

Abstract

Conversion of N-Boc-protected quaternary proline derivatives under thermal Curtius rearrangement conditions was found to afford a series of ring-opened ketone and unsaturated pyrrolidine products instead of the expected carbamate species. The nature of the substituent on the quaternary carbon thereby governs the product outcome due to the stability of a postulated N-acyliminium species. A continuous flow process with in-line scavenging was furthermore developed to streamline this transformation and safely create products on a gram scale.
Since its first reports in the late 19th century, the Curtius rearrangement1 has established itself as one of the most versatile transformations to convert ubiquitous carboxylic acids into valuable amine derivatives.2 Acyl azide species (1a) are thereby the key intermediates in this process that release nitrogen upon rearranging into isocyanates that can be trapped with various nucleophiles (Scheme 1). Mechanistic studies support a concerted reaction pathway for thermal Curtius rearrangements, whereas photochemical alternatives proceed stepwise via nitrene intermediates.3 The popularity of the thermal Curtius rearrangement is evident from its regular application in natural product syntheses and drug development programs.4 Moreover, recent years have witnessed further developments by harnessing the salient features of continuous flow5 processing to yield modern variants6 that mitigate safety concerns due to the use of toxic azides, the release of nitrogen gas, and the potential for run-away phenomena related to the inherent exothermicity of this reaction sequence. Importantly, the exploitation of continuous flow processing has enabled several medicinal chemistry studies7 culminating in the safe execution of Curtius rearrangements on scales of >40 kg.8
Scheme 1. Overview of the Curtius Rearrangement.
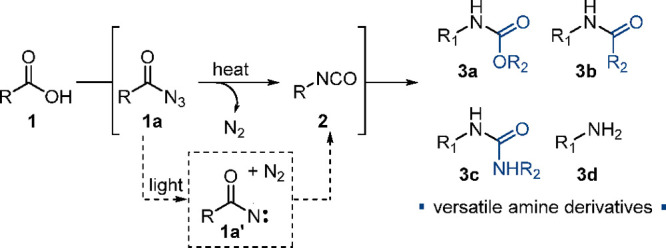
Despite these advances, essentially all studies on the Curtius rearrangement convert carboxylic acids into amines and amine derivatives such as carbamates, amides, and ureas (e.g., 3a–d), thus overlooking opportunities for new directions. To address this shortcoming, this Note reports the realization of an interrupted Curtius rearrangement process rendering a set of new products such as γ-amino ketones and unsaturated pyrrolidines from Boc-protected proline derivatives.
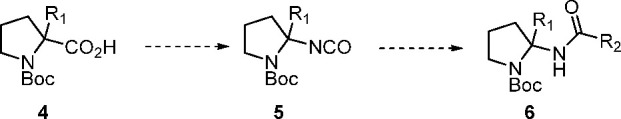
Expanding on prior studies that integrated enzymatic impurity tagging strategies with flow-based Curtius rearrangement reactions,9 this work evaluated the use of Boc-protected proline species bearing substitution on the chiral carbon (e.g., 4). Generation and subsequent trapping of the intermediate isocyanate 5 were thereby anticipated to afford a selection of versatile α-amino pyrrolidine species 6 as novel and potentially useful amine building blocks (Scheme 2).
Scheme 2. Intended Curtius Rearrangement Application for N-Boc Proline Derivatives.
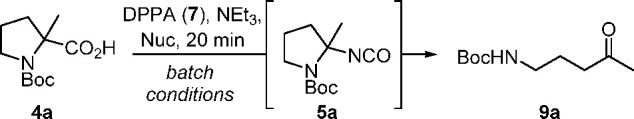
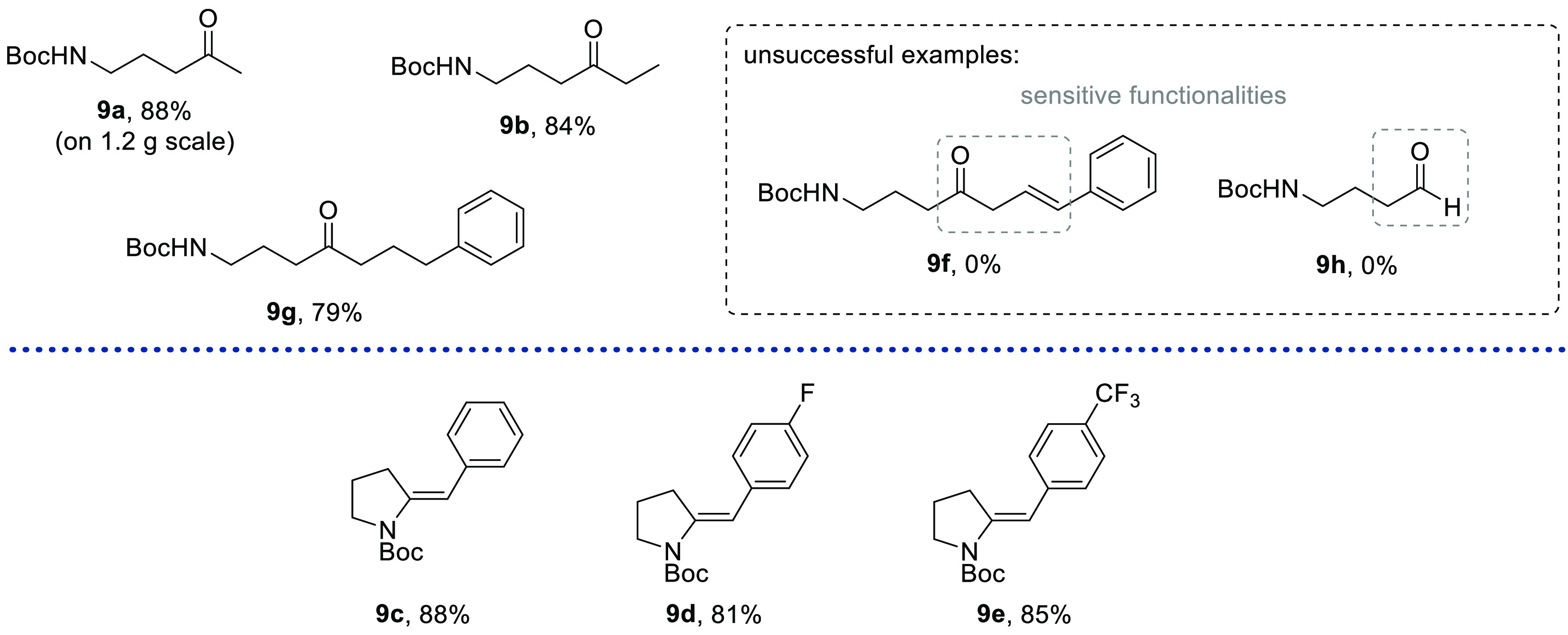
Commencing our study, the readily available proline derivative 4a (R1 = Me)10 was subjected to standard Curtius rearrangement conditions using DPPA (diphenylphosporyl azide, 7) as the azide source, triethylamine as the base, and benzyl alcohol as the trapping agent. Upon heating the reaction mixture in acetonitrile to reflux, the evolution of nitrogen gas was observed within minutes, indicating the onset of the anticipated rearrangement process. However, upon analyzing the crude reaction mixture by 1H NMR spectroscopy, not the anticipated Cbz product (R2 = OBn, 6a), but a new ring-opened product was observed (entry 1, Table 1). This material was subsequently identified as ketone 9a and confirmed by comparison to literature NMR data.11 As the evolution of nitrogen gas had indicated the generation of the anticipated isocyanate species, it was surmised that steric hindrance around the chiral center may have precluded nucleophilic attack by the modestly reactive benzyl alcohol. Reactions with other alcohols such as ethanol, methanol, and water (entries 2–4) resulted in the isolation of the same ketone product as the sole product in all three cases. Furthermore, alternative solvents such as toluene and dioxane did not alter the reaction outcome (entries 5 and 6). This outcome was reproduced even in the absence of an added alcohol nucleophile (entry 7). These results prompted consideration of an unprecedented reaction path that may be governed by the presence of the pyrrolidine ring as well as the quaternary center.
