

Abstract
Maintenance of skeletal muscle mass and function is an incredibly nuanced balance of anabolism and catabolism that can become distorted within different pathological conditions. In this paper we intend to discuss the distinct intracellular signaling events that regulate muscle protein atrophy for a given clinical occurrence. Aside from the common outcome of muscle deterioration, several conditions have at least one or more distinct mechanisms that creates unique intracellular environments that facilitate muscle loss. The subtle individuality to each of these given pathologies can provide both researchers and clinicians with specific targets of interest to further identify and increase the efficacy of medical treatments and interventions.
Keywords: Muscle Protein Degradation, Sarcopenia, Muscle Loss, Atrophy, Cachexia, Catabolism
Graphical Abstract
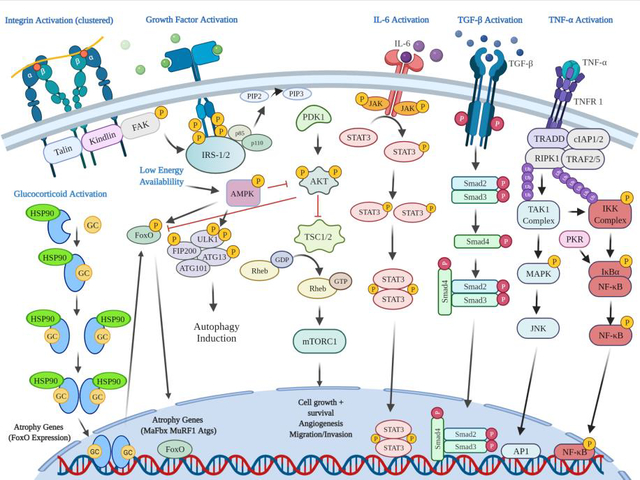
Above shows varying signaling pathways that regulate both skeletal muscle anabolism and catabolism. Focal Adhesion Kinase (FAK) is shown activate mammalian target of rapamycin (mTOR) in response to mechanical load through the phosphatidylinositol 3 kinase (PI3K), protein kinase B (AKT) pathway. Insulin or insulin-like growth factor (IGF) stimulation can activate mTOR through the same cascade. Also shown are the catabolic pathways related to inflammatory cytokines such as tumor necrosis factor-α (TNF-α), and interlukin-6 (IL-6). TNF-α receptor 1 (TNFR1) ligand binding activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and c-Jun N-terminal kinase (JNK) pathway increasing rates of proteolysis. Protein Kinase R (PKR) can also stimulate the activation of NF-kB. IL-6 receptor binding activates the Janus Kinase (JAK) Signal Transducer and Activator of Transcription (STAT) signaling that can increase rates of muscle loss. Transforming Growth Factor-beta (TGF- β) receptor binding facilitates small mothers against decapentaplegic (SMAD) homologs 2 and 3 (SMAD2/3) phosphorylation, and subsequent SMAD 4 phosphorylation, upregulating genes related to collagen synthesis and fibrosis. Additionally, glucocorticoid (GC) receptor activation, dimerization, and DNA binding subsequently facilitates increases in FOXO expression which can increase the expression of UPS related E3 ligases. During low energy availability, increased 5’ adenosine monophosphate-activated protein kinase (AMPK)- Unc-51 like autophagy activating kinase 1 (ULK1) phosphorylation induces the activation of autophagy related processes.
Introduction
Skeletal muscle is a dynamic tissue that is remodeled through a balance of anabolic and catabolic processes that are governed by a variety of factors (McCarthy and Esser, 2010). Increased rates of muscle protein synthesis (MPS) by anabolic processes maintains correct skeletal muscle architecture and contractile properties by creating new functional proteins. Conversely, catabolic processes increase muscle protein degradation (MPD), which is a normal process for removal of damaged or inadequately functioning proteins. Both anabolism and catabolism exist in a harmonious balance that is controlled by diet and exercise within normal populations (Tipton et al., 2004). Muscle loss or dysfunction is observed in various situations such as: cancer cachexia, obesity related chronic inflammation, ageing sarcopenia, immobilization, muscular dystrophy, and peripheral artery disease (PAD) (Collins et al., 2018; Dirks et al., 2016; English and Paddon-Jones, 2010; Penet and Bhujwalla, 2015; Pipinos et al., 2008a, 2008b; Rolland et al., 2008). Within each of these situations, shared and distinct signaling cascades that result in negative protein balance function to ultimately decrease the mass and function of skeletal muscle tissue. In this review, we aim to discuss and examine the varying situational mechanisms of skeletal MPD. The purpose of the review is to discuss the cellular and molecular mechanisms controlling skeletal muscle atrophy induced by various pathological conditions. Specifically, we discuss both shared pathways as well as condition-specific differences associated with muscle atrophy. We begin by providing an overview of catabolic and anabolic systems regulating MPD and MPS. This is followed by a discussion of the specific pathophysiological mechanisms that have been identified to play a role in muscle atrophy from both preclinical studies as well as human studies.
