

Abstract
Hepatoblastoma (HB) is the predominant primary liver tumor in children. While the prognosis is favorable when the tumor can be resected, the outcome is dismal for patients with progressed HB. Therefore, a better understanding of the molecular mechanisms responsible for HB is imperative for early detection and effective treatment. Sequencing analysis of human HB specimens unraveled the pivotal role of Wnt/β-catenin pathway activation in this disease. Nonetheless, β-catenin activation alone does not suffice to induce HB, implying the need for additional alterations. Perturbations of several pathways, including Hippo, Hedgehog, NRF2/KEAP1, HGF/c-Met, NK-1R/SP, and PI3K/AKT/mTOR cascades and aberrant activation of c-MYC, n-MYC, and EZH2 proto-oncogenes, have been identified in HB, although their role requires additional investigation. Here, we summarize the current knowledge on HB molecular pathogenesis, the relevance of the preclinical findings for the human disease, and the innovative therapeutic strategies that could be beneficial for the treatment of HB patients.
Keywords: hepatoblastoma, Wnt/β-catenin, Hippo, NRF2
Hepatoblastoma (HB) is the most frequent liver malignancy in childhood. HB incidence has increased from 0.61 per 1,000,000 in the period of 1973 to 19771 to 2.16 per 1,000,000 in the period of 2000 to 2015.2 The tumor usually arises in children of 0 to 4 years of age. The ratio of male/female is 1.67. HB in adulthood is extremely rare and has a dismal prognosis.3 Although most often HB is sporadic, occasionally it can be associated with other conditions, including Beckwith–Wiedemann syndrome,4 familial adenomatous polyposis,5 isolated hemihyperplasia,6 type 1a glycogen storage disease,7 and trisomy 18.8 These associations emphasize the importance of an extended genetic evaluation in children with HB, and a careful tumor screening inpatients at risk of HB. In this regard, it remains unclear whether low birth weight (i.e., less than 2,500 g) represents a higher risk of developing HB or is a marker of other exposures.9 Histologically, HB consists of immature hepatocytic cells, which encompass the different stages of liver development. HB may be epithelial or mixed epithelial–mesenchymal. Epithelial HB subtypes include pure fetal with low or high mitotic activity, embryonal, small cell, and cholangioblastic. Noticeably, different histologic subtypes often are observed within the same tumor, implying a perturbation at various steps of hepatocyte maturation.10 HB cells of the pure fetal subtype are organized in trabeculae and resemble fetal hepatocytes with a large cytoplasm, which appears clear or pink due to the different contents of glycogen and lipids. In the low mitotic subtype, less than two mitoses are present in 10 high power fields. This subtype is associated with the best postoperative prognosis.11 By contrast, the mitotically active subtype is characterized by more than two mitoses per high power field, a more amphophilic cytoplasm, and a higher nuclear/cytoplasmic ratio. This pattern should be distinguished from the pure fetal subtype, as it needs chemotherapy.12 The most frequent variant of HB is the embryonal subtype, consisting of cells characterized by prominent highly basophilic nuclei and a small cytoplasm, like those observed in the liver of the first weeks of gestation.10 The small-cell undifferentiated subtype is associated with a poor outcome. α-Fetoprotein (AFP) is normal or low in these cases. Tumors consisting exclusively of this morphological subtype are rare. However, the negative prognosis is maintained even when clusters of small undifferentiated cells are observed in large tumors of different subtype. The immunohistochemical expression of INI1 in the small-cell undifferentiated subtype is of relevance in the differential diagnosis of HB and malignant rhabdoid tumors.13 The cholangioblastic variant is instead characterized by the presence of ductal-like structures associated with embryonal or fetal cell clusters. This arrangement possibly derives from a bipotential neoplastic progenitor.14 Finally, the mixed mesenchymal–epithelial HB subtype is characterized by the presence of fibrosis, muscular and osteoid components, as well as of teratoid features that account for the embryonal origin of this tumor.10
Failure to thrive, weight loss, and an enlarging abdominal mass are the main findings of this tumor at presentation. AFP serum levels are generally high, peaking up to 1,000,000 ng/mL. On ultrasound examination, HB appears as an echogenic, well-defined mass. Areas of necrosis, hemorrhage, and calcifications can be observed within the neoplasia. Computed tomography and magnetic resonance imaging are necessary to identify vessel involvement and distant metastases.15
The current assessment of patients with HB comes from the achievements of four trial groups: the International Childhood Tumor strategy group (SIOPEL),16 the Children Oncology Group (COG),17 the German Society of Pediatric Oncology (GPOH),18 and the Japanese Study Group for Pediatric Liver Tumors (JPLT).19 The coalition of these groups led to the Children’s Hepatic tumors International Cooperation (CHIC). The unified CHIC analysis of the risk in patients with HB created a common approach to staging. This analysis is based on a pooled database of 1,605 patients treated in 25 years by the four trial groups.20 Currently, the severity of the disease takes into account the pretreatment extent of the tumor (PRETEXT stage), patient’s age, serum levels of AFP, presence of distant metastases, and the annotation factors, including the portal vein and vena cava involvement, the contiguous extrahepatic tumor spread, the multifocality, and the tumor rupture. According to the radiologic staging system for primary hepatic malignancies of childhood,15 four PRETEXT stages are identified. The PRETEXT I stage applies to conditions where the tumor is confined to one peripheral section (Couinaud segments 2 and 3 or 6 and 7). The PRETEXT II stage defines tumors involving the entire right or left lobe, or the median section (segment 4 or 1). In the PRETEXT III stage, a single section is free of tumor. The PRETEXT stage IV denotes the presence of multifocal lesions in all sections or a single large tumor extended to all sections. In PRETEXT stages I and II, the age of the patients is significantly associated to the outcome (hazard ratio 6.5 for the age group > 8 years old). In PRETEXT stage III, in addition to the extent of the tumor, the low level of AFP and the presence of annotation factors confer a higher risk. In PRETEXT stage IV, age > 3 years and annotation factors significantly increase the risk. In a metastatic disease of whatever PRETEXT stage, an additional risk factor is represented by a relatively low level of AFP.20
The current 5-year survival rate of HB ranges from 50 to 100 percent, depending on the disease stratification risk.21,22 However, the side effects of chemotherapy, including cisplatin-associated ototoxicity and nephrotoxicity, and doxorubicin-associated cardiomyopathy and secondary leukemia, impact on the quality of life and long-term outcome of these patients. In addition, the treatment of high risk and refractory cases remains an unmet need. The ongoing Pediatric Hepatic International Tumor Trial (PHITT; https://www.clinicaltrials.gov; NCT03017326) stems from the cooperation of SIOPEL, COG, GPOH, and JPLT and will address these issues. In children affected by HB, PHITT primary objectives are: (1) to evaluate whether the chemotherapy regimen of low-risk HB can be reduced; (2) to compare different regimens for HB intermediate-risk group; (3) to compare different postinduction regimens for HB high-risk group; (4) to collect samples for biological and toxicity studies. PHITT will also evaluate new therapy regimens in children with primary hepatocellular carcinoma (HCC). Accordingly, HB patients at extremely low risk and pure fetal well-defined histology will receive surgery alone. Patients with different histology will receive, in addition to surgery, two cycles of adjuvant cisplatin. In HB patients at low risk, a dose reduction of cisplatin (4 vs. 6 cycles) will be compared. In HB patients at intermediate risk, three different chemotherapy regimens will be compared: a combination of cisplatin, doxorubicin, and carboplatin (SIOPEL-3HR) versus cisplatin, 5-fluorouracil, vincristine, and doxorubicin versus cisplatin monotherapy. High-risk HB patients will receive induction therapy according to the SIOPEL4 protocol. Patients who clear metastases will then receive surgery and consolidation treatment with carboplatin and doxorubicin. Those who do not clear metastases after the SIOPEL4 protocol will be randomized to two consolidation arms consisting of carboplatin/doxorubicin and irinotecan/vincristine versus carboplatin/doxorubicin and carboplatin/etoposide.
Molecular Classification of HB
The molecular classification of human cancers has been tremendously accelerated by the Cancer Genome Atlas (TCGA) project.23–25 The TGCA characterized over 20,000 primary tumor tissues and nontumor samples among 33 cancer types.25 Unfortunately, due its rarity, HB was not included in TCGA studies. Nevertheless, recently, the genomic and transcriptomic features of human HB have been characterized (Table 1).26–30 Based on the molecular characteristics, HB was classified into distinct subtypes and gene expression signatures. These molecular classifications could be applied to stratify HB patients and provide guidance toward precision medicine.31,32
Table 1.
Molecular classification of hepatoblastoma
| Study | Levels | Study type | Characterized by | Subtypes of HB (clusters) | Features | Number of HBs |
|---|---|---|---|---|---|---|
| Cairo et al26 | RNA | Transcriptomic | 16-Gene signature gene expression profiling | C1, C2 | C1: enriched in hepatic perivenous program-related genes; β-catenin membranous and cytoplasmic accumulation. C2: enriched in cell-cycle and stem-cell-related genes; β-catenin nuclear accumulation. |
85 |
| Cairo et al27 | miRNA | Transcriptomic/epigenetic variation | 4-miR signature miRNA expression profiling | Cm1, Cm2 | Cm1:miR-100/let-7a-2/miR-125b-1 cluster overexpression Cm2: miR-371–3 cluster overexpression |
65 |
| Hooks et al28 | RNA | Transcriptomic | 4-Gene signature gene expression profiling | C1, C2A, and C2B | C1: stronger diffused ITGA6 staining. C2A: upregulation of TOP2A, FA pathway activation. C2B: VIM organized into visible spindle-shaped structures. |
39 |
| Carrillo-Reixach et al29 | DNA | Epigenetic variation | DNA hypomethylation and CpG island hypermethylation | Epi-CA and Epi-CB | Epi-CA: a weak global hypomethylation except for CpG islands compared with Epi-CB. Epi-CB: activation of Wnt/β- catenin signaling and a strong 14q32-gene signature. | 113a |
| Sumazin et al30 | DNA, RNA | Genomic and transcriptomic | Differential activation of hepatic progenitor cell markers and metabolic pathways | HB1, HB2, and HB3 | HB1 (low risk): low expression of LIN28B and let-7; high expression of HNF1A. HB2 (high risk): high expression of NFE2L2, LIN28B, HMGA2, SALL4, AFP, oncofetal proteins and stem-cell markers. HB3 (intermediate-risk): situated between HB1 and HB2. |
88 |
Abbreviations: AFP, α-fetoprotein; HMGA2, high mobility group AT-hook 2; HNF1A, hepatic nuclear factor 1 α; ITGA6, integrin α 6; let-7, lethal-7; LIN28B, lin-28 homolog B; NFE2L2, nuclear factor, erythroid 2–like 2; SALL4, spalt-like transcription factor 4; TOP2A, topoisomerase 2-α.
