

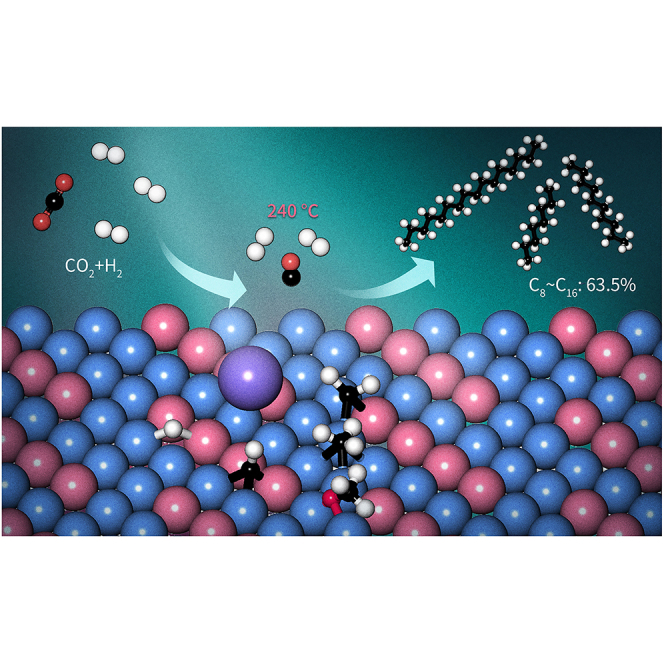
Abstract
The direct conversion of carbon dioxide (CO2) using green hydrogen is a sustainable approach to jet fuel production. However, achieving a high level of performance remains a formidable challenge due to the inertness of CO2 and its low activity for subsequent C–C bond formation. In this study, we prepared a Na-modified CoFe alloy catalyst using layered double-hydroxide precursors that directly transforms CO2 to a jet fuel composed of C8–C16 jet-fuel-range hydrocarbons with very high selectivity. At a temperature of 240°C and pressure of 3 MPa, the catalyst achieves an unprecedentedly high C8–C16 selectivity of 63.5% with 10.2% CO2 conversion and a low combined selectivity of less than 22% toward undesired CO and CH4. Spectroscopic and computational studies show that the promotion of the coupling reaction between the carbon species and inhibition of the undesired CO2 methanation occur mainly due to the utilization of the CoFe alloy structure and addition of the Na promoter. This study provides a viable technique for the highly selective synthesis of eco-friendly and carbon-neutral jet fuel from CO2.
Keywords: carbon dioxide hydrogenation, C–C coupling, heterogeneous catalysis, jet fuel, CoFe alloys
Graphical abstract
Public summary
-
•
An alloy is developed for the direct CO2 hydrogenation to jet-fuel-range hydrocarbons
-
•
The selectivity of the hydrocarbons (63.5%) exceeds the theoretical maximum value
-
•
The CoFe alloy is the active phase in the coupling reaction between surface carbons
-
•
The CoFe alloy is a highly efficient catalyst in the presence of a sodium promoter
Introduction
The increased consumption of fossil resources is responsible for the emission of large amounts of anthropogenic CO2, which results in climate change and ocean acidification. The recent popularization of electric cars has helped reduce gasoline consumption. Nonetheless, in the near future, liquid fuels consisting of long-chain hydrocarbons will remain a necessity for the transportation sector, especially the aviation, nautical, and land-based automotive industries.1 Therefore, it is imperative to develop processes and technologies for the effective hydrogenation of CO2 to liquid fuels using renewable hydrogen (H2). Despite some breakthroughs in the synthesis of gasoline (C5–C11-range hydrocarbons) directly via CO2 hydrogenation,2, 3, 4, 5, 6 there have been few reports on the selective synthesis of jet fuel (C8–C16-range hydrocarbons).7,8 The traditional approach to the direct synthesis of products with more than two carbons (C2+) through CO2 hydrogenation involves the carbon monoxide (CO) intermediate formed by cascading the reverse water-gas shift (RWGS) and Fischer-Tropsch synthesis (FTS) reactions. However, methanation of CO2 or CO can also occur, which diminishes the economic value of this process.9, 10, 11
Of the industrially relevant FTS catalysts, Fe and Co, the Fe-based catalysts are preferred, using CO2 as a carbon source, owing to their high RWGS activities.9,12, 13, 14 Typically, alkenes are the main products, but large quantities of undesired CO are also formed. The alkenes are upgraded to hydrocarbon fuels over zeolites.2,15, 16, 17, 18, 19, 20, 21 According to some recent reports, high C5+ selectivity (58%–65%) can be achieved using modified Fe catalysts. However, the hydrocarbon products obtained have a very broad distribution.13,22 When following the FTS mechanism, the hydrocarbon selectivity toward a specific fraction is mainly determined by the Anderson–Schulz–Flory (ASF) distribution, which predicts that the C8–C16 selectivity cannot exceed 41%.9,23 Therefore, it is extremely challenging to achieve a higher product selectivity during jet fuel synthesis via a “one-pot” FTS process. In traditional FTS, Co-based catalysts featuring metallic Co active sites are known to have much higher chain growth capabilities and improved catalytic stability compared with Fe-based catalysts with iron carbide active sites.24,25 However, for FTS starting from CO2, short-chain hydrocarbons are predominantly formed, with a CH4 selectivity of up to 70%.12,26, 27, 28, 29 An intuitive approach is to tune the product distribution using FeCo bimetallic catalysts. Recently, carburized CoFe catalysts have been reported to perform CO2 hydrogenation, generating lower olefins as the main products.18,30, 31, 32, 33 Since, at high temperature (>280°C), CoFe catalysts can be easily carburized by CO/syngas pre-treatment or under high CO partial pressure, as in syngas conversion or the RWGS reaction,18,30,34, 35, 36 there are few studies of C–C coupling reactions on the CoFe bimetallic alloy phase.35, 36, 37 Therefore, it is necessary to obtain CoFe bimetallic alloy catalysts with superior performance in the selective production of jet fuel via direct CO2 hydrogenation.