The Skeletal Muscle Proteolytic Systems
Calpains
There are three proteolytic systems that function to remove and breakdown protein from skeletal muscle tissue: the calpain, ubiquitin-proteasomal, and autophagy lysosomal systems. Calpains are intracellular calcium (Ca2+)- dependent cysteine proteases that belong to the papain protease superfamily (Ono and Sorimachi, 2012; Sorimachi et al., 1989). Three separate calpains exist within skeletal muscle, which include; μ- and m- (or calpain-1 and calpain-2, respectively) and muscle specific calpain-3 (formally known as CAPN3[p94]) (Ono and Sorimachi, 2012; Sorimachi et al., 1989). In order to become active, calpain-1 and calpain-2 must form a heterodimer with calpain small subunit-1 (formally known as CAPNS1[30k]) producing either calpain-1/S1 or calpain-2/S1 conformations within the cytosol, respectively (Ono and Sorimachi, 2012). Calpain-1 and calpain-2 activity is dependent on the formation of a heterodimer with calpain small subunit-1 which acts as a regulatory subunit (Ono and Sorimachi, 2012). Muscle specific calpain-3, however, forms a homodimer and interacts with connectin/titin between the Z- and M-lines within muscle (Hayashi et al., 2008; Ono and Sorimachi, 2012; Sorimachi et al., 1995). Calpain activation occurs allosterically with increased Ca2+ concentration within the cytosol of the myofibril (Byrd, 1992; Takekura et al., 2001). Increased intracellular Ca2+ concentration can occur from excitation-contraction coupling or myofibrillar damage to the t-tubule and sarcoplasmic reticulum (Byrd, 1992; Takekura et al., 2001). Calpains are activated in both rats and humans in response to exercise and are currently thought to help with the early stages of myofibrillar remodeling (Belcastro, 1993; Hsieh et al., 2008; Huang and Forsberg, 1998). Interestingly, activated calpains in vivo exhibit selective proteolysis of receptor proteins, membrane proteins such as protein kinase A and C, cytoskeletal proteins, and contractile proteins (Belcastro et al., 1998; Busch et al., 1972; Saido et al., 1994). Additionally, this selectivity to cleave specific protein sites enables calpains to cleave and release proteins inaccessible by other proteolytic systems (Goll et al., 2008; Hyatt and Powers, 2020; Neti et al., 2009). Therein, it appears that the calpain system does also appear to work in concert with the other proteolytic systems, complementing them to cleave and degrade proteins in an organized and somewhat sequential fashion.
