

Abstract
Melittin, the primary peptide component of bee venom, is a potent cytolytic anti-cancer peptide with established anti-tumor activity. However, practical application of melittin in oncology is hampered by its strong, nonspecific hemolytic activity and intrinsic instability. To address these shortcomings, delivery systems are used to overcome the drawbacks of melittin and facilitate its safe delivery. Yet, a recent study revealed that encapsulated melittin remains immunogenic and can act as an adjuvant to elicit a fatal antibody immune response against the delivery carrier. We discovered that substitution of L-amino acids with D-amino acids mitigates this problem: D-melittin nanoformulations induce significantly decreased immune response, resulting in excellent safety without compromising cytolytic potential. We now report the first application of D-melittin and its micellar formulations for cancer treatment. D-melittin was delivered by a pH-sensitive polymer carrier that (i) forms micellar nanoparticles at normal physiological conditions, encapsulating melittin, and (ii) dissociates at endosomal pH, restoring melittin activity. D-melittin micelles (DMM) exhibits significant cytotoxicity and induces hemolysis in a pH-dependent manner. In addition, DMM induce immunogenic cell death, revealing its potential for cancer immunotherapy. Indeed, in vivo studies demonstrated the superior safety profile of DMM over free peptide and improved efficacy at prohibiting tumor growth. Overall, we present the first application of micellar D-melittin for cancer therapy. These findings establish a new strategy for safe, systemic delivery of melittin, unlocking a potential pathway toward clinical translation for cytotoxic peptides as anti-cancer agents. which can revolutionize in vivo delivery of therapeutic peptides and peptide antigens.
Keywords: Anti-cancer peptide, Melittin, Drug delivery, Polymeric micelles, Polymer-peptide conjugates
Graphical Abstract
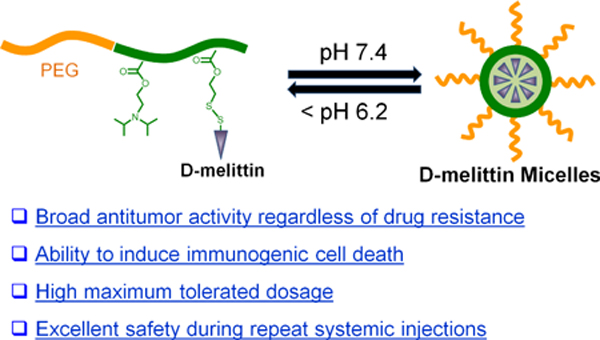
1. Introduction
Anti-cancer peptides (ACPs) have emerged as a promising class of cancer therapeutics due to their ease of synthesis, high potency, and cell selectivity [1, 2]. ACPs exert anti-tumor activity through various and distinctive mechanisms, such as specifically targeting tumor suppressive signaling pathways or inducing cell death through membranolytic activity [3–6]. Compared to conventional chemotherapeutics, ACPs possess better selectivity and lower off-target toxicity to normal cells. Importantly, the likelihood of cancer cells developing drug resistance against ACPs is low because ACPs effect critical cell pathways that cells cannot bypass [7]. The continued susceptibility of malignant cells to ACPs highlights another major advantage of ACPs, as drug resistance is increasingly responsible for the failure of chemotherapy [1, 8]. Given the attractive properties of ACPs, there has been increasing interest in the development of ACP delivery systems [2, 9, 10].
Melittin, the active 26-amino acid peptide component of honeybee venom, is a potent and natural ACP with established anti-tumor efficacy against a broad spectrum of cancers, including prostate, breast, and liver cancer[11, 12]. The cationic charge and amphipathic properties of melittin enable it to interact with and disrupt biological membranes[13, 14]. In addition to being membranolytic, melittin can also modulate multiple cell signaling pathways, such as inhibiting angiogenesis, inducing cell cycle arrest, and preventing metastasis [15]. Despite melittin’s great potential for cancer treatment, melittin also suffers from significant drawbacks that limit its clinical application, including nonspecific hemolysis, rapid clearance and degradation, and poor tumor accumulation [15]. To address these issues, melittin has been incorporated into drug delivery systems such as polymer-peptide conjugates or nanoparticle (NP) with the goal of decreasing nonspecific hemolysis to enhance safety in vivo and to increase bioavailability in tumors [16–19].
While some drug delivery platforms have facilitated melittin delivery, these systems can also introduce new challenges, as demonstrated by the generation of an adaptive immune response to the delivery carrier. Similar to the generation of anti-PEG antibodies against PEGylated platforms, systemic administration of conjugates with bioactive drugs can trigger a host immune response, eliciting antibodies against the carrier and resulting in accelerated blood clearance and hypersensitivity reactions [20, 21]. This adverse immune response is attributed to the bioactive drug, which acts as an adjuvant to activate the immune system against the carrier. Studies show that the generated immune response correlates with the immunogenicity of the loaded cargos [22]. In contrast to most small molecule chemotherapeutics, melittin is an immunogenic host defense peptide that can elicit a severe immune response [15, 16, 23]. Indeed, we recently demonstrated that repeat systemic administration of PEGylated melittin micelles was fatal in mice and that death could be attributed to the robust generation of anti-PEG antibodies [24]. By substituting the L-amino acids with D-amino acids, we attenuated the immune response, ultimately enhancing the therapeutic index of our delivery platform without compromising bioactivity of the peptide. Based on these findings, we hypothesized that D-melittin could serve as a safer alternative to L-melittin for systemic cancer delivery applications.
Herein, we report the application of D-melittin micelle (DMM) nanoformulations for treatment of solid tumors. We employed a polymer platform, virus inspired polymer for endosomal release (VIPER), to facilitate intracellular melittin delivery [25]. D-melittin was conjugated to an ultra pH-sensitive polymer that shields melittin at physiological pH and rapidly unsheathes melittin at endosomal pH. Thus, the polymer-peptide conjugate self-assembles into micellar nanoparticles at physiological pH and dissociates after entry into the cells (Scheme 1). After characterizing the polymer-peptide conjugate, we examined its activity in vitro and in vivo. First, the cytotoxic activity of free peptide and polymer-peptide conjugates was investigated against different tumor cells. The ability of these formulations to induce immunogenic cell death, as characterized by calreticulin (CRT) surface expression, ATP secretion, and HMGB1 release, was also evaluated. Next, the safety profiles of D-melittin peptide and micelles were established in normal mice, as determined by the maximum tolerated dose (MTD). Finally, the in vivo anti-tumor efficacy was evaluated in CT26 (colon cancer) and 4T1 (breast cancer) tumor-bearing mice. To the best of our knowledge, this is the first application of D-melittin for cancer therapy, which can help unveil the promising potential of melittin for in vivo therapeutic use.
Scheme 1.
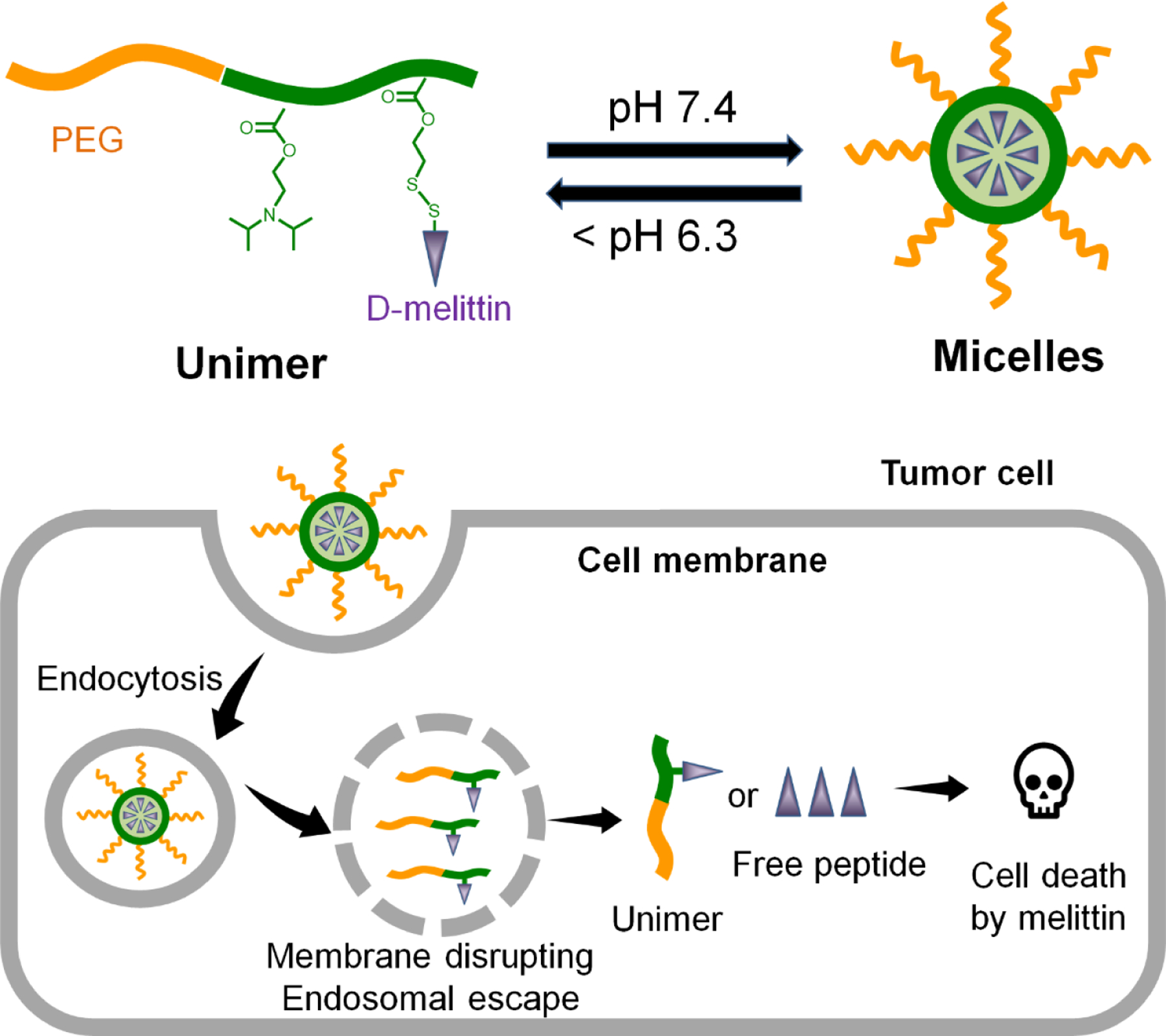
Design of D-melittin conjugate for systemic delivery of melittin for cancer therapy.
