

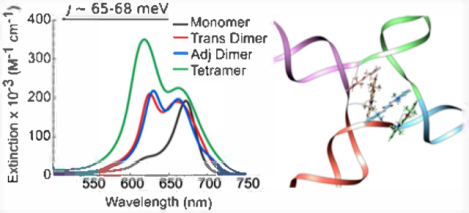
Abstract
Exciton delocalization plays a prominent role in the photophysics of molecular aggregates, ultimately governing their particular function or application. Deoxyribonucleic acid (DNA) is a compelling scaffold in which to template molecular aggregates and promote exciton delocalization. As individual dye molecules are the basis of exciton delocalization in molecular aggregates, their judicious selection is important. Motivated by their excellent photostability and spectral properties, here, we examine the ability of squaraine dyes to undergo exciton delocalization when aggregated via a DNA Holliday junction (HJ) template. A commercially available indolenine squaraine dye was chosen for the study given its strong structural resemblance to Cy5, a commercially available cyanine dye previously shown to undergo exciton delocalization in DNA HJs. Three types of DNA–dye aggregate configurations—transverse dimer, adjacent dimer, and tetramer—were investigated. Signatures of exciton delocalization were observed in all squaraine–DNA aggregates. Specifically, strong blue shift and Davydov splitting were observed in steady-state absorption spectroscopy and exciton-induced features were evident in circular dichroism (CD) spectroscopy. Strongly suppressed fluorescence emission provided additional, indirect evidence for exciton delocalization in the DNA-templated squaraine dye aggregates. To quantitatively evaluate and directly compare the excitonic Coulombic coupling responsible for exciton delocalization, the strength of excitonic hopping interactions between the dyes was obtained by simultaneously fitting the experimental steady-state absorption and CD spectra via a Holstein-like Hamiltonian, in which, following the theoretical approach of Kühn, Renger, and May, the dominant vibrational mode is explicitly considered. The excitonic hopping strength within indolenine squaraines was found to be comparable to that of the analogous Cy5 DNA-templated aggregate. The squaraine aggregates adopted primarily an H-type (dyes oriented parallel to each other) spatial arrangement. Extracted geometric details of the dye mutual orientation in the aggregates enabled a close comparison of aggregate configurations and the elucidation of the influence of dye angular relationship on excitonic hopping interactions in squaraine aggregates. These results encourage the application of squaraine-based aggregates in next-generation systems driven by molecular excitons.
Graphical Abstract
INTRODUCTION
Exciton delocalization often plays a decisive role in governing the photophysical properties of molecular aggregates, ultimately determining their suitability for particular applications. A well-established framework for understanding exciton delocalization in molecular aggregates, and associated excitonic coupling responsible for delocalization, is the molecular exciton model developed by Kasha.1 In Kasha’s molecular exciton model, exciton delocalization results from the collective excitation of the locally excited states of an array of proximate dye molecules forming a molecular aggregate.
Described in Kasha’s molecular exciton model, the excitonic hopping parameter, Jm,n, is the strength of that component of the Coulombic coupling (electrodynamic interactions2) between dye m and dye n (within a molecular aggregate) that induces a transition from the excited state of one molecule while simultaneously inducing a transition from the ground state to the electronic excited state of the other molecule. Thereby, an excitation is transferred from one molecule to the other. This is a hopping interaction and is referred to in the literature in a number of categorical ways in terms of interaction, hopping, and coupling or in the context of a parameter, shift, or potential. Relative to the former, Jm,n is referred to as an exchange interaction,3,4 instantaneous intermolecular interaction,5 resonant interaction or coupling,6–8 resonant exciton hopping,9 intermolecular Coulombic interaction,10–14 intermolecular dipole–dipole coupling,15 long-range dipole–dipole Coulombic interaction,16 excitonic coupling,17 and electronic interaction.18,19 In the context of the latter, Jm,n is called a hopping parameter,20 monoexcitonic shift,21 and point dipole interaction potential.22 Although not a complete list, the number of ways Jm,n is described suggests that a more thorough description may be useful. The excitonic hopping interaction inherent in Jm,n enables exciton delocalization and relaxation.5,7,8,11–13 Where intermolecular distances are less than ~4 Å (e.g., aggregates with π-stacking or covalently bridged), wave function overlap can induce intermolecular charge transfer (also described as super-exchange coupling or Dexter mechanism, coupling or interaction) and can contribute to the Jm,n term.11–13,16,18–20 In our work, it is assumed that intermolecular charge transfer can be neglected. It should be noted that Jm,n is often approximated as the coupling between a pair of transition point dipoles and is referred to as such. In our analysis, the point dipole approximation was deemed inadequate since the spacing between the dyes is shorter than their length. Instead, an extended dipole approximation23 was employed where the dipoles are modeled as two point charges of opposite sign separated by nearly the length of the dye core. This extended dipole approximation better accounts for the physical charge distribution that spreads out over the length of the molecule with the maxima of charge density occurring near the ends of the dye.
Exciton delocalization in molecular aggregates was first observed in the 1930s by Jelley and Scheibe, who independently noted large shifts in the absorption spectra of molecular aggregates as compared to those of the respective monomers.24–26 Exciton delocalization has also been observed in molecular aggregates present in natural light harvesting systems.27–31 Rational control of exciton delocalization in molecular aggregates in synthetic, or artificial, systems has significant implications on next-generation technologies, such as artificial light harvesting,32 nanoscale excitonic computing,33–37 solar energy conversion,32,38,39 and on fundamental studies of quantum entanglement40–43 in molecular aggregates, which may find potential applications in room-temperature quantum information systems.44,45
Rational control of exciton delocalization in molecular aggregates requires the judicious selection of the constituent molecules, or dyes, and control of their spatial arrangement. Exciton delocalization was originally observed in concentrated solutions (i.e., well above the solubility limit), which promoted the formation of large dye aggregates via their spontaneous precipitation out of solution.24–26,46–48 The ability to rationally control exciton delocalization via the number of dyes and their spatial arrangement in dye aggregates formed via spontaneous precipitation, however, has significant limitations. In natural systems, in contrast, a protein matrix is used to organize a precise number of dyes and maintain precise control of their relative distance and orientation.44,49–53 While the assembly of dye aggregates via proteins affords exquisite control over dye aggregation, the intricate design rules of protein folding and dye–protein interactions substantially complicate the rational use of protein nanostructures as scaffolds.54–57 The design rules associated with an alternative biomacromolecule, deoxyribonucleic acid (DNA), are comparatively much simpler. Because there are only four nucleic acid building blocks in DNA that associate via Watson–Crick pairing, design rules to rationally and predictably synthesize complex DNA nanostructures are increasingly mature and compelling.58–61 Critically, a controlled assembly of dye aggregates in DNA has been demonstrated. Generally, there are two approaches to control the assembly of dye aggregates via DNA, either via noncovalent interaction of dyes with DNA62–72 or via direct covalent binding of dyes to DNA73–85 While templating of dye aggregates via their noncovalent intercalation with DNA offers improved control over the spontaneous assembly,62–72 the approach still confers limited control over the number of dyes in the aggregate and their spatial arrangement since aggregation is induced in concentrated solutions and via noncovalent interactions with specific base pairs in the minor groove of long sequences of doublestranded, or duplex, DNA (dsDNA). Direct covalent binding of dyes to DNA, in contrast, provides more extensive control of the number of dyes in the aggregate and their spatial arrangement. Direct covalent binding of dyes to DNA enables (i) choice of the type of dye,75,77 (ii) choice of the number of dyes,22,83 including assembly into large two- and three-dimensional (3-D) dye aggregate arrays, and (iii) the precise positioning of the dyes along the DNA backbone,74,75,77–79,81 which confers control over their spatial arrangement. Numerous studies have demonstrated exciton delocalization in dye aggregates templated within dsDNA structures,74–82,84–96 even within more complex, higher-order DNA nanostructures.75,97
Among the many types of dyes employed in DNA-templated aggregation,74–82,84–93 cyanine dyes, a well-known group of a broader family of polymethine dyes, have established a particular prominence. Of especial note is the commercially available cyanine dye CyS (Figure 1a), which is a pentamethine dye that exhibits an intense absorption profile in the visible (specifically, the red) part of the electromagnetic spectrum. Taking advantage of the optimal photophysical properties of Cy5, whose visible electronic absorption exhibits a large peak extinction coefficient of ~250 000 cm−1 M−1 at ca. 650 nm, our group recently demonstrated exciton delocalization in dimers, trimers, and tetramers of Cy5 templated using both dsDNA and four-armed DNA Holliday Junction (HJ)98,99 scaffolds.100,101 Proximate Cy5 dyes at distances of <1 nm in the core of the DNA HJ resulted in extensive spectral shifts, evident via significant Davydov splitting102 (i.e., splitting of the monomer peak—one red-shifted and the other blue-shifted), indicative of strong exciton coupling and exciton delocalization. A benefit of employing a DNA HJ as a scaffold over lower-order DNA, such as dsDNA, is that it enables the assembly of up to 4 dyes in the core of HJ. As a result, different dimer, trimer, and tetramer dye aggregate configurations can be selectively accessed by templating via a DNA HJ. At the same time, aggregation beyond the desired number of dyes is suppressed. Additionally, Cannon et al. showed that the packing behavior of the Cy5 dyes could be controlled, i.e., either J- or H-type aggregate packing arrangements could be promoted, when two dyes were positioned on adjacent or opposite arms of the DNA HJ, respectively.101 Despite their remarkable spectral properties, concerns remain regarding the general utility of Cy5 aggregates given the dye’s strong susceptibility to photooxidation and photoisomerization.103,104
Figure 1.