Ubiquitin Proteasomal System
Another major proteolytic system used to dissemble proteins into their individual amino acid constituents is the ubiquitin proteasomal system (UPS). This system uses a series of cytosolic enzymes to tag and chaperone proteins to a large proteasome complex that subsequently degrades the protein. The tagging and chaperoning of proteins occurs through a series of steps that are regulated by a ubiquitin activating enzyme (E1), a ubiquitin conjugating enzyme (E2), and a ubiquitin ligase (E3) (Kleiger and Mayor, 2014). E1 enzymes initiate this cascade by creating a thioester bond with the ubiquitin molecule’s C-terminal glycine residue and the E1’s catalytic cysteine residue, activating the ubiquitin protein (Hann et al., 2019; Kleiger and Mayor, 2014). The activated ubiquitin (now bound to the E1 activating enzyme) is then subsequently transferred to the E2 conjugating enzyme’s catalytic cysteine residue (Hann et al., 2019; Kleiger and Mayor, 2014). The ubiquitin-bound E2 enzyme then binds a class of E3 ligases; either a Really Interesting New Gene/U-box -type (RING), Homologous to E6AP C-Terminus-type (HECT), or a RING Between RING (RBR) E3 ligase (Weber et al., 2019; Ye and Rape, 2009). Once bound to one of these E3 ligases, the ubiquitin is then subsequently transferred from the E2 to the E3, and then bound to a lysine residue on the target protein (Hann et al., 2019; Kleiger and Mayor, 2014; Weber et al., 2019; Ye and Rape, 2009). Following the initial ubiquination, formation of a poly-ubiquitin chain of at least four ubiquitin molecules must be formed before a protein will undergo degradation (Kleiger and Mayor, 2014). Once the protein reaches sufficient ubiquitin chain length, it is transported to the 26S proteasome (comprised of a core 20S subunit and two 19S regulatory subunits that form two caps on the 20S core). The ubiquitin chain on the target protein docks to the 19S subunit of the 26S proteasome by anchoring to the receptor’s proteasome regulatory particle base subunit 10 (Rpn10) and Rpn13 (Elsasser et al., 2004; Kleiger and Mayor, 2014). Once bound to the 19S receptors, the entire ubiquitin chain is cleaved which creates enough space for the target protein to enter the 20S core for degradation (Kleiger and Mayor, 2014). The 20S core is comprised of four heptameric rings, two outer α rings that function as gate keepers, and two inner β rings that carry out the proteolytic functions (Ben-Nissan and Sharon, 2014). Within the heptameric β rings, there are three β subunits that are catalytically active and contain caspase-like, trypsin-like and chymotrypsin-like proteases, which are responsible for breaking down the target protein into oligopeptides (Ben-Nissan and Sharon, 2014). These small amino acid chains are currently thought to either be recycled for the creation of other proteins, used for energy metabolism, or removed from the cell.
Glucocorticoids and proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), and various interleukins (IL) such as IL-1 are known to increase ubiquitin proteasomal activity within skeletal muscle (Lecker et al., 2006; McElhinny et al., 2002). For example, the membrane permeable glucocorticoid is able to bind its intracellular receptor within muscle, form a homodimer with another activated glucocorticoid receptor and bind the glucocorticoid response element, upregulating the expression the muscle specific E3 ligase, muscle RING finger protein 1 (MuRF1) as well as O-type forkhead transcription factors (FOXO) and kruppel-like factor-15 (KLF-15) (Bodine and Baehr, 2014). The increased expression of FOXO’s or KLF-15 then subsequently binds to the promoter region of the other muscle specific E3 ligase, muscle atrophy F-box (MaFbx), and induces its expression (Bodine and Baehr, 2014). Similarly, TNF-α binds to its transmembrane receptor and subsequently leads to the activation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), which also increases the expression of MuRF1 and MaFbx, thus increasing MPD (Glass, 2005). Although other homologs of MuRF1 exist, such as MuRF2 and MuRF3, current evidence suggests muscle atrophy is only associated with MuRF1 (Baehr et al., 2011; Bodine and Baehr, 2014). Interestingly, MuRF1, MuRF2, and MuRF3 have been shown to localize at the M-lines, while MuRF1 and MuRF3 have been found at the Z-lines of sarcomeres (Bodine and Baehr, 2014; Centner et al., 2001; McElhinny et al., 2002). This would seem to indicate that the contractile apparatus of skeletal muscle are the target proteins for these E3 ligases. However, the ubiquitin proteasome system cannot degrade intact myofibrils, and it has been speculated that calpains work synergistically to allow contractile proteins to be bound and ubiquinated for degradation (Goll et al., 2008; Jackman and Kandarian, 2004; Neti et al., 2009).