Herein, we report a Na-modified CoFe alloy catalyst for the direct hydrogenation of CO2 to jet-fuel-range hydrocarbons. This catalyst exhibits an outstanding selectivity toward C8–C16 hydrocarbons (up to 63.5%), which exceeds the maximum value predicted by the ASF model by a significant margin. In addition, it has a low combined selectivity of ∼22% toward CO and CH4. We also demonstrate that the CoFe bimetallic alloy phase is responsible for the chain propagation reaction and ensures a very high product selectivity.
Results and discussion
Composition and catalytic performance
As can be seen from Table S1, the Co/Fe atomic ratio is approximately 1.9 for the CoFe catalysts obtained by the calcination of CoFe-layered double hydroxides (LDHs), denoted as CoFe-xNa, where x represents the Na concentration in wt % (x = 0.23, 0.81, 3.54), and the catalyst without Na is labeled as CoFe. For comparison, catalysts without Fe (Co and Co-0.63Na with 0.63 wt % Na and 70.5 wt % Co) or Co (Fe and Fe-0.67Na with 0.67 wt % Na and 69.7 wt % Fe) were also prepared.
We assessed the CO2 hydrogenation performance of the various catalysts, and the results are listed in Table 1. CoFe exhibits high catalytic activity, with a conversion of 19.6%, and high CH4 selectivity of 77.3%. With increasing Na concentration, the extent of CO2 conversion decreases gradually, with a concomitant increase in CO selectivity. The CH4 selectivity drops significantly, while the C8+ selectivity increases remarkably, reaching a maximum of 64.2% at a Na concentration of 0.81 wt %. Compared with CoFe, CoFe-0.81Na achieves a 2.4-fold increase in C8+ selectivity, with a 4-fold lower CH4 selectivity, suggesting that the effect of Na in decreasing CH4 production is more pronounced than that in promoting C8+ formation. In addition, according to Figure 1A, with increasing space velocity of the feed gas, the C8+ fraction corresponding to CoFe-0.81Na increases substantially from 52.5% to 73.1%, along with a notable reduction in the CH4 selectivity and CO2 conversion, and CO formation is promoted. This indicates the suppression of CO2 methanation at a lower conversion level. Moreover, with decreasing reaction temperature, the CH4 selectivity declines remarkably from 26.5% to 5.8%, while the C8+ selectivity increases significantly from 42.2% to 81.7%, with a slight increase in CO selectivity. The catalytic activity is also decreased significantly (Figure 1B). Notably, most of the CoFe, Fe, or oxide/zeolite bifunctional catalysts developed for the hydrogenation of CO2 to higher hydrocarbons tend to produce large amounts of the by-product, CO, partly because the RWGS reaction is favored at high reaction temperatures (>300°C).9,38 The CO selectivity of the CoFe catalysts in previous reports exceeded 30% even at lower reaction temperatures.18 In this study, the CO selectivity is well below 10% for all the CoFe-xNa catalysts at 240°C. Most of the CO2 input is transformed to hydrocarbons on the CoFe-0.81Na catalyst. However, the CO selectivity is as high as 59.2% over Fe-0.67Na, indicating that the RWGS reaction is more likely to occur at the Fe sites modified by Na.
Table 1.
Catalytic hydrogenation of CO2 over various catalysts
| Entry | Catalyst | Conv. (%) | CO sel. (%) | Hydrocarbon distribution (C mol %) |
|||
|---|---|---|---|---|---|---|---|
| CH4 | C2–C4 | C5–C7 | C8+ | ||||
| 1 | CoFe-3.54Na | 7.0 | 8.8 | 12.7 | 14.9 | 9.2 | 63.1 |
| 2 | CoFe-0.81Na | 10.2 | 5.2 | 17.8 | 9.4 | 8.7 | 64.2 |
| 3 | CoFe-0.23Na | 12.6 | 5.0 | 55.1 | 5.4 | 0.5 | 39.0 |
| 4 | CoFe | 19.6 | 2.9 | 70.3 | 2.3 | 0.3 | 27.1 |
| 5 | Co-0.63Na | 8.3 | 7.2 | 76.5 | 3.6 | 2.3 | 17.5 |
| 6 | Co | 49.3 | 0.3 | 89.9 | 6.3 | 1.4 | 2.4 |
Standard reaction conditions: H2/CO2/N2 ratio 73/24/3, temperature (T) 240°C, gas hourly space velocity (GHSV) 5,500 mL·g−1·h−1, pressure (P) 3 MPa. The data were collected after 48 h on stream.
Figure 1.

The evaluation data of CO2 hydrogenation
(A and B) Effects of (A) space velocity and (B) reaction temperature on conversion of CO2, selectivity of CO, and hydrocarbon distribution.
(C) Detailed distribution of hydrocarbons without CO obtained over CoFe-0.81Na under reaction conditions shown in Table 1.
(D) Catalytic stability of CO2 hydrogenation over the CoFe-0.81Na catalyst under standard reaction conditions.
In additional to the low selectivity toward the undesired CH4 and CO, the formation of light hydrocarbons (C2–C4) is also suppressed. A C5+ selectivity of up to 71.7% is observed, with the dominant products being liquid paraffins and hydrocarbon products in accordance with a double ASF model (Figure S1A).39,40 The chain growth probability is 0.76 for C2–C7 (α1) and 0.6 for heavier C8+ hydrocarbons (α2). The catalyst exhibits a selectivity of 63.5% for C8–C16 hydrocarbons (Figure 1C), outperforming the Fe-Mn-K catalyst reported previously for the synthesis of C8–C16 hydrocarbons (47.8% of all hydrocarbons) from CO27 and exceeding the maximum fraction (41%) obtained via the ASF mechanism. This differs significantly from the catalytic performance of Fe-based13,22,41,42 or CoFe catalysts30,34,43 with carbides as the active sites for the hydrogenation of CO2 to higher hydrocarbons, producing olefins as the main products with very broad distributions (the highest carbon number typically exceeds 20) at higher temperatures (∼300°C). In addition, we tested the performance of CoFe-0.81Na for CO hydrogenation, which afforded liquid fuels with a narrow hydrocarbon distribution (Figure S1B).
