

Abstract
The high binding affinities and specificities of antibodies have led to their use as drugs and biosensors. Single-domain VHH antibodies exhibit high specificity and affinity but have higher stability and solubility than conventional antibodies as they are single-domain proteins. In this work, based on physicochemical measurements and molecular dynamics (MD) simulations, we have gained insight that will facilitate rational design of single-chain VHH antibodies. We first assessed two homologous VHH antibodies by differential scanning calorimetry (DSC); one had a high (64.8 °C) and the other a low (58.6 °C) melting temperature. We then generated a series of the variants of the low stability antibody and analyzed their thermal stabilities by DSC and characterized their structures through MD simulations. We found that a single mutation that resulted in 8.2 °C improvement in melting temperature resulted in binding affinity an order of magnitude lower than the parent antibody, likely due to a shift of conformational space explored by the single-chain VHH antibody. These results suggest that the delicate balance among conformational stability, binding capability, and conformational space explored by antibodies must be considered in design of fully functional single-chain VHH antibodies.
Subject terms: Biophysical chemistry, Proteins, Molecular biophysics, Protein design
Introduction
Antibodies are important molecules in our bodies, as they recognize foreign pathogens or antigens in a course of immune responses. The high binding affinities and specificities of antibodies also enable their use as drug candidates and biosensors. The sequences and structural features of antibodies vary depending on species1–3. For example, some antibodies from camelids have both heavy and light chain variable domains, as do conventional antibodies from humans, but camelids also have antibodies that lack the light chains that are termed single-domain VHH antibodies. Single-domain VHH antibodies combine the advantages of the specificity and affinity of conventional antibodies with high stability and solubility originating from nature of single-domain proteins4,5.
The antigen binding sites of conventional antibodies consist of six complementarity determining regions (CDRs) L1, L2, L3, H1, H2, and H3. The CDRs other than H3 adopt structures that are classified into limited conformations called canonical structures, and some residues in framework regions support these limited conformations6–12. Therefore, binding affinities can be affected by engineering not only residues in CDRs, but also residues in framework regions, as demonstrated in earlier work on antibody humanization13. CDR-H3 is located in the center of the antigen binding site, is the most diverse both in sequence and structure, and is the most critical of the CDRs to antigen recognition14–17. In contrast, the antigen binding site of single-domain VHH antibodies consists of only three CDRs. Thus, the framework regions of single-domain VHH antibodies are sometimes directly involved in recognition of antigens18–21.
Computational methods as well as in vitro library technologies are now frequently used to engineer antibodies22–31. For instance, Kiyoshi et al. computationally predicted affinity-enhancing mutations and experimentally demonstrated that some of the predicted mutations indeed improved the binding affinity of the antibody32. Olson et al. compared sequence fitness profiles, generated by computational design calculations and experimental mutagenesis, of a single-domain antibody that exhibited an unusually high melting temperature (Tm = 85.0 °C), demonstrating accuracies and limitations of current computational models33. Previous studies also demonstrated that single-domain antibodies can be engineered to improve the thermal and colloidal stability4,34,35. In one such example, based on molecular dynamics (MD) simulations, Bekker et al. showed that the fraction of native contacts, or Q-value, that had been employed in studies of protein folding36 could be used as an evaluation metric to identify residues important for thermal stability of single-domain antibodies37. In another study, Zabetakis et al. demonstrated that the stability-enhancing mutations identified by Bekker et al. led to reductions of the binding affinities to antigen38. This demonstrated the difficulty of simultaneously improving thermal stability and binding affinity and showed that our understanding of the relationships between binding capability and other physical properties is not yet sufficient to design antibodies in a rational manner.
In this study, we employed physicochemical measurements, structural modeling and MD simulations to analyze two homologous, single domain VHH antibodies that differ in melting temperatures by 6.2 °C. We sought to determine what underlies different thermal stabilities of these two antibodies that differ at a limited set of residues, to understand the tradeoff between thermal stability and function, and to identify the features that result in both higher affinity and higher thermal stability. Our results showed that there is a delicate balance among thermal stability, binding affinity, and explored conformational space that must be considered in engineering of single-domain VHH antibodies.
Results
Single-domain VHH antibodies to serum albumin with nine amino acid differences have dramatically different thermal stabilities and binding affinities
This study focused on two single-domain VHH antibodies, Z18 and Z26, that were selected in-house for affinity to human serum albumin (HSA). Pair-wise sequence comparison revealed that Z18 and Z26 are highly homologous; only nine amino acids are different between the two antibodies (Fig. 1A). Computational structure predictions based on each sequence revealed highly homologous framework structures with distinct conformations of the CDR-H3 (Fig. 1B).
Figure 1.

Comparison of amino acid sequences and predicted structures of the single-domain VHH antibodies Z18 and Z26. (A) Amino acid sequences of Z18 and Z26. CDR regions are indicated and amino acids that are not identical are indicated by asterisks. Residues are numbered according to the IMGT numbering scheme39. (B) Homology models of Z18 and Z26. The CDR-H3 and mutational positions are shown in orange and magenta, respectively. Protein images were generated with UCSF Chimera40.
An analysis by differential scanning calorimetry (DSC) showed that Z18 had lower thermostability than Z26. The difference in melting temperature (Tm) was 6.2 °C (Table 1). Assessment of the antigen binding by surface plasmon resonance (SPR) showed that the affinity of Z18 for HSA was 3 orders of magnitude lower than that of Z26 (1.75 × 10–7 M and 5.40 × 10–10 M for Z18 and Z26, respectively; Table 1 and Figure S1).
