

Abstract
The outbreak of acute respiratory disease in 2019, namely Coronavirus Disease-2019 (COVID-19), has become an unprecedented healthcare crisis. To mitigate the pandemic, there are a lot of collective and multidisciplinary efforts in facilitating the rapid discovery of protein inhibitors or drugs against COVID-19. Although many computational methods to predict protein inhibitors have been developed [ 1– 5], few systematic reviews on these methods have been published. Here, we provide a comprehensive overview of the existing methods to discover potential inhibitors of COVID-19 virus, so-called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). First, we briefly categorize and describe computational approaches by the basic algorithms involved in. Then we review the related biological datasets used in such predictions. Furthermore, we emphatically discuss current knowledge on SARS-CoV-2 inhibitors with the latest findings and development of computational methods in uncovering protein inhibitors against COVID-19.
Keywords: COVID-19, SARS-CoV-2, therapeutics, antiviral drugs, inhibitors, computer-aid drug design
Introduction
Since the first instance of the new coronavirus, Coronavirus Disease-2019 (COVID-19), was uncovered in Hubei Province, China in December 2019, there have been approximately 18 months after turning the local pandemic into the global one. As of 8 June 2021, a total of about 174 million people were infected by COVID-19, including over 3 870 000 deaths worldwide [6]. The pandemic has devastating consequences not only on humans lives but also on the global economy, including more than 8.5 trillion US dollars lost in 2020 and 2021 [7, 8]. Therefore, there is an urgent need to control the pandemic by accelerating the development or production of effective drugs against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
According to the previous studies [9, 10], SARS-CoV-2 is the single-stranded enveloped RNA virus with a symmetrical nucleocapsid. The viral genome of SARS-CoV-2 is highly similar to those of SARS-CoV and MERS-CoV [11], whose outbreaks happened within two decades in China and Saudi Arabia, respectively. Hence, the drugs or inhibitors designed for SARS-CoV and MERS-CoV were considered to be applied for SARS-CoV-2 as well. For example, SARS-CoV enters into the target cells through the structure spike (S) protein by binding to the angiotensin-converting enzyme 2 (ACE2) receptor [10]. The conservation of spike protein of SARS-CoV-2 suggests that the same interaction between the spike protein and the ACE2 receptor would be remained during the processing of inflection. In addition, other small molecules can be potential targets that play critical roles in viral genome replication and gene transcription, e.g. RNA-dependent RNA polymerase (RdRp), or cleavage and activation of the spike protein to enter the host genome and assist genome replication, e.g. type 2 transmembrane serine protease (TMPRSS2). They also keep the similar conservative characteristics among SARS-CoV, MERS-CoV and SARS-CoV-2.
Regardless of their unknown side effects, effective vaccines have been developed against the SARS-CoV-2 infection, like BioNTech vaccine (from Germany), Moderna vaccine (from the USA), Sinopharm vaccine (from China) and AstraZeneca vaccine (from Britain) [12]. Among them, messenger RNA (mRNA)-based vaccines are a relatively novel technology that remained to be further proven. Both available mRNA-based vaccines, Moderna and BioNTech, encode the spike protein of SARS-CoV-2 binding with the ACE2 receptor. But the SARS-CoV-2 virus has mutated frequently during its evolution and transmission [13, 14], resulting in genetic variations in the population of circulating viral strains throughout the COVID-19 pandemic. Until June 2021, multiple major variants of SARS-CoV-2 have dominated the world, e.g. Alpha virus (found in England), Beta virus (found in South Africa), Delta virus (found in India), [15]. Variants of SARS-CoV-2 have different characteristics, leading to unknown efficacy of the existing vaccine against the mutated virus [16]. Hence, design of inhibitors or drugs for specifically mutated SARS-CoV-2 is still necessary and challengeable. Figure 1 presents the timeline of major events related to the SARS-CoV-2 outbreak and vaccine development during 2020 and 2021 until 30 June 2021.
Figure 1.
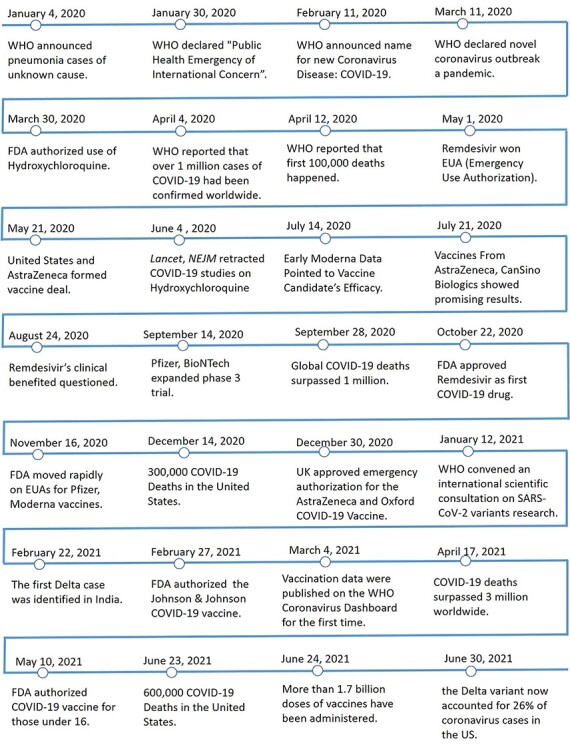
Major events about the SARS-CoV-2 outbreak, transmission, and SARS-CoV-2 vaccine development during 2020 and 2021 until 30 June 2021.
Compared with the traditional drug/inhibitor design process which is time-consuming and costly, computational methods for drug/inhibitor design are highly efficient to predict or identify potential molecules for the disease treatment [17]. Thus, the computer-aided approaches have great potentials for rapidly designing drugs or vaccines for mutated SARS-CoV-2. In the past months, there were several small molecules identified as potential inhibitors targeting SARS-CoV-2, even though more experimental validations are needed on the molecular targets. Among the molecular targets of SARS-CoV-2, main protease (Mpro) or 3-chymotrypsin-like protease (3CLpro) [18], structure proteins (e.g. spike protein), and nonstructure proteases, such as RdRp, and helicase [19], are highly conserved as well as essential to the viral life process. The structural information and functional roles of these major molecular targets against SARS-CoV-2 are summarized in Table S1 in the supplementary material.
We will start the review with diverse computational methods for drug and inhibitor design, followed by detailed descriptions and discussions on the findings of multiple enzymes as valid targets for potential inhibitors to treat coronaviruses diseases.
Insights to the Computer-Aided Drug Design
Computer-Aided Drug Design (CADD) emerged as an efficient method to uncover potential lead compounds and aiding the development of possible drugs for a wide range of diseases based on the knowledges collected by huge compound libraries [20]. Typically, CADD has three types of approaches, including structure-based drug design (SBDD), ligand-based drug design (LBDD) and virtual screening (VS). Furthermore, machine learning-based drug design (MLDD) has been widely applied with the rapid development of computer science communities [21, 22]. Herrin, we will provide a brief summary of CADD approaches and related databases as seen in Figure 2.
Figure 2.
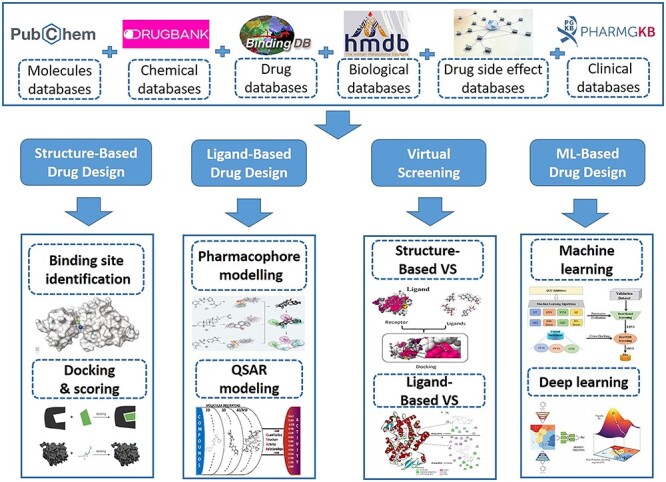
Schematic diagram of Computer-Aid Drug Design approaches (CADD).
