

Summary
Host-directed therapy (HDT) is gaining traction as a strategy to combat infectious diseases caused by viruses and intracellular bacteria, but its implementation in the context of parasitic diseases has received less attention. Here, we provide a brief overview of this field and advocate HDT as a promising strategy for antimalarial intervention based on untapped targets. HDT provides a basis from which repurposed drugs could be rapidly deployed and is likely to strongly limit the emergence of resistance. This strategy can be applied to any intracellular pathogen and is particularly well placed in situations in which rapid identification of treatments is needed, such as emerging infections and pandemics, as starkly illustrated by the current COVID-19 crisis.
Keywords: malaria, antimalarial drug discovery, Host-directed therapy
Graphical abstract
Drug resistance threatens to reverse progress achieved in malaria control. Host-directed therapy (HDT) provides a barrier to drug resistance: since the target is not under the genetic control of the parasite, resistance-conferring mutations in the target cannot be selected under treatment. Wei et al. review opportunities for HDT in the fight against malaria.
The current crisis in anti-infective drugs
In the mid-20th century, newly deployed penicillin and sulfonamides saved countless lives from bacterial infections during World War II. In the decades that followed, new classes of antibiotics regularly came on the market, until the 1990s.1 Over this period, drugs targeting non-bacterial pathogens were also deployed with tremendous effect; arguably the one with the most impact was chloroquine, discovered in 1934 and used for mass treatment of malaria in the following decades, and whose outstanding efficacy against the multiple species of malaria parasites, tolerability, and low cost made it an antimalarial wonder.
Unfortunately, this golden age is over. The first cases of bacteria with decreased susceptibility to penicillin were reported in 1940,2 and the emergence and spread of chloroquine resistance in malaria parasites started in the 1950s, ultimately rendering the drug largely useless. The same trend affected essentially all molecules deployed as antimalarials. Resistance to one particularly promising drug, atovaquone, was observed even before mass deployment,3 and was subsequently shown to have emerged through the selection of a mutation in the gene encoding its molecular target (in this case the cytochrome b of the parasite) that reduced susceptibility to the drug.4 Resistance caused by the selection of modified/altered targets under drug pressure has affected many other treatments, in the context of other infectious diseases beyond malaria.5 The rapidity with which drug-resistant pathogens can be selected through this mechanism is a grave concern in the context of global public health. The limited return on investment for research and development in infectious disease chemotherapy, in comparison to other types of indications such as cancer or chronic diseases, has exacerbated the problem by decreasing the overall investment from the pharmaceutical industry for the development of de novo anti-infectives. Today more than ever, additional strategies to combat infectious agents are urgently needed.
Host-directed therapy
It is well established that intracellular pathogens of any taxon (viruses, bacteria, eukaryotic parasites) rely on host cell functions for their survival and proliferation. As a consequence of this reliance, inhibitors of host cell enzymes that are essential for pathogen viability have anti-infective properties. Furthermore, since the target is host encoded, the emergence of resistance based on the selection of mutations in the gene of a target whose affinity for the drug is decreased, and ensuing preferential proliferation of parasites with a resistant genotype under drug pressure, cannot occur. This offers opportunities for host-directed therapy (HDT), an innovative strategy that consists of targeting host functions that mediate diseases caused by infectious agents, and is being explored in the context of many infectious diseases. For example, the host methyltransferase SETD3 was recently proposed as a target for the treatment of enterovirus and rhinovirus infections6; thus, the long-standing problem of a cure for the common cold may finally be solved through HDT. Alternatively, HDT can target host mechanisms implicated in pathogenesis, rather than enzymes directly required for pathogen survival as outlined above; studies with Leishmania7 and Mycobacterium tuberculosis8 exemplify this strategy. As a recent example, it was shown that ibrutinib, an inhibitor of the Bruton tyrosine kinase (BTK) that plays a critical role in B cell signaling, is more efficient than conventional anti-leishmanial drugs in an animal efficacy model; furthermore, the authors established that this effect is not mediated by microbicidal activity of the compounds, but by promoting protective immunity and targeting host pathways that mediate susceptibility.9 Perhaps surprisingly, HDT approaches have been underexplored for malaria, although its potential benefits have been discussed by our teams and others.10, 11, 12, 13, 14
Human toxicity potential of HDT and de-risking strategies
The advantage of HDT in not targeting parasites directly, and thus limiting the risk for resistance, comes with the caveat of potential for toxicity, which clearly needs to be addressed. The main risk is that the inhibition of human targets could lead to toxicity, translating into potentially severe side effects in treated patients, which can affect a very small proportion of patients and thus may not be detected in safety clinical trials. It is therefore essential to embed a de-risking strategy when considering HDT. HDT hit and lead compounds should be profiled in vitro on panels of human receptors and kinases, which together represent a substantial proportion of key intracellular components essential for the viability and functionality in all human cells. This profiling could assist in the identification of the key host target(s) mediating the anti-parasitic activity. If not the case, then the off-target effect would be characterized as in any other classical drug discovery programs. The potencies observed can then be compared to the antiparasitic potency of the compound in a relevant phenotypic assay, and the ratio can help to evaluate the selectivity factor and assess potential toxicity risk. It is important to note that a given inhibitory potency against a host cell target is not necessarily associated with toxicity. Should human enzymes or receptors associated with a given toxicity (e.g., cardiovascular, hepatic, reproductive) be inhibited in vitro by the HDT compound, medicinal chemistry would guide lead optimization to improve selectivity (host target versus off-target); in vivo toxicology studies should be conducted on animal models to further define the safety margin of the optimized compound. Determining the oral exposure profiles of the compound in animal toxicity and efficacy models will allow the safety margin to be evaluated. If available, human clinical, pharmacokinetic or efficacy data for the HDT compound would allow the calculation of an even more relevant therapeutic margin. In general, a safety margin of at least 10-fold is requested, but 20-fold is ideal to accommodate the diversity/variability of drug exposure profiles among different patients. Malaria being particularly severe in young children and pregnant women, juvenile and embryo fetal toxicity risk assessment will need to be conducted.15 Malaria treatments are based on 3-day dosing and ideally aim at full efficacy as a single-dose cure, which is the preferred treatment regimen in most clinical trials.16 It is therefore important to consider the difference in toxicity risks between the treatment of acute disease (which can be very brief) and the long-term treatment required for chronic disease (e.g., cancer, metabolic disorders), since the latter necessitate a repeated and sustained treatment that may lead to cumulative exposures and hence higher toxicity risk. Nevertheless, antimalarials must have long duration exposure even if the dosing regimen is infrequent. Regular infections in endemic regions also emphasize the safety required in any new antimalarial.
In summary, determining the effective exposed concentration needed to treat malaria while avoiding side effects in the host must govern the evaluation of risks. HDT can be accelerated by drug repositioning (see below); in such a framework, only the drugs displaying antimalarial potency equivalent to or below (in case of anticancer drugs) the concentration needed to treat patients of the primary indication disease can be considered.
Host cell targets during Plasmodium infection of hepatocytes and erythrocytes
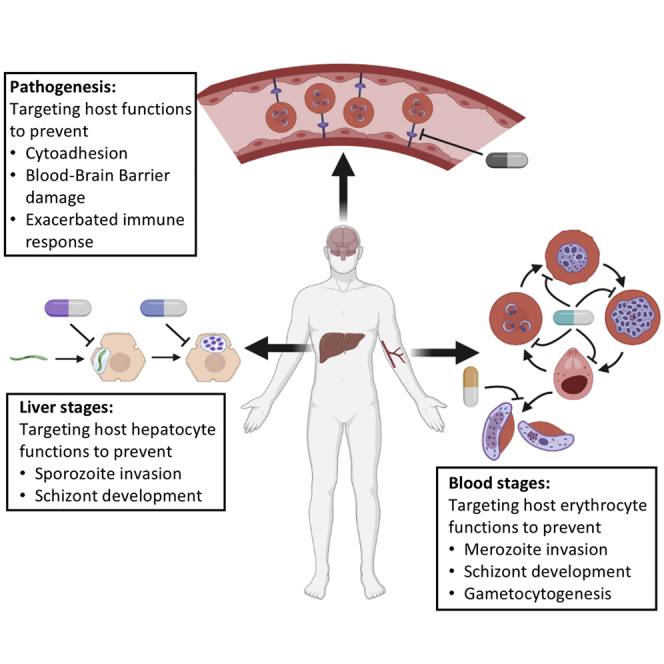
Malaria parasites have a complex life cycle (Figure 1). Human infection is initiated by the bite of an infected Anopheles mosquito, which delivers sporozoites at the bite site. These sporozoites gain access to the bloodstream and reach the liver, where they invade hepatocytes and develop into schizonts, producing tens of thousands of merozoites. After their release into the bloodstream, merozoites invade erythrocytes to produce more merozoites, causing malaria pathogenesis. Some of these merozoites develop into sexual forms (male or female gametocytes) that can be transmitted to a mosquito during a blood meal.