Table 1. Formation of Ketone 9a from N-Boc Proline Derivative 4a.
| entry | solvent | nucleophile (Nuc) | yield (%) |
|---|---|---|---|
| 1 | MeCN | BnOH | 85 |
| 2 | MeCN | EtOH | 81 |
| 3 | MeCN | MeOH | 77 |
| 4 | MeCN | water | 75 |
| 5 | 1,4-dioxane | BnOH | 71 |
| 6 | toluene | BnOH | 84 |
| 7 | MeCN | none | 81 |
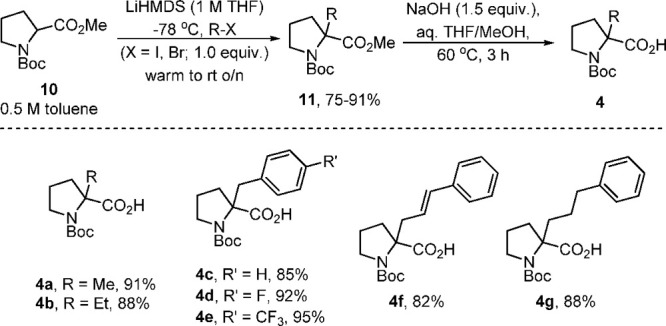
In view of the synthetic versatility of the 1,4-disubstitution pattern observed in this unexpected product and the desire to evaluate the generality of this process, a small selection of N-Boc-protected proline derivatives were subjected to these reaction conditions. Pleasingly, their syntheses were readily accomplished by lithiation of N-Boc proline methyl ester (10, 0.5 M in toluene, 1.0 equiv) with LiHMDS (1.0 M in THF, 1.1 equiv) as a base at −78 °C, followed by trapping the resulting carbanion with various electrophiles bearing alkyl and benzyl appendages (Scheme 3). Hydrolysis of the methyl ester (11) under alkaline conditions rendered the desired carboxylic acid building blocks 4 in high chemical yields.
Scheme 3. Batch Synthesis of Substrates 4 by Lithiation of N-Boc Proline Methyl Ester 10.
To streamline the following evaluation of the reaction scope, a flow reactor system was exploited to improve the process in view of safety, reproducibility, and scalability.12 In addition, toluene was preferred as a solvent in view of its higher boiling point and lower propensity to introduce water that may quench the acyl azide or isocyanate intermediate. Alcohols as nucleophiles were not added as they were found to have little effect on the reaction outcome (Table 1, entry 7). A simple flow process was devised and exploited in which streams of the substrate (4a–g, 1 M, toluene) in the presence of triethylamine as a base (1.0 equiv) and DPPA (7, 0.95 M, toluene, 0.95 equiv) were mixed in a T-piece before entering a heated flow coil (10 mL, 1/16 in id, PFA, 100 °C) with a residence time of 20 min (Scheme 4). The reaction mixture passed a back-pressure regulator (100 psi, Kinesis13) to ensure the steady release of nitrogen gas before collection in a receiving flask. A glass column containing a mixture of scavenger resins (Amberlyst A-21 and Amberlyst A-15;14 2 g each per mmol of substrate 4a–g) was optionally placed at the end of the reaction sequence to remove acidic and basic byproducts (NEt3, substrate, and diphenyl phosphonic acid).
Scheme 4. Flow Approach toward Evaluating the Reaction Scope of Interrupted Curtius Rearrangements.

The resulting flow study confirmed that the initial substrate bearing a methyl substituent (4a) smoothly underwent the rearrangement process rendering methyl ketone 9a in high chemical yield. Pleasingly, the same result was obtained when performing this process on a gram scale (Figure 1). Equally, introducing an ethyl group as a modified alkyl appendage was well tolerated and gave product 9b in high yield. An interesting case was observed when subjecting cinnamyl analogue 4f to the flow rearrangement protocol. As before, the formation of a gaseous byproduct (e.g., N2) was observed, and analysis of the crude material by 1H NMR indicated product formation in high yield; however, during purification by silica flash column chromatography, this material decomposed. It is surmised that the delicate skipped keto styryl moiety thereby tautomerized to a Michael acceptor, thus triggering decomposition by aldol or cycloaddition pathways. To support this assumption, cinnamyl ester 11f was converted under transfer hydrogenation conditions15 into its saturated counterpart, and pleasingly, the corresponding acid 4g furnished the anticipated ketone product (9g) in high yield and without any stability concerns. Furthermore, when subjecting unsubstituted N-Boc proline to the rearrangement process, decomposition of the labile aldehyde product (9h) was observed.
Figure 1.
Reaction scope rendering acyclic products and unsaturated pyrrolidines.
In addition, several benzyl-substituted substrates (e.g., 4c–e) were subjected to the flow protocol to verify if the analogous ring-opening process would take place. Formation of a gaseous byproduct was observed as before when employing the previous flow reaction conditions for this interrupted Curtius rearrangement process. However, when analyzing the purified reaction products (9c–e) by various NMR techniques, it was apparent that no ketone product was obtained. As these products were solids, single crystals were successfully grown for product 9e, enabling X-ray diffraction experiments to unambiguously determine the correct connectivity of this product.16 This clearly indicated the presence of a partially unsaturated pyrrolidine ring bearing an exocyclic, E-configured alkene instead of a ring-opened product. Comparison of the NMR data of the other benzyl-derived products (9c and 9d) indicated that the same product had formed as an exclusive E-isomer. Surprisingly relatively few mild and stereoselective methods for creating such unsaturated pyrrolidine scaffolds are reported in the literature. As these include examples that require transition metal catalysts and potentially render alternative alkene isomers or ring sizes,17 the methodology presented herein may serve as an attractive alternative.
To account for the observed reaction products, the following mechanism is proposed (Scheme 5). Activation of the carboxylic acid functionality with DPPA forms acyl azide 12, which subsequently undergoes thermal rearrangement to an isocyanate (5) accompanied by the release of nitrogen gas. This quaternary isocyanate is assumed to be too hindered to undergo nucleophilic attack as anticipated in the regular Curtius rearrangement. In the absence of a strong base, it is proposed that a unimolecular process in which (iso)cyanate anion is expelled and cyclic acyliminium18 species 13 is obtained. This highly electrophilic acyliminium ion then reacts with adventitious water to give ketones (9a,b,g). In the case of a neighboring benzylic methylene group, the corresponding acyliminium appears to tautomerize rapidly by loss of the benzylic proton giving unsaturated pyrrolidine structures 9c–e instead, which appear to be stable under the reaction conditions.19 It is believed that conjugation of the exocyclic alkene into the benzene ring for products 9c–e imparts higher stability toward attack by nucleophiles such as water and thus accounts for the observed reaction outcome.
Scheme 5. Proposed Reaction Mechanism Accounting for Bifurcated Pathway.

In conclusion, a novel reaction pathway for quaternary N-Boc proline species under thermal Curtius rearrangement conditions is reported. Fragmentation of the intermediate isocyanate species thereby renders a proposed N-acyliminium species, which facilitates ring-opening by adventitious water to give γ-amino ketone products in the case of small aliphatic substituents, whereas benzylic appendages render unsaturated pyrrolidines via tautomerization of this proposed N-acyliminium intermediate. A continuous flow protocol was successfully established to enable the safe and scaled exploration of this transformation. In view of its simplicity, high yields, and the value of the generated reaction products, this interrupted Curtius rearrangement method may find future synthetic applications in both batch and flow mode.
Experimental Section
Materials and Methods
Unless otherwise stated, all solvents were purchased from Fisher Scientific and used without further purification. Substrates and reagents were purchased from Fluorochem or Sigma-Aldrich and used as received.
The heating of reaction mixtures was achieved by using DrySyn metal heating blocks available from Asynt.
1H NMR spectra were recorded on 300, 400, and 500 MHz instruments and are reported relative to the residual solvent: CHCl3 (δ 7.26 ppm). 13C{1H} NMR spectra were recorded on the same instruments (100 and 125 MHz) and are reported relative to CHCl3 (δ 77.16 ppm). 19F NMR were recorded at 282 and 376 MHz. Data for 1H NMR are reported as follows: chemical shift (δ/ ppm) (integration, multiplicity, coupling constant (Hz)). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br s = broad singlet, app = apparent. Data for 13C{1H} NMR are reported in terms of chemical shift (δ/ppm) and multiplicity (C, CH, CH2, or CH3). COSY and HSQC experiments were used in the structural assignment.