Structure-based drug design
With the development of chemical biology and structural biology technology, the structural information of more and more drugs has been uncovered, providing essential elements for SBDD. Depending on the 3D structure of targets (proteins), such as X-ray crystallography or NMR spectroscopy, SBDD method predicts the potential interaction by evaluating the strength of the binding force between small molecule compounds and targets with the known structure. Molecular docking, a molecular modeling technique as the most basic method in SBDD, allows exhaustive search for the most suitable binding conformation of small molecules in the binding pocket of the protein. The framework of molecular docking is a search algorithm in which the ligand conformation is computed recursively until it converges to the lowest energy. It can effectively determine the ligand molecules that match the spatial and electrical characteristics of the active sites of the target receptors. At present, molecular docking plays an increasingly important role in SBDD [23]. Some common molecular docking software are listed in Table S2, including AutoDock [24], AutoDock Vina [25], AutoDockFR [26], ZDOCK [27], Glide [28], Flare [29], Induced Fit [30], MolDock [31] and M-ZDOCK [32].
Ligand-based drug design
However, the 3D structures of some drug targets have not been resolved successfully. For such cases, people developed another approach for direct drug design, LBDD, by taking advantage of existing compounds with known biological activities then establishing the relationship between query molecules and the bioactive molecules. In general, LBDD first converts the molecular structure into digital descriptors from a constructed database, e.g. molecular fragments, physiochemical properties, topology and pharmacophores, then generates the relationship between the molecular activities and constructs these descriptors by specific design models. The new drug molecules can be predicted or designed based on proper statistical methods, whereas their possible targets can be inferred from the bioactive molecules having high chemical affinities with the query. In addition to two common LBDD methods, quantitative structure–activity relationship (QSAR) and pharmacophore model [33], other popular LBDD-based software and their corresponding information are listed in Table S3, including McQSAR [34], SYBYL-X [35], TOPS-MODE [36], LigandScout [37], PLIP [38], FindSite-metal [39], CORAL [40].
Virtual screening
VS is another technique that uses a high-performance computer to analyze large databases of compounds to identify potential drug candidates that bind well to known structural targets [22]. There are two specific strategies for VS: receptor structure-based and ligand similarity-based. Despite different detailed strategies in the VS, the following four steps are essential: (i) preparing target protein and compound database; (ii) docking the molecules in the molecular library with the target one by one; (iii) obtaining a reasonable binding mode according to scores of the binding modes between small molecule and target, then evaluating the binding strength; (iv) purchasing selected preranked screened compounds followed by the activity tests.
The whole VS process can be carried out on computers by indexing the structures of compound molecules in the database instead of purchasing and testing the real compound molecules before the selection. Obviously, VS method is more convenient, cost efficient and quicker, compared to the experimental synthesis. Table S4 lists some common VS tools with brief introductions, e.g. PyRx [41], LiSiCA [42], MTiOpenScreen [43], iScreen [44], DockThor [45], GOLD [46], FlexX-Scan [47].
Machine learning-based drug design
Machine learning (ML) is an advanced data analysis method to improve the model automatically through the learning process from data and patterns [48]. ML technologies have been widely used in many fields, such as computer vision [49–51], natural language processing [52–55] and bioinformatics [56–60]. MLDD adopts various algorithms, such as recursive partitioning, support vector machine (SVM), k-nearest neighbors and neural networks [61–63], to investigate the activities of compounds against a target before the clinical trials [64, 65]. For example, Holden et al. [66] applied the SVM classification algorithm to the analysis of structure–activity relationship to predict the inhibition of dihydrofolate reductase by pyrimidines. Meng et al. [67] proposed persistent spectral-based ML models for drug design, which consist of the persistent spectral graph, persistent spectral simplicial complex and persistent spectral hypergraph based on the spectral theory. Now the integration of SARS-CoV-2 related studies with modern ML algorithms becomes a hot topic in drug repurposing models [68–70].
Databases
Drug discovery and development have initiated many databases containing essential information and knowledge of combinatorial chemistry synthesis, genomics/genetics and drugs or drug candidates. In turn, these databases enhance and improve the CADD. Generally, publicly available datasets about drugs or drug candidates’ discovery can be classified into seven scenarios (Table S5): (i) chemical molecules for activities against biological assays (e.g. PubChem [71] and ChEMBL [72]); (ii) chemical features for drug compounds (e.g. DrugBank [73] and SuperDRUG2 [74]); (iii) drugs verified by FDA (e.g. e-Drug3D [75]); (iv) drug targets with genetic and proteomic information (e.g. BindingDB [76] and BioGRID [77]); (v) metabolome or pathway related information (e.g. HMDB [78] and SMPDB [79]); (vi) drug side effects (e.g. DrugMatrix [80] and SIDER [81]); (vii) clinical databases (e.g. AACT database [82] and PharmGKB [83]).
These databases provide a variety of knowledges about drug candidates including physicochemical properties, molecule structure, in addition to diverse data in vitro, in vivo and from clinical. For example, PubChem [71] is a database of chemical molecules collected by the National Center for Biotechnology Information (NCBI). The NCBI now hosts three dynamically growing primary databases, including 111 million entries of compounds, 293 million entries of substances, and bioactivity results from 1.25 million high-throughput screening assays. Similar to PubChem, ChEMBL [72] is a publicly available database, containing information on binding, functional and ADMET for drug-like bioactive compounds. Currently, the database consists of 5.4 million bioactivity measurements for more than 1 million compounds and 5200 protein targets, which were manually abstracted from the primarily published literatures. There are some databases about drug compounds. For example, DrugBank [73] combines drug data with the information of drug targets and drug actions, which has been widely used in drug-target discovery, drug design, drug docking or screening, and drug interaction prediction. It collects approximately 4900 drug entries including 60% more FDA-approved small molecules and 10% more experimental biotech drug rugs. DrugBank has significantly improved the simplicity of its infrastructure and text query searches in the later updates. The e-Drug3D [75] is a 3D chemical structure database for drugs that provides several collections of drugs and commercial drug fragments. It currently contains 1519 annotated 3D structures of 1305 different FDA-approved drugs with molecular weight less than 2000. In the meantime, the drug databases in genetic and proteomic provide another scenario for drug design or discovery. As of September 2018, BioGRID [84] has recorded 1 598 688 biological interactions manually annotated from 55 809 publications for 71 species. BioGRID also accumulates details for over 700 000 post-translational modification sites. The recently updated BioGRID also annotates genome-wide CRISPR/Cas9-based screens with gene-phenotype and gene–gene relationships.
During the drug development phases, biological information for therapeutic or metabolism are important and valuable. For example, the HMDB [78], released in 2007, is now considered as the standard metabolomic resource for human metabolic studies including information about human metabolites, physiological concentrations, disease knowledge, chemistry associations, reference spectra and metabolic pathways. Side effects, known as adverse events to a drug, are a crucial research point in drug repurposing. DrugMatrix [80] has been developed based on drug toxicities, consisting of the comprehensive results of thousands of highly controlled and standardized toxicological experiments. It focuses on toxicities research with more than 200 compounds tested in vivo in rat tissues and 125 compounds in the in vitro rat hepatocytes. There is no doubt thought of clinical data which can provide high-quality information supporting drug design or discovery. PharmGKB [83] is an open-access database with clinically relevant information, collecting approved drug labels, gene-drug interactions and relationships between genotype and phenotype. The corresponding detailed information can be found in Table S5.
CADD against SARS-CoV-2: targeting Mpro
The main protease (Mpro, also known as 3CLpro) is recognized as a key enzyme to play a dominant role in the processing of mediating viral transcription and replication [85]. Since the binding pocket of this enzyme is highly conserved among all coronaviruses, like SARS-CoV, MERS-CoV and HCV, the antiviral drug targeting Mpro may be effective against SARS-CoV-2 as well [86]. Indeed, a lot of recent studies have been published that employed CADD to discover anti-SARS-CoV-2 agents against Mpro by different strategies, e.g. structure-based, ligand-based, VS or ML-based approaches (Figure 3).
Figure 3.
