

Streszczenie
Deficyt aktywności transaldolazy należy do wrodzonych błędów metabolizmu na szlaku przemiany pentoz, który do tej pory rozpoznano i opisano u 33 pacjentów, w tym 4 z Polski.
W artykule przedstawiono obraz kliniczny, patogenezę i diagnostykę choroby. Autorzy przedstawili ponadto własną propozycję algorytmu diagnostyki deficytu transaldolazy.
Słowa kluczowe: transaldolaza, deficyt transaldolazy, szlak przemiany pentoz, poliole
Abstract
Transaldolase deficiency is a rare inborn autosomal recessive error of the pentose phosphate pathway that, to date, has been diagnosed in 33 patients, including 4 from Poland.
The aim of this manuscript was to present the clinical presentation, pathogenesis and diagnostic process of transaldolase deficiency. The authors also present a diagnostic algorithm of transaldolase deficiency.
Key words: transaldolase, transadolase deficiency, pentose phosphate pathway, polyols
1. Wstęp
Deficyt aktywności transaldolazy (ang. transaldolase deficiency, TALDO) należy do wrodzonych błędów metabolizmu na szlaku przemiany pentoz (ang. pentose phosphate pathway, PPP). Szlak pentozofosforanowy obejmuje ciąg reakcji biochemicznych, zachodzących w cytozolu, których produktami końcowymi są rybozo-5-fosforan i NADPH. Sumaryczne równanie reakcji cyklu pentozofosforanowego jest następujące:
glukozo-6-fosforan + 2NADP+ + H2O → rybozo-5-fosforan + 2NADPH + 2H+ + CO2.
Ta droga przemian biochemicznych spełnia dwie role:
wytwarzanie NADPH (zredukowany dinukleotyd nikotynoamidoadeninowy), który spełnia rolę donora protonów i elektronów w redukcyjnych procesach biosyntezy.
dostarczanie reszt rybozy do biosystezy nukleotydowi kwasów nukleinowych.
Szczególną intensywność wykazuje on w okresie wzrastania płodu oraz w pierwszych latach życia [1, 2, 3].
Przemiany na szlaku pentozofosforanowym dzielimy na dwie fazy: nieodwracalną fazę oksydacyjną i odwracalną fazę nieoksydacyjną. Transaldolaza (EC 2.2.1.2), obok transketolazy, jest enzymem odwracalnej fazy cyklu pentozofosforanowego i katalizuje następującą reakcję:
sedoheptulozo-7-fosforan + gliceraldehydo-3-fosforan → erytrozo-4-fosforan + fruktozo-6-fosforan
W wyniku deficytu transaldolazy dochodzi do nagromadzenia polioli – erytritolu, arabitolu, rybitolu, sedoheptitolu, perseitolu oraz siedmiowęglowych cukrów − sedoheptulozy, mannoheptulozy i fosfosedoheptulozy.
Celem pracy jest analiza obrazu klinicznego, patogenezy i diagnostyki deficytu aktywności transaldolazy na podstawie danych z piśmiennictwa oraz doświadczeń własnych. Jak do tej pory, w dostępnych publikacjach nie został przedstawiony algorytm diagnostyczny, stąd autorzy przedstawili propozycję własną algorytmu diagnostyki TALDO.
2. Obraz kliniczny
Deficyt aktywności transaldolazy został po raz pierwszy opisany przez Verhoeven i wsp., w 2001 r., u kilkumiesięcznego niemowlęcia z wewnątrzmacicznym opóźnieniem wzrastania, hepatosplenomegalią i zaburzeniami krzepnięcia [4]. Biopsja wątroby, wykonana w 2. roku życia, wykazała drobnoguzkową marskość wątroby. W wieku kilkunastu lat, opisywany pacjent, rozwinął przewlekłą niewydolność wątroby i tubulopatię [4, 5].
Dotychczas, TALDO opisano u 33 pacjentów (tabela I), w tym 4 z Polski [4-18]. Większość (79%) z nich pochodziła od spokrewnionych rodziców, w tym 12 z jednego arabskiego rodu [11].
Tabela I.
Charakterystyka pacjentów z deficytem transaldolazy.