2. Materials and methods
2.1. Materials, polymer synthesis, and characterization.
Poly(ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (PEG-CTA), 2-diisopropylaminoethyl methacrylate (DIPAMA), and pyridyl disulfide ethyl methacrylate (PDSEMA) were purchased from Sigma (Saint Louis, MO, USA). Fetal bovine serum (FBS) was purchased from R&D Systems (Minneapolis, MN, USA). Anti-mouse CTLA-4 (clone 9H10) and PD-1 (clone 29F.1A12) antibodies were purchased from Bio X cell. All other chemicals were purchased from Sigma and used as received.
2.2. Synthesis and characterization.
Polymers and peptides were synthesized and characterized as previously described [24]. Briefly, D-melittin (GIGAVLKVLTTGLPALISWIKRKRQQC) peptides were synthesized on a microwave peptide synthesizer (Liberty Blue CEM) via solid phase peptide synthesis using L- or D-amino acids, respectively, and purified via reverse-phase HPLC in 0.1% TFA water and acetonitrile. Peptide molecular mass was determined by MALDI-TOF.
To synthesize the polymer, PEG-CTA was polymerized with DIPAMA, PDSEMA, and azobisisobutyronitrile (AIBN) in dimethylacetamide (DMAc), and immersed in an oil bath at 70 °C. After 24 hours, the polymerization was quenched with liquid nitrogen and the resultant polymer (PEG113-b-p(DIPAMA40-co-PDSEMA2)) was purified via dialysis against methanol and DI water. D-melittin was conjugated to PDSEMA via disulfide exchange reaction in methanol and water (5:1) and purified by dialysis against DI water. The micelles were prepared in acidic phosphate buffer (pH 4.0) and the pH was adjusted to pH 7–8, and sterile filtered using a 0.22 mm pore filter. Polymers were characterized by 1H NMR in deuterated chloroform. Peptide conjugation to PDSEMA was monitored via UV (353 nm) for the release of 2-thiopyridine. Micelle size was assessed by dynamic light scattering (0.5 mg/mL) and critical micellar concentration (CMC) was determined via Nile red method (ex/em 557/625 nm) with 0.5 mg/mL dye. The transition point of the micelles was determined using a Nile red method as previously described.[25]
2.2. Cell culture.
RAW 264.7 macrophages, CT26, and 4T1 cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco). For toxicity studies, cells were seeded at 15–20k cells/well in a 96 well plate. Cells were cultured with peptide or micelles for 24 hours and viability was determined by MTS/PMS (Promega) by plate reader. The Gal8-RAW 264.7 cell line was generated as previously described [26, 27]. Briefly, RAW cells were transfected with plasmids containing a transposable Gal8-GFP construct and PiggyBac transposon (generous gift of Prof. Jordan Green) using Lipofectamine 2000. Cells were sorted for the top 5% brightest GFP+ singlet cell events, expanded, and sorted three more time to yield a population of Gal8-GFPhigh cells.
2.3. Hemolysis assay.
Hemolysis assays were conducted as described [28]. Human blood was obtained in accordance with University of Washington Institutional Review Board (IRB) guidelines. Briefly, blood was washed twice in 150 mM NaCl and resuspended in phosphate buffered saline (PBS) at each pH value to be tested (pH 5.4, 6.4, 7.4). Blood was diluted 1:50 and plated in a V-bottom 96-well plate. Samples were incubated with peptide in appropriate pH buffer at 37 °C for 1 hour. Supernatant was collected and absorbance (541 nm) was detected on a plate reader. Triton X-100 20% (w/v) and PBS at appropriate pH were used as positive and negative controls, respectively.
2.4. Imaging.
Confocal imaging of endosomal escape was conducted as previously described.[26] Briefly, Gal8-RAW 264.7 macrophages were plated (15k) in a Greinier HalfArea 96 well plate and incubated with peptide (1 µM) or micelles (6.25 µM) for 16–18 hours. Cells were imaged in Fluorobright media supplemented with 25 mM HEPES, 10% FBS, Hoechst 33342 (2000X), and PI (1000X). Wells were imaged in a 4×4 region of interest with a 20X objective (Leica SP8X).
2.5. Immunogenic cell death (ICD) characterization.
ICD was characterized by surface calreticulin expression and ATP and HMGB1 secretion in cell supernatant. Calreticulin staining. CT26 cells were incubated with peptide (2 µΜ) or micelles (10 µΜ) for 24 hours, lifted with Accutase, and stained with anti-calreticulin primary antibody (1:20, Abcam) followed by an anti-mouse secondary antibody (1:750), and analyzed on an Attune NxT (Invitrogen) flow cytometer. ATP release. CT26 cells were incubated with peptide or micelles at indicated concentrations for 24 hours and supernatant was collected and assessed for ATP concentration via the ENLITEN ATP Assay (Promega) according to manufacturer instructions. HMGB1 release. CT26 cells were incubated with peptide (2 µΜ) or micelles (12.5 µΜ) for 48 hours and supernatant was collected. Insoluble lipids were removed via centrifugation. HMGB1 concentration was assessed via ELISA (Chondrex) according to manufacturer instructions.
2.6. Animal studies.
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Washington and were conducted in accordance with use and regulations. Female Balb/c mice (6–8 weeks) were ordered from Charles River Laboratories. Tumors (1M 4T1 or CT26 cells in 100 µl serum free RPMI) were inoculated subcutaneously under the 4th nipple (hind inguinal tumors). Treatment began on day 8 after inoculation. Mice were injected intravenously (IV) via tail vein at 5 mg peptide/kg every 4th day. Immune checkpoint blockade (ICB) antibodies (anti-PD-1 and anti-CTLA-4) were administered by intraperitoneal (IP) injection at 100 µg/mouse, on indicated days. Mice were humanely euthanized when euthanasia criteria was met (e.g. hunched, depressed respiration); tumors exceeded 10% of body weight; or tumor ulcers had discharge. For AST/ALT enzyme evaluation, serum was collected in serum separator tubes (BD), allowed to coagulate for 30 min at room temperature, and centrifuged at 1000 xg for 10 min at 4 °C. AST and ALT enzyme activity was evaluated with a kit (Sigma) following manufacturer instructions.
For tumor dissociation studies to assess TIL populations, tumor-bearing mice (100 mm3) were injected with (i) PBS, (ii) CP, (iii) D-melittin 2 mg/kg, or (iv) DMM 5 mg peptide/kg IV. Twenty four hours later, mice were sacrificed, perfused with PBS, and tumors resected. Tumors were chopped into small pieces in serum free RPMI supplemented with DNAse I (125 U/mL) and Collagenase IV (20 U/mL) in a gentleMACS dissociator. The tumor cell suspension was filtered over a 70 µm cell strainer and prepared for flow staining. Cells were stained for viability with Zombie Violet and stained in three plates for T cells (CD45-APC/Cy7, CD4-AlexaFluor647, CD4-PE, CD8-FITC), macrophages (CD45-APC/Cy7, CD11b-FITC, CD80-PE, CD206-BV604), and dendritic cells (CD45-APC/Cy7, CD11c-FITC, CD80-PE, MHCII-PE/Cy5).
For tumor biodistribution studies, CT26 tumor-bearing mice were injected IV with PBS or Cy5-DMM (2 mg peptide/kg). At indicated time points (30 min, 4 hour, 6 hour), mice were sacrificed and perfused. Organs were collected, imaged on the Xenogen, and homogenized in RIPA buffer supplemented with DNAse-1 (200 U/mL). Homogenized organs were centrifuged at 21,000g for 10 min and supernatant was assayed for fluorescence (ex/em 633/666) and normalized to protein (Pierce BCA kit) content.
2.7. Maximal tolerated dose (MTD) studies.
Peptide or micelles were injected IV at indicated doses and mice were observed for two weeks following injection. Mice were humanely euthanized if they exhibited signs of acute distress (i.e. inability to walk, moribund) or weight loss exceeding 20% of starting body weight.
2.8. Histochemical analysis.
For H&E imaging, mice were injected with peptide or micelles at indicated doses, every 4th day for a total of 3 injections. Twenty-four hours after the fourth injection, mice were sacrificed and perfused with PBS. Tissues were collection and fixed in 4% PFA for 48 hours at 4 °C, transferred to PBS, and submitted to the UW Histology and Imaging Core for tissue processing, staining, and scoring by a blinded third-party.
3. Results
3.1. Polymer synthesis of melittin micelles
The PEG-VIPER polymer was synthesized by reversible addition-fragmentation chain-transfer (RAFT) polymerization of DIPAMA and PDSEMA with PEGylated macro chain transfer agents (PEG-CTAs) (Fig. S1), as previously described [24, 25]. The DIPAMA block confers pH-sensitivity, as DIPAMA sharply transitions from hydrophobic at neutral pH to hydrophilic at acidic pH, and PDSEMA enables conjugation with thiol-containing peptides (Fig. 1A). These polymers self-assemble into micelles at neutral pH (7.4) with a diameter of 32.6 ± 13 nm (Fig. 1B), yielding D-melittin micelles (DMM). The obtained micelles had a critical micelle concentration (CMC) of 0.03 mg/mL in PBS (7.4) and were stable in PBS containing 10% FBS for over two days, indicating good colloidal stability (Fig. 1C). Additionally, we demonstrate that these micelles disassemble into polymer chains at acidic pH 6.4 (Fig. 1D), exposing conjugated peptides upon cellular internalization in endosomes.
Fig. 1.