Table 1.
Thermal stabilities and binding affinities of Z18 mutants and Z26.
| Tm (°C) | kon (1/M·S) | koff (1/s) | KD (M) | |
|---|---|---|---|---|
| Z18 | 58.6 ± 0.1 | 7.74 × 105 | 0.259 | 3.34 × 10–7 |
| Z18-V2L | 57.8 ± 0.4 | 7.54 × 105 | 0.276 | 3.66 × 10–7 |
| Z18-G32D | 57.8 ± 0.4 | 6.96 × 105 | 0.120 | 1.72 × 10–7 |
| Z18-R50L | 60.9 ± 0.4 | 6.89 × 105 | 0.284 | 4.12 × 10–7 |
| Z18-I56V | 55.1 ± 0.4 | 7.41 × 105 | 0.220 | 2.96 × 10–7 |
| Z18-T61L | 55.5 ± 0.7 | 4.33 × 106 | 0.037 | 8.63 × 10–9 |
| Z18-G78A | 66.8 ± 0.3 | 1.55 × 105 | 1.101 | 7.11 × 10–6 |
| Z18-N85S | 56.2 ± 0.9 | 6.92 × 105 | 0.238 | 3.44 × 10–7 |
| Z18-Y114F | 57.8 ± 0.8 | 7.45 × 106 | 0.096 | 1.29 × 10–8 |
| Z18-S133R | 59.2 ± 0.6 | 1.16 × 106 | 0.078 | 6.72 × 10–8 |
| Z18-R50L/G78A/S133R | 70.9 ± 0.1 | 1.76 × 106 | 0.785 | 4.47 × 10–7 |
| Z18-G32D/R50L/G78A/S133R | 70.6 ± 0.2 | 5.16 × 105 | 0.412 | 7.97 × 10–7 |
| Z26 | 64.8 ± 0.4 | 1.51 × 106 | 8.16 × 10–4 | 5.40 × 10–10 |
Unlike conventional antibodies, single-domain antibodies refold correctly after heat-induced denaturation4. Therefore, we used an enzyme-linked immunosorbent assay (ELISA) and circular dichroism (CD) to assess the binding abilities and conformational changes, respectively, after thermal stress at different temperatures (30 °C, 50 °C, 70 °C, 90 °C). Both antibodies showed similar trends in binding capabilities and CD spectra before and after the thermal stress (Fig. 2); both antibodies preserved the binding affinities and secondary structures up until 50 °C, whereas, when the temperature was elevated to 70 °C or 90 °C, the antibodies began to denature and lose their binding abilities. These results motivated us to investigate the origin of the difference in physicochemical properties between the two antibodies generated by the difference in very few amino acids.
Figure 2.
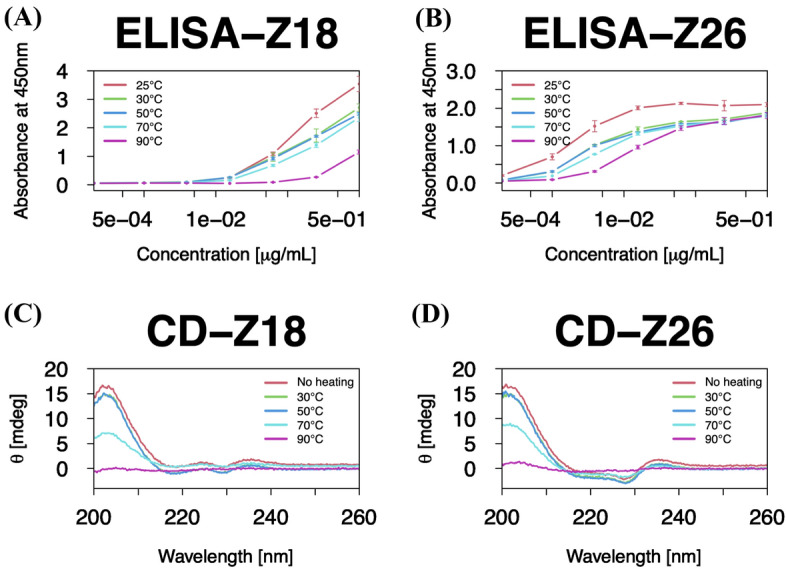
Binding capabilities and secondary structures of the single-domain VHH antibodies show similar trends between Z18 and Z26. (A,B) ELISA to quantify binding of (A) Z18 and (B) Z26 to HSA. ELISA was independently repeated three times, and the average values were plotted with the standard deviations. (C,D) CD spectra of (C) Z18 and (D) Z26. CD measurements were performed with 0.15 mg/mL of a sample in PBS. All graphs in this article were made by R packages41.
A single mutation in Z18 increases thermal stability but decreases binding affinity
To examine how the differences in sequence between the low stability Z18 and the high stability Z26 influence stability and function, we prepared nine mutants of Z18, each mutation altered a single residue to the amino acid observed in Z26. Thermal stabilities were measured by DSC (Table 1). The mutant G78A had the largest impact on the thermal stability; the Tm of this mutant was 8.2 °C higher than that of the wild-type Z18 and was 2.0 °C higher than that of wild-type Z26. When binding affinity was measured by SPR for these mutants, the most stable mutant Z18-G78A had considerably lower affinity than Z18 and Z26 (Table 1, Figures S1 and S2). Furthermore, the Z18-R50L mutant also improved the thermal stability by 2.3 °C while it had neutral effects on the binding affinity.