113 HBs (discovery set: 33 patients; validation set: 80 patients).
At the mRNA level, HB molecular classification was performed using either microarrays or RNASeq.26,28,29 In particular, Cairo et al identified two molecular HB subtypes, indicated as C1 and C2 groups, with a discriminating 16-gene signature based on tumor transcriptome profiling. Specifically, the C1 group displayed the overexpression of mature hepatocyte markers such as GLUL, RHBG, CYP2E1, and CYP1A1, and roughly recapitulated the molecular features of the fetal liver. The C2 subtype, instead, mainly consisted of tumors with an embryonal phenotype, showing the upregulation of hepatic progenitor/stem cells and proliferation markers (AFP, TACSTD1, DLG7, CDC2, BUB1, AURKB, IGF2, DLK1, PEG3, PEG10, BEX1, MEG3, NDN, BIRC5, NPM1, and HDAC2) and the activation of MYC signaling. This subtype also demonstrated high invasive and metastatic features and predicted a poor prognosis.26,33 MYC activation in aggressive HB was confirmed at the micro-RNA level, and a 4-miRNA prognostic signature composed of miRs from the miR-100/let-7a-2/miR-125b-1 tumor suppressor cluster and from the miR-371–3 oncogenic cluster identified two patient groups with different outcomes.27 C1/C2 classification based on molecular expression patterns was also associated with the histological subtypes and especially with the main epithelial component. Specifically, C1 tumors mainly displayed a mitotically inactive fetal phenotype, whereas C2 tumors had a more immature pattern that includes predominance of crowded fetal, macrotrabecular, and embryonal components.26 More recently, Hooks et al performed RNASeq analysis of 25 HB cases and matching normal liver samples; noticeably, the results largely recapitulated the transcriptomic subtypes identified by Cairo et al. Further analysis revealed a concise 4-gene signature (HSD17B6, ITGA6, TOP2A, and VIM), which allowed the further subclassification of the C2 group into two distinct clusters, C2A and C2B. In particular, the C2A group displayed an increased expression of TOP2A and activation of the DNA repair related pathway, indicating a highly proliferative and aggressive phenotype, whereas a strong VIM expression with visible spindle-shaped structures observed in the C2B group suggested a mesenchymal phenotype with intermediate risk.28
Genomic alterations, DNA methylation, histone modifications, and metabolomics analysis were also used for HB molecular classification. For instance, by epigenetic profile analysis, Carrillo-Reixach et al unraveled two distinct HB subtypes, named epigenetic cluster A and epigenetic cluster B (Epi-CA and Epi-CB). The two clusters exhibited striking differences in the degree of DNA hypomethylation and CpG island.29 DNA methylation and CpG island methylation directly affect the regulation of the transcriptome.34 Indeed, the Epi-CA/CB clusters demonstrated differential expression signatures, which were strongly associated with the C1/C2A/C2B molecular subclasses.28,29 Based on the molecular findings, the authors defined the first molecular risk stratification of HB (MRS-HB), which improves the current clinical risk stratification approach.29
Furthermore, Sumazin et al attempted to identify diagnostic, therapeutic, and prognostic biomarkers from HB molecular profiles by combining genomic and transcriptomic analysis. The data obtained revealed the existence of three prognosis-predictive molecular subtypes, namely HB1, HB2, and HB3. By analyzing the molecular variations on hepatic/progenitor cell markers, hepatobiliary, metabolic, and cancer-related pathways underlying these three HB subtypes, several predictive biomarker candidates were identified, including HNF1A, NFE2L2, SALL4, HMGA2, and LIN28B.30 Finally, the metabolic profile of HB and HCC cell lines was determined by Crippa et al.35 The authors found that numerous glycolytic enzymes, including glucose transporter type 3 (GLUT3), hexokinase 1 (HK1), phosphofructokinase, platelet type (PFKP), and lactate dehydrogenase B (LDHB), were significantly elevated in embryonal-like cells compared with fetal-like HB cells. Fetal-like cells exhibited higher levels of gluconeogenesis genes, such as PPARG coactivator 1 alpha (PPARGC1A), aquaporin-9 (AQP9), glycerol kinase (GK), and glucose-6-phosphatase (G6PC).
Molecular Pathogenesis of Hepatoblastoma: Evidence from Whole-Genome Studies
DNA sequencing studies provided the first link between genetic mutations and HB molecular pathogenesis. Indeed, it has been found that mutations of the Wnt/β-catenin cascade occur in the vast majority of human HB samples, and almost exclusively affect the CTNNB1 gene (encoding β-catenin), located at 3p21.26,29,36–38 Intriguingly, long deletions encompassing CTNNB1 exons 3 and 4 are detected only in pure fetal HB, and correlate with increased NOTCH versus Wnt pathway activation. Conversely, embryonal HB harbors small deletions or missense mutations in CTNNB1 and exhibits the preponderance of Wnt over NOTCH activation.39 Furthermore, mutant β-catenin interacts with α-catenin only in HB cells with alterations confined to CTNNB1 exon 3, whereas larger deletions spanning exon 3 to 4, which are peculiar for fetal HB, prevent the binding to α-catenin.39 Given the critical role played by α-catenin in intercellular adhesion40 and gene transcription suppression,41 the presence or absence of the β-catenin/α-catenin complexes might result in diverse molecular and histopathological outcomes. Accordingly, it has been demonstrated that overexpression of each of 14 different HB and non-HB associated β-catenin mutants as well as the wild-type form of the gene in the mouse liver by hydrodynamic tail-vein injection is oncogenic in association with an activated form of yes-associated protein (YAP). Noticeably, the tumor growth rate, histology features, intracellular localization of β-catenin, and metabolic and transcriptional characteristics were β-catenin mutation-specific.42 Besides the association between CTNNB1 aberrations and histological subtypes, it has been reported that β-catenin target gene expression is tightly linked with specific histological phenotypes. Indeed, C1 tumors with a main fetal component have an increased expression of β-catenin target genes of the perivenous program, whereas C2 tumors with a more embryonal phenotype exhibit overexpression of β-catenin target genes related to proliferation and survival.26
Overall, these data strongly suggest that β-catenin pathway activation is the driver event in HB.43 The downstream consequences of CTNNB1 mutations might differ depending on the type of mutation (Fig. 1).
Fig. 1.
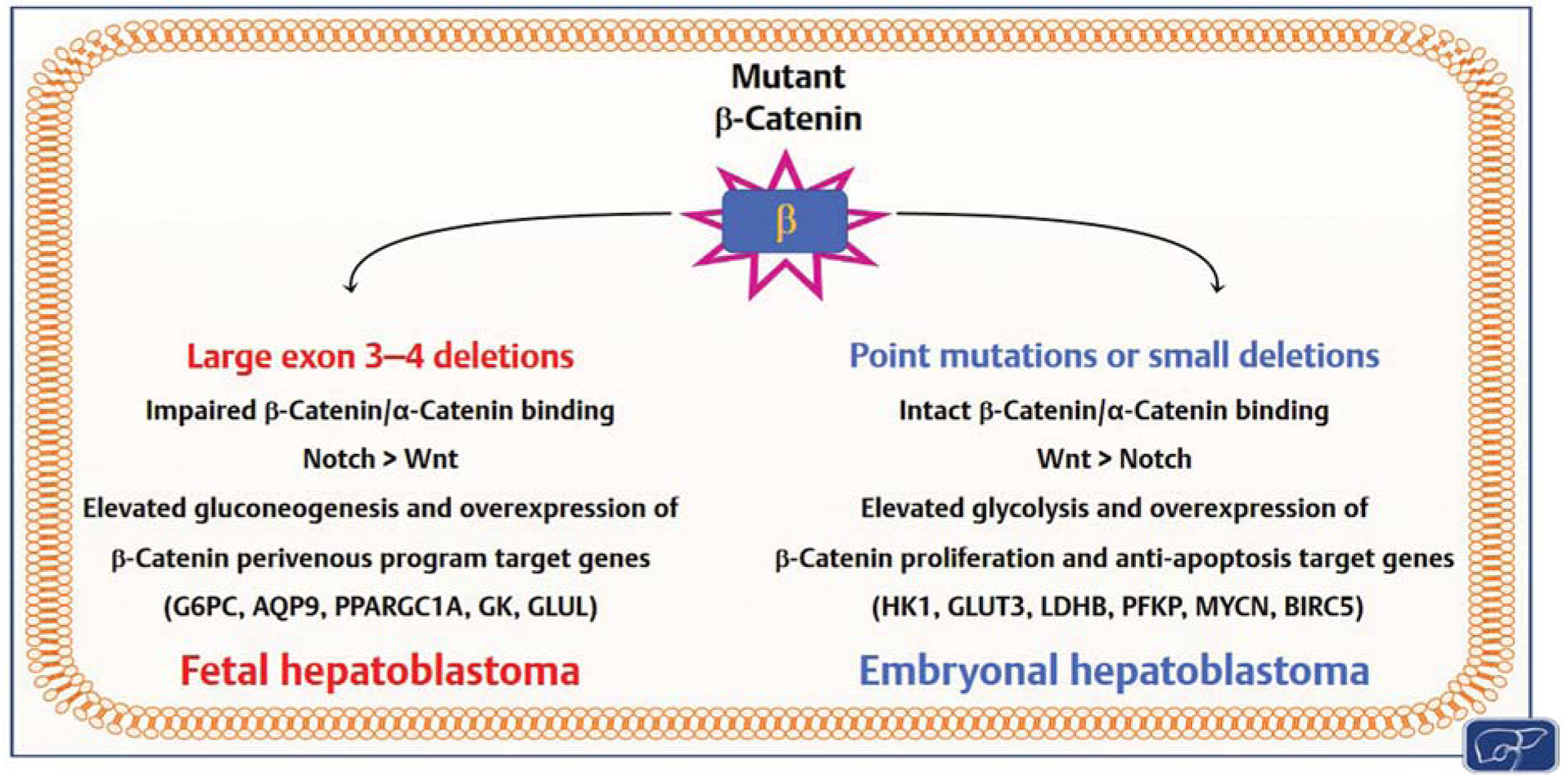
Scheme showing the distinct features of hepatoblastomas (HBs) depending on the type of β-catenin mutation. Details are reported in the main text.