Figure 1.

The life cycle of malaria parasites offers opportunities for HDT
Plasmodium-infected Anopheles mosquitoes inject sporozoites into human hosts during blood meals. Sporozoites migrate to the liver and mature into schizonts within hepatocytes, which then rupture and release merozoites into the bloodstream. For P. vivax and P. ovale, parasites can also undergo a dormant stage by forming hypnozoites in hepatocytes, which can reactivate to cause relapsing disease up to several years after the initial infection. The merozoites produced during hepatocytic schizogony access the bloodstream and undergo cyclical asexual multiplication in erythrocytes. Merozoites infect red blood cells and mature from trophozoites into schizonts, which rupture and release merozoites. In the case of P. falciparum, infected erythrocytes can adhere to the vascular endothelium, which causes endothelial barrier dysfunction. Some blood-stage parasites differentiate into sexual erythrocytic stages in the bone marrow and release gametocytes into the bloodstream after maturation. Anopheles mosquitoes ingest gametocytes during blood meals; they mature in the mosquito midgut into male gametes (microgametes) and female gametes (macrogametes) that fuse to form zygotes, which then develop into motile and elongated ookinetes. The ookinetes invade the mosquito midgut wall, where they develop into oocysts, rupture, and release sporozoites. These sporozoites then migrate to the salivary glands of the mosquito. Inoculation of sporozoites from salivary glands into new human hosts continues during the next blood meal. HDT can be implemented at both the liver and blood stages of this life cycle; potential strategies are listed in the boxes to the right (see text for details). Adapted from Nilsson et al.17
HDT can be implemented during the infection of either hepatocytes or erythrocytes (Figure 1), through targeting host cell functions that are directly required for parasite survival and proliferation at these stages. Some host targets may be relevant to both stages. Table 1 presents examples of ligands or inhibitors of host targets with known antimalarial activity.
Table 1.
List of host-targeting compounds with known anti-Plasmodium activity
| Compound | Target | Target type | Proposed mechanism | Ref. |
|---|---|---|---|---|
| K252a | c-MET and other kinases | protein kinase | inhibits kinase activity of c-Met and other tyrosine kinases | Carrolo et al.18 |
| Blockers of lipid transport (BLTs) | scavenger receptor (SR)-B1 | lipoprotein receptor | inhibits SR-B1-mediated selective uptake of lipids from high-density lipoproteins | Rodrigues et al.19 |
| Nutlin-3 | MDM2 | E3 ubiquitin-protein ligase | prevents degradation of p53, promotes lipid peroxidation in infected hepatocytes | Kaushansky et al.20 |
| Serdemetan | MDM2 | E3 ubiquitin-protein ligase | minimizes degradation of p53, eliminate Plasmodium-infected hepatocytes | Douglass et al.21 |
| SB505124 | TGF-β receptor 1 | protein kinase | inhibits the enzymatic activity of kinases involved in multiple cellular processes | Arang et al.22 |
| Auphen | AQP3 | membrane protein | selectively and irreversibly inhibits glycerol transport by AQP3; effective against both liver and blood stages and against multiple human malarias | Posfai et al.23 |
| Brefeldin A | COPI | coatomer protein complex | blocks host intracellular protein trafficking | Raphemot et al.24 |
| Golgicide A | GBF1 | guanine nucleotide exchange factor | blocks host intracellular protein trafficking | Raphemot et al.24 |
| ABT-737, obatoclax | Bcl-2 family proteins | B cell lymphoma 2 family proteins | overcomes apoptotic block placed by parasites, eliminates Plasmodium-infected hepatocytes | Douglass et al.,21 Kaushansky et al.25 |
| PP1 | Src family kinases | protein kinase | inhibits negative regulation of endothelial permeability by Src-family kinases, mediates dissociation of vascular endothelial (VE)-cadherin and redistribution of ZO-1 | Kaushansky et al.,25 Gillrie et al.26 |
| Fasudil | Rho-associated protein kinase | protein kinase | decreases nuclear factor (NF)-κB activation and endothelial cell apoptosis, restores endothelial barrier integrity via rho-kinase signaling pathway | Gillrie et al.,26 Taoufiq et al.27 |
| Imatinib | receptor tyrosine kinases | protein kinase | inhibits erythrocyte band 3 phosphorylation, preventing parasite egress | Sicard et al.,28 Kesely et al.29 |
| PHA-665752 | c-MET | protein kinase | inhibits host c-MET activation during intra-erythrocytic development | Adderley et al.30 |
| Crizotinib | c-MET/Alk | protein kinase | inhibits host c-MET activation during intra-erythrocytic development | Adderley et al.30 |
| SB-590885 | B-Raf | protein kinase | inhibits host B-Raf activation, which halts parasite development during intra-erythrocytic ring stage development | Adderley et al.30 |
| IPA-3 | PAK1 | protein kinase | inhibits host MEK1 activation during intra-erythrocytic development | Sicard et al.28 |
| U-0126 | MEK1/2 | protein kinase | inhibits MEK1/2 activation during intra-erythrocytic development | Sicard et al.28 |
| Propranolol | heterotrimeric G protein (Gs) | G proteins | blocks merozoite invasion of erythrocytes and blood-stage growth | Murphy et al.31 |
| Erastin | SLC7A11 | cystine/glutamate transporter | blocks host SLC7a11-GPX4 pathway to induce lipid peroxidation in Plasmodium-infected hepatocytes | Kain et al.32 |
It must be kept in mind that it is not trivial to assign the efficacy of any HDT compound to the inhibition of the targeted host protein, as any compound could cause parasite killing through parasite- or host-encoded off-targets; off-target effects need to be characterized as in any other classical drug discovery program. This is particularly problematic for blood stages, as erythrocytes (unlike hepatocyte lines) are not directly amenable to genetic manipulations (e.g., replacement of wild-type alleles with alleles encoding drug-resistant targets) that can be used to tackle this problem. However, recent developments in the manipulation of erythroid lineage precursors to generate genetically engineered erythrocytes that are susceptible to infection with Plasmodium33 offer a (labor-intensive) window of opportunity for ascribing pharmacology to specific host-encoded targets. In parallel to this approach, pharmacological validation with a panel of reference inhibitors (when available) is possible.
Liver stages
The liver stage of infection represents a critical target for HDT, as blocking infection during this stage could prevent blood infection, and hence malaria pathogenesis and subsequent transmission. However, prophylaxis using liver infection inhibitors would require a level of safety even greater than curative approaches. The establishment of hypnozoites, a dormant form that can cause relapsing infections, is one of the possible outcomes of hepatocyte infection with Plasmodium vivax; recent models suggest that eliminating a small fraction of hypnozoites could have a dramatic impact on P. vivax prevalence.34 Only two drugs, both belonging to the same 8-aminoquinoline chemical class, are approved for the elimination of hypnozoites.35 These two molecules, primaquine and tafenoquine, presumably have the same mechanism of action (although this is not yet fully elucidated), and both cause significant complications in G6PD-deficient individuals.36 Very few additional antihypnozoite drug candidates are in the pipeline.16 Identifying factors in the host hepatocyte that are required for hypnozoite establishment, maintenance, and/or reactivation could contribute to fill this gap.