The Z18-T61L mutant was an order of magnitude tighter binder of HSA than Z18, but its Tm was lower than that of Z18 by 3.1 °C (Table 1), suggesting that, although single-domain VHH antibodies do not always use CDR-H2 to interact with the antigens, the T61L mutation, which is located in CDR-H2, may have a role in the antigen binding. Similarity, the Z18-Y114F and Z18-S133R mutants, which are located in CDR-H3, exhibited slightly better binding affinities than Z18, implying a role of these residues in the antigen recognition. Interestingly, the S133R mutation also slightly affect the thermal stability favorably by 0.6 °C. When the thermally stabilizing mutations (R50L/G78A/S133R) were combined, the Tm was higher than that of Z18-G78A by 4.1 °C, and the binding affinity became comparable to that of Z18.
A previous study demonstrated that a combination of 2 mutations, each of which showed decreased affinity or no expression, was able to improve the binding affinity of an antibody42. Notably, those 2 mutations were spatially far from each other. To explore such corporative effects of distantly located mutations, we focused on the G32D mutation, which is located in CDR-H1, but did not affect the binding affinity (Table 1). We then combined four mutations, G32D, R50L, G78A, and S133R, which resulted in improvement of the thermal stability by 12.0 °C, but the binding affinity still remained comparable to that of Z18.
Together, these data suggest that each mutation has a distinctive role in either thermal stability or binding affinity (Table 1 and Figure S1). In most cases, mutations in CDRs influenced binding affinity and those in framework regions altered thermal stability. Some mutations (e.g., V2L and G32D) were neutral. However, it is worth noting that we focused only on thermal stability and binding affinity in this study, but other factors, such as colloidal stability and specificity, could be affected by these mutations43.
Conformational space of a CDR governs thermal stability and binding affinity
To gain molecular insights into roles of the stabilizing mutation G78A, we visually inspected the predicted structures of Z18 and Z26. The difference in chemical structure between the two amino acids is a single methyl group present in Ala but not in Gly. This methyl group appears to fill a cavity observed inside the predicted structure of Z18 (Figure S3). Core regions of protein structures are known to be well packed44, and even a small cavity can lead to destabilization. Filling such a cavity in the antibody structure could change its dynamics. Therefore, we further employed 1.1 μs MD simulations to investigate roles of mutations in conformational dynamics. Since epitope information of the antigen is not available, we limited ourselves to computational assessments of the unbound states of the wild-type antibodies and the mutants. Based on the experimental results, we computationally analyzed Z18, Z18-G78A, Z18-G32D/R50L/G78A/S133R, and Z26. We performed 5 independent simulations with different initial velocities for each antibody (total aggregated time ~ 22 μs).
The root mean square deviations (RMSDs) of Cα atoms during the simulations were determined (Figure S4). All showed standard deviations less than 1.0 Å after 600 ns, suggesting the convergence of each trajectory (Table S2). When we split each trajectory after 600 ns in half, we observed substantial overlap between the first half and last half of each trajectory (Figure S5), supporting our claim that our simulations were converged well. However, Cα-RMSD of RUN-1 of Z18 deviated largely from those of the other 4 simulations (Figure S5). Therefore, in the analyses below, we did not consider the first 600 ns of the trajectories of each antibody and the entire trajectory of RUN-1 of Z18. Antibodies recognize antigens through their CDRs, and previous studies suggested that CDRs impact thermal stability34,45. Therefore, we also computed Cα-RMSDs of each CDR to further quantify the dynamics. We first superposed the Cα-atoms of the framework region and then computed Cα-RMSDs of each CDR. Of the CDRs, CDR-H3 exhibited the largest movements, followed by CDR-H1 and CDR-H2 (Figure S6). Interestingly, the RMSDs of the CDR-H1 of Z18 and Z26 clearly differed: Z18, which has the lower Tm had larger fluctuations than Z26 (Figure S6). This difference was more evident when conformational space was described by the kernel density estimation as frequency distributions of the RMSDs (Fig. 3A). Z18 as well as the stabilized mutants exhibited broader conformational space than Z26.
Figure 3.
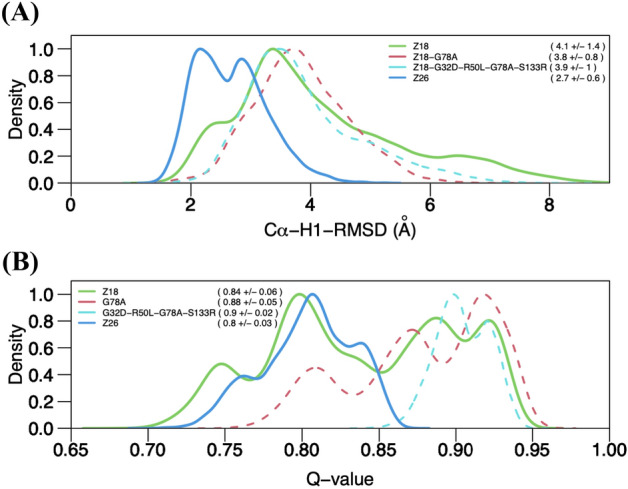
Dynamics of CDR-H1 of Z18 and Z26 differ. Kernel density estimation of (A) Cα-RMSDs of CDR-H1 and (B) Q-values computed based on the last 500 ns simulations for Z18, Z18-G78A, Z18-G32D/R50L/G78A/S133R, and Z26. The last 500 ns trajectories of each system were merged in each plot. Averages and standard deviations are indicated in parenthesis. Y-axis was normalized to a range between 0 and 1 for each system.