Although limited by the sample size, recent whole-exome sequencing studies provided a more comprehensive picture of the genetic landscape of human HB. Importantly, using this methodology, it has been demonstrated that HB is a genetically “simple” tumor entity (containing an average of 2.9 mutations per tumor). Besides the common CTNNB1 changes, the authors found additional mutations affecting the telomerase reverse-transcriptase (TERT) promoter and the nuclear factor and erythroid 2-like 2 gene (NFE2L2, also known as NRF2) as well as the presence of chromosomal instability due to deletions of the RAD17 and TP53 genome caretakers.44 In line with these observations, a comparative study of pediatric cancer genomic landscape indicated HB as the least frequently mutated.45 In a successive whole-exome sequencing analysis, additional genes harboring mutations in HB were identified, including cytoplasmic activation/proliferation-associated protein-2 (CAPRIN2), speckle-type POZ protein (SPOP), olfactory receptor-511 (OR5I1), cell division cycle 20B (CDC20B), kelch-like 22 (KLHL22), transient receptor potential cation channel subfamily C member 4 associated protein (TRPC4AP), and ring finger protein 169 (RNF169).46 Furthermore, other genes affected by mutations, amplification, or loss were identified through comprehensive genomic profiling. Among them were ERBB4, MDM4, FBXW7, SRC, and BRCA2; although the alteration frequency for each of these genes was relatively low (~3%), they are targetable candidates whose inhibition might be therapeutically relevant for some HB patients.47
HB shows a relatively low frequency of genomic mutations in canonical oncogenes. However, we cannot exclude that alterations might occur further downstream of these oncogenes. Recently, RNA editing has emerged as a widespread epigenetic mechanism conferring RNA nucleotide changes, without altering DNA sequence, in cancer cells.48 Of note, dysregulation of RNA editing together with hyper-editing of nucleotide 5 of the BLCAP gene occur in approximately 25% of HB patients.29
Collectively, whole-genome studies have unraveled the presence of few mutations in HB and identified some potential driver genes and therapeutic targets for this tumor type.
Wnt/β-Catenin Pathway
The canonical Wnt/β-catenin signaling is a highly evolution-arily conserved cascade, playing a pivotal function in hepatic proliferation and liver homeostasis.49 The key factor of this pathway is the β-catenin protein, which also contributes to epithelial cell–cell adhesion in association with E-cadherin. β-Catenin protein levels and subcellular localization are tightly controlled by a destruction complex, comprising of three scaffold proteins, namely AXIN1, AXIN2, and adenomatous polyposis coli (APC), and two kinases, casein kinase I isoform-α (CK1-α) and glycogen synthase kinase 3β (GSK-3β). In quiescence, this complex phosphorylates β-catenin, allowing its binding to the β-transducin repeat-containing E3 ubiquitin protein ligase (β-TrCP) and consequent proteasomal degradation of β-catenin. Upon activation by various growth stimuli, Wnt ligands bind to cell membrane Frizzled receptors and the coreceptor LRP 5/6 to induce the recruitment of the scaffold protein Disheveled (Dvl). Once activated, Dvl phosphorylates and relocates AXIN1 to the plasma membrane, thus disrupting the destruction complex. These series of events lead to β-catenin release from the destruction complex and its nuclear accumulation. Once in the nucleus, β-catenin promotes the transcription of target genes, such as GLUL, AXIN2, LECT2, involved in cell proliferation, survival, invasion, and migration.50,51 While a balance between the β-catenin destruction complex and Wnt ligands preserves normal tissue homeostasis, perturbations in the Wnt/β-catenin signaling lead to unrestrained growth and are responsible for various diseases, including cancer.49,52
Somatic mutations in CTNNB1 or other key components of the Wnt pathway occur in over 80% of HB samples, representing a major molecular hallmark of this tumor type. Noticeably, HB shows the highest rate of CTNNB1 mutations among the whole spectrum of human cancers.33 CTNNB1 mutations consist of in-frame deletions or missense substitutions at the exon 3 serine/threonine residues or adjacent amino acids affecting β-catenin phosphorylation and degradation. These mutations hinder the possibility of β-catenin proteolysis, resulting in the constitutive induction of β-catenin transcriptional activity and uncontrolled growth.26,53 Furthermore, loss-of-function mutations in the β-catenin destruction complex components have been found in HB. The most frequent mutated genes are APC (10–23.1%), AXIN1(1.7%), and AXIN2 (3.8%).50,54,55 These mutations disrupt the degradation complex, leading to β-catenin stabilization and nuclear translocation, thus mimicking the effects ofCTNNB1 mutations.56 In addition, two distinct gain-of-function mutations (R968-H/S969) in CAPRIN2, an oncogene-promoting β-catenin cytosolic stabilization by LRP5/6 phosphorylation,57 have been detected in HB,46 implying the existence of multiple mechanisms either triggering or reinforcing β-catenin unconstrained activity in HB. Moreover, growth regulation by estrogen in breast cancer 1 (GREB1) has been identified as a novel downstream target gene of the Wnt/β-catenin pathway that is able to promote HB in vitro and in vivo cell growth, by suppressing the transforming growth factor-β (TGF-β) signaling.58 The mechanisms leading to activation of β-catenin are summarized in Fig. 2. Altogether, the aforementioned genomic studies strongly underlined the role of the Wnt/β-catenin cascade as the driving pathway in HB development and prompted researchers to recapitulate the observed findings by overexpressing mutant forms of β-catenin in the mouse liver. Unexpectedly, however, it was found that activating mutations of CTNNB1in hepatocytes alone are not sufficient to induce HB formation in mice.59 This unpredicted discovery suggested the need for cooperation of β-catenin activation with other pathways to initiate HB development. Nonetheless, β-catenin stabilization alone in early fetal progenitor cells was found to induce liver tumor development (consisting of both HCC and HB) in mice, envisaging the possibility that β-catenin oncogenic effects in the liver depend on the cell type targeted by β-catenin mutations.59
Fig. 2.
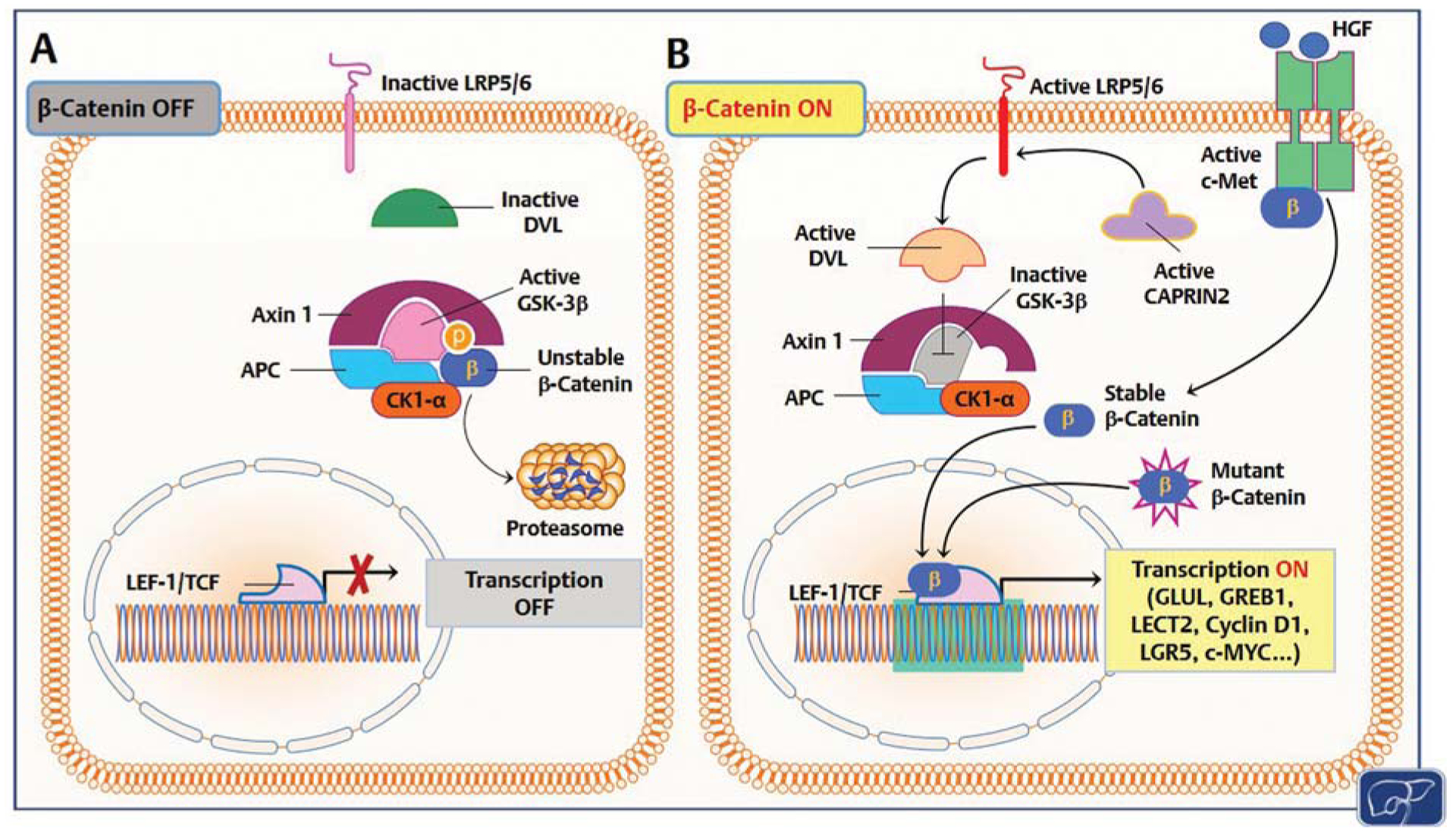
Mechanisms responsible for β-catenin activation in human hepatoblastoma (HB). (A) In quiescent hepatocytes, the β-catenin pathway is turned off (β-catenin OFF). Wild-type β-catenin (unstable β-catenin) is sequestered by a destruction complex consisting of adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK-3β), AXIN1, and casein kinase I α (CK1-α) and primed for proteolysis. (B) In HB, through somatic mutations (mutant β-catenin), β-catenin escapes proteasomal degradation and translocates into the nucleus, where it associates with T cell factor (TCF)/lymphoid enhancer 1 (LEF-1) transcription factors and induces the transcription (transcription ON) of target genes, including glutamine synthetase (GLUL), growth regulation by estrogen in breast cancer 1 (GREB1), leukocyte-cell-derived chemotaxin 2 (LECT2), cyclin D1, leucine-rich repeat containing G protein-coupled receptor 5 (LGR5), c-Myc, etc. Alternatively, β-catenin degradation is suppressed by HB tumor cells via CAPRIN2-activating mutations. Mutant CAPRIN2 activates LDL-receptor-related protein 5 and 6 (LRP5/6) than in turn activates the adaptor protein Disheveled (DVL). Consequently, DVL triggers GSK-3β inactivation and disrupts the β-catenin destruction complex. Furthermore, c-Met receptor activation by hepatocyte growth factor (HGF) induces nuclear translocation of β-catenin via tyrosine phosphorylation. In quiescence, c-Met and β-catenin physically interact at the inner surface of the hepatocyte plasma membrane.
