

Summary
Chronic kidney disease (CKD) is associated with substantial morbidity and mortality. We developed a mouse model that mimics human CKD with inflammation, extracellular matrix deposition, tubulointerstitial fibrosis, increased proteinuria, and associated reduction in glomerular filtration rate over time. Using this model, we show that genetic deficiency of SMOC2 or therapeutic silencing of SMOC2 with small interfering RNAs (siRNAs) after disease onset significantly ameliorates inflammation, fibrosis, and kidney function loss. Mechanistically, we found that SMOC2 promotes fibroblast to myofibroblast differentiation by activation of diverse cellular signaling pathways including MAPKs, Smad, and Akt. Thus, targeting SMOC2 therapeutically offers an approach to prevent fibrosis progression and CKD after injury.
Subject areas: Biological sciences, Biochemistry, Molecular biology
Graphical abstract
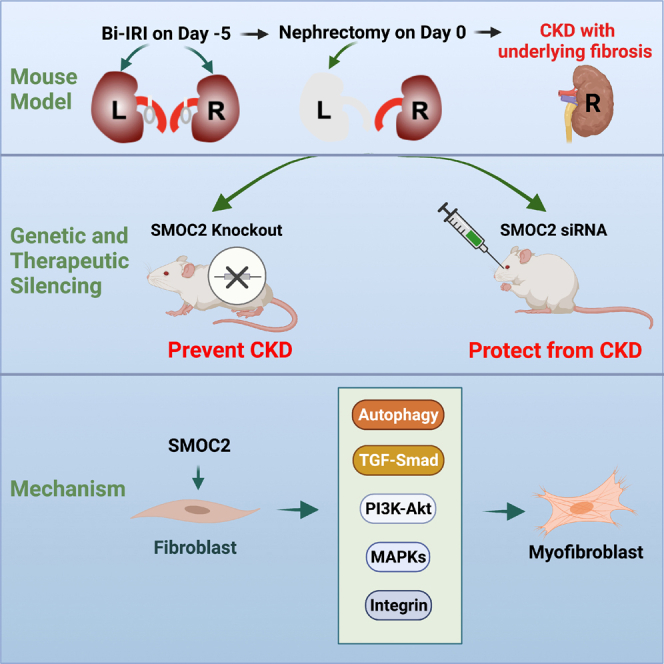
Highlights
-
•
A mouse model of CKD that mimics human disease was generated
-
•
SMOC2 is significantly expressed in CKD and colocalizes with fibroblasts
-
•
Genetic and pharmacologic inhibition of SMOC2 prevents kidney function loss
-
•
SMOC2 activates multiple signaling pathways and autophagy in vitro and in vivo
Biological sciences; Biochemistry; Molecular biology
Introduction
Chronic kidney disease (CKD) affects more than 10% of the adult population and has been recognized as a major cause of hypertension, cardiovascular death, and kidney failure requiring dialysis or transplantation to survive (Hoerger et al., 2010). CKD is a heterogenous disease that often is associated with tubulo-interstitial injury typically culminating in deposition of extracellular matrix (ECM) components such as collagen and fibronectin with irreversible fibrosis (Farris and Colvin, 2012). The resulting loss of kidney function over a period of months to years is associated with high morbidity and mortality rates including accelerated cardiovascular risk (Lysaght, 2002) as well as progression to end-stage kidney disease. Better animal models and effective therapeutic targets are critically needed to arrest progression of CKD and reduce the associated complications including end-stage kidney disease worldwide. Here, we have developed a new mouse model of CKD that mimics human CKD with inflammation, extracellular matrix deposition, and tubulo-interstitial fibrosis with associated reduction in glomerular filtration rate over time. We have used this model to characterize the role of SMOC2 in the development of fibrosis after injury and the mechanisms by which protection against development of CKD can be afforded by treatment after kidney injury to better simulate effective SMOC2 targeting after establishment of injury in humans.
SMOC2 belongs to the Secreted Protein Acidic and Rich in Cysteine (SPARC) family of matricellular proteins that are secreted into the extracellular space and interact with structural matrix proteins, cell surface receptors, growth factors, proteases, and other bioactive effectors to regulate cell-matrix interactions and cell functions (Bornstein and Sage, 2002; Wong and Rustgi, 2013). SMOC2 has mitogenic activity in fibroblasts (Liu et al., 2008) and may mediate intracellular signaling and cell type-specific differentiation during gonad and reproductive tract development (Pazin and Albrecht, 2009). As a member of the SPARC family, SMOC2 interacts with cysteine proteases, including cathepsins F, K, L1, L2, S and W. Functionally, SMOC2 is not only important in extracellular matrix assembly signaling but also as a target for controlling angiogenesis in tumor growth and myocardial ischemia (Brekken and Sage, 2000, 2001).
Previously we have shown that inhibition of SMOC2 before folic acid or unilateral ureteral obstruction (UUO) reduces kidney fibrosis (Gerarduzzi et al., 2017); however, the more clinically relevant inhibition after injury on long-term consequences of that injury remains unknown. Therefore, the objectives of this study were to (1) develop an experimental mouse model of CKD that mimics progressive dysfunction seen in humans, (2) investigate whether genetic or pharmacologic inhibition of SMOC2 protects mice from fibrogenesis and loss of kidney function over time, and (3) investigate the cellular mechanisms of SMOC2-mediated fibrosis.
Results
SMOC2 is highly expressed in a human-relevant experimental mouse model of chronic kidney disease with progressive kidney function loss
To establish a pathophysiologically relevant mouse model of human CKD, mice were subjected to bilateral ischemia-reperfusion injury (Bi-IRI) (10 min for the left kidney and 24 min for the right kidney) and 5 days later they were subjected to nephrectomy of the left kidney (Figure 1A). The glomerular filtration rate (GFR) significantly decreased at 22 days post nephrectomy and kept declining to 44 days (Figure 1B) compared with sham surgery. Blood urea nitrogen (BUN) and the total urine protein to creatinine ratio (uPCR) increased starting day 2 and remained significantly elevated until day 45 as compared with sham controls (Figures 1C and 1D). PAS-stained kidney cortex showed marked increase in proximal tubular injury and matrix accumulation at 45 days post nephrectomy compared with sham controls (Figure 1E). Morphologically, the kidneys of mice with experimentally induced CKD at 45 days post nephrectomy were pale and had significant matrix accumulation as compared with controls (Figure 1F). Consistently, there was also a significantly increased expression of the fibrotic protein collagen 1, myofibroblast differentiation marker α-SMA (Figures 1G, S1A, and S1B), pericyte marker PDGFRβ (Figure S1C), and macrophage infiltration marker F4/80 (Figure S1D) compared with sham controls. Furthermore, Picosirius Red staining showed enhanced collagen deposition in the experimental mouse CKD model at day 45 (Figures 1H and 1I) compared with sham controls and bilateral ischemia and nephrectomy controls.
Figure 1.

