair et al. 2020). On the other hand, SRSF4 and SRSF11 elicit poison exon skipping in SRSF10, while SRSF9 and hTRA2β promote its inclusion (Leclair et al. 2020).
ROLES OF SRSF10 IN CELLS AND ORGANS
SRSF10 is an atypical SR protein because it is not required for generic splicing in vitro. Moreover, the genetic ablation of SRSF10 leads to late embryonic lethality, indicating that mouse embryonic development is nearly normal and that most cells can survive the loss of SRSF10 (Feng et al. 2009). Nevertheless, SRSF10 may have specialized cellular functions. One way of identifying important processes requiring SRSF10 is by discovering genes or groups of genes that use SRSF10 as a splicing regulator. SRSF10 was initially uncovered as a splicing repressor during mitosis and during heat shock. A later study in chicken DT40 cells indicated that SRSF10 affected the alternative splicing of transcripts involved in multiple processes (Zhou et al. 2014c). Mice embryonic fibroblasts and human RKO cells show more comparable SRSF10-regulated AS events than chicken DT40 cells (Wu et al. 2018). Common orthologous SRSF10-regulated events occurring in human, mouse and chicken cells include SRSF2, ABI2, KIF23, ATXN2, NASP, BCLAF1, and ACTB. A larger set of SRSF10-regulated genes indicates that SRSF10 controls the splicing of vertebrate transcripts encoding components involved in chromosome organization, RNA processing and the cytoskeleton network (Wu et al. 2018). The functional contribution of these SRSF10-regulated splicing events is unclear given that they are unlikely to be required for survival in most cells, at least in mouse.
While several pathways are affected by the depletion of SRSF10, many of SRSF10-confirmed alternative exons have been linked to stress response and apoptosis. Consistent with this observation, cells depleted of SRSF10 are more susceptible than control cells to endoplasmic reticulum (ER) stress-induced apoptosis (Zhou et al. 2014b). Importantly, restoring SRSF10 expression in knockout cells recovers wild-type splicing patterns and rescues many of the stress-related defects. Notably, and in contrast to heat-shock, the ER-stress-induced response was not associated with the dephosphorylation of SRSF10.
Thus, while SRSF10 may not make a big impact in the daily life of a cell in most organs, it may be critical for the function of some organs or cell types, or it may accomplish specific roles when cells are subjected to stress. Cellular localization can also point to specific functions: SRSF10 shuttles between the nucleus and the cytoplasm. SRSF10 accumulates in the cytoplasm when cells are treated with the transcription inhibitor actinomycin D (Cowper et al. 2001). SRSF10 has also been identified as an important component of paraspeckles (Hennig et al. 2015), which are responsive to a variety of stresses including viral infection. After heat shock, a fraction of SRSF10 is found in cytoplasmic granules that also contain snRNPs (Shin et al. 2005). These SRSF10-containing cytoplasmic stress granules communicate with the nuclear paraspeckles (An et al. 2019). Other SR proteins like SRSF1 and SRSF3 can function as regulators of protein synthesis in the cytoplasm (Kim et al. 2014; Maslon et al. 2014). No such specific function has yet been discovered for SRSF10, although a role for SRSF10 in ribosome biogenesis or in translation has been proposed based on the observation that SRSF10 can interact with the peptidyltransferase center of 28S rRNA (Liu and Harland 2005).
Another way through which SRSF10 may affect cell function is if the SRSF10 gene sustains mutations or if its expression becomes altered. Many SRSF10 mutations have been reported but with unclear impact on function. Changes in the expression of SRSF10 have also been noted between tissues and in specific diseases (see below). While the causes for such differences in expression remain to be ascertained, the changes could be driven by transcription, or they could be mediated by alternative splicing coupled to NMD. As indicated above, SRSF4, SRSF9, SRSF11 and hTRA2β affect poison exon skipping in SRSF10 (Leclair et al. 2020). Thus, cell or disease-specific differences in their abundance or activity would be expected to impact the steady-state level of SRSF10. In addition, a change in the ratio of phosphorylated and dephosphorylated SRSF10 may also affect alternative splicing with functional consequences. To illustrate the growing functional relevance of SRSF10 in many biological systems, we will now review some of the most telling observations.
Repression of splicing during mitosis
The dephosphorylation of SRSF10 occurs in mitotic cells grown in culture and may participate in the overall splicing repression that is observed in extracts made from such cells (Shin and Manley 2002). It is unclear if the phosphorylation status of SRSF10 can be used as a marker to distinguish differentiated versus proliferative tissues. At least in differentiated human hepatocytes, SRSF10 is phosphorylated and sensitive to compounds inhibiting this post-translational modification (Chabrolles et al. 2020). Whether mitotic splicing repression by SRSF10 is essential also remains an open question since embryos lacking SRSF10 develop almost normally (Feng et al. 2009). Notably, the SRPK1 kinase is highly active during mitosis (Gui et al. 1994), raising the possibility that during mitosis CLK kinases might be suppressed or that the phosphatase PP1 might be activated. While CLK1 expression (protein not mRNA) increases at the G2/M stage of the cell cycle, it is unclear if this change correlates with activity (Dominguez et al. 2016). As for PP1, its activity is coordinately controlled during mitosis (Nasa et al. 2018). Moreover, activation of PP1 is required for mitotic exit and occurs by dissociation of the inhibitor-1 protein from PP1 (Wu et al. 2009). This dissociation and possibly that of the NIPP1 inhibitor may trigger SRSF10 dephosphorylation.
Heat-shock response
Heat stress causes a rapid inhibition of splicing. Following heat-shock, SRSF10 redistributes in the cell nuclei to colocalize with snRNPs (Shin et al. 2005). SRSF10 plays a key role in the impairment of splicing during heat-shock (Shin et al. 2004); as mentioned above, its rapid dephosphorylation allows it to interfere with U1 snRNP binding to the 5′ss. SRSF10 may act as a heat sensor throughout vertebrates, and an SR protein with related properties, RSF1, may have a similar role in flies (Labourier et al. 1999).
Following heat-shock, a splicing recovery program is initiated. Protective proteins that are induced by heat-shock include small heat-shock proteins (sHsps). One study reported that Hsp27 did not prevent splicing inhibition or SRSF10 dephosphorylation by heat-shock at 45°C but enhanced the recovery of splicing, and accelerated rephosphorylation of SRSF10 after heat-shock (Marin-Vinader et al. 2006). Another study indicated that overexpression of Hsp27 interacted with SRSF10 and inhibited its dephosphorylation in response to heat-shock at 44°C (Shi et al. 2011). A one-degree difference may be important here since 45°C leads to a more complete dephosphorylation of SRSF10, which may overwhelm Hsp27. Thus, splicing thermotolerance may be acquired through maintenance of phosphorylated SRSF10, helped in part by Hsp27. Hsp90 also appears to be required but not Hsp70 (Marin-Vinader et al. 2006).
Upon heat-shock, dephosphorylated SR proteins are sequestered in nuclear stress bodies (nSBs) formed on highly repetitive satellite III lncRNAs (Ninomiya et al. 2020). In the recovery phase, the rephosphorylation of SR proteins is mediated by the CLK kinases since exposure to stresses, such as heat-shock and osmotic stress, switches the production of splice variants of CLK1 and CLK4 to active forms that will phosphorylate SRSF10 (Fig. 4; Ninomiya et al. 2011, 2020).
FIGURE 4.

Impact of temperature on SRSF10 activity and feedback regulation of SRSF10 through CLK1 splicing. SRSF10 phosphorylated residues are protected from dephosphorylation by 14-3-3 proteins while the activity of protein phosphatase 1 (PP1) is kept repressed by a complex that contains NIPP1. Upon heat-shock, PP1 dissociates from NIPP1 and SRSF10 dissociates from 14-3-3 proteins, allowing dephosphorylation of SRSF10. CLK kinases and SRPK1 will rephosphorylate SRSF10 as part of the recovery from heat-shock. Homeostatic levels of phosphorylated SRSF10 are controlled through both CLK1 and SRSF10 pre-mRNA splicing.