A previous study suggested that fraction of native contacts, or Q-value, are correlated well with thermal stabilities of single-domain antibodies37. In the five simulations performed for each system, we observed variations of Q-value (Figure S7). Two trajectories of Z18, the antibody with the lowest stability, had higher Q-values on average than the averages of the Z26 trajectories. On the other hand, the most stable antibody, Z18-G32D/R50L/G78A/S133R, had consistently the higher Q-values (~ 0.9) throughout the simulations. When conformational space was described in terms of Q-values of all the 5 trajectories together for each antibody, Z18 exhibited broader distribution than Z26 (Fig. 3B). Notably, the stabilized mutants explored a higher range of conformational space than Z18 and Z26. These data suggest that although Q-value analysis at 298 K, conditions that approximate room temperature, is not an absolute method for assessing thermal stability, it could inform rational antibody design. Indeed, in plots of Q-value versus Cα-RMSD of CDR-H1, we observed strong correlations (Fig. 4).
Figure 4.
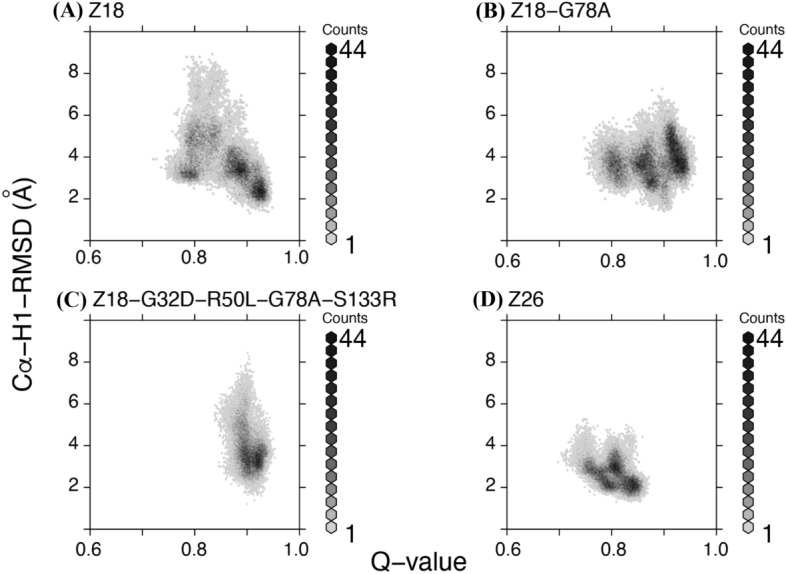
Conformational space explored correlates with thermal stability. Plots of Q-value versus Cα-RMSDs of CDR-H1 of indicated single-domain VHH antibodies. The last 500 ns trajectories of five simulations were merged in each plot.
Both Q-values and Cα-RMSDs of CDR-H1 indicate that the space explored by Z18 is larger than that explored by Z26 (Figs. 3, 4A,D). The single mutation Gly to Ala mutation at the position 78 of Z18 resulted in comparable stability to Z26 but binding affinity was sacrificed (Table 1). This observation may be rationalized by the fact that the G78A mutation shifted the conformational space explored by Z18 (Figs. 3 and 4). The Z18-G32D/R50L/G78A/S133R mutant, which was more thermodynamically stable than Z26 explored similar conformational space to Z18-G78A, with a narrower range of the Q-value (Figs. 3 and 4C). The binding affinity of the Z18-G32D/R50L/G78A/S133R mutant was tighter than that of Z18-G78A and comparable to that of Z18, but still inferior to that of Z26. Our simulations included only antibodies, and therefore the conformational spaces correspond to the conformational ensembles of unbound-state antibody structures. As the antigen binding could induce conformational changes in antibodies, understanding differences in binding affinities of Z18-G32D/R50L/G78A/S133R, Z18-G78A, and Z26 will require structural information of the antibody-antigen complexes.
Discussion and conclusions
Two single-domain VHH antibodies that have high sequence homology, but different thermal stabilities, were analyzed through physicochemical measurements, structural modeling, and MD simulations. Our comparative analyses support the hypothesis that the thermal stability of antibodies can be increased by restricting conformational space. Previous works validated this approach by introducing disulfide bonds in antibody structures46. We demonstrated here that conformational restriction resulting from filling a cavity inside the structure of the Z18 antibody by mutation of Gly to Ala enhanced stability considerably.
Starting from the Z18 antibody, we were able to design more thermally stable antibodies, but these antibodies still had lower binding affinities for HSA than the Z26 antibody. Our MD simulations suggested that these antibodies Z18-G78A and Z18-G32D/R50L/G78A/S133R explored a higher range of conformational space, in terms of Q-value, than the higher affinity Z26 antibody, and still similar conformational space of CDR-H1 to that of Z18 rather than Z26. Thus, there seems to be a tradeoff between thermal stability and binding affinity, and conformational space explored by antibodies is a factor that governs the stability-function tradeoff.