IR spectra were obtained by use of a Bruker Platinum spectrometer (neat, ATR sampling) with the intensities of the characteristic signals being reported as weak (w, <20% of the tallest signal), medium (m, 21–70% of the tallest signal), or strong (s, >71% of the tallest signal).
High-resolution mass spectrometry was performed using the indicated techniques on a micromass LCT orthogonal time-of-flight mass spectrometer with leucine-enkephalin (Tyr-Gly-Phe-Leu) as an internal lock mass. GC–MS was performed on a Waters GCT Premier Agilent 7898 system (column Macherey–Nagel; Optima 5 MS, length 15 m, diameter 0.25 mm).
Melting points were recorded on a Stuart SMP10 melting point apparatus and are uncorrected.
Continuous flow experiments were performed on a Vaportec E-series system in combination with Omnifit glass columns (6.6 mm id, 150 mm length) filled with scavenging resins (A-15/A-21).
Synthetic Procedures and Spectroscopic Data
Synthesis of Substituted N-Boc Proline Derivatives 11b–h
For a reaction on 2 mmol scale, to a solution of the N-Boc proline methyl ester (10, 1.0 equiv, 2.0 mmol) in toluene (0.5 M, held at −78 °C) was added a solution of LiHMDS (1 M, THF, 1.1 equiv, 2.2 mmol) dropwise. The resulting mixture was allowed to warm to 0 °C within 20 min before cooling to −78 °C. A solution of alkyl bromide or iodide electrophile (0.5 M, toluene, 1.0 equiv, 2.0 mmol) was subsequently added dropwise, and the reaction mixture continued stirring for 12 h, eventually warming to rt. The reaction mixture was quenched by the addition of NH4Cl solution (sat. aq) and extracted (DCM/water). The combined organic layers were dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure to give the crude product. Purification was achieved via silica gel chromatography using a cyclohexane/ethyl acetate (75:15) eluent system.
1-(tert-Butyl) 2-Methyl 2-ethylpyrrolidine-1,2-dicarboxylate (11b).20
Yield: 640 mg (3.6 mmol, 83%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (400 MHz, CDCl3): δ 3.75–3.69 (m), 3.66 (s), 3.63–3.55 (m), 3.42–3.31 (m), 2.37–2.25 (m), 2.20–2.10 (m), 2.06–1.94 (m), 1.92–1.74 (m), 1.42 (s), 1.37 (s), 0.88–0.80 (m). 13C{1H} NMR (101 MHz, CDCl3): δ 175.6, 175.4, 154.0, 153.8, 79.8, 79.3, 68.3, 67.8, 52.0, 51.9, 48.7, 48.6, 36.9, 35.6, 28.4, 28.3, 27.8, 26.6, 23.2, 22.7, 7.9. IR (neat): ν/cm–1 2974 (m), 2879 (w), 1741 (m), 1693 (s), 1455 (m), 1386 (s), 1234 (m), 1157 (s), 1130 (s), 1075 (m), 1002 (m), 772 (m). HRMS (TOF-ESI+): calcd for C13H23NO4Na, 280.1519; found, 280.1522 (M + Na+).
1-(tert-Butyl) 2-Methyl 2-benzylpyrrolidine-1,2-dicarboxylate (11c)
Yield: 870 mg (2.7 mmol, 91%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (500 MHz, CDCl3): δ 7.30–7.21 (m), 7.14 (td, J = 6.8, 5.9, 3.2 Hz), 3.77 (d, J = 14.0 Hz), 3.75 (s), 3.57 (d, J = 13.8 Hz), 3.48 (dt, J = 10.5, 7.5 Hz), 3.39 (dt, J = 10.4, 7.2 Hz), 3.05 (d, J = 13.8 Hz), 3.04 (d, J = 13.8 Hz), 2.99 (ddd, J = 10.5, 7.6, 5.3 Hz), 2.88 (ddd, J = 10.4, 7.6, 5.6 Hz), 2.12–1.97 (m), 1.58 (ddt, J = 18.2, 7.4, 5.2 Hz), 1.51 (s), 1.49 (s), 1.00–0.83 (m). 13C{1H} NMR (126 MHz, CDCl3): δ 175.3, 175.1, 154.1, 153.5, 137.3, 136.9, 130.8, 130.7, 128.2, 128.0, 126.6, 126.4, 80.3, 79.6, 68.3, 68.0, 52.3, 48.2, 39.7, 38.5, 36.6, 35.4, 28.5, 28.4, 22.8, 22.2. IR (neat): ν/cm–1 2975 (m), 2877 (w), 1741 (m), 1694 (s), 1454 (m), 1389 (s), 1366 (m), 1251 (m), 1168 (s), 1119 (m), 1020 (m), 704 (m). HRMS (TOF-ESI+): calcd for C18H25NO4Na, 342.1676; found, 342.1679 (M + Na+).
1-(tert-Butyl) 2-Methyl 2-(4-fluorobenzyl)pyrrolidine-1,2-dicarboxylate (11d)
Yield: 750 mg (2.3 mmol, 75%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (500 MHz, CDCl3): δ 7.11–7.06 (m), 6.98–6.92 (m), 3.74 (s), 3.73 (s), 3.54–3.45 (m), 3.40 (dt, J = 10.6, 7.2 Hz), 3.03 (d, J = 14.2 Hz), 3.01 (d, J = 14.2 Hz), 3.00–2.96 (m), 2.89 (ddd, J = 10.4, 7.4, 5.7 Hz), 2.09–1.98 (m), 1.65–1.55 (m), 1.49 (s), 1.47 (s), 1.05–0.87 (m). 13C{1H} NMR (126 MHz, CDCl3): δ 175.1, 175.0, 161.9 (CF, d, J = 246 Hz), 161.8 (CF, d, J = 246 Hz), 154.2, 153.5, 132.9 (d, J = 4 Hz), 132.6 (d, J = 4 Hz), 132.1 (d, J = 8 Hz), 132.0 (d, J = 8 Hz), 115.1 (d, J = 21 Hz), 114.8 (d, J = 21 Hz), 80.4, 79.7, 68.3, 67.9, 52.3, 52.3, 48.3, 48.2, 38.9, 37.7, 36.5, 35.3, 28.4, 28.4, 22.8, 22.2. IR (neat): ν/cm–1 2976 (m), 2877 (w), 1741 (s), 1693 (s), 1510 (s), 1388 (s), 1251 (m), 1221 (m), 1160 (s), 1131 (m), 1016 (m), 844 (m), 772 (m). HRMS (TOF-ESI+): calcd for C18H24FNO4Na, 360.1582; found, 360.1585 (M + Na+).
1-(tert-Butyl) 2-Methyl 2-(4-(trifluoromethyl)benzyl)pyrrolidine-1,2-dicarboxylate (11e)
Yield: 920 mg (2.4 mmol, 80%). Appearance: white solid. Melting range: 94–96 °C. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (400 MHz, CDCl3): δ 7.48 (dd, J = 8.2, 4.3 Hz), 7.22 (d, J = 7.9 Hz), 3.78 (d, J = 13.8 Hz), 3.71 (s), 3.70 (s), 3.56 (d, J = 13.8 Hz), 3.47 (dt, J = 10.6, 7.3 Hz), 3.37 (dt, J = 10.5, 7.1 Hz), 3.08 (d, J = 13.8 Hz), 3.07 (d, J = 13.8 Hz), 2.95 (ddd, J = 10.5, 7.6, 5.5 Hz), 2.86 (ddd, J = 10.4, 7.4, 5.8 Hz), 2.10–1.94 (m), 1.64–1.54 (m), 1.45 (s), 1.43 (s), 1.01–0.86 (m). 13C{1H} NMR (101 MHz, CDCl3): δ 174.8, 174.7, 154.2, 153.4, 141.5, 141.2, 129.0 (q, J = 32 Hz), 128.8 (q, J = 32 Hz), 125.0 (q, J = 4 Hz), 124.8 (q, J = 4 Hz), 124.3 (CF3, q, J = 273 Hz), 124.2 (CF3, q, J = 273 Hz), 80.5, 79.8, 68.1, 67.8, 52.3, 48.2, 39.7, 38.4, 36.6, 35.3, 28.3, 28.3, 22.7, 22.2.IR (neat): ν/cm–1 2977 (w), 2880 (w), 1742 (m), 1694 (s), 1389 (s), 1324 (s), 1251 (m), 1163 (s), 1123 (s), 1111 (s), 1067 (s), 1019 (m), 851 (m). HRMS (TOF-ESI+): calcd for C19H24NF3O4Na, 410.1550; found, 410.1551 (M + Na+).