Development and characterization of an experimental chronic kidney disease (CKD) model in mice
(A) The experimental design for the mouse CKD model.
(B) Glomerular filtration rate (GFR) in kidney of the CKD models versus shams in mice
(C) Blood urea nitrogen (BUN) in kidney of the CKD models versus shams in mice.
(D) Urine protein to creatinine ratio (uPCR) in kidney of the CKD models versus shams in mice.
(E and F) Morphology studies in kidney of the CKD models versus shams in mice.
(G) Western blot showing collagen 1 and α-SMA protein expressions in the kidney of CKD model and vehicle at the timeline indicated in the figure.
(H and I) Representative images of Picrosirius Red stain for fibrillar collagen 45 days after nephrectomy (20x magnification and scale bar, 50 μm) and quantification of interstitial fibrosis as compared with sham control, bilateral ischemia, and nephrectomy controls. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; n = 6/group unless otherwise stated. Data are represented as mean ± SEM.
We next characterized the expression patterns of SMOC2 in the CKD model. SMOC2 protein (Figures 2A, 2C, and 2D) and mRNA (Figure 2B) expression significantly increased in diseased kidneys of CKD mice as compared with sham controls. Consistent with the expression of SMOC2 in CKD kidney in vivo, NIH/3T3 fibroblasts stimulated with TGFβ in vitro showed robust induction of SMOC2 within 30 min in NIH/3T3 fibroblast cells (Figure 2E). In CKD kidney tissue, SMOC2 was detected and evaluated for co-staining with interstitial myofibroblasts (α-SMA+, Figures 2F and 2G), epithelial cells (LTL+, Figures 2H and 2I), as well as pericytes (PDGFRβ+, Figures 2J and 2K). There was no overlap between SMOC2 and LTL, whereas there was overlap between SMOC2 and both PDGFRβ and α-SMA supporting the localization of the protein in the interstitial pericytes/myofibroblasts. There was a significant increase in injury-induced inflammation in the mouse CKD model as assessed by macrophage marker F4/80; however, no co-localization was observed between SMOC2 and F4/80 suggesting the absence of its expression in activated/infiltrating macrophages (Figures 2L and 2M). Collectively, these results indicate that SMOC2 is significantly expressed and colocalized with activated fibroblasts and pericytes in an experimental mouse model of CKD that results in progressive GFR loss, interstitial inflammation, and fibrosis.
Figure 2.

SMOC2 is highly expressed in fibroblasts in the experimental CKD model in mice
(A) SMOC2 protein expression in kidney at indicated timeline by western blotting analysis.
(B) mRNA quantification at days 23, 30, and 45 in diseased kidney versus sham.
(C and D) Representative image (40x magnification and scale bar, 20 μm) and quantification showing increased SMOC2 expression in CKD kidney.
(E) TGFβ-induced SMOC2 expression in NIH/3T3 fibroblasts by western blotting analysis.
(F–M) SMOC2 expression identified in α-SMA (myofibroblasts), LTL (proximal tubule), PDGFRβ (pericytes/fibroblasts), and F4/80 (macrophage)-positives cells, the markers for myofibroblast, epithelial, fibroblast, and macrophage cells in diseased kidney. Relative quantification was normalized with sham control. Immunofluorescence staining with 40x magnification and scale bar, 20 μm. n = 6/group unless otherwise stated. Data are represented as mean ± SEM.
SMOC2 deficiency prevents inflammation, fibrosis, and gradual loss of kidney function in the experimental mouse model of CKD
To investigate the contributory role of SMOC2 to CKD, mice deficient in SMOC2 (SMOC2 KO) along with wild type (WT) were subjected to the CKD model described in Figure 1. After 29 days, the GFR was statistically significantly reduced in WT mice compared with preservation of GFR in SMOC2 KO mice (Figure 3A). Similarly, BUN levels increased to a greater extent in WT when compared with SMOC2 KO as early as day 2 (D2). BUN in both WT and SMOC2 KO decreased rapidly, but SMOC2 KO mice values returned to baseline levels by D37, whereas WT mice values remained significantly elevated (Figure 3B). The total urine protein to creatinine ratio levels in SMOC2 KO mice were significantly lower at 23–45 days post nephrectomy and when compared with WT mice (Figure 3C). The preservation of kidney function in the SMOC2 KO mice was consistently associated with significantly reduced collagen deposition and accumulation (Figures 3D and 3E) as well as a substantial reduction in matrix proteins—collagen 1, α-SMA, and fibronectin (Figures 3F–3H) and collagen 1, α-SMA, fibronectin, and PDGFRβ mRNA levels (Figures 3I, S2A, and S2B). There was also significantly less upregulation of inflammatory genes IL-1β, TNFα, IL-6, and TGFβ1 as compared with WT mice at D45 post nephrectomy (Figures 3J, S2C, and S2D). Immunofluorescent staining revealed that tubular epithelial cells were less injured and there was preservation of proximal tubule structure as indicated by LTL staining in SMOC2 KO mice (Figure 3K). Consistently, the number of PDGFRβ+ cells (Figure 3L) was markedly reduced, as was the recruitment of macrophages (Figure 3M) in KO mice. Thus, genetic elimination of SMOC2 protected mice from CKD progression and fibrosis consistent with a causal role of SMOC2 in the pathogenesis of the progressive CKD.
Figure 3.

SMOC2 knockout mice are protected from kidney function loss and fibrogenesis in the experimental CKD model
(A) Time course of glomerular filtration rate (GFR) showing the effect of SMOC2 knockout (KO) versus WT in CKD models.
(B and C) Time course of blood urea nitrogen (BUN) and urine protein creatinine ratio (uPCR) in CKD models of SMOC2 KO and WT.
(D and E) Picrosirius Red staining showing the interstitial fibrosis and quantification in the CKD models of SMOC2 KO and WT.
(F) Western blots showing the fibrotic protein levels in the mouse CKD kidney in response to SMOC2 depletion.
(G and H) Representative images and quantification of interstitial fibrosis by immunofluorescent staining of collagen 1, α-SMA and fibronectin.
(I and J) Expression of collagen 1 and α-SMA and inflammatory genes of IL-1β and TNFα mRNAs in SMOC2 KO and WT mice.
(K–M) Representative images and quantification of LTL+, PDGFRβ+, F4/80 + cells in WT versus SMOC2 KO CKD mouse model. Immunofluorescence staining with 40x magnification and scale bar, 20 μm. n = 6/group unless otherwise stated. Data are represented as mean ± SEM.