Hippo Pathway
The Hippo cascade is a central regulator of liver tissue growth and organ size.60 The core of the Hippo pathway consists of a kinase cassette composed of mammalian STE20-like kinases 1/2 (MST1/2) and large tumor suppressor 1/2 (LATS1/2), in combination with Salvador homologue (SAV1), MOB kinase activator 1A (MOB1A), and MOB1B adaptor proteins.61,62 YAP and its paralogue, transcriptional coactivator with PDZ-binding motif (TAZ), are the downstream effectors of the Hippo pathway. The activity and subcellular localization of YAP and TAZ are regulated by the Hippo tumor suppressor kinases. When the Hippo pathway is activated, YAP and TAZ are phosphorylated, sequestered in the cytoplasm, and degraded. When the Hippo cascade is impaired, YAP and TAZ translocate to the nucleus and bind to members of the TEAD family of transcription factors to activate target gene expression.63 In the liver, the Hippo pathway restrains tissue growth and controls organ size by limiting YAP/TAZ activity.64 Inhibition of Hippo and the subsequent activation of YAP and TAZ is implicated in liver carcinogenesis.65 In human HB samples, both YAP and TAZ are almost ubiquitously localized in the nucleus and therefore activated.66,67 Moreover, silencing of YAP and/or TAZ inhibits human HB cell growth in culture, with the antigrowth effects being more remarkable when the two oncoproteins are suppressed simultaneously.66,67 The role of YAP and TAZ in human HB has been even more clearly defined in animal studies. Specifically, while overexpression of either YAP or TAZ was unable to promote liver malignant transformation, the combined overexpression of YAP with either constitutive active β-catenin (β-catenin/Δ90) or β-catenin point mutants (S33Y or S45Y) caused HB development in mice.66,68 A subsequent study showed that YAP withdrawal triggers HB regression in YAP/β-catenin mice, identifying YAP as a potential therapeutic target for HB patients.69 Moreover, overexpression of β-catenin combined with TAZ induces HB development in mice, in a TEAD4-dependent manner.66,67,70 Although the molecular mechanisms whereby YAP and TAZ exert their oncogenic properties require additional investigation, our recent data indicate that TAZ but not YAP coexpression with β-catenin in the mouse liver leads to the development of HB lesions displaying epithelial and mesenchymal features. The mesenchymal component of TAZ/β-catenin tumors was found to be induced by the NOTCH pathway, as inhibition of NOTCH transcriptional activity prevented the appearance of the mesenchymal component in TAZ/β-catenin tumors, without affecting tumor incidence and onset.67
Altogether, this body of evidence implies the Hippo/YAP/TAZ cascade as a major pathway cooperating with activated Wnt/β-catenin in HB induction.
MYC Pathway
Myc is a family of transcription factors acting as oncogenes virtually in all tumor types.71 The Myc family consists of three related human genes, namely c-Myc (also referred to as MYC), l-Myc (MYCL), and n-Myc (MYCN).71 MYC genes function downstream of multiple signaling pathways, such as Wnt, receptor tyrosine kinases (RTK), and TGF-β.71,72 Among the three MYC genes, c-MYC is the most investigated in physiology and tumorigenesis. Once activated, c-MYC dimerizes with its binding partner MAX to target genes involved in cell growth, metabolism, apoptosis, and carcinogenesis.73 As we discussed previously, perturbation of the Wnt/β-catenin signaling pathways is a common oncogenic event in HB. As c-MYC is a well-known target of the Wnt/β-catenin cascade, it is not surprising that a strong nuclear immunoreactivity for c-MYC was detected in 83% of human HB biopsies.74 Furthermore, an MYC activation signature and the upregulation of both c-MYC and n-MYC genes were detected in the C2 subgroup of human HB defined by Cairo et al.26 More recently, c-Myc overexpression was observed in activated β-catenin and YAP-induced HB in mice. Further studies have shown that c-Myc is necessary to maintain rapid HB tumor growth in vivo, and c-Myc-dependent and independent pathways along HB development have been identified.75 Moreover, the importance of Myc proteins in HB was further underlined by the finding that treatment with bromodomain and extraterminal motif (BET) and Aurora inhibitors targeting Myc hinders the growth of HB cells in vitro.76
Hedgehog Pathway
The Hedgehog (Hh) pathway is a crucial cascade for the growth and differentiation of many tissues along embryonic development. Vertebrates possess three Hh proteins, namely, Sonic Hh (Shh), Indian Hh (Ihh), and Desert Hh (Dhh), all binding to the Patched 1 (Ptch1) receptor. In the absence of ligand stimulation, Ptch1 inactivates the Hh signaling by repressing the transmembrane protein Smoothened (Smo). This negative modulation can be relieved by ligand stimulation; consequently, Smo induces the Hh pathway by transcriptional activation of the downstream effectors, such as glioma-associated oncogene homologs (Gli)1, Gli2, and Gli3.77,78 Several studies have investigated and proven the relevance of the Hh pathway in human HB. For instance, it has been shown that Hh signaling is activated in this tumor type, with Shh, GLI1 and PTCH1 levels being significantly increased in HB samples when compared with normal liver tissues.79,80 Noticeably, the Hedgehog interacting protein (HHIP) gene, a tumor suppressor blocking the Hh cascade, is transcriptionally silenced by promoter hypermethylation in a HB subset. In addition, suppression of the Hh signaling using the antagonist cyclopamine strongly impaired HB cell growth in vitro by inducing apoptosis.79 Massive cell death in the Huh6 HB cell line was also triggered by the administration of another Hh pathway inhibitor, Forskolin.81 Furthermore, it has been recently demonstrated that Smo and Gli1 expressions are significantly associated with poor prognosis by univariate and multivariate analyses in HB.82 Therefore, targeting Hh might be a potentially relevant approach for the treatment of human HB.
NFE2L2/KEAP1 Pathway
Based on exome sequencing studies, besides CTNNB1, NFE2L2 was identified as a recurrent mutated gene, as it was found altered in approximately 10% of human HB cases.44 Missense mutations in the NFE2L2 gene prevent the interaction between NFE2L2 protein and the KEAP1/CUL3 degradation complex, thus hampering NFE2L2 proteolysis and allowing NFE2L2 transcriptional activity. Once activated, NFE2L2 induces a plethora of genes involved in stress response, inflammation, and survival.83 Notably, HB tumors with high expression of NFE2L2 are characterized by HCC-like features and clinical aggressiveness, suggesting that NFE2L2 activation might be of prognostic significance for HB patients.44 Also, silencing of NFE2L2 in HEP293TT human HB cells (harboring NFE2L2 gene amplification) significantly decreased HB cell growth via inducing G2/M arrest,84 further underscoring the potential relevance of NFE2L2 as a therapeutic target in HB.
HGF/c-Met Pathway
c-Met is the tyrosine kinase receptor for hepatocyte growth factor (HGF). Following ligand binding, HGF undergoes activation via autophosphorylation and in turn triggers the activation of downstream pathways involved in cellular proliferation, survival, angiogenesis, morphogenesis, and epithelial–mesenchymal transition.85 Elevated serum levels of HGF have been reported in children following resection of HB and might be responsible for the growth of the remaining tumor cells.86 In addition, it has been shown that HGF promotes the survival of HB cells to antiapoptotic stimuli induced by chemotherapeutic drugs through the activation of the PI3K/AKT cascade.87 Furthermore, elevated levels of activated/phosphorylated c-Met were found to be positively associated with nuclear accumulation of β-catenin in human HB specimens (Fig. 2),88 consistent with the notion that the HGF/c-Met pathway can activate the β-catenin signaling cascade in a Wnt-independent manner.89 The positive correlation between activated c-Met and nuclear β-catenin might explain the observed activation of β-catenin in some HB subsets that display a wild-type CTNNB1 gene.88
PI3K/AKT/mTOR Pathway
The phosphatidylinositol-3-kinase/Akt/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling regulates various cellular functions, including proliferation, survival, motility, and metabolism. Once activated, the PI3K/AKT/mTOR pathway induces the activation of many intracellular proteins mainly via phosphorylation. In particular, the mTOR protein is part of two distinct complexes: the mTORC1 and mTORC2, with mTORC1 regulating protein synthesis, lipid and glucose metabolism, and mTORC2 being involved in carcinogenesis by its ability to activate AGC serine/threonine kinases, such as AKT and SGK proteins.90,91 In human HB, gain-of-function mutations of the p110α subunit of PI3K(PI3KCA) were detected in 2% of HB cases. In addition, the authors revealed that PI3K/AKT/mTOR axis activation is required for HB survival.92 More recent data show that YAP induces the expression of the amino acid transporter SLC38A1 resulting in mTOR activation in human HB cell lines and in the YAP/β-catenin mouse model.93 Furthermore, β-catenin was found to induce the activation of mTORC1 through the upregulation of glutamine synthase.94 Thus, the mTORC1 pathway might be the converging point of YAP and β-catenin pathways in HB and could represent a valid target for HB treatment.