SRSF10 is dephosphorylated by the phosphatase PP1 (Shi and Manley 2007). Heat-shock promotes the dissociation of PP1 from its inhibitors, including NIPP1. The 14-3-3 proteins that associate with SRSF10 and protect it from dephosphorylation under nonstress conditions, dissociate upon heat-shock (Fig. 4; Shi and Manley 2007). The RS domain of SRSF10 directs the interaction with activated PP1. The level of phosphorylated SRSF10 is expected to drop preferentially following heat-shock because of the low activities of SR protein kinases (e.g., CLKs and SRPKs) for SRSF10 compared to other SR proteins. Moreover, because the CLK kinases are much less active at 37°C–38°C than at 32°C–34°C (Haltenhof et al. 2020), heat-shock at 45°C should reversibly inactivate the CLK kinases, thereby further contributing to the accumulation of dephosphorylated SRSF10 upon heat-shock.
The CLK-SRSF10 relationship revealed by heat-shock allows to underscore the multilevel connections that link SRSF10 and the CLK1 kinase. First, CLK1 phosphorylates SRSF10 in a temperature-dependent manner. Second, because the loss of SRSF10 increases the production of the exon 4-containing active form of CLK1 (Sohail et al. 2021), phosphorylated SRSF10 likely down-regulates the inclusion of exon 4 of CLK1. Third, CLK1 phosphorylation of SRSF10 likely activates exon 2a/3 splicing of Srsf10 to prevent production of full-length SRSF10 protein. CLK1 therefore controls SRSF10 activity and SRSF10 splicing, while SRSF10 phosphorylation by CLK1 controls Srsf10 and CLK1 alternative splicing (Fig. 4).
Apoptosis
In chicken DT40 cells, SRSF10 controls the splicing of transcripts that function in apoptosis, DNA repair, and cell-cycle control: alternative splicing of Bap1 (BRCA1-associated protein), Cdk13 and Casp1 were in the top 12 events controlled by SRSF10 (Zhou et al. 2014b). Likewise, transcripts encoding proteins linked to apoptosis (e.g., BCLAF1 and RAC1) form a top functional category controlled by SRSF10 in human RKO cells (Zhou et al. 2014a). The functional relationship between SRSF10 and apoptosis/cell-cycle is not unique to this SR protein as transcripts whose alternative splicing is disrupted by the depletion or mutation of SRSF1, SRSF2, SRSF3, SRSF6, or SRSF9 also reveal a similar connection (Das and Krainer 2014; Kim et al. 2015; Ajiro et al. 2016; Juan-Mateu et al. 2018; Pellagatti et al. 2018; Song et al. 2019; Ha et al. 2021).
Recent work on Bcl-x splicing suggests a contribution of SRSF10 in favoring apoptosis (Fig. 3A). The 5′ss of pro-apoptotic Bcl-xS is weakened by flanked G-quadruplexes forming elements that interfere with U1 snRNP binding (Dominguez et al. 2010; Weldon et al. 2018). hnRNP F can sequester G-tracts to keep them in a single-stranded conformation (Dominguez et al. 2010), thereby explaining why hnRNP F/H can favor the use of the 5′ss of Bcl-xS (Garneau et al. 2005). On the other hand, hnRNP K represses Bcl-xS 5′ss usage by preventing hnRNP F/H binding (Revil et al. 2009; Shkreta et al. 2016). Treating cells with oxaliplatin leads to the dephosphorylation of SRSF10 which interacts with hnRNP K to dissociate from the pre-mRNA, thereby allowing hnRNP F/H binding and production of pro-apoptotic Bcl-xS (Fig. 3A). This pro-apoptotic function of SRSF10 contrasts with a study indicating that the loss of SRSF10 promotes cell death when combined with G4-stabilizing molecules (Zyner et al. 2019).
Cancer
Cancer-associated splicing changes are caused by mutations in splice sites or splicing regulatory sequences or by mutations that affect the expression or activity of core and regulatory splicing factors (Sveen et al. 2011; Dvinge et al. 2016).
Mutations in the SRSF10 gene
The National Cancer Institute (https://portal.gdc.cancer.gov/), the cBioPortal for Cancer Genomics (https://www.cbioportal.org) and the Sanger Institute (https://cancer.sanger.ac.uk/) all report many types of SRSF10 mutations in patients’ cancer tissues from missense and splice site mutations to gene fusion. Approximately 1% of sampled cancer tissues harbor mutations in SRSF10. A systematic evaluation of the functional impact of these mutations is required to help ascertain their specific contribution to cancer.
Mutations in SRSF10 binding sites
Sequence elements that are potential binding sites for SRSF10 are often found to be mutated in cancer with an enrichment for A to G mutations in motifs located in coding sequences and introns including their 3′ss (Singh et al. 2018). Genes with significantly mutated regions harboring SRSF10 binding motifs showed association to apoptosis and immune response. Mutations in intronic SRSF10 binding motifs have been associated with increased expression of a dystrophin gene transcript in breast cancer, and a splice variant switch in MAPK10 (JNK3) in colorectal cancer (Singh et al. 2018).
Changes in SRSF10 expression
Increased SRSF10 expression is frequently observed in cancer. Among human organs, the thymus and the bone marrow have one of the highest levels of SRSF10 expression, while adult liver has one of the lowest levels (Uhlen et al. 2017). SRSF10 regulates the proliferation of chicken bursal lymphoma DT40 cells (Wu et al. 2018). Scanning cancer expression resources available on the web reveal that SRSF10 expression is generally stronger in cancer relative to normal human tissues. For example, SRSF10 RNA expression is twofold higher in ovarian cancer relative to normal stromal tissues (https://www.ebi.ac.uk/gxa/home). SRSF10 is also overexpressed in Hepatitis B virus-associated hepatocarcinoma (Tremblay et al. 2016). A high level of SRSF10 gene expression was linked with poor survival in patients suffering from prostate, liver or renal cancer (http://www.proteinatlas.org). SRSF10 expression is up-regulated in metastatic colorectal cancer (Zhou et al. 2014a), likely due to a dysregulated Wnt/β-catenin pathway (Zhao et al. 2019). SRSF10 promotes the production of the pro-tumorigenic splice variant BCLAF1-L, and both SRSF10 and BCLAF1-L expression correlate with high tumor grades (Zhou et al. 2014a). Knocking down SRSF10 or BCLAF1-L decreased in vivo tumor growth. In a xenograft model, knockdown of SRSF10 in RKO cells nearly completely eliminated tumor formation in mice (Zhou et al. 2014a). In addition to BCLAF1-L, other SRSF10-regulated splicing targets are also likely contributing to the cancer phenotypes (Zhou et al. 2014a). For example, SRSF10 regulates the alternative splicing of the cancer-associated transcripts MDM4 and SLK in HCT116 cells (Sohail et al. 2021).
SRSF10 up-regulates the production of a circular RNA (Circ-ATXN1) that plays a role in glioma angiogenesis by sequestering miR-526b-3p which normally represses expression of pro-angiogenic MMP2 and VEGFA (Liu et al. 2018).
The human papillomavirus oncoproteins E6 and E7 regulate the expression of SRSF10 in cervical cancer through the E2F1 axis (Liu et al. 2018). Knockdown of SRSF10 in a xenograft mouse model of cervical cancer decreased the tumor growth rate and tumor weight. Thus, SRSF10 promotes tumorigenesis in human papilloma virus 16/18-positive cervical cancer.
Change in SRSF10 phosphorylation
Changes in the phosphorylation of SRSF10 has not been examined in the context of cancer per se. Nevertheless, DNA damage elicited by the anticancer drug oxaliplatin leads to a partial dephosphorylation of SRSF10 at residues that contribute to its splicing regulatory function on Bcl-x, and co-opts SRSF10 into promoting splicing changes in transcripts involved in apoptosis, cell-cycle control and DNA repair (Shkreta et al. 2016). hnRNP A1/A2 and Sam68 cooperate with SRSF10 to control this broader program of splicing response to DNA damage (Cloutier et al. 2018). SRSF10 becomes similarly dephosphorylated when the CLK kinase inhibitor GPS167 is applied to HCT116 colon cancer cells. In this case, cytotoxicity is associated at least in part with changes in the splicing of the SRSF10 targets MDM4 and SYK to elicit p53-dependent apoptosis (Sohail et al. 2021).