The product selectivity of the Na-modified Co catalysts changes significantly upon alloying with Fe. Comparing CoFe-0.81Na with Co-0.63Na, an increase in CO2 conversion from 8.3% to 10.2% was observed. A more significant difference was found in the hydrocarbon distribution. For Co-0.63Na, the CH4 selectivity is remarkably high (76.5%), while the C8+ selectivity is only 17.5%. The C8+ selectivity over CoFe-0.81Na is 64.2%, which is approximately 4-fold that of Co-0.63Na. The catalytic activity is significantly enhanced upon introducing Co. The CO2 conversion over CoFe-0.81Na is more than twice that over Fe-0.67Na, which has a similar Na content (Table S3). Although the CH4 selectivity over Fe-0.67Na is as low as 17.2%, the C8+ selectivity of 44.1% is considerably lower than that over CoFe-0.81Na. A similar trend was observed for catalysts without Na. Compared with the pure Co catalyst, the CoFe catalyst exhibits significantly higher C8+ selectivity and lower CH4 selectivity. A C8+ selectivity of 6.8% is achieved over the pure Fe catalyst, with a reduced CH4 selectivity of 50.9% (Table S3), while the CO2 conversion (9.1%) is much lower than that over the Co (49.3%) or CoFe (19.6%) catalysts. The stability of the CoFe-0.81Na catalyst was also investigated. The catalyst required less than 48 h to reach a steady-state operation (Figure 1D). However, the C8–C16 selectivity increased and a similar amount of time was required for the process to stabilize on stream. The CO selectivity increased continuously and stabilized at 8.5% after approximately 110–120 h. In the stability test, the extent of CO2 conversion decreased from 14.8% to 10.6% during the initial 28 h and was maintained at 10.5% after 120 h. Similar trends were observed for CoFe-3.54Na with respect to time when the process was conducted on stream (Figure S2A). For the CoFe catalyst without Na and catalysts without Fe, the incubation time was shortened to 8 h (Figures S2B–S2D).
Structural characterization
Uncalcined CoFe-LDH precursors have lamellar structures (Figures 2A, S3, and S4). X-ray diffraction (XRD) analysis showed that the typical (001) basal reflection peaks of LDH materials were absent from the calcined samples (Figures S5A and 2C). A new phase of FeIII-substituted Co3O4 (CoIICoIIIFeIIIO4), hereafter denoted as Co2FeO4, was detected for the CoFe-xNa catalysts. The LDH-derived catalysts maintain the layered structure (Figures S3 and S6A), which promotes metal dispersion.44 In addition, compared with other samples, CoFe-0.23Na and CoFe-0.81Na exhibited much higher specific surface areas (Table S1). Further reduction of CoFe-xNa in a pure H2 atmosphere at 400°C for 6 h yielded cubic Co7Fe3 alloy nanoparticles, and no other phases were detected (Figures S5B and S6D). The CoFe alloy nanoparticle sizes of CoFe-0.81Na increased after reduction (Figures S6B and S6C). The influence of Na ions on the formation of the CoFe alloy phase was investigated using in situ XRD and H2 temperature-programmed reduction (TPR) measurements, the results of which are illustrated in Figures S7 and S8, respectively. Initially, Co2FeO4 was reduced to CoO and FeO as the reduction temperature increased from 200°C to 350°C, and this process had negligible effect on the Na content. The Co7Fe3 alloy phase was formed when the reduction temperature exceeded 400°C. The introduction of excess Na inhibited the reduction of CoO and FeO to metallic Co and Fe, respectively. The addition of Na also suppressed the reduction of CoO to Co over Co-0.63Na.
Figure 2.

Catalyst characterization
(A) High-resolution TEM image of spent CoFe-0.81Na and TEM image (inset) of the corresponding uncalcined precursors.
(B) STEM image with the corresponding elemental mapping of spent CoFe-0.81Na.
(C) XRD patterns of the samples after calcination.
(D) CO2-TPD profiles of various calcined samples.
(E and F) Fourier transforms of the k3-weighted EXAFS spectra (solid lines) and fitted curves (circles) of the calcined, reduced, and spent CoFe-0.81Na (after reaction for 48 h) and reference samples at the (E) Co K-edge and (F) Fe K-edge.
On examination of the XRD patterns of the spent catalysts (Figures S5C and S5D), highly crystallized Co and poorly crystallized CoO phases were detected for Co and Co-0.63Na after 48 h of reaction. The occurrence of solely the Co7Fe3 alloy phase for the spent CoFe-xNa catalysts indicated that the formation of the CoFe alloy structure inhibited the oxidation of metallic Co during CO2 hydrogenation. We also performed transmission electron microscopy (TEM) and scanning TEM (STEM)-energy dispersive X-ray spectrometry (EDX) characterizations to investigate the morphology and elemental distribution of the reduced and spent CoFe-xNa catalysts. No phase segregation was observed, and only the Co7Fe3 alloy phase was found after the CO2 hydrogenation reaction (Figures 2A, 2B, S9, and S10). In addition, the oxygen was primarily distributed in the outer shell region of the spent CoFe alloy nanoparticles, whereas Na was homogeneously distributed in the CoFe alloy particles of the reduced and spent catalysts (Figures 2B and S10).