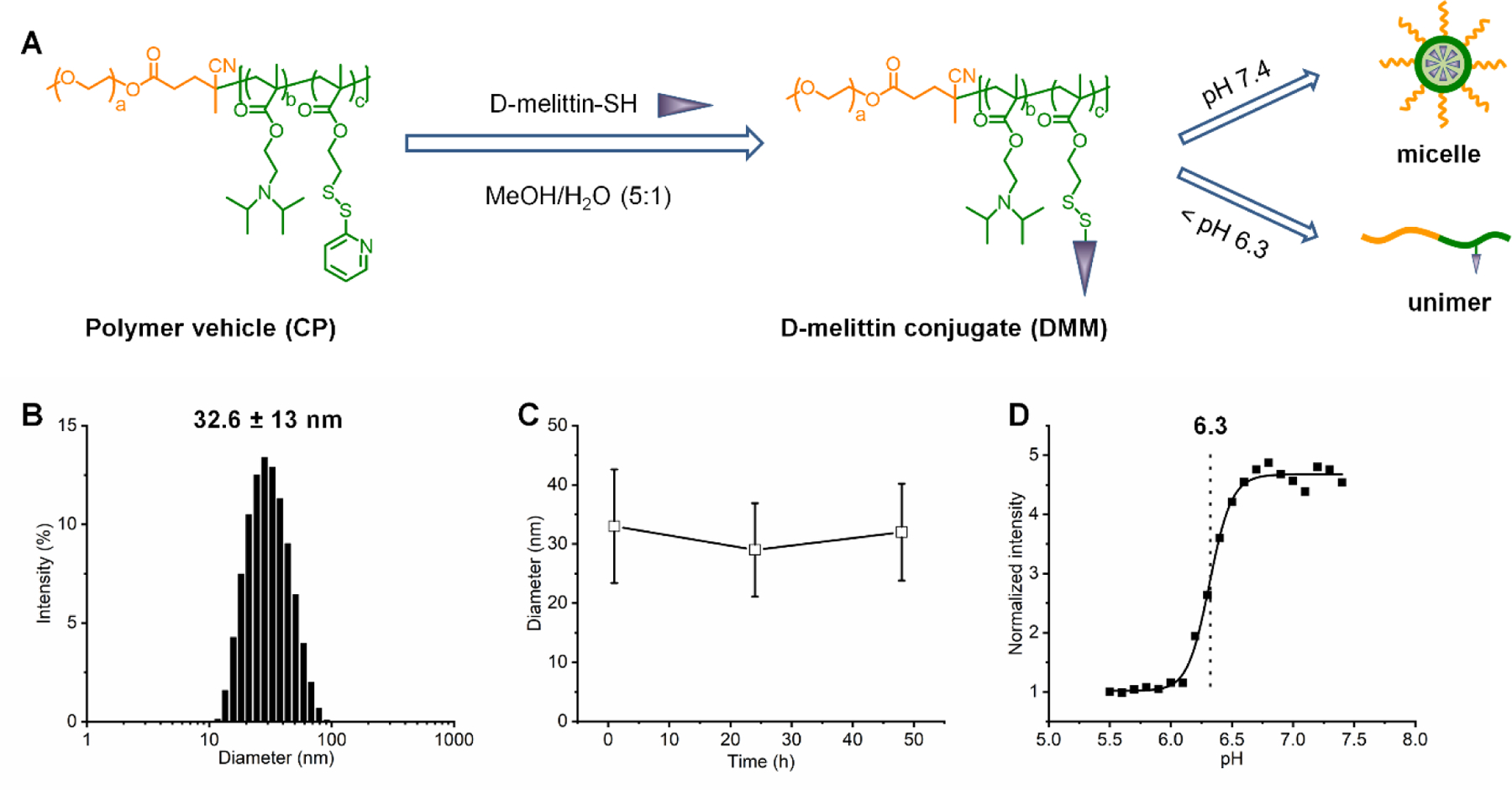
Synthesis and characterization of D-melittin conjugate. A) Synthesis route for D-melittin conjugate and its self-assembly behavior under different pH conditions. B) Size distribution of D-melittin micelles (DMM) in PBS. C) Size stability of DMM in PBS containing 10% FBS. D) pH-transition study of DMM.
3.2. Lytic activity in vitro
The lytic activity of D-melittin peptides and micelles was evaluated in vitro via cell toxicity and hemolysis assays, and visualization of endosomal escape. Peptide and micelles were potent against both human (A549, MDA-MB-435) and murine tumor cells (3T3, CT26) (Fig. 2A-D, Table 1), as indicated by similar half maximal inhibitory concentrations (IC50) across cell lines. Micelles without peptide (CP) had no cytotoxic activity (Fig. S2). Both peptide and micelles demonstrate robust toxicity in wild type (WT) and doxorubicin-resistant (DOX-R) MDA-MB-435 cells (Fig 2C-D, with confirmation of doxorubicin-resistance is reported in Fig. S3). While the IC50 of free peptide is comparable in WT and DOX-R lines (3.53 ± 0.2 and 3.44 ± 0.3 µM, respectively), the IC50 of DMM is 10-fold lower in DOX-R cells compared to WT cells (30.09 ± 3.9 and 2.98 ± 0.7 µM in WT and DOX-R, respectively). Notably, DMM is 10-fold more cytotoxic compared to free peptide in DOX-R cells. Next, hemolytic activity was evaluated against human red blood cells (RBCs) at pH 5.4, 6.4, and 7.4 (Fig. 3E-F, Table 2), a range that covers endosomal pH, micelle transition point, and physiological pH, respectively. The peptide was lytic at all pH values and exhibited increasing activity at higher pH, as indicated by decreasing concentration for 50% RBC hemolysis (HC50) with increasing pH. In contrast, the HC50 of DMM was 10–40 fold lower than free peptide at acidic conditions, yet exhibited no lytic activity at pH 7.4, confirming both the potent lytic activity of DMM at endosomal pH (5.4 – 6.4) and its safe encapsulation of D-melittin at physiological pH (7.4). Lastly, endosomal disruption was evaluated in the Gal8-GFP-RAW 264.7 reporter cell line (Fig. 2H) [26]. Cells constitutively express Gal8-GFP throughout their cytoplasm, but upon endosomal disruption, redistribute and bind to the inner face of endosomes. Thus, disrupted endosomes can be visualized as green punctae. Cells were additionally stained with propidium iodine (PI) to assess viability of cells and the percent of live cells (PI-) expressing punctae was quantified. While D-melittin did not disrupt endosomes (0.03%), DMM induced GFP+ punctate in nearly 10% of live cells. Although micelles without melittin (CP) were also capable of disrupting endosomes to some extent (0.17%), CP had no cytotoxic or hemolytic activity (Fig. S2). Overall, both D-melittin and DMM were shown to be cytotoxic and hemolytic, but DMM has no hemolytic activity at pH 7.4. Furthermore, DMM was capable of disrupting cell endosomes whereas free peptide was not.
Figure 2.
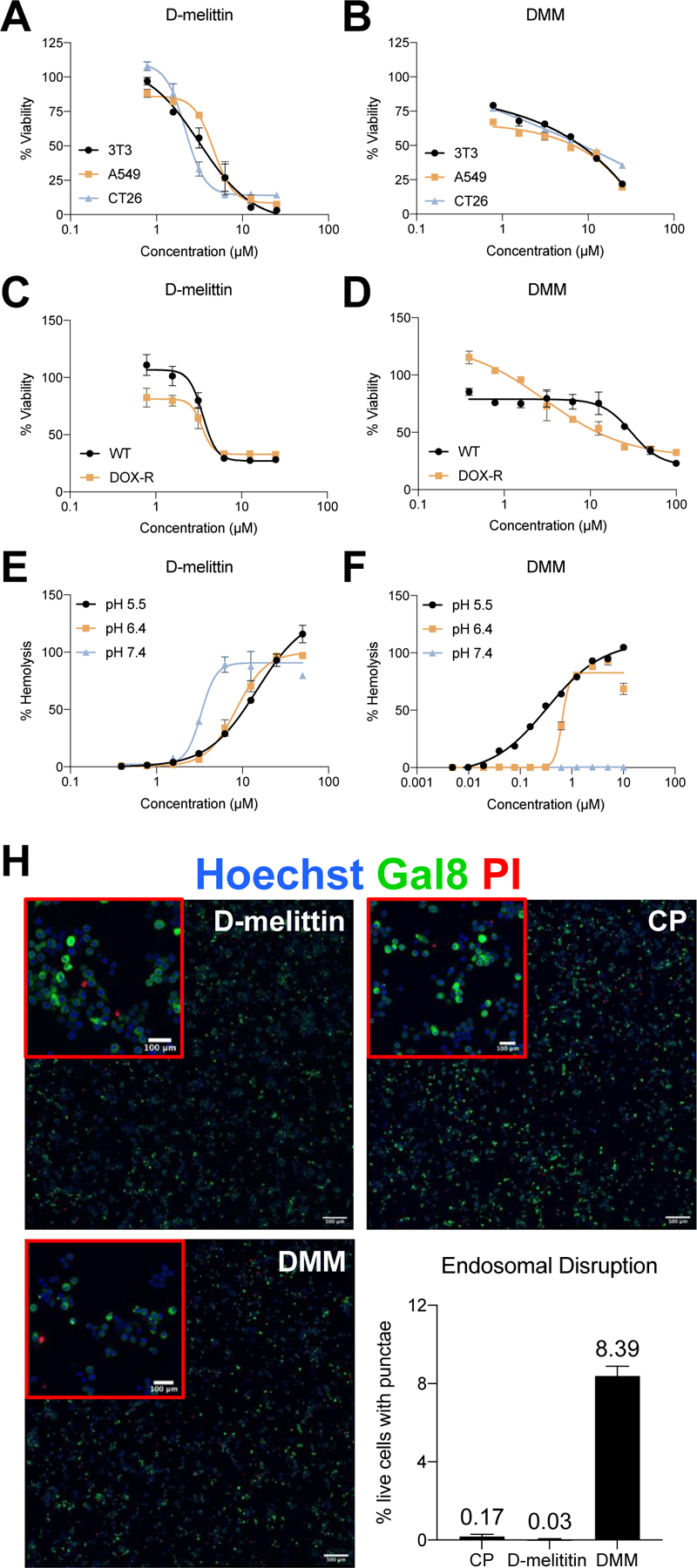
Peptide and micelle activity in vitro. A-B) D-melittin peptide (A) and micelles (B) were incubated with human and murine tumor cells (3T3, A549, and CT26) for 24 hours, and viability was assessed via MTS assay. C-D) D-melittin peptide (C) and micelles (D) were incubated with both wild type (WT) and doxorubicin-resistant (DOX-R) MDA-MB-435 cells for 24 hours, and viability was assess via MTS assay. E-F) Hemolytic activity of peptide (E) and (F) micelles was determined against red blood cells (RBCs) at pH 5.4, 6.4, and 7.4. H) Endosomal escape of micelles was demonstrated in Gal8-GFP-RAW 264.7 macrophages (500 µm scale). Endosomal disruption is observed as green punctae, as Gal8-GFP+ binds to the inner membrane of endosomes. Image insets (red) are magnification of cells (100 µm scale).
Table 1.
Toxicity of D-melittin peptide and micelles against a range of human and murine tumor cell lines. For DMM, the concentration is expressed as containing a peptide equivalent to free peptide.
| Viability IC50 (µM) | ||
|---|---|---|
| Cell line | D-melittin | DMM |
| 3T3 | 3.2 | 8.5 |
| A549 | 4.5 | 6.9 |
| CT26 | 2.2 | 11.6 |
| MDA-MB-435 WT | 3.5 | 30.1 |
| MDA-MB-435 DOX-R | 3.4 | 3.0 |
Table 2.