Metabolism
Srsf10-knockout mice have defective liver development (Feng et al. 2009). These mice also display significantly decreased glucose production that is also observed in mouse primary hepatocytes, especially during fasting conditions (Wei et al. 2015). PGC1α is a master regulator during gluconeogenesis, and its pre-mRNA is a target of SRSF10 (Fig. 5A). SRSF10 specifically binds to constitutive exon 7 and represses inclusion of downstream alternative exon 7a to produce a functional protein. SRSF10 knockdown increases exon 7a inclusion, concomitantly decreasing glucose production after fasting.
FIGURE 5.
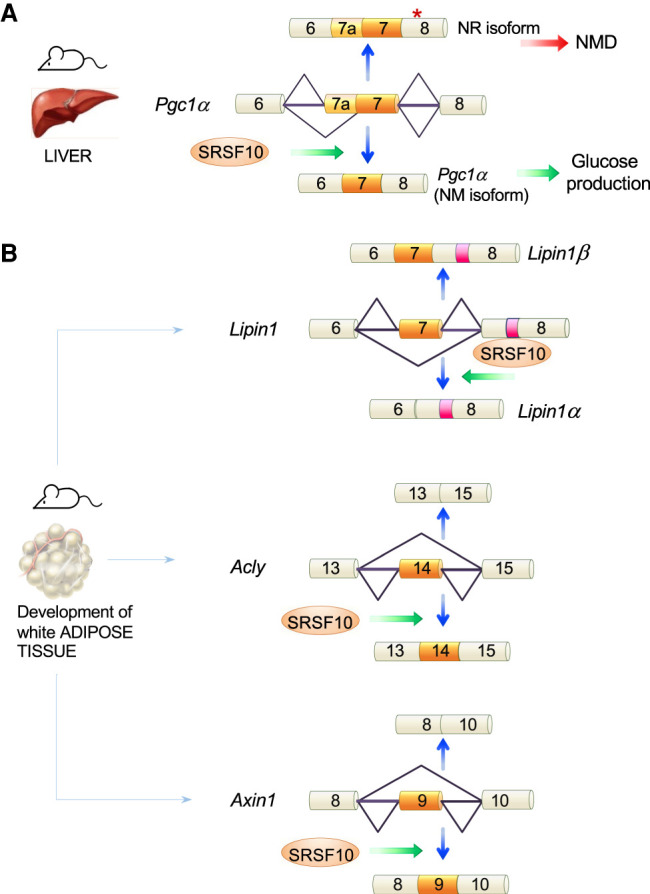
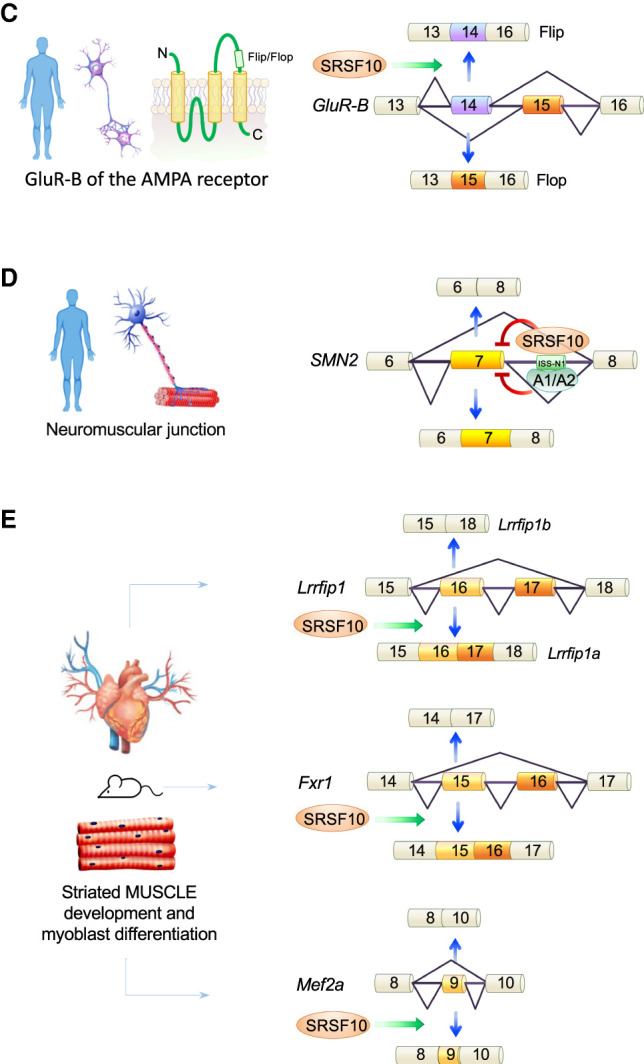
SRSF10 and the control of alternative splicing of key transcripts. SRSF10 controls splice site selection of mammalian transcripts implicated in glucose metabolism (A), in the development of adipose tissue (B), in neuronal (C), and neuromuscular (D) function as well as muscle development (E).
The transcription factor DEAF1 controls the expression of peripheral tissue antigens in lymph node stromal cells. During progression of type 1 diabetes, DEAF1 function is decreased and may contribute to the pathogenesis of disease. In the pancreatic lymph nodes of patients with type 1 diabetes, DEAF1 splicing shifts to produce more of the dominant-negative DEAF1-VAR1 isoform. SRSF10 has been implicated in this splicing regulation along with PTBP2, and overexpression of SRSF10 alone or in combination with PTBP2 increases human DEAF1-VAR1 (Yip et al. 2015).
Mice lacking SRSF10 exhibit severely impaired development of subcutaneous white adipose tissue indicative of a defect in adipogenic differentiation (Li et al. 2014). Many of the severe adipogenic defects can be reproduced in the cell line C3H10T1/2 by knocking down SRSF10, or in mouse embryonic fibroblasts lacking SRSF10 and induced into adipocyte differentiation. The lack of SRSF10 is associated with splicing defects in transcripts that are relevant to adipogenesis including Lipin1, AclY, Axin1, and Upf1 (Fig. 5B; Li et al. 2014).
SRSF10 also controls the alternative splicing of low-density lipoprotein receptor, which is a major apolipoprotein E receptor critical to cholesterol homeostasis and, possibly, Alzheimer's disease (Ling and Estus 2010). SRSF10 similarly emerged as a top candidate regulator of advanced atherosclerosis, and the silencing of SRSF10 reduced the production of cholesterol in a cell culture model (Bjorkegren et al. 2014).
Neuronal tissue differentiation
SRSF10 promotes neuronal differentiation of mouse embryonic carcinoma P19 cells (Liu et al. 2004). SRSF10 has also been implicated in the regulation of neural differentiation in Xenopus laevis (Liu and Harland 2005). Overexpression of SRSF10 inhibits primary neuronal differentiation in a Notch pathway-dependent manner. Conversely, depletion of SRSF10 results in dysregulation of neurogenesis. SRSF10 becomes more expressed in the neural plate during embryogenesis and is transcriptionally induced by the neurogenic bHLH protein neuroD, possibly providing a negative feedback to limit neurogenesis (Liu and Harland 2005).
SRSF10 expression plays a protective role in the brain by reducing neuronal injury after mouse transient global cerebral ischemia (Qi et al. 2015). Expression of SRSF10, SRSF7, SRSF3, TRA2α, as well as the kinase CLK1 are down-regulated during mouse aging in the hippocampal CA1 region. The histone deacetylase inhibitor SAHA partially rescues spatial memory function in aging mice. The almost complete recovery of normal splicing after SAHA treatment has been proposed to be linked to the restored expression of these splicing factors (Benito et al. 2015).
PINK1 gene deficiencies have been associated with Parkinson's disease, and the brain of aging Pink1-deleted mice displays a reduction in Srsf10 expression (Torres-Odio et al. 2017).
SRSF10 regulates in vivo splicing of the GluR-B subunit of the AMPA receptor (Fig. 5C; Komatsu et al. 1999). Using a minigene model in human PFSK-1 cells subjected to heat-shock, dSRSF10 was associated with a change in the alternative splicing of mutually exclusive flip/flop exons in the Glutamate receptor subunit B (GluR-B) (Zhang et al. 2014). Overexpression of SRSF10 promotes flip exon inclusion but depleting SRSF10 in non-heat shock cells has no effect. Increased flip exon inclusion occurs in kainate-induced epilepsy in rats (Pollard et al. 1993). Transfecting an SRSF10ΔRRM derivative did not affect GluR-B splicing, but deleting portions of the RS domains promoted skipping of both exons (Zhang et al. 2014).