To determine the fine structure of CoFe-0.81Na, we performed X-ray adsorption near-edge structure (XANES) and extended X-ray adsorption fine structure (EXAFS) experiments (Figures 2E, 2F, and S11). The Co and Fe K-edge XANES spectra of the reduced and spent CoFe-0.81Na catalysts were similar, and the peaks at 7,111 and 7,712 eV corresponded to the 1s to 3d transitions in Fe0 and Co0, respectively, indicating that the Co and Fe species were mainly present in the metallic form. The Co-O and Fe-O coordination shells were absent from both the reduced and the spent samples, while new Co-Fe (Fe-Co) coordination shells were observed in both (Figures 2E and 2F), which indicates that the CoFe alloy structure is formed after the reduction and remains stable during the reaction. Fitting of the Co and Fe edge EXAFS data also reveals the formation of new Co-Fe bonds after the reduction (Tables S4 and S5). In addition, the Co-Fe (Co) and Fe-Co (Fe) coordination numbers increase slightly after the reaction.
To investigate the CO2 adsorption properties of the Co, Co-0.63Na, and CoFe-xNa catalysts, their surface basicity was measured using CO2 temperature-programmed desorption (TPD). With increasing Na content, the CO2 desorption peaks were shifted to higher temperatures and the total number of basic sites increased significantly (Figure 2D). Compared with Co and Co-0.63Na, the peaks associated with weakly (50°C–200°C) and strongly (>200°C) basic sites were observed at much higher temperatures for the CoFe and CoFe-0.81Na samples, indicating that the formation of the Co7Fe3 alloy structure strengthened the chemisorption of CO2. As can be seen from the H2-TPD profiles (Figure S12A), the desorption peaks at 150°C–400°C, which are related to the hydrogenation ability of the samples and spillover or subsurface hydrogen adsorption,42,45 were shifted to higher temperatures with increasing Na content, indicating that the hydrogenation ability is weakened upon addition of Na. H2 desorption from CoFe and CoFe-0.81Na occurs at higher temperatures than from the corresponding samples without Fe, which indicates the lower reactivity of the hydrogen species adsorbed on the CoFe and CoFe-0.81Na surfaces. The enhanced CO2 adsorption and weakened hydrogenation ability inhibit CH4 production and increase the chain growth probability owing to the lower ratio of hydrogen to carbon species on the sample surface.14,46 As shown in Figure S12B, CO adsorption on CoFe in the high-temperature desorption regions (200°C–400°C) is remarkably enhanced compared with that on Co, particularly in the presence of Na, which favors the transformation of the CO intermediates in the tandem process.19 However, the degree of transformation decreases with increasing Na concentration. Therefore, the Co7Fe3 alloy structure inhibits CH4 formation and plays a crucial role in the selective hydrogenation of CO2 to the higher hydrocarbons that constitute jet fuel.
In situ DRIFTS study
The CO2 activation and hydrogenation reactions over the Co and CoFe catalysts were studied using in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). As shown in Figure S13, compared with the CO2-DRIFTS results of CoFe, the infrared (IR) peak intensities corresponding to the surface carbonate, bicarbonate, and formate species, which are important intermediates formed during the RWGS reaction,42 for the Co catalyst are markedly weakened, which is consistent with the results of the CO2-TPD analysis. This leads to a lower activity of the Co catalyst toward RWGS.
Switching from CO2 to pure H2 over the CoFe catalyst at 240°C led to the immediate formation of gaseous CO (2,178 and 2,117 cm−1), the concentration of which decreased sharply after a few minutes (Figure S14). As shown in Figures 3A and S14, along with the consumption of CO, CH4, and C2+, paraffins and olefins were detected after only 3 min. The band intensities of the higher olefins were very weak, while those of the higher paraffins increased remarkably during the initial 30 min and stabilized in the following 60 min, indicating that the C2+ paraffins were the principal hydrocarbons formed over CoFe. After 8 min of H2 flow, methoxy species (CH3O∗, 1,036 and 2,809 cm−1) were also detected, and the intensity of the band corresponding to CH4 increased. Compared with the IR bands over CoFe, the band intensity of the methoxy species was enhanced and that of CH4 was significantly lowered over CoFe-0.81Na. For the pure Co catalyst, the bands corresponding to CH4 and the higher hydrocarbons were observed after only 1 min of H2 flow (Figure 3B), and the band intensity of CH4 was much stronger than those of the higher hydrocarbons, indicating that the methanation activity of the Co catalyst was considerably higher. The intensities of these bands over Co decreased significantly upon exposure to H2 flow. In addition, the band assigned to the methoxy species disappeared after 3 min. These results indicate that the formation of CH4 may be related to the methoxy species. Compared with the pure Co catalyst, the band intensities for CH4 are much weaker over CoFe, suggesting that the CoFe catalyst has a lower activity toward CO2 methanation.
Figure 3.

In situ characterization
(A and B) DRIFTS spectra were recorded for CO2 hydrocarbon reaction over (A) CoFe and (B) Co at 240°C.
(C and D) XPS (C) Co 2p and (D) Fe 2p core level spectra of fresh CoFe-0.81Na catalyst (gray curves), after reduction in pure hydrogen (blue curves), and after CO2 hydrogenation reaction at 200°C (red curves) or 240°C (green curves).
Active sites
The XRD, STEM-EDX, and XAFS results confirm that the CoFe alloy species are the active phases participating in CO2-based FTS. However, Co2+ and Fe3+ species were detected on the spent CoFe-xNa surface through ex situ X-ray photoelectron spectroscopy (XPS) analysis (Figure S15). To reveal the sites responsible for CO2 activation and the formation of hydrocarbon products, we investigated the evolution of the surface electronic structure of CoFe-0.81Na during the CO2 activation/hydrogenation process using XPS in an ultra-high-vacuum chamber directly connected to the high-pressure reaction cell. The Co 2p spectrum of the as-prepared sample has a profile similar to that of Co3O4, with a peak at 780.2 eV for Co 2p3/2 and two shoulder peaks at 787 and 790 eV (Figure 3C).47 The Fe 2p spectrum of the as-prepared sample is consistent with that of Fe2O3 reported elsewhere (Figure 3D).48 After reduction, both the Co 2p3/2 and the Fe 2p3/2 peaks became much sharper and were shifted toward the lower binding energies of 777.6 and 706.4 eV, confirming their nearly complete reduction (Figures 3C, 3D, and S16), and demonstrating the presence of the CoFe alloy.