Therapeutic silencing of SMOC2 by small interfering RNAs administration after disease initiation protects from fibrogenesis and kidney function loss
Given the reduction of kidney injury, inflammation, and fibrosis in SMOC2 KO mice, we next investigated whether therapeutic administration of SMOC2 siRNA after the onset of injury could prevent CKD progression. The delivery and distribution of SMOC2 siRNA to the kidney was confirmed by administering fluorescent SMOC2 siRNA (30 mg/200 μl) to mice via retro-orbital vein injection and tracking the fluorescence over time in the kidneys. SMOC2 siRNA was detected in the kidney as early as 30 min (Figure S3A), peaked at 24–48 h, and then declined by 72 h. At a cellular level the fluorescently labeled SMOC2 siRNA colocalized with proximal tubules (Megalin+), endothelial cells (CD 31+), and mesangial cells or vascular smooth muscle cells in the glomerulus (PDGFRβ+) (Figure S3B). The delivery and distribution to the kidney also biologically translated into significant downregulation of SMOC2 protein by approximately 75% (Figure S3C) and mRNA by 3.5-fold (Figure S3D) following SMOC2 siRNA administration. Mice subjected to experimental CKD (Figure 1) then received either SMOC2 siRNA (30 μg/200 μl) or PBS at day 21 post nephrectomy surgery (after chronic disease was established) twice/week until day 43 for a total of seven injections (Figure 4). SMOC2 siRNA treatment further resulted in a GFR that was ∼2.5-fold higher than controls (Figure 4A) at D44, with BUN levels ∼80% lower (Figure 4B) and protein/creatinine ∼40% lower (Figure 4C) at D45. Quantitative pathological assessments indicated a dramatic reduction in interstitial collagen by Picrosirius Red detection at D45 (Figures 4D and 4E). Collagen 1, α-SMA, and fibronectin protein (Figures 4F–4H) and gene transcripts, as well as mRNA including collagen 1, α-SMA, fibronectin, and PDGFRβ (Figures 4I, S4A, and S1B) were significantly reduced in CKD mouse treated with SMOC2 siRNA as compared with PBS control mice. Correspondingly, mRNA levels of proinflammatory genes IL-1β, TNFα, IL-6, and TGFβ1 were significantly reduced in the SMOC2 siRNA-treated mice as compared with untreated mice (Figures 4J, S4C, and S4D). The tubular epithelium showed less loss of LTL (Figure 4K), fewer PDGFRβ (Figure 4L) and macrophage infiltration (Figure 4M) in the CKD mice treated with SMOC2 siRNA as compared with untreated CKD mice.
Figure 4.

Silencing SMOC2 with siRNA prevents development of tubule-interstitial inflammation, fibrosis, and kidney function loss in the experimental mouse CKD model
(A) Glomerular filtration rate (GFR) measurement in the CKD mice treated with PBS or SMOC2 siRNA (30 μg/200 μL) twice/week starting day 21 after disease onset till day 43 for a total of seven treatments.
(B and C) Time course of blood urea nitrogen (BUN) and total urine protein to creatinine ratio (uPCR) in CKD mice treated with PBS and SMOC2 siRNA.
(D and E) Picosirius Red staining showing the interstitial fibrosis and quantification.
(F) Western blots showing the effect of silencing SMOC2 on fibrotic protein of collagen 1, α-SMA, and fibronectin in whole kidney lysates.
(G and H) Representative immunofluorescent images and quantification of protein expressions of collagen 1, α-SMA, and fibronectin in the CKD kidneys after SMOC2 silencing.
(I and J) mRNA levels of genes encoding collagen 1, α-SMA, and inflammatory genes of IL-1β and TNFα in CKD mice treated with PBS or SMOC2 siRNA.
(K–M) Representative images and quantification of LTL+, PDGFRβ+, F4/80 + cells in PBS versus SMOC2 siRNA-treated CKD mice. Immunofluorescence staining with 40x magnification and scale bar, 20 μm. n = 6/group unless otherwise stated. Data are represented as mean ± SEM.
Inhibition of autophagy in fibroblasts reduces SMOC2-mediated fibrogenic signaling
To elucidate the downstream cellular mechanism by which SMOC2 activates fibroblasts, the effects of SMOC2 on the Smad signaling pathway as well as the mammalian target of rapamycin (mTOR) pathway were investigated using NIH/3T3 cells that showed increases in inflammatory and fibrogenic genes following SMOC2 stimulation (Figure S5). SMOC2 activated Smad signaling in a time-dependent manner with an increase in the phosphorylation of the Smad 2/3 as early as 5 min after exposure to SMOC2 that was maintained for 15 min but declined rapidly thereafter (Figures 5A, S6A). SMOC2 also transiently increased phosphorylation of mTOR at 5 min; however, phosphorylation of mTOR was significantly decreased at 15 min, 30 min, 1 h, and 4 h, which indicated SMOC2 inhibited mTOR signaling pathway for a long-term effect (Figures 5A, S6B). We hypothesized that inhibition of mTOR signaling could lead to an activation of autophagy in NIH/3T3 cells, which mediated the downstream of SMOC2-activated signaling pathway. When NIH/3T3 cells were treated with SMOC2 (100 ng/mL) for 30 min in the presence of the lysosome pH alkalinizer and hence autophagy inhibitor, chloroquine, a significant suppression of Smad phosphorylation was observed even at low concentrations of 10 nM of chloroquine (Figures 5B, S6C). Furthermore, chloroquine also significantly reduced SMOC2-induced collagen 1 expression dose dependently (Figures 5C, S6D). Consistent with this result, other lysosome inhibitors such as bafilomycin (Figures 5D, 5E; S6E, S6F) and alternative inhibitors of autophagy, MHY1485 and 3-MA (Figures 5F, S6G), also resulted in reduction of SMOC2-stimulated p-Smad signaling along with a decrease in collagen 1 expression. In the last, SMOC2-induced collagen 1 levels were reduced (Figures 5G, S6H) when Beclin-1 (BCN1) was knocked down using siRNA (Figures 5H, S6I) to inhibit autophagy. These data suggested that SMOC2 stimulates fibrogenic pathways by activating autophagy.
Figure 5.

Lysosome-autophagosome signaling pathways are involved in SMOC2-induced kidney fibrosis
(A) In NIH/3T3 cells, the time course of SMOC2 (100 ng/mL)-induced Smad2/3 and mTOR phosphorylation (5 min, 15 min, 30 min, 1 h, 4 h, and 24 h; TGFβ1 stimulation for 30 min was used as a positive control for Smad activation).
(B and C) The dose-dependent effect of lysosome and autophagy inhibitor chloroquine (0.01, 0.1, 10, and 100 μM) on SMOC2 (100 ng/mL)-induced Smad 2/3 phosphorylation and collagen 1 expression.
(D and E) The dose-dependent effect of lysosome inhibitor, Bafilomycin (0.01, 0.1, 10, and 100 μM) on SMOC2 (100 ng/mL)-induced Smad 2/3 phosphorylation and collagen 1 expression.
(F) The effect of mTOR activator, MHY1485 (10 μM), and autophagy inhibitor, 3-MA (10 μM), on SMOC2 (100 ng/mL)-induced collagen 1 expression in NIH/3T3 cells.
(G and H) The effect of inhibiting autophagosomes by siRNA transduction of BECN1 on SMOC2 (100 ng/mL)-induced collagen 1 expression. Blots are representative of three independent experiments.