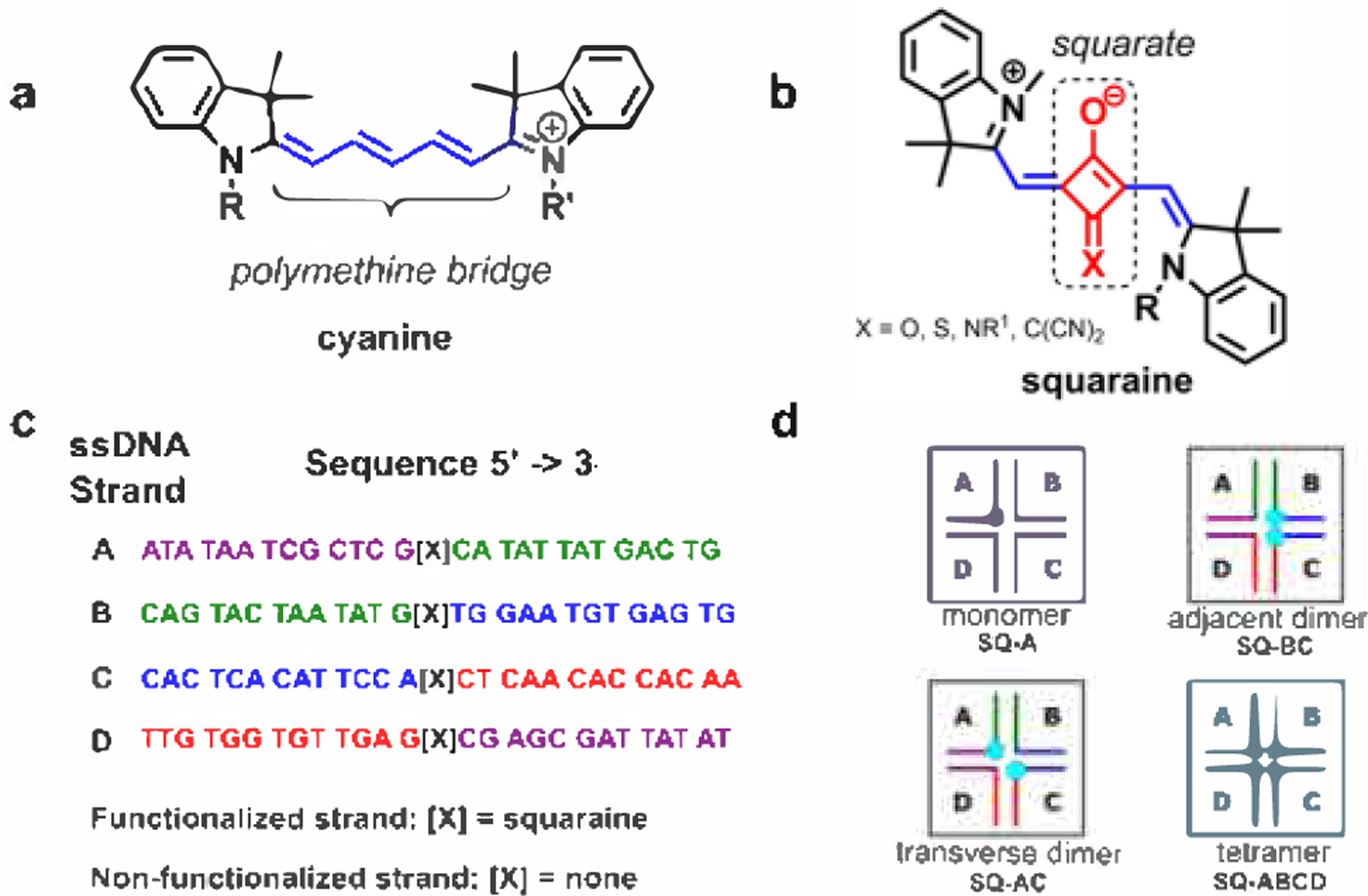
(a) Representative example of the chemical structure of cyanine dye CyS, where R and R′ are sites of attachment to DNA through phosphoramidite linkers. (b) Representative example of the chemical structure of core-substituted indolenine squaraine dye, where X = O, S, NR1, C(CN)2; R is a site of attachment to DNA through a serinol linker; and R1 is an alkyl (also see Figure S1). (c) Asymmetric DNA sequences to assemble immobile Holliday junctions; complementary regions of ssDNA are color-coded: for example, the purple region of strand A is complementary to the purple region of strand D, and the green region of strand A is complementary to the green region of strand B, and so on. (d) Schematic representation of dye monomer, dimers, and a tetramer in four-armed duplex DNA junctions (Holliday junctions) where squaraine dyes are depicted as blue dots. As discussed in Sections S3 and S10, the DNA HJ may exist primarily in a stacked conformation. ssDNA strands used to assemble DNA HJ are labeled as A, B, C, and D.
Squaraines (also known as squarylium dyes) are a family of dyes that exhibit improved photochemical stability in addition to optical properties similar to cyanines.105–108 Similar to cyanines, squaraines are characterized by an intense absorption profile in the visible and near-infrared regions of the electromagnetic spectrum, with peak extinction coefficients in the range of ~200 000 to 250 000 M−1 cm−1. While their optical properties are similar, squaraines structurally differ from cyanines due to the incorporation of a squarate moiety at the center of the polymethine bridge (Figure 1b). Though squaraines can contain various aromatic (e.g., aniline, phenol, azulene) and heterocyclic (e.g., pyrroline, indolenine, benzothiazole) rings, all derivatives of squaraines have in common the central squarate moiety. The central squarate moiety confers improved photochemical stability by inhibiting the oxidation of the polymethine bridge and limiting the ability of the polymethine bridge to undergo photoinduced rotational motion, or isomerization. Originally developed in 1965 by Treibs and Jacob,109 squaraines and squaraine aggregates have since been utilized in a variety of applications ranging from photovoltaic devices110–112 to sensors113 and fluorescent labels in biomedicine.111,112,114 Aggregation of squaraine dyes has been observed in concentrated solutions115,116 and upon conjugation to proteins.108 The excitonic coupling was studied experimentally and theoretically in the covalently linked bis-squaraine dimers exhibiting J-type behavior.117,118 However, to our knowledge, there is only one report that describes DNA-induced aggregation of squaraines studied on one form of indolenine-type (i.e., squaraines containing idolenine rings on both sides of the squaraine moiety) squaraine dimer attached to a short DNA duplex.76 The reported squaraine dimer was observed to exhibit an H-type packing with spectral signatures of strong exciton interactions; however, the exciton interactions were not evaluated quantitatively.
In this study, we examine exciton delocalization in a broader range of squaraine aggregates (i.e., -mers): two dimer aggregates and a tetramer, templated by a DNA HJ. Given the structural similarity of cyanines and squaraines—and given the excellent results observed with aggregates of Cy5 templated via DNA HJs100,101—we hypothesized that similar excitonic Coulombic coupling and delocalization could be achieved in DNA HJs of squaraine dyes. To facilitate the comparison with Cy5 containing indolenine rings at both ends of the penthamethine bridge (Figure 1a), we chose a commercially available squaraine dye featuring indolenine rings on both sides of the squarate moiety (Figure 1b), which we subsequently refer to as indolenine squaraine. As representative dye aggregates (i.e., -mers), we assembled a transverse dimer, an adjacent dimer, and a tetramer in the core of an immobile DNA HJ containing asymmetric sequences (Figure 1c) to prevent branch migration. Signatures of the exciton delocalization were observed in the steady-state absorption and circular dichroism (CD) spectra of the DNA-templated squaraine dye aggregates. The steady-state absorption spectra of the DNA-templated squaraine aggregates exhibited a significant blue shift (i.e., peak shift and redistribution of oscillator strength toward short wavelengths or higher energies) compared with that of the monomer, which is a signature of exciton delocalization and indicates an H-type aggregate packing arrangement that is adopted in all DNA-templated squaraine dye aggregates. The presence of pronounced exciton-coupled CD signals in the circular dichroism spectra represents an additional signature of exciton delocalization. Significant fluorescence quenching observed via steady-state fluorescence emission spectroscopy provided indirect evidence of exciton delocalization. The absorption and CD spectra were modeled theoretically to quantitatively evaluate the excitonic hopping parameter Jm,n, that characterizes the strengths of excitonic Coulombic coupling responsible for exciton delocalization in DNA-templated squaraine aggregates. The modeling derived appreciable Jm,n values, comparable in magnitude to Jm,n values responsible for exciton delocalization in the analogous DNA-templated Cy5 aggregate. The details of the spatial arrangement of the constituent squaraines in H-aggregates were extracted from the modeling providing insights into how the geometry parameters affect the excitonic Coulombic coupling strength.
METHODS
Sample Preparation.
DNA oligomers internally functionalized with Square 660-NHS (KS-1352, SETA BioMedicals, Urbana-Champaign, IL) via a non-nucleosidic amino serinol sequence modifier and purified via dual high-performance liquid chromatography were purchased from Bio-Synthesis, Inc. Nonfunctionalized DNA oligomers purified by standard desalting were purchased from Bio-Synthesis, Inc. All DNA oligomers were rehydrated in ultrapure water (Barnstead Nanopure, Thermo Scientific) to prepare 100 μM stock solutions. Concentrations of DNA samples were determined spectroscopically on a NanoDrop One Microvolume UV–Vis Spectrophotometer (Thermo Scientific) using calculated extinction coefficients. DNA Holliday junctions were prepared by combining equimolar amounts of complimentary oligomers in 1× TBE, 15 mM MgCl2 buffer solution to a final DNA concentration 1.5 μM. All DNA samples were annealed in a Mastercycler Nexus PCR cycler (Eppendorf) according to the following protocol: 4 min at 94 °C, followed by cooling ramps at 0.1 °C per 15 s from 94 to 64 °C and 10 °C per 1 min from 64 °C to room temperature. For the fluorescence measurements, the DNA samples were further diluted to 0.5 μM DNA concentration.
Optical Characterization.
UV–vis spectra were recorded in triplicate at room temperature on a dual-beam Cary 5000 UV–vis–NIR spectrophotometer (Agilent Technologies) in a quartz cuvette with a 1 cm path length (Starna). Absorbance spectra were monitored over a 230–800 nm wavelength range. Circular dichroism (CD) measurements were performed on a JACS0-J810 spectropolarimeter. DNA samples (120 μL) were transferred to a 1 cm path length quartz cuvette (Jasco). Spectra were recorded over the 230–800 nm wavelength range (three scans per sample were averaged) at a speed of 200 nm min−1 Steady-state fluorescence spectra were obtained using a Horiba PTI QuantaMaster 400 spectrofluorometer (Horiba Scientific) in a 1 cm path length quartz cuvette (Starna) and monitored as a function of wavelength when excited at λexc = 650 nm. The fluorescence spectra were corrected for the wavelength dependence of the detection system response using the correction curve provided by the manufacturer. The fluorescence spectra were scaled by the absorptance at the excitation wavelength.
RESULTS AND DISCUSSION
Construct Design and Synthesis.
Three types of squaraine aggregate constructs, an adjacent dimer, a transverse dimer, and a tetramer, were prepared by templating the dyes in the branch point of an immobile DNA HJ. The immobile DNA HJ with arm branch length of 13 base pairs was formed by four asymmetric single DNA strands A, B, C, and D (26 bases each) (Figure 1c). Because of its large peak molar extinction and narrow absorption bands, Square 660 was chosen as a representative commercially available indolenine squaraine dye (Figure 1b). To functionalize each DNA strand with the squaraine dye, the dyes were covalently attached through a non-nucleosidic serinol-based linker (Figure S1). Functionalized and nonfunctionalized strands were used to assemble constructs depicted in Figure 1d. A single Square 660 dye was templated in a DNA HJ to prepare a monomer construct as a control. We denote this configuration as SQ-A (Figure 1d). Aggregation of two Square 660 dyes into a dimer was performed using two different configurations. In the transverse configuration SQ-AC, two Square 660 dyes were attached to opposing DNA arms A and C, while in the adjacent configuration SQ-BC, the two dyes were attached to adjacent DNA arms B and C. Complementary oligo strands were combined in equimolar amounts followed by annealing to ensure complete hybridization. The Square 660 dye aggregates were analyzed by nondenaturing polyacrylamide gel electrophoresis for the presence of remaining single functionalized strands. The amount of unreacted functionalized single strands (i.e., A, B, C, and D) in the aggregate samples did not exceed 5% as estimated by densitometry analysis (Figure S2).