After the subsequent reaction at 200°C for 5 h, no noticeable changes were observed in the Co 2p and Fe 2p spectra, which confirms the metallic states of the elements under the above reaction conditions. However, significant changes were observed in the spectra when CO2 hydrogenation was performed at 240°C over the reduced sample. The Co 2p spectrum exhibits a Co 2p3/2 peak at 781 eV and a prominent Co2+ satellite peak, indicating the transformation of the surface metallic Co to CoO (Figure 3C). The Fe 2p spectrum after reaction at 240°C is similar to that of the fresh catalyst containing Fe3+ species (Figure 3D). These results are consistent with the STEM-EDX results of the spent CoFe-based catalysts (Figures 2B and S10). The oxidation of the metal is attributed to its exposure to oxidative products, such as water, at higher concentrations and temperatures. Previous studies suggest that the formation of iron oxide on the CoFe alloy surface due to oxidation promotes the formation of surface CO intermediates via RWGS,49 which facilitates the formation of long carbon chains during the subsequent FTS process. Thus, the C8+ selectivity over the CoFe catalysts is much higher than that over Co catalysts. Moreover, it can be seen from Figures 3C, 3D, S15, and S16 that no iron or cobalt carbides were observed when the reaction was performed at 240°C.
Reaction mechanism
To elucidate the influence of the catalyst structure on the selectivity toward long-chain hydrocarbons, density functional theory (DFT) calculations were performed to predict the energetics of hydrogenation and self- and cross-coupling of the CHx (x = 1, 2) species formed via CO activation (Table S6), which have been suggested to influence the chain growth probability during the FTS reactions.35,50,51 Although CoO species may exist on the CoFe catalyst surface, chain growth typically occurs over metallic Co surfaces. We postulate that as long as the intermediate species has a CH or CH2 moiety, it can couple with another unsaturated hydrocarbon species such as CH or CH2, leading to continued carbon chain growth until the CH or CH2 moiety is hydrogenated, which results in chain termination. Therefore, we first compared the differences in the energy barriers for the hydrogenation of the CH adsorbate (denoted as CH∗) and its coupling with the CH2 adsorbate (denoted as CH2∗) on the Co(10-11) and CoFe(110) slab models. As shown in Figure 4A, over the Co(10-11) surface, CH∗ + CH2∗ coupling involves a much lower energy barrier (0.17 eV) than CH∗ hydrogenation (0.62 eV). The coupling reaction on the CoFe(110) surface also involves a relatively low energy barrier and is thermodynamically more favorable than hydrogenation, as shown in Figure 4B, although the energy barrier of CH∗ hydrogenation (0.42 eV) is somewhat lower than that of CH∗ + CH2∗ coupling (0.61 eV). These results suggest that the Co catalyst favors chain growth over chain termination, which is important for the generation of long-chain hydrocarbons. However, Co has negligible activity toward RWGS, which restricts the formation of long-chain hydrocarbons during CO2 hydrogenation owing to the scarcity of CH∗/CH2∗ species on the Co surface. In contrast, the CoFe alloy catalyst is less efficient at promoting chain growth. The transition states for CH∗ hydrogenation and CH∗ + CH2∗ coupling reactions are shown in Figures 4C–4F. As shown in Figure S17A, CH2∗ self-coupling on the Co(10-11) surface involves a much lower energy barrier (0.38 eV) compared with that on the CoFe(110) surface (0.51 eV), and it is also lower than that of CH2∗ hydrogenation on the Co(10-11) surface (0.53 eV). These computational results suggest that the CoFe alloy phase is less favorable for the carbon chain propagation than the Co phase. Structures of the transition states involved in CH2∗ hydrogenation and self-coupling on the above two surfaces are shown in Figures S17B–S17E, whereas those of the initial and final states are presented in Figures S18 and S19. The active site identified by these studies is consistent with that obtained via charge density analysis, as shown in Figure S20.
Figure 4.

DFT prediction of the relative reactivities of hydrogenation and C–C coupling reactions
(A and B) Potential energy profiles for CH∗ hydrogenation versus CH∗ + CH2∗ coupling on the Co and CoFe alloy phases modeled by the (A) Co(10-11) and (B) CoFe(110) slabs.
(C–F) Structures of the optimized transition states for (C and E) CH∗ hydrogenation and (D and F) CH∗ + CH2∗ coupling on the (C and D) Co(10-11) and (E and F) CoFe(110) slabs, respectively (colors of surface atoms: Co, blue; Fe, magenta; C, gray; H, white).
Considering that the Na promoter significantly affects both the activity and the selectivity of the Co and CoFe alloy catalysts, we further calculated the energy barriers to hydrogenation and self- and cross-coupling of CHx over the Na-promoted Co(10-11) and CoFe(110) catalyst models. As illustrated in Figure S21 (with the additional data listed in Table S6 in parentheses), the computational results, which indicate that the addition of the Na promoter to the Co catalyst leads to a lower catalytic activity, are consistent with the experimentally observed reduced CO2 reactivity and increased CO selectivity over the Co catalyst upon Na addition. However, for the CoFe alloy catalyst, the Na promoter favors the coupling over hydrogenation, as evidenced by the significantly higher C8+ selectivity and lower CH4 selectivity.