SMOC2 activates diverse cellular signaling pathways to promote fibroblast to myofibroblast activation
SMOC2 rapidly stimulates phosphorylation of extracellular signaling-related kinase (ERK; P42 and P44), P38, c-Jun N-terminal kinase (JNK), and Akt activation within 5 mins (Figure S7). To test the significance of these activated intracellular signaling pathways as well as TGFβ/Smad signaling for SMOC2-mediated collagen 1 expression in fibroblasts we conducted a series of experiments using inhibitors of TGFβRI/II (LY2109761), TGFβRI (SB431542), or TGFβRII (GW788388) (Figure 6A). SMOC2-induced collagen 1 protein increase was significantly blocked by a TGFβRII (GW788388) inhibitor, and it was partially blocked by TGFβRI and TGFβRI/II inhibitors, indicating SMOC2 cross-activated the TGFβRI/II signaling pathways. Since SMOC2 has been shown to act via integrin interactions to stimulate migration and adhesion, we investigated the role of integrins in SMOC2-mediated collagen 1 expression. We co-stimulated cells with inhibitors of integrin α5β1 (ATN-161) and αvβ5/αvβ3 (SB273005), which partially diminished SMOC2-induced collagen 1 expression (Figure 6B). We next used small-molecule inhibitors of P42/44 (U0126), P38 (SB203580), JNK (SP600125), and Akt (LY294002), which partially inhibited SMOC2-induced collagen 1 expression (Figure 6C). This suggested that SMOC2-stimulated fibrogenesis could be inhibited partially by blocking MAPKs and PI3/Akt signaling. As α-SMA expression leads to the assembly of stress fibers in vitro, we next validated this structural formation after SMOC2 treatment of fibroblasts. SMOC2-stimulated stress fiber formation during myofibroblast differentiation was completely blocked by inhibitors of MAPKs (U0126, SB203580, and SP600125, Figure 6D), PI3K/Akt (LY294002, Figure 6D), ALK5/TGFbRI/II (LY2109761, GW788388, and SB431542, Figure 6D), integrins (SB273005, Figure 6D, and ATN-161, Figure S8), and lysosomes/autophagy (chloroquine and bafilomycin, Figure S8). As positive controls TGFβ triggered stress fiber formation and formation was blocked by inhibitors of TGFβRI and/or TGFβRII (Figure 6E).
Figure 6.

SMOC2 induces kidney fibrosis by cross-activating signaling cascades of TGFβ, Integrin, MAPKs, and Akt
(A) Western blots showing collagen 1 production in response to several stimuli at 24 h. Note that SMOC2 stimulation markedly increases collagen 1 expression. ALK5/TGFβR II inhibitor of GW788388 (10 μM), ALK5 inhibitor of SB431542 (10 μM), and TGFβR I/II inhibitor of LY2109761 (10 μM) block the SMOC2 (100 ng/mL, 24 h)-induced collagen 1 expression.
(B) Western blots show that SMOC2 (100 ng/mL, 24 h)-induced collagen 1 responses of the different inhibitors, integrin α5 antagonist of ATN-121 (100 μM) and integrin antagonist of SB273005 (1 μM).
(C) Western blots show the effects of inhibition of different signaling pathways on the SMOC2-induced collagen 1 expression with p42/44-MAPK inhibition (U0126 [10 μM]), p38-MAPK inhibition (SB203580 [10 μM]), JNK/SAPK inhibition (SP600125 [10 μM]), and PI3K/Akt inhibition (LY294002 [20 μM]).
(D and E) Immunostaining for F-Actin shows effects on NIH/3T3 of co-stimulation of SMOC2 (100 ng/mL, 24 h) with inhibitors or antagonists (U0126, SP600125, SB203580, LY294002, LY2109761, GW788388, SB431542, SB273005). Positive controls consisted of predicted modulation of TGFβ1 (10 ng/mL, 24 h) stimulation and cascades inhibitors (SB431542, LY2109761, GW788388). Immunofluorescence staining with 40x magnification and scale bar, 20 μm. Images and blots are representative of three independent experiments.
Discussion
Kidney fibrosis is a common underlying characteristic of most forms of CKDs that often leads to end-stage kidney disease. In the present study, we generated an experimental mouse model of CKD resulting in injury and maladaptive repair with inflammation, progressive reduction in GFR, elevated BUN, and interstitial fibrosis. Commonly used animal models of CKD associated with interstitial fibrosis include folic acid-induced nephropathy, cyclosporin A nephrectomy, DOCA salt nephropathy, 5/6 nephrectomy, radiation nephropathy, and UUO (Rabe and Schaefer, 2016). Each model has their own characteristics and limitations with limited applicability to translatability to humans (Yang et al., 2010a). In addition, we and others have used ischemia/reperfusion and aristolochic acid nephropathy (Yang et al., 2010b). The UUO model is the most widely used but suffers from little proteinuria and the rapidity of changes due to the irreversibility and severity of the intervention. Although these models manifest severe tubulo-interstitial fibrosis and inflammation, the use of toxicants, e.g., folic acid, introduces an added dimension that may not map to human pathobiology (Leaf and Duffield, 2017). Ischemia, either global or regional, is an important contributor to human CKD, but our standard bilateral ischemia model often provides limited functional and pathological changes over time especially in the C57Bl/6 mouse strain, which is often used because of the availability of genetic mutants on this background. The experimental mouse CKD model we developed recapitulates many features of human kidney fibrotic disease, including the gradual loss of kidney function measured as decreased GFR, and increased BUN and proteinuria. Most importantly, the timeline of this novel experimental model is prolonged over 6 weeks as opposed to an acute surgical insult such as UUO.
The matricellular protein SMOC2 is highly expressed in the CKD mouse model and its expression patterns correlate with expression of fibrotic proteins collagen, α-SMA, and fibronectin. We show that genetic and pharmacologic inhibition of SMOC2 after the initial insult protects mice from loss of kidney function, fibrosis, and CKD. Previously, we found that SMOC2 promoted kidney fibrosis with enhanced extracellular matrix accumulation (Gerarduzzi et al., 2017). Recently, the fibrogenic role of SMOC2 was extended to fibrosis in the lung and liver. Li reported in the mouse model of bleomycin-induced lung fibrosis deficiency of SMOC2 significantly decreased lung fibrosis by inhibition of the TGF-β1 signaling pathway and the reduction of the inflammatory response (Luo et al., 2018). Another study found that depletion of SMOC2 blocked high-fat-diet-induced metabolic syndrome, fibrosis, and hepatic steatosis in mice (Yuting et al., 2019). However, the detailed mechanisms of how SMOC2 leaves parent cells and activates target cells to trigger fibrosis remains largely unclear.