Multiple host receptors have been demonstrated to be required for Plasmodium entry into hepatocytes.18,19,37,38 The roles of several of these factors is context dependent,39, 40, 41 and the two most important species with respect to global disease burden, P. falciparum and P. vivax, use different receptors for the invasion of hepatocytes.40 Targeting multiple factors is therefore critical to block invasion by diverse Plasmodium isolates and species. Once the parasite is within the hepatocyte, other factors are critical for the maintenance and development of the liver stage infection. The host cell fatty acids synthesis machinery is required for parasite development in the liver,42,43 presumably through facilitating nutrient uptake and parasite membrane growth. Recent studies have also shown that a series of host-derived vesicles sequester to the parasite.24,44, 45, 46, 47, 48, 49, 50 Targeting proteins that are involved in trafficking, such as coatomer protein complex I subunits (COPB2, COPG1) or the adaptor protein GGA1, impairs parasite development in hepatocytes.24 Furthermore, the tumor suppressor p53 negatively regulates Plasmodium liver stage development20 by altering lipid peroxidation in the hepatocyte32; boosting levels of p53 results in dramatically fewer liver-stage parasites,20,21 offering additional opportunities for interference with the parasite at this stage of its life cycle. The membrane water and small-molecule channel aquaporin-3 (AQP3) has also been shown to support development in multiple stages of the life cycle of the parasite, including liver stages.23 Excitingly, targeting AQP3 was recently shown to successfully impair P. vivax liver stages,51 suggesting that small-molecule inhibitors against AQP3 may have antihypnozoite activity, and could provide long-sought-after tools for targeting relapses. Finally, several host protein kinases, a family of enzymes with proven druggability,52,53 are important for parasite development in hepatocytes.10,22 Excitingly, some of these targets overlap with kinases that are also implicated in the subsequent blood stages (see below).
Blood stages
After maturation within the hepatocyte, the parasite exits the liver and invades erythrocytes. Circulating erythrocytes comprise mostly mature normocytes, a simplified cell largely devoid of organelles and fully adapted for the transport of oxygen by hemoglobin, as well as a small number of the more complex early reticulocytes that contain several hundred additional proteins and remnant organelles.54 These host-cell differences are reflected in the tropism of some human malaria parasites, with P. vivax preferentially infecting reticulocytes, while P. falciparum can infect erythrocytes of all ages. We now understand that these host erythrocytes play an active role during Plasmodium infection and are beginning to elucidate which host factors are of specific or general importance for different human malarias. First, it was shown in the early 2000s that host erythrocyte heterotrimeric G proteins, upstream components of many signaling pathways, are activated by infection.55 Subsequently, a phospho-signaling pathway implicating host cell PAK (p21-activated kinase) and MEK1 (MAP/ERK kinase) was shown to likewise be activated by infection in the host erythrocyte. Treatment with U0126, a highly selective allosteric inhibitor of human MEK1, impaired parasite proliferation in both the hepatocyte and the erythrocyte.28 The toxicity associated with MEK inhibitors precludes their direct repurposing for malaria; however, these compounds offer new medicinal chemistry starting points to be developed, with the goal of reducing toxicity to a point that treatment of malaria infections becomes acceptable. More recently, AQP3 was found to be required for P. vivax development in reticulocytes, with an AQP3 inhibitor offering pan-malaria activity against both P. vivax in reticulocytes and P. falciparum in normocytes,23,51 in addition to its potential as a liver stage inhibitor (see above).
We recently published an antibody microarray-based global analysis of infected erythrocyte phospho-signaling pathways, which revealed that the host erythrocyte undergoes dynamic activation of numerous signaling elements.30 This strongly suggests that host cell signaling has a more central role in parasite development than previously thought. Highly selective inhibitors (that will remain selective until, as seen quite often, off-target activities are observed) targeting activated host cell enzymes identified in this study display nanomolar activity against both P. falciparum and Plasmodium knowlesi in culture and against the rodent malaria parasite P. berghei in vivo, suggesting that they constitute attractive targets for HDT against various Plasmodium clades (Table 1). These kinases are the subject of extensive drug discovery in the context of cancer chemotherapy, and highly selective inhibitors of these (and other) kinases display potent antimalarial activity in parasite proliferation.28,30,56 The fact that many of these enzymes do not have orthologs in the kinome of the parasite reduces the likelihood that their inhibitors have parasite-encoded off-targets, although this needs to be demonstrated by reverse genetics in each case (see above). The potential of inhibitors that target host kinases with no ortholog in the parasite is illustrated by the potent antimalarial activity of imatinib (Gleevec), the first kinase inhibitor to have reached the market as a treatment for chronic myelogenous leukemia and acute lymphocytic leukemia.29,57 As mentioned above, some of the host kinases required for parasite proliferation in erythrocytes are also essential for the hepatic stage of the life cycle (and likely for the development of gametocytes as well, although this needs to be investigated), offering opportunities for multi-stage, pan-malaria intervention and potentially leading to HDT compounds with additional prophylactic or transmission-blocking properties.
HDT targeting malaria pathogenesis
The infected erythrocyte evades clearance in the spleen through cytoadherence to the endothelium of the peripheral vasculature, which is a major driver of the pathogenesis of P. falciparum infections. Cytoadherence is mediated by products of the var gene family, which are expressed at the surface of the infected erythrocyte and interact with specific receptors on endothelial cells (reviewed in Bernabeu and Smith,58 Smith,59 and Wassmer et al.60). Cerebral malaria, the main cause of malaria-induced mortality, is dependent on the cytoadherence of infected erythrocytes in the brain vasculature and is strongly associated with inflammation-mediated brain swelling,61 possibly triggered by the breakdown of the blood-brain barrier. Molecules that interfere with parasite cytoadherence and/or barrier-strengthening compounds represent an opportunity to target the most severe symptoms of malaria (reviewed in Glennon et al.13). Importantly, while current antimalarials that inhibit parasite-encoded functions have facilitated major strides in reducing malaria mortality and morbidity, they are not entirely effective at eliminating death due to cerebral malaria. Fast-acting host-targeting interventions that block rapid signaling processes involved in brain inflammation or blood-brain barrier disruption may act rapidly enough to save the lives of patients suffering from cerebral malaria episodes. This is supported by studies in mouse models of cerebral malaria: for example, treatment of animals with MEK inhibitors decreases organ inflammation and immune cell recruitment, thus limiting tissue damage62; this cooperates with a direct effect of MEK inhibitors on parasite proliferation (see above) to prevent the death of treated animals. Likewise, treatment with neuregulin-1 (NRG-1, a neuronal growth factor) attenuates cerebral malaria pathogenesis in infected mice by regulating ErbB4/AKT/STAT3 signaling, leading the authors to suggest that augmenting NRG-1 may be an effective adjunctive therapy to reduce brain tissue injury during cerebral malaria.63
The opportunity for drug repurposing
Targeting the host to eliminate infection or reduce pathology associated with disease presents an opportunity for the repurposing of molecules originally developed to target non-communicable diseases. One advantage of targeting the host is that a wealth of information is available for many potential host targets. As an example, the aforementioned tumor suppressor protein p53, which has been shown to regulate both liver-stage malaria in the laboratory20,21,32 and clinical symptoms of malaria in the field,64 has prompted nearly 100,000 publications in PubMed, approximately the same number of publications as in the entirety of the malaria field. Furthermore, large collections of well-characterized small-molecule inhibitors of host targets are available, which is not the case for parasite-encoded proteins. One example is the 12,000-compounds ReFRAME (Repurposing, Focused Rescue, and Accelerated Medchem) library, an open-access drug repositioning screening set combining 3 widely used commercial drug databases (Clarivate Integrity, GVK Excelra GoStar, and Citeline Pharmaprojects). This library has been screened against parasites such as Trypanosoma cruzi,65 the malaria-related Crytposporidium,66 and pathogenic amoebae,67 as well as viruses such as Zika68 and Lassa,69 and it would be of great interest to implement a screen against malaria parasites.
