

OVERVIEW OF THE ENDOGENOUS OPIOID SYSTEM
The endogenous opioid system uses the three major neuropeptides enkephalin (ENK), dynorphin (DYN), and β-endorphin (END), although there is relatively little END in the amygdala compared with ENK and DYN. These peptides are derived from the peptide precursors proenkephalin (pENK), prodynorphin (pDYN), and proopiomelanocortin (POMC), respectively, and interact with the three subtypes of G-protein-coupled opioid receptors. Enkephalins interact with both mu opioid receptors (MOR) and delta opioid receptors (DOR), while DYN has the highest affinity for kappa opioid receptors (KOR), and END is thought to act primarily at MOR (Henry, Gendron, Tremblay, & Drolet, 2017; Lutz & Kieffer, 2013; Mansour, Fox, Akil, & Watson, 1995; Mansour, Watson, & Akil, 1995). These peptides, however, do not show a large degree of discrimination for the three opioid receptor subtypes (Clarke, Zimmer, Zimmer, Hill, & Kitchen, 2003; Schoffelmeer et al., 1990).
This review will cover the anatomy and physiological effects of these three peptides and their receptors in the amygdala and discuss the behavioral responses seen during genetic or pharmacological manipulations of the amygdalar opioid system. Since all responses mediated or affected by amygdalar opioids are beyond the scope of this review, we will focus on opioid effects in nociception, stress and anxiety-related responses, associative learning and conditioned fear, ethanol effects, and opioid dependence or withdrawal. Much of the work in the amygdala has historically focused on the ENK system and MOR-mediated effects, although interesting new data are emerging on the role of the DYN/KOR system and actions of ENK via DOR in these functional outcomes. In this review we use the broader term opioid to refer to natural or synthetic substances that bind to opioid receptors, while the term opiate refers more selectively to compounds that are naturally derived from the poppy plant, such as morphine and heroine.
BASIC AMYGDALA ANATOMY: NUCLEI AND NEURONS
The amygdala contains about a dozen nuclei, each with several subdivisions. The terminology used to describe the amygdalar nuclei differs among investigators and atlases. In this review, the terminology of the atlas by Paxinos and Watson (1997) will be used. Many of the nuclei of the amygdala can be seen in a coronal section cut through the middle of the amygdala (Fig. 1). The cortical and medial nuclei are located along the ventral and medial surfaces of the amygdala in most mammals including rodents (Fig. 1). The cortical nucleus has three main subdivisions: Copl, posterolateral; Coa, anterior; and Copm, posteromedial. There are four medial nuclear subdivisions: Mad, anterodorsal; Mav, anteroventral; Mpd, posterodorsal; Mpv, posteroventral. The central nucleus (CEA) is located dorsolateral to the medial nucleus (MEA). Three of the four main subdivisions of the central nucleus are shown in Fig. 1: CM, medial; CL, lateral; and CLC, lateral capsular. The basolateral nuclear complex (BLA) is located deep to the cortical and central nuclei. It consists of three main nuclei, the lateral (LA), basolateral (BL), and basomedial nuclei (BM), arranged from dorsal to ventral, respectively. Each nucleus has several subdivisions (LA: Ldl, dorsolateral, Lvl, ventrolateral, and Lvm, ventromedial subdivisions; BL: BLa, anterior, BLp, posterior, and BLv, ventral subdivisions; BM: BMa, anterior, and BMp posterior subdivisions of Paxinos & Watson, 1997) (Fig. 1). Surrounding the basolateral nuclear complex are clusters of small GABAergic neurons, the intercalated nuclei (IN). The posteromedial region of the amygdala, where it merges with the hippocampal formation, contains a nucleus termed the amygdalohippocampal area (AHA).
FIG. 1.
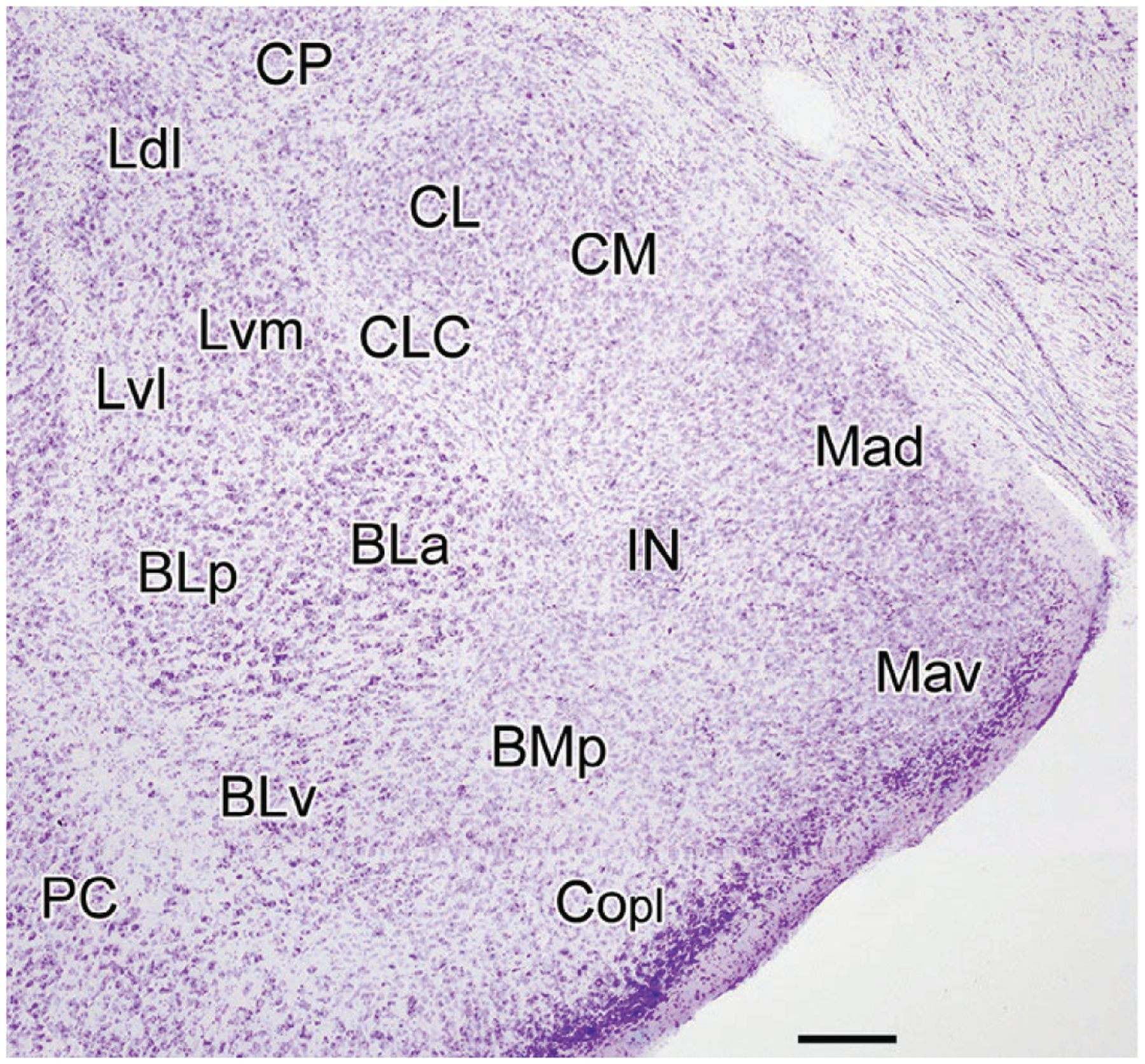
Nissl-stained coronal section through the amygdala of the rat (bregma-2.8 level; nomenclature of Paxinos & Watson, 1997). BLa, anterior basolateral nucleus; BLp, posterior basolateral nucleus; BLv, ventral basolateral nucleus; BMp, posterior basomedial nucleus; CL, lateral central nucleus; CLC, lateral capsular central nucleus; CM, medial central nucleus; Copl, posterolateral cortical nucleus; CP, caudatoputamen; IN, intercalated nucleus; Ldl, dorsolateral lateral nucleus; Lvl, ventrolateral lateral nucleus; Lvm, ventromedial lateral nucleus; Mad, anterodorsal medial nucleus; Mav, anteroventral medial nucleus; PC, piriform cortex. Scale bar=250μm.
The amygdalar nuclei can be grouped into two major regions: (1) the cortical and basolateral nuclei (CBL) which contain cortex-like neurons, and the central and medial nuclei which contain striatal-like neurons (McDonald, 1992, 2003). The principal neurons of the CBL, glutamatergic pyramidal neurons (PNs), have pyramidal or semi-pyramidal somata and spiny dendrites like cortical pyramidal neurons. The nonpyramidal neurons (NPNs) of the CBL, like cortical non-pyramidal neurons, are GABAergic neurons with spine-sparse dendrites that mainly function as interneurons. Neurons in the central and medial nuclei are predominantly GABAergic, and those in lateral portions of the central nucleus adjacent to the striatum closely resemble the GABAergic medium spiny neurons of the striatum (McDonald, 1992, 2003).
The bed nucleus of the stria terminalis (BNST) has distinct regions that closely resemble those of the central and medial amygdalar nuclei. The reason for this is that in early development the centromedial amygdala and BNST were a single structure. However, as the internal capsule develops it separates this single structure into rostral and caudal parts, each containing the same nuclear components (Johnston, 1923; Olucha-Bordonau, Fortes-Marco, Otero-García, Lanuza, & Martinez-García, 2015). The caudal part becomes the central and medial amygdalar nuclei, while the rostral part becomes the BNST. The neurons/nuclei in lateral portions of the BNST (BNSTL) closely resemble those in the CEA whereas those in medial portions of the BNST (BNSTM) closely resemble those in the MEA. Collectively, the central and medial amygdalar nuclei and the BNST have been termed the “extended amygdala” (Alheid & Heimer, 1988). The central nucleus and BNSTL are termed the “central extended amygdala,” and the medial nucleus and BNSTM are termed the “medial extended amygdala.” The “sublenticular extended amygdala,” which is located ventral to the lenticular nucleus, is interposed between the CEA/MEA caudally and the BNST rostrally. This review will use the terminology of the rat atlas by Paxinos and Watson (1997) to describe the nuclei of the BNST.
LOCALIZATION OF OPIOID PEPTIDES IN THE AMYGDALA
Enkephalin: Immunohistochemical studies of ENK localization
The first report of enkephalin-like immunoreactivity (ENKir) in the amygdala (Elde, Hokfelt, Johansson, & Terenius, 1976) was published 1 year after the discovery of the enkephalins (Hughes et al., 1975; Terenius & Wahlstrom, 1975). This immunohistochemical study utilizing antibodies to leu-enkephalin (leu-ENK), reported a high density of ENK+ (ENK immunopositive) axons in the CEA and the BNSTL (i.e., central extended amygdala), as well as scattered axons of variable density in other amygdalar nuclei (Elde et al., 1976). No ENK+ somata were seen in the amygdala or in any other brain region. These results are consistent with radioimmunoassays which demonstrated high levels of met-enkephalin (met-ENK) in the rat CEA and BNST, and lower levels in other amygdalar nuclei (Gros et al., 1978). In general, most leu-ENK and met-ENK antibodies exhibit some limited cross-reactivity for the other ENK, and staining is very similar with both types of antibodies in the amygdala (Poulin, Chevalier, Laforest, & Drolet, 2006).
Many subsequent studies of ENKir (immunoreactivity) in the rat amygdala utilized intracerebroventricular (i.c.v.) injections of colchicine to increase levels of ENK in neuronal somata, thus permitting their identification. Colchicine blocks axonal transport of ENK from somata by disrupting microtubules. A high density of ENK+ somata and axons was seen in the CEA (especially in CL and CLC), MEA, BNST, and INs, and lower densities of ENK+ somata and axons were seen in all other amygdalar nuclei (Finley, Maderdrut, & Petrusz, 1981; Gray, Cassell, & Kiss, 1984; Khachaturian, Lewis, Hollt, & Watson, 1983; Loughlin, Leslie, & Fallon, 1995; Merchenthaler, Maderdrut, Altschuler, & Petrusz, 1986; Wamsley, Young, & Kuhar, 1980). In immunohistochemical studies in monkey, without colchicine injections, many axons were seen in CEA and LA, and a few axons were seen in the basal and cortical nuclei (Haber & Elde, 1982).
Recent light microscopic studies using a sensitive nickel-intensified diaminobenzidine (DAB) immunoperoxidase technique stained many ENK+ somata and axons in the rat amygdala without the need for colchicine injections (Zhang & McDonald, 2016). Numerous ENK+ somata and axons were observed in the CEA, especially in CL and CLC (Fig. 2A), while many ENK+ axons, but just a few somata were seen in all subdivisions of the MEA. The INs had a high density of ENK+ axons and somata. ENK axonal staining in the nuclei of the basolateral nuclear complex was relatively light (Figs. 2 and 3). All portions of the basolateral nuclear complex, as well as the cortical nuclei and AHA, contained small NPNs that exhibited moderate to strong ENKir. Their small somata were usually ovoid and 8–12μm in diameter (Fig. 3A). These ENK+ NPN neurons gave rise to 3–4 thin aspiny primary dendrites that branched sparingly. Their distinctive morphology (i.e., small perikaryon and very thin dendrites) closely resembles that of small NPNs in the BL and LA that co-express vasoactive intestinal peptide (VIP), the calcium-binding protein calretinin (CR), and cholecystokinin (CCK) (Mascagni & McDonald, 2003). In fact, preliminary studies performed in our laboratory have demonstrated that ENK+ neurons constitute a subpopulation of VIP+ and CR+ interneurons. In addition to the labeling of NPNs, there was light ENKir in numerous larger neurons with pyramidal or piriform somata that were obviously PNs. The somata of PNs inventral portions of the BLa exhibited stronger ENKir than those in other portions of the basolateral nuclear complex.
FIG. 2.

Nissl-stained coronal sections through the amygdala of the rat stained for ENK (A) or MOR (B). Representative INs are indicated by asterisks in B. Scale bar=250μm.
FIG. 3.

(A) High-power photomicrograph of ENKir in the BLa in a section stained with nickel-intensified DAB (diaminobenzidine). This field contains two ENK+ nonpyramidal neurons that are in the plane of focus, and numerous small ENK+ puncta presumed to be axon terminals; an axon with large varicosities is indicated with an arrow. (B) High-power photomicrograph of ENKir in the lateral nucleus in a section stained with nickel-intensified DAB. This field contains numerous ENK+ puncta that average about 1μm in diameter that appear to be axon terminals. Scale bar=25μm. Adapted from Zhang, J., & McDonald, A. J. (2016). Light and electron microscopic analysis of enkephalin-like immunoreactivity in the basolateral amygdala, including evidence for convergence of enkephalin-containing axon terminals and norepinephrine transporter-containing axon terminals onto common targets. Brain Research, 1636, 62–73. doi:10.1016/j.brainres.2016.01.045, with permission.
In ultrastructural studies of the BLa, ENKir was seen in a variety of neuronal profiles including somata, dendrites, spines, axons, and axon terminals (Zhang & McDonald, 2016). ENKir in most large somata was sparse and confined to a few immunoparticles in the Golgi complex. These large somata appeared to represent PNs, and the localization of ENKir in the Golgi complex suggests that ENK is transported to the axons and dendrites of BLa PNs. Preliminary studies in colchicine-injected animals have demonstrated that ENKir accumulates in the axon initial segments of these PNs (personal observations of AJM), further indicating that ENK is transported to the axon terminals of these neurons. Synaptic vesicles in most ENK+ terminals were small, clear, and round or oval. Larger dense core vesicles, which were seen in a small number of ENK+ terminals, were typically located distant to the active zone of synapses. Sixty-eight percent of ENK+ terminals were observed forming synapses. ENK+ terminals mainly formed asymmetrical (excitatory) synapses (85%) and their most frequent targets were ENK+ (72%) and ENK-negative spines (28%) (Fig. 4). In addition to BLa PNs, it is also possible that some of these excitatory glutamatergic terminals could belong to cortical or thalamic afferents to the BLa (Brinley-Reed, Mascagni, & McDonald, 1995; Carlsen & Heimer, 1988; Farb & LeDoux, 1999; LeDoux & Farb, 1991; LeDoux, Farb, & Milner, 1991; Smith & Pare, 1994; Smith, Pare, & Pare, 2000). Symmetrical (inhibitory or neuromodulatory) synapses formed by ENK+ terminals constituted 15% of all synapses and their most frequent targets were thin ENK+ dendritic shafts. These terminals most likely represent axons from GABAergic ENK+ BLa interneurons and GABAergic ENK+ neurons in the INs (Asede, Bosch, Luthi, Ferraguti, & Ehrlich, 2015; Marowsky, Yanagawa, Obata, & Vogt, 2005). Interestingly, the sole targets of ENK+/VIP+ and ENK+/CR+ interneurons in the hippocampus are other interneurons, including other ENK+ interneurons (Blasco-Ibanez, Martinez-Guijarro, & Freund, 1998), and one of the main actions of ENK in the hippocampus is disinhibition of principal neurons via modulation of the somatodendritic and axonal compartments of GABAergic interneurons (Drake, Chavkin, & Milner, 2007; Zieglgansberger, French, Siggins, & Bloom, 1979). It is likely that a similar mechanism exists in the BLa.
FIG. 4.
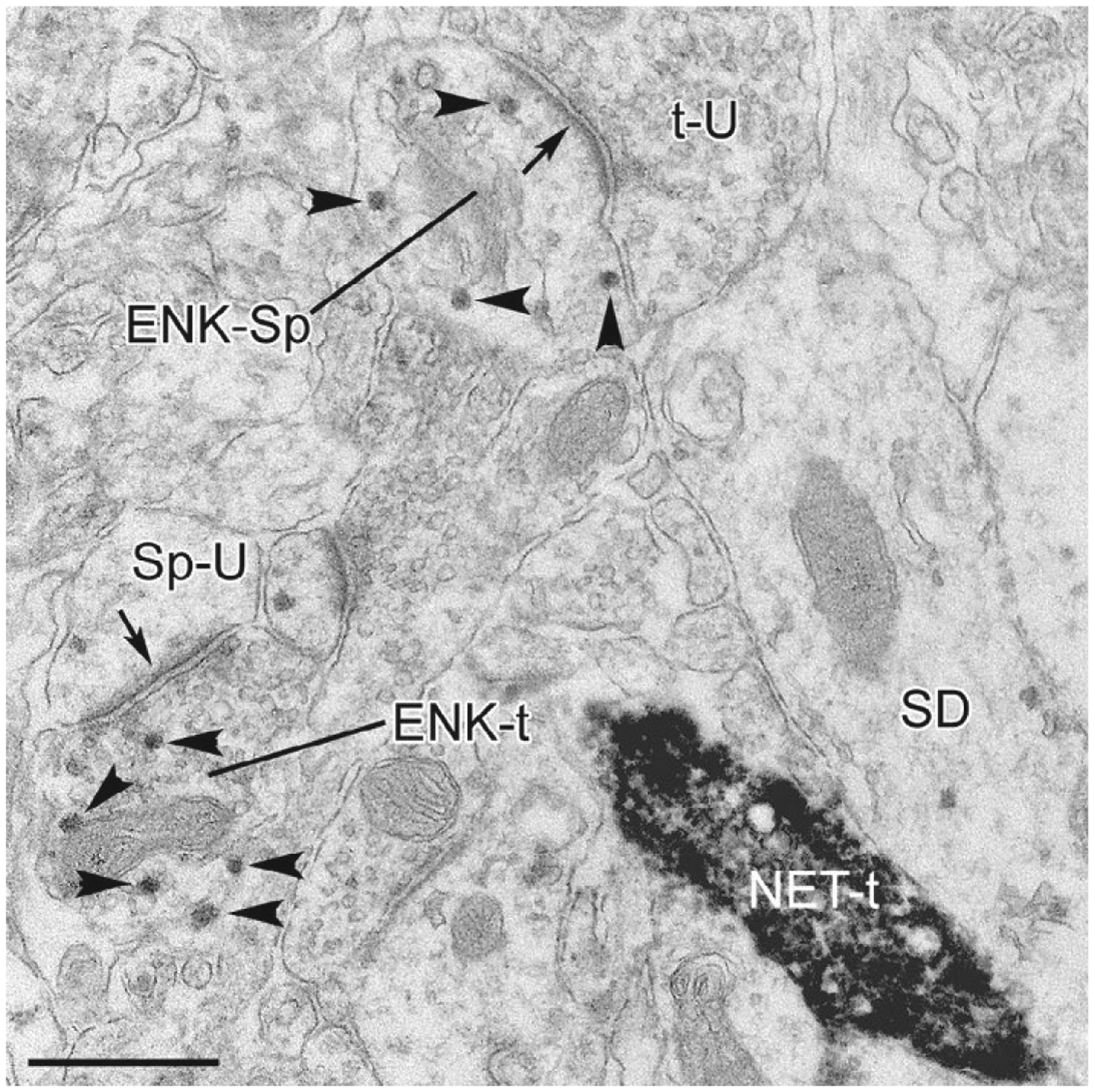
Electron micrograph of ENKir (particulate vector-VIP reaction product) and NET-ir (dense, diffuse DAB reaction product) in the BLa. Small arrowheads indicate representative vector-VIP particles in ENK+ structures. An ENK+ axon terminal (ENK-t) forms an asymmetrical synapse (arrow) with an unlabeled spine (Sp-U) in the lower left corner. This field also contains an unlabeled axon terminal (t-U) that forms an asymmetrical synapse (arrow) with an ENK+ spine (ENK-Sp). A noradrenergic axon terminal immunoreactive for the norepinephrine transporter protein (t-NET) forms an apposition with a small-caliber dendrite (SD) that is very lightly labeled for ENK. Although convergence of enkephalinergic and noradrenergic axon terminals onto dendrites is common in the locus coeruleus (Van Bockstaele, Chan, & Biswas, 1996), there was limited convergence in the BLa. Scale bar=0.5 μm. Adapted from Zhang, J., & McDonald, A. J. (2016). Light and electron microscopic analysis of enkephalin-like immunoreactivity in the basolateral amygdala, including evidence for convergence of enkephalin-containing axon terminals and norepinephrine transporter-containing axon terminals onto common targets. Brain Research, 1636, 62–73. doi:10.1016/j.brainres.2016.01.045, with permission.
ENK+ axons and axon terminals in the INs are apposed to axon terminals forming excitatory synapses and to dendritic spines, where they can, respectively, inhibit presynaptic glutamate release from BLa terminals via DORs and activate postsynaptic potassium currents via MORs (Winters et al., 2017).
Enkephalin: In situ hybridization studies of preproenkephalin (ppENK) and pENK mRNA localization
Studies of ppENK mRNA localization in the rat have demonstrated extensive expression in the amygdala (Harlan, Shivers, Romano, Howells, & Pfaff, 1987; Loughlin et al., 1995; Poulin, Arbour, Laforest, & Drolet, 2009). There are many ppENK cells in the BNST, CEA, MEA, and Coa, as well as moderate numbers of ppENK cells in the BLa, rostral LA, Copl, and Copm. Subsequent studies used dual-labeling in situ hybridization of ppENK mRNA with VGLUT1 (vesicular glutamate transporter 1), VGLUT2 (vesicular glutamate transporter 2), or GAD65 (glutamic acid decarboxylase 65) mRNAs to determine if ppENK neurons in the amygdala were glutamatergic (i.e., containing VGLUT mRNAs) or GABAergic (i.e., containing GAD65 mRNA) (Poulin et al., 2009; Poulin, Castonguay-Lebel, Laforest, & Drolet, 2008). Many ppENK/VGLUT1 neurons were seen in the ventral and ventromedial portions of BLa, consistent with the findings of Zhang and McDonald (2016) that this region contained lightly stained ENK+ PNs. The BM and cortical nuclei had a mixture of ppENK/VGLUT2 and ppENK/GAD neurons. The latter probably correspond to the small ENK+ interneurons seen by Zhang and McDonald (2016). The rat CEA and INs had a high concentration of ppENK/GAD neurons, which agrees with previous findings that these nuclei contain mostly GABAergic neurons. Very high levels of pENK mRNA were also seen in the CEA and BNST of humans (Hurd, 1996). The MEA, like the adjacent cortical nuclei and BM, contained both ppENK/GAD neurons and ppENK/VGLUT2 neurons (Poulin et al., 2008). Although Zhang and McDonald (2016) observed small ENK+ interneurons in all portions of the basolateral nuclear complex, very few neurons co-expressing ppENK and GAD65 mRNAs were observed in the basolateral nucleus by Poulin et al. (2008). The ppENK/GAD neurons in the LA seen by these investigators probably correspond to the small ENK+ interneurons seen by Zhang and McDonald (2016). Many ppENK neurons were observed in the BNST, especially in the BNSTL and the posterior BNSTM (Poulin et al., 2009). In situ hybridization studies of the BNST have shown that most ENK neurons express GAD mRNA and are GABAergic, but some in the posterior BNSTM express VGLUT2 and are glutamatergic (Poulin et al., 2009).
In the CEA of the rat, GABAergic ENK+ neurons are distinct from GABAergic corticotropin-releasing factor (CRF) neurons, but show some overlap with GABAergic neurotensin-positive (NT+) neurons (Day, Curran, Watson, & Akil, 1999; Veinante, Stoeckel, & Freund-Mercier, 1997). There is significant colocalization of CRF and NT in the rat lateral CEA and BNSTL (Shimada et al., 1989), so there appear to be two classes of NT+ neurons, some that express ENK and some that express CRF. In the CEA, the majority of ENK+ neurons also express glucocorticoid receptor immunoreactivity, which may explain their activation during stress (Honkaniemi et al., 1992).
Dynorphin: Immunohistochemical and in situ hybridization studies
Immunohistochemical and in situ hybridization studies have shown that the distribution of DYN-positive (DYN+) somata and axons closely resembles that of ENK+ somata and axons in the rat amygdala (Fallon & Leslie, 1986; Khachaturian et al., 1982; Loughlin et al., 1995; Poulin et al., 2009). Thus, many DYN+ somata and axons are found in the CEA, BNSTL, and INs. A low density of scattered DYN+ axons is seen in most other amygdalar nuclei and in the stria terminalis. Immunohistochemical studies have also shown that there is little colocalization of ENK and pDYN in the CEA, and about one-third of pDYN+ neurons in the CEL, but no pDYN+ neurons in the BNSTL, express CRF (Marchant, Densmore, & Osborne, 2007). Likewise, about one-third of CEA neurons expressing DYN mRNA also express CRF mRNA (Reyes, Drolet, & Van Bockstaele, 2008). Some CRF+ neurons in the CEA project to the locus coeruleus (LC) and EM studies have demonstrated that 35% of DYN+ axon terminals in the LC also express CRF, but very few LC DYN+ terminals express ENK (Reyes et al., 2008). The finding of a lack of colocalization of ENK and DYN in amygdalar somata and LC axon terminals in the rat is consistent with the observation that ENK and DYN precursors generally do not colocalize in neurons (Khachaturian et al., 1982, 1985). In the human amygdala, the overall levels of pDYN mRNA are significantly higher than pENK mRNA, but there is very dense pENK mRNA in the accessory basal nucleus (Hurd, 1996).
β-Endorphin
There are no somata expressing END in the amygdala. Axons arising from END+ neurons in the arcuate hypothalamic nucleus provide a sparse to moderate innervation of the BNST, MEA, CM, INs, LA, and BM (Finley, Lindstrom, & Petrusz, 1981; Gray et al., 1984; Loughlin et al., 1995).
LOCALIZATION OF OPIOID RECEPTORS IN THE AMYGDALA
Autoradiographic receptor binding studies
The first autoradiographic receptor binding study to provide detailed information about the localization of opiate receptors in discrete amygdalar nuclei in the rat used 3H-diprenorphine, a partial agonist with approximately equal affinities for MOR, DOR, and KOR (Atweh & Kuhar, 1977). The amygdala has especially high levels of binding compared with other telencephalic brain regions. Moderate to high levels of binding are found in all amygdalar nuclei (including the BNST) with the exception of the lateral nucleus.
Subsequent autoradiographic receptor binding studies utilized radioactive synthetic opioid peptides with high affinities for MORs (DAMGO [d-Ala2, NMe-Phe4, Gly-ol5]-enkephalin, dihydromorphine) or DORs (DADLE [d-Ala2, d-Leu5-enkephalin]) to localize these specific opioid receptor subtypes in the amygdala and other brain regions. The amygdalar nuclei had very high levels of both receptor subtypes compared with other brain regions in both mice (Moskowitz & Goodman, 1984) and rat (Loughlin et al., 1995; McLean, Rothman, & Herkenham, 1986). In the rat, all amygdalar nuclei had moderate to high levels of MOR, with the exception of the CEA and BNST; binding was especially dense in the INs and in the BL, LA, and Copm (Loughlin et al., 1995; McLean et al., 1986; Paden, Krall, & Lynch, 1987). There were moderate to high levels of DOR binding in all amygdalar nuclei of the rat, but the BLa had especially high DOR levels (Loughlin et al., 1995; McLean et al., 1986). Similar results were obtained in the mouse, but the level of MORs in the CEA was higher, perhaps due to the use of a different synthetic opioid peptide ligand in these studies (dihydromorphine versus DAMGO) (Moskowitz & Goodman, 1984). Assessment of mRNA expression in the latter study revealed higher ratios of DOR/MOR in the central, basolateral, and cortical nuclei.
Results similar to those of McLean et al. (1986) in the rat were obtained by Mansour, Khachaturian, Lewis, Akil, and Watson (1987) using different tritiated ligands to MORs and DORs. They also studied the localization of KORs using 3H-(−)bremazocine; KORs were found in all amygdalar nuclei, but the highest levels were in the basolateral and lateral nuclei (Mansour et al., 1987). In studies using 3H-ethylketocyclazozine as a ligand to identify KORs, extremely high levels were observed in the medial extended amygdala including the MEA and its “homolog,” the BNSTM (Lynch, Watt, Krall, & Paden, 1985; Paden et al., 1987). Autoradiograms by Loughlin et al. (1995) have shown that KOR binding is found in high concentrations in the rostral half of the amygdala. The INs had the highest concentrations, but high concentrations were also seen in the MEA, BLa, and LA.