1-(tert-Butyl) 2-Methyl 2-cinnamylpyrrolidine-1,2-dicarboxylate (11f)
Yield: 550 mg (1.6 mmol, 81%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (300 MHz, CDCl3): δ 7.40–7.18 (m), 6.48 (app d, J = 15.7 Hz), 6.27–6.08 (m), 3.76 (s, 3H), 3.73–3.52 (m), 3.48–3.32 (m), 3.26 (dd, J = 14.1, 6.9 Hz), 3.08 (dd, J = 14.3, 6.3 Hz), 2.81 (d, J = 9.1 Hz), 2.76 (d, J = 8.6 Hz), 2.23–2.05 (m), 1.96–1.73 (m), 1.48 (s), 1.46 (s). 13C{1H} NMR (126 MHz, CDCl3): δ 175.1, 174.9, 154.0, 153.6, 137.6, 137.3, 133.9, 133.7, 128.6, 128.5, 127.3, 127.1, 126.3, 126.1, 125.3, 124.9, 80.2, 79.6, 67.9, 67.3, 52.2, 52.2, 48.5, 48.5, 38.9, 37.6, 37.1, 35.9, 28.4, 28.4, 23.2, 22.7. IR (neat): ν/cm–1 2975 (m), 2876 (w), 1741 (s), 1697 (s), 1390 (s), 1249 (m), 1164 (m), 1135 (m), 748 (w), 695 (w). HRMS (TOF-ESI+): calcd for C20H27NO4Na, 368.1832; found, 368.1835 (M + Na+).
1-(tert-Butyl) 2-Methyl 2-(3-phenylpropyl)pyrrolidine-1,2-dicarboxylate (11g)
To a solution of ester 11f (1 mmol, 1.0 equiv) in EtOAc (10 mL, 0.1 M) were added ammonium formate (5 mmol, 5.0 equiv) and Pd/C (10%, ca. 100 mg). This solution was then heated under reflux for 5 h when TLC indicated full conversion of the substrate. After cooling to room temperature, the mixture was filtered through a pad of Celite, washed with EtOAc (10 mL), and concentrated in vacuo to give the saturated product 11g (quant.).
Yield: 344 mg (1.0 mmol, 99%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (500 MHz, CDCl3): δ 7.32–7.24 (m), 7.19 (app dd, J = 8.2, 4.4 Hz), 3.75–3.71 (m), 3.70 (s), 3.69 (s), 3.64–3.56 (m), 3.42–3.30 (m), 2.74–2.56 (m), 2.38 (ddd, J = 14.0, 12.2, 4.8 Hz), 2.20 (td, J = 13.3, 4.4 Hz), 2.13–1.94 (m), 1.94–1.84 (m), 1.79 (tq, J = 12.6, 6.5 Hz), 1.71–1.60 (m), 1.60–1.51 (m), 1.45 (s), 1.33 (s). 13C{1H} NMR (126 MHz, CDCl3): δ 175.5, 175.3, 154.0, 153.8, 142.6, 142.1, 128.5, 128.4, 128.3, 128.3, 125.8, 125.7, 79.8, 79.4, 67.9, 67.4, 52.1, 52.0, 48.6, 48.5, 37.5, 36.2, 36.1, 34.7, 33.9, 28.4, 28.2, 25.9, 25.4, 23.2, 22.8. IR (neat): ν/cm–1 2974 (m), 2870 (w), 1741 (m), 1697 (s), 1454 (w), 1391 (s), 1366 (m), 1235 (m), 1163 (m), 1132 (m), 700 (w). HRMS (TOF-ESI+): calcd for C20H29NO4Na, 370.1989; found, 370.1994 (M + Na+).
Synthesis of Acid Products 4a–g
For a reaction on 2 mmol scale, to a solution of ester intermediate 11 (2 mmol, 1.0 equiv, 2 M, THF/MeOH, 50:50 vol) was added a solution of NaOH (1 M aqueous, 4 mL, 2.0 equiv). The resulting mixture was heated to reflux until TLC indicated full consumption of the substrate (ca. 3 h). The resulting solution was partitioned between EtOAc and water. The aqueous layer was subsequently acidified by the addition of 1 M HCl (ca. 5 mL) and extracted with DCM, giving the target acid products after drying of the organic layer over anhydrous Na2SO4, filtration, and final evaporation under reduced pressure.
1-(tert-Butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic Acid (4a).21
Yield: 2.5 g (10.9 mmol, 91%). Appearance: colorless crystalline solid. Melting range: 129–131 °C (CHCl3). Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (500 MHz, CDCl3): δ 11.67 (br s), 3.60–3.39 (m), 2.38–2.30 (m), 2.26–2.22 (m), 1.92 (app tt, J = 10.2, 5.4 Hz), 1.86–1.79 (m), 1.55 (s), 1.48 (s), 1.42 (s), 1.38 (s). 13C{1H} NMR (126 MHz, CDCl3): δ 180.7, 179.0, 154.9, 153.7, 80.4, 80.4, 65.6, 64.8, 48.2, 47.8, 40.3, 39.0, 28.4, 28.3, 23.0, 22.8, 22.7, 22.1. IR (neat): ν/cm–1 3200–2800 (br), 2977 (m), 2884 (m), 1731 (s), 1619 (s), 1446 (m), 1417 (s), 1392 (s), 1366 (s), 1248 (m), 1159 (s), 1138 (s), 1080 (m), 892 (m), 852 (m), 772 (m), 583 (m). HRMS (TOF-ESI+): calcd for C11H20NO4, 230.1387; found, 230.1388 (M + H+).
1-(tert-Butoxycarbonyl)-2-ethylpyrrolidine-2-carboxylic Acid (4b)
Yield: 0.5 g (2.1 mmol, 88%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (400 MHz, CDCl3): δ 3.73–3.67 (m), 3.54–3.46 (m), 3.37 (dt, J = 10.7, 7.5 Hz), 3.29 (dt, J = 10.3, 8.0 Hz), 2.65–2.48 (m), 2.21–2.03 (m), 2.03–1.85 (m), 1.81–1.71 (m), 1.45 (s), 1.38 (s), 0.84 (app t, J = 7.5 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 180.9, 175.4, 156.9, 153.9, 81.8, 80.2, 70.8, 67.7, 49.4, 48.6, 36.9, 34.5, 28.3, 28.3, 27.4, 27.1, 22.7, 8.1, 7.7. IR (neat): ν/cm–1 3400–2400 (br), 2974 (m), 2880 (w), 1741 (m), 1698 (s), 1391 (s), 1367 (s), 1161 (s), 931 (m), 858 (m), 773 (m). HRMS (TOF-ESI+): calcd for C12H21NO4Na, 244.1543; found, 244.1550 (M + Na+).