Although it remains somewhat controversial, an emerging body of evidence demonstrates that autophagy plays important roles in acute kidney injury and CKD (Canaud et al., 2019; Kimura et al., 2017; Lenoir et al., 2016; Liu et al., 2016). Inhibition of autophagy by 3-MA increased tubular epithelial cell apoptosis and interstitial fibrosis in a rat model of unilateral ureteral fibrosis (UUO) (Ding et al., 2014; Kim et al., 2012). Consistently, genetic depletion of either LC3 or Beclin1 results in decrease of autophagy in the UUO model in mice with aggravation of fibrosis by enhanced collagen deposition in the interstitium (Ding et al., 2014). Studies have also shown that persistent activation of autophagy promotes renal fibrosis because of induction of fibroblast proliferation, activation, and tubular epithelial cell death (Livingston et al., 2016). By using a newly developed autophagy reporter mouse model, PT-Atg7 KO mice, and pharmacological inhibitors of autophagy, Livingston et al. showed that activation of autophagy in kidney proximal tubular cells promoted kidney fibrosis by enhancing inflammation, tubular atrophy, and the production of profibrotic factors (Livingston et al., 2016). Also, Xue et al. demonstrated that blocking autophagy by inhibition of the activation of PKCα could decrease TGFβ-induced fibrotic response in kidney fibroblasts, which confirmed the profibrotic role for the activation of autophagy (Xue et al., 2018). In this study, we found the inhibition of autophagy decreased SMOC2-induced fibrogenic signaling in fibroblasts. This suggests that the profibrotic role of SMOC2 is via autophagy in vitro. Although SMOC2 has not been previously associated with autophagy, its family member SPARC has been implicated in the induction of autophagy and, subsequently, autophagy-mediated apoptosis by elevation of cathepsin B (Bhoopathi et al., 2010). SPARC deficiency was identified to reduce autophagy-mediated apoptosis in the vascular endothelial cell in a concanavalin A-induced acute liver damage model (Peixoto et al., 2015). Overexpression of SPARC in neuroblastoma cells resulted in ER stress-induced autophagy, which finally led to apoptosis (Rabe and Schaefer, 2016; Sailaja et al., 2013).
In summary, we provide evidence that SMOC2 plays a central role in fibrogenesis and inflammation by activating multiple signaling pathways and autophagy in vitro and in vivo. Furthermore, we show that silencing SMOC2 after disease onset prevents progression of the disease and underlying fibrosis in a physiologically relevant experimental mouse model of CKD. Our investigation provides new evidence that targeting SMOC2 offers a therapeutic approach to treat kidney fibrosis and CKD.
Limitation of the study
CKD is a complex multifactorial disease with heterogenous pathology, and although the experimental mouse model of CKD that we developed in this study recapitulates many features of human kidney fibrotic disease, the surgically induced ischemia-reperfusion injury followed by nephrectomy may not encompass the breadth of pathophysiological causes of CKD in humans. The CKD model also needs to be further characterized for human relevance in terms of physiological changes, functional changes, and pathological changes underlying CKD beyond fibrosis such as inflammation, disruption of vasculature, glomerular and tubular cell perturbation, cell cycle arrest, and cellular senescence.
STAR★Methods
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SMOC2 | Santa Cruz Biotechnology Inc. | Cat# sc-67396; RRID: AB_2605425 |
| Mouse monoclonal anti-α-SMA | Sigma-Aldrich | Cat# A2547; RRID: AB_476701 |
| Rabbit polyclonal anti-Collagen 1 | Abcam | Cat# ab600408; RRID: AB_10556808 |
| Rabbit polyclonal anti-Fibronectin | Abcam | Cat# ab23750; RRID: AB_447655 |
| Rat monoclonal anti-F4/80 | Abcam | Cat# ab6640; RRID: AB_1140040 |
| Rabbit polyclonal anti-PDGFRβ | Abcam | Cat# ab32570; RRID: AB_777165 |
| Rabbit polyclonal anti-p-mTOR | Cell Signaling | Cat# 2971S; RRID: AB_330970 |
| Rabbit polyclonal anti-p-Smad2/3 | Cell Signaling | Cat# 13820S; RRID: AB_2631089 |
| Rabbit polyclonal anti-p-p42/44 | Cell Signaling | Cat# 4377S; RRID: AB_331775 |
| Rabbit polyclonal anti-p-p38 | Cell Signaling | Cat# 9211S; RRID: AB_331641 |
| Rabbit polyclonal anti-p-JNK | Cell Signaling | Cat# 9251S; RRID: AB_331659 |
| Rabbit polyclonal anti-p-Akt | Cell Signaling | Cat# 9271S; RRID: AB_329825 |
| Rabbit polyclonal anti-BCN1 (E-8) | Santa Cruz Biotechnology Inc. | Cat# sc-48341; RRID: AB_626745 |
| Chemicals, peptides, and recombinant proteins | ||
| ATN-161 (Ac-PHSCN-NH2) | Selleckchem | Cat# S8454 |
| Chloroquine diphosphate salt | Sigma | Cat# C 6628-50G |
| GW788388 | Selleckchem | Cat# S2750 |
| LY2109761 | Selleckchem | Cat# S2704 |
| LY294002 | GIBCO | Cat# PHZ1144 |
| 3-MA | Millipore Sigma | Cat# 189490-100MG |
| MHY1485 | Millipore Sigma | Cat# 5005540001 |
| SB203580 | Selleckchem | Cat# S1076 |
| SB273005 | Selleckchem | Cat# S7540 |
| SB431542 | Selleckchem | Cat# S1067 |
| SP600125 | Selleckchem | Cat# S1460 |
| U0126 | EMD Millipore | Cat# 19-147 |
| Mouse SMOC2 recombinant protein | R&D | Cat# 6075-SM-050 |
| Critical commercial assays | ||
| GFR measurement: Transdermal Mini GFR Monitor | MediBeacon | Model: Q-P10A |
| BUN measurement: Urea Liquid Stable Reagent | Infinity/ ThermoScientic | Cat# TR12421 |
| siRNA in vitro transfection: lipofectamine® RNAiMAX | Thermo Fisher Scientific | Cat# 13778075 |
| Cell differentiation assay: Phalloidin-iFluor 488 | Abcam | Cat# ab176753 |
| Experimental models: cell lines | ||
| NIH-3T3 fibroblast | ATCC | CRL-1568 |
| Experimental models: organisms/strains | ||
| Mouse: B6-Tyrc-2J/J | The Jackson Laboratory | Stock No. 000058 |
| Mouse: B6N.Cg-Tg(Sox2-cre)1Amc/J | The Jackson Laboratory | Stock No. 014094 |
| Mouse: C57Bl/6 | The Jackson Laboratory | Stock No. 000664 |
| Oligonucleotides | ||
| Accell SMOC2 siRNA | Horizon a PerkinElmer Company | Cat# A-049770-16-0050 |
| Software and algorithms | ||
| Photoshop | Adobe | N/A |
| BioRender | App.biorender.com | BioRender Harvard |
| imageJ | http://imagej.net/ZContributors | Version: 2.1.0/1.53c |
| Prism | www.graphpad.com | Version: 9.1.0 (216) |
Resource availability
Lead contact
Further information, inquiries, and request should be directed to the Lead Contact Cuiyan Xin (cxin@bwh.harvard.edu)
Materials availability
This study did not generate new reagents.