There have been fewer autoradiographic receptor binding studies in primates. A study of 3H-DAMGO binding in the monkey demonstrated that the amygdala had the highest concentration of MORs in the forebrain (Daunais et al., 2001). Binding was found in all nuclei, including the BNST, and the highest density of MOR binding was in the basal (BLa and BLp of the rat), accessory basal (BM of the rat), and medial nuclei (Daunais et al., 2001). Similar results for MOR binding in the monkey amygdala were obtained by Ragen, Freeman, Laredo, Mendoza, and Bales (2015), while KOR-binding levels in the monkey amygdala were much less than MOR binding, and were more homogeneous among amygdalar nuclei (Ragen et al., 2015). An autoradiographic receptor binding study of opioid receptors in humans did not provide densities in individual amygdalar nuclei but suggested that, collectively, KOR binding was denser than MOR binding, which was in turn denser than DOR binding. Another study of the human amygdala demonstrated that DOR binding in the amygdala as a whole was of intermediate density compared with other forebrain regions (Blackburn, Cross, Hille, & Slater, 1988).
MOR: Immunohistochemical, in situ hybridization, and knock-in studies
The results of several light microscopic immunohistochemical studies of MORs in the rat amygdala were fairly similar (Ding, Kaneko, Nomura, & Mizuno, 1996; Mansour, Fox, Burke, Akil, & Watson, 1995; Poulin et al., 2006; Wilson, Mascagni, & McDonald, 2002; Zhang, Muller, & McDonald, 2015). MORir was mainly seen in the neuropil in these investigations, but in studies by one group, a small number of lightly stained somata was seen in the basolateral and cortical nuclei (Wilson et al., 2002; Zhang et al., 2015; Zhang & McDonald, 2016). Extremely dense neuropilar staining was seen in the INs, and dense staining was seen in Mav and Copm (Fig. 2B). Moderate neuropilar staining was observed in the CEA, BNST, Mad, BL, BMa, and Coa while very light neuropilar staining was seen in most of the remaining nuclei. Similar results were seen in MOR knock-in mice expressing MOR fused to the red fluorescent mCherry protein (Erbs et al., 2015).
In situ hybridization studies suggest that many neurons in the amygdala express MOR mRNAs (Poulin et al., 2006). The density of these neurons is very high in regions that have high levels of MOR protein (INs, Mad, and Copm), and moderate in most other nuclei with the exception of the anterior pole of the BLa (Mansour, Fox, Burke, et al., 1994; Mansour, Fox, Thompson, Akil, & Watson, 1994; Poulin et al., 2006). It is of interest that the anterior pole of the BLa has very high levels of DOR (see below). There are somata expressing MOR mRNA in all portions of the BNST, but higher concentrations are seen in posterior portions of the BNSTM (Poulin et al., 2009).
In an electron microscopic (EM) study of MORs in the BLa, light to moderate MORir was seen in various structures including somata, dendritic shafts, spines, and axon terminals (Zhang et al., 2015). PN somata only had MORir in the Golgi complex, suggesting that these receptors were in the process of being transported to axons and/or dendrites. The most frequently labeled processes were dendritic structures, including small-caliber dendrites and spines, which constituted 50% and 10% of all MOR+ structures, respectively (Fig. 5). Some dendrites could be identified as belonging to PNs or interneurons based on their morphology. These data are consistent with electrophysiological studies which have shown that the selective MOR agonist DAMGO activates a voltage-dependent potassium current in PN dendrites (Faber & Sah, 2004) and activation of MORs hyperpolarizes some interneurons in the BLA (Sugita & North, 1993; Sugita, Tanaka, & North, 1993). Twenty percent of MOR+ structures in the BLa were axon terminals, most of which (80%) formed asymmetrical (excitatory) synapses; their main postsynaptic targets were spines, the great majority of which were MOR-negative (Zhang et al., 2015). These data agree with studies that found MOR-mediated modulation of glutamate release in the basolateral amygdala (Yang et al., 2014). The main targets of MOR+ terminals forming symmetrical (inhibitory or neuromodulatory) synapses were MOR+ small-caliber dendrites. It is likely that some of the MOR+ terminals forming symmetrical synapses are noradrenergic. Noradrenergic axons form symmetrical synapses with small-caliber dendrites in the BLa (Asan, 1998; Zhang, Muller, & McDonald, 2013), and the release of norepinephrine in the BLa during stress is inhibited by ENK and END, an effect blocked by naloxone (Quirarte, Galvez, Roozendaal, & McGaugh, 1998; Tanaka, Yoshida, Emoto, & Ishii, 2000).
FIG. 5.

Electron micrographs of MOR+ spines. Arrowheads indicate examples of particulate MORir. (A) A MOR+ spine (M-Sp) forms an asymmetrical synaptic contact (arrow) with an unlabeled terminal (U-t). (B) A MOR+ spine receives an asymmetrical synaptic contact from an unlabeled terminal (arrow). Three unlabeled spines (asterisks) also receive asymmetrical synaptic contacts from unlabeled terminals. Scale bars=0.5μm. From Zhang, J., & McDonald, A. J. (2016). Light and electron microscopic analysis of enkephalin-like immunoreactivity in the basolateral amygdala, including evidence for convergence of enkephalin-containing axon terminals and norepinephrine transporter-containing axon terminals onto common targets. Brain Research, 1636, 62–73. doi:10.1016/j.brainres.2016.01.045, with permission.
EM studies of the CEA have shown that MORir is expressed in somata, dendrites, and axons (Glass, Vanyo, Quimson, & Pickel, 2009). Most of the MOR+ axon terminals formed asymmetrical (excitatory) synapses. The colocalization of MOR with NMDA (NMDAR) and AMPA (AMPAR) receptors was studied using EM. Extensive colocalization of MOR with NMDARs and GLUA2-AMPARs, but not GLUA1-AMPARs, was observed in CEA somata, in dendrites and spines that were postsynaptic to axon terminals forming asymmetrical (excitatory) synapses, and, to a lesser extent, in axon terminals forming excitatory synapses (Beckerman & Glass, 2011; Glass et al., 2009; Glass, Kruzich, Colago, Kreek, & Pickel, 2005). These results provide anatomical support for MOR activation to modulate presynaptic and postsynaptic glutamate signaling in the CEA, in agreement with electrophysiological findings (Chieng, Christie, & Osborne, 2006; Zhu & Pan, 2004, 2005). Some of the CEA neurons exhibiting NMDAR/MOR colocalization in their somata and dendrites have been shown to project to the BNSTL, but their axons in the BNSTL, which mainly form symmetrical synapses, rarely exhibit NMDAR/MOR colocalization (Beckerman & Glass, 2012). It is also of interest that some neurons in the BNSTL exhibit NMDAR/MOR colocalization, which is not surprising since both the CEA and BNSTL are part of the central extended amygdala. Moreover, this colocalization of MOR with glutamate receptors may serve as the interface for opiate regulation of glutamatergic processes critical in learning, opiate addiction, and stress (Glass, 2010; Peters & De Vries, 2012; Scavone, Asan, & Van Bockstaele, 2011; Wilson, Grillo, Fadel, & Reagan, 2015).
EM studies have shown that the somatodendritic compartments of neurons in CL and the anterolateral BNSTL (i.e., “homologous” portions of the central extended amygdala) exhibit colocalization of MORs and CRF type 1 receptors (CRF-R1s) ( Jaferi & Pickel, 2009). Activation of MORs and CRFR1s have opposing effects on neurons, with the former being inhibitory and the latter excitatory, which may lead to opposing roles of ENK and CRF in the amygdala in mediating anxiety-related responses, and anxiogenic responses during ethanol or opiate withdrawal (see below).
DOR: Immunohistochemical, in situ hybridization, and knock-in studies
An immunohistochemical study demonstrated that DORir in the rat amygdala was located only in axons (Wilson et al., 2002). Immunostaining for DOR was observed in most amygdalar nuclei but was particularly dense in the CEA and Mpd. In all portions of the MEA, most DOR+ axon terminals were scattered throughout the neuropil, but some formed peri-cellular baskets that surrounded the cell bodies and proximal dendrites of unstained neurons. Moreover, in the Mpd, male rats had higher densities of DORs than females, but not in other amygdalar nuclei (Wilson et al., 2002), although the functional significance of this sex difference is not clear. Since the Mpd contains a large number of neurons containing androgen receptor mRNA or estrogen receptor mRNA (Simerly, Chang, Muramatsu, & Swanson, 1990), it will be of interest to determine if this sex difference is due to the organizational or the acute activational influences of gonadal hormones. A recent EM immunohistochemical study reported that there is extensive colocalization of DOR and CRF in both somata and axon terminals in the CEA and BL (Reyes, Kravets, Connelly, Unterwald, & Van Bockstaele, 2017). Moreover, the DOR/CRF axon terminals were in close proximity to norepinephrine (NE) terminals, demonstrating the convergence of three important systems modulating stress and anxiety in the amygdala: NE, CRF, and DOR.
In situ hybridization studies have shown that most nuclei of the amygdala contain numerous neurons expressing DOR mRNA (Mansour, Fox, Burke, et al., 1994; Poulin et al., 2006). As in autoradiographic receptor binding studies, the highest concentration was seen in the rostral pole of the BLa, with high concentrations also observed in Mpv and BMp. Most neurons in the BNST that expressed DOR mRNA were located in the posterior portions of the BNSTM (Poulin et al., 2009). Similar results were obtained in DOR knock-in mice expressing DOR fused to the green fluorescent protein eGFP (Erbs et al., 2015). In DOR-MOR double knock-in mice co-expression of DORs with MORs was virtually nonexistent in the amygdala (Erbs et al., 2015).
KOR: Immunohistochemical and in situ hybridization studies
An immunohistochemical study of KORs in the rat CNS demonstrated that KORir is found in fibers in the CEA (especially in CM), MEA (especially in Mpd), and BNST (Mansour, Burke, Pavlic, Akil, & Watson, 1996) and a few KOR+ somata were observed in the BNST in colchicine-injected rats. An in situ hybridization study found labeled somata in most amygdalar nuclei, with high levels of KOR mRNA in the CL, BL, MEA (especially its posterior portion), and the AHA (Mansour, Fox, Meng, Akil, & Watson, 1994). In the BNST most somata with KOR mRNA were in the posterior BNSTL and posterior BNSTM (with the exception of its most medial part) (Poulin et al., 2009).
OPIOID PEPTIDERGIC CIRCUITS IN THE AMYGDALA
The sources of enkephalinergic afferents to the central and medial amygdalar nuclei in the rat were identified by combining in situ hybridization for ppENK with retrograde tract tracing (Poulin et al., 2006). Injections of retrograde tracer into the CEA identified retrogradely labeled neurons in over two dozen structures, including many with ENK neurons, but very few regions contained double-labeled neurons. These studies demonstrated that the CEA receives a significant ENK input from the ventromedial hypothalamic (VMN) and parabrachial (PBN) nuclei, BMa, COa, MEA, INs, BNSTL, and BNSTM, and more sparse inputs from BL. The MEA receives a significant ENK input from the VMN, PBN, BMa, COa, and BNSTM. It is not known if the enkephalinergic inputs from the basolateral and cortical amygdalar nuclei are provided by PNs or the small ENK+ NPNs. However, since the latter are thought to be interneurons, it may be glutamatergic ppENK/VGLUT PNs that innervate the CM. If so, it is likely that these PN axons may express MORs (see above). In fact, both DAMGO and enkephalin, but not DOR or KOR agonists, inhibit glutamate release in the CEA upon BLa stimulation (Zhu & Pan, 2005).
The CEA and BNSTL project to several important autonomic and monoaminergic brainstem nuclei. Gray and coworkers studied the CEA and BNSTL cell types providing outputs to various brainstem nuclei by combining immunohistochemistry for ENK and other peptides with retrograde tract tracing. It was found that there were no enkephalinergic projections to the PBN, dorsal vagal nucleus, nucleus solitarius, or periaqueductal gray (PAG), but there were projections from CRF, somatostatin, and neurotensin neurons in the CEA and BNSTL (Gray & Magnuson, 1987; Moga & Gray, 1985; Moga, Saper, & Gray, 1989). However, using the same technique it was found that ENK neurons in the CEA project to the BNSTL (Rao, Yamano, Shiosaka, Shinohara, & Tohyama, 1987). These data suggest that ENK+ neurons in the CEA have projections to other portions of the central extended amygdala, but not to extrinsic regions.
By combining immunohistochemistry for DYN with retrograde tract tracing, it was found that some DYN+ neurons in the lateral hypothalamus, perifornical area, peripeduncular nucleus, and lateral substantia nigra project to the CEA (Fallon, Leslie, & Cone, 1985; Zardetto-Smith, Moga, Magnuson, & Gray, 1988), and some DYN+ neurons in the CEA project to the LC, ventral tegmental area, and substantia nigra (Fallon et al., 1985; Reyes et al., 2008). By combining immunohistochemistry for END with retrograde tract tracing, it was found that END+ neurons in the arcuate hypothalamic nucleus have projections to the CEA (Gray et al., 1984).
ELECTROPHYSIOLOGICAL EFFECTS OF OPIOIDS IN THE AMYGDALA
MOR or DOR activation in the lateral and basolateral amygdala (BLA)
As summarized in Table 1, early studies using in vivo electrophysiological approaches in anesthetized preparations demonstrated that the spontaneous firing rate of cat BLA neurons was increased by systemic administration of morphine, although these effects were only partially blocked by naloxone (Chou & Wang, 1977). Similarly, morphine enhanced electrographic activity in the anterior amygdala region evoked by stimulation of the olfactory bulb (Klemm & Mallari, 1979). Local iontophoretic application of morphine or DADLE, however, did not alter the glutamate-driven single-unit firing in the BLA of rats (Freedman & Aghajanian, 1985) and the MOR agonist DAMGO failed to induce postsynaptic responses of BLA PNs in brain slices (Blaesse et al., 2015). Recordings from BLA PNs, however, showed morphine and DAMGO increased spike frequency adaptation via upregulation of a voltage-dependent potassium current (Kv1.2), and these effects were specifically seen in the apical dendrites (Faber & Sah, 2004), a finding supported by the anatomical location of MOR in the BLA (Zhang et al., 2015). This MOR-induced upregulation of potassium currents would decrease the number of spikes induced by depolarizing stimuli, thereby suggesting the MOR activation would inhibit output from lateral amygdala PNs, as indicated by morphine-induced reductions in spike trains following glutamate application (Faber & Sah, 2004). Interestingly, the effects of MOR agonists opposed those induced by norepinephrine, acetylcholine, glutamate, and serotonin on spike frequency adaptation, and MOR agonists were able to block the norepinephrine-induced decreases in spike frequency adaptation. In LA, met-ENK and DAMGO also induced hyperpolarization of one type of NPN, and these effects were antagonized by the MOR antagonist CTOP [d-Pen-Cys-Tyr-d-Trp-Orn-Thr-PenThr-NH2] (Sugita et al., 1993; Sugita & North, 1993).
TABLE 1.
Electrophysiological effects of MOR, DOR, or KOR receptor activation in amygdala.
| Subregion | OR | Preparation used for recording | Summary of electrophysiological findings | Primary effect of MOR, DOR, or KOR receptor activation | Reference |
|---|---|---|---|---|---|
| Anterior amygdala | MOR | In vivo anesthetized EEG | Systemic administration of morphine enhanced evoked field potentials in anterior amygdala, and altered spontaneous firing rates of neurons in this area | Enhanced evoked field potentials | Klemm and Mallari (1979) |
| Lateral amygdala | |||||
| LA interneurons | DOR | Slices (rat) | DPDPE had no postsynaptic effects, but inhibitedGABA synaptic potentials by decreasing presynaptic GABA release. These effects were blocked by the DOR antagonist ICI174864 | Reduced presynaptic GABA release | Sugita and North (1993) |
| LA interneurons | KOR | Slices (rat) | U50488 had no postsynaptic effects and did not alter GABA synaptic potentials | No effect on inhibitory synaptic inputs | Sugita and North (1993) |
| LA interneurons | MOR | Slices (rat) | DAMGO and met-ENK hyperpolarized ~50% of cells, and inhibited GABA synaptic potentials. Latter effects were from decreased presynaptic release since there was no effect of MOR activation on effects of exogenously applied GABA. Effects were antagonized by the MOR antagonist CTOP. Met-ENK hyperpolarized only type 2 interneurons in LA | Postsynaptic inhibition and reduced presynaptic GABA release | Sugita and North (1993), Sugita et al. (1993) |
| LA PN | MOR | Slices (rat) | Morphine and DAMGO increased spike frequency adaptation in apical dendrites of LA pyramidal neurons, which decreased the number of spikes induced by depolarization and decreased PN neuron output. MOR effects were through Gi/o mechanisms to activate the arachidonic acid pathway and upregulate a voltage-dependent Kv 1.1/1.2 potassium current. Morphine reduced spike trains after glutamate pressure application suggesting MOR activation decreased glutamatergic activation in this region | Postsynaptic inhibition of spike frequency adaptation on apical dendrites | Faber and Sah (2004) |
| Basolateral amygdala | |||||
| BLA | KOR | Slices (mice) | U50,488 H (KOR agonist) decreased field potential amplitude in BLA, and the induction of LTP, induced by stimulation of the LA. These effects were blocked by norBNI, but the antagonist had no effect on its own | Inhibition of field potentials and LTP | Huge, Rammes, Beyer, Zieglgansberger, and Azad (2009) |
| BLA PN | KOR | Slices (rat) | KOR agonists U69593 and DYN-A increased spontaneous IPSC frequency in adolescent, but not adult, BLA PNs, and these effects were blocked by tetrodotoxin (TTX). U69593 did not affect spontaneous EPSC frequency | Inhibition of presynaptic GABA release | Przybysz, Werner, and Diaz (2017) |
| BLA | MOR | Slices (rat) | DAMGO decreased eIPSCs in 75% of BLA neurons that projected to CEA, as well as decreasing mIPSCs and increasing the paired pulse ratio of eIPSCs. Effects of MOR activation involved decreased presynaptic GABA release mediated through Kv 1.1/1.2 potassium channels, which was supported by immunohistochemical colocalization studies. DAMGO had minimal effects on EPSCs; >75% of the neurons did not respond to DAMGO with EPSCs | Decreased presynaptic GABA release on BLA neurons projecting to CEA | Finnegan, Chen, and Pan (2006) |
| BLA | MOR | In vivo anesthetized single unit | Systemic administration of morphine increased spontaneous firing rates of neurons in the BLA | Increased spontaneous firing rate | Chou and Wang (1977) |
| BLA (PN) | MOR | Slices (mice) | DAMGO failed to hyperpolarize BLA PN. | No postsynaptic effects | Blaesse et al. (2015) |
| Intercalated cell masses (IN) | |||||
| IN | MOR | Slices (mice; GAD67-eGFP mice) | DAMGO and endomorphin 1 induced hyperpolarization of IN neurons through an outward potassium current and increase in membrane input resistance. These effects were blocked by CTAP | Postsynaptic inhibition | Blaesse et al. (2015) |
| Central amygdala | |||||
| CEA (CM, CL) | DOR | Slices (rat) | No postsynaptic effects of DOR agonists in CEA | No postsynaptic effects | Zhu and Pan (2004) |
| CEA | DOR | Slices (rat) | DOR agonist deltorphin II induced outward potassium current in ~18% of low threshold bursting neurons of CM | Postsynaptic inhibition | Chieng et al. (2006) |
| CEA (CM) | DOR | Slices (WT and DOR KO mice) | DOR agonist DPDPE decreased mIPSC frequency in 60% of CM cells. Attenuating DOR activity with the antagonist naltrindole or using DOR KO mice enhanced the ethanol-induced increases in IPSCs evoked from CL stimulation. Results suggest a tonic DOR-mediated inhibition of GABA release | Inhibition of presynaptic GABA release | Kang-Park, Kieffer, Roberts, Siggins, and Moore (2007) |
| CEA | DOR | Slices (rat) | DOR agonist DPDPE had no effect on eEPSCs | No effect on excitatory synaptic inputs | Zhu and Pan (2005) |
| CEA (CM) | DOR | Slices (control or ethanol treated rats) | DOR agonist DPDPE had no effect on eEPSCs or eIPSCs in CMof control rats, but decreased evoked glutamate EPSCs and GABA IPSCs inCMof ethanol-treated rats (2-week CPP procedure). This induction of DOR activation effects was not seen with acute ethanol (required chronic ethanol exposure). Effects on EPSCs were through decreasing presynaptic glutamate release and results suggest an upregulation of DOR on terminals | Inhibition of presynaptic glutamate and GABA release, only in ethanol treated rats | Bie, Zhu, and Pan (2009a) |
| CEA (CM) | DOR | Slices (control or morphine-treated rats) | DOR agonist DPDPE had no effect on eEPSCs in CM of control rats, but decreased eEPSCs in CM of morphine-treated rats by decreasing presynaptic glutamate release. This was corroborated by an increase in DOR immunoreactivity in the synaptosomal fraction of morphine-treated rats compared to controls | Inhibition of presynaptic glutamate release, only in morphine-treated rats | Bie, Zhu, and Pan (2009b) |
| CEA | KOR | Slices (rat) | KOR agonist induced outward potassium current in 17% of CEA neurons which was blocked by norBNI. Only 13% of cells responded to both KOR and MOR agonists | Postsynaptic inhibition | Chieng et al. (2006) |
| CEA (CM, CL) | KOR | Slices (rat) | KOR agonist U69593 induced outward current only in ~50% of type B CEA neurons and no type A CEA neurons (this was ~6% of all CEA neurons) | Postsynaptic inhibition | Zhu and Pan (2004) |
| CEA (CM) | KOR | Slices (rat) | DYN[l-7] and U69595 both decreased evoked IPSCs in ~80 % of cells in CM, and this was blocked with norBNI and associated with a decrease in presynaptic GABA release. Both the KOR antagonist norBNI and ethanol also increased eIPSCs, but the combined effects were not synergistic suggesting there is an endogenous KOR tone and ethanol’s effects may involve antagonism at this site | Inhibition of presynaptic GABA release | Gilpin, Roberto, Koob, and Schweitzer (2014) |
| CEA (CM) | KOR | Slices (WT and KOR KO mice) | U69595 decreased eIPSCs in ~60 % of cells in CEM, and this was blocked with norBNI and associated with a decrease in presynaptic GABA release. NorBNI increased eIPSCs in ~50% of neurons in slices from WT mice, suggesting endogenous tone on these KOR. Enhanced effects of ethanol on eIPSCs were seen with norBNI and in KOR KO mice, suggesting ethanol’s effects involve KOR-mediated inhibition of GABA release | Inhibition of presynaptic GABA release | Kang-Park, Kieffer, Roberts, Siggins, and Moore (2013) |
| CEA | KOR | Slices (rat) | KOR agonist U69593 decreased spontaneous IPSC frequency in both adolescent and adults CEA neurons | Inhibition of presynaptic GABA release | Przybysz et al. (2017) |
| CEA | KOR | Slices (rat) | KOR agonist U69593 had no effect on evoked EPSCs | No effect on excitatory synaptic inputs | Zhu and Pan (2005) |
| CEA (CM) | KOR | Slices (female WT and KOR KO mice) | Diminishing KOR activation using the antagonist norBNI, or KOR KO mice, increased the effects of CRF on mIPSC frequency | Enhances CRF effects | Kang-Park, Kieffer, Roberts, Siggins, and Moore (2015) |
| CEA | MOR | Slices | ~60% of CEA neurons (all cell types and subregions) showed postsynaptic outward (potassium) currents with DAMGO. All of the neurons in CM (low-threshold bursting cells) that projected to PBN, thalamus, or BNST responded to DAMGO. There was no overlap with cells responding to the KOR agonist U69593, but cells in the CM that responded to the DOR agonist deltorphin II also responded to DAMGO | Postsynaptic inhibition | Chieng et al. (2006), Chieng and Christie (2009) |
| CEA (CM, CL) | MOR | Slices (rat) | Met-ENK and DAMGO inhibited ~ 80% of TYPE Al and 17% of A2 type CEA neurons through activating a GIRK channel, and effects were blocked by CTAP but not natlrindole. CEA B cells did not respond to MOR agonists, but some responded to a KOR agonist | Postsynaptic inhibition | Zhu and Pan (2004) |
| CEA (CM) | MOR | Slices (mice; GAD67-eGFP mice) | DAMGO decreased the eIPSCs from BLA stimulation and directly induced an outward current in ~40% of CM neurons. DAMGO also increased the failure rate of eIPSCs induced by uncaging glutamate in the IN, suggesting a decrease in probability of GABA release. CTAP enhanced plasticity seen in CM with theta burst stimulation | Postsynaptic inhibition and reduced presynaptic GABA release | Blaesse et al. (2015) |
| CEA | MOR | Slices (rat) | DAMGO decreased mIPSCs and eIPSCs in CEA neurons projecting to vlPAG, suggesting reduced presynaptic GABA release | Reduced presynaptic GABA release | Finnegan, Chen, and Pan (2005) |
| CEA (CM) | MOR | Slices (WT and MOR KO mice) | DAMGO decreased the frequency of mIPSCs, while infusion of the MOR antagonist naloxonazine increased mIPSCs suggesting tonic opioid tone. MOR KO mice also showed enhanced evoked IPSCs in CM compared to WT control mice | Reduced presynaptic GABA release | Kang-Park et al. (2009) |
| CEA | MOR | Slices (rat) | MOR activation (morphine, DAMGO) decreased evoked IPSCs in ~50% of neurons, but had no effect in ~30 % of cells. MOR agonists also decreased mIPSCs, while the MOR antagonist CTOP increased mIPSC frequency, suggesting tonic tone at MOR | Reduced presynaptic GABA release | Bajo, Roberto, Madamba, and Siggins (2011), Bajo, Madamba, Roberto, and Siggins (2014) |
| CEA | MOR | Slices (rat) | Met-ENK and DAMGO reduced evoked EPSCs following stimulation in the CEA or BLA, and these effects were blocked with CTAP. Met-ENK also increased paired pulse ratio, and decreased the frequency of mEPSCs suggesting a decrease in presynaptic glutamate release. MOR’s presynaptic effect involves 4-aminopyridine sensitive potassium channels | Reduced presynaptic glutamate release | Zhu and Pan (2005) |
| CEA, MEA | MOR, DOR | In vivo anesthetized, single unit | Morphine and DADL decreased glutamate-driven firing rate in >75% of CEA and MEA neurons (but not BLA neurons) | Inhibition of glutamate-induced firing rate | Freedman and Aghajanian (1985) |
| Bed nucleus of the stria terminalis (BNST) | |||||
| BNSTL | KOR | Slices (mice including ppDYN-IRES-Cre mice) | U69593 and DYN-A decreased both eEPSCs and mEPSCs in the dorsolateral BNST. This effect was prevented but not reversed by norBNI, and involved the p38 signaling pathway. KOR activation also decreased EPSCs evoked in BNST from optogenetic stimulation of BLA, but not PFC, inputs, as well as decreasing the fidelity of action potentials induced in the BNST with BLA stimulation. Viral deletion of amygdala KOR eliminated this inhibition with BLA stimulation, indicating KOR are expressed presynaptically on BLA neurons. Optogenetic stimulation of DYN+ neurons in the BNST led to a monosynaptic IPSC, but also decreased eEPSCs, suggesting DYN decreases local glutamate transmission in the BNST | Decreased presynaptic glutamate release from BLA inputs | Crowley et al. (2016) |
| BNSTL | KOR | Slices (mice, vGat-ires-Cre mice) | Dynorphin A and U69593 decreased the amplitude of the eIPSCs in the dorsolateral BNST (oval nucleus), including a decrease in the GABA IPSCs induced by optogenetic stimulation of the CEA inputs to the BNST. KOR activation also decreased frequency of spontaneous IPSCs and mIPSCs with TTX suggesting effects on presynaptic GABA release. These KOR effects were dependent on the extracellular signal-regulated kinase (ERK 1/2) signaling pathway | Inhibition of presynaptic GABA release from CEA inputs | Li et al. (2012) |
In BLA PNs that specifically projected to the CEA, DAMGO decreased evoked inhibitory postsynaptic currents (eIPSCs) in 75% of cells, as well as decreasing miniature IPSCs (mIPSCs). Additional studies indicated MOR activation decreased presynaptic GABA release via activating Kv1.1 and Kv1.2 potassium channels (Finnegan et al., 2006), suggesting MOR regulation of GABAergic inputs on PNs projecting to CEA. DAMGO had minimal effects on excitatory postsynaptic currents (EPSCs) in these BLA PNs that projected to the CEA (Finnegan et al., 2006), consistent with the inability of MOR agonists to alter glutamate-driven single-unit activity (Freedman & Aghajanian, 1985). Both MOR (DAMGO) and DOR (DPDPE; [d-Pen 2,5-enkephalin]) agonists also inhibited GABA synaptic potentials of NPNs by about 50% through decreased presynaptic GABA release (Sugita & North, 1993).
KOR activation in basolateral amygdala
Activation of KOR receptors using the agonist U50,488H reduced the amplitude of BLA field potentials evoked by stimulation of the LA, as well as the induction of long-term potentiation (LTP). Both effects were reversed with the KOR antagonist norBNI, but the antagonist did not influence field potential amplitude or LTP on its own (Huge et al., 2009). KOR agonists (U69593, DYN-A) also increased spontaneous IPSC frequency in adolescent, but not adult, BLA PNs through increases in GABA transmission, although KOR activation did not affect spontaneous EPSCs at either age. Remarkably, this study suggested that there was a developmental shift in KOR effects on BLA PN, without a difference in amygdalar KOR expression between adolescents (postnatal days 0–45) and adults (Przybysz et al., 2017).
MOR activation in the intercalated nuclei
The INs contain a dense concentration of MOR (see above) so it is not surprising that the MOR agonists (DAMGO, endomorphin1) induced hyperpolarization of IN neurons through an outward potassium current and an increase in membrane input resistance; these effects were blocked by CTAP (Blaesse et al., 2015).
MOR activation in central amygdala
In anesthetized rats, the local iontophoretic application of morphine or DADLE decreased glutamate-driven single-unit firing in the central and medial amygdala (Freedman & Aghajanian, 1985). Systemic morphine administration had similar effects on the firing rate in these regions. A DOR selective antagonist (ICI174864) antagonized morphine responses on some, but not all cells. Interestingly, cells inhibited by morphine were also inhibited by clonidine (the α2 adrenergic receptor agonist). As discussed below, after chronic morphine administration, both naloxone and the DOR antagonist increased the firing rate; these effects of antagonists were not seen in control animals (Freedman & Aghajanian, 1985).