Hemolytic activity of D-melittin peptide and micelles at pH 5.4, 6.4, and 7.4. For DMM, the concentration is expressed as containing a peptide equivalent to free peptide.
| Hemolysis HC50 (µM) | |||
|---|---|---|---|
| Entry | pH 5.4 | pH 6.4 | pH 7.4 |
| D-melittin | 14.8 | 8.5 | 3.3 |
| DMM | 0.33 | 0.65 | N/A |
3.3. Characterization of immunogenic cell death (ICD)
Based on prior reports of melittin-induced NLRP3 inflammasome activation, we next investigated if D-melittin peptide and micelles could induce immunogenic cell death (ICD), as evaluated by the three major hallmarks of ICD: surface expression of calreticulin (CRT) and extracellular release of high mobility group protein B1 (HMGB1) and ATP (Fig. 3A) [23, 29, 30]. These events occur during (CRT, ATP release) and after (HMGB1) cellular apoptosis, and thus were evaluated at 24 and 48 hours post-incubation[31]. To assess translocation of CRT from the nucleus to the cell surface, CT26 cells were incubated with peptide or micelles for 24 hours and stained with an anti-CRT antibody (Fig. 3B). In contrast to micelles without peptide (CP), both peptide (****p < 0.0001) and micelles (****p < 0.0001) induced significantly higher CRT surface-expression. Extracellular HMGB1 was assessed in cell supernatant via ELISA after 48 hour incubation with peptide or micelles at IC50 concentrations (Fig. 3C). Both D-melittin (**p = 0.0031) and DMM (****p < 0.0001) induced a significantly higher increase in HMGB1 release over CP-treated cells, and DMM induced significantly higher release compared to D-melittin (****p < 0.0001). Similarly, a robust increase in extracellular ATP (24 hour incubation) was observed in DMM-treated cells, while ATP release in D-melittin and CP-treated cells was negligible (Fig. 3D). Together, these results suggest that DMM is capable of inducing ICD in cancer cells.
Fig. 3.
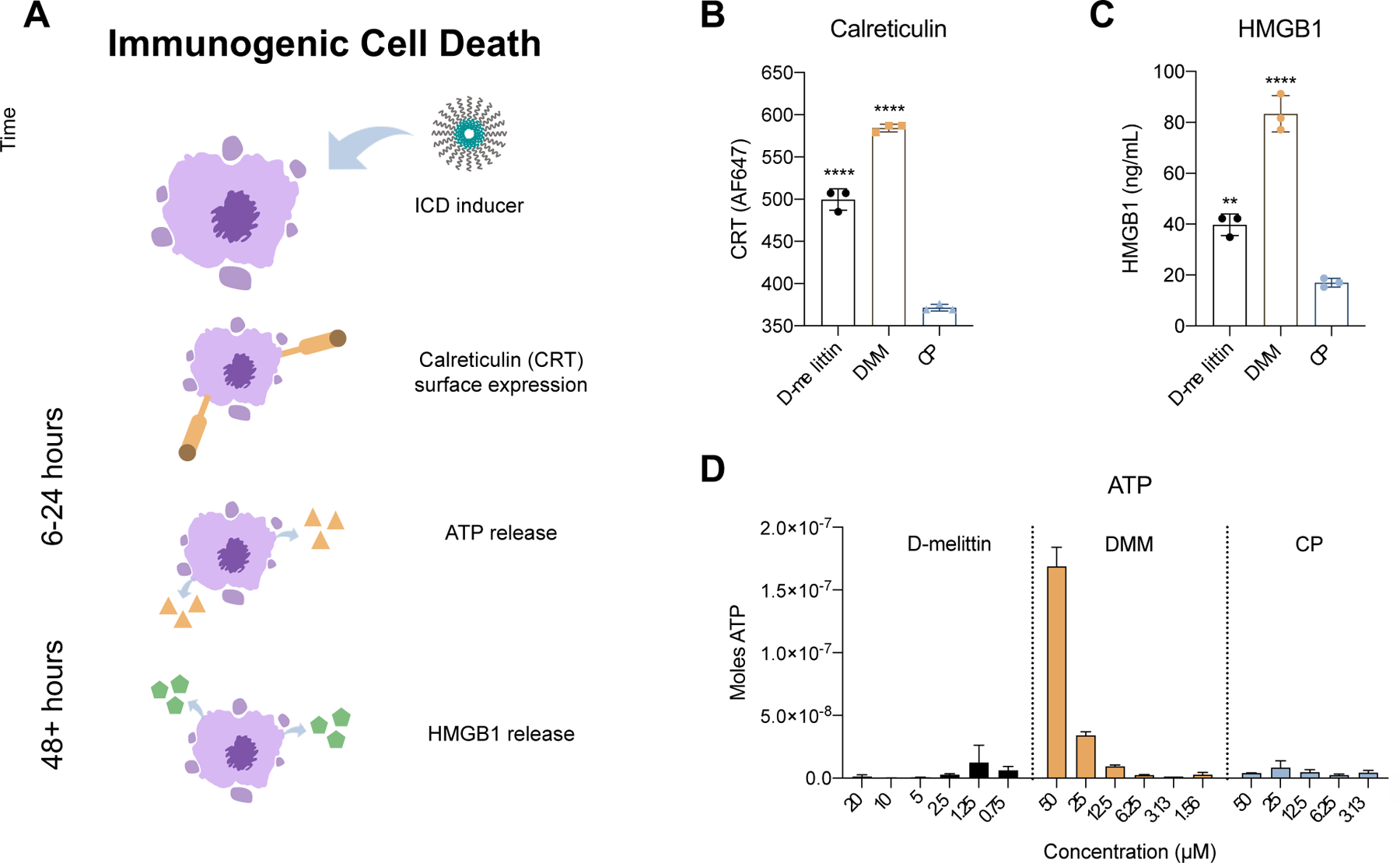
Evaluation of immunogenic cell death in CT26 cancer cells. A) A schematic of the primary hallmarks of ICD: calreticulin (CRT) surface expression and ATP and HMGB1 release. Translocation of CRT to the cell surface and ATP release occur in pre-apoptotic and dying cells, and occur over a time course of hours. HMGB1 release occurs in dying and dead cells and occurs over a time course of days. B) Cells were incubated with peptide or micelles for 24 hours and surface expression of CRT was measured via flow cytometry. C) Extracellular HMGB1 concentration was measured in cell supernatant via ELISA after 48 hour incubation with peptide or micelles. Significance is calculated via ANOVA and denoted in regards to CP. D) Extracellular ATP was measured in cell supernatant via luciferin reaction.
3.4. Safety of free peptide and micelles in vivo
We next evaluated the maximum tolerated dose (MTD) of free peptide and micelles in vivo to determine appropriate doses for anti-cancer treatment (Table 3). MTD was determined by injecting mice intravenously (IV) with peptide or micelles and monitoring weight loss, survival, and overall disposition; mice were euthanized when euthanasia criteria (i.e. difficulty breathing) were met. The MTD of micelles in normal mice was previously determined to be 20 mg peptide/kg (an equivalent of 125 mg micelles/kg) [24]. Due to the highly lytic nature of free peptide, the range of tested doses was 10-fold lower; normal mice were injected with D-melittin at 2, 4, 6, and 8 mg/kg. While all mice survived an injection of peptide at 4 mg/kg, this dose was poorly tolerated (lethargy, poor grooming), and the MTD was determined to be 2 mg/kg, which is 10-fold lower than that of micelle-encapsulated peptide.
Table 3.
MTD of D-melittin and D-melittin micelles. Normal mice were injected with a range of concentrations and sacrificed when euthanasia criteria were met. While mice survived following peptide injection at 4 mg/kg, this dose was poorly tolerated. The MTD of free peptide and micelles was determined to be 2 mg/kg and 20 mg peptide/kg, respectively. For DMM, the dose is expressed in regards to peptide amount in the formulation. (n = 4 mice/group)
| D-melittin free peptide | DMM | ||
|---|---|---|---|
|
| |||
| Dose | Deaths | Dose | Deaths |
| 2 mg/kg | 0/4 | 10 mg/kg | 0/4 |
| 4 mg/kg | 0/4 | 20 mg/kg | 0/4 |
| 6 mg/kg | 4/4 | 30 mg/kg | 2/4 |
| 8 mg/kg | 4/4 | 40 mg/kg | 4/4 |
| MTD | 2 mg/kg | MTD | 20 mg/kg |
3.5. Safety of particles upon repeat injection
Although we have previously established that the incorporation of D-amino acids abrogated the generation of anti-PEG antibodies and extended survival, we additionally sought to confirm safety of these particles for repeat injection via hematoxylin and eosin (H&E) staining and analysis of serum ALT/AST enzymes[24]. Mice were injected every 4th day with (i) PBS, (ii) CP, (iii) D-melittin 2 mg/kg, or (iv) DMM 5 mg peptide/kg for a total of 3 injections. Twenty-four hours following the final injection, blood was drawn and major organs (heart, lung, liver, kidney, spleen) were harvested and submitted for H&E processing (Fig. 4). Slides were evaluated by a blinded veterinary pathologist and changes were graded on a 1 (minimal) to 4 (severe) scale (Table S1). Overall, no clinically significant treatment-related changes were noted. This was supported by activity of ALT/AST enzymes in the serum, which was all within normal range (Fig. 5).
Figure 4.
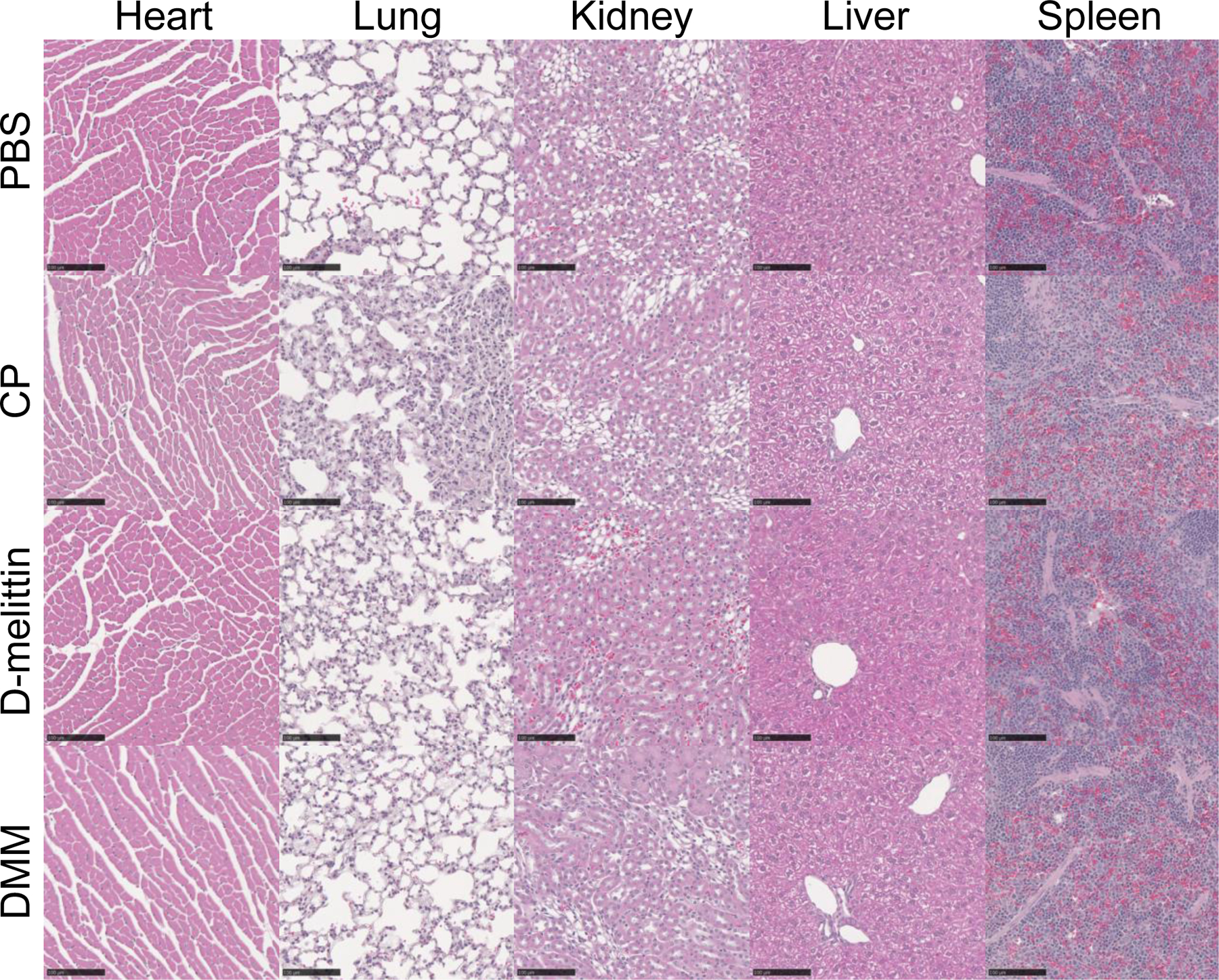
H&E staining of major organs after repeat injection of peptide and particles. Mice were injected IV with (i) PBS, (ii) CP, (iii) D-melittin 2 mg/kg or (iv) DMM 5 mg peptide/kg every 4th day for a total of 3 injections. Twenty-four hours after the 3rd injection, organs were harvested and processed for H&E staining, and evaluated by a blinded third-party. Overall, there were no identified treatment-related changes. The black scale bar represents 100 µm. (n = 2 mice/group)
Figure 5.