Table I. Characteristics of TALDO deficient patients reported in the literature.
| Pacjent Patient | Manifestacja prenatana Antenatal manifestation |
Obraz kliniczny Clinical outcome |
Wiek w momencie rozpoznania Age at diagnosis |
Wiek ostatniego badania Age at last follow-up |
badania Wynik molekularnego Molecular variant /protein effect |
Piśmiennictwo References |
|---|---|---|---|---|---|---|
| 1 | lUGR | marskość wątroby, tubulopatia, przewlekła choroba nerek, zgon w wieku 17 lat liver cirrhosis, tubulopathy, chronic kidney failure, died at 17 years of age | 10 lat 10 years |
17 lat 17 years |
c.512_514delCCT/ p.Serl71del; hmz | Verhoeven et al [4] |
| 2 | zespól HELLP HELLP syndrome |
niewydolność wątroby, niewydolność tubulopatia, krążeniowo-zgon w 18. oddechowa, dobie życia liver failure, respiratory failure, heart failure, tubulopathy, died at 18 days of age | post mortem (diagnoza pośmiertna) post mortem diagnosis | 18 dni 18 days |
c.575G>A/ p.Argl92His; hmz | Verhoeven et al [6] |
| 3 | znaczny (20 kg) przyrost masy ciała matki w ciąży excessive maternal weight gain (20 kg) during pregnancy | zgon w wieku 5 miesięcy died at 5 months of age | post mortem (diagnoza pośmiertna) post mortem diagnosis | 5 miesięcy 5 months |
c.512_514delCCT/ p.Serl71del; hmz | Valayannopoulos et al [7] |
| 4 | obrzęk płodowy hydrops fetalis | terminach ciąży termination of pregnancy | (diagnoza post mortem pośmiertna) post mortem diagnosis | |||
| 5 | prawidłowy rozwój normal | marskość wątroby, tubulopatia, przewlekła choroba nerek liver cirrhosis, tubulopathy, chronic kidney failure | 4 lata 4 years |
9 lat 9 years |
||
| 6 | prawidłowy rozwój normal | marskość wątroby, tubulopatia liver cirrhosis, tubulopathy | 3 miesiące 3 months |
4 lata 10 miesięcy 4 years 10 months |
||
| 7 | matowodzie, splenomegalia oligohydramnios, splenomegaly | marskość wątroby, tubulopatia liver cirrhosis, tubulopathy |
2 lata 2 years |
2 lata 2 years |
C.574C>T/ p.Argl92Cys; hmz | Wamelink et al [9] |
| 8 | prawidłowy rozwój normal | wielonarządowa niewydolność, zgon w wieku 4.5 miesięcy multiorgan failure, died at 4.5 months of age | post mortem (diagnoza pośmiertna) post mortem diagnosis | 4.5 miesięcy 4.5 months | c.895_897delAAC/ p.Asn299del; c. 931 G>A/ p. Gly311Arg; comphtz | Balasubramaniam et al [8] |
| 9 | prawidłowy rozwój normal | rak wątrobowokomórkowy, przeszczepienie wątroby hepatocellular carcinoma, liver transplantation |
1 rok
1 year |
16 miesięcy 16 months |
C.512C>T/ p.Ser171Phe; hmz |
LeDuc et al [12] |
| 10 | prawidłowy rozwój normal | marskość wątroby liver cirrhosis | 3 lata 3 years |
3 lata 3 years |
||
| 11 | prawidłowy rozwój normal | prawidłowy rozwój normal | 8 lat 8 years |
9 lat 9 years |
||
| 12 | prawidłowy rozwój normal | marskość wątroby liver cirrhosis | 10 lat 10 years |
10 lat 10 years |
c.793delC/ p.Gln265Argfs*56
; hmz et Eyaid al [11], Jassim et al [14] |
|
| 13 | wielowodzie polyhydramnios | zgon w wieku 5 miesięcy died at 5 months of age | post mortem (diagnoza pośmiertna) post mortem diagnosis | 5 miesięcy 5 months |
||
| 14 | prawidłowy rozwój normal | hepatomegalia hepatomegaly | 6 miesięcy 6 months |
6 miesięcy 6 months |
||
| 15 | prawidłowy rozwój normal | marskość wątroby liver cirrhosis | 4 lata 4 years |