2-Benzyl-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic Acid (4c)
Yield: 0.7 g (2.3 mmol, 85%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (500 MHz, CDCl3): δ 7.31–7.22 (m,), 7.17–7.13 (m), 3.67 (d, J = 13.9 Hz), 3.61 (d, J = 13.8 Hz), 3.51 (dt, J = 10.5, 7.3 Hz), 3.40 (ddd, J = 10.7, 7.5, 5.0 Hz), 3.10 (d, J = 13.7 Hz), 3.03 (d, J = 14.0 Hz), 3.01–2.96 (m), 2.93 (dt, J = 10.7, 7.5 Hz), 2.36 (ddd, J = 13.0, 7.2, 5.6 Hz), 2.21–2.09 (m), 2.01 (ddd, J = 13.0, 8.3, 7.1 Hz), 1.68–1.59 (m), 1.54 (s), 1.52 (s), 1.19 (tdd, J = 12.4, 7.0, 5.3 Hz), 0.93 (dp, J = 12.3, 7.5 Hz). 13C{1H} NMR (126 MHz, CDCl3): δ 180.6, 177.1, 156.1, 153.6, 136.7, 136.2, 130.7, 130.6, 128.3, 128.2, 126.8, 126.7, 81.3, 80.8, 69.8, 68.0, 49.0, 48.3, 39.4, 38.6, 36.7, 34.7, 28.5, 28.4, 22.4, 22.2. IR (neat): ν/cm–1 3300–2700 (br), 2976 (m), 2878 (m), 1737 (m), 1697 (s), 1392 (s), 1368 (m), 1167 (s), 1139 (m), 994 (m), 703 (m). HRMS (TOF-ESI+): calcd for C17H24NO4, 306.1700; found, 306.1705 (M + H+).
1-(tert-Butoxycarbonyl)-2-(4-fluorobenzyl)pyrrolidine-2-carboxylic Acid (4d)
Yield: 0.5 g (1.5 mmol, 92%). Appearance: waxy white solid. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (500 MHz, CDCl3): δ 7.16–7.09 (m), 7.01–6.94 (m), 3.67 (d, J = 13.9 Hz), 3.59–3.50 (m), 3.43 (ddd, J = 10.6, 7.5, 5.2 Hz), 3.08 (d, J = 14.0 Hz), 3.05–2.98 (m), 2.94 (dt, J = 10.7, 7.3 Hz), 2.30 (ddd, J = 13.2, 7.2, 5.9 Hz), 2.18 (dt, J = 13.5, 7.7 Hz), 2.11–1.95 (m), 1.73–1.63 (m), 1.53 (s), 1.51 (s), 1.29–1.18 (m), 0.99 (dp, J = 12.1, 7.5 Hz). 13C{1H} NMR (126 MHz, CDCl3): δ 180.8, 178.0, 161.9 (2xd, J = 247 Hz), 155.6, 153.6, 132.3 (2x), 132.0 (m), 115.2 (d, J = 21 Hz), 115.1 (d, J = 21 Hz), 81.0, 81.0, 69.2, 67.9, 48.8, 48.3, 38.6, 37.6, 36.7, 34.9, 28.5, 28.4, 22.5, 22.3. IR (neat): ν/cm–1 3300–2600 (br), 2977 (m), 2880 (w), 1737 (m), 1696 (s), 1510 (s), 1392 (s), 1368 (s), 1223 (s), 1160 (s), 1140 (m), 996 (m), 840 (m), 771 (m). HRMS (TOF-ESI+): calcd for C17H23FNO4, 324.1611; found, 324.1618 (M + H+).
1-(tert-Butoxycarbonyl)-2-(4-(trifluoromethyl)benzyl)pyrrolidine-2-carboxylic Acid (4e)
Yield: 0.6 g (1.6 mmol, 95%). Appearance: waxy solid. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (400 MHz, CDCl3): δ 7.56–7.52 (m), 7.28–7.24 (m), 3.69 (d, J = 13.7 Hz), 3.62 (d, J = 13.9 Hz), 3.53 (dt, J = 10.5, 7.3 Hz), 3.46–3.39 (m), 3.18 (d, J = 13.6 Hz), 3.08 (d, J = 13.9 Hz), 3.03–2.91 (m), 2.37 (dt, J = 12.7, 6.6 Hz), 2.24–2.15 (m), 2.08–2.02 (m), 1.98–1.88 (m), 1.73–1.58 (m), 1.52 (s), 1.48 (s), 1.31–1.21 (m), 1.00 (dt, J = 12.5, 7.3 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 179.9, 176.3, 156.1, 153.5, 140.9, 140.3, 131.0, 130.8, 125.2, 125.1, 125.1, 81.6, 81.1, 49.0, 48.3, 39.4, 38.5, 36.7, 34.7, 28.4, 28.4, 22.4, 22.3, 20.6; some resonances were not observed clearly. IR (neat): ν/cm–1 3400–2700 (br), 2977 (m), 2882 (w), 1738 (m), 1696 (s), 1392 (s), 1324 (s), 1163 (s), 1124 (s), 1112 (s), 1068 (s), 851 (m). HRMS (TOF-ESI+): calcd for C18H22F3NO4Na, 396.1393; found, 396.1393 (M + Na+).
1-(tert-Butoxycarbonyl)-2-cinnamylpyrrolidine-2-carboxylic Acid (4f)
Yield: 0.6 g (1.8 mmol, 82%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (300 MHz, CDCl3): δ 7.44–7.21 (m), 6.53 (d, J = 15.7 Hz), 6.04 (dt, J = 15.5, 7.5 Hz), 3.59–3.48 (m), 3.38–3.28 (m), 3.09 (dd, J = 13.7, 7.9 Hz), 2.88 (dd, J = 13.9, 7.1 Hz), 2.80–2.67 (m), 2.27–2.17 (m), 2.05–1.92 (m), 1.85–1.74 (m), 1.51 (app s). 13C{1H} NMR (126 MHz, CDCl3): δ 180.3, 175.9, 156.5, 153.7, 137.2, 137.2, 134.8, 134.1, 128.6, 127.5, 127.4, 126.2, 126.2, 124.7, 123.3, 81.7, 80.7, 69.6, 67.3, 49.3, 48.5, 38.5, 37.3, 37.2, 34.9, 28.4, 28.4, 22.7, 22.7. IR (neat): ν/cm–1 3300–2700 (br), 2975 (m), 2879 (w), 1733 (m), 1695 (s), 1390 (s), 1367 (s), 1247 (m), 1164 (s), 998 (m), 969 (m), 734 (s), 694 (s). HRMS (TOF-ESI+): calcd for C19H25NO4Na, 354.1676; found, 354.1677 (M + Na+).
1-(tert-Butoxycarbonyl)-2-(3-phenylpropyl)pyrrolidine-2-carboxylic Acid (4g)
Yield: 0.4 g (1.2 mmol, 88%). Appearance: clear oil. Mixtures of Boc-rotamers were observed by NMR spectroscopy; please see copies of 1H and 13C NMR spectra in Supporting Information. 1H NMR (400 MHz, CDCl3): δ 7.29–7.23 (m), 7.20–7.12 (m), 3.75–3.65 (m), 3.54–3.45 (m), 3.37 (dt, J = 10.9, 7.4 Hz), 3.32–3.22 (m), 2.74–2.54 (m), 2.25–1.98 (m), 1.94–1.72 (m), 1.69–1.50 (m,), 1.47 (s), 1.31 (s). 13C{1H} NMR (101 MHz, CDCl3): δ 180.9, 175.7, 156.7, 153.8, 142.0, 141.9, 128.4, 128.3, 128.3, 125.9, 125.8, 81.8, 80.3, 70.0, 67.3, 49.2, 48.5, 37.5, 36.0, 35.8, 35.2, 34.3, 33.9, 28.4, 28.2, 26.0, 25.3, 22.7, 22.7. IR (neat): ν/cm–1 3250–2600 (br), 2973 (m), 2932 (m), 2873 (w), 1738 (m), 1694 (s), 1453 (m), 1389 (s), 1366 (s), 1244 (m), 1162 (s), 1136 (s), 856 (m), 749 (m), 698 (s). HRMS (TOF-ESI+): calcd for C19H28NO4 334.2013; found, 334.2017 (M + H+).