Autophagy Lysosomal System
The third intracellular proteolytic pathway which coordinates protein breakdown in skeletal muscle is the autophagy lysosomal system. Autophagy is the only mechanism able to degrade large structures and, in the absence of stress, serves a house keeping function, eliminating damaged components that could become detrimental to the cell. There are three different defined types of autophagy: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy (Glick et al., 2010). All three types result in the lysosomal degradation of proteins by various acid hydrolases such as cathepsins, carboxypeptidases, and aminopeptidases (Glick et al., 2010; Kaushik and Cuervo, 2018; McEwan and Dikic, 2010; Oku and Sakai, 2018). Macro-autophagy is a process that allows larger cytosolic proteins, such as organelles, to be degraded within cells. Macro-autophagy begins with the creation of a membrane known as a phagophore, that expands to surround proteins, organelles, and ribosomes eventually creating a double-membrane autophagosome (McEwan and Dikic, 2010). The autophagosome acts as a transport vehicle that fuses with a lysosome allowing for the degradation of the encased proteins by lysosomal acid proteases (Glick et al., 2010). In micro-autophagy, cytosolic components are directly engulfed by the lysosome without requiring the fusion of an autophagosome by various mechanisms such as lysosomal wrapping, lysosomal invagination, or endosomal invagination (Oku and Sakai, 2018). Chaperone-mediated autophagy is a process where specific proteins that contain the pentapeptide KFERQ-like motif are bound to heat shock cognate 71 kDa protein (HSC70) and shuttled to the lysosome for degradation (Kaushik and Cuervo, 2018). Approximately 25–30% of all cellular proteins contain a homologous pentapeptide sequence that enables binding and translocation to the lysosome by HSC70 and its co-chaperones (Kaushik and Cuervo, 2018; Knecht and Salvador, 2013). At the lysosomal membrane, the target protein and HSC70 bind to a lysosome-associated membrane protein type 2A (LAMP2A), which initiates the unfolding of the target protein by the chaperone proteins (Kaushik and Cuervo, 2018). An isoform of HSC70 is present within the lysosome and helps facilitate the movement of the unfolded target protein from the cytosol through the translocation complex at the membrane and into the lysosome for degradation (Kaushik and Cuervo, 2018).
Macro-autophagy is suspected to be the responsible for most intracellular MPD and will be the predominant autophagy pathway discussed throughout the remainder of this review. Macro-autophagy is stimulated by energy depletion, low glycogen concentration, low intracellular amino acid content, hypoxia, and inflammation. On the contrary, macro-autophagy is inhibited by insulin signaling, as well as by activation of mammalian target of rapamycin complex 1 (mTORC1) (Rabinowitz and White, 2010). The molecular studies which characterized macro-autophagy initiation found that proteins created from autophagy-related genes (Atg), such as Atg1, coordinate the formation of autophagosomes in yeast and fungus (He and Klionsky, 2009; Rabinowitz and White, 2010). In nutrient depleted conditions, Atg1 forms a protein complex with Atg13, Atg17, Atg29, and Atg31, and is thought to stimulate autophagosome formation (Kabeya et al., 2005). In mammals, Atg1 has a homologous protein known as Unc-51 like autophagy activating kinase 1 (ULK1) that helps facilitate the autophagy cascade (He and Klionsky, 2009; Rabinowitz and White, 2010). ULK1 forms a complex with mammalian Atg13 (mAtg13), focal adhesion kinase (FAK) family-kinase interacting protein of 200 kDa (FIP200), and Atg101 (Hara et al., 2008; Hosokawa et al., 2009; Mercer et al., 2009; Park et al., 2016). Under nutrient deprivation, 5’ adenosine monophosphate-activated protein kinase (AMPK) phosphorylates ULK1 on multiple residues, resulting in ULK1 activation and the subsequent phosphorylation of both FIP200 and mAtg13 in the bound complex (Mao and Klionsky, 2011; Park et al., 2016). This complex is then thought to be translocated to the endoplasmic reticulum tubulovesicular membranes, which are tagged with Atg9-containing vesicles to help facilitate autophagosome membrane formation (Zachari and Ktistakis, 2020). The process of autophagosome formation is incredibly detailed and is thoroughly reviewed elsewhere (Galluzzi et al., 2017; Yu et al., 2018; Zachari and Ktistakis, 2020).
Apoptosis
In addition to these proteolytic systems, apoptosis, or programmed cell death, may also be an important contributing factor for muscle atrophy in certain conditions, although its role is less well-defined (Dupont-Versteegden, 2006). The main enzymes thought to be involved in apoptosis are the caspases; caspase-8, 9, and 12 are involved in the initiation process, while capase-3, 6, and 7 play a role in apoptosis execution (Dupont-Versteegden, 2006). Caspase activation occurs following changes in the ratio of pro- and anti- apoptotic proteins of the B-cell lymphoma (Bcl)-2 family. Specifically, Bcl-2 is considered anti-apoptotic and Bcl-2-associated X protein (Bax) is pro-apoptotic. High levels of calcium can initiate signaling that activates Bax and leads to its association with the mitochondrial membrane, where it is believed to play a role in promoting permeabilization and the release of important apoptotic signals such as cytochrome C (Garrido et al., 2006). However, Bax function can be inhibited by Bcl-2; thus an elevated Bax/Bcl-2 ratio can up-regulate caspase-3-induced apoptosis (Garrido et al., 2006). Caspase-induced apoptosis is reviewed in detail elsewhere (Primeau et al., 2002a).