Experimental model and subject details
SMOC2 genetic mouse models
Smoc2tm1.1 (KOMP)Vlcg was generated by the Knockout Mouse Phenotyping Program (KOMP2) at The Jackson Laboratory by our group as described previously (Gerarduzzi et al., 2017) using embryonic stem (ES) cells provided by the International Knockout Mouse Consortium. The ZEN-UB1 Velocigene cassette was inserted into the gene, and replacing all coding exons and intervening sequences. The construct was then introduced into C57BL/6N-derived VGB6 ES cells, and correctly targeted ES cells were injected into B6-Tyrc-2J/J (stock no. 000058) blastocysts. The resulting chimeric males were bred to C57BL/6J females and then to B6N.Cg-Tg(Sox2-cre)1Amc/J (stock no. 014094) to remove neo cassette. Resulting offspring were crossed to remove the cre-expressing transgene. Genetic konckout mouse models were compared with their respective wild type (WT) littermates.
Chronic kidney disease (CKD) model
C57Bl/6 mice (Stock No. 000664) were purchased from the Jackson Laboratory. Male 10-12 weeks of age C57Bl/6 mice were first subjected to bilateral ischemia reperfusion injury (Bi-IRI) (Yang et al., 2010b) with a difference in the duration of ischemia on each kidney: Mice were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and body temperature was maintained at 36–38°C. Kidneys were exposed through flank incisions. Both renal pedicles were then clamped with nontraumatic microaneurysm clamps (Roboz, RS-5420) to induce ischemia. The incisions were temporarily closed during ischemia or sham surgery. After the clamps were removed, reperfusion of the kidneys was visually confirmed. For the left kidney the perfusion was restored after 10 min of ischemia whereas it was restored after 24 min in the right kidney (Figure 1A). At day 5 post Bi-IRI, the left kidney was removed (Figure 1A). Subsequently blood and urine were collected prior to Bi-IRI and twice per week until 45 days post nephrectomy. Glomerular filtration rate (GFR) was measured prior to Bi-IRI and weekly until 45 days post nephrectomy. Four groups of mice were investigated: sham control surgery (Sham), nephrectomy only surgery on left kidney (Nx), bilateral ishemia reperfusion injury only (Bi-IRI), and double surgeries of bilateral ishemia reperfusion injury following nephrectomy on the left kidney to produce CKD. Mouse kidneys were collected at day -7 and 45 respectively post nephrectomy. All studies were carried out under approved IACUC protocols at Brigham and Women’s Hospital.
Cell line for in vitro study
NIH/3T3 cells from ATCC, which were grown as a monolayer in polystyrene culture dishes containing DMEM F12 (Corning, 10-092-CV) and supplemented with 10% FBS (Gibco, 10437-028). Cells were grown until 80% confluency before passage maintained at 37°C in a humidified 5% CO2 incubator.
Method details
In vivo siRNA administration
Male 10-12 weeks of C57Bl/6 mice received SMOC2 siRNA (Dharmacon, A-049770-16-0050) 30 μg/200μl or PBS in RNase-free PBS carriage medium by retro-orbital injection twice per week beginning at day 21 post nephrectomy surgery till day 43 for a total of 7 injections.
Histology and picrosirius red staining
Kidney tissues of CKD models from C57Bl6, SMOC2 WT, SMOC2 KO and their respective littermate controls were perfused with cold PBS before harvesting. Kidneys were then fixed in formalin, dehydrated in 70% EtOH, and sent to invivo BioService pathology core led by Sunny Lu embedded in paraffin, and stained for PAS. Paraffin-embedded tissues were also cut into 5- to 7-μm sections and stained with Picrosirius Red (Polysciences, 09400).
In vivo immunofluorescence
Kidney tissues for immunofluorescence were fixed in 4% paraformaldehyde at 4°C for 2 hours, then washed in 18% sucrose solution overnight prior to cryopreservation in Tissue-Tek O.C.T. (Avantik, EM0123). OCT-embedded mouse kidneys were cut into 5- to 7-μm sections and permeabilized in 1× PBS containing Triton X-100 (0.1%) for 10 minutes. The sections were then labeled with primary antibodies (1:200; Table S1). Slides with primary antibodies were subsequently exposed to Cy3 (1:400; Jackson ImmunoResearch Laboratories, 711-165-152)- or FITC (1:400; ThermoFisher, F-2765)-conjugated secondary antibodies. VECTASHIELD Mounting Medium with DAPI (VECTOR LABORATORIES, H-1200) was used for nuclear staining (blue).
Immunoblotting
Kidney tissues were homogenized in RIPA buffer (Boston BioProduct, BP-115-5X) containing protease and phosphatase inhibitors (Roche Applied Science, 11836153001). Samples were centrifuged for 10 min at 13000 rpm and the supernatant was taken for protein determination. Protein concentrations were determined using the Bradford protein estimation Kit (Bio-Rad, 5000006), and an equal amount of protein (10-25 μg) was loaded on either a 6% or 10% polyacrylamide gel (PAGE). Protein transfer was performed using a nitrocellulose membrane. The primary antibodies were used to detect the specific protein as listed in Table S2. Horseradish peroxidase–conjugated secondary antibodies against mouse (Agilent, P044701-2) and rabbit (Agilent, P044801-2) were used to detect the appropriate primary antibody. Bands were detected with SuperSignal West Dura Chemiluminescent Kit (Thermo Fisher Scientific, 34076) and captured with ChemiDOC MP imaging System (Bio-Rad).
Blood urea nitrogen and urine protein to creatinine ratio measurements
Mice blood were collected in Microhematocrit Heparinized Capillary Tubes (FISHER SCIENTIFIC, 22362566) and centrifuged for 10 min at 5000 rpm. 3 ul of plasma was for BUN measurement and this study followed the instruction of Urea Liquid Stable Reagent (Infinity/ ThermoScientic, TR12421). 10 ul of urine were sent to O’Brien Center Core C Resource of The University of Alabama at Birmingham (UAB/UCSD) for creatinine measurement by isotope dilution LC-MSMS. Total urine protein was detected from mouse urine by quanTest red total protein assay system (Quantimetrix, REF 2210-02).