4 lata 4 years |
||
| 16 | prawidłowy rozwój normal | hepatosplenomegalia, pancytopenia hepatosplenomegaly, pancytopenia | 2.5 lat 2.5 years |
2.5 lat 2.5 years |
||
| 17 | hiperechogenny kardiomegalia, obraz jelit cardiomegaly, hyperechogenic bowel on ultrasound | zespół marskość wątrobowo-wątroby, płucny liver cirrhosis, hepatopulmonary syndrome | 1.5 lat 1.5 years |
1.5 lat 1.5 years |
||
| 18 | prawidłowy rozwój normal | marskość wątroby liver cirrhosis | 5 lat 5 years |
5 lat 5 years |
||
| 19 | małowodzie, IUGR, całkowite odwrócenie trzew, hepatosplenomegalia IUGR, oligohydramnios, situs inversus totalis, hepatosplenomegaly | marskość wątroby, zaburzenia krzepnięcia, małopłytkowość, neutropenia liver cirrhosis, neutropenia, thrombocytopaenia, bleeding diathesis | 12 miesięcy 12 months |
12 miesięcy 12 months | c.793delC/ p.Gln265Argfs*56; hmz Eyaid et al [11], et Jassim al [14] |
|
| 20 | prawidłowy rozwój nomal | nepatomegalia, naczyniak wątroby | b.d. not known |
b.d. not known |
||
| 21 | IUGR | hepatosplenomegalia hepatosplenomegaly | 8 miesięcy 8 months |
8 miesięcy 8 months |
||
| 22 | prawidłowy rozwój normal | hepatosplenomegalia, złóg w nerkach w wieku 7 ms hepatosplenomegaly, renal calculus observed at 7 months of age | 2.5 lat 2.5 years |
2.5 lat 2.5 years |
||
| 23 | prawidłowy rozwój normal | hepatosplenomegalia, małopłytkowość hepatosplenomegaly, thrombocytopenia | 7 lat 7 years |
7 lat 7 years |
||
| 24 | b.d. not known | hepatosplenomegalia, podwyższona aktywność aminotransferaz, prawidłowy koagulogram hepatosplenomegaly, elevated serum transaminases, normal coagulation profile |
2 lata 2 years |
2 lata 2 years |
C.574C>T/ p.Arg192Cys; hmz Al-Shamsi et al [15] | |
| 25 | b.d. not known | hepatomegalia, małopłytkowość, prawidłowa aktywność aminotransferaz, prawidłowy koagulogram, rybia łuska, zez, wtórny brak miesiączki hepatomegaly, thrombocytopenia, normal serum transaminases, normal coagulation profile, ichthyosis, nystagmus, secondary amenorrhoea | 25 lat 25 years |
25 lat 25 years |
||
| 26 | b.d. not known | przeszczepienie wątroby (w 1. roku życia), obecnie podwyższona aktywność aminotransferaz, prawidłowy koagulogram liver transplantation (at 1 year of age), currently normal coagulation profile and slightly elevated serum transaminases |
1 rok
1 year |
3 lata 3 years |
C.574C>T/ p.Arg192Cys; hmz Al-Shamsi et al [15] | |
| 27 | obrzęk płodowy hydrops fetalis | niewydolność oddechowa wtórna do krwotoku płucnego severe respiratory distress due to pulmonary haemorrhage | 1 miesiąc 1 month |
3 miesiące 3 months |
||
| 28 | lUGR, hiperechogenny obraz jelit IUGR, hyperechogenic bowel on ultrasound | niedrożność smółkowa jelit, niewydolność wątroby, zgon w wieku 2 miesięcy meconium plug, liver failure, died at months of age | post mortem (diagnoza pośmiertna) post mortem diagnosis |
2 miesiące 2 months |
C.669C>G/ p.Tyr223*hmz | Banne et al [16] |
| 29 | prawidłowy rozwój normal | hepatomegalia, podwyższona aktywność aminotransferaz, prawidłowy koagulogram hepatomegaly, elevated liver transaminases, normal coagulation profile | 9 miesięcy 9 months |
16 miesięcy 16 months |
C.574C>T/ p.Argl92Cys | Rodan et al [17] |