In spinal muscular atrophy (SMA), production of a functional survival of motor neuron (SMN) protein is provided by inclusion of exon 7 in the SMN2 transcript. Abolishing the binding of hnRNP A1/A2 with an antisense oligonucleotide that hybridizes to a downstream intron silencer element (ISS-N1) increases exon 7 inclusion and has led to a treatment for SMA (Hua et al. 2008; Chiriboga et al. 2016). A recent study has shown that SRSF10 also binds to ISS-N1 and to other intronic elements to promote exon 7 skipping (Fig. 5D; Frederiksen et al. 2021).
Heart development
Given the disproportionate role of alternative splicing in brain but also in heart development (Mazin et al. 2021), SRSF10 is expected to play an important role in these organs. Srsf10-knockout mice display defective heart and liver development (Feng et al. 2009). The majority of homozygous Srsf10 null mice survive only until day 15.5 of embryonic development and display multiple cardiac defects. SRSF10 is required for myoblast differentiation and cardiac development. Embryonic cardiomyocytes from Srsf10 null mice display defects in calcium handling. SRSF10 controls the alternative splicing of the pre-mRNA encoding cardiac triadin, a protein that regulates calcium release from the sarcoplasmic reticulum during excitation-contraction coupling. This defect results in significantly reduced levels of triadin, and of its interacting protein CASQ2 (Feng et al. 2009). SRSF10 binds specifically to the regulated exon and modulates triadin splicing in vitro. SRSF10 also modulates the muscle-specific splicing of the Lrrfip1 pre-mRNA by promoting inclusion of exons 16 and 17, an essential event in mouse heart development and myoblast differentiation (Fig. 5E; Wei et al. 2015). In addition to Lrrfip1, the alternative splicing of Fxr1, Mef2a, and Nasp expressed in heart ventricles is controlled by SRSF10 (Fig. 5E). Cardiomyocytes also produce titin which is the largest protein in humans. A back-splicing event occurring on the titin pre-mRNA creates a circular RNA (cTTN1) harboring an SRSF10 binding site. The loss of cTTN1 causes abnormal splicing of the SRSF10 targets MEF2A and CASQ2 (Tijsen et al. 2021).
Germ cells
SRSF10 expression is regulated during mouse testes development where SRSF10 is found to be mostly phosphorylated and cytosolic in germ cells (Xiao et al. 2007a). This cytosolic localization persists in testicular cancer, and is also seen in liver, endometrial and urothelial cancers (Uhlen et al. 2015). Dephosphorylation of SRSF10 is induced by androgen in mouse epididymis and phosphorylated SRSF10 expression is increased during sperm maturation (Xiao et al. 2011).
Immunity
Out of over one thousand SRSF10-controlled alternative splicing events, several occur in transcripts involved in cell-cycle and immunity. For example, SRSF10 knockdown promoted the exclusion of exon E13 in the alternate terminator of interleukin-1 receptor accessory protein (IL1RAP) producing mIL1RAP, which forms a complex with interleukin IL-1/IL-1RI to initiate IL-1β signal transduction. Overexpression of SRSF10 or mIL1RAP up-regulates the expression of CD47, which in turn promotes IL1-β-induced NF-κB activation to inhibit macrophage phagocytosis (Liu et al. 2018).
SRSF10 also contributes to the IL-17 signaling pathway involved in autoimmune diseases, such as psoriasis and multiple sclerosis (He et al. 2021). Specifically, a psoriasis-susceptible variant of the lncRNA TRAF3IP2-AS1 displays an enhanced ability to interact with SRSF10 and to inhibit downstream IL-17 signaling. While SRSF10 impacts IL-17 signaling by down-regulating the expression of IRF1, a transcriptional factor for Act1, it is unclear if this regulation involves splicing modulation. Notably, lentivirus-mediated overexpression of SRSF10 had a positive therapeutic impact in mouse models of psoriasis and experimental autoimmune encephalomyelitis.
Viral replication
The assumption that an appropriate level of SRSF10 may be critical for the maturation of a group of transcripts under certain conditions is borne out in the case of viral infection. Knocking down SRSF10 affects human immunodeficiency virus type 1 (HIV-1) pre-mRNA splicing (Shkreta et al. 2017). hTRA2β regulates HIV-1 splicing specifically by binding to an exonic splicing enhancer that stimulates the 5'ss D3 (Erkelenz et al. 2013). Consistent with the view that an SRSF10/hTRA2β complex may stimulate D3/A3 splicing, the siRNA-mediated depletion of SRSF10 represses D3/A3 splicing (Shkreta et al. 2017). SRSF10 also collaborates with TRA2 proteins to activate the downstream 5′ss D2b via the exonic splicing enhancer ESE2b (Brillen et al. 2017). The small molecule 1C8 that promotes the dephosphorylation of SRSF10 and inhibits HIV-1 replication (Cheung et al. 2016) stimulates the interaction between SRSF10 and hTRA2β (Shkreta et al. 2017). 1C8 increased the production of the D3/A3 spliced product, and the depletion of SRSF10 compromised this 1C8-mediated effect (Shkreta et al. 2017).
SRSF10 is also involved in the regulation of Kaposi Sarcoma virus lytic replication by binding to the m6A-modified pre-mRNA encoding the replication transcription activator RTA together with YTHDC1 and SRSF3 (Ye et al. 2017).
More recently, SRSF10 was recovered as a strong interactor of the hepatitis B virus (HBV) Core protein (HBc) in the nucleus of human hepatocytes where this viral protein is believed to play important regulatory functions. In this case, the depletion of SRSF10 increased the level of HBV nascent RNA production without affecting their splicing (Chabrolles et al. 2020). A compound (1C8) inhibiting SRSF10 phosphorylation decreased the production of viral HBV nascent RNA, and the depletion of SRSF10 abrogated this effect (Chabrolles et al. 2020). Thus, if dSRSF10 inhibits HBV RNA replication, then depleting SRSF10 would abrogate its contribution.
By controlling the alternative splicing of ANP32A, SRSF10 also impacts the replication of the avian influenza virus (Fang et al. 2020).
Of relevance to the recent pandemic of COVID-19, it was noted that a spliceosome inhibitor targeting the splicing factor SF3B1 prevented viral replication at nontoxic concentrations (Bojkova et al. 2020), revealing splicing as an essential pathway for SARS-CoV-2 replication. The same study identified SRSF10 as part of a small list of proteins whose expression was changed by the virus and that also interacted with viral proteins.
CONCLUDING REMARKS
Dubbing SRSF10 as “atypical” is based on the ability of a dephosphorylated SRSF10 to antagonize the positive action of other SR proteins in generic splicing. While a phosphorylated SRSF10 is now associated with a positive role in splicing, the precise contribution of SRSF10 in alternative splicing may depend as much on its phosphorylation status as to where it binds relative to other sequence elements that dictate splice site selection. Moreover, not all phosphorylation events may have a similar impact. There are over 20 potential phosphorylation sites in the RS domains of SRSF10, but the contribution to splicing control of only two of these has been investigated (Shkreta et al. 2016, 2017). There is therefore a great need to assess the individual function of each of these sites, identify the modifying kinases and appreciate the dynamic patterns of phosphorylation that likely occurs during normal growth but that also could be disrupted in human diseases.
In addition to its documented role in RNA splicing, SRSF10 may possibly have additional functions in transcription and RNA stability. These functions may explain the impact that dephosphorylating SRSF10 had on the production of HBV nascent transcripts (Chabrolles et al. 2020). A potential effect on transcription could be via an interaction with PTEF-b to compete or collaborate with other SR proteins such as SRSF2 to enhance transcription elongation (Ji et al. 2013). It is also possible that SRSF10, like other regulatory factors involved in splicing, regulates the decay of viral RNAs.
The absence of immunoprecipitating antibodies against SRSF10 has created a lag in our ability to validate SRSF10 binding sites in specific transcripts and at the transcriptome level. As the ability of SRSF10 to enforce or abrogate a given splicing event often depends on its phosphorylation status, the development of antibodies that can distinguish phosphorylated from nonphosphorylated epitopes in SRSF10 would boost our understanding of how phosphorylation affects not only binding to RNA targets but also its intracellular localization in normal, stress- and disease-associated conditions.