SP/NK-1R Pathway
In the neurokinin cascade pathway, the substance P (SP) binds to the neurokinin-1 receptor (NK-1R) and triggers numerous downstream events, such as cell proliferation, survival, migration, and neoangiogenesis.95 Similar to other cancer types, HB samples and cell lines overexpress the truncated form of NK-1R and SP.96 In vitro activation of the neurokinin pathway induces HB cell proliferation, which can be severely impaired with the use of NK-1R antagonists, such as Aprepitant. At the molecular level, the NK-1R/SP cascade functionally interacts with several oncogenic pathways in HB. Specifically, suppression of NK-1R hampers β-catenin nuclear accumulation and inhibits the forkhead box M1 (FOXM1) proto-oncogene, which is crucial for β-catenin translocation to the nucleus.97
IGF Pathway
The insulin-like growth factor (IGF) signaling is also implicated in HB development. In this disease, the IGF signaling cascade is induced by the overexpression of the IGF2 ligand, which confers survival advantages to HB cells.98 While the expression of IGF2 is normally restricted to the paternal allele since the maternal copy is epigenetically silenced (an event referred to as genomic imprinting), loss of imprinting for IGF2 was detected in HB, leading to biallelic expression and IGF2 overexpression.99,100 Furthermore, levels of IGF-binding proteins 2 and 3 (IGFBP2 and IGFBP3), which inhibit this pathway by physical interaction with IGF ligands, were found to be down-regulated in HB when compared with nontumorous surrounding livers and associated with lower differentiation, vascular invasion, and distant metastasis.101,102 At the molecular level, it was found that promoter hypermethylation is responsible for the observed low levels of IGFBP3 in HB.101
Other Important Players in Hepatoblastoma Development
Polo-like kinase 1 (PLK1) is a serine/threonine protein kinase acting as a critical regulator of cell cycle progression (G2-M transition), mitosis, cytokinesis, and DNA damage response, and whose levels are often upregulated in various cancer types.103 By expression profiling, it was found that PLK1 was the only oncogene whose levels were strongly upregulated in HB tissue when compared with the nontumorous counterpart. Moreover, high levels of PLK1 were significantly associated with unfavorable outcome of HB patients.101 Subsequent studies revealed that the selective PLK1 inhibitor Volasertib effectively restrains the growth of HB cells. More importantly, when Volasertib and the relapse-related HB chemotherapeutic irinotecan were simultaneously administered, a strong antigrowth effect was observed both in vitro and in vivo. These intriguing findings deserve further preclinical exploration and a deeper investigation for a clinical trial concept.104
Enhancer of Zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase participating in histone methylation and transcriptional repression.105,106 Perturbations of EZH2 levels have been described in many forms of cancer. Through its histone methylation properties, EZH2 inhibits genes responsible for tumor suppression. Noticeably, blockade of EZH2 activity significantly slowed tumor growth in many experimental models.105,106 In human HB, EZH2 levels are higher in tumors when compared with the corresponding nontumorous livers; in addition, a more prominent nuclear immunoreactivity of EZH2 is associated with the presence of distant metastases in the primary tumor, thus implying the involvement of EZH2 both in tumor development and clinical aggressiveness.107,108 Furthermore, suppression of EZH2 inhibits cell proliferation, induces cell cycle arrest, and enhances the expression of the G1/S-phase checkpoint inhibitor p27.109 Therefore, EZH2 might represent a potential diagnostic marker and a therapeutic target for HB.
Gankyrin, a component of the 26S proteasome, regulates cellular growth, proliferation, invasion, and metastasis, and is prominently overexpressed in most human cancers.110,111 In HB, Gankyrin is robustly overexpressed and involved in carcinogenesis by promoting the degradation of tumor suppressor proteins such as the retinoblastoma protein (pRb), TP53, CCAAT enhancer binding protein α (C/EBPα), and hepatocyte nuclear factor α (HNF4α).112 Furthermore, it has been shown in experimental models that dephosphorylation of the tumor suppressor protein C/EBPα results in its shift into an oncogene, and the development of preneoplastic foci giving rise to aggressive forms of HB.113
Dipeptidase 1 (DPEP1) is a zinc-dependent metalloproteinase that regulates glutathione metabolism.114 DPEP1 promotes HB cell proliferation, migration, and invasion by activating the PI3K/Akt/mTOR pathway. Upregulation of DPEP1 was found in human HBs and was associated with poor prognosis.115
Many additional pathways were found to be involved in HB tumorigenesis, growth, and progression, such as NOTCH,116 Jak/Stat,117 Ras,118 and TGF-β.119 Further studies are needed to elucidate the specific function(s) of these cascades in human HB.
Future Perspectives
HB is the predominant hepatic neoplasm of the childood.1–5,120 While most patients can be effectively cured from HB, relapsed and metastatic HB remains an unmet clinical need. Furthermore, long-term side effects (ototoxicity, infertility, second malignancies, cardiomyopathy) and the eventual need of liver transplantation still represent major issues in about two-thirds of HB survivors. Therefore, novel and less aggressive therapeutic modalities should be developed to increase the number of HB survivors as well as to improve their quality of life after successful therapy. To achieve this goal, a better understanding of the molecular mechanisms responsible for HB development and progression is imperative. Over the last years, high-throughput approaches have unraveled the major genomic, epigenetic, and transcriptomic features of HB. A growing number of experimental in vitro and in vivo models have been also developed to investigate the molecular pathogenesis of this aggressive disease. In particular, it has been discovered that HB is a tumor type characterized by a limited number of genomic alterations, with only CTNNB1 and NFE2L2 genes being frequently mutated. Perturbation of several signaling cascades has been shown to be implicated in HB, but their specific contribution to HB tumorigenesis remains poorly defined. Indeed, the functional crosstalk of the identified pathways as well as the compensatory molecular circuits at play once one signaling cascade is inhibited should bebetter elucidated. Furthermore, the translation of some of the preclinical findings into clinical practice is hampered by the fact that many of the putative HB driver genes, such as CTNNB1, c-MYC, and YAP are considered to be “undruggable.” Alternative strategies aimed at suppressing the oncogenic activity of these genes should be developed. In this regard, encouraging preclinical data support the effectiveness of BET protein inhibitors and of Tankyrase inhibitors for the blockade of c-MYC and β-catenin/YAP pathways in cancer, respectively.121–123 Further studies are necessary to validate their usefulness, either alone or in combination, in the HB context. Nonetheless, several targetable proteins have been also identified in HB, including c-Met, Smo/Hh, NK-1R, PI3K/AKT/mTOR, IGF-1R, EZH2, and PLK1, for which selective inhibitors exist (Fig. 3). Currently, of 57 ongoing clinical trials in recurrent/refractory HB, 21 are based on molecular targeted therapies (https://www.clinicaltrials.gov/; Table 2).124–149 Some of these clinical trials are part of the NCI-COG Pediatric MATCH (Molecular Analysis for Therapy Choice), a pediatric precision medicine cancer treatment trial exploring whether targeted therapies can be effective for children and adolescents with solid tumors harboring specific gene mutations (https://www.cancer.gov/about-cancer/treatment/clinical-trials/nci-supported/pediatric-match). The results from these clinical studies, together with the mechanistic information coming from experimental models, will be highly helpful for a deeper comprehension of the molecular pathogenesis of this disease as well as for tailoring innovative therapeutic approaches against HB.
Fig. 3.
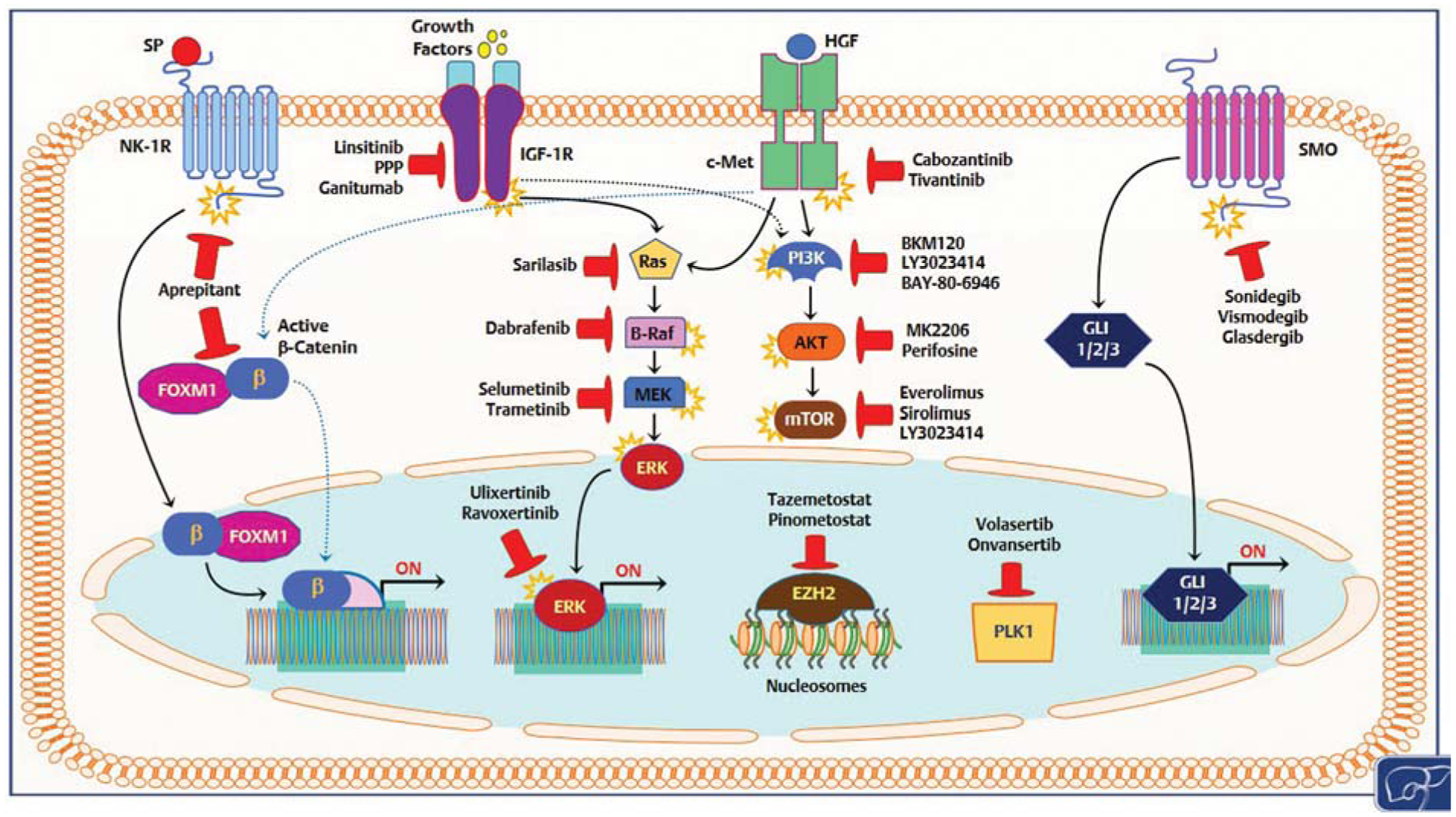
Overview of various signaling pathways deregulated in human hepatoblastoma that might be therapeutically targeted. Only drugs either approved by the Food and Drug Administration or currently in clinical trials are shown. Details are reported in the main text. Stars attached to proteins indicate protein activation; blunted red arrows indicate inhibition.
Table 2.