| 30 | znaczny (20 kg) przyrost masy ciała matki w ciąży, IUGR excessive maternal weight gain (20 kg) during pregnancy, IUGR | marskość wątroby, tubulopatia liver cirrhosis, tubulopathy | 3.5 lat 3.5 years |
13 lat 13 years |
c.575G>A/ p.Argl92His; hmz | Tylki-Szymańska et al [10,13], Lipiński et al [18] |
| 31 | znaczny (20 kg) przyrost masy ciała matki w ciąży, IUGR excessive maternal weight gain (20 kg), IUGR | marskość wątroby, tubulopatia liver cirrhosis, tubulopathy | 6 miesięcy 6 months |
10 lat 10 years |
c.575G>A/ p.Arg192His; hmz | |
| 32 | małowodzie, IUGR oligohydramnios, IUGR | marskość wątroby, tubulopatia, kamica nerkowa liver cirrhosis, tubulopathy, nephrolithiasis |
2 miesiące 2 months |
8 lat 8 years |
c.462-174_981+53del/p.?; hmz | |
| 33 | obrzęk płodowy, IUGR hydrops fetalis, IUGR | włóknienie wątroby, tubulopatia, kamica nerkowa liver fibrosis, tubulopathy, nephrolithiasis | 1.5 lat 1.5 years |
4 lata 4 years |
c.575G>A/p.Argl92His; c. 462-174 981 +53del/ p. ?; comphtz | |
| comphtz – złożona heterozygota; hmz – homozygota; b.d. – brak danych; IUGR – wewnątrzmaciczne zahamowanie rozwoju comphtz – compound heterozygote; hmz – homozygote; IUGR – intrauterine growth retardation | ||||||
Obraz kliniczny TALDO jest zróżnicowany, jednak zawsze obejmuje postępujące uszkodzenie wątroby oraz nerek (dysfunkcja cewek nerkowych) [5]. Wyróżnia się postać o wczesnym początku (ang. early-onset TALDO) – wystąpienie objawów w okresie prenatalnym lub w ciągu pierwszych 3 miesięcy życia, stanowiącą chorobę o ciężkim przebiegu i niepomyślnym rokowaniu – oraz postać objawiającą się później (ang. late-onset TALDO) – powyżej 3. miesiąca życia, stanowiącą powoli postępującą chorobę z możliwym długoletnim przeżyciem [18].
Większość przypadków opisanych w literaturze stanowiło TALDO o wczesnym początku. U noworodków i niemowląt stwierdzano małą masę urodzeniową – odnotowywano już wewnątrzmaciczne opóźnienie wzrastania, powiększenie wątroby i śledziony, niedokrwistość, małopłytkowość, zaburzenia krzepnięcia, a w kilku przypadkach obrzęk płodowy. W niektórych przypadkach przebieg ciąży charakteryzował się masywnym przyrostem masy ciała u matek oraz dużym i nieprawidłowym łożyskiem. Blisko połowa tych pacjentów zmarła w okresie niemowlęcym, najczęściej w wyniku krwawienia, jako efektu koagulopatii w przebiegu uszkodzenia wątroby [4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18].
Nieco odmiennie przedstawia się przebieg deficytu transaldolazy o wczesnym początku u polskich pacjentów, którzy mimo początkowo złego rokowania (masywne wodobrzusze, hipoalbuminemia wymagająca okresowych wlewów albumin), wkroczyli w drugą dekadę życia (3 z 4 pacjentów) z objawami skompensowanej marskości wątroby i kontrolowanej tubulopatii (poprzez leczenie substytucyjne) [18].
TALDO o późnym początku wystąpienia objawów, mimo potencjalnie lepszego rokowania, prowadzi powoli do postępującego uszkodzenia wątroby i nerek. LeDuc i wsp. opisali 8-letniego chłopca, u którego TALDO zostało wykryte przypadkowo (skrining rodzinny) [12]. Al-Shamsi i wsp. opisali TALDO u 25-letniej pacjentki, która w dzieciństwie (brak dokładnych danych) prezentowała hepatosplenomegalię i małopłytkowość, a rozpoznanie TALDO postawiono przypadkowo w toku diagnostyki wtórnego braku miesiączki [15].