The effects of MOR agonists in CEA slices have been demonstrated by several investigators, and these studies all suggest that the opioid effects in this region are dependent on the cell type (Bajo et al., 2011; Chieng et al., 2006; Chieng & Christie, 2009; Finnegan et al., 2005; Zhu & Pan, 2004, 2005). Both Zhu and Pan (2005) and Chieng et al. (2006) identified three cell types based on distinct electrophysiological properties, with the latter study demonstrating that late-firing neurons are present in the CLC and amygdalostriatal transition (AStr) area, while low-threshold bursting neurons comprised >60% of the cells in the CL and CM portions of the CEA (Chieng et al., 2006). Approximately 40%–60% of neurons of all subtypes in all CEA subregions showed a postsynaptic outward (potassium) current with DAMGO that was antagonized with CTAP (Blaesse et al., 2015; Chieng et al., 2006; Chieng & Christie, 2009). In contrast, all of the neurons in CM (low-threshold bursting cells) that projected to PBN, PAG, thalamus, or BNST responded to DAMGO (Chieng et al., 2006; Chieng & Christie, 2009). There was no overlap between cells responding to DAMGO and the KOR agonist U69593, but cells in the CM that responded to the DOR agonist, deltorphin II, also responded to DAMGO (Chieng et al., 2006). In an analogous fashion, Zhu and Pan (2004) found that DAMGO and met-ENK evoked a G-protein-coupled inwardly rectifying potassium channel (GIRK) currents in ~80% of type A1 and 17% of type A2 CEA neurons; these effects were blocked by CTAP but not naltrindole. No effects were seen with a DOR agonist, while ~50% of type B cells were inhibited by the KOR agonist U69593 (Zhu & Pan, 2004). Thus, these studies suggest that MOR and KOR agonists exert inhibitory postsynaptic effects on distinct neuronal populations and that DOR agonists have limited postsynaptic activity on most cells in the CEA, although their postsynaptic inhibitory effects are seen on the same neurons that respond to MOR.
As seen in Table 1, MOR activation also decreases both glutamatergic and GABAergic presynaptic inputs in the CEA, consistent with MOR colocalization with the presynaptic marker synaptophysin in the CEA (Finnegan et al., 2005). Both met-ENK and DAMGO reduced evoked EPSCs (eEPSCs) following stimulation in either the CEA or BLA and these effects were blocked with CTAP. Met-ENK also increased paired-pulse ratio, and decreased the frequency of mEPSCs indicating MOR activation decreased presynaptic glutamate release (Zhu & Pan, 2005). Additional studies indicate that MOR activation decreases presynaptic GABA release. MOR agonists (DAMGO, morphine) decreased the frequency of mIPSCs in CEA neurons (specifically CM), and decreased eIPSCs in ~50% of neurons, while perfusion with MOR antagonists (CTOP, naloxonazine) increased mIPSCs, suggesting tonic opioid tone (Bajo et al., 2014; Bajo et al., 2011; Kang-Park et al., 2009). MOR knockout mice also showed enhanced eIPSCs in CM compared with wild-type (WT) control mice (Kang-Park et al., 2009). Similarly, in CEA neurons that selectively project to the ventrolateral PAG (vlPAG), DAMGO decreased mIPSCs or eIPSCs in 47% and 69% of these neurons, respectively, while most neurons showed no effect of DAMGO on mEPSCs (69%) or eEPSCs (83%) (Finnegan et al., 2005). DAMGO also decreased IPSCs evoked from BLA stimulation and directly induced an outward current in ~40% of CM neurons, while the MOR antagonist CTAP enhanced plasticity in CM induced by theta burst stimulation of these inputs (Blaesse et al., 2015). Using a method to uncage glutamate and specifically activate IN inputs to CEA, DAMGO also increased the failure rate of these eIPSCs suggesting a decrease in the probability of GABA release from IN projections (Blaesse et al., 2015). Taken together, these electrophysiological studies demonstrate that distinct sets of CEA neurons respond to MOR and KOR agonists (Blaesse et al., 2015; Chieng et al., 2006; Zhu & Pan, 2004), and that MOR agonists not only induce postsynaptic inhibition but also reduce presynaptic release of both GABA and glutamate in the amygdala (Blaesse et al., 2015; Finnegan et al., 2005, 2006; Zhu & Pan, 2005). Further, MOR agonists can modulate synaptic inputs of CEA projection neurons to the vlPAG, PBN, BNST, and thalamic reticular nucleus (Chieng et al., 2006; Finnegan et al., 2005). Many of the inhibitory MOR agonist effects appear to involve the activation of potassium channels (Faber & Sah, 2004; Finnegan et al., 2006; Zhu & Pan, 2005).
DOR activation in the central amygdala
The effects of DOR activation are limited to the CM subregion and are enhanced by chronic manipulations that alter opioid tone, such as ethanol or morphine administration (see Tables 1, 6, and 7). The DOR agonist deltorphin II elicited an outward current in only ~18% of CEA neurons, which were all low-threshold bursting neurons found in the CM (Chieng et al., 2006). In CM, ~35% of all cells, and about 29% of CM neurons projecting to the PAG responded to deltorphin II (Chieng & Christie, 2009). The DOR agonist, DPDPE, also decreased mIPSC frequency in ~60% of CM cells (Kang-Park et al., 2007), but did not alter evoked IPSCs or EPSCs (Bie et al., 2009a, 2009b; Zhu & Pan, 2005). These results suggest a tonic DOR-mediated inhibition of GABA release in the CM, most likely originating from the ENK-containing neurons in the CL. Interestingly, the action of DOR in the CEA is modulated by ethanol or morphine exposure. Attenuating DOR activity either with the antagonist, naltrindole, or using slices from DOR knockout mice, enhanced the ethanol-induced increases in IPSCs evoked from CL stimulation (Kang-Park et al., 2007). Further, in rats exposed to chronic ethanol or morphine, DPDPE inhibited GABA-mediated IPSCs and eEPSCs, indicating DOR activation inhibited GABA and glutamate release in CM, while these effects were not seen in control rats. This physiological effect was associated with an increase in DOR immunoreactivity in the synaptosomal fraction (Bie et al., 2009a, 2009b). As discussed below, DOR regulation in the CM might induce a weak inhibition of GABA release in control conditions, but these DOR-related effects might be more pronounced, and include inhibition of presynaptic glutamate and GABA release, after manipulations that induce changes in the opioid regulation of the amygdala such as ethanol or morphine administration.
TABLE 6.
Amygdalar opioid regulation of ethanol effects.
| Subregion | Manipulation or injection | OR | Behavioral test or response measured | Subjects | Summary of findings | Overall opiate effect | Authors |
|---|---|---|---|---|---|---|---|
| Amygdala | |||||||
| AMY | Binge-like ethanol (2g/kg, orally) (3 days/week) | ENK | Met-ENK-Arg6Phe7 (MEAP), DYN-B, END | Rats (male, 4–9 weeks old) | Episodic binge-like exposure to ethanol in adolescent rats decreased MEAP, without altering DYN-B or END in amygdala at both 2h and 3 weeks after treatment | Binge-like ethanol administration in adolescent rats decreased MEAP, without altering DYN-B or END | Granholm, Segerstrom, and Nylander (2018) |
| AMY | Maternal stress plus ethanol self-administration | DOR | Opioid gene expression in amygdala | Rats (male, Postnatal day 1–21) | Early life stress (maternal separation, 360 min) led to high POMC gene expression in amygdala, which was decreased after ethanol drinking. There was also a correlation between voluntary ethanol intake and expression of the DOR (Oprd1) gene in amygdala | DOR (Oprd1) gene expression in amygdala was correlated with ethanol intake | Granholm et al. (2017) |
| CEA, IN | Ethanol consumption, naloxone | ENK, DYN | ppENK and ppDYN mRNA | Rats (male Fawn-hooded) | Ethanol consumption increased ppENK mRNA, but not ppDYN mRNA, in CEA and IN, but not medial Copm or BLA. This difference was not altered by systemic naloxone administration | Ethanol consumption increased ppENK mRNA, but not ppDYN mRNA, in CEA and IN | Cowen and Lawrence (2001) |
| AMY | Ethanol (1.5g/kg, intragastric) 3x/day, 1 or 5 days | KOR | Opiate gene expression | Ethanol exposure increases pDYN and KOR gene expression in amygdala. Increases in pDYN mRNA were seen after 1 day of ethanol and on the first day of withdrawal. KOR mRNA was increased after 5 days of ethanol exposure | Ethanol exposure increases pDYN and KOR gene expression in amygdala | D’Addario, Caputi, Ekstrom, et al. (2013), D’Addario, Caputi, Rimondini, et al. (2013) | |
| AMY | Systemic chronic opiate antagonist (naltrexone, nalmefene) | MOR, KOR | DAMGO (MOR) and U50488 (KOR) stimulation of GTPγS-binding; ethanol consumption with limited (2h) two bottle choice | Rats (female Alko) | Subchronic systemic administration (repeated injections or infusion) of naltrexone or nalmefene attenuated alcohol consumption in a limited-access ethanol self-administration paradigm (two-bottle choice). Antagonists also increased maximum DAMGO (MOR) stimulation of GTPγS binding in amygdala, without changes in KOR stimulation of GTPγS binding | Subchronic opiate antagonist administration with ethanol self-administration increased MOR stimulation of GTPγS binding in amygdala | Korpi et al. (2017) |
| CEA, BLA | naltrexone | MOR, DOR | EPM | Rats (male) | Naltrexone injections into either the CEA or the BLA failed to alter the anxiolytic actions of ethanol in the EPM | Opiate antagonist in CEA or BLA did not alter anxiolytic effects of ethanol | Burghardt and Wilson (2006) |
| Central amygdala | |||||||
| CEA | Oral administration of ethanol, nicotine, fat, water | ENK | cFos in ENK mRNA containing neurons, ENK mRNA | Rats (male) | Oral administration of ethanol, nicotine, and fat enhanced the density of ENK mRNA-expressing neurons in CEA, but not BLA, and activated ENKir neurons in the CL and CLC | Acute ethanol (and nicotine) increased activation of ENKir neurons in CEA | Chang, Karatayev, Barson, Liang, and Leibowitz (2014) |
| CEA | Acute ethanol (2 g/kg, i. P.) | ENK | cFos in ENK or CRF neurons | Rats (male) | Acute ethanol increased cFos in ppENK-containing GABAergic neurons in CEA, but little co-localization was seen in CRF-containing population | Acute ethanol activated ppENK-containing GABAergic CEA neurons | Criado and Morales (2000) |
| CEA | Over-expression of ENK, naltrexone | ENK | EPM | Rats (male) | Using replication deficient herpes simplex viruses, overexpression of ENK in the CEA enhanced the anxiolytic actions of ethanol in the EPM. Anxiolytic effects of ethanol were attenuated by large injections of naltrexone in the CEA | ENK over-expression in CEA enhanced the anxiolytic effects of ethanol | Wilson, Burghardt, Lugo, Primeaux, and Wilson (2003) |
| CEA | prenatal/postnatal ethanol exposure | ENK | met-ENK levels | Rats (male, female) | In a three trimester model of perinatal ethanol exposure, ethanol exposed rats showed decreased met-ENKir in the CEA compared to nontreated controls, although they were not different from intubated controls suggesting an interaction with stress | Perinatal ethanol exposure decreased met-ENKir in CEA | Lugo, Wilson, and Kelly (2006) |
| CEA | Ethanol | END, ENK, DYN | Microdialysis for END, met-ENK, DYNA[1–8] | Rats (male) | Acute systemic ethanol administration induced increases in END and DYN, but not ENK, release in the CEA | Acute ethanol administration increased END and DYN, but not ENK, release in the CEA | Lam, Marinelli, Bai, and Gianoulakis (2008) |
| CEA | Acute ethanol (0.35–2.5 g/kg, i.p.) | Spontaneous neural activity in CEA | Rats (male) | Systemic ethanol administration significantly inhibited spontaneous activity of CEA neurons, even at low (0.35 mg/kg) doses | Ethanol administration inhibits spontaneous activity of CEA neurons | Naylor et al. (2001) | |
| CEA (CM) | Acute ethanol in slices | MOR | GABA-mediated IPSCs in amygdala slices | Mice (WT and MOR KO) | Ethanol increased the eIPSCs and mIPSCs in CM, and this effect was similar in slices from WT and MOR KO mice | Ethanol-induced increases GABA transmission in CEA are not altered in MOR KO mice | Kang-Park et al. (2009) |
| CEA | Methyl-naloxonium | MOR | Operant oral ethanol self-administration | Rats (male) | The opioid antagonist methylnaloxonium in CEA dose- dependently decreased operant ethanol self-administration | Opiate antagonist in CEA decreased ethanol self-administration | Heyser, Roberts, Schulteis, and Koob (1999) |
| CEA | CTOP, naltrindole | MOR, DOR | Operant oral ethanol self-administration | Rats (male, Wistar, AA high-drinking) | Both CTOP and naltrindole decreased ethanol responding in an operant self-administration paradigm | MOR and DOR antagonists in CEA decreased ethanol self-administration | Hyytia and Kiianmaa (2001) |
| CEA (CM) | Naltrexone, naltrindole | DOR | Ethanol conditioned place preference (CPP) | Rats | Naltrexone and DOR antagonist naltrindole in CEA decreased ethanol CPP behavior without modifying baseline behaviors in the test | Naltrexone and DOR antagonist naltrindole in CEA decreased ethanol CPP | Bie et al. (2009a) |
| CEA (CM) | Acute ethanol in slices | DOR | GABA-mediated IPSCs in amygdala slices | Mice (WT and DOR KO) | Ethanol increased the eIPSCs in CM, and this effect was increased with the DOR antagonist naltrindole and in slices from DOR KO mice. The DOR inverse agonist ICI174864 had no effect alone, but increased ethanol’s effects on mIPSCs | Decreasing DOR activation enhanced ethanol-induced increases in GABA transmission in CEA | Kang-Park et al. (2007) |
| CEA (CM) | Acute ethanol in slices, norBNI | KOR | GABA-mediated IPSCs in amygdala slices | Mice (WT and KOR KO) | Ethanol increased the eIPSCs in CM through enhanced presynaptic GABA release. These effects were enhanced with KOR antagonist norBNI (WT mice) and in KOR KO mice, suggesting ethanol’s effects involve KOR-mediated inhibition of GABA release | Decreasing KOR activation enhanced ethanol-induced increases in GABA transmission in CEA | Kang-Park et al. (2013) |
| CEA (CM) | Acute ethanol in slices | KOR | GABA-mediated IPSCs in amygdala slices | Rats (male) | Ethanol increased the eIPSCs in CM through enhanced presynaptic GABA release, and reversed the effects of KOR agonists. The KOR antagonist norBNI prevented, but could not reverse, ethanol’s effects | A KOR antagonist prevented ethanol-induced increases in GABA transmission in CEA | Gilpin et al. (2014) |
| CEA | norBNI, ethanol self-administration, ethanol vapor exposure | KOR | Ethanol self-administration; Ethanol dependence (ethanol vapor exposure), physiological signs during withdrawal | Rats (male) | CEA injections of norBNI attenuated the enhanced ethanol consumption during protracted abstinence in ethanol-dependent rats, but did not attenuate the physiologic measures of withdrawal | KOR antagonists in CEA decreased withdrawal-induced ethanol consumption-independent rats | Kissler and Walker (2016) |
| CEA | norBNI, nalmefene, CTOP, naltrindole, Ethanol vapor exposure; ethanol self-administration | KOR | DYN stimulation of GTPγS binding, DYNir, ethanol self-administration during withdrawal; ethanol dependence (ethanol vapor exposure) | Rats (male) | Ethanol-dependent rats showed elevated DYNir in capsular CEA, and increased DYN stimulation of GTPγS binding in amygdala. Ethanol consumption during ethanol withdrawal was decreased in ethanol-dependent rats with CEA injections of norBNI. In contrast, CEA injections of a CTOP/naltrindole cocktail only blocked ethanol consumption in non-dependent rats, while nalmefene blocked ethanol intake in both dependent and non-dependent rats | Ethanol dependence is associated with an upregulation of DYN expression and increased KOR coupling in CEA | Kissler et al. (2014) |
| Basolateral amygdala | |||||||
| LA | Withdrawal after two-bottle choice for ethanol | MOR | MOR binding (125I-FK 33,824) | Rats (male Fawn-hooded) | Ethanol withdrawal increased MOR density in lateral amygdala, with binding levels increased over ethanol consuming groups in a two-bottle choice or non-ethanol exposed controls | Ethanol withdrawal increases MOR binding in LA | Djouma and Lawrence (2002) |
| BLA | Context-induced reinstatement of ethanol seeking, naltrexone | cFos | cFos induced by exposure to extinction paired context | Rats (male) | Reexposure to a context associated with ethanol self-administration induced cFos in the BLA, but not the CEA, and this response was attenuated with systemic administration of naltrexone | Context-induced reinstatement of ethanol self-administration induced activation in BLA which was attenuated by naloxone | Marinelli, Funk, Juzytsch, Li, and Le (2007) |
| BLA | Naloxone | OR | Context-induced reinstatement of ethanol self-administration | Rats (male) | Naloxone administration in the BLA dose dependently decreased lever presses for ethanol during reexposure to a context associated with ethanol self-administration | Naloxone in BLA decreases ethanol responding during context-induced reinstatement | Marinelli, Funk, Juzytsch, and Le (2010) |
| BNST | |||||||
| BNST | norBNI, re-instatement of ethanol self-administration | KOR | Expression of DYN and KOR genes, ethanol self-administration, physiological signs and EPM behaviors during withdrawal | Rats (male) | Reinstatement of ethanol self-administration induced by systemic injections of the KOR agonist U50,488 increased cFos in the BNST, and was blocked by intra-BNST injections of norBNI. BNST injections of U50,488 partially reinstated ethanol self-administration | KOR antagonist in BNST blocked reinstatement of ethanol self-administration induced by systemic KOR agonists | Le, Funk, Coen, Tamadon, and Shaham (2018) |
| BNSTL | norBNI, Ethanol vapor exposure, ethanol self-administration | KOR | Expression of DYN and KOR genes, ethanol self-administration, physiological signs of withdrawal, EPM during withdrawal | Rats (male) | Expression of KOR mRNA (Oprk1 gene), but not pDYN was increased in BNST in ethanol-dependent rats self-administering ethanol, compared to non-dependent rats. BNST injections of norBNI attenuated the enhanced ethanol consumption, but not physiological measures or EPM behaviors, during withdrawal | KOR antagonist in BNST decreased withdrawal-induced ethanol consumption in dependent rats | Erikson, Wei, and Walker (2018) |
TABLE 7.
Amygdala opioid regulation of opiate (morphine) dependence and withdrawal.
| Subregion | Manipulation or injection | OR | Behavioral test or response measured | Subjects | Summary of findings | Overall opiate effect | Authors |
|---|---|---|---|---|---|---|---|
| Amygdala | |||||||
| AMY | Chronic morphine administration | MOR | DAMGO stimulated GTPγS binding | Rats (male) | Neither chronic nor acute morphine administration changed amygdala MOR-stimulated GTPγS binding | Morphine administration did not change amygdala MOR-stimulated GTPγS binding | Sim, Selley, Dworkin, and Childers (1996) |
| AMY | Heroin self-administration | MOR, DOR | DOR or MOR stimulated GTPγS binding, 3H-naltrexone binding | Rats (male) | Heroin self-administration decreased MOR, but not DOR, stimulated GTPγS binding in amygdala, but did not alter receptor number (3H-naloxone binding) | Heroin self-administration decreased MOR stimulated GTPγS binding in amygdala | Sim-Selley et al. (2000) |
| CEA, BNST | Naloxone, control and morphine-dependent rats | ENK | cFos in CRFir and ENKir neurons | Rats (male) | Naloxone-precipitated withdrawal in morphine-dependent rats induced cFos in ENKir neurons, but not CRFir neurons, in the CEA and BNSTL. Naloxone in controls induced less cFos in these areas but activated both CRFir and ENKir neurons | Naloxone-precipitated withdrawal activated ENKir neurons in the CEA and BNSTL | Veinante, Stoeckel, Lasbennes, and Freund-Mercier (2003) |
| AMY | Morphine tolerance and withdrawal | DYN | DYN(1–13) levels (radioimmunoassay) | Rats (male) | Increased levels of DYN in amygdala during morphine treatment and during withdrawal (18 h) | Morphine treatment and withdrawal increased DYN levels | Rattan, Koo, Tejwani, and Bhargava (1992) |
| Central amygdala | |||||||
| CEA | Morphine microinjections | MOR | morphine CPP | Rats (male) | CEA injections of morphine did not produce CPP although the numbers of subjects was very low in this early study | CEA injections of morphine did not produce CPP | van der Kooy, Mucha, O’Shaughnessy, and Bucenieks (1982) |
| CEA, MEA, BLA | Naloxone injections in morphine tolerant rats; CEA lesions | OR | Naloxone injections in CEA, withdrawal signs | Rats (male) | Naloxone injections (unilateral) in the CEA, MEA, BLA, or lateral anterior nucleus induced withdrawal symptoms in morphine tolerant rats (jumps, wet dog shakes, paw tremor, chewing, teeth chattering, diarrhea). Jumps were only seen with injections in CEA, and were reduced during withdrawal with CEA lesions | Opioid antagonist in amygdala precipitated withdrawal in morphine-dependent rats | Lagowska, Calvino, and Ben-Ari (1978), Calvino, Lagowska, and Ben-Ari (1979) |
| CEA | Methyl-naloxonium in morphine-dependent rats (morphine pellets) | OR | Methylnaloxonium-induced conditioned place aversion (CPA) and somatic withdrawal signs | Rats (male) | Microinjections of methylnaloxonium in CEA-induced conditioned place aversion (CPA) in morphine-dependent rats, but did not induce physical abstinence signs. Injections in PAG induced both CPA and physical withdrawal (including escapes) | Microinjections of methylnaloxonium in the CEA induced CPA in morphine-dependent rats | Stinus, Le Moal, and Koob (1990) |
| CEA | Naltrexone precipitated withdrawal from morphine | MOR | Acoustic startle | Rats | DAMGO injections (unilateral) in CEA blocked the enhanced acoustic startle responses seen during naltrexone-precipitated withdrawal from acute morphine tolerance | DAMGO in CEA blocked enhanced startle during morphine withdrawal | Cabral, Ruggiero, Nobre, Brandao, and Castilho (2009) |
| CEA | Morphine, DADL, naloxone, ICI 174864, clonidine, NE, control, morphine-dependent rats | MOR, DOR | Glutamate-driven firing rate in anesthetized rats, micro-iontophoresis of drugs | Rats (male) | Iontophoretic application of morphine or DADL decreased glutamate-driven firing rate in CEA and MEA, but not BLA. All cells responding to morphine were inhibited by the α2 agonist clonidine. In morphine-dependent rats, but not controls, both naloxone and the DOR antagonist ICI174864 increased firing rates, and these effects could be blocked with clonidine | Naloxone and a DOR antagonist increased CEA firing rates morphine-dependent rats | Freedman and Aghajanian (1985) |
| CEA (CM) | Deltorphin II, DAMGO, brain slices (control, morphine-dependent rats) | MOR, DOR | Postsynaptic inhibition, morphine CPP | Rats (male) | Morphine-treated rats showed increased glutamate synaptic strength (eEPSCs) in CM. This was associated with enhanced DOR effects in morphine-treated slices, with the DOR agonist DPDPE inducing decreases in evoked EPSCs in 70% of CEA neurons (compared to no effect in controls) through inhibition of presynaptic glutamate release. Consistent with this, DOR expression was enhanced in a synaptosomal preparation | Slices from morphine-dependent rats showed increased DOR-mediated presynaptic inhibition of evoked glutamate release in CM | Bie et al. (2009b) |
| CEA (CM) | Deltorphin II, DAMGO, brain slices, control, morphine-dependent rats | MOR, DOR | Postsynaptic inhibition (GIRK-mediated) | Rats (male) | Chronic morphine increased the number of cells responding to deltorphin II (GIRK current) from ~35% (controls) to 69% (morphine dependent). In control slices DOR responsive neurons also responded to DAMGO, but chronic morphine increased the number of neurons responding to the DOR agonist alone (31%) and decreased the number of neurons responding to DAMGO alone (62% to 15%). In neurons projecting from CM to PAG, the number of DOR responsive neurons increased from 29% (controls) to 86% (morphine dependent), with a decrease in MOR responsive neurons | Morphine-dependent animals showed increased DOR responsive neurons in CM, especially in CM neurons projecting to the PAG | Chieng and Christie (2009) |
| CEA | Morphine, DAMGO, CTOP, brains slices from control, morphine-dependent rats | MOR | GABA neurotransmission (mIPSCs, epics) in the presence of morphine-independent rats | Rats (male) | Morphine and DAMGO decreased eIPSCs in ~50% of neurons (no effect in ~30%). MOR agonists decreased mIPSCs, while CTOP increased mIPSC frequency, suggesting tonic tone at MOR. Many responses were unaltered in slices from morphine-dependent rats, although the inhibitory effects of MOR agonists were blunted suggesting the development of tolerance | Slight tolerance to the inhibitory effects of MOR agonists on GABA neurotransmission in CEA | Bajo et al. (2011) |
| CEA | Morphine, brain slices after naloxone-precipitated withdrawal | MOR | GABA neurotransmission (mIPSCs) in the presence of naloxone (controls), morphine/naloxone (dependent rats) | Rats (male) | Increased mIPSCs were observed in CEA slices following naloxone-precipitated withdrawal. Morphine, in the presence of continuous naloxone perfusion, either increased (64%) or decreased (32%) mIPSCs in control rats, but this ratio shifted in naloxone-withdrawn slices | Naloxone-precipitated withdrawal in morphine-dependent rats increased GABA inhibition (mIPSCs) in CEA | Bajo et al. (2014) |
| Basolateral amygdala | |||||||
| LA | Morphine microinjections, lesions | MOR | Morphine CPP | Rats (male) | Lateral amygdala injections of morphine did not produce CPP and lesions did not block CPP induced by systemic morphine administration. Injections in VTA or PAG produced CPP and naloxone in these sites blocked CPP from systemic morphine | Lateral amygdala injections of morphine did not produce CPP and lesions did not block CPP induced by systemic morphine administration | Olmstead and Franklin (1997a, 1997b) |
KOR activation in central amygdala
Inhibitory postsynaptic effects of the KOR agonist, U69593, have been seen on some CEA neurons. Application of U69593 induced an outward potassium current in a small percentage (6%–17%) of CEA neurons, mostly type B neurons (Chieng et al., 2006; Zhu & Pan, 2004). In addition, U69593 decreased spontaneous IPSC frequency in both adolescent and adult CEA neurons (Przybysz et al., 2017). Additionally, the KOR agonists, DYN[1–7] and U69595, also decreased eIPSCs (60%–80% of neurons) in CM of mice (Kang-Park et al., 2013) and rats (Gilpin et al., 2014) indicating a mechanism of KOR-induced decreases in presynaptic GABA release. Not only did the KOR antagonist, norBNI, block the effects of exogenous KOR agonists, but perfusion with norBNI alone also increased eIPSCs, suggesting there is an endogenous KOR tone that diminishes GABA release in the CEA. Interestingly, two studies have implicated KOR-induced GABA inhibition in the CM in mediating ethanol’s effects but found different effects of norBNI when combined with ethanol (Gilpin et al., 2014; Kang-Park et al., 2013) (see below). The diminishing activity of KOR in the CM also enhanced the effects of CRF on mIPSCs in the CM (Kang-Park et al., 2015). The KOR agonist U69593 had no effect on eEPSCs in the CEA (Zhu & Pan, 2005).
KOR activation in BNST
A series of elegant studies in the BNST suggest that KOR activation has distinct effects on specific input pathways from the amygdala and can presynaptically inhibit both GABA and glutamate release in the BNST. KOR activation with DYN-A and U69593 decreased the amplitude of the eIPSCs in the dorsolateral BNST (oval nucleus), including a decrease in the GABA IPSCs induced by optogenetic stimulation of the CEA inputs to the BNST. Additional manipulations indicated that this KOR activation was due to decreased presynaptic GABA release mediated through activation of the extracellular signal-regulated kinase (ERK 1/2) signaling pathway in the BNST (Li et al., 2012). In addition, the same group showed KOR agonists (U69593, DYN-A) decreased both eEPSCs and mEPSCs in the dorsolateral BNST. This effect was prevented but not reversed by norBNI (Crowley et al., 2016). KOR activation also decreased EPSCs evoked in BNST from optogenetic stimulation of BLA, but not PFC, inputs, as well as decreasing the fidelity of action potentials induced in the BNST with BLA stimulation. Viral deletion of amygdala KOR eliminated this inhibition with BLA stimulation, indicating KOR is expressed presynaptically on BLA neurons. Using transgenic ppDYN-IRES-Cre mice, optogenetic stimulation of DYN+ neurons in the BNST led to a monosynaptic IPSC, but also decreased eEPSCs, suggesting DYN from these neurons decreases local glutamate transmission in the BNST (Crowley et al., 2016).
THE ROLE OF AMYGDALAR OPIOID SYSTEMS IN BEHAVIOR: HUMAN IMAGING STUDIES PLUS GENETIC AND PHARMACOLOGICAL MANIPULATIONS IN RODENTS
Human and animal studies have implicated amygdalar opioid systems in many behavioral or physiological processes and disorders ranging from stress and anxiety responses to pain modulation to drug or alcohol addiction (Drolet et al., 2001; Henry et al., 2017; Lutz & Kieffer, 2013; Ribeiro, Kennedy, Smith, Stohler, & Zubieta, 2005; Vaccarino & Kastin, 2000). The evidence below highlights the role of amygdalar opioids in mediating several of these responses, specifically nociception, stress and anxiety-related responses, associative learning and conditioned fear, ethanol effects, and opiate or opioid addiction/dependence. These overviews do not address the general role of the amygdala, or opioid systems, in these effects, but focus specifically on studies demonstrating the role of amygdalar ENK or DYN, and/or activation of DOR, MOR, or KOR in the amygdala, in these responses. Our overview includes human imaging studies focused on amygdalar changes, as well as genetic modulation of peptide expression using virus-mediated gene transfer or pharmacological approaches targeting amygdalar opioid systems. Although opioid processes in the amygdala are clearly involved in many other related functions, we have focused on more in-depth reviews of a few key topics. In addition, while anatomical and electrophysiological studies reviewed above clearly demonstrate distinct opioid regulation in different amygdalar subnuclei, many of the approaches utilized to elucidate the functional role of amygdalar opioids cannot easily target specific subnuclei. Therefore, effects have been generally described in the central (CEA), basolateral (BLA), or medial amygdala (MEA) regions, or the BNST, in the sections below.