Synthesis of Products 9a–g
For a reaction on a 2 mmol scale, a solution containing substrate acid 4 (1 M, toluene, 2 mmol, 1.0 equiv) and NEt3 (2 mml, 1.0 equiv) was prepared and pumped using a Vaportec E-series flow reactor at a flow rate of 0.25 mL/min. A second solution containing DPPA (0.95 M, toluene, 1.9 mmol, 0.95 equiv) was pumped at the same flow rate and mixed with the substrate solution via a T-piece (1/16 in. id). The combined mixture was then directed into a heated coil reactor (10 mL volume, PFA tubing, 1/16 in. id) held at 100 °C resulting in a residence time of 20 min. Upon exiting this reactor coil, the mixture passed through a BPR (100 psi, Kinesis) before entering an Omnifit glass column containing A-15 and A-21 scavenger resins (mixed bed, ca. 2 equiv each) at an ambient temperature. The crude mixture was collected in a flask and evaporated under reduced pressure. Purification was performed via silica gel chromatography using EtOAc/cyclohexane (10–20% EtOAc) as an eluent system.
A scaled version of this procedure was used starting from acid 4a (1.55 g, 6.78 mmol) to prepare compound 9a (1.20 g, 5.97 mmol, 88%).
tert-Butyl (4-Oxopentyl)carbamate (9a).11
Yield: 1.20 g (5.97 mmol, 88%). Appearance: colorless oil. 1H NMR (400 MHz, CDCl3): δ/ppm 4.61 (br s, 1H), 3.08 (q, J = 6.6 Hz, 2H), 2.45 (t, J = 7.1 Hz, 2H), 2.11 (s, 3H), 1.72 (p, J = 7.0 Hz, 2H), 1.40 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3): δ/ppm 208.4 (C), 156.0 (C), 79.1 (C), 40.7 (CH2), 39.9 (CH2), 29.9 (CH3), 28.4 (3CH3), 24.2 (CH2). IR (neat): ν/cm–1 3365 (br, m), 2976 (m), 2932 (m), 1688 (s), 1517 (m), 1449 (w), 1391 (m), 1365 (s), 1249 (s), 1162 (s). HRMS (TOF-ESI+): calcd for C10H19NO3Na, 224.1257; found, 224.1259 (M + Na+).
tert-Butyl (4-Oxohexyl)carbamate (9b)
Yield: 289 mg (1.3 mmol, 84%). Appearance: colorless oil. 1H NMR (500 MHz, CDCl3): δ/ppm 4.60 (s, 1H), 3.12 (q, J = 6.6 Hz, 2H), 2.47–2.41 (m, 4H), 1.77 (p, J = 7.0 Hz, 2H), 1.44 (s, 9H), 1.06 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ/ppm 211.2 (C), 156.0 (C), 79.2 (C), 40.1 (CH2), 39.4 (CH2), 36.0 (CH2), 28.4 (3CH3), 24.1 (CH2), 7.8 (CH3). IR (neat): ν/cm–1 3362 (br), 2976 (m), 2936 (m), 1689 (s), 1517 (m), 1454 (m), 1365 (m), 1248 (s), 1165 (s), 1042 (m), 1023 (m), 980 (m), 875 (m), 780 (m). HRMS (TOF-ESI+): calcd for C11H21NO3Na 238.1414; found, 238.1415 (M + Na+).
tert-Butyl (E)-2-Benzylidenepyrrolidine-1-carboxylate (9c).17a
Yield: 380 mg (1.47 mmol, 88%). Appearance: white solid. Melting range: 78–81 °C. 1H NMR (500 MHz, CDCl3): δ/ppm 7.32–7.28 (m, 2H), 7.26–7.23 (m, 2H), 7.15–7.11 (m, 2H), 3.66 (t, J = 7.0 Hz, 2H), 2.82 (td, J = 7.4, 2.0 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H), 1.56 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3): δ/ppm 152.9 (C), 140.9 (C), 139.0 (C), 128.2 (2CH), 128.0 (2CH), 125.0 (CH), 108.3 (CH), 80.6 (C), 48.9 (CH2), 30.6 (CH2), 28.5 (3CH3), 22.1 (CH2). IR (neat): ν/cm–1 2975 (w), 2883 (w), 1702 (s), 1639 m), 1383 (s), 1323 (s), 1243 (m), 1159 (s), 1136 (s), 1076 (m), 1003 (m), 856 (m), 748 (m), 694 (s). HRMS (TOF-ESI+): calcd for C16H22NO2, 260.1645; found, 260.1647 (M + H+).
tert-Butyl (E)-2-(4-Fluorobenzylidene)pyrrolidine-1-carboxylate (9d)
Yield: 223 mg (0.81 mmol, 81%). Appearance: white solid. Melting range: 65–68 °C. 1H NMR (400 MHz, CDCl3): δ/ppm 7.14 (dd, J = 8.7, 5.6 Hz, 2H), 7.06 (br s, 1H), 6.94 (t, J = 8.8 Hz, 2H), 3.62 (t, J = 7.0 Hz, 2H), 2.72 (td, J = 7.4, 2.0 Hz, 2H), 1.82 (p, J = 7.2 Hz, 2H), 1.51 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3): δ/ppm 160.6 (CF, d, J = 243 Hz), 152.8 (C), 140.7 (C), 134.9 (C, d, J = 4 Hz), 129.4 (2CH, d, J = 8 Hz), 114.8 (2CH, d, J = 21 Hz), 107.2 (CH), 80.6 (C), 48.8 (CH2), 30.4 (CH2), 28.4 (3CH3), 22.0 (CH2). 19F-NMR (376 MHz, CDCl3): δ/ppm δ −118 (s). IR (neat): ν/cm–1 2977 (m), 2934 (w), 1703 (s), 1645 (m), 1507 (s), 1386 (s), 1329 (s), 1228 (m), 1158 (m), 1140 (s), 1002 (m), 860 (m), 768 (m). HRMS (TOF-ESI+): calcd for C16H20NFO2, 278.1551; found, 278.1552 (M + H+).
tert-Butyl (E)-2-(4-(Trifluoromethyl)benzylidene)pyrrolidine-1-carboxylate (9e)
Yield: 457 mg (1.40 mmol, 85%). Appearance: white solid. Melting range: 86–88 °C. 1H NMR (500 MHz, CDCl3): δ/ppm δ 7.51 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.2 Hz, 2H), 7.16 (br s, 1H), 3.66 (t, J = 6.9 Hz, 2H), 2.81 (td, J = 7.3, 2.0 Hz, 2H), 1.87 (p, J = 7.2 Hz, 2H), 1.54 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3): δ/ppm 152.7 (C), 143.0 (C), 142.8 (C), 128.0 (2CH), 126.6 (C, q, J = 32.3 Hz), 124.9 (2CH, q, J = 3.9 Hz), 124.5 (CF3, q, J = 270 Hz), 107.0 (CH), 81.0 (C), 49.0 (CH2), 30.7 (CH2), 28.3 (3CH3), 22.0 (CH2). 19F-NMR (282 MHz, CDCl3): δ/ppm δ −62.2 (s). IR (neat): ν/cm–1 2978 (w), 2889 (w), 1706 (m), 1637 (m), 1610 (m), 1386 (m), 1314 (s), 1244 (m), 1159 (s), 1140 (s), 1107 (s), 1066 (s), 1002 (m), 859 (s). HRMS (TOF-ESI+): calcd for C17H20F3NO2Na, 350.1338; found, 350.1338 (M + Na+). Crystal data: CCDC 2083011: C17H20F3NO2; monoclinic, a = 15.3119(2) Å, b = 10.2080(2) Å, c = 21.3592(3) Å, α = 90°, β = 92.8390(10)°, γ = 90°; Z = 8; space group P21/c; T = 100 K; R1 = 0.0872.
tert-Butyl (4-Oxo-7-phenylheptyl)carbamate (9g)
Yield: 238 mg (0.79 mmol, 79%). Appearance: clear oil. 1H NMR (500 MHz, CDCl3): δ/ppm 7.28 (t, J = 7.7 Hz, 2H), 7.22–7.14 (m, 3H), 4.58 (s, 1H), 3.10 (q, J = 6.7 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.41 (m, 4H), 1.90 (p, J = 7.4 Hz, 2H), 1.73 (p, J = 7.0 Hz, 2H), 1.43 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3): δ/ppm 210.3 (C), 156.0 (C), 141.5 (C), 128.5 (2CH), 128.4 (2CH), 125.9 (CH), 79.2 (C), 42.0 (CH2), 40.0 (CH2), 39.9 (CH2), 35.1 (CH2), 28.4 (3CH3), 25.2 (CH2), 24.0 (CH2). IR (neat): ν/cm–1 3368 (br, m), 2975 (m), 2931 (m), 1701 (s), 1513 (m), 1453 (m), 1391 (m), 1365 (m), 1248 (m), 1165 (s), 748 (m), 700 (s). HRMS (TOF-ESI+): calcd for C18H27NO3Na, 328.1883; found, 328.1886 (M + Na+).