Thermal denaturation experiments were performed to examine the conformation and stability of the DNA HJ templating the squaraine aggregates (Section S3). DNA denaturation was monitored by the absorption in the nucleobase region (260 nm). The resulting sigmoidal curves indicated the cooperative melting of the stacked DNA HJ conformation, templating all of the squaraine aggregates in the presence of MgCl2 (Figure S3). The formation of the stacked DNA HJ conformation was further supported with the comparative melting experiments in the presence of NaCl, promoting the open DNA HJ conformation (Figure S4). A reference unmodified DNA HJ melted at 60.0 °C in 1× TBE, 15 mM MgCl2. The insertion of a single squaraine dye in the DNA HJ through a serinol linker resulted in a slight decrease in the melting point of the SQ-A monomer (−1.7 °C) compared to the unmodified DNA HJ, which is equivalent to the dissociation of one canonical AT pair. This result suggests that electronic interactions between a squaraine dye and surrounding nucleobases are repulsive in nature and rather weak. The squaraine dimers melted at approximately the same temperature as the unmodified DNA HJ, indicating that dye–dye interactions compensate for the insertion of two serinol linkers (Table S2). The presence of the four dyes in the SQ-ABCD tetramer increased the melting point to 4.2 °C compared to the unmodified DNA HJ, suggesting strong attractive interaction between the four squaraine molecules with an overall stabilizing effect on the dye–DNA construct.
Steady-State Optical Characterization.
As an individual dye molecule forms the basis of a molecular aggregate, a DNA HJ containing a single Square 660 dye was first prepared and characterized. As our dye monomer reference, we studied Square 660 attached to the A strand and hybridized with all other unlabeled (B–D) strands. The electronic absorption spectrum of SQ-A exhibited two prominent absorption spectral bands with peak maxima at 672 and 630 nm (Figure 2a), respectively assigned to the 0–0 and 0–1 vibronic transitions of the individual Square 660 dye. The 0–0 band exhibits a peak molar extinction of ca. 192 000 M−1 cm−1, indicative of a strongly allowed electronic transition. Both the minimal vibronic structure and large extinction coefficient (which suggests a large transition dipole moment) are conducive to exciton delocalization and strong excitonic Coulombic coupling, suggesting Square 660 is an excellent candidate for DNA-templated dye aggregates. Strong fluorescence of monomer SQ-A was observed with a 0–0 band peaking at 687 nm (Figure S5). The difference in energy between the 0–0 band in absorption and the 0–0 band in fluorescence, i.e., the Stokes shift, is 325 cm−1 (40 meV). This value is on the smaller end than that of Cy5 dye monomers attached to DNA (300–600 cm−1 [37–77 meV]),100,101,119 indicating a smaller change in the geometry between the ground state and the excited state in indolenine squaraine, which might result from the more rigid structure of squaraine compared to that of Cy5 dye. The optical characteristics of SQ-A are similar to those of the “free” Square 660 dye not attached to DNA, which in a pH 7.0 phosphate buffer exhibits an absorption 0–0 band peaking at 658 nm (182 000 M−1 cm−1 extinction) and fluorescence 0–0 band peaking at 676 nm (Figure S6a). The difference in spectral characteristics between the monomer SQ-A and the “free” dye in solution may be attributable to solvatochromic effects, such as differences in the buffer conditions or the local DNA environment, or other effects such as electronic interactions with nucleobases. However, alternating the functionalized strand in the monomer construct (i.e., a squaraine is attached to strand B, C, or D instead of strand A) did not influence the absorption peak maxima indicating that a squaraine does not exhibit strong electronic interactions with the surrounding nucleobases (Figure S6b). The SQ-A monomer in DNA HJ exhibited high fluorescence quantum yield (ΦF) measured to be 0.37 (Section S5), which is slightly higher than that of Cy5 in ssDNA reported in the range 0.29–0.33.119 The lower quantum yield in Cy5 may be related to torsion motion around the polymethine bridge. Squaraines are less susceptible to torsional motion due to the squarate moiety incorporated into the polymethine bridge.
Figure 2.
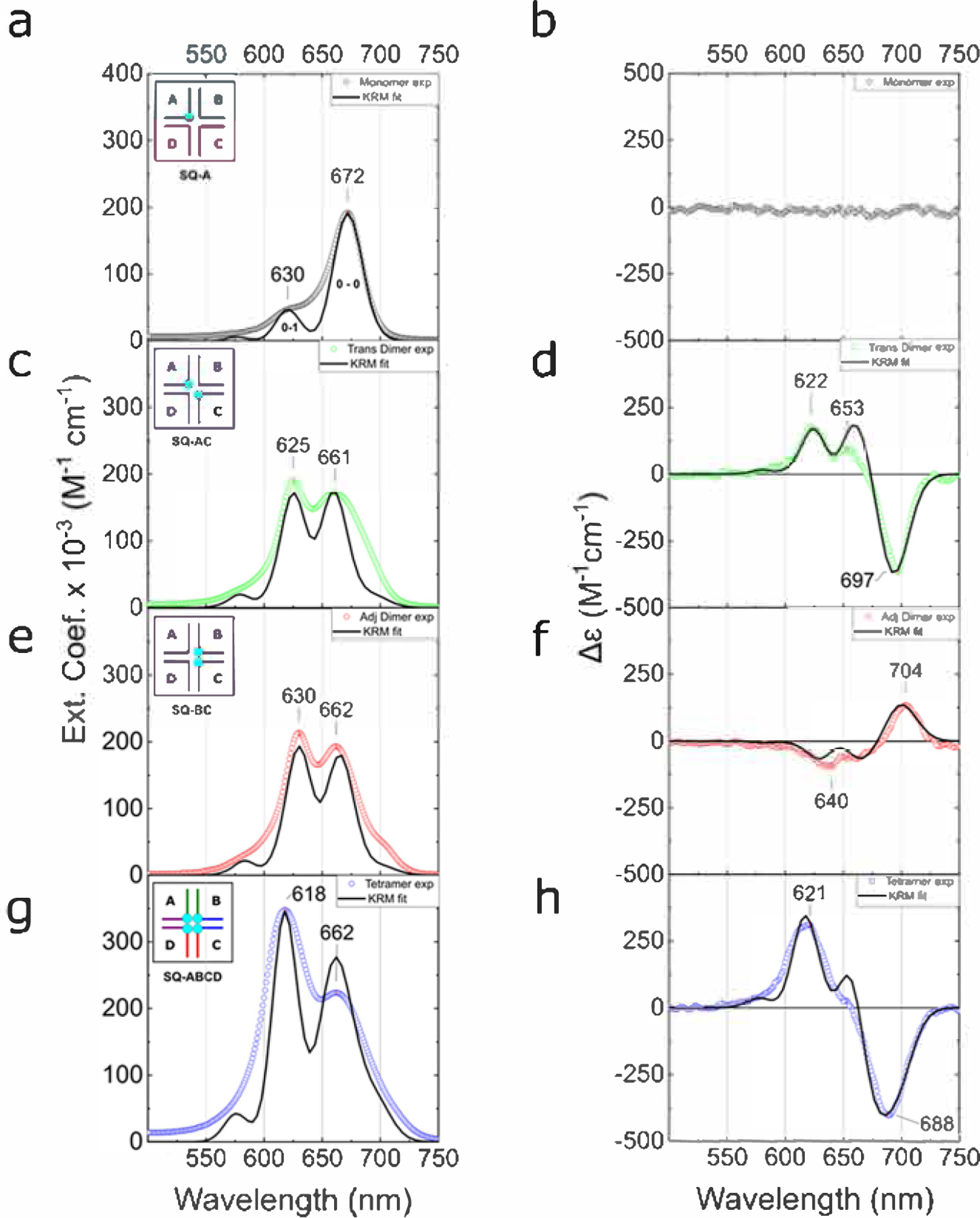
(a, c, e, g) Acquired steady-state absorption spectra of the DNA–Square 660 dye constructs in 1× TBE, 15 mM MgCl2 at room temperature (dotted lines) and theoretical absorption spectra derived from KRM modeling (solid lines). The DNA–dye construct concentration was 1.5 μM. The insets show a schematic representation of dye monomer, dimers, and tetramer constructs in DNA HJs. (b, d, f, h) Acquired CD spectra of the DNA–Square 660 dye constructs in 1× TBE, 15 mM MgCl2 at room temperature (dotted lines) and theoretical CD spectra derived from KRM modeling (solid lines). The DNA–dye construct concentration was 1.5 μM.