The overexpression of SRSF10 in many diseases and its contribution to the splicing of transcripts implicated in clinically relevant pathways suggest that SRSF10 could become a target of therapeutic potential. One potentially important role for SRSF10 is in the alternative splicing of Pgc-1α in mouse liver, although it is unclear if this occurs in humans given that SRSF10 expression is low in the adult human liver. PGC-1α is expressed and alternatively spliced in human muscle. If SRSF10 performs the same function in human, strategies that would aim to inhibit SRSF10 in diabetic patients may provide therapeutic benefits given that blocking PGC-1α gluconeogenic activity improves type 2 diabetes (Sharabi et al. 2017).
Another health relevant application would be to inhibit SRSF10 in SMA patients to improve SMN2 exon 7 inclusion (Frederiksen et al. 2021). In addition, the fact that SRSF10 controls the splicing of MDM4 offers an opportunity to inhibit ferroptosis and potentially reduce the impact of degenerative brain disorders, including Parkinson's, Huntington's, and Alzheimer's diseases, as well as other forms of neurodegeneration and traumatic and hemorrhagic brain injury (Stockwell et al. 2017; Klein et al. 2021).
The documented contribution of SRSF10 in colorectal cancer and potentially other types of cancers (e.g., glioma, prostate, liver) also warrant testing interventions directed at reducing the expression or compromising the activity of SRSF10. As was tested recently, the CLK inhibitor GPS167 alters SRSF10 activity and is preferentially cytotoxic to colorectal cancer cells and organoids relative to normal colonocytes and organoids (Sohail et al. 2021).
Targeting SRSF10 activity can be achieved in different ways. Abrogating SRSF10 regulatory function could be achieved with antisense oligonucleotides that cover SRSF10 binding sites to prevent binding. In reverse, imposing SRSF10 regulatory function could be achieved by delivering SRSF10 at specific locations. Given that recruiting a MS2-tagged SRSF10 protein to an SMN2 exon or an intron with a MS2 RNA affinity element yields modulatory activity (Frederiksen et al. 2021), tethering SRSF10 at specific locations using a bifunctional oligonucleotide (Brosseau et al. 2014) containing a nonhybridizing tail with SRSF10 binding sites is an option that may be worth exploring. Blocking SRSF10 expression using RNA interference or CRISPR-Cas technologies can also be envisioned for some applications. In cancer cells however, a recent study has shown that while the loss of SRSF10 has little consequence on the growth of HCT116 cancer cells, the CLK inhibitor GPS167 can be cytotoxic for the colorectal cancer cell line HCT116 in an SRSF10-dependent manner (Sohail et al. 2021). These results suggest that dephosphorylation may be an important way to co-opt SRSF10 into promoting cell death in cancer cells. Although specifically targeting SRSF10 phosphorylation may be challenging because SR kinases phosphorylate several other SR proteins, the fact that SRSF10 is an intrinsically poor substrate for SR kinases compared to other SR proteins (Shi and Manley 2007) suggests that SRSF10 may be the first SR protein affected at low concentrations of SR kinase inhibitors.
The fact that a requirement for SRSF10 during mouse embryonic development appears restricted to the developing heart, adipose tissues, striated muscle, and liver (Feng et al. 2009; Wei et al. 2015) is also a possible indicator that interfering with SRSF10 in normal adult tissues may have limited cytotoxicity, and thus that SRSF10-based approaches to combat diseases may lead to potentially useful therapeutic outcomes with limited cytotoxicity for normal tissues.
Finally, targeting SRSF10 activity using inhibitory compounds or other strategies may be interesting to counteract viral replication. Indeed, SRSF10, like other SR and proteins, may regulate the replication of several viruses either directly by interfering with the production of viral RNAs or indirectly by modulating the synthesis of cellular factors able to restrict or promote viral replication. This could be particularly interesting in the case of oncogenic viruses for which compounds inhibiting SRSF10 activity may result in both antiviral and anti-tumoral effects.
ACKNOWLEDGMENTS
Work in the laboratory of B.C. is supported by grant PJT-165966 from the Canadian Institutes of Health Research. Work in the laboratory of A.S. is supported by funding from the Institut National de la Santé et de la Recherche Médicale (INSERM) and grants from the Agence Nationale de la Recherche sur le Sida et les Hépatites virales (ANRS).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078879.121.
Freely available online through the RNA Open Access option.
REFERENCES
- Ajiro M, Jia R, Yang Y, Zhu J, Zheng ZM. 2016. A genome landscape of SRSF3-regulated splicing events and gene expression in human osteosarcoma U2OS cells. Nucleic Acids Res 44: 1854–1870. 10.1093/nar/gkv1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Tan JT, Shelkovnikova TA. 2019. Stress granules regulate stress-induced paraspeckle assembly. J Cell Biol 218: 4127–4140. 10.1083/jcb.201904098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ, Fu XD, Adams JA. 2016. Release of SR proteins from CLK1 by SRPK1: a symbiotic kinase system for phosphorylation control of pre-mRNA splicing. Mol Cell 63: 218–228. 10.1016/j.molcel.2016.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BS. 1989. Sex in flies: the splice of life. Nature 340: 521–524. 10.1038/340521a0 [DOI] [PubMed] [Google Scholar]
- Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, Awasthi A, Jain G, Capece V, Burkhardt S, Navarro-Sala M, et al. 2015. HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest 125: 3572–3584. 10.1172/JCI79942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkegren JL, Hagg S, Talukdar HA, Foroughi Asl H, Jain RK, Cedergren C, Shang MM, Rossignoli A, Takolander R, Melander O, et al. 2014. Plasma cholesterol-induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet 10: e1004201. 10.1371/journal.pgen.1004201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, Cinatl J, Munch C. 2020. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583: 469–472. 10.1038/s41586-020-2332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillen AL, Walotka L, Hillebrand F, Muller L, Widera M, Theiss S, Schaal H. 2017. Analysis of competing HIV-1 splice donor sites uncovers a tight cluster of splicing regulatory elements within exon 2/2b. J Virol 91: e00389-17. 10.1128/JVI.00389-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosseau JP, Lucier JF, Lamarche AA, Shkreta L, Gendron D, Lapointe E, Thibault P, Paquet E, Perreault JP, Abou Elela S, et al. 2014. Redirecting splicing with bifunctional oligonucleotides. Nucleic Acids Res 42: e40. 10.1093/nar/gkt1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot B. 1996. Directing alternative splicing: cast and scenarios. Trends Genet 12: 472–478. 10.1016/0168-9525(96)10037-8 [DOI] [PubMed] [Google Scholar]
- Chabot B, Shkreta L. 2016. Defective control of pre-messenger RNA splicing in human disease. J Cell Biol 212: 13–27. 10.1083/jcb.201510032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrolles H, Auclair H, Vegna S, Lahlali T, Pons C, Michelet M, Coute Y, Belmudes L, Chadeuf G, Kim Y, et al. 2020. Hepatitis B virus core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog 16: e1008593. 10.1371/journal.ppat.1008593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung PK, Horhant D, Bandy LE, Zamiri M, Rabea SM, Karagiosov SK, Matloobi M, McArthur S, Harrigan PR, Chabot B, et al. 2016. A parallel synthesis approach to the identification of novel diheteroarylamide-based compounds blocking HIV replication: potential inhibitors of HIV-1 pre-mRNA alternative splicing. J Med Chem 59: 1869–1879. 10.1021/acs.jmedchem.5b01357 [DOI] [PubMed] [Google Scholar]
- Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM. 2016. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 86: 890–897. 10.1212/WNL.0000000000002445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier A, Shkreta L, Toutant J, Durand M, Thibault P, Chabot B. 2018. hnRNP A1/A2 and Sam68 collaborate with SRSF10 to control the alternative splicing response to oxaliplatin-mediated DNA damage. Sci Rep 8: 2206. 10.1038/s41598-018-20360-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowper AE, Caceres JF, Mayeda A, Screaton GR. 2001. Serine-arginine (SR) protein-like factors that antagonize authentic SR proteins and regulate alternative splicing. J Biol Chem 276: 48908–48914. 10.1074/jbc.M103967200 [DOI] [PubMed] [Google Scholar]
- Damianov A, Ying Y, Lin CH, Lee JA, Tran D, Vashisht AA, Bahrami-Samani E, Xing Y, Martin KC, Wohlschlegel JA, et al. 2016. Rbfox proteins regulate splicing as part of a large multiprotein complex LASR. Cell 165: 606–619. 10.1016/j.cell.2016.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Krainer AR. 2014. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res 12: 1195–1204. 10.1158/1541-7786.MCR-14-0131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Hu Z, French CE, Lloyd JPB, Brenner SE. 2020. Networks of splice factor regulation by unproductive splicing coupled with nonsense mediated mRNA decay. bioRxiv 10.1101/2020.05.20.107375 [DOI] [Google Scholar]
- Dominguez C, Fisette JF, Chabot B, Allain FH. 2010. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat Struct Mol Biol 17: 853–861. 10.1038/nsmb.1814 [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tsai YH, Weatheritt R, Wang Y, Blencowe BJ, Wang Z. 2016. An extensive program of periodic alternative splicing linked to cell cycle progression. Elife 5: e10288. 10.7554/eLife.10288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. 2016. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16: 413–430. 10.1038/nrc.2016.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eperon IC, Ireland DC, Smith RA, Mayeda A, Krainer AR. 1993. Pathways for selection of 5′ splice sites by U1 snRNPs and SF2/ASF. EMBO J 12: 3607–3617. 10.1002/j.1460-2075.1993.tb06034.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkelenz S, Poschmann G, Theiss S, Stefanski A, Hillebrand F, Otte M, Stuhler K, Schaal H. 2013. Tra2-mediated recognition of HIV-1 5' splice site D3 as a key factor in the processing of vpr mRNA. J Virol 87: 2721–2734. 10.1128/JVI.02756-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang A, Bi Z, Ye H, Yan L. 2020. SRSF10 inhibits the polymerase activity and replication of avian influenza virus by regulating the alternative splicing of chicken ANP32A. Virus Res 286: 198063. 10.1016/j.virusres.2020.198063 [DOI] [PubMed] [Google Scholar]
- Feng Y, Chen M, Manley JL. 2008. Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nat Struct Mol Biol 15: 1040–1048. 10.1038/nsmb.1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Valley MT, Lazar J, Yang AL, Bronson RT, Firestein S, Coetzee WA, Manley JL. 2009. SRp38 regulates alternative splicing and is required for Ca2+ handling in the embryonic heart. Dev Cell 16: 528–538. 10.1016/j.devcel.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen SB, Holm LL, Larsen MR, Doktor TK, Andersen HS, Hastings ML, Hua Y, Krainer AR, Andresen BS. 2021. Identification of SRSF10 as a regulator of SMN2 ISS-N1. Hum Mutat 42: 246–260. 10.1002/humu.24149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. 10.1038/nrg3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O'Hanlon D, Sung HK, Alvarez M, Talukder S, Pan Q, Mazzoni EO, et al. 2011. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147: 132–146. 10.1016/j.cell.2011.08.023 [DOI] [PubMed] [Google Scholar]
- Garneau D, Revil T, Fisette JF, Chabot B. 2005. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem 280: 22641–22650. 10.1074/jbc.M501070200 [DOI] [PubMed] [Google Scholar]
- Ge H, Manley JL. 1990. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell 62: 25–34. 10.1016/0092-8674(90)90236-8 [DOI] [PubMed] [Google Scholar]
- Gueroussov S, Weatheritt RJ, O'Hanlon D, Lin ZY, Narula A, Gingras AC, Blencowe BJ. 2017. Regulatory expansion in mammals of multivalent hnRNP assemblies that globally control alternative splicing. Cell 170: 324–339 e323. 10.1016/j.cell.2017.06.037 [DOI] [PubMed] [Google Scholar]
- Gui JF, Lane WS, Fu XD. 1994. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 369: 678–682. 10.1038/369678a0 [DOI] [PubMed] [Google Scholar]
- Ha J, Jang H, Choi N, Oh J, Min C, Pradella D, Jung DW, Williams DR, Park D, Ghigna C, et al. 2021. SRSF9 regulates cassette exon splicing of caspase-2 by interacting with its downstream exon. Cells 10: 679. 10.3390/cells10030679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltenhof T, Kotte A, De Bortoli F, Schiefer S, Meinke S, Emmerichs AK, Petermann KK, Timmermann B, Imhof P, Franz A, et al. 2020. A conserved kinase-based body-temperature sensor globally controls alternative splicing and gene expression. Mol Cell 78: 57–69.e4. 10.1016/j.molcel.2020.01.028 [DOI] [PubMed] [Google Scholar]
- Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, et al. 2013. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498: 241–245. 10.1038/nature12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Braunschweig U, Gonatopoulos-Pournatzis T, Weatheritt RJ, Hirsch CL, Ha KCH, Radovani E, Nabeel-Shah S, Sterne-Weiler T, Wang J, et al. 2017. Multilayered control of alternative splicing regulatory networks by transcription factors. Mol Cell 65: 539–553.e7. 10.1016/j.molcel.2017.01.011 [DOI] [PubMed] [Google Scholar]
- He R, Wu S, Gao R, Chen J, Peng Q, Hu H, Zhu L, Du Y, Sun W, Ma X, et al. 2021. Identification of a long noncoding RNA TRAF3IP2-AS1 as key regulator of IL-17 signaling through the SRSF10-IRF1-Act1 axis in autoimmune diseases. J Immunol 206: 2353–2365. 10.4049/jimmunol.2001223 [DOI] [PubMed] [Google Scholar]
- Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knott GJ, Iyer KS, Ho D, Newcombe EA, et al. 2015. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol 210: 529–539. 10.1083/jcb.201504117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg-Laufer H, Neufeld N, Brody Y, Nadav-Eliyahu S, Ben-Yishay R, Shav-Tal Y. 2019. Availability of splicing factors in the nucleoplasm can regulate the release of mRNA from the gene after transcription. PLoS Genet 15: e1008459. 10.1371/journal.pgen.1008459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JM, Sanford JR. 2015. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA 6: 93–110. 10.1002/wrna.1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. 2008. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82: 834–848. 10.1016/j.ajhg.2008.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S. 2017. SR proteins: binders, regulators, and connectors of RNA. Mol Cells 40: 1–9. 10.14348/molcells.2017.2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD. 2013. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153: 855–868. 10.1016/j.cell.2013.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan-Mateu J, Alvelos MI, Turatsinze JV, Villate O, Lizarraga-Mollinedo E, Grieco FA, Marroqui L, Bugliani M, Marchetti P, Eizirik DL. 2018. SRp55 regulates a splicing network that controls human pancreatic β-cell function and survival. Diabetes 67: 423–436. 10.2337/db17-0736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilchert C, Wittmann S, Vasiljeva L. 2016. The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol 17: 227–239. 10.1038/nrm.2015.15 [DOI] [PubMed] [Google Scholar]
- Kim J, Park RY, Chen JK, Kim J, Jeong S, Ohn T. 2014. Splicing factor SRSF3 represses the translation of programmed cell death 4 mRNA by associating with the 5′-UTR region. Cell Death Differ 21: 481–490. 10.1038/cdd.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617–630. 10.1016/j.ccell.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, de Queiroz RM, Venkatesh D, Prives C. 2021. The roles and regulation of MDM2 and MDMX: it is not just about p53. Genes Dev 35: 575–601. 10.1101/gad.347872.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kominami E, Arahata K, Tsukahara T. 1999. Cloning and characterization of two neural-salient serine/arginine-rich (NSSR) proteins involved in the regulation of alternative splicing in neurones. Genes Cells 4: 593–606. 10.1046/j.1365-2443.1999.00286.x [DOI] [PubMed] [Google Scholar]