AMYGDALAR OPIOID REGULATION OF NOCICEPTION
Human imaging and nociception
As reviewed in Table 2, the administration of MOR agonists in conjunction with functional magnetic resonance imaging (fMRI) or cerebral blood flow (rCBF) analysis suggested MOR agonist administration activated the amygdala. Morphine administration enhanced the BOLD signal in the sublenticular extended amygdala (SLEA) when compared with saline (Becerra et al., 2006), while the short-acting MOR agonist, remifentanil, activated (bilaterally) the amygdala (Leppa et al., 2006). In individuals with a history of opioid abuse, the MOR agonist, hydromorphone, increased rCBF in the amygdala; these changes were not seen with the kappa-like opioid, butorphanol (Schlaepfer et al., 1998).
TABLE 2.
Opioid-related effects in human amygdala: Imaging studies.
| Response examined | Imaging technique (patient population) | Summary of major findings | Authors |
|---|---|---|---|
| Age-gender | PET with [11C]carfentanil (males and females) | MOR-binding potential decreased with age in amygdala, but only in women, yielding an age × sex interaction but no overall gender difference in amygdala MOR | Zubieta, Dannals, and Frost (1999) |
| MOR agonist administration | fMRI in humans with BOLD (healthy volunteers) | Morphine administration (small i.v. dose) in healthy human volunteers activated the sublenticular extended amygdala compared with saline injection in the same subjects | Becerra, Harter, Gonzalez, and Borsook (2006) |
| MOR agonist administration | fMRI with BOLD (healthy males and females) | Administration of the very short acting MOR agonist remifentanil to healthy subjects induced bilateral activation of the amygdala, when compared to saline injection in the same session | Leppa et al. (2006) |
| MOR agonist administration | rCBF (opioid abusers) | In individuals with a history of opioid abuse, the MOR agonist hydromorphone increased rCBF in amygdala; changes were not seen with the kappa-like opioid butorphanol | Schlaepfer et al. (1998) |
| Opioid dependence | MRI, fMRI, and DTI (prescription opioid-dependent patients vs. controls) | Opioid-dependent individuals showed reduced amygdala volumes and changes in functional connectivity in the amygdala compared with healthy controls | Upadhyay et al. (2010) |
| Affective state | PET with [11C]carfentanil and MRI (females with major depressive disorder (MDD) or controls) | Women with MDD (un-medicated) showed greater MOR activation in amygdala during a shift from a neutral to sad affective state, induced by autobiographical recall, than controls. This was seen as lower MOR-binding potential bilaterally in the amygdala during the sad vs. neutral state | Kennedy, Koeppe, Young, and Zubieta (2006) |
| Affective state | PET and rCBF (male volunteers) | Aversive visual stimuli activated the left amygdala, and there was a negative correlation between rCBF in the left inferior temporal pole in response to aversive emotional visual stimuli and MOR-binding potential, consistent with an anxiolytic role for endogenous opioids in limbic areas projecting to the amygdala | Liberzon et al. (2002) |
| Affective state | PET with [11C]carfentanil (volunteers) | Increases in MOR-binding potential with sad versus neutral affective recall of autobiographical event suggest a decrease in opioid transmission in amygdala, especially left amygdala. In amygdala, there was also a correlation between the change in MOR binding and the decrease in positive affect using the Positive and Negative Affectivity Scale (PANAS). This regional deactivations suggest there is tonic opioid tone (release) during the neutral condition | Zubieta et al. (2003) |
| Affective state | PET with [11C]carfentanil and MRI (females) | Amygdala showed similar opioid de-activation, indicated by increased MOR-binding potential, with both sadness (autobiographical recall) and pain (induced by saline infusion into the masseter muscle), although sadness induced bilateral deactivation of the opioid system. Both manipulations induced similar negative affect using the PANAS instrument | Ribeiro et al. (2005) |
| Traumatic stress | PET with [11C]carfentanil (male veterans with PTSD, combat exposed veterans without PTSD, controls) | Even at rest (baseline), combat-exposed veterans without PTSD showed decreased MOR-binding potential than controls or veterans with PTSD, suggesting that patients with PTSD failed to downregulate opioid receptors in amygdala during exposure to traumatic stress. Both trauma exposed groups showed decreased MOR-binding potential compared to controls in the sublenticular extended amygdala | Liberzon et al. (2007) |
| Pain (nociception) | PET with [11C]carfentanil (males and females) | A pain challenge in health volunteers, induced by saline infusion in the masseter muscle, activated the MOR system in amygdala, and individual variations in amygdala activation were negatively correlated with the suppression in the sensory ratings of pain (McGill Pain Questionnaire) | Zubieta et al. (2001) |
| Pain (nociception) | PET with [11C]carfentanil and MRI (males and females) | A pain challenge in health volunteers, induced by saline infusion in the masseter muscle, activated the MOR system in amygdala to a greater degree in men compared to women (early follicular phase of menstrual cycle), and these differences were associated with sex differences in the suppression of sensory pain ratings | Zubieta et al. (2002) |
| Pain (nociception) | PET with [11C]carfentanil (females with fibromyalgia vs. controls) | Women with fibromyalgia showed reduced MOR-binding potential compared with healthy controls in the amygdala. There was a significant negative correlation between amygdalar MOR-binding potential and patient depression symptoms, but not pain ratings | Harris et al. (2007) |
| Pain (nociception) | PET with [11C]carfentanil (males and females) | Placebo-induced activation of amygdalar MOR was associated with different personality traits | Pecina, Love, Stohler, Goldman, and Zubieta (2015) |
| Pain (nociception), conditioned responses | fMRI and skin conductance responses (SCRs) during a conditioning procedure (male volunteers) | A conditioning protocol consisting of CS+ presentation paired with a thermal pain stimulus induced conditioned hypoalgesia, as indicated by the decrease in pain rating, decreased SCRs, and decreased reaction times to the CS +. The CS+ induced amygdala activation decreased during the protocol, and naloxone administration attenuated this response suggesting that habituation to the CS+ in the amygdala was opioid dependent. Naloxone also enhanced behavioral conditioned responses | Eippert, Bingel, Schoell, Yacubian, and Buchel (2008) |
| Alcohol abstinence | PET with [11C]carfentanil (MOR) or [11C] methylnaltrindole (DOR) (males and females in alcohol withdrawal vs. controls) | MOR, but not DOR, binding potential in the amygdala was increased during alcohol abstinence (day 5) compared to healthy controls | Weerts et al. (2011) |
| MOR gene polymorphism OPRM1 A118G (rs1799971) | PET with [11C]carfentanil (male and female smokers and nonsmoker controls with A vs. G OPRM1 genotype) | A common single nucleotide polymorphism (SNP) in the MOR gene (OPRM1 A118G (rs1799971)) shifts MOR expression and drug dependence phenotypes. The G allele carriers showed lower MOR-binding potential in amygdala compared with wild-type A allele carriers. In the G allele carriers, the difference in self-reported reward between a nicotine-containing cigarette and a de-nicotinized cigarette was also correlated to the difference in MOR-binding potential in the amygdala | Ray et al. (2011) |
| MOR gene polymorphism OPRM1 A118G (rs1799971) | PET with [11C]carfentanil (males smokers with A vs. G OPRM1 genotype) | The G allele carriers showed less MOR in the amygdala compared to A allele carriers. Tobacco smoking increased MOR-binding potential (decreased opioid release) in amygdala, but differences between cigarettes with and without nicotine were seen in A allele carriers | Domino, Hirasawa-Fujita, Ni, Guthrie, and Zubieta (2015) |
A series of studies using positron emission tomography (PET) imaging with the MOR ligand, [11C]carfentanil to determine MOR-binding potential, have implicated the endogenous amygdala opioid system in human pain processing. Using this technique, a reduction in binding potential in vivo suggests that the endogenous opioid system has been activated (see Ribeiro et al., 2005; Zubieta et al., 2003 for explanation). These studies demonstrated that a pain challenge in healthy human volunteers (males and females) induced by saline infusion in the masseter muscle activates the amygdalar MOR system (reduces MOR-binding potential), and that individual variations in amygdala activation negatively correlated with the suppression in the sensory ratings of pain using the McGill Pain Questionnaire (Zubieta etal.,2001). Further, the MOR system in the amygdala was activated to a greater degree by this noxious stimulusin men compared with women in the early follicular phase of their menstrual cycle, and these differences were associated with sex differences in the suppression of sensory pain ratings (Zubieta et al., 2002). Women with fibromyalgia also showed reduced MOR-binding potential compared with healthy controls, suggesting greater MOR activation in the amygdala with chronic pain (Harris et al., 2007). Curiously, there was a significant negative correlation between MOR-binding potential in the amygdala and patient depression symptoms but not pain ratings. Further, the amygdala showed similar opioid deactivation, indicated by increased MOR-binding potential, with both sadness (autobiographical recall) and pain (induced by saline infusion into the masseter muscle), although sadness induced bilateral deactivation of the opioid system (Ribeiro et al., 2005). Activation of the amygdalar MOR system has also been associated with placebo-induced analgesia based on different personality traits (Pecina et al., 2015). Using fMRI and skin conductance responses (SCRs), a conditioning protocol consisting of the presentation of a conditioned stimulus (CS+) paired with a thermal pain stimulus-induced conditioned hypoalgesia evidenced by the decrease in pain ratings, decreased SCRs, and decreased reaction times to the CS+. Naloxone administration attenuated the amygdala activation induced by the CS+ decreased during the protocol, suggesting that habituation to the CS+ in the amygdala was opioid dependent. Naloxone also enhanced behavioral conditioned hypoalgesia responses (Eippert et al., 2008).
CEA and nociception
Endogenous levels of ENK and DYN, as well as MOR and KOR receptors, are high in the CEA, and particularly the CL and CLC (see above). Several studies implicate this endogenous opioid system with analgesia induced by systemic opiates or opioids, supraspinal aspects of nociception, and plasticity associated with changes in nociceptive behaviors (see Table 3). Administration of morphine into the CEA was antinociceptive and increased jump thresholds with footshock (File & Rodgers, 1979a; Rodgers, 1977). Enhancing endogenous opioid levels using the enkephalinase inhibitor SCH-32615 injected bilaterally in the CEA also induced antinociception (increased latencies) in the hot-plate test, but not the tail-flick test, and these effects were naloxone reversible (al-Rodhan et al., 1990). Similarly, overexpression of ENK in the CEA using virus-mediated gene transfer reduced phase two flinching behavior in the formalin test that was reversed with naloxone, suggesting enhanced ENK expression reduced the supra-spinal modulation of nociception in this test (Kang et al., 1998, 1999). Interestingly, these viral injections targeted the CL/CLC region of the CEA, which is not only where ENK and MOR expressions are high but also the region receiving inputs from the PBN, including ENK-containing projections (Bernard & Besson, 1990; Poulin et al., 2006). Interestingly, all of the neurons in CM (low-threshold bursting cells) that projected to PBN and PAG showed postsynaptic outward (potassium) currents with DAMGO (Chieng et al., 2006; Chieng & Christie, 2009), suggesting opioid modulation of reciprocal projections between CM and brainstem nuclei (PBN, PAG) might mediate nociceptive responses. Injections of the MOR selective antagonist methylnaloxonium into the CEA also attenuated the systemic analgesic effects of both morphine and the enkephalinase inhibitor RB101 on the motor response (tail-flick) and vocalizations (tailshock vocalizations and after-discharge vocalization) with tailshock (Valverde et al., 1996).
TABLE 3.
Amygdalar opioid regulation of nociception.
| Subregion | Manipulation or injection | OR | Behavioral test or response measured | Subjects | Summary of findings | Overall opiate effect | Authors |
|---|---|---|---|---|---|---|---|
| Amygdala | |||||||
| AMY | Morphine, plantar formalin injections | MOR | fMRI in rats | Rats (male) | In anesthetized rats, both morphine and intraplantar formalin injections activated the BOLD signal in the amygdala, but pretreatment with morphine attenuated the formalin-induced amygdala activation | Morphine activates amygdala | Shah et al. (2005) |
| AMY | Morphine, END, naltrexone, naltrindole, βFNA | MOR, DOR | Tail-flick latencies to heat, jump thresholds with footshock; data pooled from CEA, MEA, and BLA | Rats (male) | Bilateral injections of morphine and END in amygdala increased latencies in the tail-flick test and jump thresholds to footshock. Effects or amygdala opioids were blocked by injections of naltrexone, naltrindole, and βFNA in the PAG | MOR agonist in amygdala is antinociceptive | Pavlovic, Cooper, & Bodnar (1996) |
| AMY | Chronic pain models | MOR, DOR | GTPγS binding, LD Box, EPM | Mice | Chronic pain (4 weeks) in both sciatic nerve ligation and CFA models induced anxiogenic effects in the LD Box and EPM. GTPγS binding (amygdala homogenates) showed decreased stimulation with DAMGO (MOR) and SNC80 (DOR), but increased stimulation with the KOR agonist ICI 199,441. KOR changes were seen in the CFA model but not the nerve ligation model | Chronic pain increases MOR and DOR, but decreases KOR, receptor coupling in amygdala | Narita et al. (2006) |
| Central amygdala | |||||||
| CEA | ENK overexpression | ENK | Formalin-induced nociceptive behaviors | Rats (male) | Overexpression of ENK in the CEA-reduced phase two flinching behavior in the formalin test that was reversed with naloxone | ENK overexpression in CEA is antinociceptive in supra-spinal pain | Kang, Wilson, Bender, Glorioso, and Wilson (1998), Kang, Wilson, and Wilson (1999) |
| CEA | Enkephalinase inhibitor SCH-32615, naloxone | ENK | Licking in hot-plate, tail-flick to thermal heat | Rats (male) | The enkephalinase inhibitor SCH-32615 injected bilaterally in the CEA induced antinociception (increased latencies) in the hot-plate test, but not the tail-flick test, and these effects were naloxone reversible | Enkephalinase inhibitor in CEA is antinociceptive | al-Rodhan, Chipkin, and Yaksh (1990) |
| CEA (CMA) BLA | Morphine | MOR | Jump thresholds with footshock | Rats (male) | Morphine injections into the centromedial amygdala, but not the BLA, increased jump thresholds to footshock. No effects on activity were observed | MOR agonist in centromedial amygdala is antinociceptive | Rodgers (1977) |
| CEA, MEA | Morphine, naloxone | MOR | Jump-flinch thresholds with footshock | Rats (male) | Morphine injections in CEA and MEA both increased thresholds for jumping/flinching in response to footshock, but these were only reversed by naloxone in the MEA | MOR agonist in CEA and MEA is antinociceptive | Rodgers and File (1979) |
| CEA, BLA | Morphine, naloxone | MOR | Conditioned hypoalgesia in hot-plate and formalin tests of nociception | Rats (male) | Unilateral morphine injections into CEA, but not BLA decreased acquisition of hypoalgesia induced by prior hotplate exposure, in both hotplate and formalin-induced responses. Morphine in the CEA attenuated the expression of formalin-induced hypoalgesia, but not hotplate paw lick latencies | Morphine in CEA impairs acquisition and retention of conditioned hypoalgesia | Good and Westbrook (1995) |
| CEA | Enkephalinase inhibitor RB101, morphine, methylnaloxonium | MOR | Tail-flick and vocalizations to tail shock | Rats (male) | Injections of methylnaloxonium into the CEA attenuated the systemic analgesic effects of both morphine and the enkephalinase inhibitor RB101 on motor response (tail-flick) and vocalizations (tail-shock vocalizations and after-discharge vocalization) with tail-shock | MOR antagonist in CEA blocked the analgesic effects of morphine and an enkephalinase inhibitor | Valverde, Fournie-Zaluski, Roques, and Maldonado (1996) |
| CEA | nor-BNI, DYN | KOR | Stress (bright light exposure), morphine-induced hyperalgesia, paw-withdrawal thresholds in Randall-Selitto paw pressure test after capsaicin conditioning stimulus, DYN levels | Rats (male) | In rats with opiate-induced hyperalgesia, stress (bright light exposure), re-instated tactile allodynia and caused a loss of inhibitory control after capsaicin that was attenuated by nor-BNI injections in the right, but not left, CEA. DYN-A levels in the right CEA were also increased. These effects were only seen with the combination of morphine-induced hyperalgesia and stress | Increased KOR activity in CEA is associated with nociceptive inhibitory control induced by morphine-induced hyperalgesia and stress | Nation et al. (2018) |
| Basolateral amygdala | |||||||
| BLA | Morphine | MOR | Tail-flick latencies to heat | Rats (male) | Bilateral morphine injections increased tail-flick latencies to radiant heat in anesthetized rats. The injection sites that were most effective were the BLA | MOR agonist in BLA is antinociceptive | Helmstetter, Bellgowan, and Tershner (1993) |
| BLA | DAMGO, DPDPE, U50,488 | MOR | Tail-flick latencies to heat | Rats (male) | Bilateral DAMGO injections into BLA dose dependently increased tail-flick latencies to radiant heat in anesthetized rats. No effects of DPDPE (DOR) or U50,488H (KOR) were seen | MOR agonist in BLA is antinociceptive | Helmstetter, Bellgowan, and Poore (1995) |
| BLA | DAMGO | MOR | Tail-flick latencies, HR | Rats (male) | DAMGO injected bilaterally in BLA decreased tail-flick latencies in anesthetized rats, but did not change HR. This effect was attenuated by decreasing activity in the vlPAG or RVM using lidocaine or electrolytic lesions | MOR agonist in BLA is antinociceptive | Helmstetter, Tershner, Poore, and Bellgowan (1998) |
| BLA | DAMGO | MOR | Tail-flick latencies, HR | Rats (male) | DAMGO injected bilaterally in BLA increased tail-flick latencies. This effect was attenuated by injections of CTAP, but not naltriben, in the vlPAG | MOR agonist in BLA is antinociceptive | Tershner and Helmstetter (2000) |
| BLA | DAMGO, CTAP, βFNA, naltrexone | MOR | Tail-flick latencies to heat | Rats (male) | DAMGO injected bilaterally in BLA increased tail-flick latencies. These effects were blocked by naltrexone and βFNA, but not CTAP. From injection autoradiograms, some parts of injection might have influenced IN or CLC | MOR agonist in BLA is antinociceptive | Shin and Helmstetter (2005) |
| BLA, MEA | Morphine, naloxone | MOR | Tail-flick latencies to heat, RVM on and off cell activity | Rats (male) | Bilateral morphine injections in BLA increased tail-flick latencies with heat in a naloxone-reversible manner. Injections into the cortical nuclei had a smaller effect. Injections in CEA, MEA, LA had no effect. Injections of morphine in the BLA, cortical nuclei and MEA all influenced RVM on and off cell activity, including increasing off-cell firing and reducing on-cell firing | MOR agonist in BLA is antinociceptive | McGaraughty and Heinricher (2002) |
| BLA, MEA | Morphine, naloxone | MOR | Tail-flick latencies to heat, RVM on and off cell activity | Rats (male) | Bilateral morphine injections in the BLA increased tail-flick latencies with heat. Injections in MEA had no effect on tail-flick latencies. Injections of morphine in the BLA and MEA increased off-cell firing and reduced on-cell firing in RVM. All of these effects were blocked by PAG lesions, suggesting antinociceptive effects of morphine in the BLA require projections through PAG to RVM | MOR agonist in BLA is antinociceptive | McGaraughty, Farr, and Heinricher (2004) |
| BLA | Morphine, methylnaloxonium | MOR | Tail-shock induced spinal reflexes and vocalizations | Rats (male) | Bilateral morphine injections into the BLA increased thresholds for inducing vocalizations during tail-shock and vocalization after discharges, but not the thresholds for spinal motor reflexes, suggesting morphine attenuated the affective component of pain. Effects of morphine were blocked by the MOR selective antagonist methylnaloxonium and were specific to injection sites in the BLA | MOR agonist in BLA is antinociceptive | Nandigama and Borszcz (2003) |
| BLA | Morphine, naloxone | MOR | Formalin-induced behaviors; glutamate release in BLA | Rats (male) | Morphine injections into the BLA blocked formalin-induced conditioned place aversion, but not formalin-induced nociceptive behaviors (licking/biting/flinching). Perfusion through the microdialysis probe also blocked formalin-induced increases in glutamate efflux in the BLA | MOR agonist in BLA blocks formalin-induced conditioned place aversion and glutamate efflux | Deyama et al. (2007) |
The effects of manipulating endogenous opioid tone suggest that antinociception induced by modulating endogenous opioids is more specific to certain tests involving supraspinal mechanisms (e.g., the second phase in the formalin test) and/or some type of plasticity such as that induced by chronic shifts in nociceptive states. Studies suggest that chronic pain decreases MOR and DOR, but increases KOR, receptor function in the amygdala. In both the sciatic nerve ligation and or complete Freund’s adjuvant (CFA) models of chronic pain (4 weeks), there was a decreased stimulation of GTPγS binding in the amygdala homogenates with the MOR agonist DAMGO and the DOR agonist SNC80. An increase in GTPγS stimulation with the KOR agonist ICI 199,441 was also seen in the CFA model but not the nerve ligation model (Narita et al., 2006). These changes in G-protein coupling were associated with the induction of anxiogenic behaviors. In a model examining the loss of inhibitory nociceptive control, rats were exposed to repeated morphine injections (systemically) to produce opiate-induced hyperalgesia, and stress (bright light exposure) reinstated tactile allodynia and caused a loss of inhibitory control evidenced by reduced maximal responses in paw withdrawal thresholds after capsaicin. This loss of inhibitory control was attenuated by nor-BNI injections in the right, but not left, CEA and associated with an increase in DYN-A levels in the right CEA, supporting a role for KOR in these changes. These effects are only seen with the combination of morphine-induced hyperalgesia and stress (Nation et al., 2018). These latter studies suggest that the amygdalar opioid system may play a role in the interactions between stress and nociception.
BLA and nociception
Opioid mechanisms in the BLA have been implicated in mediating a variety of nociceptive responses using different rodent models (see Table 3) (Helmstetter et al., 1993, 1995, 1998; McGaraughty et al., 2004; McGaraughty & Heinricher, 2002; Nandigama & Borszcz, 2003; Pavlovic et al., 1996; Shin & Helmstetter, 2005; Tershner & Helmstetter, 2000). Bilateral injections of morphine, DAMGO, and END in the BLA produced antinociceptive effects and increased latencies in the tail-flick responses to heat or jump thresholds with footshock (Helmstetter et al., 1993, 1995, 1998; McGaraughty et al., 2004; McGaraughty & Heinricher, 2002; Pavlovic et al., 1996; Shin & Helmstetter, 2005; Tershner & Helmstetter, 2000), although one study failed to see this effect on jump thresholds (Rodgers, 1977). These effects in amygdala appear to be mediated via MOR, since the DOR agonist DPDPE and the KOR agonist U50,488H failed to alter tail-flick latencies with heat (Helmstetter et al., 1995), and the effects of DAMGO in the BLA were blocked by the MOR antagonist β-funaltrexamine (βFNA) (Shin & Helmstetter, 2005). Interestingly, morphine injections in BLA also increased thresholds for tailshock-induced vocalizations, but not the spinal motor reflex to shock, and the effects on vocalizations were blocked with methylnaloxonium indicating MOR involvement (Nandigama & Borszcz, 2003). Morphine injections into the BLA blocked formalin-induced conditioned place aversion, but not formalin-induced nociceptive behaviors, and local perfusion of morphine attenuated formalin-induced increases in glutamate efflux in the BLA as measured by microdialysis (Deyama et al., 2007). This latter effect might be related to MOR receptors on apical dendrites of glutamatergic PNs that can modulate spike frequency adaptation (as is seen in LA) to decrease local glutamate release from BLA neurons (Faber & Sah, 2004) and/or decrease release from glutamatergic inputs into the BLA. Since drug injections into the BLA cannot eliminate drug actions in the INs, this effect could be related to MOR-induced decreases in presynaptic glutamate release in the main IN island induced by BLA stimulation (Winters et al., 2017). Although some studies show effects of opioid agonists in the MEA on nociceptive responses (File & Rodgers, 1979a; Pavlovic et al., 1996; Rodgers, 1977), others fail to see such effects (McGaraughty et al., 2004; McGaraughty & Heinricher, 2002).
Several studies have indicated that the effects of MOR activation in the BLA on antinociception involve indirect projections through the PAG to the rostral ventromedial medulla (RVM; Table 3). Antinociceptive responses induced by morphine or DAMGO injections in the amygdala, and especially the BLA, are blocked by lesion or lidocaine inactivation of the PAG or RVM (Helmstetter et al., 1998; McGaraughty et al., 2004). The antinociceptive effects of BLA MOR receptor activation on tail-flick latencies and jump thresholds are also blocked with opioid antagonists into the vlPAG, including the nonselective antagonist naltrexone and the MOR antagonists CTAP or βFNA (Pavlovic et al., 1996; Tershner & Helmstetter, 2000). The role of DOR in the PAG is less clear, since one study failed to see attenuation of effects with the DOR antagonist naltriben in the PAG (Helmstetter et al., 1998), but another showed that the DOR antagonist naltrindole in the PAG was highly effective in attenuating morphine effects on jump thresholds (Pavlovic et al., 1996). These results are consistent with the release of ENK in the PAG induced by morphine injections into the amygdala (Ma & Han, 1991). Bilateral morphine injections in the BLA increased tail-flick latencies, and injections into the BLA or MEA increased off-cell firing and reduced on-cell firing in the RVM (McGaraughty et al., 2004; McGaraughty & Heinricher, 2002). All of these effects were blocked by lesions of the PAG, suggesting that the antinociceptive effects of morphine in the BLA require projections through PAG to RVM. Interestingly, projections from the CEA to the PAG, particularly those inducing inhibitory responses in PAG with CEA stimulation, involve MOR or DOR receptors (da Costa Gomez & Behbehani, 1995; Rizvi, Ennis, Behbehani, & Shipley, 1991). What remains unclear is if these antinociceptive effects in the BLA are mediated via MOR-induced inhibition of presynaptic GABA release to disinhibit BLA stimulation of CEA neurons that could then project to the PAG (Finnegan et al., 2006), since BLA does not have significant projections to PAG.
AMYGDALAR OPIOIDS AND STRESS, ANXIETY, AND ANXIOLYTICS
Human imaging and affective state
In humans, changes in MOR-binding potential in the amygdala have been associated with different affective states and responses to aversive emotional stimuli (Table 2). PET imaging with the MOR ligand [11C]carfentanil, identified a negative correlation between MOR-binding potential and rCBF in the left inferior temporal pose in response to aversive emotional stimuli, suggesting endogenous opiates in limbic areas might serve an anxiolytic role (Liberzon et al., 2002). Increases in MOR-binding potential with sad versus neutral affective recall of autobiographical events suggest a decrease in opioid transmission in amygdala, which was especially strong in the left amygdala. Correlations were identified between the change in amygdalar MOR binding and the decrease in positive affect using the Positive and Negative Affectivity Scale. This regional deactivation suggests that there is a tonic opioid tone (release) during the neutral condition (Zubieta et al., 2003). Interestingly, this change in amygdalar MOR-binding potential was similar to the induction of a negative affective state (sadness) and a pain stimulus (Ribeiro et al., 2005). Changes in MOR-binding potential have also been seen in posttraumatic stress disorder (PTSD) and women with depression (Kennedy et al., 2006; Liberzon et al., 2007). Thus, greater MOR activation was observed bilaterally in the amygdala with a shift from a neutral to sad state in unmedicated women with major depressive disorder (MDD) compared with controls (Kennedy et al., 2006). Even at rest (baseline), combat-exposed veterans without PTSD showed decreased MOR-binding potential compared with controls orveterans with PTSD, suggesting that perhaps patients with PTSD failed to downregulate opioid receptors in the amygdala during exposure to traumatic stress. Both trauma-exposed groups (with and without PTSD) showed decreased MOR-binding potential compared with controls in the sublenticular extended amygdala (Liberzon et al., 2007).
Amygdala opioids and anxiety-related responses
In rodents, studies have suggested a role of amygdalar opioids in anxiety-related responses, as well as in the responses to benzodiazepines or antidepressants (see Table 4). A clear picture of opioid regulation of anxiety-related responses, however, is complicated by the different manipulations or anxiety tests used to examine opioid effects, the three opioid receptors and two densely expressed endogenous peptides (ENK, DYN) with overlapping distributions in different amygdalar subnuclei, and the fact that the endogenous opioid ENK can activate both MOR or DOR. Further, several different models examining stress- or anxiety-related responses have been used that are likely to engage distinct amygdalar circuits in producing behavioral or physiological outcomes.
TABLE 4.