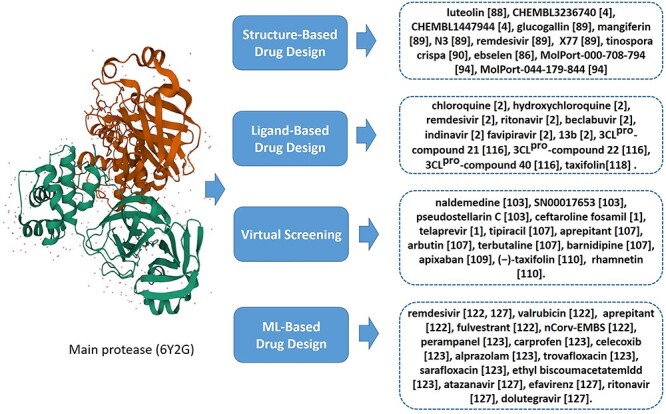
Summary of literatures using CADD to target Mpro of SARS-CoV-2.
For example, people used the structure-based docking approaches to predict the inhibitory activity and help drug design against SARS-CoV-2 Mpro [87]. Yu et al. [88] screened potential drugs by molecular docking to examine the effects of some common antiviral drugs like ribavirin, remdesivir, chloroquine and honeysuckle (a traditional Chinese medicine) as shown in Figure 4. Importantly, they recognized the luteolin as the control molecule is the main flavonoid in honeysuckle (Figure 3), which had a high binding affinity to the same sites of the main protease of SARS-CoV-2. Motonori Tsuji [4] performed structural refinement and energy calculations in the presence of peptidomimetic α-ketoamide inhibitors (PDB ID: 6Y2G, shown in Figure 5A). They found 28 bioactive compounds, including CHEMBL3236740, CHEMBL1447944 and others, were identified as effective anti-SARS-CoV-2 drug candidates (Figure 3). Singh et al. [89] identified several compounds, glucogallin, mangiferin, N3, remdesivir and X77 which had stronger binding affinities with Mpro. Furthermore, the results suggest that the phlorizin had the lowest binding free energy toward Mpro (Figure 4), followed by glucogallin and mangiferin.
Figure 4.
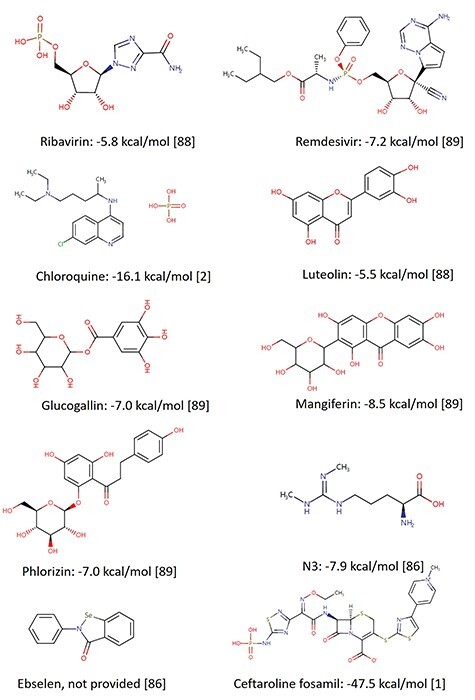
Two-dimensional (2D) chemical structures of some drugs and drug candidates with corresponding available binding energies to Mpro. All structures were collected from the DrugBank database.
Figure 5.
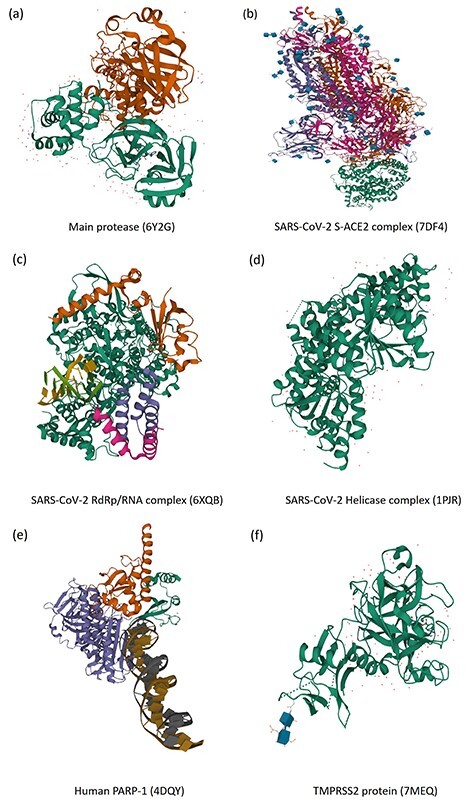
Crystal structures (orthorhombic form) of target proteins and formed complexes. (A) SARS-CoV-2 (2019-nCoV) main protease and potential inhibitors. (B) SARS-CoV-2 S-ACE2 complex. (C) SARS-CoV-2 RdRp/RNA complex. (D) SARS-CoV-2 Helicase complex. (E) Human PARP-1 bound to a DNA double-strand break. (F) Human TMPRSS2 with Nafamostat. All structures were collected from the protein data bank database.
However, long-range interactions have not been discussed as often as the short-range interactions during the selection of candidate inhibitors. Sencanski et al. [90] used the protocol with both long-range and short-range interactions to select inhibitor candidates. They applied the informational spectrum method and molecular docking for small molecules to search the DrugBank database. Interestingly, 57 drugs were identified as potential SARS-CoV-2 Mpro inhibitors. Additionally, tinospora crispa (Figure 3) was recognized as one potential COVID-19 Mpro inhibitor based on another independent molecular docking study [91].
To rapidly discover lead compounds for clinical treatments, Jin et al. [86] investigated a mechanism-based inhibitor (N3) by CADD and the crystal structure of Mpro of SARS-CoV-2 with complex N3. They built a predicted model by integrating structure-based virtual and high-throughput screening, which assayed over 10 000 compounds as inhibitor candidates of Mpro. One of these compounds, named ebselen, also had potential antiviral ability in cell-based assays (Figures 3 and 4).
Besides small molecules, some researchers have tried much effort to find potential candidates from natural products (NPs) against SARS-CoV-2 [92] [93]. For example, Ibrahim et al. [94] screened the MolPort database with molecular docking techniques. The top 5000 natural-like products (NLPs) were chosen according to the corresponding docking scores, like MolPort-000-708-794 and MolPort-044-179-844 (Figure 3). They found that the most promising NPs shared the same binding mode with key amino acid residues including HIS164, HIS163 and GLU166 based on molecular docking and molecular dynamics. The findings of these studies are expected to provide insight into the field of COVID-19 drug discovery [95–102].
Some recent studies have shown the feasibility of employing VS in inhibitor design of targeting Mpro. For example, Abel et al. [103] developed a VS method with both ligand- and structure-based approaches. The proposed VS was performed for two NPs databases, Super Natural II [104] and Traditional Chinese Medicine [105]. Additionally, they used an integrated drug repurposing approach to identify potential inhibitors against SARS-CoV-2 Mpro. Some drugs, like naldemedine, SN00017653, and pseudostellarin C, were identified as potential inhibitors for the first time (Figure 3). Lee et al. [1] identified potential inhibitors against COVID-19 from the Korea Chemical Bank drug repurposing (KCB-DR) database [106]. The results suggest ceftaroline fosamil (Figure 4) and the hepatitis C virus (HCV) protease inhibitor telaprevir as potential inhibitors against Mpro.
Although some drugs, such as remdesivir, favipiravir or dexamethasone, have been known beneficial for COVID-19 treatment, they have limitations clinically for different reasons. Hence, Nayak et al. [107] accomplished the VS of a variety of US-FDA-approved drugs using computer-aided tools. The US-FDA-approved drug structures were selected from DrugBank. Among them, arbutin, terbutaline, barnidipine, tipiracil and aprepitant were identified as potential hits. Moreover, tipiracil and aprepitant bound to the Mpro consistently, demonstrating potentially promising effects in pharmacologic treatments for COVID-19.
Structure-based VS is adopted to predict the best interaction between a ligand and a molecular target by scoring function. For example, Kumar et al. [108] utilized structure-based VS to identify hit molecules binding with the highest affinity to Mpro. The results indicated that the hydrogen bonding and hydrophobic interactions are the major contributing factors in binding pocket of COVID-19 Mpro. In addition, Hage-Melim et al. [109] used VS approaches based on the structure of the enzyme and two compound libraries to identify apixaban as a potential drug for future treatment of COVID-19. Fischer et al. [110] used shape screening and two docking protocols relevant for pharmacokinetics to narrow down commercially available compounds, leading to the natural compounds (−)-taxifolin and rhamnetin as potential inhibitors of Mpro (Figure 3). These new findings may bring insight into our further understanding and discovery of inhibitor candidates targeting Mpro [5, 111–113].