Compared to Z18, Z26 had higher thermal stability and better binding affinity. Of the nine residues that differ between these two single-domain VHH antibodies, only a mutation in Z18 to the amino acid in Z26 (S133R) improved both of the thermal stability and the binding affinity. Mutations at two sites (R50L, G78A) had favorable effects on the thermal stability whereas mutations at two other sites (T61L, Y114F) contributed favorably to the binding affinity. Two mutations (I56V, N85S) decreased thermal stability with marginal effects on binding affinity. The other two mutations (V2L, G32D) were neutral. The locations and the effects of the mutations were summarized in Fig. 5. These results indicated that combination of mutations yields more than a simple additive effect.
Figure 5.
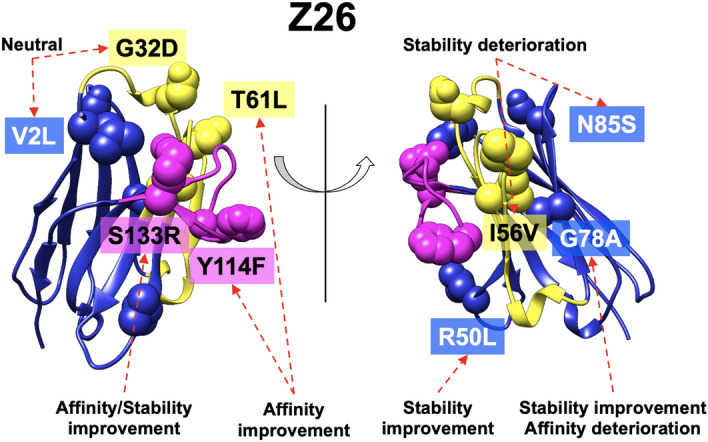
Correlations between the location and effect of the mutations. Residues that differ between Z18 and Z26 are shown as spheres. CDR-H1 and H2 are colored in yellow whereas CDR-H3 are in magenta.
Antibodies evolve in specific response to antigens through somatic hypermutation of their germline genes, resulting in the gradual accumulation of mutations in CDRs as well as in framework regions47. As demonstrated in this study, mutations in CDRs could direct affinity maturation whereas those in framework regions may improve physical stability. In this study, we further showed that mutations inside an antibody structure or in a framework region could modulate conformational space explored by antibodies. Previous studies showed that somatic mutations restrict conformational space48–52, which leads to better binding affinities by minimizing entropy loss upon antigen binding. On the other hand, a recent large-scale repertoire analysis suggest that somatic mutations do not typically result in rigidification53. In this context, we showed that rigidification of an antibody could stabilize an antibody, but instead scarify binding affinity, and there is a complex interplay between binding affinity and physical stability, in agreement with previous observations from the directed evolution of a single domain antibody54,55.
In summary, based on experimental measurements and computational techniques, we revealed that there is a delicate balance among thermal stability, binding affinity, and conformational space explored by single-domain VHH antibodies. Our results suggest that consideration of antibody structural dynamics is an important step in rational antibody design.
Methods
Expression and purification of antibodies
The DNAs encoding VHH antibodies Z18 and Z26 were incorporated into pRA2 vector56 with N-terminal pelB signal peptide and a C-terminal hexa-histidine tag. E. coli strain BL21 (DE3) cells (Novagen) were transformed with the expression vectors and grown at 28 °C in LB medium. Protein expression was induced by addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside when the optical density at 600 nm reached a value of 1.0. Cells were allowed to grow for an additional 16 h at 20 °C. The cells were harvested by centrifugation at 6500 × g for 10 min, and the cell pellet was resuspended in buffer A (20 mM Tris–HCl, 500 mM NaCl, pH 8.0) supplemented with 5 mM imidazole, after which cells were lysed with an ultrasonic disruptor (UD-201, TOMY) for 20 min. The cell lysate was centrifuged (40,000×g for 30 min) at 4 °C. The supernatant was filtered through an 0.8-μm pore-size filter and subsequently loaded onto a 1-mL Ni-NTA agarose column (Qiagen) equilibrated with buffer A. After washing with buffer A containing 20 mM imidazole, VHH antibodies were eluted from the column with buffer A supplemented with 300 mM imidazole. The eluate was subjected to size-exclusion chromatography using a HiLoad 26/600 Superdex 75 pg column (GE Healthcare) equilibrated with a buffer containing 20 mM TRIS–HCl, 200 mM NaCl.
Antigen
The antigen, HSA, was purchased from Sigma-Aldrich Japan (Tokyo, Japan). The HSA was dissolved in a phosphate buffer and the HSA stock solution was prepared by extensive overnight dialysis at 4 °C.
Surface plasmon resonance assays
The interactions between antibodies and HSA were analyzed by SPR on a Biacore T200 instrument (GE Healthcare). A research-grade CM5 Biacore sensor chip (GE Healthcare) was activated by a short treatment with N-hydroxysuccinimide/N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide hydrochloride, followed by immobilization of antigen HSA at a surface density of approximately 1000 RU. The activated groups on the surface of the sensor were subsequently blocked by injecting 1 M ethanolamine as previously described57. Kinetic data were obtained by injecting increasing concentrations of antibodies over the sensor chip at a flow rate of 30 μl/min. The measurements were carried out in PBS containing 0.05% (v/v) Tween-20. Contact time and dissociation time were 4 min and 10 min, respectively. Data analysis was performed with the BIAevaluation software (GE Healthcare). Association (kon) and dissociation (koff) constants were calculated by a global fitting analysis assuming a Langmuir binding model and a stoichiometry of (1:1).