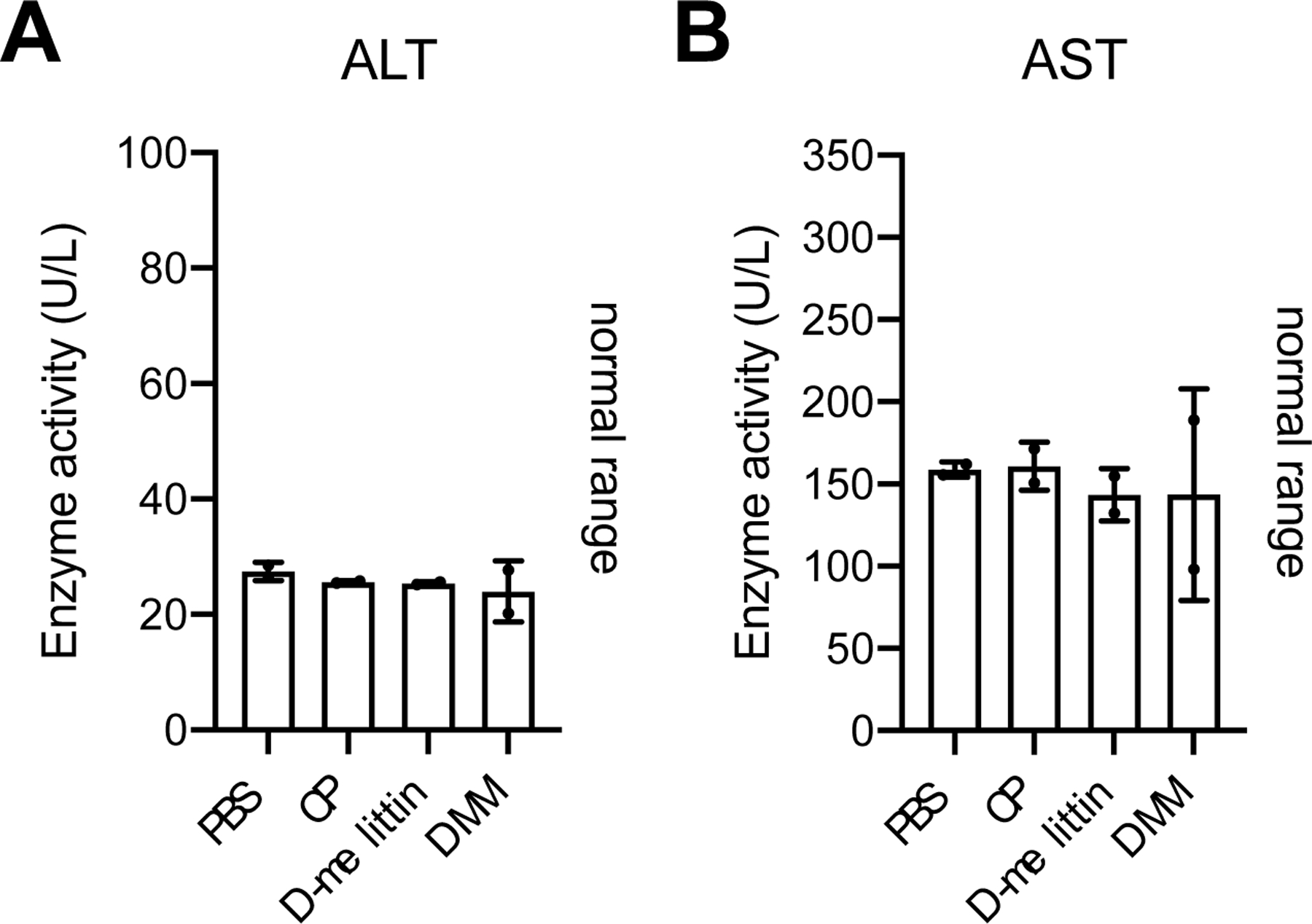
Serum ALT/AST activity. The activity of ALT (A) and AST (B) enzymes in the serum was evaluated via ELISA. All values were within normal range. (n = 2 mice/group)
3.6. Anti-tumor efficacy of D-melittin peptide and micelles in vivo
After determining safe, injectable doses of free peptide and micelles, we next compared anti-tumor efficacy of each in a CT26 colon cancer model. Eight days after subcutaneous tumor inoculation (1M cells) in the right inguinal flank, mice were injected IV every 4th day with (i) PBS, (ii) D-melittin 2 mg/kg, or (iii) DMM 5 mg peptide/kg for a total of 3 treatments (Fig. 6A). We have previously observed that polymer without melittin (CP) has no anti-tumor efficacy (Fig. S4) in vivo, so we did not include CP-treatment in this study. While D-melittin-treated mice had smaller tumors than PBS-treated mice (***p = 0.0009), there was no difference in survival between the groups (p = 0.2265) (Fig. 6B-C). In contrast, DMM-treated mice had significantly smaller tumors than both PBS (****p < 0.0001) and peptide- (****p < 0.0001) treated mice, as well as extended survival (**p = 0.045). D-melittin was dosed at its MTD, which was largely ineffective at inhibiting tumor growth. On the other hand, polymer encapsulation of D-melittin permitted higher peptide dosing (5 mg peptide/kg), which was sufficient to slow tumor growth and thus enhance anti-tumor efficacy. Together, this data demonstrates the poor anti-cancer efficacy (tumor reduction, survival) of free peptide and the potential of DMM for cancer treatment.
Figure 6.
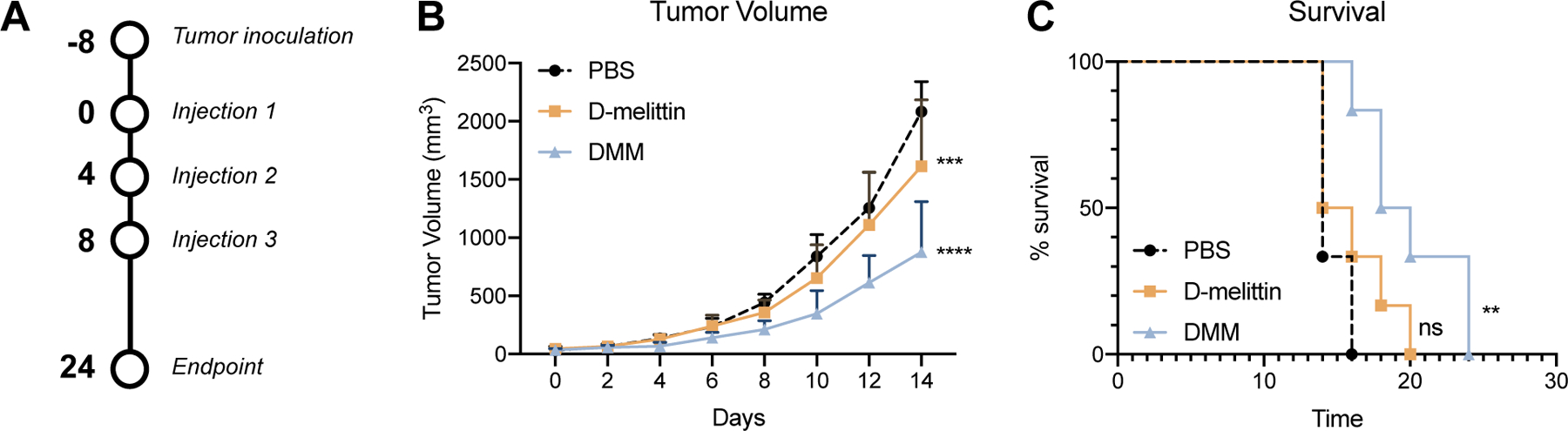
Evaluation of anti-tumor effects of free peptide and micelles in a CT26 tumor model. A) Mice were inoculated subcutaneously with CT26 tumor cells (1M). Eight days later, mice were injected IV with (i) PBS, (ii) D-melittin 2 mg/kg, or (iii) DMM 5 mg peptide/kg every 4th day for a total of 3 injections. B) Tumor volume was measured every 2nd day. Significance is denoted in comparison to PBS-treated mice, calculated with a two-way ANOVA. D-melittin-treated mice had significantly smaller (***p = 0.0009) tumors than PBS mice, and DMM-treated mice had significantly smaller tumors than PBS-treated (****p < 0.0001) and D-melittin-treated (****p < 0.0001) mice. C) Survival of mice after treatment. Mice were sacrificed when euthanasia criteria were met (e.g., tumor volume > 10% of body weight). Significance is denoted in comparison to PBS-treated mice and was calculated with a Gehan-Breslow-Wilcoxon test. There was no significance (ns) in survival between PBS- and D-melittin treated groups, whereas DMM mice survived significantly longer PBS- (**p = 0.0029) and D-melittin (*p = 0.045) treated mice. (n = 6 mice/group)
3.7. Anti-tumor efficacy of combinatorial DMM&ICB treatment
We next investigated anti-tumor efficacy of DMM in combination with immune checkpoint blockade (ICB) therapy, anti-CTLA-4 and anti-PD-1 antibodies, in murine breast (4T1) and colon (CT26) cancer models. In this work (section 3.3), we demonstrated that DMM induced ICD in vitro, which provided a beneficial anti-tumor effect. 4T1 and CT26 tumor models were used because they have high immune infiltration compared to other syngeneic murine models and have moderate to low mutational burden, and thus respond to ICB therapy to different extents [32]. ICB therapy has marked response in CT26 tumors, whereas 4T1 tumors are more resistant to treatment [33]. These two tumor models enabled us to differentiate synergistic efficacy of our treatment from tumor-specific responses. Overall, we hypothesized that DMM could induce ICD of tumor cells which could subsequently activate tumor infiltrating leukocytes (TIL) and synergize with ICB to disrupt negative immune regulatory checkpoints, ultimately resulting in robust immune activation and tumor growth inhibition.