W przebiegu TALDO w wątrobie stwierdza się charakterystyczne drobnoguzkowe włóknienie, a ostatecznie marskość wątroby. Z czasem u chorych z dłuższym okresem przeżycia pojawiają się objawy tubulopatii – hiperkalciuria, białkomocz cewkowy (kłębuszkowo-cewkowy) – zwykle jako pierwsze objawy, aminoaciduria, glukozuria, kwasica kanalikowa – oraz stopniowo rozwija się przewlekła choroba nerek [5]. W obrazie klinicznym stwierdza się ponadto osteopenię/osteoporozę oraz niskorosłość (skutki tubulopatii i uszkodzenia wątroby).
Charakterystyczne dla TALDO są zmiany skórne – poszerzona siatka naczyń i teleangiektazje na skórze tułowia (zwłaszcza pleców) oraz tendencja do tworzenia naczyniaków jamistych (haemangioma cavernosum). W literaturze znajdują się ponadto opisy pacjentów z cutis laxa oraz hipertrychozą [4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18].
Ponadto występują zaburzenia endokrynologiczne − u ok. 1/3 wszystkich pacjentów w okresie niemowlęcym stwierdzano nieprawidłowości w zakresie zewnętrznych narządów płciowych (wnętrostwo, kliteromegalia), a badania hormonalne wykazały u części z nich hipogonadyzm hipergonadotropowy.
W niektórych przypadkach opisywane są wrodzone wady serca, do najczęstszych należą drożny przewód tętniczy oraz ubytek przegrody międzyprzedsionkowej [4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18]. Część autorów opisuje cechy dysmorfii, czego nie stwierdzono u polskich pacjentów, jednakże należy mieć na uwadze, że cechy te nie zostały dokładnie sprecyzowane i powinny być podobne w opisywanych przypadkach.
Rozwój psychoruchowy i umysłowy pacjentów są prawidłowe [4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18].
3. Patogeneza, diagnostyka
Deficyt aktywności transaldolazy prowadzi do kumulacji w płynach ustrojowych polioli − erytritolu, arabitolu, rybitolu, sedoheptitolu, perseitolu i siedmiowęglowych cukrów − sedoheptulozy, mannoheptulozy i fosfosedoheptulozy [2, 19, 20, 21, 22].
Diagnostyka opiera się na stwierdzeniu wydalania z moczem, jak i kumulacji w surowicy (metodą spektrometrii mas) rybitolu, arabitolu, erytritolu, oraz o najwyższej czułości sedoheptulozy i sedoheptulozo-7-fosforanu [2, 19, 20, 21, 22]. Wstępna metoda diagnostyczna (stosowana w IP-CZD), opiera się określeniu stosunku D-/L- arabinitolu w moczu (normy przedstawiono w tabeli II [22]).
Tabela II.
Poziom D-/L-arabinitolu w moczu w zależności od wieku [22].
Table II. Urinary D-/L-arabitol ratio depending on age [22].
| Wiek [lata] Age [years] |
D-/L-Aarabinitol D-/L- arabinitol |
|---|---|
| 0-1 | <3,6 |
| >1-3 | <3,3 |
| >3-10 | <3,0 |
| >10 | <2,5 |
Potwierdzenie rozpoznania wymaga badania aktywności transaldolazy w leukocytach krwi obwodowej lub hodowanych fibroblastach skóry. Metodę weryfikacji rozpoznania może stanowić również badanie molekularne. Badania enzymatyczne mogą być pominięte na rzecz badań molekularnych (sekwencjonowanie genu TALDO1) (rycina 1).