The reliability and accuracy of the ligand-based CADD method have been proven [114]. Han et al. [2] utilized the ligand–protein docking and molecular dynamic simulation for ab initio study to explore the binding mechanism or inhibitory ability by comparing two types of drugs: (i) clinically approved drugs including chloroquine, hydroxychloroquine, remdesivir, ritonavir, beclabuvir, indinavir and favipiravir, and (ii) a designed α-ketoamide inhibitor (13b) (Figure 3). The results suggested chloroquine had the strongest binding affinity with Mpro/3CLpro. Meanwhile, inhibitor 13b has a higher research priority to treat the SARS-CoV-2 since its improved inhibition efficiency. Eleftheriou et al. [115] uncovered that anticoagulant therapy has been proposed for the treatment of severe SARS-CoV-2 caused pneumonia, particularly, DPP-4 inhibitors may be more effective for SARS-CoV-2-infected diabetic patients.
QSAR model, the classical ligand-based CADD method, was also utilized in recent inhibitor design studies. For example, Ishola et al. [116] selected SARS coronavirus 3C-like protease (3CLpro) inhibitors data from the CHEMBL database. They constructed a QSAR model using the data with high correlations, which made the model statistically significant. The analysis revealed that 3CLpro-compound 21, 3CLpro-compound 22, 3CLpro-compound 40 complexes (Figure 3) were steadier than the baseline complex (3CLpro-X77). Alves et al. [3] developed QSAR models of these inhibitors then applied these models in VS with drugs in the DrugBank by conducting similarity searching and molecular docking in parallel. As a result, 42 compounds were identified as consensus computational hits. They were reported coincidentally in subsequent experimental screening studies (https://opendata.ncats.nih.gov/covid19/). Kumar et al. [117] developed a 2D-QSAR model based on multiple linear regression (MLR) with 3CLpro inhibitors. The proposed model clearly exhibited the structural features which enhanced the inhibitory activity against the 3CLpro enzyme. Additionally, the most and least active molecules were investigated using molecular docking tools to explore the molecular interactions involved in binding. Gogoi et al. [118] screened a library of 44 citrus flavonoids using molecular docking. The nontoxic compounds were further investigated with molecular dynamics simulation and predicted activity (IC50 value) with the 3D-QSAR model. They suggested taxifolin (Figure 3) as a potential inhibitor against SARS-CoV-2 Mpro which can be further analyzed by subsequent experiments for treatment of COVID -19. There are more literatures about ligand-based CADD in inhibitor candidates designing targeting Mpro [119, 120].
As ML techniques can be applied to the predictive scenario based on previous knowledges and well-known patterns, some recent studies have contributed to the development of ML-based CADD methods targeting Mpro. For example, Huang et al. [121] developed a biological activity-based modeling (BABM) approach, by which the compound activity can be predicted for a new target or other assays by using profiles across multiple well-defined assays. This model obtained 311 compounds against SARS-CoV-2, 32% of which showed antiviral activity in a cell culture live virus assay. More importantly, the most potent compounds presented nanomolar concentration levels for a half-maximal inhibitory. Nayarisseri et al. [122] proposed a shape-based ML method, which generates the 3D shaped pharmacophoric features of the seed compound. Furthermore, molecular docking was performed with optimized potential for liquid simulations (OPLS) algorithms to recognize high affinity compounds targeting Mpro. The shape-based ML reported that remdesivir, valrubicin, aprepitant and fulvestrant were the best therapeutic drugs (Figure 3) since the highest affinities with the target protein. They also found a novel compound ‘nCorv-EMBS’, which is not included in public chemical databases (PubChem, ZINC or ChEMBL) so far. The results of toxicity analysis suggested nCorv-EMBS was valuable to further research as the main protease inhibitor in COVID-19 [122].
Inspired by ensemble learning, Gimeno et al. [123] first applied molecule docking against the structure of Mpro using three popular tools: Glide [28], FRED [124] and AutoDock Vina [25]. Then, they proposed a hybrid ensemble approach to generate hypothetic binding modes replying on three score functions. Seven possible SARS-CoV-2 Mpro inhibitors were predicted including perampanel, carprofen, celecoxib, alprazolam, trovafloxacin, sarafloxacin and ethyl biscoumacetate (Figure 3). Battisti et al. [125] also proposed an inhibitor predicting framework, which not only combines molecular dynamics simulations with molecular docking but also focuses on the feature information of pharmacophore modeling and the flexibility of molecular dynamics simulations simultaneously. The proposed approach identified 10 compounds with high coronavirus inhibition potential.
In addition to the traditional data-driven ML modeling, some studies used deep learning-based approaches to predict potential inhibitors of SARS-CoV-2 Mpro [126]. For example, Park et al. [127] recognize some potentially drugs against SARS-CoV-2 using the pretrained deep learning drug-target interaction model called Molecule Transformer-Drug Target Interaction. They found that atazanavir, remdesivir, efavirenz, ritonavir and dolutegravir were the chemical compounds, showing an inhibitory potency against the SARS-CoV-2 3CLpro. Interestingly, they found that lopinavir, ritonavir and darunavir, which were designed to target viral proteinases, also bound to the replication complex components of SARS-CoV-2. Bung et al. [128] employed deep generative and predictive models to discover small molecules targeting inhibiting Mpro. The transfer learning and reinforcement learning was applied to optimize the proposed deep learning model, which learned chemical space around the protease inhibitors. Other features, including multiple physicochemical property filters and VS scores, were used for the final screening as well. Finally, they proposed 33 potential compounds for further synthesis and testing against SARS-CoV-2. Based on the structural model, Zhang et al. [129] performed a deep learning-based VS method to rank and identify protein–ligand interactions. The summary of drugs or inhibitors targeting SARS-CoV-2 Mpro/3CLpro can be found in Table S6.
CADD against SARS-CoV-2: targeting the structure protein
SARS-CoV-2 contains four structural proteins, including membrane protein (M), spike protein (S), envelope protein (E) and nucleocapsid protein (N), in addition to 16 nonstructural proteins (NSP1–16 as seen in the next section) [130]. Among them, the S protein can mediate the process of coronaviruses entering into host cells, so it becomes an attractive antiviral target for COVID-19 treatment.
Computational approaches have been developed to predict potential SARS-CoV-2 inhibitors targeting S protein. Previous studies demonstrated ACE2 as the key factor for SARS-CoV-2 to enter the host cells being bound by the spike protein of SARS-CoV-2 (Figure 5B). Hence, ACE2 becomes another common target of drug intervention. Wen et al. [131] investigated the existing drugs according to their abilities to block the binding of S protein to ACE2. According to the pathogenesis of SARS-CoV-2 from the perspective of S protein and ACE2 binding, they found some substances, including peptide P6, griffithsin, EK1 and extracts from traditional chinese medicine, which fought against SARS-CoV-2 through binding ACE2 receptor, S protein, or inhibiting the host and virus. Faria et al. [132] also focused on the molecules that can inhibit the interaction between the S protein and human ACE2. They discovered some molecules at the interaction sites: four molecules in Tyr-491(Spike)-Glu-37(ACE2) and one in Gly-488(Spike)-Lys-353(ACE2). Furthermore, they found that the molecule 1629 and the molecule 2542 had significant inhibitory effects on the site of Gly488-Ly353 and Tyr491-Glu37, respectively, suggesting further laboratory tests on the combination of these molecules that can work at two interaction sites simultaneously. Additionally, the human furin protease, cleaving the S1-S2 domains involved in entering the host cell, may become the third target. CUBUK et al. [133] docked five drug molecules, favipiravir, hydroxychloroquine, remdesivir, lopinavir and ritonavir, on not only S protein and main protease but also human furin protease. The results of molecular docking revealed that the human furin protease can be a potential target of SARS-CoV-2, whereas remdesivir, a nucleic acid derivative, can be used as a template for designing novel furin protease inhibitors to fight against the disease. Taking advantage of the DrugBank and PubChem, Unni et al. [134] identified Bisoxatin (DB09219), a laxative drug, as a promising repurposable drug to develop a new chemical compound for inhibiting SARS-CoV-2 entry into the host, even though Bisoxatin was used to treat constipation and preparation. GR 127935 hydrochloride hydrate, GNF-5, RS504393, and eptifibatide acetate were found to connect to viral binding motifs of ACE2 receptor by Tomar et al. [135]. Table S6 presents the summary of drugs or inhibitors targeting SARS-CoV-2 S protein and ACE2.