Amygdalar opioid regulation in stress, anxiety-related responses, and anxiolytic actions.
| Subregion | Manipulation or injection | OR | Behavioral test or response measured | Subjects | Summary of findings | Overall opiate effect | Authors |
|---|---|---|---|---|---|---|---|
| Amygdala | |||||||
| AMY | Morphine, END, met-ENK | MOR, DOR | Norepinephrine (NE) activity using MHPG-SO4 levels following 1-h immobilization stress | Rats (male) | Systemic administration of morphine, END, and met-ENK attenuated NE release in amygdala induced by 1-h immobilization stress, as assessed by MHPG-SO4 metabolite levels. Effect was similar to that seen with a benzodiazepine. Naloxone increased NE release. Opioid agonists also decreased behavioral stress responses (struggling, vocalizations, defecation) | Opiate agonists decrease stress-induced norepinephrine release in amygdala | Tanaka et al. (2000) |
| AMY | Antidepressant administration | MOR | DAMGO stimulation of GTPγS binding; FK33,824 binding for high-affinity MOR state | Rats (male) | Subchronic (10-day) treatment with desmethyl-imipramine or sertraline decreased MOR binding. Both I125-FK33,834 agonist binding (MOR high affinity state) and DAMGO-stimulation of GTPγS binding were decreased at 6 h after injection | Antidepressant treatments reduce MOR binding in amygdala | Chen and Lawrence (2004) |
| AMY | Acute restraint stress | ENK | ENK-degrading aminopeptidase activity | Rats (male) | Acute restraint stress decreases ENK degrading enzyme activity in amygdala | Restraint stress decreases ENK-degrading enzyme activity | Hernandez et al. (2015) |
| AMY | Single housing | ENK | Met-ENK-Arg6Phe7 (MEAP), DYN-B | Rats (male) | Single housing (7 days) in adolescent rats decreases ENKir compared to group housed controls or rats exposed to brief social isolation. No changes in DYN were seen | Single housing stress decreases ENK in amygdala | Granholm, Roman, and Nylander (2015) |
| AMY | Predator odor (TMT) stress, individual differences in freezing responses | ENK | ENK mRNA, cFos in ENK neurons; LD Box; defensive behaviors (freezing) with predator stress | Mice (male) | Predator stress (10-min TMT exposure) decreased ENK mRNA expression in CEA, MEA, and BLA, and decreased the number of ENKir neurons in CEA in mice showing enhanced freezing responses to TMT compared to low responders. Overall there were no differences in cFos activation in ENK-containing neurons in amygdala due to predator stress | Predator stress decreased ENK mRNA expression in amygdala in high responders | Hebb et al. (2004) |
| Central amygdala | |||||||
| CEA | Naloxone or naloxone-methiodide (peripheral only) | cFos | cFos activation, rearing, grooming, eye twitches | Rats (male) | Systemic administration of naloxone, but not naloxone-methiodide which does not cross the blood brain barrier, increased cFos in the CEA (especially the capsular and lateral regions in caudal CEA), with only a few cells activated in MEA and none in lateral or basolateral amygdala. Naloxone also decreased rearing and grooming behaviors, and increased eye twitches | Naloxone increased cFos in CEA and MEA | Gestreau, Le Guen, and Besson (2000) |
| CEA | Naltrexone (systemic) | cFos | cFos activation | Rats (male) | Systemic administration of naltrexone increased cFos in CEA and BNST | Naloxone increased cFos in CEA and BNST | Carr, Kutchukhidze, and Park (1999) |
| CEA | Overexpression of ENK, naltrexone | ENK | EPM | Rats (male) | Using replication deficient herpes simplex viruses, overexpression of ENK in the CEA enhanced the anxiolytic actions of the benzodiazepine agonist diazepam in the EPM, but did not affect baseline open arm time. These effects were naloxone reversible, and not seen with injections in other brain sites or at time points after the transient overexpression had returned to baseline (7–9 days). Naltrexone injections into the CEA also blocked the anxiolytic effects of diazepam in the EPM | Overexpression of ENK in CEA enhanced the anxiolytic actions of benzodiazepines, but had no effect in vehicle-treated rats in the EPM | Kang, Wilson, and Wilson (2000) |
| CEA | ENK knockdown using virus-mediated gene transfer | ENK | EPM, social interaction, contextual fear conditioning | Rats (male) | Decreased ENK mRNA expression in CEA using lentivirus-mediated gene transfer of shRNA increased open arm entries in the EPM, although this was confounded by increased total arm entries and a lack of change in open arm time. No effects of ENK knockdown in CEA were seen in social interaction or contextual fear, although ENK knockdown in CEA reduced freezing during acquisition of fear conditioning | Decreased CEA ENK expression had mild anxiolytic effects in EPM, and no effects on SI or contextual fear expression | Poulin, Berube, Laforest, and Drolet (2013) |
| CEA, MEA | morphine, naloxone | MOR | Head-dips and activity in holeboard test | Rats (male) | Morphine injections in CEA and MEA decreased exploration (head-dips) and locomotion in the holeboard test, but these effects were only reversed by naloxone in the MEA | MOR agonist in CEA is anxiogenic | File and Rodgers (1979b) |
| CEA | Morphine | MOR, DOR | EPM | Rats (male) | Morphine injections in CEA had no effect on EPM behaviors | Morphine in CEA had no effects in EPM | Zarrindast, Babapoor-Farrokhran, Babapoor-Farrokhran, and Rezayof (2008) |
| CEA | Morphine | MOR | Social interaction, OF | Rats (male) | Morphine injections into CEA, not MEA, produced partial anxiolytic effects in the SI test, but not the OF. Although overall differences in social interaction were not seen, morphine seemed to equalize differences between low-high light/familiar-unfamiliar conditions. Morphine also increased overall activity | Morphine in CEA had weak anxiolytic effects in social interaction | File and Rodgers (1979b) |
| CEA | DAMGO, CTAP, βFNA | MOR | EPM, defensive burying with predator stress | Rats (male) | Injections of the MOR agonist DAMGO decreased open arm time in the EPM, while the MOR antagonist CTAP had an anxiolytic effect. DAMGO decreased burying duration, increased the latency to bury, and increased rears in the defensive burying task, while the MOR antagonists CTAP and βFNA had the opposite effects. MOR agonists and antagonists failed to alter the anxiolytic effects of systemically administered diazepam. The opposing effects of MOR agonists in these two tasks, plus the increase in rearing and escape behaviors with DAMGO in the defensive behavior test, suggest opioids in the CEA might induce a shift in behavioral outputs toward escape/avoidance behaviors | MOR agonists had opposite anxiety-related effects in EPM and defensive burying tasks, likely due to enhanced avoidance behaviors | Wilson and Junor (2008) |
| CEA | DPDPE, naltrindole | DOR | EPM, OF | Rats (male) | Injections of the DOR agonist DPDPE into the CEA dose-dependently decreased anxiety-related measures (increased open arm time/entries) in the EPM. Increased locomotor activity was seen with the highest dose in the OF. These effects were blocked by naltrindole, but the DOR antagonist had no effect on EPM behaviors alone. CEA administration of DPDPE blocked the anxiogenic effects of swim stress | DOR agonists in CEA are anxiolytic and block anxiogenic effects of acute stress | Randall-Thompson, Pescatore, and Unterwald (2010) |
| CEA | naltrindole, βFNA | DOR | EPM, defensive prod burying | Rats (male) | Systemic administration of the DOR antagonist naltrindole attenuated the ability of ENK overexpression in the CEA to enhance the anxiolytic effects of diazepam | Effects of ENK overexpression in CEA are mediated through DOR | Primeaux, Wilson, McDonald, Mascagni, and Wilson (2006) |
| Basolateral amygdala | |||||||
| BLA | MOR OPRM1 A112G SNP | cFos | Social interaction (SI), social defeat, cFos induced by social defeat | OPRM1 A112G mice | The Oprm1 A112G SNP increased SI, decreased response to social defeat, and enhanced BLA neuronal activation in response to an aggressor | The Oprm1 A112G SNP enhanced BLA neuronal activation in response to an aggressor | Briand et al. (2015) |
| BLA (BLp) | Vulnerable and susceptible phenotypes in social defeat | ENK | ENK and DYN levels | Rats (male) | Rats were divided into resilient and susceptible phenotypes based on latencies to be defeated during a 7-day resident-intruder social defeat paradigm, and compared with handled controls. Levels of ENK mRNA were decreased only in the BLp in susceptible rats, compared to controls and resilient groups. No changes were seen in DYN in amygdala | Rats susceptible to social defeat showed decreased ENK mRNA expression in BLA | Berube, Laforest, Bhatnagar, and Drolet (2013) |
| BLA | ENK knockdown using virus-mediated gene transfer | ENK | EPM, social interaction: resilient and vulnerable rats with chronic unpredictable stress | Rats (male) | Decreased ENK mRNA expression was seen in the BLp of rats vulnerable to chronic unpredictable stress, compared with resilient or handled control groups (characterized by decreased SI, decreased sucrose intake, and EPM). Decreasing ENK mRNA expression in BLp using lentivirus-mediated gene transfer decreased SI and decreased open arm time in the EPM, but did not alter sucrose intake | Decreased BLA ENK expression produced anxiogenic effects in EPM and social interaction, and might be a marker of vulnerability | Berube, Poulin, Laforest, and Drolet (2014) |
| BLA (BLp) | Vulnerable and susceptible phenotypes in social defeat | ENK | ENK and DYN levels | Mice (male) | Mice were divided into resilient and vulnerable phenotypes based on avoidance during a SI test after 10 days of social defeat, and compared with handled controls. Levels of ENK mRNA were decreased in the BLA of vulnerable rats, compared to controls and resilient groups | Rats susceptible to social defeat showed decreased ENK mRNA expression in BLA | Henry et al. (2018) |
| BLA, CEA | Naltrexone | MOR, DOR | EPM | Rats (male) | Naltrexone injections into the CEA did not alter baseline anxiety-related behaviors in the EPM, while naltrexone in the BLA led to a slight reduction in open arm time. Naltrexone injections in the CEA, but not the BLA, attenuated the anxiolytic effects of systemic administration of the benzodiazepine diazepam, although neither injections in CEA or BLA reduced the anxiolytic actions of ethanol | Naltrexone in the CEA did not alter baseline anxiety measures, but attenuated anxiolytic effects of diazepam | Burghardt and Wilson (2006) |
| BLA | CTOP, naltrindole, DYN | MOR, DOR, KOR | LD Box | Rats (male) | BLA injections of the MOR antagonist CTOP and the DOR antagonist naltrindole decreased time in the lit compartment of the LD Box | MOR and DOR antagonists in BLA are anxiogenic | Narita et al. (2006) |
| BLA | KNT-127, naltrindole | DOR | EPM, OF, conditioning (context) | Rats (male) | Injections of the DOR agonist KNT-127 into the BLA dose-dependently decreased anxiety-related measures (open arm time/entries) in the EPM, and increased center time in the OF, without affecting locomotion. The DOR antagonist naltrindole had no effect on EPM behaviors. KNT-127 blocked the expression of contextual fear, and this effect was antagonized by naltrindole although the antagonist alone did not alter conditioned fear responses | DOR agonists in BLA are anxiolytic and block contextual fear | Sugiyama et al. (2018) |
| BLA, CEA | JDTic | KOR | EPM | Rats (male) | Injection of the KOR antagonist JDTic into the BLA, but not the CEA, increased open arm time in the EPM | A KOR antagonist in the BLA is anxiolytic in the EPM | Knoll et al. (2011) |
| BLA | DYN | KOR | LD Box | Rats (male) | BLA injections of the DYN decreased time in the lit compartment of the LD Box | DYN in BLA is anxiogenic | Narita et al. (2006) |
| BLA | CRF administration (i.c.v.) | KOR | Activation of KOR (phosphor-KORir) | Mice (WT, pdyn KO) | DYN release and KOR activation was assessed using immunohistochemistry with an antibody to the phosphorylated KOR protein. CRF administration (i.c.v.) increased KOR activation significantly in BLA, slightly in BNST, but not at all in the CEA | CRF induced activation of KOR receptors, suggesting DYN release, in BLA and BNST, but not CEA | Land et al. (2008) |
| BLA | norBNI, conditional KOR KO in BLA | KOR | Stress- or yohimbine-induced nicotine CPP | Mice (WT, BLA KOR KO) | Both norBNI injections and cre-induced conditional KOR knockdown in the BLA blocked yohimbine or stress-induced reinstatement of nicotine CPP and associated cFos expression in the BLA. Similar effects using M4 inhibitory DREADDs and the CAMKII promoter suggested this was mediated via decreases in BLA PN activity | Decreased KOR activity in BLA blocked reinstatement of nicotine CPP | Nygard, Hourguettes, Sobczak, Carlezon, and Bruchas (2016) |
| BLA | norBNI, swim stress or CRF | KOR | EPM; KOR activation (phosphor-KORir) | Mice (WT, PDYN KO) | BLA injections of the KOR antagonist norBNI blocked the anxiogenic effects of CRF (i.c.v.) and a swim stress in the EPM, as well as KOR activation in the BLA assessed using immunohistochemistry for phosphorylated KOR | norBNI in BLA blocked anxiogenic effects of stress and CRF in EPM | Bruchas, Land, Lemos, and Chavkin (2009) |
| BNST | |||||||
| BNSTL | Viral deletion of presynaptic KOR in BLA | KOR | EPM, OF, optogenetic stimulation of BLA-BNST projection | Mice (PDYN-IRES-Cre mice) | Viral approaches to delete presynaptic KOR in the BLA increased EPM open time. Optogenetic stimulation of the BLA-BNST pathway-induced anxiolytic effects in OF, and reduced the anxiogenic effects of systemic administration of the KOR agonist U50,455 | BLA KOR knockdown decreased anxiety in EPM. Stimulating the BLA projection to BNST reduced anxiogenic effects of a KOR agonist | Crowley et al. (2016) |
Amygdalar lesions, peptide administration, and anxiolytic drugs can show divergent results in various animal tests of anxiety or unconditioned fear, and it has been suggested that these models assess different aspects of fear or anxiety responses (see Henry et al., 2017; Rodgers, 1997; Wilson & Junor, 2008). The elevated plus maze (EPM), light:dark box (LD box), or open field (OF) take advantage of the natural tendencies of animals to avoid brightly lit, open, or elevated spaces, but relies on a passive avoidance response to detect anxiety behavior (e.g., avoidance of open arms, light compartment, center of OF); results of these tests can be confounded by changes in activity levels. In contrast, the defensive burying model is less affected by locomotor changes and (more importantly) the index of anxiety involves an active behavioral response, e.g., burying of a discrete object (the shock probe, predator odor). Social interactions (SI), or changes induced by social defeat, involve interactions with a social partner and psychosocial stress. Other models examine the effects of acute stressors such as restraint, immobilization, or predator stress, or models that induce enduring changes in anxiety-related responses such as chronic unpredictable stress (see Henry etal., 2017; Rodgers, 1997; Wilson & Junor, 2008).
Several stress paradigms modulate opiate systems in the amygdala and in some cases these methodologies did not dissociate effects in specific subnuclei. Acute immobilization stress decreases aminopeptidases in the amygdala (Hernandez et al., 2009, 2015) which would prevent degradation of ENK, thereby enhancing endogenous ENK levels. This could be consistent with more severe or longer-term stressors decreasing ENK expression. Thus, prolonged single housing in adolescent rats decreases ENKir compared with group-housed controls or rats exposed to brief social isolation in the amygdala, while no changes in DYN were seen (Granholm et al., 2015). Predator stress [10min 2,5-dihydro-2,4,5-trimethylthiazoline (TMT) exposure] decreased ENK mRNA expression in CEA, MEA, and BLA but did not shift activation of cFos in ENKir neurons in any subregion (Hebb et al., 2004). This study also showed increases in ENK-containing neurons induced by predator stress, but only in the CEA. Systemic administration of morphine, END, and met-ENK all attenuated norepinephrine release in amygdala induced by 1 h immobilization stress, as assessed by MHPG-SO4 metabolite levels; this effect was similar to that seen with the benzodiazepine, diazepam. Naloxone not only reversed these effects but also increased NE release. Opioid agonists also attenuated the behavioral stress responses, including struggling, vocalizations, defecation, and loss of body weight associated with this acute stressor (Tanaka et al., 2000).
CEA and anxiety-related responses
In examining the role of opioid systems in the CEA in unconditioned anxiety-like responses, early studies using microinjection of the MOR agonist, morphine, produced partial anxiolytic effects in the social interaction (SI) test, but not the OF (Table 4). Although overall differences in SI were not seen, morphine seemed to equalize differences between low-high light/familiar-unfamiliar conditions suggesting effects were dependent on testing conditions. Morphine in the CEA and MEA also decreased exploration (head-dips) and locomotion in the holeboard test, but these effects were only reversed by naloxone in the MEA (File & Rodgers, 1979a, 1979b) and other labs have failed to see effects of morphine in the CEA using the EPM (Zarrindast et al., 2008). Injections of the nonselective antagonist naltrexone into the CEA did not alter anxiety-related behaviors in the EPM but did attenuate the anxiolytic influences of the benzodiazepine diazepam in the EPM (Burghardt & Wilson, 2006). We have also shown that virus-mediated overexpression of ENK in the CEA does not alter baseline anxiety-like responses in the EPM but potentiates the anxiolytic effects of diazepam (Kang et al., 2000). Although decreasing ENK mRNA expression in CEA increased open-arm entries in the EPM (an anxiolytic effect), this effect was confounded by increased total arm entries and a lack of change in open-arm time. No effects of ENK knockdown in CEA were seen in SI or contextual fear, although ENK knockdown in CEA reduced freezing during the acquisition of fear conditioning (Poulin et al., 2013).
Studies using selective agents have shown opposing effects of MOR and DOR agonists on anxiety-related behaviors. CEA injections of the MOR agonist DAMGO were anxiogenic (decreased open-arm time) in the EPM, while the MOR antagonist CTAP, but not β FNA, had a slight anxiolytic effect (Wilson & Junor, 2008). In contrast, CEA DAMGO injections decreased burying duration, increased the latency to bury, and increased rears in the defensive burying task, suggesting anxiolytic actions of MOR agonists, while the MOR antagonists, CTAP and βFNA, had the opposite effects (Wilson & Junor, 2008). The opposing effects of MOR agonists in these two tasks, and the increase in rearing and escape behaviors with DAMGO in the defensive behavior test, suggest activation of MOR receptors in the CEA might induce a shift in behavioral stress responses toward escape/avoidance behaviors. Injections of the DOR agonist, DPDPE, into the CEA dose dependently decreased anxiety-related measures (increased open-arm time/entries) in the EPM, and blocked the anxiogenic effects of swim stress, without affecting locomotion. These effects were blocked by naltrindole, although the DOR antagonist in the CEA had no effect on EPM behaviors alone (Randall-Thompson et al., 2010). The relative lack of effect of antagonists in the EPM is somewhat surprising, since studies using systemic administration of naloxone or naltrexone have shown increases in neuronal activation using cFos in the CEA and BNST suggesting some endogenous opioid tone (Carr et al., 1999; Gestreau et al., 2000; Veinante et al., 2003). Nevertheless, the opposing effects of MOR and DOR agonists on anxiety-related behaviors might explain the lack of effects using nonselective antagonists and/or changes in ENK expression in the EPM and similar tests (since ENK can act through both DOR and MOR).
Our results demonstrated that DAMGO injections into the CEA reduced in burying behavior, increased the latency to bury, reduced the number of animals showing burying behavior, and increased rearing during exposure to predator (ferret) odor (Wilson & Junor, 2008). Although DAMGO reduced burying behavior in the defensive burying task in a manner similar to the effects of anxiolytic compounds, this interpretation fails to encompass a potential shift in the full behavioral profile elicited during this test, including the increases in rearing and escape-type behaviors observed with DAMGO injections in the CEA. Similar behavioral shifts have been seen using perfusion of DAMGO into the CEA with microdialysis during exposure to a predator odor stress (Carrero, Kaigler, Hartshorn, Fadel, & Wilson, 2019). Thus, one potential explanation of the result in the defensive burying test is direct MOR activation in the CEA, perhaps via interactions with a distinct set of output neurons in the CEA, induced a shift toward more escape-like or avoidance behaviors rather than burying. This might also explain the decrease in open-arm time in the plus maze with DAMGO injections in the CEA (Wilson & Junor, 2008), since a shift in behavioral response toward avoidance might not only activate escape-type behavior to a predator (discrete) threat, but also avoidance of an unpredictable threatening environment (open arms of the maze). Perhaps MOR more directly activates rearing or escape-like behavioral outputs, rather than burying responses, and/or with endogenous ENK release both MOR and DOR might be activated with effects on different neuronal populations leading to different behavioral outcomes. Electrophysiological studies in amygdala slices (Table 1) have suggested that the effects of MOR agonists in CEA are dependent on the characterization of the cell type in this area (Chieng et al., 2006; Chieng & Christie, 2009; Finnegan et al., 2005; Zhu & Pan, 2004, 2005). Recordings from CEA neurons that selectively project to the vlPAG demonstrated that DAMGO decreased the frequency of IPSCs in approximately half of these neurons (Finnegan et al., 2005), and the PAG is known to be important for shaping the behavioral defensive responses to predator threat (see Assareh, Sarrami, Carrive, & McNally, 2016). Interestingly, injections of opioid antagonists into the CEA of morphine-dependent rats also induce a withdrawal profile that includes escapes and jumps (Calvino et al., 1979; Lagowska et al., 1978). Alternatively, inhibition of one population of CEA neurons with DAMGO could offset the balance of ENK-mediated versus CRF-mediated influences, since these two peptides have opposing effects on many behavioral outputs and are localized in distinct neurons in CEA (Day et al., 1999; Veinante et al., 1997), and MOR and CRF R1 receptors are colocalized in many CEA neurons (Jaferi & Pickel, 2009).
BLA and anxiety-related responses
In the BLA, several studies have demonstrated that chronic stressors can decrease the expression of ENK, but this effect shows considerable individual variation (Table 4). Thus, both chronic unpredictable stress and social defeat showed decreased ENK mRNA expression in BLA, and particularly posterior BLA, after stress, but only in the vulnerable population of rodents (Berube et al., 2013, 2014; Henry et al., 2017, 2018). Rats or mice divided into resilient and susceptible phenotypes based on behaviors associated with a chronic social defeat showed decreased ENK mRNA expression in BLA (specifically BLp), but only in susceptible rats, compared with controls and resilient groups (Berube et al., 2013; Henry et al., 2017, 2018). No changes were seen in DYN mRNA in the amygdala (Berube et al., 2013). Decreased ENK mRNA expression was also seen in the posterior BLA (BLp) of rats vulnerable to chronic unpredictable stress characterized by decreased social interaction, decreased sucrose intake, and differential EPM behaviors (Berube et al., 2014). Decreasing ENK mRNA expression in BLp using lentivirus-mediated gene transfer decreased social interaction and decreased open-arm time ratios in the EPM but did not alter sucrose intake, suggesting the changes in BLA ENK could be responsible for the individual differences in stress susceptibility (Berube et al., 2014). These changes in ENK might induce behavioral changes through either MOR or DOR, since BLA injections of the MOR antagonist, CTOP, and the DOR antagonist, naltrindole, decreased time in the lit compartment of the LD box suggesting an anxiogenic effect (Narita et al., 2006), and the nonselective antagonist naltrexone induced avoidance of the open arms in the EPM (Burghardt & Wilson, 2006), much like ENK downregulation by stressors. Similarly, injections of the DOR agonist, KNT-127, into the BLA dose-dependently decreased anxiety-related measures (open-arm time/entries) in the EPM, and increased center time in the OF, without affecting locomotion in either test. These effects were blocked by naltrindole, although the DOR antagonist in the BLA had no effect on EPM behaviors alone (Sugiyama et al., 2018). However, since ENK mRNA is located in glutamatergic projection neurons of the BLA (Poulin et al., 2008; Zhang & McDonald, 2016), many of which project to CEA, this might suggest that effects of this decreased ENK mRNA expression are seen as reduced ENK release in the projection sites of BLA PNs to modulate stress-related behaviors. Thus, as proposed by Drolet and colleagues, it appears that a decrease in ENK transmission from the BLA is a maladaptive mechanism that may help determine individual susceptibility or resilience during chronic stress (Henry et al., 2017).
Cells of the BLA express KOR mRNA, but a low density of DYN+ axons (Mansour et al., 1996; Mansour, Fox, Meng, et al., 1994). However, the effects of KOR agonists and antagonists in the BLA are consistent with the notion that the DYN/KOR system induces an anxiogenic state, and the BLA is a critical site in mediating these anxiogenic effects. BLA injections of DYN decreased time in the lit compartment of the LD box, suggesting an anxiogenic effect (Narita et al., 2006), while injection of the KOR antagonist, JDTic, into the BLA, but not the CEA, produced anxiolytic effects (increased open-arm time) in the EPM (Knoll, Meloni, Thomas, Carroll, & Carlezon, 2007). Similarly, deleting presynaptic KOR in the BLA using viral approaches increased EPM open time, without a change in distance traveled (Crowley et al., 2016). BLA injections of the KOR antagonist, norBNI, also blocked the anxiogenic effects of both CRF (i.c.v. administration) and swim stress in the EPM (Bruchas et al., 2009). Using immunohistochemistry with an antibody to the phosphorylated KOR protein to assess in vivo DYN release and KOR activation, CRF administration (i.c.v.) increased KOR activation significantly in BLA, slightly in BNST, but not at all in the CEA, (Land et al., 2008), and this effect in the BLA was blocked by norBNI (Bruchas et al., 2009). Both norBNI injections and cre-induced conditional KOR knockdown in the BLA blocked yohimbine or stress-induced reinstatement of nicotine conditioned place preference (CPP) and associated cFos expression in the BLA. Similar attenuation of these reinstatement effects using M4 inhibitory DREADDs under the CAMKII promoter suggested that this stress effect might have been mediated via changes in BLA PN activity (Nygard et al., 2016). KOR agonists in the BLA inhibit field potentials and increase IPSCs on BLA PNs via increases in presynaptic GABA release (Huge et al., 2009; Przybysz et al., 2017), and KOR activation decreased the EPSCs in BNST evoked by optogenetically stimulating the BLA (Crowley et al., 2016). Thus, it is possible that some anxiogenic effects of DYN and KOR activation might be mediated via regulation of the BLA projections to BNST. This is supported by the fact that optogenetic stimulation of the BLA-BNST pathway induced anxiolytic effects in the open field and reduced the anxiogenic effects of systemic administration of the KOR agonist U50,455 (Crowley et al., 2016).
AMYGDALAR OPIOID REGULATION OF ASSOCIATIVE LEARNING PROCESSES AND CONDITIONED FEAR
Human imaging and conditioned responses
Amygdalar opioid processes have also been shown to mediate associative learning processes, including conditioned fear responses. As mentioned earlier, in humans, a conditioning protocol consisting of CS+ presentation paired with a thermal pain stimulus showed that naloxone administration attenuated the amygdalar changes in activation to the repeated CS+ presentation, suggesting that habituation to the CS+ in the amygdala was opioid dependent and associated with behavioral conditioned responses (Eippert et al., 2008).
CEA: Associative learning and conditioned fear
Studies in rodents and rabbits have similarly consistently suggested that activation of the amygdalar opioid system, particularly MOR activation in the CEA, impairs the acquisition and retention of conditioned responses (Table 5) (Gallagher et al., 1981, 1982; Hernandez et al., 1990). Injections of the mixed opioid agonist levorphanol, morphine, or MOR agonists impaired acquisition of Pavlovian conditioned fear and heart rate (HR) responses (Gallagher et al., 1981, 1982; Westbrook et al., 1997) and also impaired both acquisition and retention in passive avoidance or inhibitory avoidance paradigms (Gallagher & Kapp, 1978; Good & Westbrook, 1995; Ragozzino & Gold, 1994). Levorphanol, but not its inactive enantiomer, injected into the CEA before training attenuated acquisition of conditioned HR responses (Gallagher et al., 1981), while post-training amygdalar injections decreased retention of passive avoidance (Gallagher & Kapp, 1978). Additional studies showed similar attenuation of acquisition of conditioned HR deceleration with the MOR agonist DALA (d-ALa2 Met5-enkephalinamide), but not DADLE, injected into the CEA suggesting greater involvement of MOR, compared with DOR, in this effect (Gallagher et al., 1982). Bilateral amygdala injections of END after training also dose dependently impaired retention 1 week later in a footshock avoidance paradigm (Flood et al., 1992). Pretraining injections of morphine in the CEA, but not the BLA, decreased acquisition of conditioned freezing (Westbrook et al., 1997), as well as both passive avoidance in a step-down paradigm and conditioned hypoalgesia following hotplate exposure (Good & Westbrook, 1995). Interestingly, the effects on conditioned freezing were seen with unilateral injections into either the left or right CEA (Westbrook et al., 1997). In many of the learning paradigms the effects of opiate agonists were blocked by naloxone (Gallagher et al., 1981, 1982; Gallagher & Kapp, 1978; Good & Westbrook, 1995), but naloxone alone was also able to increase retention in a passive avoidance task (Gallagher & Kapp, 1978) and enhance the acquisition of conditioned HR responses (Gallagher et al., 1981), suggesting endogenous opioid release during these tasks. Systemic administration of naloxone similarly enhanced acquisition of conditioned HR responses (bradycardia) as well as increased the multiunit CEA responses (decreased activity) during CS+ presentation and conditioning. The magnitude in the shifts in multiunit activity in the CEA and deceleration of HR in the naloxone group were correlated, suggesting opioid regulation of CEA activity might control vagal outputs inducing HR deceleration (Hernandez et al., 1990).
TABLE 5.