Muscle Protein Synthesis
PI3K-AKT-mTOR Pathway
In addition to enhanced MPD, muscle breakdown can also be potentially explained by reduced MPS. Research has demonstrated that signaling by mTOR plays a central role in the regulation of MPS (Laplante and Sabatini, 2012; Sengupta et al., 2010). Specifically, mTOR-mediated phosphorylation of the eukaryotic initiation factor 4E binding protein 1 (4E-BP1) results in recruitment of the initiation factor eIF4G to the 5’ end of mRNA, promoting the initiation of protein synthesis (Haghighat et al., 1995). In addition, mTOR can phosphorylate and activate the p70 ribosomal protein S6 kinase (p70S6K), which can promote an increase in the activity of another initiation factor, eIF4A, providing an additional stimulus for initiation (Raught et al., 2004). The mTOR pathway can be activated by insulin binding to its receptor, triggering tyrosine kinase activity via insulin receptor substrate-1 (IRS-1), and subsequent activation of phosphatidylinositol 3 kinase (PI3K), which phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-triphosphate (PIP3). This results in the recruitment of phosphoinositide-dependent protein kinase-1 (PDK1), which phosphorylates and activates protein kinase B (Akt), which further activates mTOR (Goodman et al., 2010). Akt can also phosphorylate glycogen synthase kinase 3β (GSK-3β), which can increase protein synthesis by increasing eukaryotic initiation factor 2B (eIF2B) activity (Welsh et al., 1998). Likewise, insulin-like growth factor-1 (IGF-1) can also bind to the insulin receptor or the IGF-1 receptor; however, the anabolic effects of IGF-1 are not demonstratable except in the presence of insulin (which mediates increased glucose and amino acid transport), consistent with the role for IGFs as permissive factors that augment signals of other factors (Biagetti and Simó, 2021; Hakuno and Takahashi, 2018; Musaro, 2010).
Sarcopenia Age-Related Loss of Muscle
The term sarcopenia, coined by Irwin Rosenberg in 1989, refers to the age-associated loss of muscle mass. Sarcopenia is thought to affect up to 29% of older adults, with some estimates as high as 60%, depending on the definition and measure of muscle mass used (Mayhew et al., 2019). This natural loss of muscle mass and concomitant function can occur at a rate of 0.64–0.7% per year and 0.8–0.98% per year, in women and men over the age of 75, respectively (Mitchell et al., 2012). Epidemiological studies suggest that total lean body mass declines by ~18% in men and ~27% in women from the second to the eighth decade of life (Janssen et al., 2000). Reductions in muscle strength and general function are more rapid than the loss of muscle mass, occurring at a rate between 2.5–4% per year (Mitchell et al., 2012). This decrease in both size and strength increases the morbidity risk of older individuals, while decreasing their quality of life and independence. Interestingly, in sarcopenia, both a decrease in muscle fiber size as well as fiber number may be evident. Specifically, some studies have observed a preferential atrophy of Type II fibers with ageing, which results in a greater ratio of Type I to Type II fiber area (Klein et al., 2003; Larsson, 1978). This may result from a loss of myosin heavy chain (MHC)IIa and IIx isoform expression, which contributes to weaker, slower, and less powerful whole muscle contractile properties (D’Antona et al., 2007). A higher relative MHCI content and lower relative MHCIIa and IIx content was also shown in muscles from elderly individuals who were physically active, removing the confounder of impaired physical function (Brocca et al., 2017a). Despite this shift in MHC isoform, however, myosin content was not different between younger and older groups. Thus, it was suggested that while a fast- to- slow shift may be occurring, there may be no down-regulation of myofibrillar proteins (i.e., myosin) in healthy ageing without disuse atrophy. Researchers also saw no difference in the myosin: actin ratio. Instead, there was evidence of MHC oxidation, which may be a factor in reduced force generation (Brocca et al., 2017a). A study by Marx et al. also supports the findings of the previous study at both the mRNA and protein level. In this investigation, there were no differences in myosin gene or protein expression between younger and older healthy individuals (Marx et al., 2002). In fact, the expression of the MHC isoforms was also not different between groups, and both younger and older muscles were predominantly type II (Marx et al., 2002). Some researchers have proposed that these inconsistencies in the findings related to myosin content during ageing may be due to other factors that may influence muscle loss. For example, it appears that when individuals are physically active throughout their life, myosin content may not be affected. Data such as these have led to the characterization of sarcopenia as either primary (due to no other factors other than ageing itself) or secondary (due to multiple factors associated with the ageing process, such as inactivity) (Cruz-Jentoft et al., 2010).