Acknowledgments
We gratefully acknowledge support from Science Foundation Ireland through the SFI Industry Fellowship Program for the project entitled “Development of Continuous Biocatalysed Processes, Continuous Biocatalysed Chemicals (CATCH)” (19/IFA/7420 to M.B.) as well as the Infrastructure Call 2018 (18/RI/5702) and the European Regional Development Fund (12/RC2275_P2). We thank Conor Kelly and Helge Müller-Bunz (both UCD, School of Chemistry) for solving the X-ray structure reported herein.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01133.
Copies of 1H and 13C NMR spectra (PDF)
Accession Codes
CCDC 2083011 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Curtius T. Ueber Stickstoffwasserstoffsäure (Azoimid) N3H. Ber. Dtsch. Chem. Ges. 1890, 23, 3023–3033. 10.1002/cber.189002302232. [DOI] [Google Scholar]; b Curtius T. 20. Hydrazide und Azide organischer Säuren. J. Prakt. Chem. 1894, 50, 275–294. 10.1002/prac.18940500125. [DOI] [Google Scholar]
- For selected reports on complementary approaches, please see:; a Thomas M.; Alsarraf J.; Araji N.; Tranoy-Opalinski I.; Renoux B.; Papot S. The Lossen rearrangement from free hydroxamic acids. Org. Biomol. Chem. 2019, 17, 5420–5427. 10.1039/C9OB00789J. [DOI] [PubMed] [Google Scholar]; b Wallis E. S.; Lane J. F. The Hofmann Reaction. Organic Reactions 2011, 267. 10.1002/0471264180.or003.07. [DOI] [Google Scholar]; c Zagulyaeva A. A.; Banek C. T.; Yusubov M. S.; Zhdankin V. V. Hofmann Rearrangement of Carboxamides Mediated by Hypervalent Iodine Species Generated in Situ from Iodobenzene and Oxone: Reaction Scope and Limitations. Org. Lett. 2010, 12, 4644–4647. 10.1021/ol101993q. [DOI] [PubMed] [Google Scholar]; d Huang X.; Seid M.; Keillor J. W. A Mild and Efficient Modified Hofmann Rearrangement. J. Org. Chem. 1997, 62, 7495–7496. 10.1021/jo9708553. [DOI] [PubMed] [Google Scholar]; e Wolff H. The Schmidt Reaction. Organic Reactions 2011, 307–336. 10.1002/0471264180.or003.08. [DOI] [Google Scholar]; f Lang S.; Murphy J. A. Azide rearrangements in electron-deficient systems. Chem. Soc. Rev. 2006, 35, 146–156. 10.1039/B505080D. [DOI] [PubMed] [Google Scholar]
- a Wentrup C.; Bornemann H. The Curtius Rearrangement of Acyl Azides Revisited - Formation of Cyanate (R-O-CN). Eur. J. Org. Chem. 2005, 2005, 4521–4524. 10.1002/ejoc.200500545. [DOI] [Google Scholar]; b Eibler E.; Sauer J. Ein Beitrag zur Isocyanatbildung bei der Photolyse von Acylaziden. Tetrahedron Lett. 1974, 15, 2569–2572. 10.1016/S0040-4039(01)92295-6. [DOI] [Google Scholar]
- a Ghosh A. K.; Brindisi M.; Sarkar A. The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry. ChemMedChem 2018, 13, 2351–2373. 10.1002/cmdc.201800518. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ghosh A. K.; Sarkar A.; Brindisi M. The Curtius rearrangement: mechanistic insight and recent applications in natural product syntheses. Org. Biomol. Chem. 2018, 16, 2006–2027. 10.1039/C8OB00138C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Guidi M.; Seeberger P. H.; Gilmore K. How to approach flow chemistry. Chem. Soc. Rev. 2020, 49, 8910–8932. 10.1039/C9CS00832B. [DOI] [PubMed] [Google Scholar]; b Baumann M.; Moody T. S.; Smyth M.; Wharry S. A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry. Org. Process Res. Dev. 2020, 24, 1802–1813. 10.1021/acs.oprd.9b00524. [DOI] [Google Scholar]; c Hartman R. L. Flow chemistry remains an opportunity for chemists and chemical engineers. Curr. Opin. Chem. Eng. 2020, 29, 42–50. 10.1016/j.coche.2020.05.002. [DOI] [Google Scholar]; d Gutmann B.; Kappe C. O. Forbidden Chemistries - Paths to a Sustainable Future Engaging Continuous Processing. J. Flow Chem. 2017, 7, 65–71. 10.1556/1846.2017.00009. [DOI] [Google Scholar]; e Gutmann B.; Cantillo D.; Kappe C. O. Continuous-Flow Technology - A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients. Angew. Chem., Int. Ed. 2015, 54, 6688–6729. 10.1002/anie.201409318. [DOI] [PubMed] [Google Scholar]; f Jensen K. F. Flow chemistry - Microreaction technology comes of age. AIChE J. 2017, 63, 858–869. 10.1002/aic.15642. [DOI] [Google Scholar]; g Ley S. V.; Chen Y.; Robinson A.; Otter B.; Godineau E.; Battilocchio C. A Comment on Continuous Flow Technologies within the Agrochemical Industry. Org. Process Res. Dev. 2021, 25, 713–720. 10.1021/acs.oprd.0c00534. [DOI] [Google Scholar]; h Britton J.; Raston C. L. Multi-step continuous-flow synthesis. Chem. Soc. Rev. 2017, 46, 1250–1271. 10.1039/C6CS00830E. [DOI] [PubMed] [Google Scholar]
- a Sahoo H. R.; Kralj J. G.; Jensen K. F. Multistep Continuous-Flow Microchemical Synthesis Involving Multiple Reactions and Separations. Angew. Chem., Int. Ed. 2007, 46, 5704–5708. 10.1002/anie.200701434. [DOI] [PubMed] [Google Scholar]; b Baumann M.; Baxendale I. R.; Ley S. V.; Nikbin N.; Smith C. D. Azide monoliths as convenient flow reactors for efficient Curtius rearrangement reactions. Org. Biomol. Chem. 2008, 6, 1587–1593. 10.1039/b801634h. [DOI] [PubMed] [Google Scholar]; c Baumann M.; Baxendale I. R.; Ley S. V.; Nikbin N.; Smith C. D.; Tierney J. P. A modular flow reactor for performing Curtius rearrangements as a continuous flow process. Org. Biomol. Chem. 2008, 6, 1577–1586. 10.1039/b801631n. [DOI] [PubMed] [Google Scholar]; d Phung Hai T. A.; De Backer L. J. S.; Cosford N. D. P.; Burkart M. D. Preparation of Mono- and Diisocyanates in Flow from Renewable Carboxylic Acids. Org. Process Res. Dev. 2020, 24, 2342–2346. 10.1021/acs.oprd.0c00167. [DOI] [Google Scholar]
- a Huard K.; Bagley S. W.; Menhaji-Klotz E.; Preville C.; Southers J. A. Jr; Smith A. C.; Edmonds D. J.; Lucas J. C.; Dunn M. F.; Allanson N. M.; Blaney E. L.; Garcia-Irizarry C. N.; Kohrt J. T.; Griffith D. A.; Dow R. L. Synthesis of spiropiperidine lactam acetyl-CoA carboxylase inhibitors. J. Org. Chem. 2012, 77, 10050–10057. 10.1021/jo3014808. [DOI] [PubMed] [Google Scholar]; b Guetzoyan L.; Ingham R. J.; Nikbin N.; Rossignol J.; Wolling M.; Baumert M.; Burgess-Brown N. A.; Strain-Damerell C. M.; Shrestha L.; Brennan P. E.; Fedorov O.; Knapp S.; Ley S. V. Machine-assisted synthesis of modulators of the histone reader BRD9 using flow methods of chemistry and frontal affinity chromatography. MedChemComm 2014, 5, 540–546. 10.1039/C4MD00007B. [DOI] [Google Scholar]; c Filipponi P.; Ostacolo C.; Novellino E.; Pellicciari R.; Gioiello A. Continuous Flow Synthesis of Thieno[2,3-c]isoquinolin-5(4H)-one Scaffold: A Valuable Source of PARP-1 Inhibitors. Org. Process Res. Dev. 2014, 18, 1345–1353. 10.1021/op500074h. [DOI] [Google Scholar]
- Marsini M. A.; Buono F. G.; Lorenz J. C.; Yang B.-S.; Reeves J. T.; Sidhu K.; Sarvestani M.; Tan Z.; Zhang Y.; Li N.; Lee H.; Brazzillo J.; Nummy L. J.; Chung J. C.; Luvaga I. K.; Narayanan B. A.; Wei X.; Song J. J.; Roschangar F.; Yee N. K.; Senanayake C. H. Development of a concise, scalable synthesis of a CCR1 antagonist utilizing a continuous flow Curtius rearrangement. Green Chem. 2017, 19, 1454–1461. 10.1039/C6GC03123D. [DOI] [Google Scholar]
- a Leslie A.; Moody T. S.; Smyth M.; Wharry S.; Baumann M. Coupling biocatalysis with high-energy flow reactions for the synthesis of carbamates and β-amino acid derivatives. Beilstein J. Org. Chem. 2021, 17, 379–384. 10.3762/bjoc.17.33. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Baumann M.; Leslie A.; Moody T. S.; Smyth M.; Wharry S. Tandem Continuous Flow Curtius Rearrangement and Subsequent Enzyme-Mediated Impurity Tagging. Org. Process Res. Dev. 2021, 25, 452–456. 10.1021/acs.oprd.0c00420. [DOI] [Google Scholar]
- Substrate 4a was obtained from alkaline hydrolysis of methyl ester 11a, which was kindly provided by Almac Sciences.