Likewise, as alluded to above, host protein kinases carry more tremendous potential in repurposing approved drugs for infectious diseases: a large proportion of the human kinome is already targeted by drugs that have been used in humans, with >60 kinase inhibitors approved by the US Food and Drug Administration (FDA), mostly for the treatment of various cancers.70,71 In addition, there are >200 kinase inhibitors in clinical trials.72 Many of the host kinases shown to be essential for parasite survival in hepatocytes and/or erythrocytes are the targets of approved inhibitors, as exemplified above for the tyrosine kinase inhibitor imatinib, whose potential in antimalarial drug development could be rapidly tested in clinical trials.29 Table 2 provides examples of FDA-approved medicines that inhibit some of the host kinases (MEK1, SYK, MET, BRAF, RET, JAK2) implicated in malaria progression and pathogenesis. As these drugs have been approved and thus extensively studied in both preclinical animal models and humans, the requisite data are available to facilitate evaluation in malaria models and guide dosing in humans, should they prove effective in malaria preclinical studies. The availability of pharmacokinetic and safety data will allow for careful delineation of dosing regimens to establish an appropriate therapeutic window. It is important to note that kinase inhibitors are not approved solely for cancer, and there is increasing interest in non-oncology indications,73,74 reflecting the ability to design selective kinase inhibitors and the accumulating evidence that suitable safety margins for these molecules can be achieved in various contexts. In addition to the potential utility of approved medicines, it would also be of particular interest to identify compounds that passed early-phase clinical trials, and hence are supposedly tolerated enough for use in humans, but failed for efficacy against their original target indication. This would provide an incentive to the industry for repurposing dead-end compounds for clinical trials in an infectious disease context.
Table 2.
FDA-approved kinase inhibitors of potential kinase targets for antimalarial HDT
| Host kinase | FDA-approved kinase inhibitor, with year of approval and original indication | Reference linking host target to malaria life cycle |
|---|---|---|
| MEK1 | trametinib (2013; melanoma) | Sicard et al.28 |
| cobimetinib (2015; melanoma) | ||
| binimetinib (2018; melanoma) | ||
| selumetinib (2020; neurofibromatosis type I) | ||
| SYK | fostamitinib (2018; chronic immune thrombocytopenia) | Pantaleo et al.,56 Kesely et al.57 |
| MET | crizotinib (2011; non-small cell lung carcinoma) | Carrolo et al.,18 Arang et al.,22 Adderley et al.,30 Leirião et al.,75 Prudêncio et al.76 |
| capmatinib (2020; non-small cell lung carcinoma) | ||
| tepotinib (2021; non-small cell lung carcinoma) | ||
| BRAF | sorafenib (2005; hepatocellular, thyroid, and advanced renal cell carcinomas) | Kain et al.,32 Adderley et al.30 |
| vemurafenib (2011; metastatic melanoma) | ||
| regorafenib (2012; metastatic colorectal cancer) | ||
| dabrafenib (2013; cancers associated with a mutated version of the gene BRAF) | ||
| encorafenib (2018; cancers associated with a mutated version of the gene BRAF) | ||
| RET | pralsetinib (2020; thyroid and lung carcinomas) | Adderley et al.30 |
| JAK2 | ruxolitinib (2011; myelofibrosis) | Adderley et al.30 |
| tofacitinib (2012; rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis) | ||
| baricitinib (2018; rheumatoid arthritis) | ||
| fedratinib (2019) | ||
| upadacitinib (2019) | ||
| CSK | dasatinib (2006; chronic myelogenous leukemia) | Arang et al.22 |
| ibrutinib (2013; mantle cell lymphoma) | ||
| FGFR4 | nintedanib (2014; idiopathic pulmonary fibrosis) | Arang et al.22 |
| erdafitinib (2019; urothelial carcinoma) | ||
| FLT1 | sunitinib (2006; advanced renal cell carcinoma and gastrointestinal stromal tumors) | Arang et al.22 |
| sorafenib (2005; advanced renal cell carcinoma) | ||
| nintedanib (2014; idiopathic pulmonary fibrosis) | ||
| vandetanib (2011; thyroid cancer) | ||
| cabozantinib (2012; medullary thyroid cancer) | ||
| pazopanib (2009; advanced renal cell cancer and advanced soft tissue sarcoma) | ||
| axitinib (2012; advanced renal cell carcinoma) | ||
| tivozanib (2021; advanced renal cell carcinoma) | ||
| lenvatinib (2015; progressive, differentiated thyroid cancer) | ||
| pexidartinib (2019; tenosynovial giant cell tumor) | ||
| FLT3 | sunitinib (2006; advanced renal cell carcinoma and gastrointestinal stromal tumors) | Arang et al.22 |
| sorafenib (2005; advanced renal cell carcinoma) | ||
| nintedanib (2014; idiopathic pulmonary fibrosis) | ||
| midostaurin (2017; acute myeloid leukemia) | ||
| cabozantinib (2012; medullary thyroid cancer) | ||
| ponatinib (2012; chronic myeloid leukemia and Philadelphia chromosome+ acute lymphoblastic leukemia) | ||
| fedratinib (2019; myelofibrosis) | ||
| ceritinib (2014; non-small cell lung cancer) | ||
| gilteritinib (2018; acute myeloid leukemia) | ||
| pexidartinib (2019; tenosynovial giant cell tumor) | ||
| brigatinib (2017; non-small cell lung cancer) | ||
| IRAK1 | sunitinib (2006; advanced renal cell carcinoma and gastrointestinal stromal tumors) | Arang et al.22 |
| STK35 | bosutinib (2012; chronic myelogenous leukemia) | Prudêncio et al.76 |
In silico-guided drug repurposing
In silico-guided drug development strategies can capture the important features of both the drug and the target space, and enable down-selection and accurate identification of target candidates for further validations.77 Computational methods for drug repurposing include signature matching, molecular docking, network-based strategies, and machine learning methods (Table 3).
Signature matching infers drug-disease associations based on the transcriptomic, proteomic, or metabolic characteristics of host cells in healthy and disease conditions.78 One application is to identify drug candidates that alter gene expression signatures.79 The logic is that if a drug can reverse the gene expression signatures for the disease phenotype, it may restore a healthy cellular state. Studies using human colon cancer cells showed that signature matching can effectively predict drugs to treat colorectal cancer.80 The integration of signature-based in silico tools with patient-specific disease signatures could facilitate the establishment of therapeutic pipelines for treating infectious diseases, including malaria.
Molecular docking predicts drug-target binding using three-dimensional (3D) structure-based modeling and computational simulation,81 and can be target centered (in which drugs are screened against the disease-associated protein) or ligand centered (in which compounds are docked into an array of protein targets).82 This approach requires the 3D structural information on both compounds and protein targets, as well as the validation of the host target for the primary disease indication. While molecular docking is a prohibitive limitation for pathogen-encoded targets whose structure has not been solved, it offers the opportunity to facilitate host-directed drug discovery and has been applied to various infectious agents such as human immunodeficiency virus (HIV), Mycobacterium tuberculosis (MTB), malaria, and severe acute respiratory syndrome-coronaviruses-1 and -2 (SARS-CoV-1 and -2).83, 84, 85, 86, 87
Network-based approaches for drug repurposing and discovery have been facilitated by the expansion of genomic, proteomic, transcriptomic, and metabolomic data.88 Disease-specific protein-protein interactions (PPIs) or gene regulatory networks are constructed,88 allowing the comparison of the molecular underpinnings of a comparatively understudied disease with the molecular basis of a well-studied disease with effective therapeutics. In addition to their therapeutic applications in cancers and coronary artery disease,88,89 network-based approaches have facilitated drug discovery and repurposing to treat infectious diseases such as pandemic and seasonal respiratory infections by redirecting the usage of known drugs to target host protein regulators of viral infection-related processes.90 As our knowledge of host-Plasmodium molecular interactions grows, network-based methods could facilitate the development of HDT for malaria.
Machine learning aims to facilitate pattern exploration and extract the most informative relationships between drugs, targets, and disease phenotypes through learning from diverse, large-scale data.91 Machine learning has been used to identify novel disease-relevant targets based on drug polypharmacology and cell phenotype data collected from small-scale drug screens. For instance, applying an elastic net regression algorithm on a small-scale drug screen dataset led to the identification of host kinases that regulate Plasmodium liver stage infection and the prediction of kinase inhibitors that inhibit parasite growth in the hepatocyte.22
Table 3.