Many computational approaches also focused on potential SARS-CoV-2 inhibitors targeting M protein, N protein and E protein, which were believed to be useful for further structure-based VS and other CADD drug and vaccine design. Dong et al. [136] searched the homologous templates of all structural proteins of SARS-CoV-2, including S, E and N proteins. Banerjee et al. [137] recognized micromolecules of inhibitors targeting M protein and E proteins of SARS-CoV-2 by integrating docking and simulation methods. They investigated some compounds from an Indian medicinal plant source (Azadirachta indica or Neem) and found 70 compounds against these two proteins. With molecular dynamics simulations, a few common compounds binding to both M and E proteins were recognized as potentially inhibit their functions. Table S6 lists drugs or inhibitors targeting SARS-CoV-2 proteins with essential information.
CADD against SARS-CoV-2: Targeting the nonstructure protein
SARS-CoV-2 nonstructure proteins can be potential targets to inhibit SARS-CoV-2 as well. For example, RdRp, as shown in Figure 5C, plays a crucial role in the viral cycle of coronaviruses, particularly the replication of the viral genome, with the assistance of nonstructure proteins, NSP7 and NSP8, in a polymerase complex. It is not surprising to see that RdRp has been recognized as an important coronavirus target for drug design. Since SARS-CoV-2 has high similarity with other SARS viruses, target-based VS and molecular docking on antiviral molecules of the SARS explored that the antiviral galidesivir had promise against SARS-CoV-2 as well [138]. Quinupristin was identified as one candidate which can bind in the RNA tunnel of RdRP and block the path and access on both sides with potentials to prevent viral replication and RNA synthesis [139]. Wu et al. [140] systematically compared SARS-CoV-2 genes encoding proteins with that from other coronaviruses, then predicted and built 19 structures with homology modeling. Based on ZINC drug database and their own NPs database, they found 78 antiviral drugs for SARS-CoV-2, which are currently on the market or undergoing clinical trials.
Helicase is another macromolecule viral replication enzyme, responsible for separating DNA and RNA into two single-stranded nucleic acids in the coronaviruses viral cycle unwinding (Figure 5D). Some studies have also suggested drugs and NPs as potential SARS-CoV-2 helicase inhibitors. For example, one study suggests that vapreotide and atazanavir, two approved drugs for treating AIDS-related diarrhea and HIV infection, are observed to interrupt the activities of the SARS-CoV-2 helicase significantly [141]. Mirza et al. [142] have proposed an integrative VS and molecular dynamics simulations approach for targeting the main protease, RdRp and helicase, which warrants in vitro testing to evaluate compound efficacy.
Iftikhar et al. [143] focused on a small molecule that specifically binds to three essential proteins (RdRp, 3CLpro and helicase). They found three FDA-approved drugs binding to 3CLpro, one drug-like molecule binding to RdRp, and two drug-like molecules specifically interacting with helicase.
The poly-ADP-ribose polymerase 1 (PARP1, shown in Figure 5E) is also critical for viral replication [144–146]. Ge et al. [147] developed a data-driven drug repositioning framework combining ML and statistical analysis approaches to explore potential drug candidates against SARS-CoV-2, by integrating their large-scale data including knowledge graphs and transcriptome data from public domain and literatures. Based on the model, CVL218, a PARP1 inhibitor, was recognized as the repurposed therapeutic agent for COVID-19.
The host serine protease TMPRSS2 has a pivotal role in the viral entry of SARS-CoV-2 (Figure 5F). In the study conducted by Singh et al. [89], they uncovered the strong binding affinity between TMPRSS2 and compounds, glucogallin, mangiferin, N3, remdesivir and X77. Among them, mangiferin showed the lowest binding free energy, followed by phlorizin and glucogallin.
Additionally, more studies focused on other nonstructure proteases in the viral replication/transcription process, such as NSP15 protein [148] (belongs to the EndoU enzyme family), C3 (complement component 3) [149] and N7-MTase [150] (Guanine-N7 Methyltransferase). According to the findings, glisoxepide, and idarubicin, used to treat diabetes and leukemia, respectively, were identified as the stronger binder of EndoU enzyme [151]. We summarized drugs or inhibitors targeting SARS-CoV-2 macromolecules as seen in Table S6.
Conclusion
Since the outbreak of COVID-19, people around the world have put much effort into investing vaccines and drugs against SARS-CoV-2. CADD and ML techniques have been employed in many studies to target SARS-CoV-2 macromolecules, which are considered as feasible options to speed up the processes for drug design and discovery. Our paper reviewed the theory and applications of these approaches with specific databases from these studies. We explored the new findings of inhibitors as potential interventions and treatments of COVID-19.
However, considering the variations of SARS-CoV-2, we are still facing big challenges to make sure that developed vaccines and drugs can keep efficient for different viral strains with specific mutations. It is known that structural variations on or even close to the binding sites could dramatically impact ligand binding properties. Gossen et al. [152] redefined the druggability of the proteins as an integrated chemical space generated by multiple conformations of binding sites when ligand binding. This process revealed the unique blueprint of SARS-CoV-2 Mpro, leading to a definition of a pharmacophore based on the specific structure, which provides a strong foundation for rational drug design for SARS-CoV-2 Mpro. Ugurel et al. [153] analyzed 3458 SARS-CoV-2 genome sequences isolated from 58 countries. They found the incidence of C17747T and A17858G mutations on helicase (NSP13) were significantly higher than others. However, four drugs, including cangrelor, fludarabine, folic acid and polydatin, interrupted both the wild type and mutant SARS-CoV-2 helicase, suggesting that they can be the most potent drugs. We expect that our review can bring insight to identify antiviral inhibitors and potential drug candidates against diverse SARS-COV-2 variants.
Key Points
Discovering potential inhibitors or drugs of SARS-CoV-2 is critical in mitigating the pandemic impact of COVID-19.
We give a brief overview of existing computer-aid drug design methods and biological databases used in predicting drugs or inhibitors.
We provide a systematic review of current knowledge, latest findings using computational methods to discover protein inhibitors of SARS-CoV-2.
Supplementary Material
Qiaoming Liu is currently a joint PhD student at the Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University. His research interests are machine learning and bioinformatics.
Dr Jun Wan is an assistant professor in the Department of Medical and Molecular Genetics, Indiana University School of Medicine with an adjunct appointment at Department of BioHealth Informatics, School of Informatics and Computing, Indiana University–Purdue University Indianapolis. His current research interests include epigenetics, proteomics and artificial intelligence.
Dr Guohua Wang is a principal investigator at Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University. He received the master’s and PhD degrees in computer science and technology from Harbin Institute of Technology, in 2003 and 2009, respectively. He has been working at the Johns Hopkins University as a postdoctoral fellow from 2014 to 2016. His current research interests include bioinformatics, epigenetics, genomics and machine learning.
Contributor Information
Qiaoming Liu, Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University, Harbin, Heilongjiang 150001, China.
Jun Wan, Department of BioHealth Informatics, School of Informatics and Computing, Indiana University–Purdue University Indianapolis, Indianapolis, IN, USA; Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA.
Guohua Wang, Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University, Harbin, Heilongjiang 150001, China; Information and Computer Engineering College, Northeast Forestry University, Harbin, Heilongjiang 150001, China.
Funding
This work has been partially supported by the National Natural Science Foundation of China (62072095, 61771165), the National Key R&D Program of China (2021YFC2100100), the Innovation Project of State Key Laboratory of Tree Genetics and Breeding (Northeast Forestry University) (2019A04) to Dr. Guohua Wang.