Enzyme-linked immunosorbent assay
Microtiter plate wells were coated with HSA at 10 μg/ml overnight at 4 °C. The wells were washed and blocked with 3% skim milk/PBS supplemented with 0.05% Tween-20 (PBS-T) at room temperature for 1 h. Following three washes with PBS-T, VHH samples were added into wells and incubated at room temperature for 1 h. The wells were washed three times with PBS-T and bound antibody was detected with anti-His-tag mAb-HRP-DirecT (MBL) after incubation at room temperature for 1 h. The wells were washed three times with PBS-T and developed with TMB substrate mixture (Cosmobio). The reaction was stopped with Stop buffer (Cosmobio) after 20 min and the absorbance at 450 nm was measured.
Circular dichroism spectra
CD spectra were recorded on a model J-820 CD spectrometer (JASCO). Far-UV CD measurements were performed with 0.15 mg/mL of a sample in PBS using a 1-mm cell and a bandwidth of 1 nm. Spectra were recorded five times for each sample.
Differential scanning calorimetry
DSC measurements of samples prepared in PBS were carried out on a MicroCal PEAQ-DSC (Malvern) at a heating rate of 60 °C per h. To evaluate thermal stability, the temperature where heat capacity was maximum was determined as apparent Tm using the software MicroCal PEAQ-DSC software (Malvern).
Modeling of VHH antibody structures
To model structures of the VHH antibodies, we divided the sequences into four parts (CDR-H1, CDR-H2, CDR-H3, and framework region) and used BLAST58 to search for each structural template in the RosettaAntibody database59,60, which was originally derived from the Protein Data Bank61. The sequences with the highest bit-scores in the database were chosen as templates (Table S1). These templates were brought together to generate a crude model of the single-domain antibodies. Next, we used the Relax optimization with harmonic constraints to minimize deviation from the template crystal structures as previously described62. The resulting VHH model structures were then used for the MD simulations to computationally assess the stabilities and dynamics of the antibodies.
Molecular dynamics simulations
MD simulations of the four antibodies were performed using GROMACS 2019.463 with the CHARMM36m force field64. The structures were solvated with TIP3P water65 in a rectangular box such that the minimum distance to the edge of the box was 15 Å under periodic boundary conditions. The protein charge was neutralized with added Na or Cl, and additional ions were added to imitate a salt solution of concentration 0.14 M. Each system was energy-minimized for 5000 steps with the steepest descent algorithm and equilibrated with position restraints of protein heavy atoms and NVT ensemble, where the temperature was increased from 50 to 298 K during 500 ps. Further non-restrained simulations were performed with the NPT ensemble at 298 K for 1.1 μs. The time step was set to 2 fs throughout the simulations. A cutoff distance of 12 Å was used for Coulomb and van der Waals interactions. Long-range electrostatic interactions were evaluated by means of the particle mesh Ewald method66. Covalent bonds involving hydrogen atoms were constrained by the LINCS algorithm67. A snapshot was saved every 100 ps. We repeated the same calculations 5 times with difference initial velocities.
Trajectory analysis was performed based on the last 500 ns trajectories. RMSDs were computed with the GROMACS package63. Q-value was computed with the MDTraj Python library68 using the equation below36:
where the sum runs over the N pairs of native contacts (i,j), rij(X) is the distance between i and j in configuration X, is the distance between i and j in the native state, β is a smoothing parameter taken to be 5 Å−1, and the factor λ accounts for fluctuations when the contact is formed, taken to be 1.8 for the all-atom simulations36.
Supplementary Information
Acknowledgements
The supercomputing resources in this study were provided in part by the Human Genome Center at the Institute of Medical Science, The University of Tokyo, and by Research Center for Computational Science, Okazaki, Japan. This work was funded in part by the Japan Society for the Promotion of Science (JP19H04202 to D.K. and JP16H02420, JP19H05766, and JP20H02531 to K.T.), the Japan Agency for Medical Research and Development (JP20wm0325002s, JP19ak0101117h, and JP20ak0101139h to D.K., JP18ak0101100h to M.N., JP18am0101094j, JP18dm0107064h, JP18mk0101081h, JP18fm0208030h, JP18fk0108073h, and JP18ak0101100h to K.T.) and JST CREST (JPMJCR20H8 to K.T.). D.K. also thanks Okawa Foundation for Information and Telecommunications for supporting his computational work on antibodies (grant number 20-10).
Author contributions
K.T. and D.K. conceived the study. E.I. and D.K. contributed equally to the execution of this work. E.I. performed all the experimental measurements. D.K. designed and conducted the computational study. E.I., D.K., and M.N. analyzed the data. D.K. and A.M. provided new tools and reagents. D.K. wrote the manuscript with the help of M.N. and E.I. All the authors approved the final version of the manuscript.
Competing interests
The Panasonic Corporation provided support in the form of salaries for E.I. The Panasonic Corporation has also filed a patent application that refers to the antibodies used in this study (16/372506, 2018-110552). This does not alter our adherence to the policies of the journal on sharing data and materials. The other authors declare no potential conflict of interest.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Emina Ikeuchi and Daisuke Kuroda.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-98977-8.