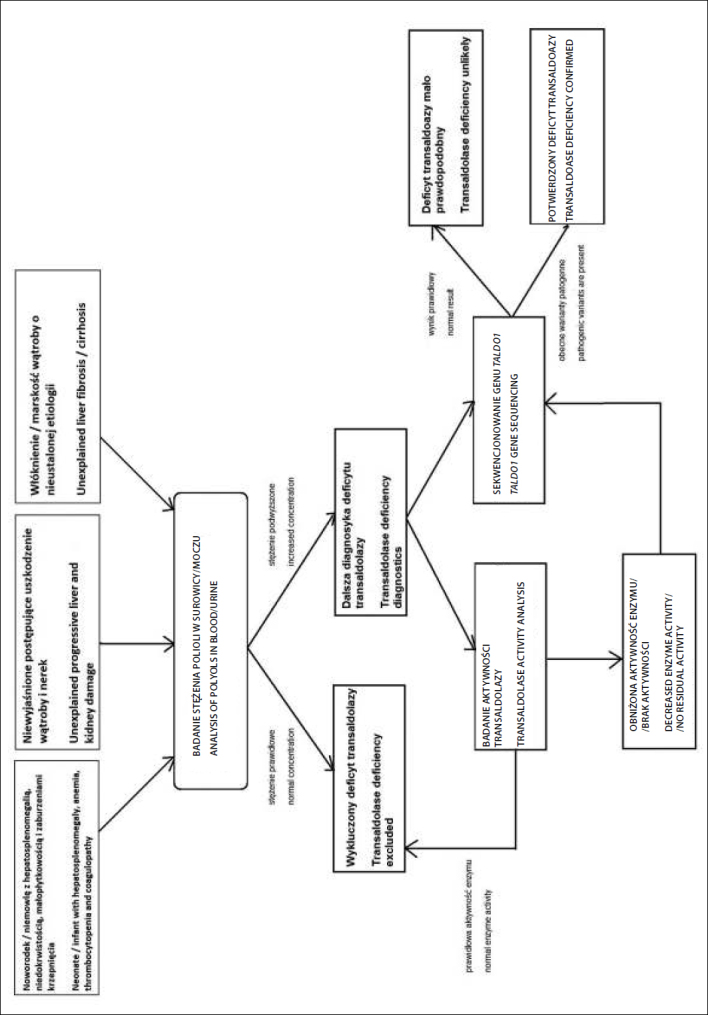
W patogenezie schorzenia podkreśla się toksyczny efekt działania gromadzonych metabolitów (ufosforylowanych cukrów) na hepatocyty i kanaliki nerkowe. Inna hipoteza podkreśla znaczenie stresu oksydacyjnego, który jest efektem braku NADPH koniecznego dla odtwarzania zredukowanej formy glutationu – kofaktora peroksydazy glutationowej przeciwdziałającej stresowi oksydacyjnemu [2, 4]. Reduktaza glutationowa, tworząca układ antyoksydacyjny w mitochondriach, także wykorzystuje NADPH jako źródło elektronów, katalizując reakcję odtwarzania zredukowanej formy glutationu kosztem utlenienia NADPH. Tym samym zapewnia równowagę peroksydacyjno-antyoksydacyjną, zapobiegając stresowi oksydacyjnemu.
4. Leczenie
Leczenie pacjentów z TALDO jest wyłącznie objawowe. Upośledzenie czynności syntetycznej wątroby może wiązać się z koniecznością okresowej podaży albumin czy osoczowych czynników krzepnięcia. Podobnie, upośledzenie czynności cewek nerkowych może wymagać suplementacji nieorganicznych fosforanów, podaży alfa-kalcydiolu czy wyrównywania zaburzeń elektrolitowych.
Dyskusyjne są wskazania do ewentualnego przeszczepienia wątroby. Niektórzy autorzy sugerują możliwość uszkodzenia przeszczepionej wątroby [3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16]. W dwóch opublikowanych przypadkach dokonano przeszczepienia wątroby w okresie niemowlęcym, jednakże przed rozpoznaniem TALDO dotychczas nie opublikowano efektów terapeutycznych.
Przeszczepienie wątroby u polskich pacjentów wobec względnie stabilnego ich stanu i równoczesnego uszkodzenia nerek nie jest obecnie brane pod uwagę [17]. Ze względu na rzadkość występowania, wskazane jest monitorowanie i prowadzenie pacjentów z TALDO w jednym, mającym doświadczenie oraz odpowiednie zasoby do diagnostyki, ośrodku.