Computational methods for host-based drug repurposing
| Approach | Input data | Methodology |
|---|---|---|
| Signature matching | transcriptomic, proteomic, or metabolomic characteristics in healthy and disease conditions | a negative correlation between drug-treated characteristics and characteristics in disease condition indicates that the drug may drive cells from disease state back to healthy state |
| Molecular docking | 3D structural information on proteins and drugs | compute the energetic binding likelihood between each drug and the target, followed by scoring of the drugs and selecting top candidates for further validations |
| Network-based method | integration of multiple data types (i.e., gene expression, protein-protein interactions, metabolic reactions, and disease pathology) | compile different information sources and expand dimension of biological systems; capture relationships between modules/elements in the networks; discover novel drug-target-disease interactions or perform quantitative analysis on these interactions |
| Machine learning | known drug-target interactions, phenotypic measurements upon drug treatment | learn features that drive the phenotypic state of cells based on known drug-target interactions; predict drug candidates that can reverse the cellular state based on the selected features |
In silico-guided drug repurposing holds great promise for identifying the next generation of host-targeted anti-malarial drugs.
Concluding remarks
Many major pharmaceutical companies (Novartis, Sanofi, GSK, Takeda, Merck KGaA, Zydus Cadila) have collaborated, or are engaged in discovery and development, with the Medicines for Malaria Venture to provide the antimalarial community with a strong pipeline of drug candidates, focusing on whole-cell actives and compounds acting on parasite-encoded targets (for a recent overview of the MMV pipeline, see Hooft van Huijsduijnen et al.92). In particular, it is possible that some of the “irresistible” compounds identified in whole-cell screens may have host targets. It is tempting to propose an evolution of this collaborative work by inviting the pharmaceutical industry to consider some of their advanced compounds for repositioning in the HDT approach. Similar to the modus operandi when companies provide their compound libraries for phenotypic screening,93, 94, 95 the repositioning exercise could be performed on the small sets of clinically relevant candidate drugs available within each company. Drugs for protecting endemic populations of low- to middle-income countries against Plasmodium infection must be distributed with a price tag that will be much lower than the one for travelers coming from high-income countries. As such, a mechanism to ensure affordability in endemic regions for those most in need would need to be established for any such repurposed drug.
As detailed above, safety assessment will be a crucial element in applying HDT to the malaria context, and it is essential to take into account the specifics of malaria (versus cancer) when addressing this problem. Cancer drugs are allowed to have a relatively aggressive profile, especially in cases in which the specific disease has a high mortality rate, such as pancreatic, liver, or lung cancers, whose 5-year survival rate are as low as 10%, 20%, or 21%, respectively.96 The much lower mortality rate for malaria (in 2019, there was an estimated 409,000 deaths for 229 million cases, or a >0.2% mortality rate) makes any risk associated with antimalarial drugs deployed for mass treatment essentially unacceptable, even more so if one considers liver stage-targeting prophylactics. Severe malaria complications, such as cerebral malaria (which has a mortality rate of ∼20% and can have very serious sequelae in survivors97), may arguably justify the use of drugs with higher toxicity; however, here again, tolerability should be very high, as exemplified by the current treatment with artesunate98; furthermore, these drugs would need to act very quickly, as death can occur very rapidly after the onset of severe malaria symptoms.
Finally, multiple pathogens may have comparable needs from the host, or initiate symptoms using similar mechanisms. As an example, most intracellular pathogens, including viruses, bacteria, and eukaryotic parasites, rely on the host cell cytoskeleton machinery to permit invasion. Likewise, many pathogens are dependent on the same set of kinases in their host cells.99 For example, receptor tyrosine kinases have been implicated not only in the infection of hepatocytes18,38 and erythrocytes30 by Plasmodium spp but also in the infection of their cognate cell types by the bacterium Listeria and by several viruses, including Ebola, hepatitis C, and influenza viruses100 (reviewed in Haqshenas and Doerig99). As a further and highly topical example, the possibility of repurposing kinase inhibitors for the treatment of COVID-19 was recently reviewed.101 Thus, targeting the host provides an opportunity to develop interventions that could be useful to treat various infectious diseases, including co-infections. This is particularly the case for well-studied infections that are challenging to address pharmacologically understudied or neglected infections, or even novel outbreaks such as the crisis caused by the SARS-CoV-2 coronavirus, where little is known about the specific host-pathogen interactions at the time treatments need to be developed. This strategy would require testing on pathogens beyond malaria, and would fill in the dangerous gap in new anti-infectives that has developed in recent decades. A major advantage of repurposing approved drugs is the rapidity with which new treatments can be deployed, a feature whose crucial importance is dramatically demonstrated by the coronavirus disease 2019 (COVID-19) pandemic.
Acknowledgments
This work was supported by funding from the National Health and Medical Research Council, Australia, to D.W.W., J.A., C.D., and D.H.D. (APP2003712), the Hospital Research Foundation (fellowship to D.W.W.) and grants from the US National Institutes of Health to A.K. (R01AI158719, R21 AI 151344, R01GM101183, R01 AI 148802). Work in the C.D. laboratory is also supported by the Victorian Government, with funding provided through Round 4 of the Victorian Medical Research Acceleration Fund for the project entitled “Host cell kinome as a target for the treatment of SARS-CoV-2 infection.”. We are grateful to Dr. Jeremy Burrows and Dr. Tim Wells (Medicines for Malaria Venture, Geneva) for their critical and insightful comments on the manuscript. Novel approach to management of viral infections by targeting host cell processes: host cell kinome as a target for the treatment of SARS-CoV-2 infection.
Contributor Information
Alexis Kaushansky, Email: alexis.kaushansky@seattlechildrens.org.
Christian Doerig, Email: christian.doerig@rmit.edu.au.
References
- 1.Silver L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham E.P., Chain E. An enzyme from bacteria able to destroy penicillin. 1940. Rev. Infect. Dis. 1988;10:677–678. [PubMed] [Google Scholar]
- 3.Looareesuwan S., Viravan C., Webster H.K., Kyle D.E., Hutchinson D.B., Canfield C.J. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 1996;54:62–66. doi: 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- 4.Korsinczky M., Chen N., Kotecka B., Saul A., Rieckmann K., Cheng Q. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob. Agents Chemother. 2000;44:2100–2108. doi: 10.1128/aac.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson D.S., Walliker D., Wellems T.E. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc. Natl. Acad. Sci. USA. 1988;85:9114–9118. doi: 10.1073/pnas.85.23.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diep J., Ooi Y.S., Wilkinson A.W., Peters C.E., Foy E., Johnson J.R., Zengel J., Ding S., Weng K.F., Laufman O. Enterovirus pathogenesis requires the host methyltransferase SETD3. Nat. Microbiol. 2019;4:2523–2537. doi: 10.1038/s41564-019-0551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varikuti S., Jha B.K., Volpedo G., Ryan N.M., Halsey G., Hamza O.M., McGwire B.S., Satoskar A.R. Host-Directed Drug Therapies for Neglected Tropical Diseases Caused by Protozoan Parasites. Front. Microbiol. 2018;9:2655. doi: 10.3389/fmicb.2018.02655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baindara P. Host-directed therapies to combat tuberculosis and associated non-communicable diseases. Microb. Pathog. 2019;130:156–168. doi: 10.1016/j.micpath.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Varikuti S., Volpedo G., Saljoughian N., Hamza O.M., Halsey G., Ryan N.M., Sedmak B.E., Seidler G.R., Papenfuss T.L., Oghumu S. The Potent ITK/BTK Inhibitor Ibrutinib Is Effective for the Treatment of Experimental Visceral Leishmaniasis Caused by Leishmania donovani. J. Infect. Dis. 2019;219:599–608. doi: 10.1093/infdis/jiy552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prudencio M., Mota M.M. Targeting host factors to circumvent anti-malarial drug resistance. Curr. Pharm. Des. 2013;19:290–299. doi: 10.2174/138161213804070276. [DOI] [PubMed] [Google Scholar]