Eight days after inoculation, tumor-bearing mice were treated with (i) PBS, (ii) ICB 100 µg/mouse, (iii) DMM 5 mg peptide/kg or (iv) DMM 5 mg peptide/kg + ICB 100 µg/mouse (Fig. 7A). Micelles were injected every 4th day IV and ICB treatment was administered intraperitoneally (IP) on days following DMM treatment. In a 4T1 tumor model, ICB and DMM alone had no efficacy in slowing tumor growth, while combinatorial treatment of DMM+ICB significantly reduced tumor growth compared to PBS (****p < 0.0001), ICB (****p < 0.0001), and DMM (****p < 0.0001) (Fig. 7B).
Figure 7.
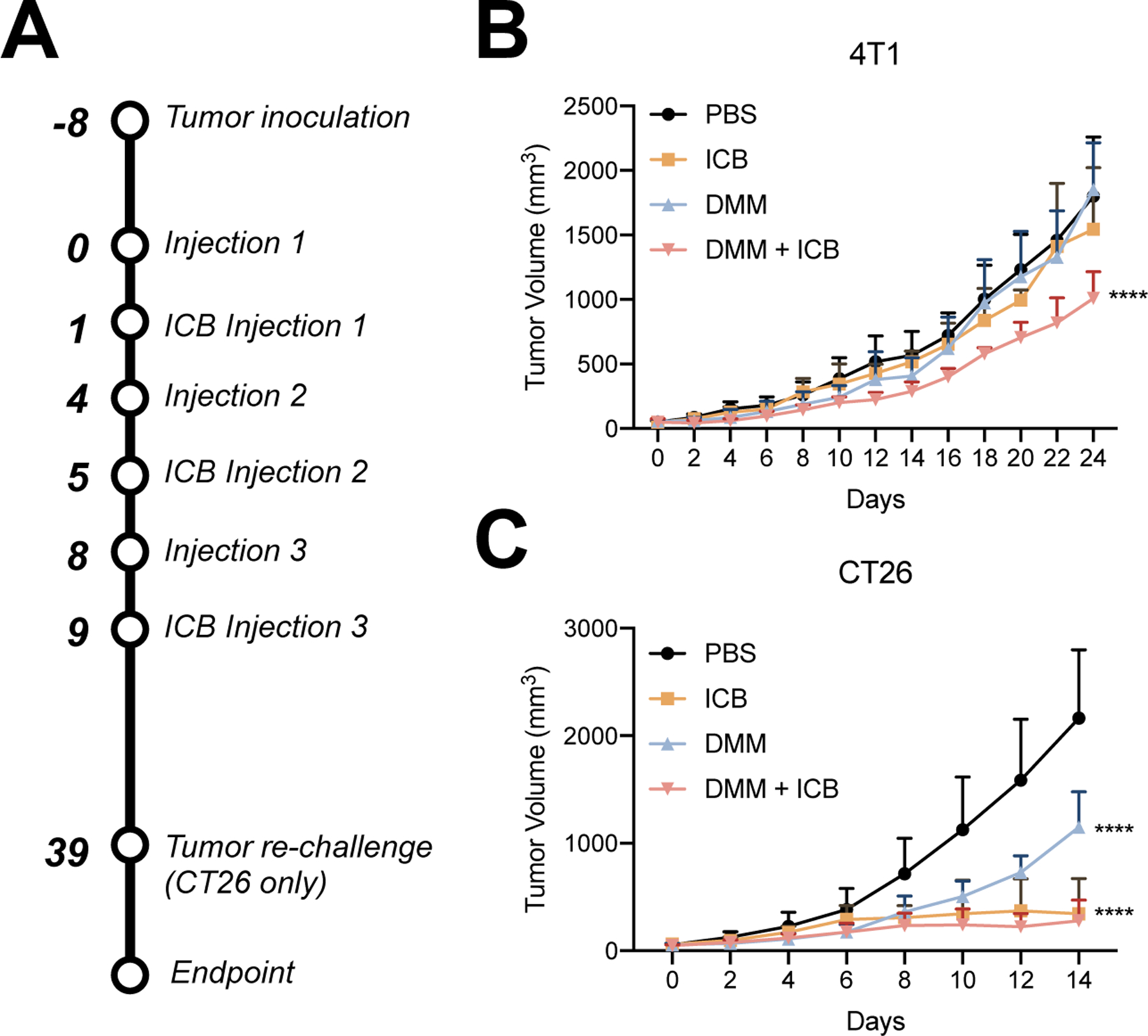
Tumor reduction with combinatorial ICB in 4T1 and CT26 tumors. A) Time line of tumor inoculation and treatment. B) 4T1 tumor growth in mice treated with (i) PBS, (ii) ICB, (iii) DMM, or (iv) DMM+ICB. Combinatorial treatment of DMM+ICB significantly reduced tumor growth compared to PBS (****p < 0.0001), ICB (****p < 0.0001), and DMM (****p < 0.0001). Significance was calculated via two-way ANOVA and is denoted in regards to PBS. C) CT26 tumor growth in mice treated with (i) PBS, (ii) ICB, (iii) DMM, or (iv) DMM+ICB for the first 14 days after treatment. Mice receiving ICB and DMM+ICB treatment had significantly smaller tumors than mice treated with PBS- (****p < 0.0001) or DMM- (****p < 0.0001). There was no significance (ns) between ICB and DMM+ICB mice. Significance was calculated via two-way ANOVA and is denoted in regards to PBS. (n = 6 mice/group)
In CT26 tumor-bearing mice, DMM showed mild efficacy in reducing tumor growth (****p < 0.0001) compared to PBS, while ICB and DMM+ICB were robustly able to halt tumor growth and in some mice, completely eliminate tumors (Fig. 7C). Mice in the ICB and DMM+ICB cohorts had significantly smaller tumors than PBS- (****p < 0.0001) and DMM- (****p < 0.0001) treated mice. However, there was no difference in tumor growth between ICB and DMM+ICB treated mice (p = 0.9619). Statistical significance was evaluated at 14 days following treatment; shortly after this timepoint, mice in PBS and DMM groups were euthanized due to increasing tumor burden and statistical tests could not be performed. In the ICB and DMM&ICB cohorts, 3 and 2 mice, respectively, were cured entirely of tumors. To investigate if treatment resulted in anti-tumor immune memory, mice were re-challenged with CT26 cells (1M) under the opposing inguinal nipple. None of these mice grew additional tumors, suggesting that ICB treatment, alone or in combination with DMM, can induce anti-tumor memory.
4. Discussion
Here, we report the first development of D-melittin nanoformulations for anti-tumor applications. We previously reported that replacement of L-amino acids with D-amino acids in polymer-peptide delivery systems abrogates the generation of anti-polymer antibodies with minimal effect on activity of the polymer-peptide platform [24]. This work opened up the possibility of evaluation D-melittin as an anti-cancer agent in a polymer-drug conjugate formulation that minimizes non-specific cytolysis without immunogenicity resulting from repeated dosing. Here, we show that D-melittin micelles (DMM) exert broad anti-tumor activity against various types of tumor cells, even cells that have developed resistance against traditional chemotherapeutics. This enhanced efficacy could be attributed to increased susceptibility of chemoresistant cells to compounds that physically disrupt the plasma membrane; the pumps that confer drug resistance also confer greater fragility to the cell membrane.[34] In normal mice, DMM was safe for repeat systemic administration and could be administered at 10-fold higher dose compared to free peptide. Furthermore, DMM synergized with immune checkpoint blockade (ICB) and significantly inhibited tumor growth in tumor-bearing mice. These findings have broad applicability for all polymer-peptide conjugates, as it demonstrates that D-amino acid peptides can enhance platform safety while retaining tumoricidal activity in vivo.
We have pursued the use of D-melittin due to enhanced safety in vivo, despite exhibiting slightly reduced lytic capacity compared to L-melittin in vitro [24]. Conjugation of peptide to the polymer endows the peptide with acute pH-responsive behavior while also enhancing the lytic activity of the peptide at acidic conditions. Furthermore, D-amino acids can potentially enhance endosomal escape, as D-peptides persist longer than L-peptides within the cell [35]. Najjar, et al. demonstrated the enhanced endosomolytic behavior with D- and L- analogs of dimeric fluorescent (df) TAT, a cell penetrating peptide, and showed that the heightened protease stability of D-dfTAT prolonged intracellular retention and accumulation within late endosomes, ultimately resulting in more severe endosomal leakage and disruption of transcriptional programming[35]. Furthermore, the authors discuss how endocytosis and endosomal escape were impacted in opposite ways by peptide chirality: D-dfTAT was less prone to cellular internalization than L-dfTAT but exhibited increased endosomal escape. This reduced propensity of D-peptides for uptake can likely be attributed to reduced capacity to trigger endocytosis rather than a difference in binding to heparan sulfates [36]. However, our drug delivery system bypasses this entirely, as the polymer is in micellar form during cellular internalization and transitions to a linear form in the endosome, unsheathing the peptide and capitalizing on the amplified endosomolytic activity of D-peptides.
Furthermore, incorporation of D-melittin enabled application of our platform for systemic injections. Although the potential of melittin as a cancer therapeutic has been established, its severe nonspecific lytic activity poses significant challenges for clinical translation. Our work here adds to the repertoire of strategies to systemically deliver melittin and other lytic peptides for cancer treatment [15, 37]. This vastly expands the scope of use of lytic ACPs, which are often constrained to intra-tumoral delivery to avoid related toxicity from systemic delivery [16, 38]. This could be particularly impactful to treatment of metastatic cancers, which are disseminated in distant locations from the primary tumor and do not benefit from enhanced permeation and retention (EPR) within large tumors [39]. Due to the controversial relevance of the EPR effect in humans and high variability of its effect between patients and cancer types, safe systemic delivery of lytic peptides is also valuable for treatment of primary tumors [40].