Ongoing clinical trials using targeted therapies in hepatoblastoma
| ClinicalTrials.gov identifier | Drug name | HB participants (enrollment) | Phase | Study status | Targets | Relative signaling pathway(s) |
|---|---|---|---|---|---|---|
| NCT01125800 | Sonidegib | 1 (76) | Ph I/II | Completed | Smoothened | Hedgehog pathway119 |
| NCT02867592 | Cabozantinib S-malate | ? (146) | Ph II | Recruiting | RET, MET, VEGFR-1, -2 and -3, KIT, TRKB, FLT-3, AXL, and TIE-2 | VEGFR2, c-MET pathways, etc.120,121 |
| NCT04195555 | Ivosidenib | ? (49) | Ph II | Not yet recruiting | IDH1 | IDH pathway122 |
| NCT04308330 | Vorinostat | ? (30) | Ph I | Recruiting | HDAC | VEGF, ATM, androgen, and estrogen pathways123–125 |
| NCT04320888 | Selpercatinib | ? (49) | Ph II | Not yet recruiting | RET kinase | RET pathway126 |
| NCT03213652 | Ensartinib | ? (98) | Ph II | Recruiting | ALK | EGFR, KRAS, PIK3CA, ERBB2 pathways127 |
| NCT04284774 | Tipifarnib | ? (49) | Ph II | Not yet recruiting | Farnesyltransferase | RAS and PI3K/Akt pathways128 |
| NCT03526250 | Palbociclib | ? (49) | Ph II | Recruiting | CDK4/6 | CDK-RB-E2F pathway129 |
| NCT03698994 | Ulixertinib | ? (49) | Ph II | Recruiting | ERK2 | MAPK/ERK pathway130 |
| NCT03213704 | Larotrectinib | ? (49) | Ph II | Recruiting | TrkA, TrkB, TrkC | TRK pathway131 |
| NCT03233204 | Olaparib | ? (49) | Ph II | Recruiting | PARP-1/2 | HR, BER/SSBR pathways132 |
| NCT01154816 | Alisertib | 6 (118) | Ph II | Completed | AAK | PI3K/Akt/mTOR pathway133 |
| NCT03220035 | Vemurafenib | ? (49) | Ph II | Recruiting | B-Raf | MAPK/ERK pathway134 |
| NCT01331135 | Sirolimus | ? (24) | Ph I | Active, not recruiting | mTOR | mTOR pathway135 |
| NCT03213678 | Samotolisib | ? (144) | Ph II | Recruiting | PI3K/mTOR | PI3K/Akt/mTOR pathway136 |
| NCT03213665 | Tazemetostat | ? (49) | Ph II | Recruiting | EZH2 | EZH2 histone methyltransferase137 |
| NCT03210714 | Erdafitinib | ? (49) | Ph II | Recruiting | pan-FGFR | FGFR1–4 pathway138 |
| NCT02390843 | Simvastatin | ? (13) | Ph I | Completed | HMG-CoA reductase | IL6/STAT3 pathway139,140 |
| NCT00831844 | Cixutumumab | 10 (116) | Ph II | Completed | IGF1R | IGF and PI3K/Akt/mTOR pathways141 |
| NCT02085148 | Regorafenib | ? (60) | Ph I | Active, not recruiting | VEGFR1-3, c-KIT, TIE-2, PDGFR-β, FGFR-1, RET, RAF-1, BRAF, p38 MAP kinase | VEGF, RAF/MEK/ERK pathways, etc.142,143 |
| NCT03155620 | Samotolisib, | ? (1,500) | Ph II | Recruiting | PI3K/mTOR | PI3K/AKT/mTOR pathway136 |
| Selpercatinib, Selumetinib sulfate, | RET kinase MEK | RET pathway126 MAPK/ERK pathway144 | ||||
| Tazemetostat, | EZH2 | EZH2 histone methyltransferase137 | ||||
| Tipifarnib, | Farnesyltransferase | Ras and PI3K pathways128 | ||||
| Ulixertinib, | ERK2 | MAPK/ERK pathway130 | ||||
| Vemurafenib | B-Raf | MAPK/ERK pathway134 |
Abbreviations: AAK, Aurora A kinase; ALK, anaplastic lymphoma kinase; BER/SSBR, base excision repair/single strand break repair; CDKs, cyclin-dependent kinases; EGFR, endothelial growth factor receptor; ERK, extracellular signal-related kinases; EZH2, enhancer of zeste homolog 2; FGFR, fibroblast growth factor receptor; FLT-3,: FMS-like receptor tyrosine kinase-3; HDAC, histone deacetylases; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; HR, homologous recombination; IDH1, isocitrate dehydrogenase-1; IGF1R, insulin-like growth factor 1 receptor; IL-6, interleukin-6; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; PARP, poly (ADP-ribose) polymerase; PDGFR-β, platelet-derived growth factor receptor β; PI3K, phosphatidylinositol 3-kinase; STAT, signal transducer and activator of transcription; TRK, tropomyosin receptor kinase; TRKB, tropomyosin receptor kinase B; VEGFR, vascular endothelial growth factor receptor.
Main Concepts and Learning Points.
Hepatoblastoma is a highly aggressive liver tumor of the infancy. In most cases, HB can be cured, although the quality of life of the patients can be severely affected afterwards by the toxicity of the treatments.
The genetic and epigenetic landscape of human HB has been unraveled and key signaling pathways involved in HB development and progression have been identified.
Subsequent in vitro and in vivo studies have unraveled the role(s) and the functional crosstalk of these molecular cascades in HB.
Many of these pathways are targetable with specific inhibitors, and their suppression as a novel therapeutic strategy against HB is currently under evaluation in several ongoing clinical trials.
Funding
This work was supported by NIH grant R01CA204586 (to X.C.), P30DK026743 for UCSF Liver Center, and a China Scholarship Council PhD fellowship (to Y.Z.; 201806 050132).
Footnotes
Conflicts of Interest
Dr. Cairo reports patent issued for “Molecular signature of liver tumor grade and use to evaluate prognosis and therapeutic regimen.” The other authors have no conflicts of interest to declare.
References
- 1.Darbari A, Sabin KM, Shapiro CN, Schwarz KB. Epidemiology of primary hepatic malignancies in U.S. children. Hepatology 2003; 38(03):560–566 [DOI] [PubMed] [Google Scholar]
- 2.Feng J, Polychronidis G, Heger U, Frongia G, Mehrabi A, Hoffmann K. Incidence trends and survival prediction of hepatoblastoma in children: a population-based study. Cancer Commun (Lond) 2019;39(01):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Celotti A, D’Amico G, Ceresoli M, et al. Hepatoblastoma of the adult: a systematic review of the literature. Surg Oncol 2016;25(03):339–347 [DOI] [PubMed] [Google Scholar]
- 4.Mussa A, Ferrero GB. Screening hepatoblastoma in Beckwith-Wiedemann syndrome: a complex issue. J Pediatr Hematol Oncol 2015;37(08):627. [DOI] [PubMed] [Google Scholar]
- 5.Trobaugh-Lotrario AD, López-Terrada D, Li P, Feusner JH. Hepatoblastoma in patients with molecularly proven familial adenomatous polyposis: clinical characteristics and rationale for surveillance screening. Pediatr Blood Cancer 2018;65(08):e27103. [DOI] [PubMed] [Google Scholar]
- 6.Clericuzio CL, Martin RA. Diagnostic criteria and tumor screening for individuals with isolated hemihyperplasia. Genet Med 2009;11(03):220–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito E, Sato Y, Kawauchi K, et al. Type 1a glycogen storage disease with hepatoblastoma in siblings. Cancer 1987;59(10):1776–1780 [DOI] [PubMed] [Google Scholar]
- 8.Farmakis SG, Barnes AM, Carey JC, Braddock SR. Solid tumor screening recommendations in trisomy 18. Am J Med Genet A 2019;179(03):455–466 [DOI] [PubMed] [Google Scholar]
- 9.Spector LG, Feusner JH, Ross JA. Hepatoblastoma and low birth weight. Pediatr Blood Cancer 2004;43(06):706. [DOI] [PubMed] [Google Scholar]
- 10.Sharma D, Subbarao G, Saxena R. Hepatoblastoma. Semin Diagn Pathol 2017;34(02):192–200 [DOI] [PubMed] [Google Scholar]
- 11.Qiao GL, Chen Z, Wang C, et al. Pure fetal histology subtype was associated with better prognosis of children with hepatoblastoma: a Chinese population-based study. J Gastroenterol Hepatol 2016;31(03):621–627 [DOI] [PubMed] [Google Scholar]
- 12.López-Terrada D, Alaggio R, de Dávila MT, et al. Towards an international pediatric liver tumor consensus classification: Proceedings of the Los Angeles COG Liver Tumors Symposium. Mod pathology 2014;27(03):472–491 [DOI] [PubMed] [Google Scholar]
- 13.Fazlollahi L, Hsiao SJ, Kochhar M, Mansukhani MM, Yamashiro DJ, Remotti HE. Malignant rhabdoid tumor, an aggressive tumor often misclassified as small cell variant of hepatoblastoma. Cancers (Basel) 2019;11(12):E1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmermann A Hepatoblastoma with cholangioblastic features (‘cholangioblastic hepatoblastoma’) and other liver tumors with bimodal differentiation in young patients. Med Pediatr Oncol 2002;39(05):487–491 [DOI] [PubMed] [Google Scholar]
- 15.Towbin AJ, Meyers RL, Woodley H, et al. 2017 PRETEXT: radiologic staging system for primary hepatic malignancies of childhood revised for the Paediatric Hepatic International Tumour Trial (PHITT). Pediatr Radiol 2018;48(04):536–554 [DOI] [PubMed] [Google Scholar]
- 16.Maibach R, Roebuck D, Brugieres L, et al. Prognostic stratification for children with hepatoblastoma: the SIOPEL experience. Eur J Cancer 2012;48(10):1543–1549 [DOI] [PubMed] [Google Scholar]
- 17.Trobaugh-Lotrario AD, Meyers RL, Feusner JH. Outcomes of patients with relapsed hepatoblastoma enrolled on Children’s Oncology Group (COG) phase I and II studies. J Pediatr Hematol Oncol 2016;38(03):187–190 [DOI] [PubMed] [Google Scholar]
- 18.Fuchs J, Rydzynski J, Von Schweinitz D, et al. ;Study Committee of the Cooperative Pediatric Liver Tumor Study Hb 94 for the German Society for Pediatric Oncology and Hematology. Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer 2002;95(01):172–182 [DOI] [PubMed] [Google Scholar]
- 19.Hiyama E, Hishiki T, Watanabe K, et al. Resectability and tumor response after preoperative chemotherapy in hepatoblastoma treated by the Japanese Study Group for Pediatric Liver Tumor (JPLT)-2 protocol. J Pediatr Surg 2016;51(12):2053–2057 [DOI] [PubMed] [Google Scholar]
- 20.Meyers RL, Maibach R, Hiyama E, et al. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children’s Hepatic Tumors International Collaboration. Lancet Oncol 2017;18(01):122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czauderna P Hepatoblastoma throughout SIOPEL trials - clinical lessons learnt. Front Biosci (Elite Ed) 2012;4:470–479 [DOI] [PubMed] [Google Scholar]
- 22.Malogolowkin MH, Katzenstein HM, Meyers RL, et al. Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology: a report from the Children’s Oncology Group. J Clin Oncol 2011;29(24):3301–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013;45(10):1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorsson V, Gibbs DL, Brown SD, et al. ;Cancer Genome Atlas Research Network. The immune landscape of cancer. Immunity 2019;51(02):411–412 [DOI] [PubMed] [Google Scholar]
- 25.Zanfardino M, Pane K, Mirabelli P, Salvatore M, Franzese M. TCGA-TCIA impact on radiogenomics cancer research: a systematic review. Int J Mol Sci 2019;20(23):E6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cairo S, Armengol C, De Reyniès A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008;14(06):471–484 [DOI] [PubMed] [Google Scholar]