There are many potential mechanisms underlying sarcopenia and the loss of muscle function. For example, a recent review lists increased fat deposition in skeletal muscle, dysregulation of proteasomal pathways, mitochondrial dysfunction, reduced satellite cell number, increased ROS production, and increased inflammation as possible associated mechanisms (McCormick and Vasilaki, 2018). One of the major hypotheses surrounding sarcopenia is age-associated anabolic resistance, or the idea that the normal exercise and nutrient responses to increase MPS and inhibit MPD are blunted with age (Cuthbertson et al., 2005; Kumar et al., 2009; Wilkinson et al., 2018). Studies have shown that, although basal protein synthesis and degradation do not seem to be greatly affected, there are major differences in MPS in response to anabolic stimuli such as feeding and exercise between young and older individuals (Volpi et al., 2001; Volpi and Rasmussen, 2000). For example, the increase in MPS is blunted by ~40% in response to acute amino acid feeding and ~30% in response to acute exercise (Cuthbertson et al., 2006; Volpi and Rasmussen, 2000). Likewise, reduced MPS and blunted muscle mass accretion was found in older individuals in response to 6 weeks of unilateral resistance exercise when compared to younger individuals, but a clear cause was not determined (Matthew S. Brook et al., 2016; M. S. Brook et al., 2016). The anabolic resistance to exercise that is associated with ageing is likely a factor of altered signaling responses. For example, Akt is a serine/threonine protein kinase involved with regulating anabolic and catabolic processes related to growth factors, nutrients, cytokines, and mechanical tension (Bodine et al., 2001b; Miyazaki et al., 2008; Wu et al., 2011). In aged mice, Akt phosphorylation (Ser473 and Thr308) was significantly greater in old mice compared to younger mice in response to exercise but did not equate to similar elevations of mTOR phosphorylation (Ser2448) and subsequent formation of eukaryotic initiation factor-4 (eIF4E) eukaryotic initiation factor-4G (eIF4G) complexes post exercise (Hwee and Bodine, 2009; Wu et al., 2009). Additionally, higher quantities of Akt phosphorylation in aged mice did not relate to GSK-3 activity (phosphorylation of Ser9), indicating there may be impaired Akt kinase function with age (Hwee and Bodine, 2009; Wu et al., 2009). The higher amount of Akt signaling that is occurring in older mice may be an ineffective compensatory mechanism in an attempt to elevate downstream signaling proteins. In humans, the activation of Akt and subsequent inhibition of proteolysis and MPD after feeding is also blunted in older compared to younger participants (12% decrease vs. 47% decrease in MPD, respectively) (Wilkes et al., 2009). In addition to its roles with intracellular nutrient regulation and mTOR activation, Akt also serves as an inhibitor of UPS and autophagy by phosphorylation of FOXO transcriptions factors and beclin 1, respectively (Brunet et al., 1999a; R. C. Wang et al., 2012). Interestingly, even in organs other than skeletal muscle, such as liver, adipose, or heart, reduced catalytic activity of Akt does not explain decrease in proteolytic activity found in aged muscle (Altun et al., 2010; Fernando et al., 2019; Höhn et al., 2016; Strucksberg et al., 2010). Despite direct evidence of anabolic resistance, it is not readily apparent that the UPS is the most influential proteolytic system that facilitates muscle tissue loss during sarcopenia. Finally, there does not seem to be a major role for the activation of the ubiquitin ligases (MuRF1 or MAFbx) in ageing independent of other conditions or illness (Greenhaff et al., 2008).