Glomerular filtration rate (GFR) measurement
This was studied using modifications of a protocol described from MediBeacon (Scarfe et al., 2018). Briefly, FITC-sinistrin solution was prepared as 40 mg/mL in phosphate buffered saline (PBS) and recommended dose of 0.15 mg FITC-sinistrin per gram body weight. Mouse was anesthetized with 3% isoflurane and shaved from the top of the hind legs up to the neck, and across the ribs. A thin layer of depilation cream was applied to the shaved area and then removed after 1- 3 min by washing it off with cotton swabs and warm water. The Transdermal GFR Monitor was firmly attached to the back of mouse by the tape. Connect the battery to the device (the device is ready to use, and data acquisition starts when the blue light emitting diodes (LEDs) start blinking), leave the device untouched for 3 minutes before the FITC-sinistrin injection to allow a steady background reading to be taken. Administer 10 ul FITC-sinistrin by retro-orbital injection using an insulin syringe. Place the mouse in a cage on its own to recover from isoflurane anesthesia and for the duration of the measurement period. Observe the mouse in the cage for 1.5 h and then remove the device. Pull off the white plaster tape from underneath the belly and remove the device and black plaster from the skin, carefully disconnect the battery from the device. Connect the device to the USB cable and then connect the cable to the computer and open the reading software (Sensor_ctrl_app.exe), Process and evaluate data in the analysis software as described in the respective manual.
In vitro inhibition experiments
For in vitro experiments, 80% confluent NIH/3T3 cells were preincubated with kinases inhibitors and receptor antagonists (Table S3) in DMEM 1x (Corning, 10-013-CV) containing 0.1 mg/ml BSA (Sigma, A7906) prior to being treated with 100 ng/ml SMOC2 recombinant protein (R&D, 6075-SM-050).
In vitro gene silencing
siRNA for BCN1 and control siRNA (Life technology) were resuspended in RNase free water and stored (−80°C, 10 μM). 24h before transfection, NIH/3T3 were seeded into a six-well plate with 300,000 cells/well. siRNA (final concentration of 25 nM/well) was resuspended in Opti-MEM® Medium, lipofectamine® RNAiMAX (Thermo Fisher Scientific, 13778075) was added, gently mixed, and further incubated for 5 min at room temperature. Medium containing siRNA was added to the wells and plates were swirled gently. Cells were incubated under normal conditions for 16 hours, followed by substitution of additional culture medium. After 48 hours, cells were evaluated for gene silencing and harvested for Western blot.
Cell differentiation assay
NIH/3T3 cells were cultured in chamber slides (Thermo Fisher Scientific, 154941) till 60-80% confluent, starved and stimulated in DMEM 1x containing 0.1 mg/ml BSA (Sigma-Aldrich, A7906-100G). Cells were then removed the media and fixed with 3.7% paraformaldehyde (Sigma-Aldrich, P6148-500G) in PBS, permeabilized with 0.5% Triton X-100 (Thermo Fisher Scientific, BP151-500) in PBS, and then blocked in 10% mouse serum in PBS (Sigma-Aldrich, M5905-10ML) for 30 min. Cytoskeletal F-Actin was visualized using Phalloidin-iFluor 488 (Abcam, ab176753) at 1:500 in PBS at 4C for overnight. Cells then mounted with Prolong Gold antifade with DAPI (Invitrogen, P36931) mounting media and sealed with nail polish.
Cell culture western blot analysis
Cells were lysed in RIPA buffer (Boston BioProduct, BP-115-5X) containing protease and phosphatase inhibitors (Roche Applied Science, 11836153001) and homogenized by 10 passes through 1 ml syringe with 27 G needle. Cell lysates were performed western blot as described in “Immunoblotting” and investigated the proteins expression as listed in Table S2.
In vitro immunofluorescence
NIH/3T3 cells were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) in PBS, permeabilized with 0.1% Triton X-100 (Thermo Fisher Scientific) in PBS, and then blocked in 3% BSA (Sigma-Aldrich). Cytoskeletal F-Actin was visualized using Alexa Fluor 564–conjugated Rhodamine Phalloidin (Thermo Fisher Scientific) at 1:500 in PBS for 1 hour. 4,6-Diamidino-2-phenylindole (Sigma-Aldrich) was used for nuclear staining (blue).
Quantitative PCR
Total RNA was isolated from cell cultures or tissue samples using Trizol reagent (ambion, 15596018) according to the manufacturer's protocol. RNA concentration was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific) and purity was determined by A260 to A280. Isolated RNA (1 μg) was reverse transcribed into cDNA using a iScript Reverse Transcription Kit from Bio-Rad (1708841). Quantitative PCR (qPCR) was performed using a iTaq Universal SYBR Green Supermix (Bio-Rad, 1725121) on a CFX96 Real-Time System (Bio-Rad). All samples were measured with technical duplicates and normalized against GAPDH. Changes in the mRNA expression were calculated using the ΔΔCt method relative to a control. Forward and reverse primer sequences for mouse-specific genes are listed in Table S4.
Imaging
Light microscopic images were captured on a Carl Zeiss AxioImager M2 with a Plan Apochromat 20×/0.8 objective using AxioVision SE64 software. Confocal images were acquired in the Nikon Imaging Center in the Renal Division, Brigham and Women's Hospital.
Quantification and statistical analysis
Data are expressed as the average ±SEM. Number of smaples assayed in each experiment is indicated in “Main figures titles and Legends”. Statistical significance for multiple comparisons was evaluated by one-way ANOVA with Tukey post-hoc analysis (p < 0.05), using GraphPad Prism (GraphPad software). Statistical significance for single comparisons was calculated by two-tailed Student's t test (p < 0.05), using Microsoft Excel. The sample size was predetermined based on the effect size and variability observed previously from similar readouts in our laboratory.
Additional resources
None.
Acknowledgments
Work in the Vaidya laboratory is supported by Outstanding New Environmental Sciences (ONES) award from NIH/NIEHS (R01ES017543). J.V.B. was supported by grants from the National Institute of Health/NIDDK R01DK072381 and UH3 TR002155.
Author contributions
Conceptualization, C.X., V.S.V, and J.V.B.; Methodology, C.X.; Formal Analysis and Validation, C.X., J.L., Q.W., Y.Y., X.Y., J.A.M.G.; Investigation, C.X., J.L., Q.W., Y.Y., X.Y., J.A.M.G., V.S., and X.S.; Writing – Original Draft, C.X., V.S.V, and J.V.B.; Writing – Review & Editing, C.X., V.S.V, and J.V.B.; Resources, C.X., V.S.V, and J.V.B.; Supervision, C.X., V.S.V, and J.V.B.; Funding Acquisition, V.S.V. and J.V.B.
Declaration of interests
V.S.V. is an employee of Pfizer, Inc., co-inventor on SMOC2 patents assigned to Mass General Brigham (formerly known as Partners Healthcare) and is a co-founder of Mediar Therapeutics. J.V.B. is co-founder and holds equity in Goldfinch Bio. J.V.B.is co-inventor on KIM-1 patents assigned to Mass General Brigham (formerly known as Partners Healthcare), holds equity in Dicerna, Goldilocks, Inniviva, Medibeacon, Medssenger, VeriNano, Renal Tracker, Rubius, Sensor-Kinesis, Sentien, and Theravance and has received consulting income from Biomarin, Aldeyra, Angion, PTC, Praxis, and Sarepta. J.V.B. and V.S.V.'s interests were reviewed and are managed by BWH and Partners HealthCare in accordance with their conflict-of-interest policies. V.S. has patents on KIM-1 assigned to Mass General Brigham (formerly known as Partners Healthcare).