We next characterized if D-melittin induced immunogenic cell death (ICD), a form of inflammatory cell death that releases danger associated molecular patterns (DAMPs) which can elicit an anti-cancer immune response[41]. We expected melittin peptide and micelles to induce ICD, as L-melittin has been reported to induce NLRP3 inflammasome activation and production of IL-1β [23]. While melittin bypasses pyroptosis, we hypothesized that melittin could induce other forms of ICD. In this report, we verified that D-melittin and DMM induced surface expression of calreticulin (CRT) and extracellular release of ATP and HMGB1. Apoptotic cells traffic CRT from the endoplasmic reticulum to the cell surface, where the exposed CRT serves as an “eat me” signal to antigen presenting cells (APCs), which can induce subsequent tumor antigen presentation and tumor-specific responses [42]. This step usually precedes morphological signs of apoptosis and occurs in the early stages of ICD [31, 41]. Dying cells also release ATP, which serves as a “find me” signal to APCs and triggers NLRP3-inflamamsome-based cytokine secretion. Similarly, HMGB1 release acts as an attractant for various immune cells and can induce dendritic cell maturation and stimulate the production of pro-inflammatory cytokines from innate immune cells. HMGB1 is released by cells during the later stages of ICD [31, 41]. Here, we observed increased CRT expression, ATP secretion, and HMGB1 in melittin-treated groups. In fact, DMM induced significantly higher markers of ICD compared to free peptide; this effect is not attributed to the polymer, as CP did not induce hardly any indication of these events. Rather, ICD is likely initiated by cytosolic delivery of melittin. These results synergize with our aforementioned conclusions that polymer-conjugation enhanced the lytic activity of the peptide, and that the use of D-amino acids contributes to its robust capacity for intracellular delivery. While the extent of ICD was rather modest, we nonetheless concluded that DMM can induce ICD in cancer cells. This modest efficacy could potentially be linked with the timing of endosomal disruption. Rupture of early endosomes facilitates improved cytosolic delivery and minimal NLRP3 inflammasome activation, whereas rupture of late endosomes/lysosomes resulted in reduced cytosolic delivery and strong NLRP3 inflammasome activation [43]. As the VIPER platform was initially designed to optimize endosomal escape for nucleic acid delivery, the modified DMM platform may have highest activity in early endosomes, minimizing inflammasome activity. Polymer incorporation of specific amino acids (e.g. pre-defined ratio of histidine:tryptophan) has been reported to increase the degree of lysosome rupture and thus the degree of inflammasome activation [44]. Such design considerations can be applied to our delivery platform to promote both cytosolic delivery and inflammasome activation for ICD. We could also employ a different pH-sensitive monomer that has a pH-transition point a lower pH and thus is activated in late endosomes, promoting inflammasome activation [45]. However, the exact correlation between the timing and minimal stimuli requirements for inflammasome activation is unclear, so the modified platform would require optimization.
Melittin-induced death is classically characterized by cell membrane disruption and pore formation, resulting in osmotic cell lysis and death [15]. However, the mechanism of death has also been connected with disruption of the mitochondrial membrane potential, similar to the KLA peptide, and with the toxic intracellular accumulation of Ca2+, either via calmodulin inhibition or L-type Ca2+ activation [46]. The exact mechanism by which melittin mediates the increase of intracellular calcium concentrations has yet to be confirmed.
After characterizing our platform in vitro, we measured the safety of free peptide and micelles in vivo after single and repeat (total of 3, every 4th day) injections. The MTD of D-melittin was determined to be 2 mg/kg, which is 10-fold lower than the MTD of DMM (20 mg peptide/kg), further emphasizing how polymer encapsulation remarkably expanded the therapeutic index of D-melittin. Polymer conjugation increased the maximum injectable dose by 10-fold (the top of the therapeutic window), while only affecting the efficacious drug concentration by 1.5- to 5-fold (the bottom of the therapeutic window), as shown for the cancer cell lines tested in Table 1.
Upon characterizing activity in vitro and safety in vivo, we compared activity of D-melittin and DMM in CT26 tumor-bearing mice and demonstrated that free peptide had poor anti-tumor efficacy. Although the MTD of DMM is 10-fold higher, we selected a lower dose 5 mg/kg due to increased efficacy of the particles compared to free peptide. While D-melittin had modest reduction of tumor growth, DMM significantly reduced tumor growth and extended survival compared to PBS- and D-melittin-treated mice. The dose-limiting toxicity of free-peptide likely prevented sufficient amounts of peptide from reaching tumor cells. Furthermore, peptides suffer from poor pharmacokinetics in vivo; while D-amino acids can prolong circulation by reducing degradation by proteases, the small size (~2 kD) of the peptide means it is readily filtered by the kidney[47]. On the other hand, polymer encapsulation enhances the dose that can safely be administered while also increasing the molecular weight to avoid renal filtration [48]. This is well demonstrated in this study, as we can safely deliver a sufficiently high dose of D-melittin to inhibit tumor growth in mice. Application of neutral PEGylated micelles for tumor targeting is widely reported, as the PEG coating enhances biodistribution and the neutral charge reduces macrophage uptake and increases tumor accumulation [49]. We observed that particles primarily accumulated in the lungs, liver, and spleen, (as expected, as they are major clearance organs), with minor accumulation in tumors that decreased over 6 hours (Fig. S6). While the exact uptake mechanism of the PEGylated micelles by tumor cells is not elucidated here, we can make inferences about cell-particle interactions based on prior reports. PEG is historically employed to prolong in vivo circulation by shielding particles from the reticuloendothelial system, facilitating tumor accumulation and uptake [50]. Still, not all PEG-coatings are equivalent, and factors such as chain length and density differentially influence cellular uptake. In micelles with mixed PEG chain lengths, a higher proportion of short chains increased protein adsorption and nanoparticle coalescence, prompting cellular uptake [51]. In another study, similar neutral, PEG-micelles were internalized by cells within 2 hours, and these particles were transported through cell monolayers [52]. Further investigation revealed that these interactions occurred through various uptake mechanisms, including clathrin-, energy-, and cholesterol-mediated endocytosis.
Lastly, we investigated if DMM synergized with ICB treatment in 4T1 and CT26 tumor-bearing mice. These tumors have varying mutational burden, which means that they respond to ICB therapy to differing degrees[32, 33]. While the robust efficacy of ICB in CT26 tumors could perhaps dampen some enthusiasm for the DMM platform, we argue that DMM holds greatest potential in tumor models that are resistant to ICB treatment or traditional chemotherapy treatments. Perhaps melittin is more cytotoxic against cells that are transformed or mutated; this could be an interesting avenue for additional investigation as it has not yet been reported. We also characterized tumor-infiltrating leukocyte (TIL) populations after treatment with 3 doses of peptide and conjugates, but did not detect substantial differences in TILs between groups under the current dosing conditions (Fig. S5). Perhaps iteration on the particle design to enhance inflammasome activation, as well as optimization of dosing conditions, could significantly modulate TIL populations.
Additional future work on this project includes incorporation of targeting ligands into the hydrophilic block of the polymer to further promote specific targeting and uptake. While the nanoparticle’s physical particles largely drive its accumulation in tumors, conjugation with targeting ligands can promote interactions with and uptake by tumor cells and enhance cell killing. We will also evaluate antitumor efficacy at higher DMM peptide concentrations, closer to its MTD (20 mg peptide/kg).
5. Conclusion
In summary, we report the first application of D-melittin for cancer therapy, emphasizing the promising potential of D-amino acids for in vivo therapeutic use. Melittin has drawn extensive attention as a potent natural ACP with broad anti-tumor activity, but its nonspecific hemolytic activity severely limited its in vivo applications. And though various delivery systems curb melittin’s nonspecific toxicity, the peptide’s potent immunogenicity facilitates the generation of a robust immune response against the carrier, further hampering its in vivo applications. Our lab addressed this hurdle by (1) employing VIPER, a pH-sensitive platform to facilitate controlled intracellular peptide delivery, and (2) replacing L-amino acids with D-amino acids, which transforms our technology into a safe delivery platform. Ultimately, we have designed a system that dually overcomes the challenge of peptide toxicity and immunogenicity, allowing us to recognize the full potential of melittin for cancer treatment.
In the context of ACPs, D-amino acid peptides are a largely unexplored territory[53]. There is an overall dearth of research on D-amino acids for cancer therapy, although the benefit of D-amino acids to markedly improve peptide stability and protease resistance in vivo has already been established[54]. Our findings first demonstrate that D-amino acids significantly enhance safety of peptide-polymer conjugates in vivo without compromising anti-cancer efficacy, which will benefit further development of D-amino acid peptides for cancer treatment.
Supplementary Material
Highlights:
D-melittin peptide and micelles demonstrate a broad spectrum of antitumor activity against various types of transformed cells regardless of drug resistance.
D-melittin micelles induce immunogenic cell death, as characterized by calreticulin exposure, ATP secretion, and HMGB1 release
Polymer-mediated delivery of D-melittin inhibits CT26 and 4T1 tumor growth in mice with minimal side effects
D-melittin micelles synergizes with immune checkpoint blockade to provide lasting tumor elimination and anti-tumor immune memory in mice
Acknowledgements
This work was supported by the U.S. National Institutes of Health (NIH R01CA17727, R01CA257563, and U54CA199090). We thank Nathanial Peters and W. M. Keck Microscopy Center (S10 OD016240) for confocal microscopy support. We thank Kim Woodrow (University of Washington) for use of her plate reader. We thank Nora Disis (University of Washington) for her generous donation of CT26 cells and Jordan Green (Johns Hopkins University) for kindly providing the Gal8-GFP and PiggyBac transposon plasmids.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
S.L., M.S., and S.H.P. conceived of this work, S.L., M.S., S.H.P. designed experiments, S.L., M.S., K.S. conducted experiments, S.L., M.S., analyzed data, S.L. and M.S. wrote the manuscript with feedback from all authors, S.H.P. acquired funding.