References
- 1.Könning D, et al. Camelid and shark single domain antibodies: Structural features and therapeutic potential. Curr. Opin. Struct. Biol. 2017;45:10–16. doi: 10.1016/j.sbi.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 2.de los Rios M, Criscitiello MF, Smider VV. Structural and genetic diversity in antibody repertoires from diverse species. Curr. Opin. Struct. Biol. 2015;33:27–41. doi: 10.1016/j.sbi.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muyldermans S, Smider VV. Distinct antibody species: Structural differences creating therapeutic opportunities. Curr. Opin. Immunol. 2016;40:7–13. doi: 10.1016/j.coi.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldman ER, Liu JL, Zabetakis D, Anderson GP. Enhancing stability of camelid and shark single domain antibodies: An overview. Front. Immunol. 2017;8:865. doi: 10.3389/fimmu.2017.00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Sapienza G, Rossotti MA, Tabares-da Rosa S. Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front. Immunol. 2017 doi: 10.3389/fimmu.2017.00977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 7.Chothia C, et al. Conformations of immunoglobulin hypervariable regions. Nature. 1989;342:877–883. doi: 10.1038/342877a0. [DOI] [PubMed] [Google Scholar]
- 8.Tramontano A, Chothia C, Lesk AM. Framework residue 71 is a major determinant of the position and conformation of the second hypervariable region in the VH domains of immunoglobulins. J. Mol. Biol. 1990;215:175–182. doi: 10.1016/S0022-2836(05)80102-0. [DOI] [PubMed] [Google Scholar]
- 9.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997;273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda D, Shirai H, Kobori M, Nakamura H. Systematic classification of CDR-L3 in antibodies: Implications of the light chain subtypes and the VL-VH interface. Proteins Struct. Funct. Bioinform. 2009;75:139–146. doi: 10.1002/prot.22230. [DOI] [PubMed] [Google Scholar]
- 11.North B, Lehmann A, Dunbrack RL. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 2011;406:228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincke C, et al. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009;284:3273–3284. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 13.Almagro JC, Fransson J. Humanization of antibodies. Front. Biosci. 2008;13:1619–1633. doi: 10.2741/2786. [DOI] [PubMed] [Google Scholar]
- 14.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 15.Shirai H, Kidera A, Nakamura H. Structural classification of CDR-H3 in antibodies. FEBS Lett. 1996;399:1–8. doi: 10.1016/S0014-5793(96)01252-5. [DOI] [PubMed] [Google Scholar]
- 16.Kuroda D, Shirai H, Kobori M, Nakamura H. Structural classification of CDR-H3 revisited: A lesson in antibody modeling. Proteins Struct. Funct. Bioinform. 2008;73:608–620. doi: 10.1002/prot.22087. [DOI] [PubMed] [Google Scholar]
- 17.Weitzner BD, Dunbrack RL, Gray JJ. The origin of CDR H3 structural diversity. Structure. 2015;23:302–311. doi: 10.1016/j.str.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zavrtanik U, Lukan J, Loris R, Lah J, Hadži S. Structural basis of epitope recognition by heavy-chain camelid antibodies. J. Mol. Biol. 2018;430:4369–4386. doi: 10.1016/j.jmb.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell LS, Colwell LJ. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. 2018;31:267–275. doi: 10.1093/protein/gzy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al Qaraghuli MM, Ferro VA. Analysis of the binding loops configuration and surface adaptation of different crystallized single-domain antibodies in response to various antigens. J. Mol. Recognit. 2017;30:e2592. doi: 10.1002/jmr.2592. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell LS, Colwell LJ. Comparative analysis of nanobody sequence and structure data. Proteins Struct. Funct. Bioinform. 2018;86:697–706. doi: 10.1002/prot.25497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouet R, Lowe D, Christ D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014;588:269–277. doi: 10.1016/j.febslet.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 23.Adams JJ, Sidhu SS. Synthetic antibody technologies. Curr. Opin. Struct. Biol. 2014;24:1–9. doi: 10.1016/j.sbi.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng. Des. Sel. 2012;25:507–521. doi: 10.1093/protein/gzs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuroda, D. & Tsumoto, K. Antibody affinity maturation by computational design. In Methodsin Molecular Biology 15–34 (2018). 10.1007/978-1-4939-8648-4_2. [DOI] [PubMed]
- 26.Kuroda D, Tsumoto K. Engineering stability, viscosity, and immunogenicity of antibodies by computational design. J. Pharm. Sci. 2020;109:1631–1651. doi: 10.1016/j.xphs.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Baran D, et al. Principles for computational design of binding antibodies. Proc. Natl. Acad. Sci. 2017;114:10900–10905. doi: 10.1073/pnas.1707171114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warszawski S, et al. Optimizing antibody affinity and stability by the automated design of the variable light-heavy chain interfaces. PLoS Comput. Biol. 2019;15:e1007207. doi: 10.1371/journal.pcbi.1007207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiba H, Tamura H, Caaveiro JMM, Tsumoto K. Computer-guided library generation applied to the optimization of single-domain antibodies. Protein Eng. Des. Sel. 2019;32:423–431. doi: 10.1093/protein/gzaa006. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida K, et al. Exploring designability of electrostatic complementarity at an antigen–antibody interface directed by mutagenesis, biophysical analysis, and molecular dynamics simulations. Sci. Rep. 2019;9:4482. doi: 10.1038/s41598-019-40461-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soler MA, et al. A consensus protocol for the in silico optimisation of antibody fragments. Chem. Commun. 2019;55:14043–14046. doi: 10.1039/C9CC06182G. [DOI] [PubMed] [Google Scholar]