Guided by the inspiring results of Cannon et al. on DNA-templated Cy5 aggregates,100,101 we next investigated aggregates of Square 660 in an immobile DNA HJ in the form of adjacent and transverse dimers. The electronic absorption spectrum of the transverse dimer SQ-AC exhibited its most prominent absorption band at 625 nm along with an additional broad feature peaking at 661 nm, both blue-shifted with respect to the monomer 0–0 band of 672 nm (Figure 2c). The absorption spectrum of the adjacent dimer SQ-BC was overall very similar, characterized by a primary blue-shifted band at 630 nm with another major band at 662 nm (Figure 2e). Such a blue shift of the electronic absorption spectrum of the aggregate compared with the monomer is indicative of a dominant face-to-face, or H-aggregate, orientation of the two Square 660 dyes in both adjacent and transverse configurations. Similar optical behavior was previously observed with the Cy5 transverse dimer templated by an immobile DNA HJ.101 Similar to the squaraine transverse dimer (i.e., SQ-AC), the Cy5 transverse dimer formed an H-aggregate with a strong excitonic Coulombic coupling. However, the Cy5 adjacent dimer assembled into a J-type aggregate. Consistent with these observations, Markova et al. also observed a propensity of indolenine squaraines for H-aggregate formation. In their studies, two indolenine squaraines, dually tethered to duplex DNA, formed an H-aggregate,76 while two Cy5 dyes formed a J-aggregate in the same duplex configuration.74
Having identified the aggregate type of the transverse and adjacent Square 660 dimers, we next proceeded to more incisively evaluate signatures of exciton delocalization in the electronic absorption spectra. To aid in evaluating signatures of exciton delocalization, the absorption spectra were fit with four Gaussian features. The four-component Gaussian fit was justified mathematically (Section S7), while observations in the CD spectra and theoretical modeling provided additional support for the four-component fit (described in subsequent sections). The four absorption bands associated with the fit, labeled as A1, A2, A3 and A4, exhibit peak maxima for the transverse dimer at 690, 660, 624, and 599 nm, respectively, and for the adjacent dimer exhibit peak maxima at 700, 663, 629, and 603 nm, respectively (Figure S7a,b and Table 1). We attribute the weak, red-shifted absorption band (A1) to the lowest-energy excitonic state of the H-dimer, with the finite absorption intensity arising from a slight oblique (i.e., nonideal) orientation of the H-dimer. The Gaussian fitting additionally reveals that while the intensity of the absorption bands of the monomer varies in the order A1 (0–0) > A2 (1–0), the intensity of the absorption bands for both dimers varies in the order A2 > A1 and A3 > A1. Such redistribution of oscillator strength in the form of a decreased An/An+1 ratio17 (that contributes to the blue shift in the absorption spectrum) is consistent with a large value for the excitonic hopping parameter J1,2, and exciton delocalization. Finally, the magnitude of the energy difference between the low- and high-energy excitonic states, i.e., the so-called Davydov splitting, can provide an estimate of the value of the excitonic hopping parameter since for dimers its value is approximately one half the Davydov splitting energy. As noted above, the transition to the low-energy excitonic state can be readily assigned to the lowest-energy absorption band (A1). Assigning the transition to the high-energy excitonic state to a specific absorption band, in contrast, is complicated by the strong vibronic coupling in the squaraine dyes. As such, and to gain qualitative insight into the excitonic Coulombic coupling strengths, we report the Davydov splitting energies (and corresponding excitonic hopping parameters) associated with A1 and the next two higher-lying absorption bands (A2 and A3) identified via the Gaussian fitting. For the transverse dimer, the A2 – A1 and A3 – A1 Davydov splitting energies are 82 meV (658 cm−1) and 190 meV (1532 cm−1), respectively; for the adjacent dimer, the A2 – A1 and A3 – A1 Davydov splitting energies are 99 meV (797 cm−1) and 200 meV (1610 cm−1), respectively. The corresponding excitonic hopping parameters estimated from these values for the transverse dimer are 41 meV (329 cm−1) and 95 meV (766 cm−1), respectively; for the adjacent dimer, they are 49 meV (398 cm−1) and 100 meV (806 cm−1), respectively. For comparison, Röhr et al. reported very similar coupling strengths (ca. 800 cm−1 or 99 meV) in the J-aggregates of indolenine squaraine dimers created by covalent coupling of two squaraine molecules.117 While a quantitative analysis of the excitonic hopping parameter in this manner is complicated by the strong vibronic coupling in the squaraine dyes and the associated ambiguity in assigning the transition to the high-energy exciton state to a particular high-energy absorption band, the magnitude of the estimated excitonic hopping parameters are clearly large and indicative of exciton delocalization in both dimers.
Table 1.
Steady-State Optical Properties of DNA-Templated Square 660 Constructs
| construct | observed Abs maximaa (nm) | calculated Abs maximab (nm) | observed FL maximac (nm) | FL suppressiond (%) | ΦFe |
|---|---|---|---|---|---|
| monomer | 630; 672 | 630; 672 | 687 | n/a | 0.37 |
| trans dimer | 625; 661 | 599; 624; 660; 690 | 688 | 80 | 0.038 |
| adj dimer | 630; 662 | 603; 629; 663; 700 | 690 | 92 | 0.023 |
| tetramer | 618; 662 | 578; 618; 663; 694 | 689 | 98 | 0.006 |
Measurements were carried out in 1× TBE, 15 mM MgCl2 containing 1.5 μM DNA construct at room temperature.
Values for the monomer are obtained from fitting the data with two Gaussians, and the values for the aggregates are obtained from fitting the data with four Gaussians.
Measurements were carried out in 1× TBE, 15 mM MgCl2 containing 0.5 μM DNA construct at room temperature. Samples were excited at λexc = 650 nm.
Fluorescence suppression relative to the monomer was calculated for 665–740 nm range as described in the Supporting Information.
Fluorescence quantum yield measured in 1× TBE, 15 mM MgCl2.
To further elucidate the excitonic delocalization in the squaraine dimers, CD spectroscopy measurements were performed. CD spectra of the monomer, dimer, and tetramer constructs showed a positive bisignate signal in the UV region between 230 and 280 nm, confirming a well-folded B-form DNA duplex of HJ (Figure S8). The achiral monomer SQ-A did not show an induced CD signal in the visible region (Figure 2b), indicating that the monomer retained conformational freedom in the core of the DNA HJ. The transverse dimer SQ-AC exhibited a negative exciton-induced CD couplet (down-up shape from right to left) in the visible part of the spectrum (Figure 2d), indicating the chiral disposition of coupled transition dipole moments. This bisignate character of the exciton-induced CD couplet and its intensity indicate coherent exciton hopping between the dyes.120 The cross point of the CD couplet at ~670 nm corresponds to the absorption maximum of the squaraine monomer SQ-A With an intense negative Cotton effect at 697 nm (Δε = −360 M−1 cm−1), the obliqueness in dye face-to-face orientation is evident from the splitting of the positive Cotton effect at 622 nm (Δε = 175 M−1 cm−1) and 653 nm (Δε = 95 M−1 cm−1). This asymmetry of the CD couplet is also predicted by the theoretical model in one configuration of coupled dipole moments (discussed below). In analogy, the adjacent dimer SQ-BC exhibits a CD couplet with positive sign (i.e., up-down shape from right to left) and asymmetric shape (Figure 2f) but of opposite chirality compared to the transverse dimer SQ-AC. The Cotton effects of the adjacent dimer SQ-BC at 704 nm (Δε = +135 M−1 cm−1), 660 nm (Δε = −70 M−1 cm−1), and 640 nm (Δε = −100 M−1 cm−1) appeared to be less pronounced, possibly due to conformational flexibility. Thus, the CD spectra provide additional confirmation of excitonic delocalization in both squaraine dimers.
As one of the most promising DNA-templated Cy5 aggregates reported by Cannon et al.100,101 was the tetramer configuration, we proceeded to prepare and characterize the Square 660 tetramer aggregates templated by DNA HJs. The electronic absorption spectrum of tetramer SQ-ABCD exhibited major bands most noticeable at 618 and 662 nm (Figure 2g). The most prominent band at 618 nm is further blue-shifted and intensified compared with both adjacent and transverse dimers. Analogous to dimer fitting, a four-component Gaussian fitting confirmed two strong spectral bands A2 and A3 with peak positions of 663 and 618 nm and an additional, weaker band A1 peaking at 694 nm (Figure S7d). The energy difference A1 – A2 in SQ-ABCD was determined to be 84 meV (673 cm–1) and the energy difference A1 – A3 to be 220 meV (1771 cm–1), which, as per above, correspond to large estimated excitonic hopping parameter values of 42 meV (337 cm–1) and 110 meV (886 cm–1), respectively. We also observed continued redistribution of the oscillator strength toward higher-energy absorption bands as seen in the increase of A2/A1 and A3/A1 intensities. In the CD spectrum, a tetramer SQ-ABCD exhibited a strikingly intense exciton-induced negative couplet signal with a couplet amplitude A = 710 M−1 cm−1 (Figure 2h). The symmetry of tetramer CD couplet with similarly intense Cotton effects at 621 nm (Δε = 310 M−1 cm−1) and at 688 nm (Δε = −400 M−1 cm−1), indicates the presence of a single chiral system composed of strongly coupled Square 660 dyes packing in the form of an H-aggregate. SQ-ABCD clearly exhibits signatures of strong excitonic Coulombic coupling.
In addition to strong perturbations to the electronic absorption spectrum and strong CD signals, significant fluorescence quenching was observed in the DNA HJ Square 660 dye dimers and a tetramer (Figure S5). Specifically, the relative fluorescence intensity for the different DNA–Square 660 dye constructs dropped in the order SQ-A ≫ SQ-AC > SQ-BC > SQ-ABCD (Figure S5) with ΦF values of 0.038, 0.023, and 0.006 measured for SQ-AC, SQ-BC, and SQ-ABCD, respectively (Table 1 and Section S5). The significantly reduced fluorescence intensity indicates that fluorescence emission is strongly quenched in SQ-AC, SQ-BC, and SQ-ABCD. Fluorescence quenching, which is typically due to drastically shortened excited-state lifetimes, has been reported previously in squaraine dimers covalently bound via methylene bridge linkers.121 While not a direct indicator of excitonic Coulombic coupling strength, this pioneering report by Liang et al. showed that the most strongly coupled squaraine dimers exhibited the most significant excited-state quenching. More recently, similar observations were made in strongly coupled Cy3 and Cy5 aggregates templated in the form of dsDNA and DNA HJs, respectively.84,119
Theoretical Spectral Modeling.
To determine the values of the excitonic hopping parameters between dye pairs in each aggregate and obtain quantitative information about the spatial arrangement of the dye molecules in their aggregates, we performed theoretical modeling based on the Kühn–Renger–May (KRM) model,122 following the previously described procedure100,101 with some modifications (Section S9). This method takes into account the effects of the dominant vibronic mode of each dye in a nonperturbative manner. In brief, the model generates theoretical absorbance and CD spectra of various dye configurations and compares them with experimental absorption and CD spectra (Figure 2). The theoretical dye configuration that yields best-fit spectra to the experimental absorbance and CD spectra is found by a stochastic gradient search. Since the spacing between dyes is typically on the order of or smaller than the dye length, an extended dipole approximation was employed in which the charge distribution is approximated by point charges separated by a distance that is comparable to the dye length.23 The Hilbert space included all configuration basis states for which the number of quanta of vibration on any dye belongs to the set {0, 1, 2}. Computed eigenvalues and eigenvectors of the Holstein-like Hamiltonian showed electronic versus vibronic contribution to energy eigenstates (Figures S9 and S10), confirming the importance of considering vibronic interactions. It is important to note that the KRM model treats an aggregate system as a pure state system. However, it is possible that some structural heterogeneity might be present in squaraine aggregates due to the weak van der Waals forces binding the dyes or DNA structure effects such as conformational isomers of stacked DNA HJ. Consequently, the KRM model results correspond to the dominant average configuration of the dye aggregate. An examination of the alternative transverse dimer SQ-BD (Section S10) revealed that conformational isomers of DNA HJ are, however, not a primary source of structural heterogeneity and that dye–dye interactions play a predominant role in the aggregate packing. The goodness of the fit was evaluated with several parameters: integral overlap between the corresponding absorbance and CD spectra, a mean-square deviation (MSD), and the weighted average of the MSD of the absorbance and CD spectra (Table S8). The dye positions were extracted from the fit in terms of the zenith and azimuthal angles and Cartesian coordinates (Tables S9 and S10). The dye orientation vectors (Figures S11 and S12) were visualized by means of Avogadro and UCSF Chimera software124 (Figures 3 and 4). To describe dyes’ mutual orientation within an aggregate, such geometric parameters as a center-to-center distance R, a slip angle θs, and an oblique angle α were extracted from the polar coordinates.