Competing Interests
S.H.P. has submitted a patent with the U.S. Patent and Trademark Office (WO2018027164A1) related to VIPER. S.H.P., S.L., and M.S. have filed a provisional patent on the DMM technology.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
References
- [1].Kurrikoff K, Aphkhazava D, Langel U. The future of peptides in cancer treatment. Curr Opin Pharmacol 2019;47:27–32. [DOI] [PubMed] [Google Scholar]
- [2].Aaghaz S, Gohel V, Kamal A. Peptides as Potential Anticancer Agents. Curr Top Med Chem 2019;19:1491–511. [DOI] [PubMed] [Google Scholar]
- [3].Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today 2015;20:122–8. [DOI] [PubMed] [Google Scholar]
- [4].Ren LF, Lv J, Wang H, Cheng YY. A Coordinative Dendrimer Achieves Excellent Efficiency in Cytosolic Protein and Peptide Delivery. Angew Chem Int Edit 2020. [DOI] [PubMed]
- [5].Qiao ZY, Lai WJ, Lin YX, Li D, Nan XH, Wang Y, et al. Polymer-KLAK Peptide Conjugates Induce Cancer Cell Death through Synergistic Effects of Mitochondria Damage and Autophagy Blockage. Bioconjugate Chem 2017;28:1709–21. [DOI] [PubMed] [Google Scholar]
- [6].Gianneschi NC, Sun H, Cao W, Zang N, Clemons T, Scheutz G, et al. Proapoptotic Peptide Brush Polymer Nanoparticles via Photoinitiated Polymerization-Induced Self-Assembly. Angew Chem Int Ed Engl 2020. [DOI] [PMC free article] [PubMed]
- [7].Felicio MR, Silva ON, Goncalves S, Santos NC, Franco OL. Peptides with Dual Antimicrobial and Anticancer Activities. Front Chem 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Srairi-Abid N, Othman H, Aissaoui D, BenAissa R. Anti-tumoral effect of scorpion peptides: Emerging new cellular targets and signaling pathways. Cell Calcium 2019;80:160–74. [DOI] [PubMed] [Google Scholar]
- [9].Araste F, Abnous K, Hashemi M, Taghdisi SM, Ramezani M, Alibolandi M. Peptide-based targeted therapeutics: Focus on cancer treatment. J Control Release 2018;292:141–62. [DOI] [PubMed] [Google Scholar]
- [10].Lv S, Sylvestre M, Prossnitz AN, Yang LF, Pun SH. Design of Polymeric Carriers for Intracellular Peptide Delivery in Oncology Applications. Chemical reviews 2021. [DOI] [PubMed]
- [11].Liu CC, Hao DJ, Zhang Q, An J, Zhao JJ, Chen B, et al. Application of bee venom and its main constituent melittin for cancer treatment. Cancer Chemoth Pharm 2016;78:1113–30. [DOI] [PubMed] [Google Scholar]
- [12].Duffy C, Sorolla A, Wang E, Golden E, Woodward E, Davern K, et al. Honeybee venom and melittin suppress growth factor receptor activation in HER2-enriched and triple-negative breast cancer. Npj Precis Oncol 2020;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Memariani H, Memariani M, Moravvej H, Shahidi-Dadras M. Melittin: a venom-derived peptide with promising anti-viral properties. Eur J Clin Microbiol 2020;39:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Peeler DJ, Yen A, Luera N, Stayton PS, Pun SH. Lytic Polyplex Vaccines Enhance Antigen-Specific Cytotoxic T Cell Response through Induction of Local Cell Death. Advanced Therapeutics
- [15].Rady I, Siddiqui IA, Rady M, Mukhtar H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett 2017;402:16–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yu X, Dai YF, Zhao YF, Qi SH, Liu L, Lu LS, et al. Melittin-lipid nanoparticles target to lymph nodes and elicit a systemic anti-tumor immune response. Nat Commun 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu HJ, Hu Y, Sun YJ, Wan C, Zhang ZJ, Dai XM, et al. Co-delivery of Bee Venom Melittin and a Photosensitizer with an Organic-Inorganic Hybrid Nanocarrier for Photodynamic Therapy and Immunotherapy. Acs Nano 2019;13:12638–52. [DOI] [PubMed] [Google Scholar]
- [18].Cao J, Zhang Y, Shan YK, Wang JG, Liu F, Liu HR, et al. A pH-dependent Antibacterial Peptide Release Nano-system Blocks Tumor Growth in vivo without Toxicity. Scientific Reports 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cheng B, Xu PS. Redox-Sensitive Nanocomplex for Targeted Delivery of Melittin. Toxins 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mima Y, Hashimoto Y, Shimizu T, Kiwada H, Ishida T. Anti-PEG IgM Is a Major Contributor to the Accelerated Blood Clearance of Polyethylene Glycol-Conjugated Protein. Mol Pharmaceut 2015;12:2429–35. [DOI] [PubMed] [Google Scholar]
- [21].Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm 2008;354:56–62. [DOI] [PubMed] [Google Scholar]
- [22].Shiraishi K, Yokoyama M. Toxicity and immunogenicity concerns related to PEGylated-micelle carrier systems: a review. Sci Technol Adv Mat 2019;20:324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Martin-Sanchez F, Martinez-Garcia JJ, Munoz-Garcia M, Martinez-Villanueva M, Noguera-Velasco JA, Andreu D, et al. Lytic cell death induced by melittin bypasses pyroptosis but induces NLRP3 inflammasome activation and IL-1 beta release. Cell Death Dis 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sylvestre M, Lv SX, Yang LF, Luera N, Peeler DJ, Chen BM, et al. Replacement of L-amino acid peptides with D-amino acid peptides mitigates anti-PEG antibody generation against polymer-peptide conjugates in mice. J Control Release 2021;331:142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheng YL, Yumul RC, Pun SH. Virus-Inspired Polymer for Efficient In Vitro and In Vivo Gene Delivery. Angew Chem Int Edit 2016;55:12013–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kilchrist KV, Dimobi SC, Jackson MA, Evans BC, Werfel TA, Dailing EA, et al. Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. Acs Nano 2019;13:1136–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rui Y, Wilson DR, Choi J, Varanasi M, Sanders K, Karlsson J, et al. Carboxylated branched poly(beta-amino ester) nanoparticles enable robust cytosolic protein delivery and CRISPR-Cas9 gene editing. Sci Adv 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li HM, et al. Ex Vivo Red Blood Cell Hemolysis Assay for the Evaluation of pH-responsive Endosomolytic Agents for Cytosolic Delivery of Biomacromolecular Drugs. Jove-J Vis Exp 2013. [DOI] [PMC free article] [PubMed]
- [29].Ma S, Song WT, Xu YD, Si XH, Lv SX, Zhang Y, et al. Rationally Designed Polymer Conjugate for Tumor-Specific Amplification of Oxidative Stress and Boosting Antitumor Immunity. Nano Lett 2020;20:2514–21. [DOI] [PubMed] [Google Scholar]
- [30].Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Menger L, Vacchelli E, Adjemian S, Martins I, Ma YT, Shen SS, et al. Cardiac Glycosides Exert Anticancer Effects by Inducing Immunogenic Cell Death. Sci Transl Med 2012;4. [DOI] [PubMed] [Google Scholar]
- [32].Zhong WY, Myers JS, Wang F, Wang K, Lucas J, Rosfjord E, et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. Bmc Genomics 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kim K, Skora AD, Li ZB, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. P Natl Acad Sci USA 2014;111:11774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Callaghan R, Riordan JR. Collateral Sensitivity of Multidrug-Resistant Cells to Narcotic Analgesics Is Due to Effects on the Plasma-Membrane. Bba-Biomembranes 1995;1236:155–62. [DOI] [PubMed] [Google Scholar]
- [35].Najjar K, Erazo-Oliveras A, Brock DJ, Wang TY, Pellois JP. An L- to D-Amino Acid Conversion in an Endosomolytic Analog of the Cell-penetrating Peptide TAT Influences Proteolytic Stability, Endocytic Uptake, and Endosomal Escape. J Biol Chem 2017;292:847–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Verdurmen WPR, Bovee-Geurts PH, Wadhwani P, Ulrich AS, Hallbrink M, van Kuppevelt TH, et al. Preferential Uptake of L- versus D-Amino Acid Cell-Penetrating Peptides in a Cell Type-Dependent Manner. Chem Biol 2011;18:1000–10. [DOI] [PubMed] [Google Scholar]
- [37].Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, et al. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest 2009;119:2830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Russell PJ, Hewish D, Carter T, Sterling-Levis K, Ow K, Hattarki M, et al. Cytotoxic properties of immunoconjugates containing melittin-like peptide 101 against prostate cancer: in vitro and in vivo studies. Cancer Immunol Immun 2004;53:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer 2012;12:39–50. [DOI] [PubMed] [Google Scholar]
- [40].Bjornmalm M, Thurecht KJ, Michael M, Scott AM, Caruso F. Bridging Bio-Nano Science and Cancer Nanomedicine. Acs Nano 2017;11:9594–613. [DOI] [PubMed] [Google Scholar]
- [41].Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012;12:860–75. [DOI] [PubMed] [Google Scholar]
- [42].Zhou JY, Wang GY, Chen YZ, Wang HX, Hua YQ, Cai ZD. Immunogenic cell death in cancer therapy: Present and emerging inducers. J Cell Mol Med 2019;23:4854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baljon JJ, Dandy A, Wang-Bishop L, Wehbe M, Jacobson ME, Wilson JT. The efficiency of cytosolic drug delivery using pH-responsive endosomolytic polymers does not correlate with activation of the NLRP3 inflammasome (vol 7, pg 1888, 2019). Biomater Sci-Uk 2019;7:2200-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Manna S, Howitz WJ, Oldenhuis NJ, Eldredge AC, Shen JJ, Nihesh FN, et al. Immunomodulation of the NLRP3 Inflammasome through Structure-Based Activator Design and Functional Regulation via Lysosomal Rupture. Acs Central Sci 2018;4:982–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Feng Q, Wilhelm J, Gao JM. Transistor-like Ultra-pH-Sensitive Polymeric Nanoparticles. Accounts Chem Res 2019;52:1485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ceremuga M, Stela M, Janik E, Gorniak L, Synowiec E, Sliwinski T, et al. Melittin-A Natural Peptide from Bee Venom Which Induces Apoptosis in Human Leukaemia Cells. Biomolecules 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Torchilin VP, Lukyanov AN. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov Today 2003;8:259–66. [DOI] [PubMed] [Google Scholar]
- [48].Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharmaceut 2008;5:505–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Xiao K, Li YP, Luo JT, Lee JS, Xiao WW, Gonik AM, et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 2011;32:3435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Suk JS, Xu QG, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliver Rev 2016;99:28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gao HJ, Liu JJ, Yang CH, Cheng TJ, Chu LP, Xu HY, et al. The impact of PEGylation patterns on the in vivo biodistribution of mixed shell micelles. Int J Nanomed 2013;8:4229–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hu X, Yang FF, Liu CY, Ehrhardt C, Liao YH. In vitro uptake and transport studies of PEG-PLGA polymeric micelles in respiratory epithelial cells. Eur J Pharm Biopharm 2017;114:29–37. [DOI] [PubMed] [Google Scholar]
- [53].Orafaie A, Sadeghian H, Bahrami AR, Rafatpanah H, Matin MM. Design, synthesis and evaluation of PD-L1 peptide antagonists as new anticancer agents for immunotherapy. Bioorgan Med Chem 2021;30. [DOI] [PubMed] [Google Scholar]
- [54].Hong SY, Oh JE, Lee KH. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem Pharmacol 1999;58:1775–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.