- Krainer AR, Conway GC, Kozak D. 1990. The essential pre-mRNA splicing factor SF2 influences 5′ splice site selection by activating proximal sites. Cell 62: 35–42. 10.1016/0092-8674(90)90237-9 [DOI] [PubMed] [Google Scholar]
- Labourier E, Bourbon HM, Gallouzi IE, Fostier M, Allemand E, Tazi J. 1999. Antagonism between RSF1 and SR proteins for both splice-site recognition in vitro and Drosophila development. Genes Dev 13: 740–753. 10.1101/gad.13.6.740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclair NK, Brugiolo M, Urbanski L, Lawson SC, Thakar K, Yurieva M, George J, Hinson JT, Cheng A, Graveley BR, et al. 2020. Poison exon splicing regulates a coordinated network of SR protein expression during differentiation and tumorigenesis. Mol Cell 80: 648–665.e9. 10.1016/j.molcel.2020.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Cheng Y, Wu W, Liu Y, Wei N, Feng X, Xie Z, Feng Y. 2014. SRSF10 regulates alternative splicing and is required for adipocyte differentiation. Mol Cell Biol 34: 2198–2207. 10.1128/MCB.01674-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling IF, Estus S. 2010. Role of SFRS13A in low-density lipoprotein receptor splicing. Hum Mutat 31: 702–709. 10.1002/humu.21244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, Harland RM. 2005. Inhibition of neurogenesis by SRp38, a neuroD-regulated RNA-binding protein. Development 132: 1511–1523. 10.1242/dev.01703 [DOI] [PubMed] [Google Scholar]
- Liu L, Chen XH, Huang J, Lin JJ, Lin WM, Xu P. 2004. NSSR1 promotes neuronal differentiation of mouse embryonic carcinoma P19 cells. Neuroreport 15: 823–828. 10.1097/00001756-200404090-00017 [DOI] [PubMed] [Google Scholar]
- Liu F, Dai M, Xu Q, Zhu X, Zhou Y, Jiang S, Wang Y, Ai Z, Ma L, Zhang Y, et al. 2018. SRSF10-mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP-NF-κB-CD47 axis. Oncogene 37: 2394–2409. 10.1038/s41388-017-0119-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Jensen TH. 2015. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16: 665–677. 10.1038/nrm4063 [DOI] [PubMed] [Google Scholar]
- Marin-Vinader L, Shin C, Onnekink C, Manley JL, Lubsen NH. 2006. Hsp27 enhances recovery of splicing as well as rephosphorylation of SRp38 after heat shock. Mol Biol Cell 17: 886–894. 10.1091/mbc.e05-07-0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. 2007. hnRNP proteins and splicing control. Adv Exp Med Biol 623: 123–147. 10.1007/978-0-387-77374-2_8 [DOI] [PubMed] [Google Scholar]
- Maslon MM, Heras SR, Bellora N, Eyras E, Caceres JF. 2014. The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife 3: e02028. 10.7554/eLife.02028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin PV, Khaitovich P, Cardoso-Moreira M, Kaessmann H. 2021. Alternative splicing during mammalian organ development. Nat Genet 53: 925–934. 10.1038/s41588-021-00851-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke S, Goldammer G, Weber AI, Tarabykin V, Neumann A, Preussner M, Heyd F. 2020. Srsf10 and the minor spliceosome control tissue-specific and dynamic SR protein expression. Elife 9: e56075. 10.7554/eLife.56075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelle L, Barbier J, Chabot B. 2011. hnRNP and hnRNP-like proteins in splicing control: molecular mechanisms and implication in human pathologies. In RNA binding proteins (ed. Zdravko L), pp. 1–27. Landes Bioscience and Springer Science+Business Media. [Google Scholar]
- Moumen A, Masterson P, O'Connor MJ, Jackson SP. 2005. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 123: 1065–1078. 10.1016/j.cell.2005.09.032 [DOI] [PubMed] [Google Scholar]
- Muller-McNicoll M, Rossbach O, Hui J, Medenbach J. 2019. Auto-regulatory feedback by RNA-binding proteins. J Mol Cell Biol 11: 930–939. 10.1093/jmcb/mjz043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naftelberg S, Schor IE, Ast G, Kornblihtt AR. 2015. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem 84: 165–198. 10.1146/annurev-biochem-060614-034242 [DOI] [PubMed] [Google Scholar]
- Nasa I, Rusin SF, Kettenbach AN, Moorhead GB. 2018. Aurora B opposes PP1 function in mitosis by phosphorylating the conserved PP1-binding RVxF motif in PP1 regulatory proteins. Sci Signal 11: eaai8669. 10.1126/scisignal.aai8669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann A, Meinke S, Goldammer G, Strauch M, Schubert D, Timmermann B, Heyd F, Preussner M. 2020. Alternative splicing coupled mRNA decay shapes the temperature-dependent transcriptome. EMBO Rep 21: e51369. 10.15252/embr.202051369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. 10.1101/gad.1525507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya K, Kataoka N, Hagiwara M. 2011. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol 195: 27–40. 10.1083/jcb.201107093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya K, Adachi S, Natsume T, Iwakiri J, Terai G, Asai K, Hirose T. 2020. LncRNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. EMBO J 39: e102729. 10.15252/embj.2019102729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, Sanchi A, Radujkovic A, Horn P, Dolatshad H, et al. 2018. Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood 132: 1225–1240. 10.1182/blood-2018-04-843771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard H, Heron A, Moreau J, Ben-Ari Y, Khrestchatisky M. 1993. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience 57: 545–554. 10.1016/0306-4522(93)90004-Y [DOI] [PubMed] [Google Scholar]
- Qi Y, Li Y, Cui SC, Zhao JJ, Liu XY, Ji CX, Sun FY, Xu P, Chen XH. 2015. Splicing factor NSSR1 reduces neuronal injury after mouse transient global cerebral ischemia. Glia 63: 826–845. 10.1002/glia.22787 [DOI] [PubMed] [Google Scholar]
- Ray D, Kazan H, Chan ET, Pena Castillo L, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR. 2009. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol 27: 667–670. 10.1038/nbt.1550 [DOI] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. 2013. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177. 10.1038/nature12311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revil T, Pelletier J, Toutant J, Cloutier A, Chabot B. 2009. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J Biol Chem 284: 21458–21467. 10.1074/jbc.M109.019711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharabi K, Lin H, Tavares CDJ, Dominy JE, Camporez JP, Perry RJ, Schilling R, Rines AK, Lee J, Hickey M, et al. 2017. Selective chemical inhibition of PGC-1α gluconeogenic activity ameliorates type 2 diabetes. Cell 169: 148–160.e15. 10.1016/j.cell.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon G, Cruz NJ, Kang DW, Gandal MJ, Wang B, Kim YM, Zink EM, Casey CP, Taylor BC, Lane CJ, et al. 2019. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 177: 1600–1618.e17. 10.1016/j.cell.2019.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manley JL. 2007. A complex signaling pathway regulates SRp38 phosphorylation and pre-mRNA splicing in response to heat shock. Mol Cell 28: 79–90. 10.1016/j.molcel.2007.08.028 [DOI] [PubMed] [Google Scholar]
- Shi Y, Nishida K, Campigli Di Giammartino D, Manley JL. 2011. Heat shock-induced SRSF10 dephosphorylation displays thermotolerance mediated by Hsp27. Mol Cell Biol 31: 458–465. 10.1128/MCB.01123-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Manley JL. 2002. The SR protein SRp38 represses splicing in M phase cells. Cell 111: 407–417. 10.1016/S0092-8674(02)01038-3 [DOI] [PubMed] [Google Scholar]
- Shin C, Feng Y, Manley JL. 2004. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature 427: 553–558. 10.1038/nature02288 [DOI] [PubMed] [Google Scholar]
- Shin C, Kleiman FE, Manley JL. 2005. Multiple properties of the splicing repressor SRp38 distinguish it from typical SR proteins. Mol Cell Biol 25: 8334–8343. 10.1128/MCB.25.18.8334-8343.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkreta L, Bell B, Revil T, Venables JP, Prinos P, Abou Elela S, Chabot B. 2013. Cancer-associated perturbations in alternative pre-messenger RNA splicing. Cancer Treat Res 158: 41–94. 10.1007/978-3-642-31659-3_3 [DOI] [PubMed] [Google Scholar]
- Shkreta L, Cloutier A, Toutant J, Vennin Rendos H, Chabot B. 2015. Regulation of alternative splicing and the case of Bcl-x. Pak J Biochem Mol Biol 48: 27–38. [Google Scholar]