Figure 3.
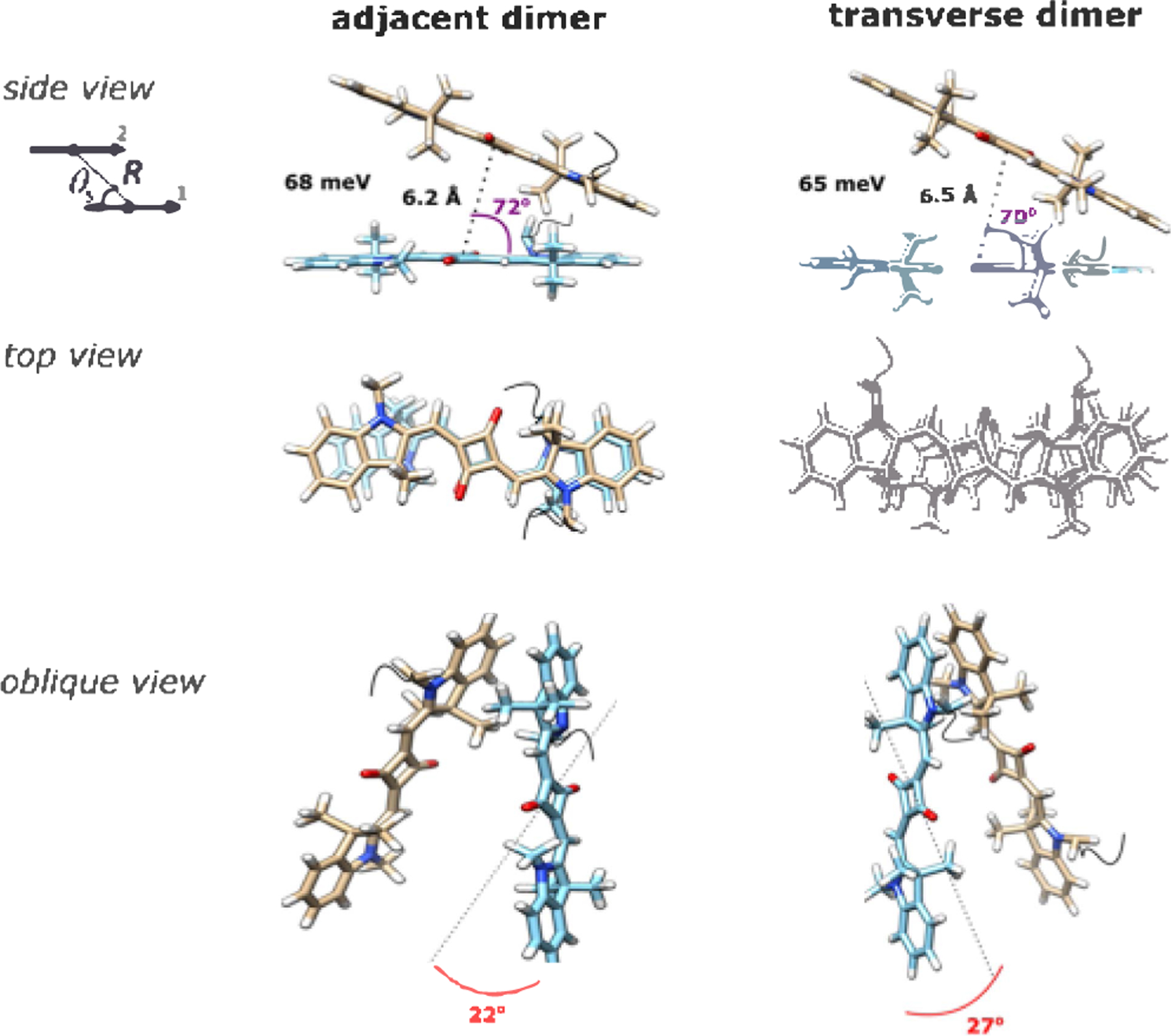
Molecular models of the Square 660 core region adjacent dimer SQ-BC and transverse dimer SQ-AC. The side view shows a J1,2 parameter, in meV, a center-to-center distance R, in Å, and a slip angle θs, in degree. The oblique view shows oblique angle α, in degree, as an angle between vectors 1 and 2 if their centers are superimposed. Note that the fitting procedure determines the position and orientation of the long axes of the Square 660 dyes but not the rotation of the dye core around its long axis. As such, the dye core rotations were arbitrarily chosen.
Figure 4.
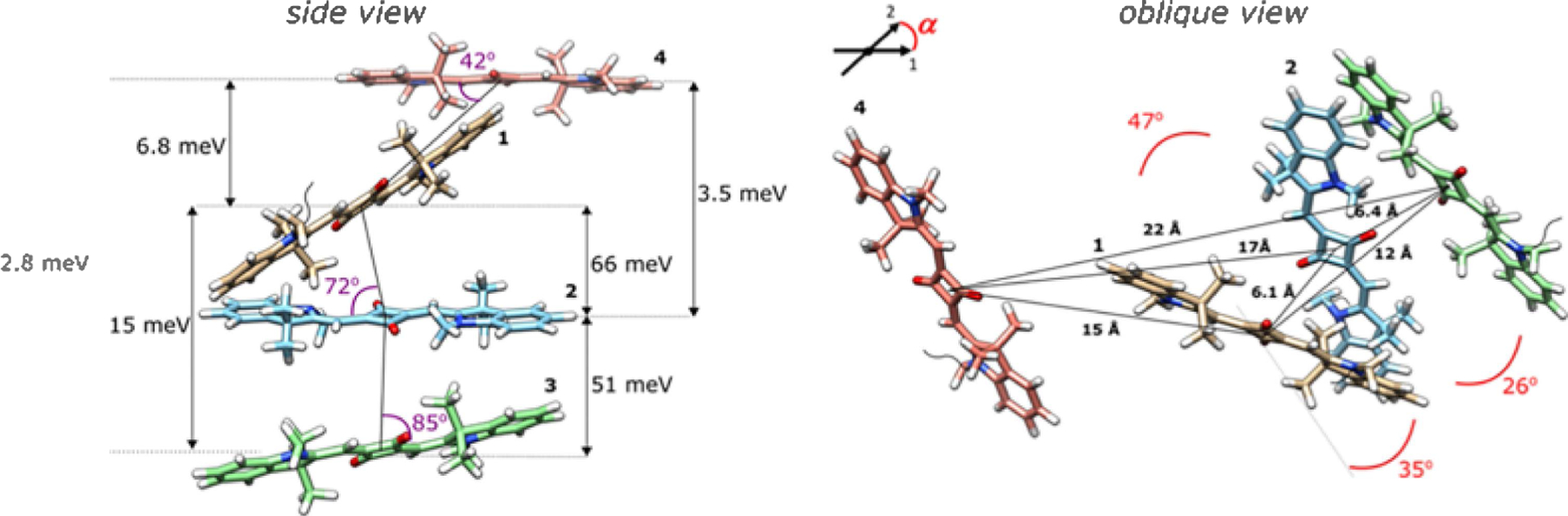
Molecular model of the Square 660 core region tetramer SQ-ABCD. The side view shows Jm,n parameter between each pair of dyes, in meV, and slip angles θs in degree. The oblique view shows a center-to-center distance R, in Å, and oblique angle α, in degree.
In this manner, we first determined a transition dipole moment of monomer SQ-A to be 11.7 D by fitting its absorption spectrum, thus accounting for the possible electronic interactions between the squaraine dyes and neighboring bases. Next, we calculated the characteristic hopping parameter constant J0 = 48 meV·nm3 derived from the transition dipole moment of the monomer SQ-A following eq S5. The constant J0 as a numerical coefficient defines the distances between the molecules extracted by fitting the absorbance and CD spectra in KRM model predictions for dye aggregates. Since J0 eliminates the degeneracy between the excitonic hopping parameter and spacing between the dyes, more definite predictions for the mutual orientation of the dye molecules are obtained.
By applying the constant J0 as an input parameter in the theoretical fitting of absorption and CD spectra, excitonic hopping parameter values J1,2 between dyes in dimer aggregates were obtained (Table S11). The excitonic hopping parameters J1,2 between squaraines were found to be 65 and 68 meV in the transverse and adjacent dimers, respectively. Theoretical fitting of the transverse SQ-AC and adjacent SQ-BC dimers revealed that the dyes are oriented with the predominant H-aggregate character, exhibiting slip angles of 70 and 72°, respectively (Figure 3). The center-to-center distance between squaraine molecules in transverse SQ-AC and adjacent SQ-BC dimers were calculated to be 6.5 and 6.2 Å, respectively. The transverse dimer exhibited a slightly larger oblique angle α of 27°, likely resulting in an experimentally observed more profound CD couplet. Interestingly, the adjacent and transverse dimers show nearly mirror symmetry of their 3-D structures. This observation of a chiral relationship between two dimer configurations is supported by the similarity in absorption spectra and, particularly, in circular dichroism, showing opposite handedness of analogical spectral line shapes. The optimized modeling procedure was also applied to the H-type Cy5 transverse dimer101 to obtain its J1,2 value for direct comparison with excitonic Coulombic coupling in squaraine dimers. The excitonic hopping parameter in the H-type Cy5 dimer was determined to be 70 meV. The Cy5 dimer was found to adopt a very similar H-aggregate geometry (Figure S13) compared to the geometry of squaraine dimers SQ-AC and SQ-BC. The Cy5 molecules in the Cy5 dimer were found to be 5.3 Å apart and characterized by a 76° slip angle and an 18° oblique angle. A slightly higher J1,2 value in Cy5 dimer is attributed to a shorter center-to-center distance between the Cy5 molecules. These results indicate that squaraines excitonically couple as strong as Cy5 dyes within a comparable dimer configuration.