Reference
- 1. Kumar R, Kumar V, Lee KW. A computational drug repurposing approach in identifying the cephalosporin antibiotic and anti-hepatitis C drug derivatives for COVID-19 treatment. Comput Biol Med 2021;130:104186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han Y, Wang Z, Ren J, et al. Potential inhibitors for the novel coronavirus (SARS-CoV-2). Brief Bioinform 2021;22(2):1225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alves VM, Bobrowski T, Melo-Filho CC, et al. QSAR Modeling of SARS-CoV M(pro) inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Mol Inform 2021;40(1):e2000113. [DOI] [PubMed] [Google Scholar]
- 4. Tsuji M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio 2020;10(6):995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh S, Florez H. Bioinformatic study to discover natural molecules with activity against COVID-19. F1000Res 2020;9:1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Senger MR, Evangelista TCS, Dantas RF, et al. COVID-19: molecular targets, drug repurposing and new avenues for drug discovery. Mem Inst Oswaldo Cruz 2020;115:e200254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tisdell CA. Economic, social and political issues raised by the COVID-19 pandemic. Economic analysis and policy 2020;68:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. He H, Harris L. The impact of Covid-19 pandemic on corporate social responsibility and marketing philosophy. J Bus Res 2020;116:176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vivekanandhan K, Shanmugam P, Barabadi H, et al. Emerging therapeutic approaches to combat COVID-19: present status and future perspectives. Front Mol Biosci 2021;8:604447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu J, Liu X. Structural biology aids the research of new anti-COVID-19 drugs. Current Organic Synthesis 2020;17(3):162–3. [DOI] [PubMed] [Google Scholar]
- 11. Vougogiannopoulou K, Corona A, Tramontano E, et al. Natural and nature-derived products targeting human coronaviruses. Molecules 2021;26(2):448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forni G, Mantovani A. COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ 2021;28(2):626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang S, Liu S, Shen J, et al. Updated SARS-CoV-2 single nucleotide variants and mortality association. J Med Virol 2021;jmv.27191. 10.1101/2021.01.29.21250757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu S, Shen J, Fang S, et al. Genetic spectrum and distinct evolution patterns of SARS-CoV-2. Front Microbiol 2020;11:593548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Starr TN, Greaney AJ, Addetia A, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021;371(6531):850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yadav PD, Sapkal GN, Abraham P, et al. Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccinees. bioRxiv 2021April 23, 2021. 10.1101/2021.04.23.441101 preprint: not peer reviewed. [DOI] [PubMed] [Google Scholar]
- 17. Mishra AK, Tewari SP. In silico screening of some naturally occurring bioactive compounds predicts potential inhibitors against SARS-COV-2 (COVID-19) protease. arXiv preprint arXiv:2004.01634 2020. [Google Scholar]
- 18. Illian DN, Siregar ES, Sumaiyah S, et al. Potential compounds from several Indonesian plants to prevent SARS-CoV-2 infection: a mini-review of SARS-CoV-2 therapeutic targets. Heliyon 2021;7(1):e06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lima WG, Brito JCM, Overhage J, et al. The potential of drug repositioning as a short-term strategy for the control and treatment of COVID-19 (SARS-CoV-2): a systematic review. Arch Virol 2020;165(8):1729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keum YS, Jeong YJ. Development of chemical inhibitors of the SARS coronavirus: viral helicase as a potential target. Biochem Pharmacol 2012;84(10):1351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Z, Sun H, Yao X, et al. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys 2016;18(18):12964–75. [DOI] [PubMed] [Google Scholar]
- 22. Zhao L, Ciallella HL, Aleksunes LM, et al. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov Today 2020;25(9):1624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adasme MF, Parisi D, Sveshnikova A, et al. Structure-based drug repositioning: potential and limits. Semin Cancer Biol 2021;68:192–8. [DOI] [PubMed] [Google Scholar]
- 24. Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit 1996;9(1):1–5. [DOI] [PubMed] [Google Scholar]
- 25. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31(2):455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ravindranath PA, Forli S, Goodsell DS, et al. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol 2015;11(12):e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins 2003;52(1):80–7. [DOI] [PubMed] [Google Scholar]
- 28. Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47(7):1739–49. [DOI] [PubMed] [Google Scholar]
- 29. Paranjpe RS, Cusenza A. FLARE: An integrated software package for friction and lubrication analysis of automotive engines-Part II: Experimental validation. In: SAE Technical Paper, 1992. 10.4271/920488. [DOI]
- 30. Bosshard HR. Molecular recognition by induced fit: how fit is the concept? Phys Ther 2001;16(4):171–3. [DOI] [PubMed] [Google Scholar]
- 31. Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 2006;49(11):3315–21. [DOI] [PubMed] [Google Scholar]
- 32. Pierce B, Tong W, Weng Z. M-ZDOCK: a grid-based approach for C n symmetric multimer docking. Bioinformatics 2005;21(8):1472–8. [DOI] [PubMed] [Google Scholar]
- 33. Yang SQ, Ye Q, Ding JJ, et al. Current advances in ligand-based target prediction. WIREs Comput Molecular Sci 2020;11(3):e1504. [Google Scholar]
- 34. Vainio MJ, Johnson MS. McQSAR: a multiconformational quantitative structure− activity relationship engine driven by genetic algorithms. J Chem Inf Model 2005;45(6):1953–61. [DOI] [PubMed] [Google Scholar]
- 35. Sybyl X. Molecular modeling software packages (Version 2.0). TRIPOS Associates, 2012. [Google Scholar]
- 36. González MP, Helguera AM, Díaz HG. A TOPS-MODE approach to predict permeability coefficients. Polymer 2004;45(6):2073–9. [Google Scholar]
- 37. Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 2005;45(1):160–9. [DOI] [PubMed] [Google Scholar]
- 38. Salentin S, Schreiber S, Haupt VJ, et al. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res 2015;43(W1):W443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brylinski M, Skolnick J. FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins 2011;79(3):735–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toropova AP, Toropov AA. CORAL software: prediction of carcinogenicity of drugs by means of the Monte Carlo method. Eur J Pharm Sci 2014;52:21–5. [DOI] [PubMed] [Google Scholar]
- 41. Dallakyan, S. and A.J. Olson, Small-molecule library screening by docking with PyRx, in Chemical Biology. 2015, Springer. p. 243–50 1263 New York, NY: Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 42. Lešnik S, Štular T, Brus B, et al. LiSiCA: a software for ligand-based virtual screening and its application for the discovery of butyrylcholinesterase inhibitors. J Chem Inf Model 2015;55(8):1521–8. [DOI] [PubMed] [Google Scholar]
- 43. Labbé CM, Rey J, Lagorce D, et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res 2015;43(W1):W448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsai T-Y, Chang K-W, Chen CY-C. iScreen: world’s first cloud-computing web server for virtual screening and de novo drug design based on TCM database@ Taiwan. J Comput Aided Mol Des 2011;25(6):525–31. [DOI] [PubMed] [Google Scholar]
- 45. Guedes IA, Krempser E, Dardenne LE. DockThor 2.0: a free web server for protein-ligand virtual screening. XIX SBQT–Simpósio Brasileiro de Química Teórica 2017. [Google Scholar]
- 46. Annamala MK, Inampudi KK, Guruprasad L. Docking of phosphonate and trehalose analog inhibitors into M. tuberculosis mycolyltransferase Ag85C: comparison of the two scoring fitness functions GoldScore and ChemScore, in the GOLD software. Bioinformation 2007;1(9):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schellhammer I, Rarey M. FlexX-scan: fast, structure-based virtual screening. PROTEINS 2004;57(3):504–17. [DOI] [PubMed] [Google Scholar]
- 48. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521(7553):436–44. [DOI] [PubMed] [Google Scholar]
- 49. Ji Q, Sun Y, Gao J, et al. Nonlinear subspace clustering via adaptive graph regularized autoencoder. IEEE Access 2019;7:74122–33. [Google Scholar]
- 50. Xie Y, Gong M, Qin AK, et al. TPNE: topology preserving network embedding. Inform Sci 2019;504:20–31. [Google Scholar]
- 51. Zhao J, Jiao L. Fast sparse deep neural networks: theory and performance analysis. IEEE Access 2019;7:74040–55. [Google Scholar]
- 52. Wold S, Esbensen K, Geladi P. Principal component analysis. Chemom Intel Lab Syst 1987;2(1–3):37–52. [Google Scholar]
- 53. Lee DD, Seung HS. Learning the parts of objects by non-negative matrix factorization. Nature 1999;401(6755):788. [DOI] [PubMed] [Google Scholar]
- 54. Roweis ST, Saul LK. Nonlinear dimensionality reduction by locally linear embedding. Science 2000;290(5500):2323–6. [DOI] [PubMed] [Google Scholar]
- 55. Ng AY, Jordan MI, Weiss Y. On spectral clustering: Analysis and an algorithm. In: Advances in Neural Information Processing Systems, 2002;849–856. https://dl.acm.org/doi/10.5555/2980539.2980649.