It can be concluded from the above theoretical analysis that Co is a stronger promoter of chain growth, with a significantly higher energy barrier to CH∗ hydrogenation, while the main product formed during CO2 hydrogenation over both the Co and the Co-0.63Na catalysts is CH4. Therefore, we suggest two pathways for CO2 hydrogenation, namely CO2 methanation and RWGS + FTS, with the latter pathway facilitating the formation of both CH4 and long-chain hydrocarbons. For an improved understanding of the significantly lower CH4 selectivity over the CoFe and CoFe-0.81Na catalysts compared with the corresponding Co-based catalysts, we performed additional DFT calculations to study the methanation activity. Previous studies suggest that the dissociation of the CH3O∗ species into CH3∗ + O∗ is rate limiting during CO2 methanation.52 For the catalysts used in this study, three possible phases can be relevant, namely the Co, CoFe, and CoO phases. Moreover, the CoFe alloy was found to suppress CH4 formation via CO2 methanation. The structures of the Co(10-11), CoFe(110), and CoO(100) slab models and the corresponding structures of the CH3O∗ adsorbates on these surfaces are presented in Figures S22 and S23, respectively. As can be seen in Figure 5A, the energy barrier to CH3O∗ dissociation on the CoO(100) surface with a surface O vacancy is the lowest at 1.43 eV, whereas it is slightly higher at 1.49 eV on the Co(10-11) surface, and much higher at 2.39 eV on the CoFe(110) surface. The corresponding transition states are shown in Figures 5B–5D. Therefore, according to our calculations, both the Co and the CoO phases are more favorable for CO2 methanation than the CoFe phase, which is in agreement with our experimental findings. Moreover, the gradual increase in CO2 methanation activity over CoFe-0.81Na with increasing reaction temperatures may also be attributed to the CoO phase formed in situ during the reaction. Notably, the reaction energies corresponding to CH3O∗ dissociation also follow the same trend, which is consistent with the correlation between the energy barriers and the corresponding reaction energies in accordance with the Brønsted-Evans-Polanyi (BEP) relationship.53 Furthermore, as shown in Figures 5E–5G, the differences in reactivity of the above three surfaces toward CH3O∗ dissociation were rationalized by performing charge density difference (Δρ) analyses. Therefore, our DFT calculations and theoretical analyses suggest that the CoFe alloy surface has a remarkable inhibitory effect on CH4 production via CO2 methanation.
Figure 5.

DFT calculations on the rate-determining step of the CO2 methanation reaction
(A) Potential energy profiles for CH3O dissociation on the CoO, Co, and CoFe alloy phases modeled by the CoO(100), Co(10-11), and CoFe(110) slab models.
(B–D) Structures of the transition states on (B) CoO(100), (C) Co(10-11), and (D) CoFe(110) (additional surface atomic color: O, red).
(E–G) Charge density difference plots upon CH3O adsorption on (E) CoO(100), (F) Co(10-11), and (G) CoFe(110), where the light blue and light yellow regions represent charge depletion and charge accumulation.
Conclusions
Herein, we report on a Na-modified CoFe alloy catalyst that enables the efficient production of jet-fuel-range hydrocarbons via direct CO2 hydrogenation. The selectivity toward C8–C16 hydrocarbons is as high as 63.5% at 10.2% CO2 conversion. The catalyst demonstrates a high carbon efficiency, with a combined selectivity of approximately 22% toward undesired CH4 and CO. The combined spectroscopic and computational studies suggest that the metallic CoFe alloy is the active phase responsible for producing C2+ hydrocarbons from the CO intermediate, whose formation is facilitated by the iron oxide surface sites generated in situ during the CO2 hydrogenation reaction. The Na-modified CoFe alloy phase has an intermediate chain propagation activity, which promotes the C–C coupling reaction and enables high C8–C16 selectivity. In addition, the introduction of Na and formation of the CoFe alloy structure effectively suppress CO2 methanation. Therefore, our knowledge of the intricate reaction network involved in CO2-based FTS is improved by these experimental and theoretical findings, which can potentially facilitate the rational development of efficient materials for the direct hydrogenation of CO2 to advanced liquid fuels.
Acknowledgments
We thank Dr. Alexander van der Made and Dr. Joost Smits for helpful discussions. X-Ray absorption studies were performed at the BL11B beamline at the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, PR China. This work was financially supported by the “Frontier Science” program of Shell Global Solutions International B.V. (PT65197), the National Natural Science Foundation of China (21773286, U1832162), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA21090204), the Youth Innovation Promotion Association CAS (2018330), and the Shanghai Rising Star Program, China (19QA1409900).
Author contributions
P.G. and Y.S. conceived the project. P.G. and S.L. analyzed the data and wrote the paper. P.G., L.Z., Y.D., X.Z., A.B., and H.W. drafted the manuscript. L.Z. prepared various materials. L.Z. and H.W. performed sample characterization and catalytic evaluation. X.Z., E.V., and Y.Y. did the in situ XPS experiments and analysis. Y.D., L.S., Y.G., and S.L. performed the DFT calculations. All authors discussed the experimental and theoretical results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published Online: September 29, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xinn.2021.100170.
Contributor Information
Peng Gao, Email: gaopeng@sari.ac.cn.
Shenggang Li, Email: lisg@sari.ac.cn.
Yuhan Sun, Email: sunyh@sari.ac.cn.