- 11.Doerig C., Abdi A., Bland N., Eschenlauer S., Dorin-Semblat D., Fennell C., Halbert J., Holland Z., Nivez M.P., Semblat J.P. Malaria: targeting parasite and host cell kinomes. Biochim. Biophys. Acta. 2010;1804:604–612. doi: 10.1016/j.bbapap.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 12.Carvalho T.G., Morahan B., John von Freyend S., Boeuf P., Grau G., Garcia-Bustos J., Doerig C. The ins and outs of phosphosignalling in Plasmodium: parasite regulation and host cell manipulation. Mol. Biochem. Parasitol. 2016;208:2–15. doi: 10.1016/j.molbiopara.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Glennon E.K.K., Dankwa S., Smith J.D., Kaushansky A. Opportunities for Host-targeted Therapies for Malaria. Trends Parasitol. 2018;34:843–860. doi: 10.1016/j.pt.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adderley J., Williamson T., Doerig C. Parasite and host erythrocyte kinomics of Plasmodium infection. Trends Parasitol. 2021;37:508–524. doi: 10.1016/j.pt.2021.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Barrow P., Clemann N. Review of embryo-fetal developmental toxicity studies performed for pharmaceuticals approved by FDA in 2018 and 2019. Reprod. Toxicol. 2021;99:144–151. doi: 10.1016/j.reprotox.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Burrows J.N., Duparc S., Gutteridge W.E., Hooft van Huijsduijnen R., Kaszubska W., Macintyre F., Mazzuri S., Möhrle J.J., Wells T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017;16:26. doi: 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsson S.K., Childs L.M., Buckee C., Marti M. Targeting Human Transmission Biology for Malaria Elimination. PLoS Pathog. 2015;11:e1004871. doi: 10.1371/journal.ppat.1004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrolo M., Giordano S., Cabrita-Santos L., Corso S., Vigário A.M., Silva S., Leirião P., Carapau D., Armas-Portela R., Comoglio P.M. Hepatocyte growth factor and its receptor are required for malaria infection. Nat. Med. 2003;9:1363–1369. doi: 10.1038/nm947. [DOI] [PubMed] [Google Scholar]
- 19.Rodrigues C.D., Hannus M., Prudêncio M., Martin C., Gonçalves L.A., Portugal S., Epiphanio S., Akinc A., Hadwiger P., Jahn-Hofmann K. Host scavenger receptor SR-BI plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe. 2008;4:271–282. doi: 10.1016/j.chom.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Kaushansky A., Ye A.S., Austin L.S., Mikolajczak S.A., Vaughan A.M., Camargo N., Metzger P.G., Douglass A.N., MacBeath G., Kappe S.H. Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell Rep. 2013;3:630–637. doi: 10.1016/j.celrep.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Douglass A.N., Kain H.S., Abdullahi M., Arang N., Austin L.S., Mikolajczak S.A., Billman Z.P., Hume J.C.C., Murphy S.C., Kappe S.H.I., Kaushansky A. Host-based Prophylaxis Successfully Targets Liver Stage Malaria Parasites. Mol. Ther. 2015;23:857–865. doi: 10.1038/mt.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arang N., Kain H.S., Glennon E.K., Bello T., Dudgeon D.R., Walter E.N.F., Gujral T.S., Kaushansky A. Identifying host regulators and inhibitors of liver stage malaria infection using kinase activity profiles. Nat. Commun. 2017;8:1232. doi: 10.1038/s41467-017-01345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posfai D., Sylvester K., Reddy A., Ganley J.G., Wirth J., Cullen Q.E., Dave T., Kato N., Dave S.S., Derbyshire E.R. Plasmodium parasite exploits host aquaporin-3 during liver stage malaria infection. PLoS Pathog. 2018;14:e1007057. doi: 10.1371/journal.ppat.1007057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raphemot R., Toro-Moreno M., Lu K.Y., Posfai D., Derbyshire E.R. Discovery of Druggable Host Factors Critical to Plasmodium Liver-Stage Infection. Cell Chem. Biol. 2019;26:1253–1262.e5. doi: 10.1016/j.chembiol.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaushansky A., Metzger P.G., Douglass A.N., Mikolajczak S.A., Lakshmanan V., Kain H.S., Kappe S.H. Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis. 2013;4:e762. doi: 10.1038/cddis.2013.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillrie M.R., Krishnegowda G., Lee K., Buret A.G., Robbins S.M., Looareesuwan S., Gowda D.C., Ho M. Src-family kinase dependent disruption of endothelial barrier function by Plasmodium falciparum merozoite proteins. Blood. 2007;110:3426–3435. doi: 10.1182/blood-2007-04-084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taoufiq Z., Gay F., Balvanyos J., Ciceron L., Tefit M., Lechat P., Mazier D. Rho kinase inhibition in severe malaria: thwarting parasite-induced collateral damage to endothelia. J. Infect. Dis. 2008;197:1062–1073. doi: 10.1086/528988. [DOI] [PubMed] [Google Scholar]
- 28.Sicard A., Semblat J.P., Doerig C., Hamelin R., Moniatte M., Dorin-Semblat D., Spicer J.A., Srivastava A., Retzlaff S., Heussler V. Activation of a PAK-MEK signalling pathway in malaria parasite-infected erythrocytes. Cell. Microbiol. 2011;13:836–845. doi: 10.1111/j.1462-5822.2011.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kesely K.R., Pantaleo A., Turrini F.M., Olupot-Olupot P., Low P.S. Inhibition of an Erythrocyte Tyrosine Kinase with Imatinib Prevents Plasmodium falciparum Egress and Terminates Parasitemia. PLoS ONE. 2016;11:e0164895. doi: 10.1371/journal.pone.0164895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adderley J.v.F.S., Jackson S.A., Bird M.J., Burns A.L., Anar B., Metcalf T., Semblat J.P., Billker O., Wilson D.W. Analysis of erythrocyte signalling pathways during Plasmodium falciparum infection identifies targets for host-directed antimalarial intervention. Nat. Commun. 2020;11:4015. doi: 10.1038/s41467-020-17829-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy S.C., Harrison T., Hamm H.E., Lomasney J.W., Mohandas N., Haldar K. Erythrocyte G protein as a novel target for malarial chemotherapy. PLoS Med. 2006;3:e528. doi: 10.1371/journal.pmed.0030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kain H.S., Glennon E.K.K., Vijayan K., Arang N., Douglass A.N., Fortin C.L., Zuck M., Lewis A.J., Whiteside S.L., Dudgeon D.R. Liver stage malaria infection is controlled by host regulators of lipid peroxidation. Cell Death Differ. 2020;27:44–54. doi: 10.1038/s41418-019-0338-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egan E.S., Jiang R.H., Moechtar M.A., Barteneva N.S., Weekes M.P., Nobre L.V., Gygi S.P., Paulo J.A., Frantzreb C., Tani Y. Malaria. A forward genetic screen identifies erythrocyte CD55 as essential for Plasmodium falciparum invasion. Science. 2015;348:711–714. doi: 10.1126/science.aaa3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White M., Amino R., Mueller I. Theoretical Implications of a Pre-Erythrocytic Plasmodium vivax Vaccine for Preventing Relapses. Trends Parasitol. 2017;33:260–263. doi: 10.1016/j.pt.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baird J.K. 8-Aminoquinoline Therapy for Latent Malaria. Clin. Microbiol. Rev. 2019;32 doi: 10.1128/CMR.00011-19. e00011-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hounkpatin A.B., Kreidenweiss A., Held J. Clinical utility of tafenoquine in the prevention of relapse of Plasmodium vivax malaria: a review on the mode of action and emerging trial data. Infect. Drug Resist. 2019;12:553–570. doi: 10.2147/IDR.S151031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silvie O., Rubinstein E., Franetich J.F., Prenant M., Belnoue E., Renia L., Hannoun L., Eling W., Levy S., Boucheix C. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat. Med. 2003;9:93–96. doi: 10.1038/nm808. [DOI] [PubMed] [Google Scholar]