- 32.Kiyoshi M, et al. Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody–antigen complex. PLoS One. 2014;9:e87099. doi: 10.1371/journal.pone.0087099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olson MA, et al. Sequence tolerance of a single-domain antibody with a high thermal stability: Comparison of computational and experimental fitness profiles. ACS Omega. 2019;4:10444–10454. doi: 10.1021/acsomega.9b00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akiba H, et al. Structural and thermodynamic basis for the recognition of the substrate-binding cleft on hen egg lysozyme by a single-domain antibody. Sci. Rep. 2019;9:15481. doi: 10.1038/s41598-019-50722-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soler MA, de Marco A, Fortuna S. Molecular dynamics simulations and docking enable to explore the biophysical factors controlling the yields of engineered nanobodies. Sci. Rep. 2016;6:34869. doi: 10.1038/srep34869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Best RB, Hummer G, Eaton WA. Native contacts determine protein folding mechanisms in atomistic simulations. Proc. Natl. Acad. Sci. 2013;110:17874–17879. doi: 10.1073/pnas.1311599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bekker G-J, Ma B, Kamiya N. Thermal stability of single-domain antibodies estimated by molecular dynamics simulations. Protein Sci. 2019;28:429–438. doi: 10.1002/pro.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zabetakis D, Shriver-Lake LC, Olson MA, Goldman ER, Anderson GP. Experimental evaluation of single-domain antibodies predicted by molecular dynamics simulations to have elevated thermal stability. Protein Sci. 2019 doi: 10.1002/pro.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lefranc M-P, et al. IMGT, the international ImMunoGeneTics information system 25 years on. Nucleic Acids Res. 2015;43:D413–D422. doi: 10.1093/nar/gku1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 41.Ihaka R, Gentleman R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996;5:299–314. [Google Scholar]
- 42.Chiba S, et al. Structure-based design and discovery of novel anti-tissue factor antibodies with cooperative double-point mutations, using interaction analysis. Sci. Rep. 2020;10:17590. doi: 10.1038/s41598-020-74545-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabia LA, Desai AA, Jhajj HS, Tessier PM. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem. Eng. J. 2018;137:365–374. doi: 10.1016/j.bej.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards FM. The interpretation of protein structures: Total volume, group volume distributions and packing density. J. Mol. Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 45.Zabetakis D, Anderson GP, Bayya N, Goldman ER. Contributions of the complementarity determining regions to the thermal stability of a single-domain antibody. PLoS One. 2013;8:e77678. doi: 10.1371/journal.pone.0077678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagihara Y, Saerens D. Engineering disulfide bonds within an antibody. Biochim. Biophys. Acta Proteins Proteomics. 2014;1844:2016–2023. doi: 10.1016/j.bbapap.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 47.Clark LA, Ganesan S, Papp S, van Vlijmen HWT. Trends in antibody sequence changes during the somatic hypermutation process. J. Immunol. 2006;177:333–340. doi: 10.4049/jimmunol.177.1.333. [DOI] [PubMed] [Google Scholar]
- 48.Wong SE, Sellers BD, Jacobson MP. Effects of somatic mutations on CDR loop flexibility during affinity maturation. Proteins Struct. Funct. Bioinform. 2011;79:821–829. doi: 10.1002/prot.22920. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt AG, et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc. Natl. Acad. Sci. 2013;110:264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thorpe IF, Brooks CL. Molecular evolution of affinity and flexibility in the immune system. Proc. Natl. Acad. Sci. 2007;104:8821–8826. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li T, et al. Rigidity emerges during antibody evolution in three distinct antibody systems: Evidence from QSFR analysis of fab fragments. PLoS Comput. Biol. 2015;11:e1004327. doi: 10.1371/journal.pcbi.1004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Willis JR, Briney BS, DeLuca SL, Crowe JE, Meiler J. Human germline antibody gene segments encode polyspecific antibodies. PLoS Comput. Biol. 2013;9:e1003045. doi: 10.1371/journal.pcbi.1003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeliazkov JR, et al. Repertoire analysis of antibody CDR-H3 loops suggests affinity maturation does not typically result in rigidification. Front. Immunol. 2018 doi: 10.3389/fimmu.2018.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Julian MC, et al. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 2015;28:339–350. doi: 10.1093/protein/gzv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Julian MC, Li L, Garde S, Wilen R, Tessier PM. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci. Rep. 2017;7:45259. doi: 10.1038/srep45259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makabe K, et al. Tumor-directed lymphocyte-activating cytokines: Refolding-based preparation of recombinant human interleukin-12 and an antibody variable domain-fused protein by additive-introduced stepwise dialysis. Biochem. Biophys. Res. Commun. 2005;328:98–105. doi: 10.1016/j.bbrc.2004.12.141. [DOI] [PubMed] [Google Scholar]
- 57.Johnsson B, Löfås S, Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal. Biochem. 1991;198:268–277. doi: 10.1016/0003-2697(91)90424-R. [DOI] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: Grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins Struct. Funct. Bioinform. 2014;82:1611–1623. doi: 10.1002/prot.24534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weitzner BD, et al. Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 2017;12:401–416. doi: 10.1038/nprot.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003;10:980–980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 62.Nivón LG, Moretti R, Baker D. A pareto-optimal refinement method for protein design scaffolds. PLoS One. 2013;8:e59004. doi: 10.1371/journal.pone.0059004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abraham MJ, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 64.Huang J, et al. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 66.Darden T, York D, Pedersen L. Particle mesh Ewald: An N ⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 67.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 68.McGibbon RT, et al. MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015;109:1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.