5. Podsumowanie
U pacjentów z niewyjaśnionym postępującym uszkodzeniem wątroby i nerek wskazane jest wykonanie badania kumulacji polioli. Potwierdzenia/wykluczenia choroby wymaga również każdy przypadek noworodka/niemowlęcia z hepatosplenomegalią, niedokrwistością, małopłytkowością i zaburzeniami krzepnięcia, zwłaszcza urodzonego z małą urodzeniową masą ciała (oraz dużym przyrostem masy ciała matki w trakcie trwania ciąży).
Włóknienie, marskość wątroby o nieustalonej etiologii zawsze wymaga badania stężenia polioli.
Footnotes
Wkład Autorów/Author’s contributions
Według kolejności/According to the order of the Authorship
Conflicts of interest
Konflikt interesu/Conflicts of interest
Autorzy pracy nie zgłaszają konfliktu interesów.
The Authors declare no conflict of interest.
Piśmiennictwo
- 1.Perl A, Qian Y, Chohan KR, Shirley CR, Amidon W, Banerjee S, Middleton FA, Conkrite KL, Barcza M, Gonchoroff N, Suarez SS, Banki K. Transaldolase is essential for maintenance the mitochondrial transmembrane potential and fertility of spermatozoa. Proc Natl Acad Sci USA. 2006;103:1481314818. doi: 10.1073/pnas.0602678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perl A. The pathogenesis of transaldolase deficiency. IUBMB Life. 2007;59:365–373. doi: 10.1080/15216540701387188. [DOI] [PubMed] [Google Scholar]
- 3.Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A, Tauqeer Alam M, Keller M, Breitenbach M, Brindle KM, Rabinowitz Jd, Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90:927–963. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhoeven NM, Huck JH, Roos B, Struys EA, Salomons GS, Douwes AC, Jakobs C. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am J Hum Genet. 2001;68:1086–1092. doi: 10.1086/320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeffen YG, Biebuyck N, Wamelink MM, Jakobs C, Mulder MF, Tylki-Szymańska A, Bökenkamp A. Nephrological abnormalities in patients with transaldolase deficiency. Nephrol Dial Transplant. 2012;27:3224–3227. doi: 10.1093/ndt/gfs061. [DOI] [PubMed] [Google Scholar]
- 6.Verhoeven NM, Wallot M, Huck JH, Dirsch O, Ballauf A, Neudorf U, Jakobs C. A newborn with severe liver failure, cardiomyopathy and transaldolase deficiency. J Inherit Metab Dis. 2005;28:169–179. doi: 10.1007/s10545-005-5261-6. [DOI] [PubMed] [Google Scholar]
- 7.Valayonnopoulos V, Verhoeven NM, Mention K, Salomons GA, Sommelet D, Gonzales M, Touati G, de Lyonalay P, Jakobs C, Saudubray JM. Transaldolase deficiency: a new cause of hydrops fetalis and neonatal multi-organ disease. Pediatrics. 2006;149:713–717. doi: 10.1016/j.jpeds.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Balasubramaniam S, Wamelink MM, Ngu LH, Talib A, Salomons GS, Jakobs C, Keng WT. Novel heterozygous mutations in TALDO1 gene causing transaldolase deficiency and early infantile liver failure. J Pediatr Gastroenterol Nutr. 2011;52:113–116. doi: 10.1097/MPG.0b013e3181f50388. [DOI] [PubMed] [Google Scholar]
- 9.Wamelink MM, Struys EA, Salomons GS, Fowler D, Jakobs C, Clayton PT. Transaldolase deficiency in a two-year-old boy with cirrhosis. Mol Genet Metab. 2008;94:255–258. doi: 10.1016/j.ymgme.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Tylki-Szymanska A, Stradomska TJ, Wamelink MM, Salomons GS, Taybert J, Pawłowska J, Jakobs C. Transaldolase deficiency in two new patients with a relative mild phenotype. Mol Genet Metab. 2009;97:15–17. doi: 10.1016/j.ymgme.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 11.Eyaid W, Al Harbi T, Anazi S, Wamelink MM, Jakobs C, Al Salammah M, Alkuraya FS. Transaldolase deficiency: report of 12 new cases and further delineation of the phenotype. J Inherit Metab Dis. 2013;36:997–1004. doi: 10.1007/s10545-012-9577-8. [DOI] [PubMed] [Google Scholar]