- 27.Cairo S, Wang Y, de Reyniès A, et al. Stem cell-like micro-RNA signature driven by Myc in aggressive liver cancer. Proc Natl Acad Sci U S A 2010;107(47):20471–20476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hooks KB, Audoux J, Fazli H, et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology 2018;68(01):89–102 [DOI] [PubMed] [Google Scholar]
- 29.Carrillo-Reixach J, Torrens L, Simon-Coma M, et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 2020;73(02):328–341 [DOI] [PubMed] [Google Scholar]
- 30.Sumazin P, Chen Y, Treviño LR, et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017;65(01):104–121 [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa H, Fujita M, Fujimoto A. Genome sequencing analysis of liver cancer for precision medicine. Semin Cancer Biol 2019; 55:120–127 [DOI] [PubMed] [Google Scholar]
- 32.Liotta L, Petricoin E. Molecular profiling of human cancer. Nat Rev Genet 2000;1(01):48–56 [DOI] [PubMed] [Google Scholar]
- 33.Mavila N, Thundimadathil J. The emerging roles of cancer stem cells and Wnt/beta-catenin signaling in hepatoblastoma. Cancers (Basel) 2019;11(10):E1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011;25(10):1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crippa S, Ancey PB, Vazquez J, et al. Mutant CTNNB1 and histological heterogeneity define metabolic subtypes of hepatoblastoma. EMBO Mol Med 2017;9(11):1589–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 1999;59(02):269–273 [PubMed] [Google Scholar]
- 37.Jeng YM, Wu MZ, Mao TL, Chang MH, Hsu HC. Somatic mutations of beta-catenin play a crucial role in the tumorigenesis of sporadic hepatoblastoma. Cancer Lett 2000;152(01):45–51 [DOI] [PubMed] [Google Scholar]
- 38.Morcrette G, Hirsch TZ, Badour E, et al. APC germline hepatoblastomas demonstrate cisplatin-induced intratumor tertiary lymphoid structures. OncoImmunology 2019;8(06):e1583547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.López-Terrada D, Gunaratne PH, Adesina AM, et al. Histologic subtypes of hepatoblastoma are characterized by differential canonical Wnt and Notch pathway activation in DLK+ precursors. Hum Pathol 2009;40(06):783–794 [DOI] [PubMed] [Google Scholar]
- 40.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004;303(5663):1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCrea PD, Gottardi CJ. Beyond β-catenin: prospects for a larger catenin network in the nucleus. Nat Rev Mol Cell Biol 2016;17(01):55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Meyfeldt J, Wang H, et al. β-Catenin mutations as determinants of hepatoblastoma phenotypes in mice. J Biol Chem 2019;294(46):17524–17542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell D, Ranganathan S, Tao J, Monga SP. Novel advances in understanding of molecular pathogenesis of hepatoblastoma: a Wnt/β-catenin perspective. Gene Expr 2017;17(02):141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eichenmüller M, Trippel F, Kreuder M, et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 2014;61(06):1312–1320 [DOI] [PubMed] [Google Scholar]
- 45.Gröbner SN, Worst BC, Weischenfeldt J, et al. ;ICGC PedBrain-Seq Project ICGC MMML-Seq Project. The landscape of genomic alterations across childhood cancers. Nature 2018;555 (7696):321–327 [DOI] [PubMed] [Google Scholar]
- 46.Jia D, Dong R, Jing Y, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 2014;60(05):1686–1696 [DOI] [PubMed] [Google Scholar]
- 47.Lee H, El Jabbour T, Ainechi S, et al. General paucity of genomic alteration and low tumor mutation burden in refractory and metastatic hepatoblastoma: comprehensive genomic profiling study. Hum Pathol 2017;70:84–91 [DOI] [PubMed] [Google Scholar]
- 48.Paz-Yaacov N, Bazak L, Buchumenski I, et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep 2015;13(02):267–276 [DOI] [PubMed] [Google Scholar]
- 49.Wang W, Smits R, Hao H, He C. Wnt/β-catenin signaling in liver cancers. Cancers (Basel) 2019;11(07):E926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perugorria MJ, Olaizola P, Labiano I, et al. Wnt-β-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol 2019;16(02):121–136 [DOI] [PubMed] [Google Scholar]
- 51.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009;17(01):9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shang S, Hua F, Hu ZW. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget 2017; 8(20):33972–33989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monga SP. β-Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015;148(07):1294–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang A, Sisson R, Gupta A, Tiao G, Geller JI. Germline APC mutations in hepatoblastoma. Pediatr Blood Cancer 2018;65(04): [DOI] [PubMed] [Google Scholar]
- 55.Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45(D1):D777–D783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017;169(06):985–999 [DOI] [PubMed] [Google Scholar]
- 57.Ding Y, Xi Y, Chen T, et al. Caprin-2 enhances canonical Wnt signaling through regulating LRP5/6 phosphorylation. J Cell Biol 2008;182(05):865–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsumoto S, Yamamichi T, Shinzawa K, et al. GREB1 induced by Wnt signaling promotes development of hepatoblastoma by suppressing TGFβ signaling. Nat Commun 2019;10(01):3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nejak-Bowen KN, Monga SP. Beta-catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol 2011;21(01):44–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maugeri-Saccà M, De Maria R. The Hippo pathway in normal development and cancer. Pharmacol Ther 2018;186:60–72 [DOI] [PubMed] [Google Scholar]
- 61.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 2015;15(02):73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu C, Li L, Zhao B. The regulation and function of YAP transcription co-activator. Acta Biochim Biophys Sin (Shanghai) 2015;47(01):16–28 [DOI] [PubMed] [Google Scholar]
- 63.Lin KC, Park HW, Guan KL. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem Sci 2017;42(11):862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005;122(03):421–434 [DOI] [PubMed] [Google Scholar]
- 65.Li H, Wolfe A, Septer S, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int 2012;32(01):38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao J, Calvisi DF, Ranganathan S, et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcino-genesis in mice. Gastroenterology 2014;147(03):690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang S, Zhang J, Evert K, et al. The Hippo effector transcriptional coactivator with PDZ-binding motif cooperates with oncogenic β-catenin to induce hepatoblastoma development in mice and humans. Am J Pathol 2020;190(07):1397–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Min Q, Molina L, Li J, et al. β-Catenin and Yes-associated protein 1 cooperate in hepatoblastoma pathogenesis. Am J Pathol 2019; 189(05):1091–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith JL, Rodríguez TC, Mou H, et al. YAP1 withdrawal in hepatoblastoma drives therapeutic differentiation of tumor cells to functional hepatocyte-like cells. Hepatology 2020. Doi: 10.1002/hep.31389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Liu P, Tao J, et al. TEA domain transcription factor 4 is the major mediator of Yes-associated protein oncogenic activity in mouse and human hepatoblastoma. Am J Pathol 2019;189(05):1077–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dang CV. MYC on the path to cancer. Cell 2012;149(01):22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levens D You don’t muck with MYC. Genes Cancer 2010;1(06):547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 1999;19(01):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ranganathan S, Tan X, Monga SP. Beta-catenin and met deregulation in childhood hepatoblastomas. Pediatr Dev Pathol 2005;8(04):435–447 [DOI] [PubMed] [Google Scholar]
- 75.Wang H, Lu J, Edmunds LR, et al. Coordinated activities of multiple Myc-dependent and Myc-independent biosynthetic pathways in hepatoblastoma. J Biol Chem 2016;291(51):26241–26251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eberherr C, Beck A, Vokuhl C, et al. Targeting excessive MYCN expression using MLN8237 and JQ1 impairs the growth of hepatoblastoma cells. Int J Oncol 2019;54(05):1853–1863 [DOI] [PubMed] [Google Scholar]
- 77.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001;15(23):3059–3087 [DOI] [PubMed] [Google Scholar]
- 78.Ruiz i Altaba A, Sánchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2002;2(05):361–372 [DOI] [PubMed] [Google Scholar]
- 79.Eichenmüller M, Gruner I, Hagl B, et al. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009;49(02):482–490 [DOI] [PubMed] [Google Scholar]
- 80.Oue T, Yoneda A, Uehara S, Yamanaka H, Fukuzawa M. Increased expression of the hedgehog signaling pathway in pediatric solid malignancies. J Pediatr Surg 2010;45(02):387–392 [DOI] [PubMed] [Google Scholar]
- 81.Yamanaka H, Oue T, Uehara S, Fukuzawa M. Forskolin, a hedgehog signal inhibitor, inhibits cell proliferation and induces apoptosis in pediatric tumor cell lines. Mol Med Rep 2010;3(01):133–139 [DOI] [PubMed] [Google Scholar]
- 82.Li YC, Deng YH, Guo ZH, et al. Prognostic value of hedgehog signal component expressions in hepatoblastoma patients. Eur J Med Res 2010;15(11):468–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med 2016;22(07):578–593 [DOI] [PubMed] [Google Scholar]
- 84.Comerford SA, Hinnant EA, Chen Y, et al. Hepatoblastoma modeling in mice places Nrf2 within a cancer field established by mutant β-catenin. JCI Insight 2016;1(16):e88549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res 2006;12(12):3657–3660 [DOI] [PubMed] [Google Scholar]
- 86.von Schweinitz D, Faundez A, Teichmann B, et al. Hepatocyte growth-factor-scatter factor can stimulate post-operative tumor-cell proliferation in childhood hepatoblastoma. Int J Cancer 2000;85(02):151–159 [DOI] [PubMed] [Google Scholar]
- 87.Grotegut S, Kappler R, Tarimoradi S, Lehembre F, Christofori G, Von Schweinitz D. Hepatocyte growth factor protects hepatoblastoma cells from chemotherapy-induced apoptosis by AKT activation. Int J Oncol 2010;36(05):1261–1267 [DOI] [PubMed] [Google Scholar]
- 88.Purcell R, Childs M, Maibach R, et al. HGF/c-Met related activation of β-catenin in hepatoblastoma. J Exp Clin Cancer Res 2011; 30:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Monga SP, Mars WM, Pediaditakis P, et al. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res 2002;62(07):2064–2071 [PubMed] [Google Scholar]