- 56. MacParland SA, Liu JC, Ma XZ, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kiselev VY, Kirschner K, Schaub MT, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods 2017;14(5):483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eraslan G, Avsec Ž, Gagneur J, et al. Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics 2019;20(7):389–403. 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- 59. Tian T, Wan J, Song Q, et al. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nature Machine Intelligence 2019;1(4):191–8. [Google Scholar]
- 60. Zhavoronkov A, Ivanenkov YA, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019;37(9):1038–40. [DOI] [PubMed] [Google Scholar]
- 61. Dimitri GM, Lió P. DrugClust: a machine learning approach for drugs side effects prediction. Comput Biol Chem 2017;68:204–10. [DOI] [PubMed] [Google Scholar]
- 62. Shoombuatong W, Schaduangrat N, Pratiwi R, et al. THPep: a machine learning-based approach for predicting tumor homing peptides. Comput Biol Chem 2019;80:441–51. [DOI] [PubMed] [Google Scholar]
- 63. Alsenan S, Al-Turaiki I, Hafez A. A recurrent neural network model to predict blood–brain barrier permeability. Comput Biol Chem 2020;89:107377. [DOI] [PubMed] [Google Scholar]
- 64. Gertrudes J, Maltarollo VG, Silva RA, et al. Machine learning techniques and drug design. Curr Med Chem 2012;19(25):4289–97. [DOI] [PubMed] [Google Scholar]
- 65. Serafim MSM, dos Santos Júnior VS, Gertrudes JC, et al. Machine learning techniques applied to the drug design and discovery of new antivirals: a brief look over the past decade. Expert Opin Drug Discovery 2021;16(9):961–75. [DOI] [PubMed] [Google Scholar]
- 66. Burbidge R, Trotter M, Buxton B, et al. Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput Chem 2001;26(1):5–14. [DOI] [PubMed] [Google Scholar]
- 67. Meng Z, Xia K. Persistent spectral–based machine learning (PerSpect ML) for protein-ligand binding affinity prediction. Science Advances 2021;7(19):eabc5329. 10.1126/sciadv.abc5329. February 11, 2020; preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Loucera C, Esteban-Medina M, Rian K, et al. Drug repurposing for COVID-19 using machine learning and mechanistic models of signal transduction circuits related to SARS-CoV-2 infection. Signal Transduct Target Ther 2020;5(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Islam M, Poly TN, Alsinglawi B, et al. A state-of-the-art survey on artificial intelligence to fight COVID-19. J Clin Med 2021;10(9):1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lucchetta M, Pellegrini M. Drug repositioning by merging active subnetworks validated in cancer and COVID-19. medRxiv 2021. May 18, 2021. 10.1101/2021.05.13.21257140 preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res 2021;49(D1):D1388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gaulton A, Bellis LJ, Bento AP, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 2012;40(D1):D1100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wishart DS, Knox C, Guo AC, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 2008;36(suppl_1):D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Siramshetty VB, Eckert OA, Gohlke BO, et al. SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res 2018;46(D1):D1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pihan E, Colliandre L, Guichou JF, et al. E-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics 2012;28(11):1540–1. [DOI] [PubMed] [Google Scholar]
- 76. Liu T, Lin Y, Wen X, et al. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res 2007;35(Database):D198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stark C, Breitkreutz BJ, Reguly T, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006;34(90001):D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wishart DS, Tzur D, Knox C, et al. HMDB: the human metabolome database. Nucleic Acids Res 2007;35(Database):D521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Frolkis A, Knox C, Lim E, et al. SMPDB: the small molecule pathway database. Nucleic Acids Res 2010;38(suppl_1):D480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Svoboda DL, Saddler T, Auerbach SS. An overview of National Toxicology Program’s Toxicogenomic applications: DrugMatrix and ToxFX. Adv Comput Toxicol 2019;30:141–57. [Google Scholar]
- 81. Kuhn M, Letunic I, Jensen LJ, et al. The SIDER database of drugs and side effects. Nucleic Acids Res 2016;44(D1):D1075–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tasneem A, Aberle L, Ananth H, et al. The database for aggregate analysis of ClinicalTrials. Gov (AACT) and subsequent regrouping by clinical specialty. PLoS One 2012;7(3):e33677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hewett M, Oliver DE, Rubin DL, et al. PharmGKB: the pharmacogenetics knowledge base. Nucleic acids research 2002;30(1):163–65. 10.1093/nar/30.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Oughtred R, Stark C, Breitkreutz BJ, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res 2019;47(D1):D529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu X, Wang XJ. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J Genet Genomics 2020;47(2):119–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jin Z, du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020;582(7811):289–93. [DOI] [PubMed] [Google Scholar]
- 87. Singh S, Florez H. Coronavirus disease 2019 drug discovery through molecular docking. F1000Res 2020;9:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yu R, Chen L, Lan R, et al. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int J Antimicrob Agents 2020;56(2):106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Singh R, Gautam A, Chandel S, et al. Protease inhibitory effect of natural polyphenolic compounds on SARS-CoV-2: an in silico study. Molecules 2020;25(20):4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sencanski M, Perovic V, Pajovic SB, et al. Drug repurposing for candidate SARS-CoV-2 main protease inhibitors by a novel in silico method. Molecules 2020;25(17):3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rakib A, Paul A, Chy MNU, et al. Biochemical and computational approach of selected phytocompounds from Tinospora crispa in the management of COVID-19. Molecules 2020;25(17):3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kumar A, Choudhir G, Shukla SK, et al. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J Biomol Struct Dyn 2021;39(10):3760–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Islam R, Parves MR, Paul AS, et al. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J Biomol Struct Dyn 2021;39(9):3213–24. 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ibrahim MAA, Abdeljawaad KAA, Abdelrahman AHM, et al. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: in-silico drug discovery. J Biomol Struct Dyn 2021;39(15):5722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Han Y, Král P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 2020;14(4):5143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Deeks HM, Walters RK, Barnoud J, et al. Interactive molecular dynamics in virtual reality is an effective tool for flexible substrate and inhibitor docking to the SARS-CoV-2 main protease. J Chem Inf Model 2020;60(12):5803–14. [DOI] [PubMed] [Google Scholar]
- 97. Das S, Sarmah S, Lyndem S, et al. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn 2021;39(9):3347–57. 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chakraborti S, Bheemireddy S, Srinivasan N. Repurposing drugs against the main protease of SARS-CoV-2: mechanism-based insights supported by available laboratory and clinical data. Mol Omics 2020;16(5):474–91. [DOI] [PubMed] [Google Scholar]
- 99. al-Sehemi AG, Pannipara M, Parulekar RS, et al. Potential of NO donor furoxan as SARS-CoV-2 main protease (M(pro)) inhibitors: in silico analysis. J Biomol Struct Dyn 2021;39(15):5804–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yang Y, Zhu Z, Wang X, et al. Ligand-based approach for predicting drug targets and for virtual screening against COVID-19. Brief Bioinform 2021;22(2):1053–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Aanouz I, Belhassan A, el-Khatabi K, et al. Moroccan medicinal plants as inhibitors against SARS-CoV-2 main protease: computational investigations. J Biomol Struct Dyn 2021;39(8):2971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Alamri MA, Tahir ul Qamar M, Mirza MU, et al. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CL(pro). J Biomol Struct Dyn 2021;39(13):4936–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Abel R, Paredes Ramos M, Chen Q, et al. Computational prediction of potential inhibitors of the main protease of SARS-CoV-2. Front Chem 2020;8:590263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Banerjee P, Erehman J, Gohlke BO, et al. Super natural II—a database of natural products. Nucleic Acids Res 2015;43(D1):D935–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. He M, Yan X, Zhou J, et al. Traditional Chinese medicine database and application on the web. J Chem Inf Comput Sci 2001;41(2):273–7. [DOI] [PubMed] [Google Scholar]
- 106. Kim, J.-Y., Kim, Dong-Wook, Kim, Sun-Woo et al. Diversity of compounds in Korea Chemical Bank. in Proceedings of the PSK Conference. 2002. The Pharmaceutical Society of Korea, 189.2–189. [Google Scholar]