Amygdalar opioid regulation of associative learning and conditioned fear.
| Subregion | Manipulation or injection | OR | Behavioral test or response measured | Subjects | Summary of findings | Overall opiate effect | Authors |
|---|---|---|---|---|---|---|---|
| Amygdala | |||||||
| AMY | Morphine sulfate, glucose injections | MOR | Inhibitory avoidance, footshock sensitivity, spontaneous alternation in Y maze | Rats (male) | Bilateral amygdala injections of morphine before training decreased latencies in an inhibitory avoidance task (impaired learning). No effects of morphine on spontaneous alternation in a Y maze task | Morphine in amygdala impairs acquisition of inhibitory avoidance | Ragozzino and Gold (1994) |
| AMY | END | END | Footshock avoidance in T-maze | Mice (male) | Bilateral amygdala injections of END after training dose-dependently impaired retention in a footshock avoidance paradigm | END impairs consolidation of footshock avoidance | Flood, Garland, and Morley (1992) |
| Central amygdala | |||||||
| CEA | Conditioned fear | ENK | mRNA for ENK and CRF | Rats (male) | Return to a conditioned context increased mRNA for ENK in the CL and CLC, but not CM, compared to conditioned group without re-exposure or non-shocked control group. No differences were seen in CRF mRNA | Re-exposure to conditioned context increases ENK mRNA in CL/CLC | Petrovich, Scicli, Thompson, and Swanson (2000) |
| CEA | ENK knockdown (virus-mediated gene transfer) | ENK | Contextual fear conditioning | Rats (male) | Decreased ENK mRNA expression in CEA using lentivirus-mediated gene transfer reduced freezing during acquisition, but not the expression of contextual fear | Decreased CEA ENK expression decreased acquisition, but not expression, of contextual fear | Poulin et al. (2013) |
| CEA | Levorphanol | OR | Passive avoidance | Rats | The mixed opioid agonist levorphanol in the amygdala after training decreased retention in a passive avoidance task, while naloxone alone increased retention and blocked the effects of levorphanol | Opioid agonist in CEA impairs retention of passive avoidance | Gallagher and Kapp (1978) |
| CEA | Levorphanol, levorphanol dextrorphan (inactive), naloxone | OR | Conditioned heart rate (HR) | Rabbits | The opioid agonist levorphanol, but not the inactive enantiomer levorphanol dextrorphan, injected into the CEA before training attenuated acquisition of conditioned HR. Naloxone enhanced acquisition of conditioned HR responses and attenuated the effects of the agonist | Opioid agonist in CEA impairs acquisition of conditioned responses | Gallagher, Kapp, McNall, and Pascoe (1981) |
| CEA | DADLE, DALA | MOR, DOR | Conditioned heart rate (HR) | Rabbits | DALA, but not DADLE (DOR), in CEA attenuated acquisition of conditioned HR in rabbits. Results suggest greater involvement of MOR, compared with DOR, in this effect | MOR agonist in CEA impairs acquisition of conditioned heart rate responses | Gallagher, Kapp, and Pascoe (1982) |
| CEA | Naloxone (i.v.) vs. saline | Conditioned heart rate (HR), CEA unit activity | Rabbits | Naloxone increased baseline HR and decreased the initial orienting response to the conditioned stimulus (CS), but enhanced acquisition of conditioned HR responses (bradycardia) and increased the multiunit CEA responses (decreased activity) during CS+ presentation and conditioning. The shifts in CEA multiunit activity and HR deceleration with naloxone were correlated, suggesting opioid regulation of CEA activity might control vagal outputs inducing HR deceleration | Naloxone enhanced acquisition of conditioned responses | Hernandez, Powell, and Gibbs (1990) | |
| CEA, BLA | Morphine, naloxone (unilateral) | MOR | Passive avoidance (step-down), hot-plate and formalin nociception | Rats (male) | Unilateral morphine injections into CEA, but not BLA, before hotplate exposure decreased acquisition of passive avoidance (step-down) and hypoalgesia. Morphine in the CEA attenuated the expression of step-down passive avoidance and formalin-induced hypoalgesia, but not hotplate paw lick latencies, when administered before the retention tests | Morphine in CEA impairs acquisition and retention of passive avoidance | Good and Westbrook (1995) |
| CEA | Morphine | MOR | Fear conditioning (context, cue) | Rats (male) | Pretraining morphine injections in the CEA decreased freezing in the conditioned context and in response to a conditioned cue | Morphine in CEA impairs acquisition of conditioned fear | Westbrook, Good, and Kiernan (1997) |
| Basolateral amygdala | |||||||
| BLA | KNT-127, naltrindole | DOR | Contextual fear conditioning | Rats (male) | Injections of the DOR agonist KNT-127 into the BLA before re-exposure to the context blocked the expression of contextual fear and reduced recall 24h later. This effect was antagonized by naltrindole, but the DOR antagonist alone did not alter conditioned fear responses | DOR agonist in BLA blocks contextual fear | Sugiyama et al. (2018) |
| BLA | JDTic | KOR | Fear-potentiated startle (FPS), EPM, KOR and pDYN mRNA | Rats (male) | Injection of the KOR antagonist JDTic into either BLA or CEA decreased fear potentiated startle, without altering baseline startle. The light-shock pairing used for FPS also increased KOR mRNA in the BLA, but not the CEA, compared with light-alone levels, and better fear extinction was associated with reduced KOR mRNA expression compared to extinction resistant rats | A KOR antagonist decreased fear-potentiated startle, and enhanced fear extinction was associated with decreased KOR mRNA expression | Knoll et al. (2011) |
| Intercalated nuclei | |||||||
| IN | Lesioning of MOR-containing ITC neurons | MOR | Extinction learning and retention | Rats (male) | Lesions of MOR-containing ITC neurons using dermorphin-conjugated saporin toxin decreased the expression of extinction, but not within-trial extinction learning. No changes were seen in acquisition of conditioned freezing or in OF behaviors | Lesions of MOR-containing IN neurons impaired expression of extinction learning | Likhtik, Popa, Apergis-Schoute, Fidacaro, and Pare (2008) |
Opioid involvement in conditioned fear responses is further suggested by increases in mRNA for ENK in the CL and CLC, but not the CM, upon return to a conditioned context while no differences were seen in CRF mRNA (Petrovich et al., 2000). This is consistent with the anatomical distinction between ENK- and CRF-containing neurons in the CEA (Day et al., 1999; Veinante et al., 1997). Although these effects could be more related to the anxiogenic effects of re-exposure to the context, they reinforce the role of the CL and CLC enkephalinergic systems in learned fear responses. Surprisingly, however, knockdown of ENK expression in the CEA using virus-mediated gene transfer decreased freezing during acquisition, but not the expression, of contextual fear responses (Poulin et al., 2013). This might suggest that ENK systems in CEA might be more responsive to the aversive nature of the initial shock presentations, and/or adaptive responses in the CEA influence downstream targets such as PBN or PAG associated with freezing and responses to nociceptive stimuli. Additional studies have also demonstrated that lesions of MOR-containing IN neurons using dermorphin-conjugated saporin toxin decreased the expression of extinction, but not within-trial extinction learning. No changes were seen in the acquisition of conditioned freezing or in open field behaviors in animals with lesions of MOR-containing IN neurons in this study (Likhtik et al., 2008).
BLA: Associative learning and conditioned fear
In contrast to CEA, influences of KOR and DOR in the BLA have been implicated in learned fear responses (Table 5). Injections of the DOR agonist, KNT-127, into the BLA before reexposure to the context blocked the expression of contextual fear and reduced recall 24h later. This DOR-induced reduction in conditioned fear was antagonized by naltrindole, although the DOR antagonist alone did not alter conditioned fear responses (Sugiyama et al., 2018). In contrast, changes in KOR mRNA expression in the BLA have been associated with fear-potentiated startle responses, with higher levels of fear-potentiated startle being associated with higher KOR mRNA expression (Knoll et al., 2011). Injections of the KOR antagonist, JDTic, into the BLA or CEA decreased fear-potentiated startle without affecting baseline startle (Knoll et al., 2011), potentially suggesting a role of DYN acting through KOR in the BLA (or CEA) in modulating conditioned startle responses. These results are consistent with the localization of both DYN and KOR in neurons and fibers in the BLA and CEA (Mansour et al., 1996; Mansour, Fox, Burke, et al., 1994; Mansour, Fox, Meng, et al., 1994), although DYN-containing neurons in the CL are denser than in BLA.
AMYGDALAR OPIOIDS AND ETHANOL EFFECTS
Amygdala opioids and ethanol
Human imaging studies using PET with [11C]carfentanil (MOR) or [11C]methylnaltrindole (DOR), demonstrated that MOR, but not DOR, binding potential in the amygdala was increased during alcohol abstinence (day 5) compared with healthy controls (Weerts et al., 2011) (Table 2). A variety of preclinical studies also suggest that many ethanol effects involve central opioid systems, including the opioid circuits in CEA, BLA, or BNST (Table 6). Studies have demonstrated that the systemic administration of ethanol via different routes, either acutely or sub-chronically (<7 days), leads to neuronal activation in ENK-containing neurons (Chang et al., 2014; Criado & Morales, 2000), enhanced density of ENK mRNA-expressing neurons in CEA (Chang et al., 2014), and altered expression of opioid peptides or receptors in the amygdala (D’Addario, Caputi, Ekstrom, et al., 2013; Granholm et al., 2018; Korpi et al., 2017); these effects occur predominantly in the CEA (especially in CL and CLC). Interestingly, acute intragastric ethanol administration increased activation of ENKir neurons in CEA in a manner similar to both nicotine and fat (Chang et al., 2014). Voluntary ethanol consumption also increased ppENK mRNA (but not ppDYN mRNA), in CEA and IN (but not Copm or BLA). In a microdialysis study, acute ethanol administration increased the release of both END and DYN, but not ENK, in the CEA (Lam et al., 2008). Ethanol also induced increases in pDYN and KOR gene expression in the amygdala, perhaps via epigenetic mechanisms (D’Addario, Caputi, Ekstrom, et al., 2013; D’Addario, Caputi, Rimondini, et al., 2013). A longer episodic binge-like exposure to ethanol in adolescent rats, however, decreased Met-ENK-Arg6Phe7 (MEAP) content in the amygdala, without altering DYN or END levels (Granholm et al., 2018), while a perinatal (three-trimester model) ethanol exposure increased met-ENKir in the CEA (Lugo et al., 2006).
CEA opioids and ethanol
Electrophysiological studies recording from brain slices in the CM have demonstrated that ethanol enhances mIPSCs and IPSCs evoked by stimulation of the CL through presynaptic increases in GABA transmission (Gilpin et al., 2014; Kang-Park et al., 2007, 2009, 2013). This is consistent with studies showing that acute systemic ethanol administration inhibits CEA single-unit activity (Naylor et al., 2001). Studies in slices suggest that these ethanol effects on evoked GABA-mediated neurotransmission are enhanced by blocking DOR activation using either the antagonist, naltrindole, or DOR knockout mice. Interestingly, the DOR inverse agonist, ICI174864, had no effect alone but increased the effect of ethanol on mIPSCs (Kang-Park et al., 2007). The same group demonstrated that MOR knockout mice showed similar ethanol effects (Kang-Park et al., 2009), suggesting that ethanol effects on the ENK-containing GABAergic projection from CL to CM are likely to involve DOR, not MOR. Further, in rats exposed to chronic intermittent ethanol administration in a conditioned place preference procedure (CPP), DPDPE produced inhibition of GABA IPSCs and eEPSCs, indicating enhanced DOR-mediated inhibition of GABA and glutamate release that was not seen in control rats, or with single acute ethanol administration (Bie et al., 2009a). This suggests that DOR regulation in the CM might induce a weak inhibition of GABA release in control conditions, but that DOR related effects might be more pronounced and include inhibition of presynaptic glutamate and GABA release after chronic ethanol (or morphine) administration.
In addition, two groups have demonstrated that ethanol effects in this same CL to CM projection are influenced by KOR manipulations. The first study demonstrated that decreasing KOR activation with norBNI perfusion or using KOR knockout mice both enhanced ethanol-induced increases in evoked GABA transmission in CM (Kang-Park et al., 2013), suggesting ethanol’s effects might involve KOR-mediated inhibition of GABA release. A second study, however, indicated that not only did norBNI prevent ethanol’s effects on evoked IPSCs in CM, but that ethanol could also antagonize the ability of KOR agonists to decrease IPSCs (Gilpin et al., 2014). The inconsistent findings between these two reports might have been due to differences in the magnitude of ethanol effects between the studies, and/or potential differences between rats and mice (for discussion see Gilpin et al., 2014). Interestingly, both studies suggested that there was a tonic inhibitory GABA tone induced by KOR activation in this region (demonstrated by effects of norBNI alone) and that ethanol modulated KOR-induced inhibition induced by DYN in this amygdalar projection.
Opioid agonists or antagonists in the CEA can modulate various ethanol-induced behaviors, but these effects are predominantly seen during self-administration protocols that have a learning component (such as operant responding for ethanol) and/or withdrawal-induced reinstatement of ethanol self-administration. This is perhaps not surprising given the role of opioid processes in amygdala (see above) in associative learning. Given that systemic ethanol injections activate ENK-containing neurons in the CL (Chang et al., 2014; Criado & Morales, 2000), it is not surprising that ENK-overexpression in the CEA enhanced the anxiolytic effects of systemically administered ethanol in the EPM (Wilson et al., 2003), similar to the influences of ENK overexpression on anxiolytic actions of benzodiazepines (Kang et al., 2000). Although large amygdalar injection volumes of naltrexone also attenuated the anxiolytic effects of ethanol in the EPM (Wilson et al., 2003), this inhibition was likely the result of a reduction in overall open arm behaviors due to effects in the BLA, rather than a selective inhibition of the ability of ethanol to enhance open-arm time. Subsequent studies demonstrated that more discrete injections of naltrexone either in the CEA or the BLA failed to block the anxiolytic actions of ethanol in the EPM (Burghardt & Wilson, 2006). Given that the effects of ENK overexpression in CEA on diazepam-induced anxiolysis were mediated via DOR activation (Primeaux et al., 2006), and that both DOR and KOR influenced ethanol-induced increases in GABA neurotransmission in the projection from CL to CM (Gilpin et al., 2014; Kang-Park et al., 2013; Kang-Park et al., 2007), the lack of naltrexone effects in the CEA could have been due to nonselective antagonism of all CEA opioid receptors. This is supported by the fact that both naltrexone and the DOR antagonist naltrindole in the CEA decreased ethanol conditioned place preference (CPP) without modifying baseline behaviors in the test (Bie et al., 2009a). There is also a correlation between DOR (Oprd1) gene expression in the amygdala and voluntary ethanol intake, although this is influenced by early-life maternal separation stress (Granholm et al., 2017).
In ethanol self-administration paradigms, both MOR antagonists (CTOP, methylnaloxonium) and DOR antagonists (naltrindole) in the CEA decreased operant responding for ethanol (Heyser et al., 1999; Hyytia & Kiianmaa, 2001). A cocktail of CTOP/naltrindole or the newer antagonist, nalmefene, in CEA also blocked ethanol consumption, although the effects of CTOP/naltrindole were only seen in nondependent rats (Kissler et al., 2014). Ethanol dependence induced with forced exposure to ethanol vapor was associated with increased DYNir in the CLC and increased DYN stimulation of GTPγS binding in the amygdala (Kissler et al., 2014). In addition, the enhanced ethanol self-administration seen during withdrawal in dependent rats was attenuated by CEA injections of the KOR antagonist norBNI, although this treatment did not block the physiological signs of ethanol withdrawal (Kissler et al., 2014; Kissler & Walker, 2016). This suggests that at least some of the motivational effects induced by ethanol withdrawal might be mediated by the upregulation of DYN/KOR systems in the CEA (see Anderson & Becker, 2017).
BLA opioids and ethanol
A role of opioid-dependent mechanisms in both the BLA (Djouma & Lawrence, 2002; Marinelli et al., 2007, 2010) and the BNST (Erikson et al., 2018; Le et al., 2018) has also been implicated during ethanol withdrawal in rodents, including the reinstatement of ethanol self-administration processes during abstinence (Table 6). Ethanol withdrawal increased MOR density in the lateral amygdala, with 125I-FK 33,824-binding levels increased in animals during ethanol withdrawal compared to ethanol-consuming groups in a two-bottle choice or non-ethanol exposed controls (Djouma & Lawrence, 2002). In addition, reexposure to a context associated with ethanol self-administration induced cFos in the BLA, but not the CEA. Not only was this BLA activation attenuated with systemic administration of naltrexone, but naloxone administration in the BLA dose dependently decreased lever presses for ethanol during reexposure to a context associated with ethanol self-administration (Marinelli et al., 2007, 2010).
BNST opioids and ethanol
In the BNST, expression of KOR mRNA (Oprk1 gene), but not pDYN, was increased in ethanol-dependent rats self-administering ethanol, compared with nondependent rats. BNST injections of norBNI attenuated the enhanced ethanol consumption during withdrawal but did not alter physiologic measures or anxiety-related behaviors in the EPM (Erikson et al., 2018). Further, reinstatement of ethanol self-administration induced by systemic injections of the KOR agonist, U50,488, increased cFos in the BNST. Not only was neuronal activation induced by KOR-induced reinstatement blocked by intra-BNST injections of norBNI, but BNST injections of the KOR agonist partially reinstated ethanol self-administration (Le et al., 2018). An intriguing possibility is that the anxiogenic effects of DYN and KOR activation during ethanol withdrawal might be mediated via regulation of the BLA projections to BNST, since optogenetic stimulation of the BLA-BNST pathway can reduce the anxiogenic effects of systemic administration of the KOR agonist U50,455 (Crowley et al., 2016). Moreover, whether changes in BLA during ethanol withdrawal are related to the decreases in ENK expression (which could result in receptor upregulation), such as those seen during chronic stressors in susceptible individuals (Henry et al., 2017), remains to be assessed.
AMYGDALAR OPIOIDS IN OPIATE DEPENDENCE AND WITHDRAWAL
Amygdala and opiate dependence
Not surprisingly, amygdalar opioid systems are involved in several aspects of opiate or opioid dependence and withdrawal. In human imaging studies (Table 2), opioid-dependent individuals showed reduced amygdala volumes as well as changes in functional connectivity when compared with healthy controls (Upadhyay et al., 2010). A common single nucleotide polymorphism (SNP) in the MOR gene [OPRM1 A118G (rs1799971)] shifts MOR expression and drug dependence phenotypes, and PET studies with [11C]carfentanil indicate that G allele carriers showed lower MOR-binding potential in amygdala compared with wild-type A allele carriers (Domino et al., 2015; Ray et al., 2011). Human and animal studies are linking this genetic polymorphism in amygdalar MOR not only to stress susceptibility (Briand et al., 2015) but also to the behavioral responses induced by other addictive drugs including nicotine (Domino et al., 2015; Ray et al., 2011).
A variety of studies using markers of neuronal activation (cFos, arc) in animals have indicated that naloxone-precipitated withdrawal or conditioned place aversion in morphine-dependent animals can activate the central and basolateral amygdala, especially the CM, IN, MEA, and both the dorsal and ventral BNST. None of these studies, however, identified the neuronal populations activated, although BLA activation was seen in GAD-negative neurons, suggesting this was specific to glutamatergic PNs (Frenois, Cador, Caille, Stinus, & Le Moine, 2002; Frenois, Stinus, Di Blasi, Cador, & Le Moine, 2005; Jin et al., 2005; Lucas et al., 2008; Lucas, Frenois, Cador, & Le Moine, 2012; Stornetta, Norton, & Guyenet, 1993). The most consistent activation during withdrawal is seen in the CM, where naloxone-precipitated withdrawal in morphine-dependent rats induced cFos specifically in ENKir neurons, but not CRFir neurons, in the CEA and the lateral BNST. Naloxone administration in controls induced a lower level of neuronal activation in these areas in both CRFir and ENKir neurons (Veinante et al., 2003) (see Table 7). Heroin self-administration decreased MOR-, but not DOR-, stimulated GTPγS binding in amygdala, but did not alter the number of receptors (3H-naloxone binding) (Sim-Selley, Selley, Vogt, Childers, & Martin, 2000), although another study failed to see changes in amygdalar MOR stimulated GTPγS binding after chronic morphine administration (Sim et al., 1996). Increased levels of DYN were seen in the amygdala following morphine treatment and withdrawal (Rattan et al., 1992).
CEA and opiate dependence
Microinjection studies have further implicated the opioid systems in the amygdala in morphine dependence and withdrawal (Table 7). Although morphine injections in the CEA (van der Kooy et al., 1982) and LA (Olmstead & Franklin, 1997a, 1997b) failed to induce CPP, injections of the opioid antagonist, methylnaloxonium, induced conditioned place aversion in morphine-dependent rats (Stinus et al., 1990). Although animals in this study did not exhibit physical signs of withdrawal, earlier studies suggested that injections of naloxone into the amygdala could induce withdrawal symptoms in morphine tolerant rats, including wet dog shakes, chewing, teeth chattering, and diarrhea, although jumps were only seen with injections in the CEA (Calvino et al., 1979; Lagowska et al., 1978). DAMGO injections (unilateral) in the CEA also attenuated the enhanced acoustic startle responses seen during naltrexone-induced withdrawal from acute morphine administration (Cabral et al., 2009).
Finally, several physiological studies have demonstrated changes in opioid regulation of GABA- or glutamate-mediated responses in CEA following the induction of morphine tolerance or withdrawal. In control slices, MOR activation (acute morphine, DAMGO) decreased eIPSCs in ~50% of neurons and decreased mIPSCs, while the MOR antagonist, CTOP, increased mIPSC frequency, suggesting tonic opioid tone at MOR. Many of these responses were unaltered in slices from morphine-dependent rats, although the inhibitory effects of MOR agonists were blunted suggesting the development of tolerance to some MOR effects (Bajo et al., 2011). In contrast, in slices from morphine-dependent rats during naloxone-precipitated withdrawal, GABA inhibition (mIPSCs) was increased in CEA compared with controls (Bajo et al., 2014). In rats exposed to morphine-induced conditioned place preference, morphine treated rats also showed increased glutamate synaptic strength (eEPSCs) and enhanced glutamate release in CEA compared with controls. Interestingly, this effect was associated with enhanced DOR effects in morphine-treated slices, with the DOR agonist, DPDPE, decreasing eEPSCs in 70% of CEA neurons (compared with no effect in control slices) through inhibition of presynaptic glutamate release. Consistent with this effect, DOR expression was enhanced in a synaptosomal preparation (Bie et al., 2009b), perhaps suggesting DOR-mediated adaptations to morphine-induced increases in glutamate synaptic strength. Similarly, chronic morphine administration increased the number of cells inhibited by the DOR agonist deltorphin II by activating a GIRK current from ~35% of neurons (controls) to 69% of neurons (morphine-dependent). In control slices, DOR-responsive neurons also responded to the MOR agonist DAMGO, but chronic morphine increased the number of neurons responding to the DOR agonist alone (from none to 31%), and decreased the number of neurons responding to DAMGO alone (from 62% to 15%) (Chieng & Christie, 2009). In neurons projecting from CM to PAG, the number of DOR responsive neurons increased dramatically from 29% to 86%, with a corresponding decrease in MOR responsive neurons (Chieng & Christie, 2009). In an early study in anesthetized rats, the DOR antagonist, ICI174864, had minimal effects on glutamate-driven firing rats, but in morphine-dependent rats, systemic administration of the DOR antagonist, ICI174864, increased firing rates suggesting an enhancement of DOR-mediated effects (Freedman & Aghajanian, 1985). These studies also indicated that opiate dependence/withdrawal involved the noradrenergic projections from the brainstem since cells that responded to morphine also responded to the α2 agonist clonidine and the effects of opioid antagonists on CEA firing rates could be blocked with the local iontophoretic application of clonidine. Noradrenergic mechanisms in the BNST have also been implicated in opiate withdrawal (Aston-Jones, Delfs, Druhan, & Zhu, 1999; Delfs, Zhu, Druhan, & Aston-Jones, 2000; Van Bockstaele, Qian, Sterling, & Page, 2008), although the role of opioid receptors in BNST in mediating these effects has not been elucidated.
Overall, however, these studies suggest that aversive effects of morphine withdrawal may involve adaptations in the opioid system in the CEA, and potentially BLA or BNST, including activation of ENKir neurons in the CEA and a switch in physiological effects of ENK from MOR to DOR dependent. This might be accomplished by enhanced terminal trafficking of DOR to presynaptic sites to inhibit presynaptic glutamate release and enhanced postsynaptic DOR inhibition of CM neurons projecting to PAG. Adaptations in the CM projections to PAG likely play a critical role in abstinence signs associated with morphine dependence. The aversive and dysphoric effects of morphine (or ethanol) abstinence may also involve interactions between opioid systems and CRF neurons since they represent different neuronal populations in the CEA (Day et al., 1999; Veinante et al., 1997). Neurons in CL and the anterolateral BNSTL exhibit colocalization of MORs and CRF-R1 receptors (Jaferi & Pickel, 2009) and have opposing physiological effects on these cells. Opioid withdrawal may include upregulation of the CRF system in neurons that have been chronically suppressed by opioids (Iredale et al., 2000; Skelton et al., 2007). Further, many CEA neurons co-express DYN and CRF mRNA, and many of these cells project to the LC (Reyes et al., 2008), suggesting that there is convergence between ENK and CRF systems on some neurons that may enhance DYN/CRF projections to LC and modulate noradrenergic processes associated with opioid withdrawal (Aston-Jones et al., 1999; Delfs et al., 2000; Van Bockstaele et al., 2008).
SUMMARY: THE AMYGDALAR OPIOID SYSTEM
As summarized above, ENK and DYN are expressed in multiple subregions of the amygdala, as are MOR, DOR, and KOR. As summarized in Fig. 6, physiological studies have suggested that activation of opioid receptors can induce both postsynaptic inhibition of neurons, as well as presynaptically inhibiting both GABA and glutamate release in specific subnuclei of the amygdala. More critically, the ability of MOR, DOR, and KOR to regulate these presynaptic inputs and postsynaptic responses are dependent on the cell types in these regions, and particularly in the CM where MOR and KOR effects are generally seen in distinct neuronal populations. Further, under control conditions, most of the responses are associated with MOR, rather than DOR, and are seen in most of the cells projecting to brainstem centers such as PBN or PAG, as well as BNST. Emerging evidence, however, suggests that situations such as chronic ethanol or morphine administration enhance the responses to DOR and KOR in the extended amygdala.
FIG. 6.

Overview of opioid mediated effects in the amygdala. Central figure shows main electrophysiological presynaptic and postsynaptic effects induced by activation of mu (MOR), delta (DOR), or kappa (KOR) receptors in the basolateral/lateral amygdala (BLA), central amygdala (CEA), or intercalated nuclei (IN). Primary functional effects of receptor activation in these regions are summarized, implicating the role of amygdalar opioids in nociception, stress and anxiety-related responses, associative learning and conditioned fear, ethanol consumption or withdrawal, and opiate dependence. GLUT, glutamate.
The effects of opiods on distinct populations of neurons in amygdala subregions seen in rodents may explain the ability of pharmacological or genetic manipulations to have distinct effects in different behavioral paradigms testing nociceptive, anxiety-related or fear responses, and drug effects (Fig. 6). Human imaging studies have suggested the role of amygdalar opioids in affective states, pain, and ethanol or opiate dependence as well as related disorders such as PTSD and depression. As seen in Fig. 6, rodent studies have similarly implicated opioids in regulation of nociceptive responses, unconditioned and conditioned fear or anxiety-related responses, and changes seen in ethanol and opiate dependence. Overall, these data suggest that the MOR activation in the CEA seems to help shift the behavioral responses seen in certain anxiogenic conditions, including nociceptive challenges and ethanol/opiate withdrawal, likely through effects on selected CEA neurons that project to brainstem centers such as PBN and PAG that are modulated by MOR activation. These responses may also be influenced by an enhanced influence of ENK effects through DOR, as well as increased DYN/KOR effects that may be in unique cell populations. Since ENK and CRF are also found in distinct populations of CEA, with some cells expressing both MOR and CRFR1, this suggests that these two peptide systems converge onto neurons in the CEA, and the balance between ENK and CRF systems likely regulate CEA outputs. Moreover, DYN is colocalized with CRF in many cells, which may suggest the dysphoric state during nociceptive stimuli, stress, or opiate/ethanol abstinence is related to enhanced activity in these neurons; many of these CRF/DYN cells project to the LC and provide a link between noradrenergic regulation and opiate or opioid abstinence. ENK expression in the BLA has also been tied to susceptibility to chronic stress, and opioid receptors in this region regulate conditioned responses and antinociception; opioid regulation via MOR, DOR, or KOR of BLA projections to both CEA and BNST may play important roles in these responses. Nevertheless, what remains unknown is how opioid peptides, and their receptors, are localized in specific neuronal phenotypes in the amygdala subregions, and how receptor activation regulates these specific circuits within the amygdala to drive functional outcomes. Utilizing the new tools available to drive specific neuronal phenotypes and circuits, such as optogenetics and chemogenetics, will help resolve the complex opioid regulation in the amygdala circuitry. The emerging roles of DOR and KOR, as well as the amygdala opioid system in susceptibility to chronic stress, however, open new avenues for understanding and treating human disorders ranging from stress-related disorders to chronic pain and alcohol or opiate/opioid abuse.
Acknowledgments
We have received funding support for MAW from VA Merit Awards I01 BX001374, IO1 BX001804 and I21 BX002664 as well as funding support for AJM from R01 MH104638.