- 90.Vogt PK, Hart JR, Gymnopoulos M, et al. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol 2010; 347:79–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 2013;497(7448):217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hartmann W, Küchler J, Koch A, et al. Activation of phosphatidylinositol-3′-kinase/AKT signaling is essential in hepatoblastoma survival. Clin Cancer Res 2009;15(14):4538–4545 [DOI] [PubMed] [Google Scholar]
- 93.Liu P, Calvisi DF, Kiss A, et al. Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget 2017;8(43):73433–73447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Adebayo Michael AO, Ko S, Tao J, et al. Inhibiting glutamine-dependent mTORC1 activation ameliorates liver cancers driven by β-catenin mutations. Cell Metab 2019;29(05):1135.e6–1150 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muñoz M, Rosso M, Coveñas R. Neurokinin-1 receptor antagonists against hepatoblastoma. Cancers (Basel) 2019;11(09):E1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Berger M, Neth O, Ilmer M, et al. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J Hepatol 2014;60(05):985–994 [DOI] [PubMed] [Google Scholar]
- 97.Garnier A, Ilmer M, Kappler R, Berger M. Therapeutic innovations for targeting hepatoblastoma. Anticancer Res 2016;36(11):5577–5592 [DOI] [PubMed] [Google Scholar]
- 98.Gray SG, Eriksson T, Ekström C, et al. Altered expression of members of the IGF-axis in hepatoblastomas. Br J Cancer 2000;82(09):1561–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rainier S, Dobry CJ, Feinberg AP. Loss of imprinting in hepatoblastoma. Cancer Res 1995;55(09):1836–1838 [PubMed] [Google Scholar]
- 100.Honda S, Arai Y, Haruta M, et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer 2008;99(11):1891–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Regel I, Eichenmüller M, Joppien S, et al. IGFBP3 impedes aggressive growth of pediatric liver cancer and is epigenetically silenced in vascular invasive and metastatic tumors. Mol Cancer 2012;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Akmal SN, Yun K, MacLay J, Higami Y, Ikeda T. Insulin-like growth factor 2 and insulin-like growth factor binding protein 2 expression in hepatoblastoma. Hum Pathol 1995;26(08):846–851 [DOI] [PubMed] [Google Scholar]
- 103.Liu Z, Sun Q, Wang X. PLK1, a potential target for cancer therapy. Transl Oncol 2017;10(01):22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kats D, Ricker CA, Berlow NE, et al. Volasertib preclinical activity in high-risk hepatoblastoma. Oncotarget 2019;10(60):6403–6417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Viré E, Brenner C, Deplus R, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006;439 (7078):871–874 [DOI] [PubMed] [Google Scholar]
- 106.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med 2016;22(02):128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hajósi-Kalcakosz S, Dezső K, Bugyik E, et al. Enhancer of zeste homologue 2 (EZH2) is a reliable immunohistochemical marker to differentiate malignant and benign hepatic tumors. Diagn Pathol 2012;7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schlachter K, Gyugos M, Halász J, et al. High tricellulin expression is associated with better survival in human hepatoblastoma. Histopathology 2014;65(05):631–641 [DOI] [PubMed] [Google Scholar]
- 109.Wang Y, Xiao Y, Chen K, et al. Enhancer of zeste homolog 2 depletion arrests the proliferation of hepatoblastoma cells. Mol Med Rep 2016;13(03):2724–2728 [DOI] [PubMed] [Google Scholar]
- 110.Lozano G, Zambetti GP. Gankyrin: an intriguing name for a novel regulator of p53 and RB. Cancer Cell 2005;8(01):3–4 [DOI] [PubMed] [Google Scholar]
- 111.Higashitsuji H, Itoh K, Nagao T, et al. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat Med 2000;6(01):96–99 [DOI] [PubMed] [Google Scholar]
- 112.Valanejad L, Lewis K, Wright M, et al. FXR-Gankyrin axis is involved in development of pediatric liver cancer. Carcinogenesis 2017;38(07):738–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cast A, Valanejad L, Wright M, et al. C/EBPα-dependent preneoplastic tumor foci are the origin of hepatocellular carcinoma and aggressive pediatric liver cancer. Hepatology 2018;67(05):1857–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Toiyama Y, Inoue Y, Yasuda H, et al. DPEP1, expressed in the early stages of colon carcinogenesis, affects cancer cell invasiveness. J Gastroenterol 2011;46(02):153–163 [DOI] [PubMed] [Google Scholar]
- 115.Cui X, Liu X, Han Q, et al. DPEP1 is a direct target of miR-193a-5p and promotes hepatoblastoma progression by PI3K/Akt/mTOR pathway. Cell Death Dis 2019;10(10):701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gil-García B, Baladrón V. The complex role of NOTCH receptors and their ligands in the development of hepatoblastoma, cholangiocarcinoma and hepatocellular carcinoma. Biol Cell 2016; 108(02):29–40 [DOI] [PubMed] [Google Scholar]
- 117.Nagai H, Naka T, Terada Y, et al. Hypermethylation associated with inactivation of the SOCS-1 gene, a JAK/STAT inhibitor, in human hepatoblastomas. J Hum Genet 2003;48(02):65–69 [DOI] [PubMed] [Google Scholar]
- 118.Honda S, Miyagi H, Suzuki H, et al. RASSF1A methylation indicates a poor prognosis in hepatoblastoma patients. Pediatr Surg Int 2013;29(11):1147–1152 [DOI] [PubMed] [Google Scholar]
- 119.Gray SG, Hartmann W, Eriksson T, et al. Expression of genes involved with cell cycle control, cell growth and chromatin modification are altered in hepatoblastomas. Int J Mol Med 2000;6(02):161–169 [PubMed] [Google Scholar]
- 120.Trobaugh-Lotrario AD, Venkatramani R, Feusner JH. Hepatoblastoma in children with Beckwith-Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol 2014;36(05):369–373 [DOI] [PubMed] [Google Scholar]
- 121.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146(06):904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ma L, Wang X, Jia T, Wei W, Chua MS, So S. Tankyrase inhibitors attenuate WNT/β-catenin signaling and inhibit growth of hepatocellular carcinoma cells. Oncotarget 2015;6(28):25390–25401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jia J, Qiao Y, Pilo MG, et al. Tankyrase inhibitors suppress hepatocellular carcinoma cell growth via modulating the Hippo cascade. PLoS One 2017;12(09):e0184068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kieran MW, Chisholm J, Casanova M, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-oncol 2017;19(11):1542–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grüllich C Cabozantinib: multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res 2018;211:67–75 [DOI] [PubMed] [Google Scholar]
- 126.Osanto S, van der Hulle T. Cabozantinib in the treatment of advanced renal cell carcinoma in adults following prior vascular endothelial growth factor targeted therapy: clinical trial evidence and experience. Ther Adv Urol 2018;10(03):109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dhillon S Ivosidenib: first global approval. Drugs 2018;78(14):1509–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett 2009;280(02):201–210 [DOI] [PubMed] [Google Scholar]
- 129.Deroanne CF, Bonjean K, Servotte S, et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002;21(03):427–436 [DOI] [PubMed] [Google Scholar]
- 130.Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem 1999; 274(44):31127–31130 [DOI] [PubMed] [Google Scholar]
- 131.Li AY, McCusker MG, Russo A, et al. RET fusions in solid tumors. Cancer Treat Rev 2019;81:101911. [DOI] [PubMed] [Google Scholar]
- 132.Yang Y, Zhou J, Zhou J, et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell lung cancer: a multicentre, phase 2 trial. Lancet Respir Med 2020;8(01):45–53 [DOI] [PubMed] [Google Scholar]
- 133.Mesa RA. Tipifarnib: farnesyl transferase inhibition at a crossroads. Expert Rev Anticancer Ther 2006;6(03):313–319 [DOI] [PubMed] [Google Scholar]
- 134.Liu M, Liu H, Chen J. Mechanisms of the CDK4/6 inhibitor palbociclib (PD 0332991) and its future application in cancer treatment (Review). Oncol Rep 2018;39(03):901–911 [DOI] [PubMed] [Google Scholar]
- 135.Roskoski R Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol Res 2019;142:151–168 [DOI] [PubMed] [Google Scholar]
- 136.Scott LJ. Larotrectinib: first global approval. Drugs 2019;79(02):201–206 [DOI] [PubMed] [Google Scholar]
- 137.Chen Y, Du H. The promising PARP inhibitors in ovarian cancer therapy: from Olaparib to others. Biomed Pharmacother 2018; 99:552–560 [DOI] [PubMed] [Google Scholar]
- 138.Niu NK, Wang ZL, Pan ST, et al. Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther 2015;9:1555–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garbe C, Eigentler TK. Vemurafenib. Recent Results Cancer Res 2018;211:77–89 [DOI] [PubMed] [Google Scholar]
- 140.Duvoux C, Toso C. mTOR inhibitor therapy: does it prevent HCC recurrence after liver transplantation? Transplant Rev (Orlando) 2015;29(03):168–174 [DOI] [PubMed] [Google Scholar]
- 141.Zheng L, Li H, Mo Y, Qi G, Liu B, Zhao J. Autophagy inhibition sensitizes LY3023414-induced anti-glioma cell activity in vitro and in vivo. Oncotarget 2017;8(58):98964–98973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Makita S, Tobinai K. Targeting EZH2 with tazemetostat. Lancet Oncol 2018;19(05):586–587 [DOI] [PubMed] [Google Scholar]
- 143.Markham A Erdafitinib: first global approval. Drugs 2019;79(09):1017–1021 [DOI] [PubMed] [Google Scholar]
- 144.Hattinger CM, Patrizio MP, Magagnoli F, Luppi S, Serra M. An update on emerging drugs in osteosarcoma: towards tailored therapies? Expert Opin Emerg Drugs 2019;24(03):153–171 [DOI] [PubMed] [Google Scholar]
- 145.Al-Rasheed NM, Al-Oteibi MM, Al-Manee RZ, et al. Simvastatin prevents isoproterenol-induced cardiac hypertrophy through modulation of the JAK/STAT pathway. Drug Des Devel Ther 2015;9:3217–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McKian KP, Haluska P. Cixutumumab. Expert Opin Investig Drugs 2009;18(07):1025–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ettrich TJ, Seufferlein T. Regorafenib. Recent Results Cancer Res 2018;211:45–56 [DOI] [PubMed] [Google Scholar]
- 148.Strumberg D, Schultheis B. Regorafenib for cancer. Expert Opin Investig Drugs 2012;21(06):879–889 [DOI] [PubMed] [Google Scholar]
- 149.Metro G, Chiari R, Baldi A, De Angelis V, Minotti V, Crinò L Selumetinib: a promising pharmacologic approach for KRAS-mutant advanced non-small-cell lung cancer. Future Oncol 2013; 9(02):167–177 [DOI] [PubMed] [Google Scholar]