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103193.
Contributor Information
Cuiyan Xin, Email: cxin@bwh.harvard.edu.
Vishal S. Vaidya, Email: vvaidya@bwh.harvard.edu.
Joseph V. Bonventre, Email: jbonventre@bwh.harvard.edu.
Supplemental information
Data and code availability
This study did not report standardized datatype and original code. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
References
- Bhoopathi P., Chetty C., Gujrati M., Dinh D.H., Rao J.S., Lakka S. Cathepsin B facilitates autophagy-mediated apoptosis in SPARC overexpressed primitive neuroectodermal tumor cells. Cell Death Differ. 2010;17:1529–1539. doi: 10.1038/cdd.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P., Sage E.H. Matricellular proteins: extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- Brekken R.A., Sage E.H. SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biol. 2000;19:569–580. doi: 10.1016/s0945-053x(00)00105-0. [DOI] [PubMed] [Google Scholar]
- Brekken R.A., Sage E.H. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- Canaud G., Brooks C.R., Kishi S., Taguchi K., Nishimura K., Magassa S., Scott A., Hsiao L.L., Ichimura T., Terzi F. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Kim S., Lee S.Y., Koo J.K., Wang Z., Choi M.E. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014;25:2835–2846. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris A.B., Colvin R.B. Renal interstitial fibrosis: mechanisms and evaluation. Curr. Opin. Nephrol. Hypertens. 2012;21:289–300. doi: 10.1097/MNH.0b013e3283521cfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerarduzzi C., Kumar R.K., Trivedi P., Ajay A.K., Iyer A., Boswell S., Hutchinson J.N., Waikar S.S., Vaidya V.S. Silencing SMOC2 ameliorates kidney fibrosis by inhibiting fibroblast to myofibroblast transformation. JCI Insight. 2017;2:e90299. doi: 10.1172/jci.insight.90299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoerger T.J., Wittenborn J.S., Segel J.E., Burrows N.R., Imai K., Eggers P., Pavkov M.E., Jordan R., Hailpern S.M., Schoolwerth A.C. A health policy model of CKD: 1. Model construction, assumptions, and validation of health consequences. Am. J. Kidney Dis. 2010;55:452–462. doi: 10.1053/j.ajkd.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Kim W.Y., Nam S.A., Song H.C., Ko J.S., Park S.H., Kim H.L., Choi E.J., Kim Y.S., Kim J., Kim Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology (Carlton) 2012;17:148–159. doi: 10.1111/j.1440-1797.2011.01541.x. [DOI] [PubMed] [Google Scholar]
- Kimura T., Isaka Y., Yoshimori T. Autophagy and kidney inflammation. Autophagy. 2017;13:997–1003. doi: 10.1080/15548627.2017.1309485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaf I.A., Duffield J.S. What can target kidney fibrosis? Nephrol. Dial Transpl. 2017;32:i89–i97. doi: 10.1093/ndt/gfw388. [DOI] [PubMed] [Google Scholar]
- Lenoir O., Tharaux P.L., Huber T.B. Autophagy in kidney disease and aging: lessons from rodent models. Kidney Int. 2016;90:950–964. doi: 10.1016/j.kint.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Liu N., Shi Y., Zhuang S. Autophagy in chronic kidney diseases. Kidney Dis. (Basel) 2016;2:37–45. doi: 10.1159/000444841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Lu J., Cardoso W.V., Vaziri C. The SPARC-related factor SMOC-2 promotes growth factor-induced cyclin D1 expression and DNA synthesis via integrin-linked kinase. Mol. Biol. Cell. 2008;19:248–261. doi: 10.1091/mbc.E07-05-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston M.J., Ding H.F., Huang S., Hill J.A., Yin X.M., Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy. 2016;12:976–998. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L., Wang C.C., Song X.P., Wang H.M., Zhou H., Sun Y., Wang X.K., Hou S., Pei F.Y. Suppression of SMOC2 reduces bleomycin (BLM)-induced pulmonary fibrosis by inhibition of TGF-beta1/SMADs pathway. Biomed. Pharmacother. 2018;105:841–847. doi: 10.1016/j.biopha.2018.03.058. [DOI] [PubMed] [Google Scholar]
- Lysaght M.J. Maintenance dialysis population dynamics: current trends and long-term implications. J. Am. Soc. Nephrol. 2002;13:S37–S40. [PubMed] [Google Scholar]
- Pazin D.E., Albrecht K.H. Developmental expression of Smoc1 and Smoc2 suggests potential roles in fetal gonad and reproductive tract differentiation. Dev. Dyn. 2009;238:2877–2890. doi: 10.1002/dvdy.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto E., Atorrasagasti C., Aquino J.B., Militello R., Bayo J., Fiore E., Piccioni F., Salvatierra E., Alaniz L., Garcia M.G. SPARC (secreted protein acidic and rich in cysteine) knockdown protects mice from acute liver injury by reducing vascular endothelial cell damage. Gene Ther. 2015;22:9–19. doi: 10.1038/gt.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe M., Schaefer F. Non-transgenic mouse models of kidney disease. Nephron. 2016;133:53–61. doi: 10.1159/000445171. [DOI] [PubMed] [Google Scholar]
- Sailaja G.S., Bhoopathi P., Gorantla B., Chetty C., Gogineni V.R., Velpula K.K., Gondi C.S., Rao J.S. The secreted protein acidic and rich in cysteine (SPARC) induces endoplasmic reticulum stress leading to autophagy-mediated apoptosis in neuroblastoma. Int. J. Oncol. 2013;42:188–196. doi: 10.3892/ijo.2012.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarfe L., Schock-Kusch D., Ressel L., Friedemann J., Shulhevich Y., Murray P., Wilm B., de Caestecker M. Transdermal measurement of glomerular filtration rate in mice. J. Vis. Exp. 2018;140:e58520. doi: 10.3791/58520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G.S., Rustgi A.K. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br. J. Cancer. 2013;108:755–761. doi: 10.1038/bjc.2012.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Ren J., Sun X., Gui Y., Feng Y., Shu B., Wei W., Lu Q., Liang Y., He W. Protein kinase Calpha drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J. Biol. Chem. 2018;293:11119–11130. doi: 10.1074/jbc.RA118.002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.C., Zuo Y., Fogo A.B. Models of chronic kidney disease. Drug Discov. Today Dis. Models. 2010;7:13–19. doi: 10.1016/j.ddmod.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Besschetnova T.Y., Brooks C.R., Shah J.V., Bonventre J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010;16:535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuting Y., Lifeng F., Qiwei H. Secreted modular calcium-binding protein 2 promotes high fat diet (HFD)-induced hepatic steatosis through enhancing lipid deposition, fibrosis and inflammation via targeting TGF-beta1. Biochem. Biophys. Res. Commun. 2019;509:48–55. doi: 10.1016/j.bbrc.2018.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not report standardized datatype and original code. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.