- 38.Kaushansky A., Douglass A.N., Arang N., Vigdorovich V., Dambrauskas N., Kain H.S., Austin L.S., Sather D.N., Kappe S.H. Malaria parasites target the hepatocyte receptor EphA2 for successful host infection. Science. 2015;350:1089–1092. doi: 10.1126/science.aad3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langlois A.C., Marinach C., Manzoni G., Silvie O. Plasmodium sporozoites can invade hepatocytic cells independently of the Ephrin receptor A2. PLoS ONE. 2018;13:e0200032. doi: 10.1371/journal.pone.0200032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manzoni G., Marinach C., Topçu S., Briquet S., Grand M., Tolle M., Gransagne M., Lescar J., Andolina C., Franetich J.F. Plasmodium P36 determines host cell receptor usage during sporozoite invasion. eLife. 2017;6:e25903. doi: 10.7554/eLife.25903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaushansky A., Kappe S.H. The crucial role of hepatocyte growth factor receptor during liver-stage infection is not conserved among Plasmodium species. Nat. Med. 2011;17:1180–1181. doi: 10.1038/nm.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikolajczak S.A., Jacobs-Lorena V., MacKellar D.C., Camargo N., Kappe S.H. L-FABP is a critical host factor for successful malaria liver stage development. Int. J. Parasitol. 2007;37:483–489. doi: 10.1016/j.ijpara.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Itoe M.A., Sampaio J.L., Cabal G.G., Real E., Zuzarte-Luis V., March S., Bhatia S.N., Frischknecht F., Thiele C., Shevchenko A., Mota M.M. Host cell phosphatidylcholine is a key mediator of malaria parasite survival during liver stage infection. Cell Host Microbe. 2014;16:778–786. doi: 10.1016/j.chom.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopes da Silva M., Thieleke-Matos C., Cabrita-Santos L., Ramalho J.S., Wavre-Shapton S.T., Futter C.E., Barral D.C., Seabra M.C. The host endocytic pathway is essential for Plasmodium berghei late liver stage development. Traffic. 2012;13:1351–1363. doi: 10.1111/j.1600-0854.2012.01398.x. [DOI] [PubMed] [Google Scholar]
- 45.Niklaus L., Agop-Nersesian C., Schmuckli-Maurer J., Wacker R., Grünig V., Heussler V.T. Deciphering host lysosome-mediated elimination of Plasmodium berghei liver stage parasites. Sci. Rep. 2019;9:7967. doi: 10.1038/s41598-019-44449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vijayan K., Cestari I., Mast F.D., Glennon E.K.K., McDermott S.M., Kain H.S., Brokaw A.M., Aitchison J.D., Stuart K., Kaushansky A. Plasmodium Secretion Induces Hepatocyte Lysosome Exocytosis and Promotes Parasite Entry. iScience. 2019;21:603–611. doi: 10.1016/j.isci.2019.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Risco-Castillo V., Topçu S., Marinach C., Manzoni G., Bigorgne A.E., Briquet S., Baudin X., Lebrun M., Dubremetz J.F., Silvie O. Malaria Sporozoites Traverse Host Cells within Transient Vacuoles. Cell Host Microbe. 2015;18:593–603. doi: 10.1016/j.chom.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 48.Labaied M., Jayabalasingham B., Bano N., Cha S.J., Sandoval J., Guan G., Coppens I. Plasmodium salvages cholesterol internalized by LDL and synthesized de novo in the liver. Cell Microbiol. 2011;13:569–586. doi: 10.1111/j.1462-5822.2010.01555.x. [DOI] [PubMed] [Google Scholar]
- 49.Real E., Rodrigues L., Cabal G.G., Enguita F.J., Mancio-Silva L., Mello-Vieira J., Beatty W., Vera I.M., Zuzarte-Luis V., Figueira T.N. Plasmodium UIS3 sequesters host LC3 to avoid elimination by autophagy in hepatocytes. Nat. Microbiol. 2018;3:17–25. doi: 10.1038/s41564-017-0054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen W., Stenzel W., Silvie O., Blanz J., Saftig P., Matuschewski K., Ingmundson A. Sequestration of cholesterol within the host late endocytic pathway restricts liver-stage Plasmodium development. Mol. Biol. Cell. 2017;28:726–735. doi: 10.1091/mbc.E16-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Posfai D., Maher S.P., Roesch C., Vantaux A., Sylvester K., Péneau J., Popovici J., Kyle D.E., Witkowski B., Derbyshire E.R. Plasmodium vivax Liver and Blood Stages Recruit the Druggable Host Membrane Channel Aquaporin-3. Cell Chem. Biol. 2020;27:719–727.e5. doi: 10.1016/j.chembiol.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roskoski R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019;144:19–50. doi: 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 53.Ferguson F.M., Gray N.S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 54.Gautier E.F., Leduc M., Cochet S., Bailly K., Lacombe C., Mohandas N., Guillonneau F., El Nemer W., Mayeux P. Absolute proteome quantification of highly purified populations of circulating reticulocytes and mature erythrocytes. Blood Adv. 2018;2:2646–2657. doi: 10.1182/bloodadvances.2018023515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harrison T., Samuel B.U., Akompong T., Hamm H., Mohandas N., Lomasney J.W., Haldar K. Erythrocyte G protein-coupled receptor signaling in malarial infection. Science. 2003;301:1734–1736. doi: 10.1126/science.1089324. [DOI] [PubMed] [Google Scholar]
- 56.Pantaleo A., Kesely K.R., Pau M.C., Tsamesidis I., Schwarzer E., Skorokhod O.A., Chien H.D., Ponzi M., Bertuccini L., Low P.S., Turrini F.M. Syk inhibitors interfere with erythrocyte membrane modification during P falciparum growth and suppress parasite egress. Blood. 2017;130:1031–1040. doi: 10.1182/blood-2016-11-748053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kesely K., Noomuna P., Vieth M., Hipskind P., Haldar K., Pantaleo A., Turrini F., Low P.S. Identification of tyrosine kinase inhibitors that halt Plasmodium falciparum parasitemia. PLoS ONE. 2020;15:e0242372. doi: 10.1371/journal.pone.0242372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bernabeu M., Smith J.D. EPCR and Malaria Severity: The Center of a Perfect Storm. Trends Parasitol. 2017;33:295–308. doi: 10.1016/j.pt.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith J.D. The role of PfEMP1 adhesion domain classification in Plasmodium falciparum pathogenesis research. Mol. Biochem. Parasitol. 2014;195:82–87. doi: 10.1016/j.molbiopara.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wassmer S.C., Taylor T.E., Rathod P.K., Mishra S.K., Mohanty S., Arevalo-Herrera M., Duraisingh M.T., Smith J.D. Investigating the Pathogenesis of Severe Malaria: A Multidisciplinary and Cross-Geographical Approach. Am. J. Trop. Med. Hyg. 2015;93(3, Suppl):42–56. doi: 10.4269/ajtmh.14-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seydel K.B., Kampondeni S.D., Valim C., Potchen M.J., Milner D.A., Muwalo F.W., Birbeck G.L., Bradley W.G., Fox L.L., Glover S.J. Brain swelling and death in children with cerebral malaria. N. Engl. J. Med. 2015;372:1126–1137. doi: 10.1056/NEJMoa1400116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu X., Dayanand K.K., Thylur R.P., Norbury C.C., Gowda D.C. Small molecule-based inhibition of MEK1/2 proteins dampens inflammatory responses to malaria, reduces parasite load, and mitigates pathogenic outcomes. J. Biol. Chem. 2017;292:13615–13634. doi: 10.1074/jbc.M116.770313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu M., Solomon W., Cespedes J.C., Wilson N.O., Ford B., Stiles J.K. Neuregulin-1 attenuates experimental cerebral malaria (ECM) pathogenesis by regulating ErbB4/AKT/STAT3 signaling. J. Neuroinflammation. 2018;15:104. doi: 10.1186/s12974-018-1147-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tran T.M., Guha R., Portugal S., Skinner J., Ongoiba A., Bhardwaj J., Jones M., Moebius J., Venepally P., Doumbo S. A Molecular Signature in Blood Reveals a Role for p53 in Regulating Malaria-Induced Inflammation. Immunity. 2019;51:750–765.e10. doi: 10.1016/j.immuni.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernatchez J.A., Chen E., Hull M.V., McNamara C.W., McKerrow J.H., Siqueira-Neto J.L. High-Throughput Screening of the ReFRAME Library Identifies Potential Drug Repurposing Candidates for Trypanosoma cruzi. Microorganisms. 2020;8:472. doi: 10.3390/microorganisms8040472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janes J., Young M.E., Chen E., Rogers N.H., Burgstaller-Muehlbacher S., Hughes L.D., Love M.S., Hull M.V., Kuhen K.L., Woods A.K. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. USA. 2018;115:10750–10755. doi: 10.1073/pnas.1810137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rice C.A., Colon B.L., Chen E., Hull M.V., Kyle D.E. Discovery of repurposing drug candidates for the treatment of diseases caused by pathogenic free-living amoebae. PLoS Negl. Trop. Dis. 2020;14:e0008353. doi: 10.1371/journal.pntd.0008353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morales Vasquez D., Park J.G., Ávila-Pérez G., Nogales A., de la Torre J.C., Almazan F., Martinez-Sobrido L. Identification of Inhibitors of ZIKV Replication. Viruses. 2020;12:1041. doi: 10.3390/v12091041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim Y.J., Cubitt B., Chen E., Hull M.V., Chatterjee A.K., Cai Y., Kuhn J.H., de la Torre J.C. The ReFRAME library as a comprehensive drug repurposing library to identify mammarenavirus inhibitors. Antiviral Res. 2019;169:104558. doi: 10.1016/j.antiviral.2019.104558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roskoski R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol. Res. 2020;152:104609. doi: 10.1016/j.phrs.2019.104609. [DOI] [PubMed] [Google Scholar]
- 71.Roskoski R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2021 update. Pharmacol. Res. 2021;165:105463. doi: 10.1016/j.phrs.2021.105463. [DOI] [PubMed] [Google Scholar]
- 72.Carles F., Bourg S., Meyer C., Bonnet P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules. 2018;23:908. doi: 10.3390/molecules23040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krahn A.I., Wells C., Drewry D.H., Beitel L.K., Durcan T.M., Axtman A.D. Defining the Neural Kinome: Strategies and Opportunities for Small Molecule Drug Discovery to Target Neurodegenerative Diseases. ACS Chem. Neurosci. 2020;11:1871–1886. doi: 10.1021/acschemneuro.0c00176. [DOI] [PubMed] [Google Scholar]
- 74.Xie Z., Yang X., Duan Y., Han J., Liao C. Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. J. Med. Chem. 2021;64:1283–1345. doi: 10.1021/acs.jmedchem.0c01511. [DOI] [PubMed] [Google Scholar]
- 75.Leirião P., Albuquerque S.S., Corso S., van Gemert G.J., Sauerwein R.W., Rodriguez A., Giordano S., Mota M.M. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell. Microbiol. 2005;7:603–609. doi: 10.1111/j.1462-5822.2004.00490.x. [DOI] [PubMed] [Google Scholar]
- 76.Prudêncio M., Rodrigues C.D., Hannus M., Martin C., Real E., Gonçalves L.A., Carret C., Dorkin R., Röhl I., Jahn-Hoffmann K. Kinome-wide RNAi screen implicates at least 5 host hepatocyte kinases in Plasmodium sporozoite infection. PLoS Pathog. 2008;4:e1000201. doi: 10.1371/journal.ppat.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 78.Lussier Y.A., Chen J.L. The emergence of genome-based drug repositioning. Sci. Transl. Med. 2011;3:96ps35. doi: 10.1126/scitranslmed.3001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iorio F., Rittman T., Ge H., Menden M., Saez-Rodriguez J. Transcriptional data: a new gateway to drug repositioning? Drug Discov. Today. 2013;18:350–357. doi: 10.1016/j.drudis.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hsieh Y.Y., Chou C.J., Lo H.L., Yang P.M. Repositioning of a cyclin-dependent kinase inhibitor GW8510 as a ribonucleotide reductase M2 inhibitor to treat human colorectal cancer. Cell Death Discov. 2016;2:16027. doi: 10.1038/cddiscovery.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meng X.Y., Zhang H.X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hodos R.A., Kidd B.A., Shameer K., Readhead B.P., Dudley J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016;8:186–210. doi: 10.1002/wsbm.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gowthaman U., Jayakanthan M., Sundar D. Molecular docking studies of dithionitrobenzoic acid and its related compounds to protein disulfide isomerase: computational screening of inhibitors to HIV-1 entry. BMC Bioinformatics. 2008;9(Suppl 12):S14. doi: 10.1186/1471-2105-9-S12-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maga G., Falchi F., Garbelli A., Belfiore A., Witvrouw M., Manetti F., Botta M. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency virus-1 replication targeting the human cellular aspartic acid-glutamic acid-alanine-aspartic acid box polypeptide 3. J. Med. Chem. 2008;51:6635–6638. doi: 10.1021/jm8008844. [DOI] [PubMed] [Google Scholar]
- 85.Jain R., Gupta S., Munde M., Pati S., Singh S. Development of novel anti-malarial from structurally diverse library of molecules, targeting plant-like CDPK1, a multistage growth regulator of P. falciparum. Biochem. J. 2020;477:1951–1970. doi: 10.1042/BCJ20200045. [DOI] [PubMed] [Google Scholar]
- 86.Fatoki T.H., Ibraheem O., Ogunyemi I.O., Akinmoladun A.C., Ugboko H.U., Adeseko C.J., Awofisayo O.A., Olusegun S.J., Enibukun J.M. Network analysis, sequence and structure dynamics of key proteins of coronavirus and human host, and molecular docking of selected phytochemicals of nine medicinal plants. J. Biomol. Struct. Dyn. 2021;39:6195–6217. doi: 10.1080/07391102.2020.1794971. [DOI] [PubMed] [Google Scholar]
- 87.Leukes V., Walzl G., du Plessis N. Myeloid-Derived Suppressor Cells as Target of Phosphodiesterase-5 Inhibitors in Host-Directed Therapeutics for Tuberculosis. Front. Immunol. 2020;11:451. doi: 10.3389/fimmu.2020.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vitali F., Cohen L.D., Demartini A., Amato A., Eterno V., Zambelli A., Bellazzi R. A Network-Based Data Integration Approach to Support Drug Repurposing and Multi-Target Therapies in Triple Negative Breast Cancer. PLoS ONE. 2016;11:e0162407. doi: 10.1371/journal.pone.0162407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng F., Desai R.J., Handy D.E., Wang R., Schneeweiss S., Barabási A.L., Loscalzo J. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 2018;9:2691. doi: 10.1038/s41467-018-05116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ekins S., Puhl A.C., Zorn K.M., Lane T.R., Russo D.P., Klein J.J., Hickey A.J., Clark A.M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019;18:435–441. doi: 10.1038/s41563-019-0338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hooft van Huijsduijnen R., Wells T., Tanner M., Wittlin S. Two successful decades of Swiss collaborations to develop new anti-malarials. Malar. J. 2019;18:94. doi: 10.1186/s12936-019-2728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gamo F.J., Sanz L.M., Vidal J., de Cozar C., Alvarez E., Lavandera J.L., Vanderwall D.E., Green D.V., Kumar V., Hasan S. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 94.Guiguemde W.A., Shelat A.A., Garcia-Bustos J.F., Diagana T.T., Gamo F.J., Guy R.K. Global phenotypic screening for antimalarials. Chem. Biol. 2012;19:116–129. doi: 10.1016/j.chembiol.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Plouffe D., Brinker A., McNamara C., Henson K., Kato N., Kuhen K., Nagle A., Adrián F., Matzen J.T., Anderson P. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. USA. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 97.Patel H., Dunican C., Cunnington A.J. Predictors of outcome in childhood Plasmodium falciparum malaria. Virulence. 2020;11:199–221. doi: 10.1080/21505594.2020.1726570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shibeshi W., Alemkere G., Mulu A., Engidawork E. Efficacy and safety of artemisinin-based combination therapies for the treatment of uncomplicated malaria in pediatrics: a systematic review and meta-analysis. BMC Infect. Dis. 2021;21:326. doi: 10.1186/s12879-021-06018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haqshenas G., Doerig C. Targeting of host cell receptor tyrosine kinases by intracellular pathogens. Sci. Signal. 2019;12:eaau9894. doi: 10.1126/scisignal.aau9894. [DOI] [PubMed] [Google Scholar]
- 100.Lupberger J., Zeisel M.B., Xiao F., Thumann C., Fofana I., Zona L., Davis C., Mee C.J., Turek M., Gorke S. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weisberg E., Parent A., Yang P.L., Sattler M., Liu Q., Liu Q., Wang J., Meng C., Buhrlage S.J., Gray N., Griffin J.D. Repurposing of Kinase Inhibitors for Treatment of COVID-19. Pharm. Res. 2020;37:167. doi: 10.1007/s11095-020-02851-7. [DOI] [PMC free article] [PubMed] [Google Scholar]