Abbreviations
- AHA
amygdalohippocampal area
- AMG
amygdala
- BL
basolateral amygdala nucleus
- BLA
basolateral amygdala complex
- BLa
anterior basolateral nucleus
- BLp
posterior basolateral nucleus
- BLv
ventral basolateral nucleus
- BM
basomedial nucleus
- BMa
anterior basomedial nucleus
- BMp
posterior basomedial nucleus
- BNST
bed nucleus of the stria terminalis
- BNSTL
lateral BNST
- BNSTM
medial BNST
- BZ
benzodiazepine
- rCBF
cerebral blood flow
- CCK
cholecystokinin
- CEA
central part of amygdala
- CL
lateral subdivision of central nucleus
- CLC
lateral capsular subdivision of central nucleus
- CM
medial subdivision of central nucleus
- Coa
anterior cortical nucleus
- Copl
posterolateral cortical nucleus
- Copm
posteromedial cortical nucleus
- CPA
conditioned place aversion
- CPP
conditioned place preference
- CR
calretinin
- CRF
corticotropin releasing factor
- CTAP
Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2
- CTOP
d-Pen-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2
- DAB
diaminobenzidine
- DADLE
d-Ala2, d-Leu5-enkephalin
- DALA
d-ALa2 Met5-enkephalinamide
- DAMGO
[d-Ala2, NMe-Phe4, Gly-ol5]-enkephalin
- DOR
delta opioid receptor
- DPDPE
[d-Pen 2,5]-enkephalin
- DYN
dynorphin
- DYNir
dynorphin immunoreactivity
- eEPSC
evoked excitatory postsynaptic currents
- eIPSC
evoked inhibitory postsynaptic currents
- END
β-endorphin
- ENK
enkephalin
- ENKir
enkephalin immunoreactivity
- EPM
elevated plus maze
- EPSC
excitatory postsynaptic current
- ERK1/2
extracellular signal-regulated kinase
- fMRI
functional magnetic resonance imaging
- βFNA
β-funaltrexamine
- GABA
gamma aminobutyric acid
- GAD
glutamic acid decarboxylase
- GIRK
G protein-coupled inwardly-rectifying potassium channel
- GLUT
glutamate
- HR
heart rate
- i.c.v.
intracerebroventricular
- IN
intercalated nuclei of amygdala
- IPSC
inhibitory postsynaptic currents
- KOR
kappa opioid receptor
- LA
lateral nucleus
- Ldl
dorsolateral lateral nucleus
- Lvl
ventrolateral lateral nucleus
- Lvm
ventromedial lateral nucleus
- LC
locus ceuruleus
- LD Box
light-dark box
- Leu-ENK
leucine-enkephalin
- LTP
long-term potentiation
- Mad
anterodorsal medial amygdala
- Mav
anteroventral medial amygdala
- Mpd
posterodorsal medial amygdala
- Mpv
posteroventral medial amygdala
- MEA
medial nucleus of amygdala
- MEAP
Met-ENK-Arg6Phe7
- MHPG-SO4
3-methoxy-4-hydroxyphenylethyleneglycol sulfate
- MOR
mu opioid receptor
- NPN
non-pyramidal neurons
- NT
neurotensin
- OR
opioid receptor
- MEAP
Met-ENK-Arg6Phe7
- Met-ENK
methionine-enkephalin
- mIPSC
miniature inhibitory postsynaptic currents
- NAc
nucleus accumbens
- norBNI
nor-binaltorphimine
- OF
open-field
- PAG
periaqueductal grey
- PANAS
positive and negative affectivity scale
- PBN
parabrachial nucleus
- pDYN
prodynorphin
- pENK
proenkephalin
- PET
positron emission tomography
- PFC
prefrontal cortex
- PN
pyramidal neuron
- POMC
proopiomelanocortin
- ppENK
preproenkephalin
- ppDYN
preprodynorphin
- PTSD
posttraumatic stress disorder
- RVM
rostral ventromedial medulla
- SI
social interaction
- SLEA
sublenticular extended amygdala
- TMT
2,5-dihydro-2,4,5-trimethylthiazoline
- VMN
ventromedial hypothalamus
- VGLUT1
vesicular glutamate transporter
- VIP
vasoactive intestinal peptide
References
- Alheid GF, & Heimer L (1988). New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: The striatopallidal, amygdaloid, and corticopetal components of substantia innominata. Neuroscience, 27(1), 1–39. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3059226. [DOI] [PubMed] [Google Scholar]
- al-Rodhan N, Chipkin R, & Yaksh TL (1990). The antinociceptive effects of SCH-32615, a neutral endopeptidase (enkephalinase) inhibitor, micro-injected into the periaqueductal, ventral medulla and amygdala. Brain Research, 520, 123–130. [DOI] [PubMed] [Google Scholar]
- Anderson RI, & Becker HC (2017). Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol. Alcoholism, Clinical and Experimental Research, 41(8), 1402–1418. 10.1111/acer.13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asan E (1998). The catecholaminergic innervation of the rat amygdala. Advances in Anatomy, Embryology, and Cell Biology, 142, 1–118. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9586282. [DOI] [PubMed] [Google Scholar]
- Asede D, Bosch D, Luthi A, Ferraguti F, & Ehrlich I (2015). Sensory inputs to intercalated cells provide fear-learning modulated inhibition to the basolateral amygdala. Neuron, 86(2), 541–554. 10.1016/j.neuron.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Assareh N, Sarrami M, Carrive P, & McNally GP (2016). The organization of defensive behavior elicited by optogenetic excitation of rat lateral or ventrolateral periaqueductal gray. Behavioral Neuroscience, 130(4), 406–414. 10.1037/bne0000151. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Delfs JM, Druhan J, & Zhu Y (1999). The bed nucleus of the stria terminalis. A target site for noradrenergic actions in opiate withdrawal. Annals of the New York Academy of Sciences, 877, 486–498. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10415666. [DOI] [PubMed] [Google Scholar]
- Atweh SF, & Kuhar MJ (1977). Autoradiographic localization of opiate receptors in rat brain. III. The telencephalon. Brain Research, 134, 393–405. [DOI] [PubMed] [Google Scholar]
- Bajo M, Madamba SG, Roberto M, & Siggins GR (2014). Acute morphine alters GABAergic transmission in the central amygdala during naloxone-precipitated morphine withdrawal: Role of cyclic AMP. Frontiers in Integrative Neuroscience, 8, 45. 10.3389/fnint.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Roberto M, Madamba SG, & Siggins GR (2011). Neuroadaptation of GABAergic transmission in the central amygdala during chronic morphine treatment. Addiction Biology, 16(4), 551–564. 10.1111/j.1369-1600.2010.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Harter K, Gonzalez RG, & Borsook D (2006). Functional magnetic resonance imaging measures of the effects of morphine on central nervous system circuitry in opioid-naive healthy volunteers. Anesthesia and Analgesia, 103(1), 208–216, table. [DOI] [PubMed] [Google Scholar]
- Beckerman MA, & Glass MJ (2011). Ultrastructural relationship between the AMPA-Glu R2 receptor subunit and the mu-opioid receptor in the mouse central nucleus of the amygdala. Experimental Neurology, 227(1), 149–158. 10.1016/j.expneurol.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman MA, & Glass MJ (2012). The NMDA-NR1 receptor subunit and the mu-opioid receptor are expressed in somatodendritic compartments of central nucleus of the amygdala neurons projecting to the bed nucleus of the stria terminalis. Experimental Neurology, 234(1), 112–126. 10.1016/j.expneurol.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JF, & Besson JM (1990). The spinal(trigemino)pontoamygdala pathway: Electrophysiological evidence for an involvement in pain process. Journal of Neurophysiology, 63, 473–490. [DOI] [PubMed] [Google Scholar]
- Berube P, Laforest S, Bhatnagar S, & Drolet G (2013). Enkephalin and dynorphin mRNA expression are associated with resilience or vulnerability to chronic social defeat stress. Physiology & Behavior, 122, 237–245. S0031–9384 (13)00126–1 [pii] 10.1016/j.physbeh.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Berube P, Poulin JF, Laforest S, & Drolet G (2014). Enkephalin knockdown in the basolateral amygdala reproduces vulnerable anxiety-like responses to chronic unpredictable stress. Neuropsychopharmacology, 39(5), 1159–1168. npp 2013316 [pii] 10.1038/npp.2013.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie B, Zhu W, & Pan ZZ (2009a). Ethanol-induced delta-opioid receptor modulation of glutamate synaptic transmission and conditioned place preference in central amygdala. Neuroscience, 160(2), 348–358. 10.1016/j.neuroscience.2009.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie B, Zhu W, & Pan ZZ (2009b). Rewarding morphine-induced synaptic function of delta-opioid receptors on central glutamate synapses. The Journal of Pharmacology and Experimental Therapeutics, 329(1), 290–296. 10.1124/jpet.108.148908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn TP, Cross AJ, Hille C, & Slater P (1988). Autoradiographic localization of delta opiate receptors in rat and human brain. Neuroscience, 27(2), 497–506. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2851117. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Goedecke L, Bazelot M, Capogna M, Pape HC, & Jungling K (2015). mu-Opioid receptor-mediated inhibition of intercalated neurons and effect on synaptic transmission to the central amygdala. The Journal of Neuroscience, 35(19), 7317–7325. 10.1523/JNEUROSCI.0204-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco-Ibanez JM, Martinez-Guijarro FJ, & Freund TF (1998). Enkephalin-containing interneurons are specialized to innervate other interneurons in the hippocampal CA1 region of the rat and guinea-pig. The European Journal of Neuroscience, 10(5), 1784–1795. [DOI] [PubMed] [Google Scholar]
- Briand LA, Hilario M, Dow HC, Brodkin ES, Blendy JA, & Berton O (2015). Mouse model of OPRM1 (A118G) polymorphism increases sociability and dominance and confers resilience to social defeat. The Journal of Neuroscience, 35(8), 3582–3590. 10.1523/JNEUROSCI.4685-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinley-Reed M, Mascagni F, & McDonald AJ (1995). Synaptology of prefrontal cortical projections to the basolateral amygdala: An electron microscopic study in the rat. Neuroscience Letters, 95(1–2), 45–48. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Lemos JC, & Chavkin C (2009). CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One, 4(12), e8528. 10.1371/journal.pone.0008528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghardt PR, & Wilson MA (2006). Microinjection of naltrexone into the central, but not the basolateral, amygdala blocks the anxiolytic effects of diazepam in the plus maze. Neuropsychopharmacology, 31(6), 1227–1240. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16123750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral A, Ruggiero RN, Nobre MJ, Brandao ML, & Castilho VM (2009). GABA and opioid mechanisms of the central amygdala underlie the withdrawal-potentiated startle from acute morphine. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 33(2), 334–344. 10.1016/j.pnpbp.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Calvino B, Lagowska J, & Ben-Ari Y (1979). Morphine withdrawal syndrome: Differential participation of structures located within the amygdaloid complex and striatum of the rat. Brain Research, 177(1), 19–34. [DOI] [PubMed] [Google Scholar]
- Carlsen J, & Heimer L (1988). The basolateral amygdaloid complex as a cortical-like structure. Brain Research, 441(1–2), 377–380. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2451985. [DOI] [PubMed] [Google Scholar]
- Carr KD, Kutchukhidze N, & Park TH (1999). Differential effects of mu and kappa opioid antagonists on Fos-like immunoreactivity in extended amygdala. Brain Research, 822(1–2), 34–42. [DOI] [PubMed] [Google Scholar]
- Carrero JP, Kaigler KF, Hartshorn GH, Fadel JR, & Wilson MA (2019). Mu opioid receptor regulation of glutamate efflux in the central amygdala in response to predator odor. Neurobiology of Stress, 11, 100197. 10.1016/j.ynstr.2019.100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GQ, Karatayev O, Barson JR, Liang SC, & Leibowitz SF (2014). Common effects of fat, ethanol, and nicotine on enkephalin in discrete areas of the brain. Neuroscience, 277, 665–678. 10.1016/j.neuroscience.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, & Lawrence AJ (2004). Chronic antidepressant treatment causes a selective reduction of mu-opioid receptor binding and functional coupling to G Proteins in the amygdala of fawn-hooded rats. Journal of Pharmacology and Experimental Therapeutics, 310(3), 1020–1026. [DOI] [PubMed] [Google Scholar]
- Chieng B, & Christie M (2009). Chronic morphine treatment induces functional delta-opioid receptors in amygdala neurons that project to periaqueductal grey. Neuropharmacology, 57(4), 430–437. 10.1016/j.neuropharm.2009.06.034. [DOI] [PubMed] [Google Scholar]
- Chieng B, Christie M, & Osborne P (2006). Characterization of neurons in the rat central nucleus of the amygdala: Cellular physiology, morphology, and opioid sensitivity. The Journal of Comparative Neurology, 497(6), 910–927. [DOI] [PubMed] [Google Scholar]
- Chou DT, & Wang SC (1977). Unit activity of amygdala and hippocampal neurons: Effects of morphine and benzodiazepines. Brain Research, 126 (3), 427–440. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/861730. [DOI] [PubMed] [Google Scholar]
- Clarke S, Zimmer A, Zimmer AM, Hill RG, & Kitchen I (2003). Region selective up-regulation of micro-, delta- and kappa-opioid receptors but not opioid receptor-like 1 receptors in the brains of enkephalin and dynorphin knockout mice. Neuroscience, 122(2), 479–489. [DOI] [PubMed] [Google Scholar]
- Cowen MS, & Lawrence AJ (2001). Alterations in central preproenkephalin mRNA expression after chronic free-choice ethanol consumption by fawn-hooded rats. Alcoholism: Clinical and Experimental Research, 25(8), 1126–1133. [PubMed] [Google Scholar]
- Criado JR, & Morales M (2000). Acute ethanol induction of c-Fos immunoreactivity in pre-pro-enkephalin expressing neurons of the central nucleus of the amygdala. Brain Research, 861, 173–177. [DOI] [PubMed] [Google Scholar]
- Crowley NA, Bloodgood DW, Hardaway JA, Kendra AM, McCall JG, Al-Hasani R, … Kash TL (2016). Dynorphin controls the gain of an amygdalar anxiety circuit. Cell Reports, 14(12), 2774–2783. 10.1016/j.celrep.2016.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa Gomez TM, & Behbehani MM (1995). An electrophysiological characterization of the projection from the central nucleus of the amygdala to the periaqueductal gray of the rat: The role of opioid receptors. Brain Research, 689(1), 21–31. [DOI] [PubMed] [Google Scholar]
- D’Addario C, Caputi FF, Ekstrom TJ, Di Benedetto M, Maccarrone M, Romualdi P, & Candeletti S (2013). Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. Journal of Molecular Neuroscience, 49(2), 312–319. 10.1007/s12031-012-9829-y. [DOI] [PubMed] [Google Scholar]
- D’Addario C, Caputi FF, Rimondini R, Gandolfi O, Del Borrello E, Candeletti S, & Romualdi P (2013). Different alcohol exposures induce selective alterations on the expression of dynorphin and nociceptin systems related genes in rat brain. Addiction Biology, 18(3), 425–433. 10.1111/j.1369-1600.2011.00326.x. [DOI] [PubMed] [Google Scholar]
- Daunais JB, Letchworth SR, Sim-Selley LJ, Smith HR, Childers SR, & Porrino LJ (2001). Functional and anatomical localization of mu opioid receptors in the striatum, amygdala, and extended amygdala of the nonhuman primate. The Journal of Comparative Neurology, 433(4), 471–485. [DOI] [PubMed] [Google Scholar]
- Day H, Curran E, Watson S, & Akil H (1999). Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria teminalis: Evidence fr their selective activation by interleukin-1beta. The Journal of Comparative Neurology, 413, 113–128. [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, & Aston-Jones G (2000). Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature, 403(6768), 430–434. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10667795. [DOI] [PubMed] [Google Scholar]
- Deyama S, Yamamoto J, Machida T, Tanimoto S, Nakagawa T, Kaneko S, … Minami M (2007). Inhibition of glutamatergic transmission by morphine in the basolateral amygdaloid nucleus reduces pain-induced aversion. Neuroscience Research, 59(2), 199–204. [DOI] [PubMed] [Google Scholar]
- Ding YQ, Kaneko T, Nomura S, & Mizuno N (1996). Immunohistochemical localization of mu-opioid receptors in the central nervous system of the rat. The Journal of Comparative Neurology, 367(3), 375–402, 377. [DOI] [PubMed] [Google Scholar]
- Djouma E, & Lawrence AJ (2002). The effect of chronic ethanol consumption and withdrawal on mu-opioid and dopamine D(1) and D(2) receptor density in Fawn-Hooded rat brain. The Journal of Pharmacology and Experimental Therapeutics, 302(2), 551–559. 10.1124/jpet.102.035915. [DOI] [PubMed] [Google Scholar]
- Domino EF, Hirasawa-Fujita M, Ni L, Guthrie SK, & Zubieta JK (2015). Regional brain [(11)C]carfentanil binding following tobacco smoking. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 59, 100–104. 10.1016/j.pnpbp.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Chavkin C, & Milner TA (2007). Opioid systems in the dentate gyrus. Progress in Brain Research, 163, 245–263. 10.1016/S0079-6123(07)63015-5. [DOI] [PubMed] [Google Scholar]
- Drolet G, Dumont EC, Gosselin I, Kinkead R, Laforest S, & Trottier JF (2001). Role of endogenous opioid system in the regulation of the stress response. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 25(4), 729–741. [DOI] [PubMed] [Google Scholar]
- Eippert F, Bingel U, Schoell E, Yacubian J, & Buchel C (2008). Blockade of endogenous opioid neurotransmission enhances acquisition of conditioned fear in humans. The Journal of Neuroscience, 28(21), 5465–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde R, Hokfelt T, Johansson O, & Terenius L (1976). Immunohistochemical studies using antibodies to leucine-enkephalin: Initial observations on the nervous system of the rat. Neuroscience, 1(4), 349–351. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11370520. [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, … Massotte D (2015). A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Structure & Function, 220(2), 677–702. 10.1007/s00429-014-0717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson CM, Wei G, & Walker BM (2018). Maladaptive behavioral regulation in alcohol dependence: Role of kappa-opioid receptors in the bed nucleus of the stria terminalis. Neuropharmacology, 140, 162–173. 10.1016/j.neuropharm.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, & Sah P (2004). Opioids inhibit lateral amygdala pyramidal neurons by enhancing a dendritic potassium current. The Journal of Neuroscience, 24(12), 3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JH, & Leslie FM (1986). Distribution of dynorphin and enkephalin peptides in the rat brain. The Journal of Comparative Neurology, 249(3), 293–336. 10.1002/cne.902490302. [DOI] [PubMed] [Google Scholar]
- Fallon JH, Leslie FM, & Cone RI (1985). Dynorphin-containing pathways in the substantia nigra and ventral tegmentum: A double labeling study using combined immunofluorescence and retrograde tracing. Neuropeptides, 5(4–6), 457–460. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2860605. [DOI] [PubMed] [Google Scholar]
- Farb CR, & LeDoux JE (1999). Afferents from rat temporal cortex synapse on lateral amygdala neurons that express NMDA and AMPA receptors. Synapse, 33(3), 218–229. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10420169. [DOI] [PubMed] [Google Scholar]
- File SE, & Rodgers RJ (1979a). Exploratory behaviour and aversive thresholds in rats following microinjection of morphine into central and medial nuclei of the amygdala. British Journal of Pharmacology, 66, 145P–146P. [PMC free article] [PubMed] [Google Scholar]
- File SE, & Rodgers RJ (1979b). Partial anxiolytic action of morphine sulphate following microinjection into the central nucleus of the amygdala in rats. Pharmacology, Biochemistry, and Behavior, 11, 313–318. [DOI] [PubMed] [Google Scholar]
- Finley JC, Lindstrom P, & Petrusz P (1981). Immunocytochemical localization of beta-endorphin-containing neurons in the rat brain. Neuroendocrinology, 33(1), 28–42. [DOI] [PubMed] [Google Scholar]
- Finley JC, Maderdrut JL, & Petrusz P (1981). The immunocytochemical localization of enkephalin in the central nervous system of the rat. The Journal of Comparative Neurology, 198(4), 541–565. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7019273. [DOI] [PubMed] [Google Scholar]
- Finnegan TF, Chen SR, & Pan HL (2005). Effect of the {mu} opioid on excitatory and inhibitory synaptic inputs to periaqueductal gray-projecting neurons in the amygdala. The Journal of Pharmacology and Experimental Therapeutics, 312(2), 441–448. [DOI] [PubMed] [Google Scholar]
- Finnegan TF, Chen SR, & Pan HL (2006). Mu opioid receptor activation inhibits GABAergic inputs to basolateral amygdala neurons through Kv1.1/1.2 channels. Journal of Neurophysiology, 95(4), 2032–2041. [DOI] [PubMed] [Google Scholar]
- Flood JF, Garland JS, & Morley JE (1992). Evidence that cholecystokinin-enhanced retention is mediated by changes in opioid activity in the amygdala. Brain Research, 585(1–2), 94–104. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1511338. [DOI] [PubMed] [Google Scholar]
- Freedman JE, & Aghajanian GK (1985). Opiate and alpha 2-adrenoceptor responses of rat amygdaloid neurons: Co-localization and interactions during withdrawal. The Journal of Neuroscience, 5(11), 3016–3024. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2997411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenois F, Cador M, Caille S, Stinus L, & Le Moine C (2002). Neural correlates of the motivational and somatic components of naloxone-precipitated morphine withdrawal. The European Journal of Neuroscience, 16(7), 1377–1389. [DOI] [PubMed] [Google Scholar]
- Frenois F, Stinus L, Di Blasi F, Cador M, & Le Moine C (2005). A specific limbic circuit underlies opiate withdrawal memories. The Journal of Neuroscience, 25(6), 1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M, & Kapp BS (1978). Manipulation of opiate activity in the amygdala alters memory processes. Life Sciences, 23(19), 1973–1977. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Kapp BS, McNall CL, & Pascoe JP (1981). Opiate effects in the amygdala central nucleus on heart rate conditioning in rabbits. Pharmacology, Biochemistry, and Behavior, 14(4), 497–505. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Kapp BS, & Pascoe JP (1982). Enkephalin analogue effects in the amygdala central nucleus on conditioned heart rate. Pharmacology, Biochemistry, and Behavior, 17(2), 217–222. [DOI] [PubMed] [Google Scholar]
- Gestreau C, Le Guen S, & Besson JM (2000). Is there tonic activity in the endogenous opioid systems? A c-Fos study in the rat central nervous system after intravenous injection of naloxone or naloxone-methiodide. The Journal of Comparative Neurology, 427(2), 285–301. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11054694. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Roberto M, Koob GF, & Schweitzer P (2014). Kappa opioid receptor activation decreases inhibitory transmission and antagonizes alcohol effects in rat central amygdala. Neuropharmacology, 77, 294–302. 10.1016/j.neuropharm.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ (2010). The role of functional postsynaptic NMDA receptors in the central nucleus of the amygdala in opioid dependence. Vitamins and Hormones, 82, 145–166. 10.1016/S0083-6729(10)82008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Kruzich PJ, Colago EE, Kreek MJ, & Pickel VM (2005). Increased AMPA GluR1 receptor subunit labeling on the plasma membrane of dendrites in the basolateral amygdala of rats self-administering morphine. Synapse, 58(1), 1–12. 10.1002/syn.20176. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Vanyo L, Quimson L, & Pickel VM (2009). Ultrastructural relationship between N-methyl-d-aspartate-NR1 receptor subunit and mu-opioid receptor in the mouse central nucleus of the amygdala. Neuroscience, 163(3), 857–867. 10.1016/j.neuroscience.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good AJ, & Westbrook RF (1995). Effects of a microinjection of morphine into the amygdala on the acquisition and expression of conditioned fear and hypoalgesia in rats. Behavioral Neuroscience, 109(4), 631–641. [DOI] [PubMed] [Google Scholar]
- Granholm L, Roman E, & Nylander I (2015). Single housing during early adolescence causes time-, area- and peptide-specific alterations in endogenous opioids of rat brain. British Journal of Pharmacology, 172(2), 606–614. 10.1111/bph.12753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm L, Segerstrom L, & Nylander I (2018). Episodic ethanol exposure in adolescent rats causes residual alterations in endogenous opioid peptides. Frontiers in Psychiatry, 9, 425. 10.3389/fpsyt.2018.00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm L, Todkar A, Bergman S, Nilsson K, Comasco E, & Nylander I (2017). The expression of opioid genes in non-classical reward areas depends on early life conditions and ethanol intake. Brain Research, 1668, 36–45. 10.1016/j.brainres.2017.05.006. [DOI] [PubMed] [Google Scholar]
- Gray TS, Cassell MD, & Kiss JZ (1984). Distribution of pro-opiomelanocortin-derived peptides and enkephalins in the rat central nucleus of the amygdala. Brain Research, 306(1–2), 354–358. [DOI] [PubMed] [Google Scholar]
- Gray TS, & Magnuson DJ (1987). Neuropeptide neuronal efferents from the bed nucleus of the stria terminalis and central amygdaloid nucleus to the dorsal vagal complex in the rat. The Journal of Comparative Neurology, 262(3), 365–374. [DOI] [PubMed] [Google Scholar]
- Gros C, Pradelles P, Humbert J, Dray F, LeGal La Salle G, & Ben-Ari Y (1978). Regional distribution of met-enkephalin within the amygdaloid complex and bed nucleus of the stria terminalis. Neuroscience Letters, 10, 193–196. [DOI] [PubMed] [Google Scholar]
- Haber S, & Elde R (1982). The distribution of enkephalin immunoreactive fibers and terminals in the monkey central nervous system: An immunohistochemical study. Neuroscience, 7(5), 1049–1095. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7050764. [DOI] [PubMed] [Google Scholar]
- Harlan RE, Shivers BD, Romano GJ, Howells RD, & Pfaff DW (1987). Localization of preproenkephalin mRNA in the rat brain and spinal cord by in situ hybridization. The Journal of Comparative Neurology, 258, 159–184. [DOI] [PubMed] [Google Scholar]
- Harris RE, Clauw DJ, Scott DJ, McLean SA, Gracely RH, & Zubieta JK (2007). Decreased central mu-opioid receptor availability in fibromyalgia. The Journal of Neuroscience, 27(37), 10000–10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb AL, Zacharko RM, Gauthier M, Trudel F, Laforest S, & Drolet G (2004). Brief exposure to predator odor and resultant anxiety enhances mesocorticolimbic activity and enkephalin expression in CD-1 mice. The European Journal of Neuroscience, 20(9), 2415–2429. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, Bellgowan PS, & Poore LH (1995). Microinfusion of mu but not delta or kappa opioid agonists into the basolateral amygdala results in inhibition of the tail flick reflex in pentobarbital-anesthetized rats. The Journal of Pharmacology and Experimental Therapeutics, 275(1), 381–388, 313. [PubMed] [Google Scholar]
- Helmstetter FJ, Bellgowan PS, & Tershner SA (1993). Inhibition of the tail flick reflex following microinjection of morphine into the amygdala. NeuroReport, 4(5), 471–474, 428. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, Tershner SA, Poore LH, & Bellgowan PS (1998). Antinociception following opioid stimulation of the basolateral amygdala is expressed through the periaqueductal gray and rostral ventromedial medulla. Brain Research, 779(1–2), 104–118. [DOI] [PubMed] [Google Scholar]
- Henry MS, Bisht K, Vernoux N, Gendron L, Torres-Berrio A, Drolet G, & Tremblay ME (2018). Delta opioid receptor signaling promotes resilience to stress under the repeated social defeat paradigm in mice. Frontiers in Molecular Neuroscience, 11, 100. 10.3389/fnmol.2018.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry MS, Gendron L, Tremblay ME, & Drolet G (2017). Enkephalins: Endogenous analgesics with an emerging role in stress resilience. Neural Plasticity. 2017, 1546125. 10.1155/2017/1546125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J, Prieto I, Segarra AB, de Gasparo M, Wangensteen R, Villarejo AB, … Ramirez-Sanchez M (2015). Interaction of neuropeptidase activities in cortico-limbic regions after acute restraint stress. Behavioural Brain Research, 287, 42–48. 10.1016/j.bbr.2015.03.036. [DOI] [PubMed] [Google Scholar]
- Hernandez J, Segarra AB, Ramirez M, Banegas I, de Gasparo M, Alba F, … Prieto I (2009). Stress influences brain enkephalinase, oxytocinase and angiotensinase activities: A new hypothesis. Neuropsychobiology, 59(3), 184–189. 10.1159/000219306. [DOI] [PubMed] [Google Scholar]
- Hernandez LL, Powell DA, & Gibbs CM (1990). Amygdaloid central nucleus neuronal activity accompanying pavlovian cardiac conditioning: Effects of naloxone. Behavioural Brain Research, 41(1), 71–79. [DOI] [PubMed] [Google Scholar]
- Heyser CJ, Roberts AJ, Schulteis G, & Koob GF (1999). Central administration of an opiate antagonist decreases oral ethanol self-administration in rats. Alcoholism, Clinical and Experimental Research, 23(9), 1468–1476. [PubMed] [Google Scholar]
- Honkaniemi J, Pelto-Huikko M, Rechardt L, Isola J, Lammi A, Fuxe K, … Hokfelt T (1992). Colocalization of peptide and glucocorticoid receptor immunoreativities in rat central amygdaloid nucleus. Neuroendocrinology, 55, 451–459. [DOI] [PubMed] [Google Scholar]
- Huge V, Rammes G, Beyer A, Zieglgansberger W, & Azad SC (2009). Activation of kappa opioid receptors decreases synaptic transmission and inhibits long-term potentiation in the basolateral amygdala of the mouse. European Journal of Pain, 13(2), 124–129. 10.1016/j.ejpain.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, & Morris HR (1975). Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature, 258(5536), 577–580. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1207728. [DOI] [PubMed] [Google Scholar]
- Hurd YL (1996). Differential messenger RNA expression of prodynorphin and proenkephalin in the human brain. Neuroscience, 72(3), 767–783. [DOI] [PubMed] [Google Scholar]
- Hyytia P, & Kiianmaa K (2001). Suppression of ethanol responding by centrally administered CTOP and naltrindole in AA and Wistar rats. Alcoholism, Clinical and Experimental Research, 25(1), 25–33. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11198711. [DOI] [PubMed] [Google Scholar]
- Iredale PA, Alvaro JD, Lee Y, Terwilliger R, Chen YL, & Duman RS (2000). Role of corticotropin-releasing factor receptor-1 in opiate withdrawal. Journal of Neurochemistry, 74(1), 199–208. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10617121. [DOI] [PubMed] [Google Scholar]
- Jaferi A, & Pickel VM (2009). Mu-opioid and corticotropin-releasing-factor receptors show largely postsynaptic co-expression, and separate presynaptic distributions, in the mouse central amygdala and bed nucleus of the stria terminalis. Neuroscience, 159(2), 526–539. 10.1016/j.neuroscience.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Araki H, Nagata M, Shimosaka R, Shibata K, Suemaru K, … Gomita Y (2005). Expression of c-Fos in the rat central amygdala accompanies the acquisition but not expression of conditioned place aversion induced by withdrawal from acute morphine dependence. Behavioural Brain Research, 161(1), 107–112. [DOI] [PubMed] [Google Scholar]
- Johnston JB (1923). Evolution of the forebrain. The Journal of Comparative Neurology, 35, 337–481. [Google Scholar]
- Kang W, Wilson MA, Bender MA, Glorioso JC, & Wilson SP (1998). Herpes virus-mediated preproenkephalin gene transfer to the amygdala is antinociceptive. Brain Research, 792, 133–135. [DOI] [PubMed] [Google Scholar]
- Kang W, Wilson MA, & Wilson SP (2000). Overexpression of proenkephalin in the amygdala potentiates the anxiolytic effects of benzodiazepines. Neuropsychopharmacology, 22, 77–88. [DOI] [PubMed] [Google Scholar]
- Kang W, Wilson SP, & Wilson MA (1999). Changes in nociceptive and anxiolytic responses following herpes virus-mediated preproenkephalin overexpression in rat amygdala are naloxone-reversible and transient. Annals of the New York Academy of Sciences, 877, 751–755. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10415698. [DOI] [PubMed] [Google Scholar]
- Kang-Park M, Kieffer BL, Roberts AJ, Siggins GR, & Moore SD (2013). kappa-Opioid receptors in the central amygdala regulate ethanol actions at presynaptic GABAergic sites. The Journal of Pharmacology and Experimental Therapeutics, 346(1), 130–137. 10.1124/jpet.112.202903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Park M, Kieffer BL, Roberts AJ, Siggins GR, & Moore SD (2015). Interaction of CRF and kappa opioid systems on GABAergic neurotransmission in the mouse central amygdala. The Journal of Pharmacology and Experimental Therapeutics, 355(2), 206–211. 10.1124/jpet.115.225870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Park MH, Kieffer BL, Roberts AJ, Roberto M, Madamba SG, Siggins GR, & Moore SD (2009). Mu-opioid receptors selectively regulate basal inhibitory transmission in the central amygdala: Lack of ethanol interactions. The Journal of Pharmacology and Experimental Therapeutics, 328(1), 284–293. 10.1124/jpet.108.140749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Park MH, Kieffer BL, Roberts AJ, Siggins GR, & Moore SD (2007). Presynaptic {delta} opioid receptors regulate ethanol actions in central amygdala. The Journal of Pharmacology and Experimental Therapeutics, 320(2), 917–925. [DOI] [PubMed] [Google Scholar]
- Kennedy SE, Koeppe RA, Young EA, & Zubieta JK (2006). Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Archives of General Psychiatry, 63(11), 1199–1208. [DOI] [PubMed] [Google Scholar]