- 12.LeDuc CA, Crouch EE, Wilson A, Lefkowitch J, Wamelink MMC, Jakobs C, Salomons GS, Sun X, Shen Y, Chung WK. Novel association of early onset hepatocellular carcinoma with transaldolase deficiency. JIMD Rep. 2014;12:121–127. doi: 10.1007/8904_2013_254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tylki-Szymanska A, Wamelink MM, Stradomska TJ, Salomons GS, Taybert J, Dąbrowska-Leonik N, Rurarz M. Clinical and molecular characteristics of two transaldolase-deficient patients. Eur J Pediatr. 2014;173:1679–1682. doi: 10.1007/s00431-014-2261-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jassim N, Alghaihab M, Saleh SA, Alfadhel M, Wamelink MM, Eyaid W. Pulmonary manifestations in a patient with transaldolase deficiency. JIMD Rep. 2014;12:47–50. doi: 10.1007/8904_2013_243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Shamsi AM, Ben-Salem S, Hertecant J, Al-Jasmi F. Transaldolase deficiency caused by the homozygous p.R192C mutation of the TALDO1 gene in four Emirati patients with considerable phenotypic variability. Eur J Pediatr. 2015;174:661–668. doi: 10.1007/s00431-014-2449-5. [DOI] [PubMed] [Google Scholar]
- 16.Banne E, Meiner V, Shaag A, Katz-Brull R, Gamliel A, Korman S, Cederboim SH, Duvdevani MP, Frumkin A, Zilkha A, Kapuller V, Arbell D, Cohen E, Eventov-Friedman S. Transaldolase Deficiency: A New Case Expands the Phenotypic Spectrum. JIMD Rep. 2016;26:31–36. doi: 10.1007/8904_2015_474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodan LH, Berry GT. N-Acetylcysteine therapy in an infant with transaldolase deficiency is well tolerated and associated with normalization of alpha fetoprotein levels. JIMD Rep. 2017;31:73–77. doi: 10.1007/8904_2016_555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipiński P, Pawłowska P, Stradomska TJ, Ciara E, Jankowska I, Socha P, Tylki-Szymańska A. Long-term systematic monitoring of four Polish transaldolase deficient patients. JIMD Rep. 2018. (in press). doi. [DOI] [PMC free article] [PubMed]
- 19.Wamelink MM, Smith DE, Jansen EE, Verhoeven NM, Struys EA, Jakobs C. Detection of transaldolase deficiency by quantification of novel seven carbon chain carbohydrate biomarkers in urine. J Inherit Metab Dis. 2007;30:735–742. doi: 10.1007/s10545-007-0590-2. [DOI] [PubMed] [Google Scholar]
- 20.Wamelink MM, Smith DE, Jakobs C, Verhoeven NM. Analysis of polyols in urine by liquid chromatographytandem mass spectrometry: a useful tool for recognition of inborn errors affecting polyol metabolism. J Inherit Metab Dis. 2005;28:951–963. doi: 10.1007/s10545-005-0233-4. [DOI] [PubMed] [Google Scholar]
- 21.Wamelink MM, Struys EA, Huck JH, Roos B, van der Knaap MS, Jakobs C, Verhoeven NM. Quantification of sugar phosphate intermediates of the pentose phosphate pathway by LC-MS/MS: application to two new inherited defects of metabolism. J Chromatogr B Anal Technol Biomed Life Sci. 2005;823:18–25. doi: 10.1016/j.jchromb.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Stradomska TJ, Mileniczuk Z. Gas chromatographic determination of D-/L-arabinitol ratio in healthy Polish children. J Chromatogr B Nal Technol Biomed Life Sci. 2002;773:175–181. doi: 10.1016/s1570-0232(02)00180-0. [DOI] [PubMed] [Google Scholar]