Theoretical modeling of squaraine tetramer SQ-ABCD afforded a matrix of Jm,n values (Table S12 and Figure S14) and revealed geometric details of four molecule positions within the tetramer. Three squaraine molecules were found to form an H-stack with a slightly zigzag character, while the fourth squaraine positioned out of the H-stack (Figure 4). A mutual arrangement of dyes 1 and 2 closely resembles the adjacent dimer configuration SQ-BC as an internal dimer unit within the tetramer. Dyes 1 and 2 in the tetramer are positioned 6.1 Å apart, exhibiting the excitonic hopping parameter of 66 meV, a value that is similar to the excitonic hopping parameter in the squaraine dimers. In contrast, while a mutual orientation of dyes 2 and 3 in the tetramer resembles the transverse dimer configuration SQ-AC, the excitonic hopping parameter for the dye pair 2–3 is less than in SQ-AC. The center-to-center distance and the oblique angle in the dye pair 2–3 correspond to the transverse dimer configuration SQ-AC, but the slip angle in the dye pair 2–3 is 15° larger than that in SQ-AC (85 vs 70°). The difference in the slip angle might account for the smaller J1,2 value observed between dyes 2 and 3 within the tetramer. This observation demonstrates how geometric parameters, in particular the slip angle, affect the excitonic Coulombic coupling between two dyes. Being positioned further from the three-dye stack (average 18 Å), dye 4 weakly couples with the other three dyes (2.8–6.8 meV). The observed exclusion of dye 4 from the stack is presumably due to steric constraints. The nature of these steric constraints as well as the position of the tetramer relative to the DNA HJ cavity has not yet been identified. For comparison purposes, we determined the values of the excitonic hopping parameters between the dyes in the Cy5 tetramer (Table S13 and Figure S15). While the excitonic hopping strengths between equidistant Square 660 dyes and Cy5 dyes are comparable, the proximity of all four H-stacked Cy5 dyes in a parallelogram-like arrangement101 results in very intense blue-shifted absorption of the Cy5 tetramer. Perhaps the parallelogram-like arrangement observed in the Cy5 tetramer could also be observed in the Square 660 tetramer if Square 660 dyes were tethered to the DNA with dual phosphoramidite linkers.
CONCLUSIONS
In this study, we demonstrate that Square 660, a commercially available indolenine squaraine dye, exhibits exciton delocalization when aggregated via a DNA HJ. DNA HJs were used to template the aggregation of Square 660 in three different configurations: a transverse dimer, an adjacent dimer, and a tetramer. Exciton delocalization was evident in all configurations via significant blue shifts in the electronic absorption spectra, intense couplets in the CD spectra, and strong fluorescence emission suppression. Matrices of the excitonic hopping parameter between the dye molecules in dimers and a tetramer were obtained via simultaneous fitting of the absorption and CD spectra. Large values of the excitonic hopping parameters characterized the strength of the exciton Coulombic interactions responsible for exciton delocalization in indolenine squaraine aggregates. The strength of excitonic hopping interactions between indolenine squaraines was found to be comparable to the strength of the analogous DNA HJ-templated aggregate of the more ubiquitous cyanine dye Cy5, previously shown to be highly promising for exciton-based applications. An interesting finding of the present work is that DNA HJ-templated Square 660 aggregates appear to generally favor an H-type packing configuration regardless of an aggregate configuration, which contrasts with DNA HJ-templated aggregates of Cy5 that are capable of adopting both H- and J-type packing arrangements. Based on their superior photostability, structural diversity, and promising aggregate optical properties, we conclude that indolenine squaraine dyes are viable candidates for next-generation technologies wherein strong excitonic Coulombic coupling and exciton delocalization are desirable, such as artificial light harvesting, and potentially even room-temperature quantum information systems.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported wholly by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Materials Sciences and Engineering Division, and DOE’s Established Program to Stimulate Competitive Research (EPSCoR) (Award DE-SC0020089), except the following: initial work on squaraine transverse dimer modeling was sponsored by the National Science Foundation INSPIRE (Award no. 1648655). The circular dichroism spectrometer was made available by the Biomolecular Research Center (BRC) at Boise State, which is supported in part by the Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences (Award no. P20GM103408) and the National Institutes of Health (Award no. P20GM109095), the National Science Foundation (Award nos. 0619793 and 0923535), the MJ Murdock Charitable Trust, and the Idaho State Board of Education. We acknowledge Natalie Magnus and Taylor Farmer and Professor Amit Jain for their assistance in KRM Model Simulation Tool software development.
Footnotes
The authors declare the following competing financial interest(s): Ewald A. Terpetschnig is the managing director of SETA BioMedicals, which has an interest in this project.
Contributor Information
Olga A. Mass, Micron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, United States;.
Christopher K. Wilson, Micron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, United States
Simon K. Roy, Micron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, United States;.
Matthew S. Barclay, Micron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, United States
Lance K. Patten, Micron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, United States;.
Ewald A. Terpetschnig, SETA BioMedicals, LLC, Urbana, Illinois 61801, United States
Jeunghoon Lee, Micron School of Materials Science & Engineering and Department of Chemistry and Biochemistry, Boise State University, Boise, Idaho 83725, United States;.
Ryan D. Pensack, Micron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, United States;.
Bernard Yurke, Micron School of Materials Science & Engineering and Department of Electrical & Computer Engineering, Boise State University, Boise, Idaho 83725, United States;.
William B. Knowlton, Micron School of Materials Science & Engineering and Department of Electrical & Computer Engineering, Boise State University, Boise, Idaho 83725, United States;.
REFERENCES
- (1).Kasha M Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res 1963, 20, 55–70. [PubMed] [Google Scholar]
- (2).Andrews DL A Unified Theory of Radiative and Radiationless Molecular Energy Transfer. Chem. Phys 1989, 135, 195–201. [Google Scholar]
- (3).Walczak PB; Eisfeld A; Briggs JS Exchange Narrowing of the J Band of Molecular Dye Aggregates. J. Chem. Phys 2008, 128, No. 044505. [DOI] [PubMed] [Google Scholar]
- (4).Roden J; Eisfeld A Anomalous Strong Exchange Narrowing in Excitonic Systems. J. Chem. Phys 2011, 134, No. 034901. [DOI] [PubMed] [Google Scholar]
- (5).Fidder H; Knoester J; Wiersma DA Optical Properties of Disordered Molecular Aggregates: A Numerical Study. J. Chem. Phys 1991, 95, 7880–7890. [Google Scholar]
- (6).Vaitekonis S; Trinkunas G; Valkunas L Red Chlorophylls in the Exciton Model of Photosystem I. Photosynth. Res 2005, 86, 185–201. [DOI] [PubMed] [Google Scholar]
- (7).Abramavicius D; Mukamel S Exciton Delocalization and Transport in Photosystem I of Cyanobacteria Synechococcus Elongates: Simulation Study of Coherent Two-Dimensional Optical Signals. J. Phys. Chem. B 2009, 113, 6097–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Abramavicius D; Palmieri B; Mukamel S Extracting Single and Two-Exciton Couplings in Photosynthetic Complexes by Coherent Two-Dimensional Electronic Spectra. Chem. Phys 2009, 357, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Abramavicius D; Palmieri B; Voronine DV; Šanda F; Mukamel S Coherent Multidimensional Optical Spectroscopy of Excitons in Molecular Aggregates; Quasiparticle Versus Supermolecule Perspectives. Chem. Rev 2009, 109, 2350–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).McRae EG; Kasha M The Molecular Exciton Model. In Physical Processes in Radiation Biology; Augenstein L; Mason R; Rosenberg B, Eds.; Academic Press: New York, 1964; pp 23–42. [Google Scholar]
- (11).Hestand NJ; Spano FC Determining the Spatial Coherence of Excitons from the Photoluminescence Spectrum in Charge-Transfer J-Aggregates. Chem. Phys 2016, 481, 262–271. [Google Scholar]
- (12).Hestand NJ; Spano FC Molecular Aggregate Photophysics Beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res 2017, 50, 341–350. [DOI] [PubMed] [Google Scholar]
- (13).Hestand NJ; Spano FC Expanded Theory of H- and j-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev 2018, 118, 7069–7163. [DOI] [PubMed] [Google Scholar]
- (14).Zhong C; Bialas D; Collison CJ; Spano FC Davydov Splitting in Squaraine Dimers. J. Phys. Chem. C 2019, 123, 18734–18745. [Google Scholar]
- (15).Abramavicius D; Mukamel S Exciton Dynamics in Chromophore Aggregates with Correlated Environment Fluctuations. J. Chem. Phys 2011, 134, 174504–174510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mirkovic T; Ostroumov EE; Anna JM; van Grondelle R; Govindjee; Scholes GD Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms. Chem. Rev 2017, 117, 249–293. [DOI] [PubMed] [Google Scholar]
- (17).Spano FC The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res 2010, 43, 429–439. [DOI] [PubMed] [Google Scholar]
