
Figure 2. Parallel evolution between viral RNA (vRNA) segments varies in H3N2 viruses from 2005 to 2014.
Seven replicate maximum-likelihood trees were reconstructed for each vRNA gene segment from human H3N2 virus sequences (2005 to 2014) as described in Figure 1. (A–B) Highly similar (PB1 and PA gene segments) (A) or dissimilar (PB1 and HA gene segments) trees (B) from replicate one were plotted as tanglegrams with discrepancies in branch topology highlighted in green. Robinson-Foulds distances (RF) and clustering information distances (CID) are shown above the tanglegram. Intersecting lines map leaves on the left tree to the corresponding leaves on the right. Strains are coded by cluster number; strain identities can be found in Supplementary file 2. Bootstrap values greater than 70 are shown in red. Scale bars indicate substitutions per site. (C) Pairwise RF were calculated between each pair of trees in each replicate. Mean tree distances were visualized in a heatmap. Refer to Figure 2—figure supplement 1 for the standard error of the mean RF of each pair of trees.

Figure 2—figure supplement 1. The standard error of the mean (SEM) of replicate Robinson-Foulds distances (RF).
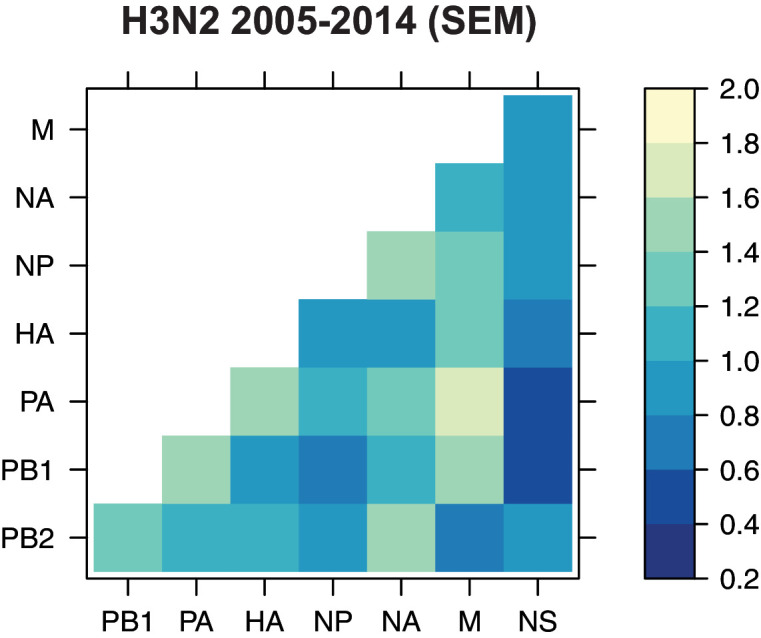
Figure 2—figure supplement 2. The mean clustering information distance (CID) of replicate viral RNA (vRNA) trees.

Figure 2—figure supplement 3. Null distribution of Robinson-Foulds distances (RF).

Figure 2—figure supplement 4. Networks determined from pairwise tree distances.