- Lima F.; Sharma U. K.; Grunenberg L.; Saha D.; Johannsen S.; Sedelmeier J.; Van der Eycken E. V.; Ley S. V. A Lewis Base Catalysis Approach for the Photoredox Activation of Boronic Acids and Esters. Angew. Chem., Int. Ed. 2017, 56, 15136–15140. 10.1002/anie.201709690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wegner J.; Ceylan S.; Kirschning A. Ten key issues in modern flow chemistry. Chem. Commun. 2011, 47, 4583–4592. 10.1039/c0cc05060a. [DOI] [PubMed] [Google Scholar]; b Baumann M.; Moody T. S.; Smyth M.; Wharry S. Overcoming the Hurdles and Challenges Associated with Developing Continuous Industrial Processes. Eur. J. Org. Chem. 2020, 2020, 7398–7406. 10.1002/ejoc.202001278. [DOI] [Google Scholar]; c Pieber B.; Gilmore K.; Seeberger P. H. Integrated Flow Processing - Challenges in Continuous Multistep Synthesis. J. Flow Chem. 2017, 7, 129–136. 10.1556/1846.2017.00016. [DOI] [Google Scholar]; d Akwi F. M.; Watts P. Continuous flow chemistry: where are we now? Recent applications, challenges and limitations. Chem. Commun. 2018, 54, 13894–13928. 10.1039/C8CC07427E. [DOI] [PubMed] [Google Scholar]; e Gérardy R.; Emmanuel N.; Toupy T.; Kassin V.-E.; Tshibalonza N. N.; Schmitz M.; Monbaliu J.-C. M. Continuous Flow Organic Chemistry: Successes and Pitfalls at the Interface with Current Societal Challenges. Eur. J. Org. Chem. 2018, 2018, 2301–2351. 10.1002/ejoc.201800149. [DOI] [Google Scholar]; f Nöel T. A Personal Perspective on the Future of Flow Photochemistry. J. Flow Chem. 2017, 7, 87–93. 10.1556/1846.2017.00022. [DOI] [Google Scholar]
- Backpressure regulators (100 psi) were purchased from Kinesis (https://kinesis.co.uk/).
- Amberlyst A-15 and Amberlyst A-21 resins were purchased from Sigma-Aldrich and used after washing with water and methanol.
- Paryzek Z.; Koenig H.; Tabaczka B. Ammonium Formate/Palladium on Carbon: A Versatile System for Catalytic Hydrogen Transfer Reductions of Carbon-Carbon Double Bonds. Synthesis 2003, 2023–2026. 10.1055/s-2003-41024. [DOI] [Google Scholar]
- This X-ray structure has been deposited as CCDC 2083011 with the Cambridge Crystallographic Data Centre and is freely available from https://www.ccdc.cam.ac.uk/.
- For selected examples, please see:; a Costello J. P.; Ferreira E. M. Regioselectivity Influences in Platinum-Catalyzed Intramolecular Alkyne O-H and N-H Additions. Org. Lett. 2019, 21, 9934–9939. 10.1021/acs.orglett.9b03557. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Couture A.; Deniau E.; Lebrun S.; Grandclaudon P.; Carpentier J.-F. A new route to ene carbamates, precursors to benzoindolizinones through sequential asymmetric hydrogenation and cyclization. J. Chem. Soc., Perkin Trans. 1 1998, 8, 1403–1408. 10.1039/a709053f. [DOI] [Google Scholar]; c Fairfax D.; Stein M.; Livinghouse T.; Jensen M. Scope of the Intramolecular Imidotitanium-Alkyne [2 + 2] Cycloaddition-Azatitanetine Acylation Sequence. An Efficient Procedure for the Synthesis of 2-(2-Keto-1-alkylidene)tetrahydropyrroles and Related Compounds. Organometallics 1997, 16, 1523–1525. 10.1021/om961074f. [DOI] [Google Scholar]; d Lee H. K.; Kim J.; Pak C. S. Reaction of Thioamides with Zinc Enolate: Synthesis of Vinylogous Carbamates. Tetrahedron Lett. 1999, 40, 2173–2174. 10.1016/S0040-4039(99)00141-0. [DOI] [Google Scholar]; e Hazelden I. R.; Carmona R. C.; Langer T.; Pringle P. G.; Bower J. F. Pyrrolidines and Piperidines by Ligand-Enabled Aza-Heck Cyclizations and Cascades of N-(Pentafluorobenzoyloxy)carbamates. Angew. Chem., Int. Ed. 2018, 57, 5124–5128. 10.1002/anie.201801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Maryanoff B. E.; Zhang H.-C.; Cohen J. H.; Turchi I. J.; Maryanoff C. A. Cyclizations of N-Acyliminium Ions. Chem. Rev. 2004, 104, 1431–1628. 10.1021/cr0306182. [DOI] [PubMed] [Google Scholar]; b Wu P.; Nielsen T. E. Scaffold Diversity from N-Acyliminium Ions. Chem. Rev. 2017, 117, 7811–7856. 10.1021/acs.chemrev.6b00806. [DOI] [PubMed] [Google Scholar]
- Subsequent experiments showed that hydrolysis of products 9c–e proceeds slowly in refluxing in MeOH (containing small amounts of HOAc) with ca. 20% conversion over 3 h; longer reaction times led to partial decomposition.
- Omelian T. V.; Dobrydnev A. V.; Ostapchuk E. N.; Volovenko Y. M. Synthesis of Novel 3a-Substituted Tetrahydro-1H-1λ6-pyrrolo[1,2-b]isothiazole-1,1,3(2H)-triones through the CSIC Reaction. ChemistrySelect 2019, 4, 4933–4937. 10.1002/slct.201900650. [DOI] [Google Scholar]
- Kelleher F.; Kelly S.; Watts J.; McKee V. Structure-reactivity relationships of l-proline derived spirolactams and α-methyl prolinamide organocatalysts in the asymmetric Michael addition reaction of aldehydes to nitroolefins. Tetrahedron 2010, 66, 3525–3536. 10.1016/j.tet.2010.03.002. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.