- (18).Renger T; May V; Kühn O Ultrafast Excitation Energy Transfer Dynamics in Photosynthetic Pigment–Protein Complexes. Phys. Rep 2001, 343, 137–254. [Google Scholar]
- (19).Schulze J; Kühn O Explicit Correlated Exciton-Vibrational Dynamics of the FMO Complex. J. Phys. Chem. B 2015, 119, 6211–6216. [DOI] [PubMed] [Google Scholar]
- (20).Tretiak S; Mukamel S Density Matrix Analysis and Simulation of Electronic Excitations in Conjugated and Aggregated Molecules. Chem. Rev 2002, 102, 3171–3212. [DOI] [PubMed] [Google Scholar]
- (21).Richter M; Ahn KJ; Knorr A; Schliwa A; Bimberg D; Madjet ME-A; Renger T Theory of Excitation Transfer in Coupled Nanostructures – from Quantum Dots to Light Harvesting Complexes. Phys. Status Solidi B 2006, 243, 2302–2310. [Google Scholar]
- (22).Asanuma H; Fujii T; Kato T; Kashida H Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol, C 2012, 13, 124–135. [Google Scholar]
- (23).Czikklely V; Forsterling HD; Kuhn H Extended Dipole Model for Aggregates of Dye Molecules. Chem. Phys. Lett 1970, 6, 207–210. [Google Scholar]
- (24).Jelley EE Spectral Absorption and Fluorescence of Dyes in the Molecular State. Nature 1936, 138, 1009–1010. [Google Scholar]
- (25).Jelley EE Molecular, Nematic and Crystal States of I: I-Diethyl-Cyanine Chloride. Nature 1937, 139, 631. [Google Scholar]
- (26).Scheibe G ÜBer Die VeräNderlichkeit Der Absorptionss-pektren in LöSungen Und Die Nebenvalenzen Als Ihre Ursache. Angew. Chem 1937, 50, 212–219. [Google Scholar]
- (27).Koepke J; Hu X; Muenke C; Schulten K; Michel H The Crystal Structure of the Light-Harvesting Complex II (B800–850) from Rhodospirillum Molischianum. Structure 1996, 4, 581–597. [DOI] [PubMed] [Google Scholar]
- (28).Chachisvilis M; Kuhn O; Pullerits T; Sundstrom V Excitons in Photosynthetic Purple Bacteria: Wavelike Motion or Incoherent Hopping? J. Phys. Chem. B 1997, 101, 7275–7283. [Google Scholar]
- (29).Hu X; Damjanovic A; Ritz T; Schulten K Architecture and Mechanism of the Light-Harvesting Apparatus of Purple Bacteria. Proc. Natl. Acad. Sci. U.SA 1998, 95, 5935–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jonas DM; Lang MJ; Nagasawa Y; Joo T; Fleming GR Pump-Probe Polarization Anisotropy Study of Femtosecond Energy Transfer within the Photosynthetic Reaction Center of Rhodobacter Sphaeroides R26. J. Phys. Chem. A 1996, 100, 12660–12673. [Google Scholar]
- (31).Arpin PC; Turner DB; McClure SD; Jumper CC; Mirkovic T; Challa JR; Lee J; Teng CY; Green BR; Wilk KE; et al. Spectroscopic Studies of Cryptophyte Light Harvesting Proteins: Vibrations and Coherent Oscillations. J. Phys. Chem. B 2015, 119, 10025–10034. [DOI] [PubMed] [Google Scholar]
- (32).Brixner T; Hildner R; Kohler J; Lambert C; Wurthner F Exciton Transport in Molecular Aggregates - from Natural Antennas to Synthetic Chromophore Systems. Adv. Energy Mater 2017, 7, No. 1700236. [Google Scholar]
- (33).Graugnard E; Kellis DL; Bui H; Bames S; Kuang W; Lee J; Hughes WL; Knowlton WB; Yurke B DNA-Controlled Excitonic Switches. Nano Lett 2012, 12, 2117–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cannon BL; Kellis DL; Davis PH; Lee J; Kuang W; Hughes WL; Graugnard E; Yurke B; Knowlton WB Excitonic and Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kellis DL; Rehn SM; Cannon BL; Davis PH; Graugnard E; Lee J; Yurke B; Knowlton WB DNA-Mediated Excitonic Upconversion FRET Switching. New J. Phys 2015, 17, No. 115007. [Google Scholar]
- (36).Wang SY; Lebeck AR; Dwyer C Nanoscale Resonance Energy Transfer-Based Devices for Probabilistic Computing. IEEE Micro 2015, 35, 72–84. [Google Scholar]
- (37).Sawaya NPD; Rappoport D; Tabor DP; Aspuru-Guzik A Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12, 6410–6420. [DOI] [PubMed] [Google Scholar]
- (38).Ostroverkhova O Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev 2016, 116, 13279–13412. [DOI] [PubMed] [Google Scholar]
- (39).Bardeen CJ The Structure and Dynamics of Molecular Excitons. Annu. Rev. Phys. Chem 2014, 65, 127–148. [DOI] [PubMed] [Google Scholar]
- (40).Eisfeld A; Briggs JS The J-Band of Organic Dyes: Lineshape and Coherence Length. Chem. Phys 2002, 281, 61–70. [Google Scholar]
- (41).Thilagam A Entanglement Dynamics of J-Aggregate Systems. J. Phys. A: Math. Theor 2011, 44, No. 135306. [Google Scholar]
- (42).Sarovar M; Ishizaki A; Fleming GR; Whaley KB Quantum Entanglement in Photosynthetic Light-Harvesting Complexes. Nat. Phys 2010, 6, 462–467. [Google Scholar]
- (43).Thilagam A Quantum Information Processing Attributes of J-Aggregates; World Scientific Publishing Company: Singapore, 2012; Vol. 2. [Google Scholar]
- (44).Ball P Physics of Life: The Dawn of Quantum Biology. Nature 2011, 474, 272–274. [DOI] [PubMed] [Google Scholar]
- (45).Lloyd S Quantum Coherence in Biological Systems. J. Phys.: Conf. Ser 2011, 302, No. 012037. [Google Scholar]
- (46).Vickerstaff T; Lemin DR Aggregation of Dyes in Aqueous Solution. Nature 1946, 157, 373. [Google Scholar]
- (47).Dickinson HO The Aggregation of Cyanine Dyes in Aqueous Solution. Trans. Faraday Soc 1947, 43, 486–491. [Google Scholar]
- (48).Heyne B Self-Assembly of Organic Dyes in Supramolecular Aggregates. Photochem. Photobiol. Sci 2016, 15, 1103–1114. [DOI] [PubMed] [Google Scholar]
- (49).Engel GS; Calhoun TR; Read EL; Ahn TK; Mancal T; Cheng YC; Blankenship RE; Fleming GR Evidence for Wavelike Energy Transfer through Quantum Coherence in Photosynthetic Systems. Nature 2007, 446, 782–786. [DOI] [PubMed] [Google Scholar]
- (50).Lee H; Cheng YC; Fleming GR Coherence Dynamics in Photosynthesis: Protein Protection of Excitonic Coherence. Science 2007, 316, 1462–1465. [DOI] [PubMed] [Google Scholar]
- (51).Scholes GD; Fleming GR; Olaya-Castro A; van Grondelle R Lessons from Nature About Solar Light Harvesting. Nat. Chem 2011, 3, 763–774. [DOI] [PubMed] [Google Scholar]
- (52).Scholes GD Quantum Biology Coherence in Photosynthesis. Nat. Phys 2011, 7, 448–449. [Google Scholar]
- (53).Fassioli F; Dinshaw R; Arpin PC; Scholes GD Photosynthetic Light Harvesting: Excitons and Coherence. J. R Soc., Interface 2014, 11, No. 20130901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Dill KA; MacCallum JL The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- (55).Yeates TO Geometric Principles for Designing Highly Symmetric Self-Assembling Protein Nanomaterials. Annu. Rev. Biophys 2017, 46, 23–42. [DOI] [PubMed] [Google Scholar]
- (56).Huang PS; Boyken SE; Baker D The Coming of Age of De Novo Protein Design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (57).Bale JB; Gonen S; Liu Y; Sheffler W; Ellis D; Thomas C; Cascio D; Yeates TO; Gonen T; King NP; et al. Accurate Design of Megadalton-Scale Two-Component Icosahedral Protein Complexes. Science 2016, 353, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wei B; Dai M; Yin P Complex Shapes Self-Assembled from Single-Stranded DNA Tiles. Nature 2012, 485, 623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ke Y; Ong LL; Shih WM; Yin P Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Rothemund PWK Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- (61).Seeman NC DNA in a Material World. Nature 2003, 421, 427–431. [DOI] [PubMed] [Google Scholar]
- (62).Seifert JL; Connor RE; Kushon SA; Wang M; Armitage B A Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc 1999, 121, 2987–2995. [Google Scholar]
- (63).Smith JO; Olson DA; Armitage B A Molecular Recognition of PNA-Containing Hybrids: Spontaneous Assembly of Helical Cyanine Dye Aggregates on PNA Templates. J. Am. Chem. Soc 1999, 121, 2686–2695. [Google Scholar]
- (64).Wang MM; Silva GL; Armitage BA DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc 2000, 122, 9977–9986. [Google Scholar]
- (65).Chowdhury A; Wachsmann-Hogiu S; Bangal PR; Raheem I; Peteanu LA Characterization of Chiral H and J Aggregates of Cyanine Dyes Formed by DNA Templating Using Stark and Fluorescence Spectroscopies. J. Phys. Chem. B 2001, 105, 12196–12201. [Google Scholar]
- (66).Garoff RA; Litzinger EA; Connor RE; Fishman I; Armitage BA Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo- and Heteroaggregates. Langmuir 2002, 18, 6330–6337. [Google Scholar]
- (67).Chowdhury A; Yu LP; Raheem I; Peteanu L; Liu LA; Yaron DJ Stark Spectroscopy of Size-Selected Helical H-Aggregates of a Cyanine Dye Templated by Duplex DNA. Effect of Exciton Coupling on Electronic Polarizabilities. J. Phys. Chem. A 2003, 107, 3351–3362. [Google Scholar]
- (68).Hannah KC; Armitage BA DNA-Templated Assembly of Helical Cyanine Dye Aggregates: A Supramolecular Chain Polymerization. Acc. Chem. Res 2004, 37, 845–853. [DOI] [PubMed] [Google Scholar]
- (69).Tomlinson A; Frezza B; Kofke M; Wang MM; Armitage BA; Yaron D A Structural Model for Cyanine Dyes Templated into the Minor Groove of DNA. Chem. Phys 2006, 325, 36–47. [Google Scholar]
- (70).Stadler AL; Renikuntla BR; Yaron D; Fang AS; Armitage B A Substituent Effects on the Assembly of Helical Cyanine Dye Aggregates in the Minor Groove of a DNA Template. Langmuir 2011, 27, 1472–1479. [DOI] [PubMed] [Google Scholar]
- (71).Banal JL; Kondo T; Veneziano R; Bathe M; Schlau-Cohen GS Photophysics of J-Aggregate-Mediated Energy Transfer on DNA. J. Phys. Chem. Lett 2017, 8, 5827–5833. [DOI] [PubMed] [Google Scholar]
- (72).Boulais É; Sawaya NPD; Veneziano R; Andreoni A; Banal JL; Kondo T; Mandal S; Lin S; Schlau-Cohen GS; Woodbuury NW; et al. Programmed Coherent Coupling in a Synthetic DNA-Based Excitonic Circuit. Nat. Mater 2018, 17, 159–166. [DOI] [PubMed] [Google Scholar]