Lead contact website
Supplemental information
References
- 1.Patterson B.D., Mo F., Borgschulte A., et al. Renewable CO2 recycling and synthetic fuel production in a marine environment. Proc. Natl. Acad. Sci. U S A. 2019;116:12212–12219. doi: 10.1073/pnas.1902335116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei J., Ge Q., Yao R., et al. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017;8:15174. doi: 10.1038/ncomms15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao P., Li S.G., Bu X.N., et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017;9:1019–1024. doi: 10.1038/nchem.2794. [DOI] [PubMed] [Google Scholar]
- 4.Wang X.X., Zeng C.Y., Gong N.N., et al. Effective suppression of CO selectivity for CO2 hydrogenation to high-quality gasoline. ACS Catal. 2021;11:1528–1547. [Google Scholar]
- 5.Noreen A., Li M.Q., Fu Y.J., et al. One-pass hydrogenation of CO2 to multibranched isoparaffins over bifunctional zeolite-based catalysts. ACS Catal. 2020;10:14186–14194. [Google Scholar]
- 6.Gao P., Zhang L.N., Li S.G., et al. Novel heterogeneous catalysts for CO2 hydrogenation to liquid fuels. ACS Cent. Sci. 2020;6:1657–1670. doi: 10.1021/acscentsci.0c00976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao B., Xiao T., Makgae O.A., et al. Transforming carbon dioxide into jet fuel using an organic combustion-synthesized Fe-Mn-K catalyst. Nat. Commun. 2020;11:6395. doi: 10.1038/s41467-020-20214-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogt C., Monai M., Kramer G.J., Weckhuysen B.M. The renaissance of the sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019;2:188–197. [Google Scholar]
- 9.Zhou W., Cheng K., Kang J.C., et al. New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 2019;48:3193–3228. doi: 10.1039/c8cs00502h. [DOI] [PubMed] [Google Scholar]
- 10.Yang D.X., Zhu Q.G., Han B.X. Electroreduction of CO2 in ionic liquid-based electrolytes. Innovation. 2020;1:100016. doi: 10.1016/j.xinn.2020.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao J., Yang G.H., Yoneyama Y., Tsubaki N. Significant advances in C1 catalysis: highly efficient catalysts and catalytic reactions. ACS Catal. 2019;9:3026–3053. [Google Scholar]
- 12.Rodemerck U., Holena M., Wagner E., et al. Catalyst development for CO2 hydrogenation to fuels. ChemCatChem. 2013;5:1948–1955. [Google Scholar]
- 13.Choi Y.H., Jang Y.J., Park H., et al. Carbon dioxide Fischer-Tropsch synthesis: a new path to carbon-neutral fuels. Appl. Catal. B. 2017;202:605–610. [Google Scholar]
- 14.Guo L.S., Sun J., Ge Q.J., Tsubaki N. Recent advances in direct catalytic hydrogenation of carbon dioxide to valuable C2+ hydrocarbons. J. Mater. Chem. A. 2018;6:23244–23262. [Google Scholar]
- 15.Ramirez A., Gevers L., Bavykina A., et al. Metal organic framework-derived iron catalysts for the direct hydrogenation of CO2 to short chain olefins. ACS Catal. 2018;8:9174–9182. [Google Scholar]
- 16.Dorner R.W., Hardy D.R., Williams F.W., Willauer H.D. Heterogeneous catalytic CO2 conversion to value-added hydrocarbons. Energy Environ. Sci. 2010;3:884–890. [Google Scholar]
- 17.Guo L., Sun J., Ji X., et al. Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts. Commun. Chem. 2018;1:11. [Google Scholar]
- 18.Gnanamani M.K., Jacobs G., Hamdeh H.H., et al. Hydrogenation of carbon dioxide over Co-Fe bimetallic catalysts. ACS Catal. 2016;6:913–927. [Google Scholar]
- 19.Zhu J., Zhang G., Li W., et al. Deconvolution of the particle size effect on CO2 hydrogenation over iron-based catalysts. ACS Catal. 2020;10:7424–7433. [Google Scholar]
- 20.Xu Y., Zhai P., Deng Y., et al. Highly selective olefin production from CO2 hydrogenation on iron catalysts: a Subtle Synergy between Manganese and Sodium Additives. Angew. Chem. Int. Ed. Engl. 2020;59:21736–21744. doi: 10.1002/anie.202009620. [DOI] [PubMed] [Google Scholar]
- 21.Li Z.H., Liu J.J., Shi R., et al. Fe-based catalysts for the direct photohydrogenation of CO2 to value-added hydrocarbons. Adv. Energy Mater. 2021;11:2002783. [Google Scholar]
- 22.Choi Y.H., Ra E.C., Kim E.H., et al. Sodium-containing spinel zinc ferrite as a catalyst precursor for the selective synthesis of liquid hydrocarbon fuels. ChemSusChem. 2017;10:4764–4770. doi: 10.1002/cssc.201701437. [DOI] [PubMed] [Google Scholar]
- 23.Li J., He Y.L., Tan L., et al. Integrated tuneable synthesis of liquid fuels via Fischer-Tropsch technology. Nat. Catal. 2018;1:787–793. [Google Scholar]
- 24.Khodakov A.Y., Chu W., Fongarland P. Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007;107:1692–1744. doi: 10.1021/cr050972v. [DOI] [PubMed] [Google Scholar]
- 25.de Smit E., Weckhuysen B.M. The renaissance of iron-based Fischer-Tropsch synthesis: on the multifaceted catalyst deactivation behaviour. Chem. Soc. Rev. 2008;37:2758–2781. doi: 10.1039/b805427d. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y.Q., Jacobs G., Sparks D.E., et al. CO and CO2 hydrogenation study on supported cobalt Fischer-Tropsch synthesis catalysts. Catal. Today. 2002;71:411–418. [Google Scholar]
- 27.Owen R.E., Plucinski P., Mattia D., et al. Effect of support of Co-Na-Mo catalysts on the direct conversion of CO2 to hydrocarbons. J. CO2 Util. 2016;16:97–103. [Google Scholar]