- Shkreta L, Toutant J, Durand M, Manley JL, Chabot B. 2016. SRSF10 connects DNA damage to the alternative splicing of transcripts encoding apoptosis, cell-cycle control, and DNA repair factors. Cell Rep 17: 1990–2003. 10.1016/j.celrep.2016.10.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkreta L, Blanchette M, Toutant J, Wilhelm E, Bell B, Story BA, Balachandran A, Cochrane A, Cheung PK, Harrigan PR, et al. 2017. Modulation of the splicing regulatory function of SRSF10 by a novel compound that impairs HIV-1 replication. Nucleic Acids Res 45: 4051–4067. 10.1093/nar/gkw1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Trincado JL, Tatlow PJ, Piccolo SR, Eyras E. 2018. Genome sequencing and RNA-motif analysis reveal novel damaging noncoding mutations in human tumors. Mol Cancer Res 16: 1112–1124. 10.1158/1541-7786.MCR-17-0601 [DOI] [PubMed] [Google Scholar]
- Sohail M, Shkreta L, Toutant J, Rabea S, Babeu J-P, Huard C, Huntington-Coulombe J, Delannoy A, Placet M, Geha S, et al. 2021. A novel class of inhibitors that preferentially target SRSF10 and promote p53-dependent cytotoxicity on human colorectal cancer cells. NAR Cancer 3: 1–22. 10.1093/narcan/zcab019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Wan X, Huang T, Zeng C, Sastry N, Wu B, James CD, Horbinski C, Nakano I, Zhang W, et al. 2019. SRSF3-regulated RNA alternative splicing promotes glioblastoma tumorigenicity by affecting multiple cellular processes. Cancer Res 79: 5288–5301. 10.1158/0008-5472.CAN-19-1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. 2017. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273–285. 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CH, Dhananjaya D, Tarn WY. 2018. Alternative splicing in neurogenesis and brain development. Front Mol Biosci 5: 12. 10.3389/fmolb.2018.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Bao Y, Han W, Song F, Shen X, Zhao J, Zuo J, Saffen D, Chen W, Wang Z, et al. 2017. Autoregulation of RBM10 and cross-regulation of RBM10/RBM5 via alternative splicing-coupled nonsense-mediated decay. Nucleic Acids Res 45: 8524–8540. 10.1093/nar/gkx508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveen A, Agesen TH, Nesbakken A, Rognum TO, Lothe RA, Skotheim RI. 2011. Transcriptome instability in colorectal cancer identified by exon microarray analyses: associations with splicing factor expression levels and patient survival. Genome Med 3: 32. 10.1186/gm248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijsen AJ, Cocera Ortega L, Reckman YJ, Zhang X, van der Made I, Aufiero S, Li J, Kamps SC, van den Bout A, Devalla HD, et al. 2021. Titin circular RNAs create a back-splice motif essential for SRSF10 splicing. Circulation 143: 1502–1512. 10.1161/CIRCULATIONAHA.120.050455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Odio S, Key J, Hoepken HH, Canet-Pons J, Valek L, Roller B, Walter M, Morales-Gordo B, Meierhofer D, Harter PN, et al. 2017. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation 14: 154. 10.1186/s12974-017-0928-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MP, Armero VE, Allaire A, Boudreault S, Martenon-Brodeur C, Durand M, Lapointe E, Thibault P, Tremblay-Letourneau M, Perreault JP, et al. 2016. Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma. BMC Genomics 17: 683. 10.1186/s12864-016-3029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. 2017. A pathology atlas of the human cancer transcriptome. Science 357: eaan2507. 10.1126/science.aan2507 [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen JY, Cody NAL, Dominguez D, et al. 2020. A large-scale binding and functional map of human RNA-binding proteins. Nature 583: 711–719. 10.1038/s41586-020-2077-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Lapasset L, Gadea G, Fort P, Klinck R, Irimia M, Vignal E, Thibault P, Prinos P, Chabot B, et al. 2013. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat Commun 4: 2480. 10.1038/ncomms3480 [DOI] [PubMed] [Google Scholar]
- Wei N, Cheng Y, Wang Z, Liu Y, Luo C, Liu L, Chen L, Xie Z, Lu Y, Feng Y. 2015. SRSF10 plays a role in myoblast differentiation and glucose production via regulation of alternative splicing. Cell Rep 13: 1647–1657. 10.1016/j.celrep.2015.10.038 [DOI] [PubMed] [Google Scholar]
- Weldon C, Dacanay JG, Gokhale V, Boddupally PVL, Behm-Ansmant I, Burley GA, Branlant C, Hurley LH, Dominguez C, Eperon IC. 2018. Specific G-quadruplex ligands modulate the alternative splicing of Bcl-X. Nucleic Acids Res 46: 886–896. 10.1093/nar/gkx1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Guo JY, Tang W, Yang CS, Freel CD, Chen C, Nairn AC, Kornbluth S. 2009. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat Cell Biol 11: 644–651. 10.1038/ncb1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Zong J, Wei N, Cheng J, Zhou X, Cheng Y, Chen D, Guo Q, Zhang B, Feng Y. 2018. CASH: a constructing comprehensive splice site method for detecting alternative splicing events. Brief Bioinform 19: 905–917. 10.1093/bib/bbx034 [DOI] [PubMed] [Google Scholar]
- Xiao PJ, Hu L, Li J, Lin W, Chen X, Xu P. 2007a. NSSR1 is regulated in testes development and cryptorchidism and promotes the exon 5-included splicing of CREB transcripts. Mol Reprod Dev 74: 1363–1372. 10.1002/mrd.20719 [DOI] [PubMed] [Google Scholar]
- Xiao R, Sun Y, Ding JH, Lin S, Rose DW, Rosenfeld MG, Fu XD, Li X. 2007b. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol 27: 5393–5402. 10.1128/MCB.00288-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao PJ, Peng ZY, Huang L, Li Y, Chen XH. 2011. Dephosphorylated NSSR1 is induced by androgen in mouse epididymis and phosphorylated NSSR1 is increased during sperm maturation. PLoS One 6: e25667. 10.1371/journal.pone.0025667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. 2016. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519. 10.1016/j.molcel.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Liu L, Lazarev D, Al-Zain A, Fomin V, Yeung PL, Chambers SM, Lu CW, Studer L, Manley JL. 2018. TCF3 alternative splicing controlled by hnRNP H/F regulates E-cadherin expression and hESC pluripotency. Genes Dev 32: 1161–1174. 10.1101/gad.316984.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen ER, Nilsen TW. 2017. Kaposi's sarcoma-associated herpesvirus utilizes and manipulates RNA N6-adenosine methylation to promote lytic replication. J Virol 91: e00466-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip L, Fuhlbrigge R, Taylor C, Creusot RJ, Nishikawa-Matsumura T, Whiting CC, Schartner JM, Akter R, von Herrath M, Fathman CG. 2015. Inflammation and hyperglycemia mediate Deaf1 splicing in the pancreatic lymph nodes via distinct pathways during type 1 diabetes. Diabetes 64: 604–617. 10.2337/db14-0803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Li LL, Chen XH, Peng ZY. 2014. Dephosphorylation of NSSR1 regulates alternative splicing of the GluR-B minigene. Genet Mol Res 13: 1753–1763. 10.4238/2014.March.17.3 [DOI] [PubMed] [Google Scholar]
- Zhao H, He L, Yin D, Song B. 2019. Identification of β-catenin target genes in colorectal cancer by interrogating gene fitness screening data. Oncol Lett 18: 3769–3777. 10.3892/ol.2019.10724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q, Han J, Wu G, Fang J, Feng Y. 2014a. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun 5: 4581. 10.1038/ncomms5581 [DOI] [PubMed] [Google Scholar]
- Zhou X, Wu W, Li H, Cheng Y, Wei N, Zong J, Feng X, Xie Z, Chen D, Manley JL, et al. 2014b. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res 42: 4019–4030. 10.1093/nar/gkt1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wu W, Wei N, Cheng Y, Xie Z, Feng Y. 2014c. Genome-wide analysis of SRSF10-regulated alternative splicing by deep sequencing of chicken transcriptome. Genom Data 2: 20–23. 10.1016/j.gdata.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyner KG, Mulhearn DS, Adhikari S, Martinez Cuesta S, Di Antonio M, Erard N, Hannon GJ, Tannahill D, Balasubramanian S. 2019. Genetic interactions of G-quadruplexes in humans. Elife 8: e46793. 10.7554/eLife.46793 [DOI] [PMC free article] [PubMed] [Google Scholar]