- (73).Nicoli F; Roos MK; Hemmig EA; Di Antonio M; de Vivie-Riedle R; Liedl T. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes. J. Phys. Chem. A 2016, 120, 9941–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Markova LI; Malinovskii VL; Patsenker LD; Häner R J-Vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a near-IR Chiroptical Reporter. Chem. Commun 2013, 49, 5298–5300. [DOI] [PubMed] [Google Scholar]
- (75).Probst M; Langenegger SM; Häner R A Modular LHC Built on the DNA Three-Way Junction. Chem. Commun 2014, 50, 159–161. [DOI] [PubMed] [Google Scholar]
- (76).Markova LI; Malinovskii VL; Patsenker LD; Häner R Synthesis and Properties of Squaraine-Modified DNA. Org. Biomol. Chem 2012, 10, 8944–8947. [DOI] [PubMed] [Google Scholar]
- (77).Malinovskii VL; Wenger D; Häner R Nucleic Acid-Guided Assembly of Aromatic Chromophores. Chem. Soc. Rev 2010, 39, 410–422. [DOI] [PubMed] [Google Scholar]
- (78).Li S; Langenegger SM; Häner R Control of Aggregation-Induced Emission by DNA Hybridization. Chem. Commun 2013, 49, 5835–5837. [DOI] [PubMed] [Google Scholar]
- (79).Häner R; Samain F; Malinovskii VL DNA-Assisted Self-Assembly of Pyrene Foldamers. Chem. - Eur. J 2009, 15, 5701–5708. [DOI] [PubMed] [Google Scholar]
- (80).Garo F; Häner R A DNA-Based Light-Harvesting Antenna. Angew. Chem., Int. Ed 2012, 51, 916–919. [DOI] [PubMed] [Google Scholar]
- (81).Adeyemi OO; Malinovskii VL; Biner SM; Calzaferri G; Häner R Photon Harvesting by Excimer-Forming Multichromo-phores. Chem. Commun 2012, 48, 9589–9591. [DOI] [PubMed] [Google Scholar]
- (82).Asanuma H; Fujii T; Kato T; Kashida H Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol., C 2012, 13, 124–135. [Google Scholar]
- (83).Kashida H; Asanuma H Preparation of Supramolecular Chromophoric Assemblies Using a DNA Duplex. Phys. Chem. Chem. Phys 2012, 14, 7196–7204. [DOI] [PubMed] [Google Scholar]
- (84).Cunningham PD; Km YC; Diaz SA; Buckhout-White S; Mathur D; Medintz IL; Melinger JS Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. [DOI] [PubMed] [Google Scholar]
- (85).Mazuski RJ; Díaz SA; Wood RE; Lloyd LT; Klein WP; Mathur D; Melinger JS; Engel GS; Medintz IL Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett 2020, 4163–4172. [DOI] [PubMed] [Google Scholar]
- (86).Asanuma H; Shirasuka K; Takarada T; Kashida H; Komiyama M DNA-Dye Conjugates for Controllable H Aggregation(l). J. Am. Chem. Soc 2003, 125, 2217–2223. [DOI] [PubMed] [Google Scholar]
- (87).Kashida H; Asanuma H; Komiyama M Alternating Hetero H Aggregation of Different Dyes by Interstrand Stacking from Two DNA-Dye Conjugates. Angew. Chem., Int. Ed 2004, 43, 6522–6525. [DOI] [PubMed] [Google Scholar]
- (88).Kashida H; Tanaka M; Baba S; Sakamoto T; Kawai G; Asanuma H; Komiyama M Covalent Incorporation of Methyl Red Dyes into Double-Stranded DNA for Their Ordered Clustering. Chem. - Eur. J 2006, 12, 777–784. [DOI] [PubMed] [Google Scholar]
- (89).Ikeda S; Okamoto A Hybridization-Sensitive On-Off DNA Probe: Application of the Exciton Coupling Effect to Effective Fluorescence Quenching. Chem. - Asian J 2008, 3, 958–968. [DOI] [PubMed] [Google Scholar]
- (90).Fujii T; Kashida H; Asanuma H Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory. Chem. - Eur. J 2009, 15, 10092–10102. [DOI] [PubMed] [Google Scholar]
- (91).Hara Y; Fujii T; Kashida H; Sekiguchi K; Liang X; Niwa K; Takase T; Yoshida Y; Asanuma H Coherent Quenching of a Fluorophore for the Design of a Highly Sensitive in-Stem Molecular Beacon. Angew. Chem., Int. Ed 2010, 49, 5502–5506. [DOI] [PubMed] [Google Scholar]
- (92).Ruedas-Rama MJ; Alvarez-Pez JM; Orte A Formation of Stable BOBO-3 H-Aggregate Complexes Hinders DNA Hybridization. J. Phys. Chem. B 2010, 114, 9063–9071. [DOI] [PubMed] [Google Scholar]
- (93).Ruedas-Rama MJ; Orte A; Martin-Domingo MC; Castello F; Talavera EM; Alvarez-Pez JM Interaction ofYOYO-3 with Different DNA Templates to Form H-Aggregates. J. Phys. Chem. B 2014, 118, 6098–6106. [DOI] [PubMed] [Google Scholar]
- (94).Kringle L; Sawaya NPD; Widom J; Adams C; Raymer MG; Aspuru-Guzik A; Marcus AH Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys 2018, 148, No. 085101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Heussman D; Kittell J; Kringle L; Tamimi A; von Hippel PH; Marcus AH Measuring Local Conformations and Conformational Disorder of (Cy3)(2) Dimer Labeled DNA Fork Junctions Using Absorbance, Circular Dichroism and Two-Dimensional Fluorescence Spectroscopy. Faraday Discuss 2019, 216, 211–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Albinsson B; Hannestad JK; Börjesson K Functionalized DNA Nanostructures for Light Harvesting and Charge Separation. Coord. Chem. Rev 2012, 256, 2399–2413. [Google Scholar]
- (97).Probst M; Wenger D; Biner SM; Häner R The DNA Three-Way Junction as a Mould for Tripartite Chromophore Assembly. Org. Biomol. Chem 2012, 10, 755–759. [DOI] [PubMed] [Google Scholar]
- (98).Seeman NC Nucleic Acid Junctions and Lattices. J. Theor. Biol 1982, 99, 237–247. [DOI] [PubMed] [Google Scholar]
- (99).Kallenbach NR; Ma R-I; Seeman NC An Immobile Nucleic Acid Junction Constructed from Oligonucleotides. Nature 1983, 305, 829–831. [Google Scholar]
- (100).Cannon BL; Kellis DL; Patten LK; Davis PH; Lee J; Graugnard E; Yurke B; Knowlton WB Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Cannon BL; Patten LK; Kellis DL; Davis PH; Lee J; Graugnard E; Yurke B; Knowlton WB Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Davydov A Theory of Absorption Spectra of Molecular Crystals. Ukr. J. Phys 2008, 53, 69–70. [Google Scholar]
- (103).Stennett EMS; Ciuba MA; Levitus M Photophysical Processes in Single Molecule Organic Fluorescent Probes. Chem. Soc. Rev 2014, 43, 1057–1075. [DOI] [PubMed] [Google Scholar]
- (104).Demchenko AP Photobleaching of Organic Fluorophores: Quantitative Characterization, Mechanisms, Protection. Methods Appl. Fluoresc 2020, 8, No. 022001. [DOI] [PubMed] [Google Scholar]
- (105).Yagi S; Nakazumi H Heterocyclic Polymethine Dyes. Topics in Heterocyclic Chemistry; Strekowski L, Ed.; Springer-Verlag: Berlin, Heidelberg, 2008; Vol. 14, pp 133–181. [Google Scholar]
- (106).Terpetsching E; Szmacinski H; Lakowicz J R Synthesis, Spectral Properties and Photostabilities of Symmetrical and Unsymmetrical Squaraines - a New Class of Fluorophores with Long-Wavelength Excitation and Emission. Anal. Chim. Acta 1993, 282, 633–641. [Google Scholar]
- (107).Terpetschnig E; Lakowicz J R Synthesis and Characterization of Unsymmetrical Squaraines - a New Class of Cyanine Dyes. Dyes Pigm 1993, 21, 227–234. [Google Scholar]
- (108).Markova LI; Terpetschnig EA; Patsenker LD Comparison of a Series of Hydrophilic Squaraine and Cyanine Dyes for Use as Biological Labels. Dyes Pigm 2013, 99, 561–570. [Google Scholar]
- (109).Treibs A; Jacob K Cyclotrimethine Dyes Derived from Squaric Acid. Angew. Chem., Int. Ed 1965, 4, 694. [Google Scholar]
- (110).Jiang JQ; Sun CL; Shi ZF; Zhang HL Squaraines as Light-Capturing Materials in Photovoltaic Cells. RSC Adv 2014, 4, 32987–32996. [Google Scholar]
- (111).Khopkar S; Shankarling G Synthesis, Photophysical Properties and Applications of NIR Absorbing Unsymmetrical Squaraines: A Review. Dyes Pigm 2019, 170, No. 107645. [Google Scholar]
- (112).Sreejith S; Carol P; Parayalil C; Ayyappanpillai A Squaraine Dyes: A Mine of Molecular Materials. J. Mater. Chem 2008, 18, 264–274. [Google Scholar]
- (113).Xia GM; Wang HM Squaraine Dyes: The Hierarchical Synthesis and Its Application in Optical Detection. J. Photochem. Photobiol, C 2017, 31, 84–113. [Google Scholar]
- (114).Ilina K; MacCuaig WM; Laramie M; Jeouty JN; McNally LR; Henary M Squaraine Dyes: Molecular Design for Different Applications and Remaining Challenges. Bioconjugate Chem 2020, 31, 194–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Würthner F; Kaiser TE; Saha-Moller C R J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials. Angew. Chem., Int. Ed 2011, 50, 3376–3410. [DOI] [PubMed] [Google Scholar]
- (116).Mayerhöffer U; Wurthner F Cooperative Self-Assembly of Squaraine Dyes. Chem. Sci 2012, 3, 1215–1220. [Google Scholar]
- (117).Röhr MIS; Marciniak H; Hoche J; Schreck MH; Ceymann H; Mitric R; Lambert C Exciton Dynamics from Strong to Weak Coupling Limit Illustrated on a Series of Squaraine Dimers. J. Phys. Chem. C 2018, 122, 8082–8093. [Google Scholar]
- (118).Malý P; Lüttig J; Mueller S; Schreck M; Lambert C; Brixner T Coherently and Fluorescence-Detected Two-Dimensional Electronic Spectroscopy: Direct Comparison on Squaraine Dimers. Phys. Chem. Chem. Phys 2020, 21222–21237. [DOI] [PubMed] [Google Scholar]
- (119).Huff JS; Davis PH; Christy A; Kellis DL; Kandadai N; Toa ZSD; Scholes GD; Yurke B; Knowlton WB; Pensack R D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett 2019, 10, 2386–2392. [DOI] [PubMed] [Google Scholar]
- (120).Spano FC Analysis of the Uv/Vis and Cd Spectral Line Shapes of Carotenoid Assemblies: Spectral Signatures of Chiral H-Aggregates. J. Am. Chem. Soc 2009, 131, 4267–4278. [DOI] [PubMed] [Google Scholar]
- (121).Liang KN; Farahat MS; Perlstein J; Law KY; Whitten DG Exciton Interactions in Nonconjugated Squaraine Dimers. Mechanisms for Coupling and Consequences for Photophysics and Photochemistry. J. Am. Chem. Soc 1997, 119, 830–831. [Google Scholar]
- (122).Kühn O; Renger T; May V Theory of Exciton-Vibrational Dynamics in Molecular Dimers. Chem. Phys 1996, 204, 99–114. [Google Scholar]
- (123).Holstein T Studies of Polaron Motion: Part I. The Molecular-Crystal Model. Ann. Phys 1959, 8, 325–342. [Google Scholar]
- (124).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera - a Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.