- 28.Shi Z.B., Yang H.Y., Gao P., et al. Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons. Chin. J. Catal. 2018;39:1294–1302. [Google Scholar]
- 29.He Z.H., Cui M., Qian Q.L., et al. Synthesis of liquid fuel via direct hydrogenation of CO2. P. Natl. Acad. Sci. U S A. 2019;116:12654–12659. doi: 10.1073/pnas.1821231116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim K.Y., Lee H., Noh W.Y., et al. Cobalt ferrite nanoparticles to form a catalytic Co–Fe alloy carbide phase for selective CO2 hydrogenation to light olefins. ACS Catal. 2020;10:8660–8671. [Google Scholar]
- 31.Gnanamani M.K., Hamdeh H.H., Jacobs G., et al. Hydrogenation of carbon dioxide over K-promoted FeCo bimetallic catalysts prepared from mixed metal oxalates. ChemCatChem. 2017;9:1303–1312. [Google Scholar]
- 32.Numpilai T., Witoon T., Chanlek N., et al. Structure activity relationships of Fe-Co/K-Al2O3 catalysts calcined at different temperatures for CO2 hydrogenation to light olefins. Appl. Catal. A. 2017;547:219–229. [Google Scholar]
- 33.Satthawong R., Koizumi N., Song C.S., Prasassarakich P. Light olefin synthesis from CO2 hydrogenation over K-promoted Fe-Co bimetallic catalysts. Catal. Today. 2015;251:34–40. [Google Scholar]
- 34.Jiang F., Liu B., Geng S.S., et al. Hydrogenation of CO2 into hydrocarbons: enhanced catalytic activity over Fe-based Fischer-Tropsch catalysts. Catal. Sci. Technol. 2018;8:4097–4107. [Google Scholar]
- 35.Hwang S.M., Han S.J., Park H.G., et al. Atomically Alloyed Fe–Co Catalyst derived from a N-coordinated Co single-atom structure for CO2 hydrogenation. ACS Catal. 2021;11:2267–2278. [Google Scholar]
- 36.Ismail A.S.M., Casavola M., Liu B.Y., et al. Atomic-scale investigation of the structural and electronic properties of cobalt-iron bimetallic Fischer-Tropsch catalysts. ACS Catal. 2019;9:7998–8011. [Google Scholar]
- 37.Chen G.B., Gao R., Zhao Y.F., et al. Alumina-Supported CoFe Alloy Catalysts derived from layered-double-hydroxide nanosheets for efficient photothermal CO2 hydrogenation to hydrocarbons. Adv. Mater. 2018;30:1704663. doi: 10.1002/adma.201704663. [DOI] [PubMed] [Google Scholar]
- 38.Yang H.Y., Zhang C., Gao P., et al. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017;7:4580–4598. [Google Scholar]
- 39.Pour A.N., Khodabandeh H., Izadyar M., Housaindokht M.R. Mechanistic double ASF product distribution study of Fischer-Tropsch synthesis on precipitated iron catalyst. J. Nat. Gas. Sci. Eng. 2013;15:53–58. [Google Scholar]
- 40.Gaube J., Klein H.F. Studies on the reaction mechanism of the Fischer-Tropsch synthesis on iron and cobalt. J. Mol. Catal. A Chem. 2008;283:60–68. [Google Scholar]
- 41.Hwang S.M., Zhang C.D., Han S.J., et al. Mesoporous carbon as an effective support for Fe catalyst for CO2 hydrogenation to liquid hydrocarbosns. J. CO2 Util. 2020;37:65–73. [Google Scholar]
- 42.Khan M.K., Butolia P., Jo H., et al. Selective conversion of carbon dioxide into liquid hydrocarbons and long-chain α-olefins over fe-amorphous AlOx bifunctional catalysts. ACS Catal. 2020;10:10325–10338. [Google Scholar]
- 43.Satthawong R., Koizumi N., Song C.S., Prasassarakich P. Bimetallic Fe-Co catalysts for CO2 hydrogenation to higher hydrocarbons. J. CO2 Util. 2013;3-4:102–106. [Google Scholar]
- 44.He S., An Z., Wei M., et al. Layered double hydroxide-based catalysts: nanostructure design and catalytic performance. Chem. Commun. 2013;49:5912–5920. doi: 10.1039/c3cc42137f. [DOI] [PubMed] [Google Scholar]
- 45.del Arco M., Trujillano R., Rives V. Cobalt-iron hydroxycarbonates and their evolution to mixed oxides with spinel structure. J. Mater. Chem. 1998;8:761–767. [Google Scholar]
- 46.Wang W., Wang S.P., Ma X.B., Gong J.L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011;40:3703–3727. doi: 10.1039/c1cs15008a. [DOI] [PubMed] [Google Scholar]
- 47.Biesinger M.C., Payne B.P., Grosvenor A.P., et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011;257:2717–2730. [Google Scholar]
- 48.Mcintyre N.S., Zetaruk D.G. X-Ray photoelectron spectroscopic studies of iron-oxides. Anal. Chem. 1977;49:1521–1529. [Google Scholar]
- 49.Zhu M.H., Wachs I.E. Iron-based catalysts for the high-temperature water gas shift (HT-WGS) reaction: a Review. ACS Catal. 2016;6:722–732. [Google Scholar]
- 50.Yao Z.H., Guo C.X., Mao Y., Hu P. Quantitative determination of C-C coupling mechanisms and detailed analyses on the activity and selectivity for Fischer-Tropsch synthesis on Co(0001): Microkinetic modeling with coverage effects. ACS Catal. 2019;9:5957–5973. [Google Scholar]
- 51.Chen C., Wang Q., Wang G., et al. Mechanistic insight into the C2 hydrocarbons formation from syngas on fcc-Co(111) surface: a DFT study. J. Phsy. Chem. C. 2016;120:9132–9147. [Google Scholar]
- 52.Yang C., Liu S., Wang Y., et al. The interplay between structure and product selectivity of CO2 hydrogenation. Angew. Chem. Int. Ed. Engl. 2019;58:11242–11247. doi: 10.1002/anie.201904649. [DOI] [PubMed] [Google Scholar]
- 53.Wang S.G., Temel B., Shen J.A., et al. Universal bronsted-evans-polanyi relations for C-C, C-O, C-N, N-O, N-N, and O-O dissociation reactions. Catal. Lett. 2011;141:370–373. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.