- 107. Baby K, Maity S, Mehta CH, et al. Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Arch Med Res 2021;52(1):38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mishra SS, Ranjan S, Sharma CS, et al. Computational investigation of potential inhibitors of novel coronavirus 2019 through structure-based virtual screening, molecular dynamics and density functional theory studies. J Biomol Struct Dyn 2021;39(12):4449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hage-Melim L, Federico LB, de Oliveira NKS, et al. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci 2020;256:117963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fischer A, Sellner M, Neranjan S, et al. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. Int J Mol Sci 2020;21(10):3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rahman MM, Saha T, Islam KJ, et al. Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment. J Biomol Struct Dyn 2021;39(16):6231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gentile D, Patamia V, Scala A, et al. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: a virtual screening and molecular Modeling study. Mar Drugs 2020;18(4):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Arun KG, Sharanya CS, Abhithaj J, et al. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J Biomol Struct Dyn 2021;39(13):4647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Culletta G, Gulotta MR, Perricone U, et al. Exploring the SARS-CoV-2 proteome in the search of potential inhibitors via structure-based pharmacophore modeling/docking approach. Comput Secur 2020;8(3):77. [Google Scholar]
- 115. Eleftheriou P, Amanatidou D, Petrou A, et al. In silico evaluation of the effectivity of approved protease inhibitors against the main protease of the novel SARS-CoV-2 virus. Molecules 2020;25(11):2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ishola AA, Adedirin O, Joshi T, et al. QSAR modeling and Pharmacoinformatics of SARS coronavirus 3C-like protease inhibitors. Comput Biol Med 2021;134:104483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kumar V, Roy K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ Res 2020;31(7):511–26. [DOI] [PubMed] [Google Scholar]
- 118. Gogoi N, Chowdhury P, Goswami AK, et al. Computational guided identification of a citrus flavonoid as potential inhibitor of SARS-CoV-2 main protease. Mol Divers 2021;25(3):1745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ngo ST, Quynh Anh Pham N, Thi le L, et al. Computational determination of potential inhibitors of SARS-CoV-2 main protease. J Chem Inf Model 2020;60(12):5771–80. [DOI] [PubMed] [Google Scholar]
- 120. Andrianov AM, Kornoushenko YV, Karpenko AD, et al. Computational discovery of small drug-like compounds as potential inhibitors of SARS-CoV-2 main protease. J Biomol Struct Dyn 2021;39(15):5779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Huang R, Xu M, Zhu H, et al. Biological activity-based modeling identifies antiviral leads against SARS-CoV-2. Nat Biotechnol 2021;39(6):747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nayarisseri A, Khandelwal R, Madhavi M, et al. Shape-based machine learning models for the potential novel COVID-19 protease inhibitors assisted by molecular dynamics simulation. Curr Top Med Chem 2020;20(24):2146–67. [DOI] [PubMed] [Google Scholar]
- 123. Gimeno A, Mestres-Truyol J, Ojeda-Montes MJ, et al. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int J Mol Sci 2020;21(11):3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. OEChem TK. OpenEye Scientific Software. Software (Inc.). NM, USA: Santa Fe, 2012. [Google Scholar]
- 125. Battisti V, Wieder O, Garon A, et al. A computational approach to identify potential novel inhibitors against the coronavirus SARS-CoV-2. Mol Inform 2020;39(10):e2000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ivanenkov Y, Aladinskiy V, Zhebrak A, et al. Potential COVID-2019 3C-like protease inhibitors designed using generative deep learning approaches. ChemRxiv Preprint posted online on February 2020;11. [Google Scholar]
- 127. Beck BR, Shin B, Choi Y, et al. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J 2020;18:784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bung NAK, Krishnan SR, Bulusu G, et al. De novodesign of new chemical entities for SARS-CoV-2 using artificial intelligence. Future Med Chem 2021;13(6):575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang H, Saravanan KM, Yang Y, et al. Deep learning based drug screening for novel coronavirus 2019-nCov. Interdiscip Sci 2020;12(3):368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wang M-Y, Zhao R, Gao LJ, et al. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol 2020;10:587269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Jiang YP, Zhao XX, Lv HQ, et al. Drug screening and development from the affinity of S protein of new coronavirus with ACE2. Eur J Clin Microbiol Infect Dis 2021;40(4):715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Faria S, Teleschi JG. Computational search for drug repurposing to identify potential inhibitors against SARS-COV-2 using molecular docking, QTAIM and IQA methods in viral spike protein - human ACE2 interface. J Mol Struct 2021;1232:130076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cubuk H, OzbIl M. Comparison of clinically approved molecules on SARS-CoV-2 drug target proteins: a molecular docking study. Turk J Chem 2021;45(1):35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Unni S, Aouti S, Thiyagarajan S, et al. Identification of a repurposed drug as an inhibitor of spike protein of human coronavirus SARS-CoV-2 by computational methods. J Biosci 2020;45(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Choudhary S, Malik YS, Tomar S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front Immunol 2020;11:1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dong S, Sun J, Mao Z, et al. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J Med Virol 2020;92(9):1542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Borkotoky S, Banerjee M. A computational prediction of SARS-CoV-2 structural protein inhibitors fromAzadirachta indica(neem). J Biomol Struct Dyn 2021;39(11):4111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Aftab SO, Ghouri MZ, Masood MU, et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J Transl Med 2020;18(1):275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pokhrel R, Chapagain P, Siltberg-Liberles J. Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. J Med Microbiol 2020;69(6):864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B 2020;10(5):766–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Borgio JF, Alsuwat HS, al Otaibi WM, et al. State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch Med Sci 2020;16(3):508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Mirza MU, Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J Pharm Anal 2020;10(4):320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Iftikhar H, Ali HN, Farooq S, et al. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput Biol Med 2020;122:103848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bortz E, Westera L, Maamary J, et al. Host- and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. MBio 2011;2(4):e00151–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Grady SL, Hwang J, Vastag L, et al. Herpes simplex virus 1 infection activates poly (ADP-ribose) polymerase and triggers the degradation of poly (ADP-ribose) glycohydrolase. J Virol 2012;86(15):8259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Liu L, Lear Z, Hughes DJ, et al. Resolution of the cellular proteome of the nucleocapsid protein from a highly pathogenic isolate of porcine reproductive and respiratory syndrome virus identifies PARP-1 as a cellular target whose interaction is critical for virus biology. Vet Microbiol 2015;176(1–2):109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ge Y, Tian T, Huang S, et al. A data-driven drug repositioning framework discovered a potential therapeutic agent targeting COVID-19. BioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Motwalli O, Alazmi M. Analysis of natural compounds against the activity of SARS-CoV-2 NSP15 protein towards an effective treatment against COVID-19: a theoretical and computational biology approach. J Mol Model 2021;27(6):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Devaurs D, Antunes DA, Kavraki LE. Computational analysis of complement inhibitor compstatin using molecular dynamics. J Mol Model 2020;26(9):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Selvaraj C, Dinesh DC, Panwar U, et al. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J Biomol Struct Dyn 2021;39(13):4582–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chandra A, Gurjar V, Qamar I, et al. Identification of potential inhibitors of SARS-COV-2 endoribonuclease (EndoU) from FDA approved drugs: a drug repurposing approach to find therapeutics for COVID-19. J Biomol Struct Dyn 2021;39(12):4201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gossen J, Albani S, Hanke A, et al. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from activity-based compound library screening guided by analysis of protein dynamics. ACS Pharmacol Translat Sci 2021;4(3):1079–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ugurel OM, Mutlu O, Sariyer E, et al. Evaluation of the potency of FDA-approved drugs on wild type and mutant SARS-CoV-2 helicase (Nsp13). Int J Biol Macromol 2020;163:1687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.