- Khachaturian H, Lewis ME, Haber SN, Houghten RA, Akil H, & Watson SJ (1985). Prodynorphin peptide immunocytochemistry in rhesus monkey brain. Peptides, 6(Suppl 2), 155–166. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3909123. [DOI] [PubMed] [Google Scholar]
- Khachaturian H, Lewis ME, Hollt V, & Watson SJ (1983). Telencephalic enkephalinergic systems in the rat brain. The Journal of Neuroscience, 3 (4), 844–855. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian H, Watson SJ, Lewis ME, Coy D, Goldstein A, & Akil H (1982). Dynorphin immunocytochemistry in the rat central nervous system. Peptides, 3(6), 941–954. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6132365. [DOI] [PubMed] [Google Scholar]
- Kissler JL, Sirohi S, Reis DJ, Jansen HT, Quock RM, Smith DG, & Walker BM (2014). The one-two punch of alcoholism: Role of central amygdala dynorphins/kappa-opioid receptors. Biological Psychiatry, 75(10), 774–782. 10.1016/j.biopsych.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissler JL, & Walker BM (2016). Dissociating motivational from physiological withdrawal in alcohol dependence: Role of central amygdala kappa-opioid receptors. Neuropsychopharmacology, 41(2), 560–567. 10.1038/npp.2015.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm WR, & Mallari CG (1979). Morphine and naloxone effects on olfactory evoked electrographic activity in the amygdala. Archives Internationales de Pharmacodynamie et de Th[notdef]erapie, 237(2), 237–250. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/485690. [PubMed] [Google Scholar]
- Knoll AT, Meloni EG, Thomas JB, Carroll FI, & Carlezon WA Jr. (2007). Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. The Journal of Pharmacology and Experimental Therapeutics, 323(3), 838–845. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, … Carlezon WA Jr. (2011). Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biological Psychiatry, 70(5), 425–433. 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER, Linden AM, Hytonen HR, Paasikoski N, Vashchinkina E, Dudek M, … Hyytia P (2017). Continuous delivery of naltrexone and nalmefene leads to tolerance in reducing alcohol drinking and to supersensitivity of brain opioid receptors. Addiction Biology, 22(4), 1022–1035. 10.1111/adb.12393. [DOI] [PubMed] [Google Scholar]
- Lagowska J, Calvino B, & Ben-Ari Y (1978). Intra-amygdaloid applications of naloxone elicits severe withdrawal signs in morphine dependent rats. Neuroscience Letters, 8(3), 241–245. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19605166. [DOI] [PubMed] [Google Scholar]
- Lam MP, Marinelli PW, Bai L, & Gianoulakis C (2008). Effects of acute ethanol on opioid peptide release in the central amygdala: An in vivo microdialysis study. Psychopharmacology, 201(2), 261–271. 10.1007/s00213-008-1267-8. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, & Chavkin C (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. The Journal of Neuroscience, 28(2), 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AD, Funk D, Coen K, Tamadon S, & Shaham Y (2018). Role of kappa-opioid receptors in the bed nucleus of stria terminalis in reinstatement of alcohol seeking. Neuropsychopharmacology, 43(4), 838–850. 10.1038/npp.2017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, & Farb CR (1991). Neurons of the acoustic thalamus that project to the amygdala contain glutamate. Neuroscience Letters, 134(1), 145–149. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1687697. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Farb CR, & Milner TA (1991). Ultrastructure and synaptic associations of auditory thalamo-amygdala projections in the rat. Experimental Brain Research, 85(3), 577–586. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1717305. [DOI] [PubMed] [Google Scholar]
- Leppa M, Korvenoja A, Carlson S, Timonen P, Martinkauppi S, Ahonen J, … Kalso E (2006). Acute opioid effects on human brain as revealed by functional magnetic resonance imaging. NeuroImage, 31(2), 661–669. [DOI] [PubMed] [Google Scholar]
- Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB, … Kash TL (2012). Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biological Psychiatry, 71(8), 725–732. 10.1016/j.biopsych.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon I, Taylor SF, Phan KL, Britton JC, Fig LM, Bueller JA, … Zubieta JK (2007). Altered central micro-opioid receptor binding after psychological trauma. Biological Psychiatry, 61(9), 1030–1038. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Zubieta JK, Fig LM, Phan KL, Koeppe RA, & Taylor SF (2002). mu-Opioid receptors and limbic responses to aversive emotional stimuli. Proceedings of the National Academy of Sciences of the United States of America, 99(10), 7084–7089. 10.1073/pnas.102174799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, & Pare D (2008). Amygdala intercalated neurons are required for expression of fear extinction. Nature, 454(7204), 642–645. nature07167 [pii]. 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin S, Leslie F, & Fallon J (1995). Endogenous opioid systems. In Paxinos G (Ed.), The rat nervous system (pp. 975–1001). New York: Academic Press, Inc. [Google Scholar]
- Lucas M, Frenois F, Cador M, & Le Moine C (2012). Remodeling of the neuronal circuits underlying opiate-withdrawal memories following remote retrieval. Neurobiology of Learning and Memory, 97(1), 47–53. 10.1016/j.nlm.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Lucas M, Frenois F, Vouillac C, Stinus L, Cador M, & Le Moine C (2008). Reactivity and plasticity in the amygdala nuclei during opiate withdrawal conditioning: Differential expression of c-fos and arc immediate early genes. Neuroscience, 154(3), 1021–1033. 10.1016/j.neuroscience.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Lugo JN Jr., Wilson MA, & Kelly SJ (2006). Perinatal ethanol exposure alters met-enkephalin levels of male and female rats. Neurotoxicology and Teratology, 28(2), 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz PE, & Kieffer BL (2013). Opioid receptors: Distinct roles in mood disorders. Trends in Neurosciences, 36(3), 195–206. 10.1016/j.tins.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch WC, Watt J, Krall S, & Paden CM (1985). Autoradiographic localization of kappa opiate receptors in CNS taste and feeding areas. Pharmacology, Biochemistry, and Behavior, 22(5), 699–705. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2989940. [DOI] [PubMed] [Google Scholar]
- Ma QP, & Han JS (1991). Naloxone blocks the release of opioid peptides in periaqueductal gray and N. accumbens induced by intra-amygdaloid injection of morphine. Peptides, 12, 1235–1238. [DOI] [PubMed] [Google Scholar]
- Mansour A, Burke S, Pavlic RJ, Akil H, & Watson SJ (1996). Immunohistochemical localization of the cloned kappa 1 receptor in the rat CNS and pituitary. Neuroscience, 71(3), 671–690. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8867040. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, & Watson SJ (1995). Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends in Neurosciences, 18, 22–29. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Akil H, & Watson SJ (1995). Immunohistochemical localization of the cloned mu opioid receptor in the rat CNS. Journal of Chemical Neuroanatomy, 8(4), 283–305. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, & Watson SJ (1994). Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: An in situ hybridization study. The Journal of Comparative Neurology, 350(3), 412–438. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Meng F, Akil H, & Watson SJ (1994). Kappa 1 receptor mRNA distribution in the rat CNS: Comparison to kappa receptor binding and prodynorphin mRNA. Molecular and Cellular Neurosciences, 5(2), 124–144. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Thompson RC, Akil H, & Watson SJ (1994). mu-Opioid receptor mRNA expression in the rat CNS: Comparison to mu-receptor binding. Brain Research, 643(1–2), 245–265. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, & Watson SJ (1987). Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. The Journal of Neuroscience, 7(8), 2445–2464. [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Watson SJ, & Akil H (1995). Opioid receptors: Past, present and future. Trends in Neurosciences, 18(2), 69–70. [PubMed] [Google Scholar]
- Marchant NJ, Densmore VS, & Osborne PB (2007). Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: Evidence of two distinct endogenous opioid systems in the lateral division. The Journal of Comparative Neurology, 504(6), 702–715. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Funk D, Juzytsch W, & Le AD (2010). Opioid receptors in the basolateral amygdala but not dorsal hippocampus mediate context-induced alcohol seeking. Behavioural Brain Research, 211(1), 58–63. 10.1016/j.bbr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli PW, Funk D, Juzytsch W, Li Z, & Le AD (2007). Effects of opioid receptor blockade on the renewal of alcohol seeking induced by context: Relationship to c-fos mRNA expression. The European Journal of Neuroscience, 26(10), 2815–2823. [DOI] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K, & Vogt KE (2005). A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron, 48(6), 1025–1037. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16364905. [DOI] [PubMed] [Google Scholar]
- Mascagni F, & McDonald AJ (2003). Differential expression of GABAa and GABAb receptor subunits in discrete interneuronal subpopulations iof the rat basolateral amygdala. Society for Neuroscience – Abstracts, 568, 511. [Google Scholar]
- McDonald AJ (1992). Neuroanatomical labeling with biocytin: A review. NeuroReport, 3(10), 821–827. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (2003). Is there an amygdala and how far does it extend? An anatomical perspective. Annals of the New York Academy of Sciences, 985, 1–21. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Farr DA, & Heinricher MM (2004). Lesions of the periaqueductal gray disrupt input to the rostral ventromedial medulla following microinjections of morphine into the medial or basolateral nuclei of the amygdala. Brain Research, 1009(1–2), 223–227. 10.1016/j.brainres.2004.02.048. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, & Heinricher MM (2002). Microinjection of morphine into various amygdaloid nuclei differentially affects nociceptive responsiveness and RVM neuronal activity. Pain, 96(1–2), 153–162. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11932071. [DOI] [PubMed] [Google Scholar]
- McLean S, Rothman RB, & Herkenham M (1986). Autoradiographic localization of mu- and delta-opiate receptors in the forebrain of the rat. Brain Research, 378(1), 49–60. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3017503. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Maderdrut JL, Altschuler RA, & Petrusz P (1986). Immunocytochemical localization of proenkephalin-derived peptides in the central nervous system of the rat. Neuroscience, 17(2), 325–348. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3517687. [DOI] [PubMed] [Google Scholar]
- Moga MM, & Gray TS (1985). Evidence for corticotropin-releasing factor, neurotensin, and somatostatin in the neural pathway from the central nucleus of the amygdala to the parabrachial nucleus. The Journal of Comparative Neurology, 241(3), 275–284. [DOI] [PubMed] [Google Scholar]
- Moga MM, Saper CB, & Gray TS (1989). Bed nucleus of the stria terminalis: Cytoarchitecture, immunohistochemistry, and projection to the parabrachial nucleus in the rat. The Journal of Comparative Neurology, 283(3), 315–332. [DOI] [PubMed] [Google Scholar]
- Moskowitz AS, & Goodman RR (1984). Light microscopic autoradiographic localization of mu and delta opioid binding sites in the mouse central nervous system. The Journal of Neuroscience, 4(5), 1331–1342. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6327936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandigama P, & Borszcz GS (2003). Affective analgesia following the administration of morphine into the amygdala of rats. Brain Research, 959(2), 343–354. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12493624. [DOI] [PubMed] [Google Scholar]
- Narita M, Kaneko C, Miyoshi K, Nagumo Y, Kuzumaki N, Nakajima M, … Suzuki T (2006). Chronic pain induces anxiety with concomitant changes in opioidergic function in the amygdala. Neuropsychopharmacology, 31(4), 739–750. [DOI] [PubMed] [Google Scholar]
- Nation KM, De Felice M, Hernandez PI, Dodick DW, Neugebauer V, Navratilova E, & Porreca F (2018). Lateralized kappa opioid receptor signaling from the amygdala central nucleus promotes stress-induced functional pain. Pain, 159(5), 919–928. 10.1097/j.pain.0000000000001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor JC, Simson PE, Gibson B, Schneider AM, Wilkins E, Firestone A, & Choy M (2001). Ethanol inhibits spontaneous activity of central nucleus of the amygdala neurons but does not impair retention in the passive-avoidance task. Alcoholism, Clinical and Experimental Research, 25 (11), 1683–1688. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11707643. [PubMed] [Google Scholar]
- Nygard SK, Hourguettes NJ, Sobczak GG, Carlezon WA, & Bruchas MR (2016). Stress-induced reinstatement of nicotine preference requires dynorphin/kappa opioid activity in the basolateral amygdala. The Journal of Neuroscience, 36(38), 9937–9948. 10.1523/JNEUROSCI.0953-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmstead MC, & Franklin KB (1997a). The development of a conditioned place preference to morphine: Effects of lesions of various CNS sites. Behavioral Neuroscience, 111, 1313–1323. [DOI] [PubMed] [Google Scholar]
- Olmstead MC, & Franklin KB (1997b). The development of a conditioned place preference to morphine: Effects of microinjections into various CNS sites. Behavioral Neuroscience, 111(6), 1324–1334. [DOI] [PubMed] [Google Scholar]
- Olucha-Bordonau FE, Fortes-Marco L, Otero-García M, Lanuza E, & Martinez-García F (2015). Amygdala: Structure and function. In Paxinos G (Ed.), The rat nervous system (pp. 441–490). (4th ed.). San Diego: Academic Press. [Google Scholar]
- Paden CM, Krall S, & Lynch WC (1987). Heterogeneous distribution and upregulation of mu, delta and kappa opioid receptors in the amygdala. Brain Research, 87(2), 349–355. [DOI] [PubMed] [Google Scholar]
- Pavlovic ZW, Cooper ML, & Bodnar RJ (1996). Opioid antagonists in the periaqueductal gray inhibit morphine and beta-endorphin analgesia elicited from the amygdala of rats. Brain Research, 741(1–2), 13–26. [DOI] [PubMed] [Google Scholar]
- Paxinos G, & Watson C (1997). The rat brain in stereotaxic coordinates (3rd ed.). San Diego: Academic Press. [Google Scholar]
- Pecina M, Love T, Stohler CS, Goldman D, & Zubieta JK (2015). Effects of the Mu opioid receptor polymorphism (OPRM1 A118G) on pain regulation, placebo effects and associated personality trait measures. Neuropsychopharmacology, 40(4), 957–965. 10.1038/npp.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, & De Vries TJ (2012). Glutamate mechanisms underlying opiate memories. Cold Spring Harbor Perspectives in Medicine. 2(9) a012088. 10.1101/cshperspect.a012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovich GD, Scicli AP, Thompson RF, & Swanson LW (2000). Associative fear conditioning of enkephalin mRNA levels in central amygdalar neurons. Behavioral Neuroscience, 114(4), 681–686. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Arbour D, Laforest S, & Drolet G (2009). Neuroanatomical characterization of endogenous opioids in the bed nucleus of the stria terminalis. Progress in Neuropsychopharmacology and Biological Psychiatry, 33(8), 1356–1365. S0278–5846(09)00211–5 [pii] 10.1016/j.pnpbp.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Berube P, Laforest S, & Drolet G (2013). Enkephalin knockdown in the central amygdala nucleus reduces unconditioned fear and anxiety. The European Journal of Neuroscience, 37(8), 1357–1367. 10.1111/ejn.12134. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Castonguay-Lebel Z, Laforest S, & Drolet G (2008). Enkephalin co-expression with classic neurotransmitters in the amygdaloid complex of the rat. The Journal of Comparative Neurology, 506(6), 943–959. 10.1002/cne.21587. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Chevalier B, Laforest S, & Drolet G (2006). Enkephalinergic afferents of the centromedial amygdala in the rat. The Journal of Comparative Neurology, 496(6), 859–876. [DOI] [PubMed] [Google Scholar]
- Primeaux SD, Wilson SP, McDonald AJ, Mascagni F, & Wilson MA (2006). The role of delta opioid receptors in the anxiolytic actions of benzodiazepines. Pharmacology, Biochemistry, and Behavior, 85, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybysz KR, Werner DF, & Diaz MR (2017). Age-dependent regulation of GABA transmission by kappa opioid receptors in the basolateral amygdala of Sprague-Dawley rats. Neuropharmacology, 117, 124–133. 10.1016/j.neuropharm.2017.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirarte GL, Galvez R, Roozendaal B, & McGaugh JL (1998). Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Research, 808(2), 134–140. [DOI] [PubMed] [Google Scholar]
- Ragen BJ, Freeman SM, Laredo SA, Mendoza SP, & Bales KL (2015). mu and kappa opioid receptor distribution in the monogamous titi monkey (Callicebus cupreus): Implications for social behavior and endocrine functioning. Neuroscience, 290, 421–434. 10.1016/j.neuroscience.2015.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino ME, & Gold PE (1994). Task-dependent effects of intra-amygdala morphine injections: Attenuation by intra-amygdala glucose injections. The Journal of Neuroscience, 14(12), 7478–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall-Thompson JF, Pescatore KA, & Unterwald EM (2010). A role for delta opioid receptors in the central nucleus of the amygdala in anxiety-like behaviors. Psychopharmacology, 212(4), 585–595. 10.1007/s00213-010-1980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao ZR, Yamano M, Shiosaka S, Shinohara A, & Tohyama M (1987). Origin of leucine-enkephalin fibers and their two main afferent pathways in the bed nucleus of the stria terminalis in the rat. Experimental Brain Research, 65(2), 411–420. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3549348. [DOI] [PubMed] [Google Scholar]
- Rattan AK, Koo KL, Tejwani GA, & Bhargava HN (1992). The effect of morphine tolerance dependence and abstinence on immunoreactive dynorphin (1–13) levels in discrete brain regions, spinal cord, pituitary gland and peripheral tissues of the rat. Brain Research, 584(1–2), 207–212, 202. [DOI] [PubMed] [Google Scholar]
- Ray R, Ruparel K, Newberg A, Wileyto EP, Loughead JW, Divgi C, … Lerman C (2011). Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proceedings of the National Academy of Sciences of the United States of America, 108(22), 9268–9273. 10.1073/pnas.1018699108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Drolet G, & Van Bockstaele EJ (2008). Dynorphin and stress-related peptides in rat locus coeruleus: Contribution of amygdalar efferents. The Journal of Comparative Neurology, 508(4), 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Kravets JL, Connelly KL, Unterwald EM, & Van Bockstaele EJ (2017). Localization of the delta opioid receptor and corticotropin-releasing factor in the amygdalar complex: Role in anxiety. Brain Structure & Function, 222(2), 1007–1026. 10.1007/s00429-016-1261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro SC, Kennedy SE, Smith YR, Stohler CS, & Zubieta JK (2005). Interface of physical and emotional stress regulation through the endogenous opioid system and mu-opioid receptors. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 29(8), 1264–1280. 10.1016/j.pnpbp.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM, & Shipley MT (1991). Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: Topography and reciprocity. The Journal of Comparative Neurology, 303(1), 121–131. [DOI] [PubMed] [Google Scholar]
- Rodgers RJ (1977). Elevation of aversive threshold in rats by intra-amygdaloid injection of morphine sulphate. Pharmacology, Biochemistry, and Behavior, 6(4), 385–390. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/882576. [DOI] [PubMed] [Google Scholar]
- Rodgers RJ, & File SE (1979). Exploratory behaviour and aversive thresholds following intra-amygdaloid application of opiates in rats. Pharmacology Biochemistry and Behavior, 11(5), 505–511. 10.1016/0091-3057(79)90033-9. [DOI] [PubMed] [Google Scholar]
- Rodgers RJ (1997). Animal models of ànxiety’: Where next? Behavioural Pharmacology, 8(6–7), 477–496. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9832964. [DOI] [PubMed] [Google Scholar]
- Scavone JL, Asan E, & Van Bockstaele EJ (2011). Unraveling glutamate-opioid receptor interactions using high-resolution electron microscopy: Implications for addiction-related processes. Experimental Neurology, 229(2), 207–213. 10.1016/j.expneurol.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer TE, Strain EC, Greenberg BD, Preston KL, Lancaster E, Bigelow GE, … Pearlson GD (1998). Site of opioid action in the human brain: Mu and kappa agonists’ subjective and cerebral blood flow effects. The American Journal of Psychiatry, 155(4), 470–473. [DOI] [PubMed] [Google Scholar]
- Schoffelmeer AN, Yao YH, Gioannini TL, Hiller JM, Ofri D, Roques BP, & Simon EJ (1990). Cross-linking of human [125I]beta-endorphin to opioid receptors in rat striatal membranes: Biochemical evidence for the existence of a mu/delta opioid receptor complex. The Journal of Pharmacology and Experimental Therapeutics, 253(1), 419–426. [PubMed] [Google Scholar]
- Shah YB, Haynes L, Prior MJ, Marsden CA, Morris PG, & Chapman V (2005). Functional magnetic resonance imaging studies of opioid receptor-mediated modulation of noxious-evoked BOLD contrast in rats. Psychopharmacology (Berl), 180(4), 761–773. [DOI] [PubMed] [Google Scholar]
- Shimada S, Inagaki S, Kubota Y, Ogawa N, Shibasaki T, & Takagi H (1989). Coexistence of peptides (corticotropin releasing factor/neurotensin and substance P somatostatin) in the bed nucleus of the stria terminalis and central amygdaloid nucleus of the rat. Neuroscience, 30, 377–383. [DOI] [PubMed] [Google Scholar]
- Shin MS, & Helmstetter FJ (2005). Antinociception following application of DAMGO to the basolateral amygdala results from a direct interaction of DAMGO with Mu opioid receptors in the amygdala. Brain Research, 1064(1–2), 56–65. [DOI] [PubMed] [Google Scholar]
- Sim LJ, Selley DE, Dworkin SI, & Childers SR (1996). Effects of chronic morphine administration on mu opioid receptor-stimulated [35S] GTPgammaS autoradiography in rat brain. The Journal of Neuroscience, 16(8), 2684–2692. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8786444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simerly RB, Chang C, Muramatsu M, & Swanson LW (1990). Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: An in situ hybridization study. The Journal of Comparative Neurology, 294, 76–95. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Selley DE, Vogt LJ, Childers SR, & Martin TJ (2000). Chronic heroin self-administration desensitizes [micro] opioid receptor-activated G-proteins in specific regions of rat brain. The Journal of Neuroscience, 20(12), 4555–4562. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10844025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton KH, Oren D, Gutman DA, Easterling K, Holtzman SG, Nemeroff CB, & Owens MJ (2007). The CRF1 receptor antagonist, R121919, attenuates the severity of precipitated morphine withdrawal. European Journal of Pharmacology, 571(1), 17–24. 10.1016/j.ejphar.2007.05.041. [DOI] [PubMed] [Google Scholar]
- Smith Y, & Pare D (1994). Intra-amygdaloid projections of the lateral nucleus in the cat: PHA-L anterograde labeling combined with postembedding GABA and glutamate immunocytochemistry. The Journal of Comparative Neurology, 342(2), 232–248. 10.1002/cne.903420207. [DOI] [PubMed] [Google Scholar]
- Smith Y, Pare JF, & Pare D (2000). Differential innervation of parvalbumin-immunoreactive interneurons of the basolateral amygdaloid complex by cortical and intrinsic inputs. The Journal of Comparative Neurology, 416(4), 496–508. [PubMed] [Google Scholar]
- Stinus L, Le Moal M, & Koob GF (1990). Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience, 37(3), 767. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Norton FE, & Guyenet PG (1993). Autonomic areas of rat brain exhibit increased fos-like immunoreactivity during opiate withdrawal in rats. Brain Research, 624(1–2), 19–28, 24. [DOI] [PubMed] [Google Scholar]
- Sugita S, & North RA (1993). Opioid actions on neurons of rat lateral amygdala in vitro. Brain Research, 612, 151–155. [DOI] [PubMed] [Google Scholar]
- Sugita S, Tanaka E, & North RA (1993). Membrane properties and synaptic potentials of three types of neurone in rat lateral amygdala. The Journal of Physiology, 460, 705–718. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8487215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama A, Yamada M, Saitoh A, Nagase H, Oka JI, & Yamada M (2018). Administration of a delta opioid receptor agonist KNT-127 to the basolateral amygdala has robust anxiolytic-like effects in rats. Psychopharmacology, 235(10), 2947–2955. 10.1007/s00213-018-4984-7. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Yoshida M, Emoto H, & Ishii H (2000). Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: Basic studies. European Journal of Pharmacology, 405(1–3), 397–406. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11033344. [DOI] [PubMed] [Google Scholar]
- Terenius L, & Wahlstrom A (1975). Search for an endogenous ligand for the opiate receptor. Acta Physiologica Scandinavica, 94(1), 74–81. 10.1111/j.1748-1716.1975.tb05863.x. [DOI] [PubMed] [Google Scholar]
- Tershner SA, & Helmstetter FJ (2000). Antinociception produced by mu opioid receptor activation in the amygdala is partly dependent on activation of mu opioid and neurotensin receptors in the ventral periaqueductal gray. Brain Research, 865, 17–26. [DOI] [PubMed] [Google Scholar]
- Upadhyay J, Maleki N, Potter J, Elman I, Rudrauf D, Knudsen J, … Borsook D (2010). Alterations in brain structure and functional connectivity in prescription opioid-dependent patients. Brain, 133(Pt 7), 2098–2114. 10.1093/brain/awq138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino AL, & Kastin AJ (2000). Endogenous opiates: 1999. Peptides, 21(12), 1975–2034. [DOI] [PubMed] [Google Scholar]
- Valverde O, Fournie-Zaluski MC, Roques BP, & Maldonado R (1996). Similar involvement of several brain areas in the antinociception of endogenous and exogenous opioids. European Journal of Pharmacology, 312(1), 15–25. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Chan J, & Biswas A (1996). Ultrastructural evidence for convergence of enkephalin and adrenaline-containing axon terminals on common targets and their presynaptic associations in the rat nucleus locus coeruleus. Brain Research, 718, 61–75. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Qian Y, Sterling RC, & Page ME (2008). Low dose naltrexone administration in morphine dependent rats attenuates withdrawal-induced norepinephrine efflux in forebrain. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 32(4), 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kooy D, Mucha RF, O’Shaughnessy M, & Bucenieks P (1982). Reinforcing effects of brain microinjections of morphine revealed by conditioned place preference. Brain Research, 243(1), 107–117. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7116146. [DOI] [PubMed] [Google Scholar]
- Veinante P, Stoeckel ME, & Freund-Mercier MJ (1997). GABA- and peptide-immunoreactivies co-localize in the rat central extended amygdala. NeuroReport, 8(13), 2985–2989. [DOI] [PubMed] [Google Scholar]
- Veinante P, Stoeckel ME, Lasbennes F, & Freund-Mercier MJ (2003). c-Fos and peptide immunoreactivities in the central extended amygdala of morphine-dependent rats after naloxone-precipitated withdrawal. The European Journal of Neuroscience, 18(5), 1295–1305. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12956728. [DOI] [PubMed] [Google Scholar]
- Wamsley JK, Young WS 3rd, & Kuhar MJ (1980). Immunohistochemical localization of enkephalin in rat forebrain. Brain Research, 190(1), 153–174. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6247008. [DOI] [PubMed] [Google Scholar]
- Weerts EM, Wand GS, Kuwabara H, Munro CA, Dannals RF, Hilton J, … McCaul ME (2011). Positron emission tomography imaging of mu- and delta-opioid receptor binding in alcohol-dependent and healthy control subjects. Alcoholism, Clinical and Experimental Research, 35(12), 2162–2173. 10.1111/j.1530-0277.2011.01565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook RF, Good AJ, & Kiernan MJ (1997). Microinjection of morphine into the nucleus accumbens impairs contextual learning in rats. Behavioral Neuroscience, 111(5), 996–1013. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Burghardt PR, Lugo JN Jr., Primeaux SD, & Wilson SP (2003). Effect of amygdalar opioids on the anxiolytic properties of ethanol. Annals of the New York Academy of Sciences, 985, 472–475. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12724179. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Grillo CA, Fadel JR, & Reagan LP (2015). Stress as a one-armed bandit: Differential effects of stress paradigms on the morphology, neurochemistry and behavior in the rodent amygdala. Neurobiology of Stress, 1, 195–208. 10.1016/j.ynstr.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, & Junor L (2008). The role of amygdalar mu-opioid receptors in anxiety-related responses in two rat models. Neuropsychopharmacology, 33, 2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Mascagni F, & McDonald AJ (2002). Sex differences in delta opioid receptor immunoreactivity in rat medial amygdala. Neuroscience Letters, 328(2), 160–164. [DOI] [PubMed] [Google Scholar]
- Winters BL, Gregoriou GC, Kissiwaa SA, Wells OA, Medagoda DI, Hermes SM, … Bagley EE (2017). Endogenous opioids regulate moment-to-moment neuronal communication and excitability. Nature Communications. 8, 14611. 10.1038/ncomms14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yang H, Du X, Ma Q, Song J, Chen M, … Zheng P (2014). Morphine and DAMGO produce an opposite effect on presynaptic glutamate release via different downstream pathways of mu opioid receptors in the basolateral amygdala. Neuropharmacology, 86, 353–361. 10.1016/j.neuropharm.2014.08.021. [DOI] [PubMed] [Google Scholar]
- Zardetto-Smith AM, Moga MM, Magnuson DJ, & Gray TS (1988). Lateral hypothalamic dynorphinergic efferents to the amygdala and brainstem in the rat. Peptides, 9(5), 1121–1127. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2469062. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Babapoor-Farrokhran S, Babapoor-Farrokhran S, & Rezayof A (2008). Involvement of opioidergic system of the ventral hippocampus, the nucleus accumbens or the central amygdala in anxiety-related behavior. Life Sciences, 82(23–24), 1175–1181. [DOI] [PubMed] [Google Scholar]
- Zhang J, & McDonald AJ (2016). Light and electron microscopic analysis of enkephalin-like immunoreactivity in the basolateral amygdala, including evidence for convergence of enkephalin-containing axon terminals and norepinephrine transporter-containing axon terminals onto common targets. Brain Research, 1636, 62–73. 10.1016/j.brainres.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Muller JF, & McDonald AJ (2013). Noradrenergic innervation of pyramidal cells in the rat basolateral amygdala. Neuroscience, 228, 395–408. 10.1016/j.neuroscience.2012.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Muller JF, & McDonald AJ (2015). Mu opioid receptor localization in the basolateral amygdala: An ultrastructural analysis. Neuroscience, 303, 352–363. 10.1016/j.neuroscience.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, & Pan ZZ (2004). Synaptic properties and postsynaptic opioid effects in rat central amygdala neurons. Neuroscience, 127(4), 871–879. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15312899. [DOI] [PubMed] [Google Scholar]
- Zhu W, & Pan ZZ (2005). Mu-opioid-mediated inhibition of glutamate synaptic transmission in rat central amygdala neurons. Neuroscience, 133 (1), 97–103. [DOI] [PubMed] [Google Scholar]
- Zieglgansberger W, French ED, Siggins GR, & Bloom FE (1979). Opioid peptides may excite hippocampal pyramidal neurons by inhibiting adjacent inhibitory interneurons. Science, 205(4404), 415–417. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/451610. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Dannals RF, & Frost JJ (1999). Gender and age influences on human brain mu-opioid receptor binding measured by PET. American Journal of Psychiatry, 156(6), 842–848. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Ketter TA, Bueller JA, Xu Y, Kilbourn MR, Young EA, & Koeppe RA (2003). Regulation of human affective responses by anterior cingulate and limbic mu-opioid neurotransmission. Archives of General Psychiatry, 60(11), 1145–1153. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, … Stohler CS (2001). Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science, 293(5528), 311–315. 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, … Stohler CS (2002). mu-Opioid receptor-mediated antinociceptive responses differ in men and women. The Journal of Neuroscience, 22(12), 5100–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]