Other mechanisms attempting to explain the effects of sarcopenia stem from age-related alterations to the neuromuscular system. The major noticeable change to the neuromuscular system with ageing is the loss of contractile strength and muscle mass, some of which occurs via loss of motor units (MUs) (Campbell et al., 1973). Age related MU loss has been hypothesized to occur via distinct mechanisms known as the “dying-back” or “dying-forward” phenomena. The “dying-back” hypothesis states that the integrity of the MU is lost first at the distal end near the neuromuscular junction (NMJ). The “dying-forward” hypothesis suggests that pathological MU loss is initiated at the proximal sites of the motor neuron (MN) near the ventral root. Comparisons of the distal end of the MU between old and young mice show unconventional acetylcholine receptor localization without the formation of NMJs and fragmented motor endplates in older mice. This aberrant NMJ structure is coupled with significant reductions in relative limb strength (Chung et al., 2017). Additionally, the ventral roots on the proximal end of the MU did not show significant differences in axon diameter, total axon count per root, or G-ratio (ratio of axon diameter to fiber diameter) between old and young mice. These data in animals indicate that MU loss and early decreases in strength may therefore be occurring by the “dying-back” phenomenon (Chung et al., 2017). Similarly, studies have shown that beyond the 7th decade of life, there is a clear loss of MNs, and it seems that the loss of functioning MUs precedes the death of the MN at the spinal cord (Tomlinson and Irving, 1977). This loss of the MUs at the distal NMJ site is referred to as denervation. Collateral sprouting and regeneration of nerve fibers from surviving MUs can remodel the MU, increase MU territory, and increase the number of muscle fibers per MU, seen as fiber grouping (Gordon, 2017; Hepple and Rice, 2016; McComas, 1977). Skeletal muscle is known to undergo repeated cycles of denervation and reinnervation during adulthood to old age (Hepple and Rice, 2016). The cyclical process of denervation and reinnervation can occur earlier in the development of sarcopenia, resulting in decreases in strength with little changes in muscle mass. Notably, these changes in NMJ structure also occur at an earlier age than myofiber atrophy in rats (Hepple and Rice, 2016). However, once the process of denervation outpaces re-innervation in old age, complete loss of MU integrity and death of MUs at the NMJ occurs (Gordon et al., 2004). Following the death of MN in the spinal cord, or axonal degeneration, muscle fibers that are no longer part of an operating MU will atrophy and become non-functional unless they are reinnervated (Doherty and Brown, 1997). Without reinnervation, the muscles experience atrophy and the mechanisms responsible have only partly been elucidated. For example, in denervated mice, inhibition of NF-κB leads to protective effects against denervation-induced loss of muscle mass and tetanic force production (Mourkioti et al., 2006). Similarly, mice deficient in MuRF1 or MaFbx show significant muscle preservation in response to denervation (Bodine et al., 2001a). In addition, muscle fibers positive for a denervation-specific sodium channel demonstrate higher levels of MuRF1 and MaFbx (Rowan et al., 2012). Furthermore, mice that are deficient in small mothers against decapentaplegic (SMAD) homologs 2 and 3 (SMAD2/3) show resistance to denervation induced muscle atrophy (Tando et al., 2016). Thus, it appears that skeletal muscle catabolism in response to MU loss is globally increased through several signaling cascades in a nonspecific manner.
It is currently thought that one of the factors that may be associated with the initiation of denervation may be enhanced ROS production. Ageing has been linked to elevated production of ROS that may overwhelm intracellular antioxidant defense mechanisms (Fulle et al., 2004; Jackson and McArdle, 2011). Superoxide dismutase (SOD) knockout mice have been shown to display both denervation and axonal sprouting between one and four months of age (Fischer et al., 2012, 2011). Similarly, SOD-null mice also display large amounts of intramuscular protein carbonylation and altered excitation-contraction coupling mechanisms that mirror old wild-type mice (Ivannikov and Van Remmen, 2015). The similarities between the SOD-null and aged mice indicate uncontrolled ROS production may play a large role in mediating the initial steps of denervation with age (Fischer et al., 2012, 2011; Ivannikov and Van Remmen, 2015). However, it is still unclear if NMJ deterioration is facilitated primarily by the muscle, axon or some synergistic mechanism involving both. The amount ROS produced by the muscle is likely far greater than that produced from the axon due to the sheer size and metabolic activity of the tissue. However, other mechanisms primarily involving the function of the MU have been recently reported as well, which suggest that apparent loss of homeostatic functions of the MU itself may play a role in the acceleration of muscle catabolism (Cisterna et al., 2020). For example, increased proteolysis may involve connexin hemichannels, which are non-selective membrane channels that decrease resting membrane potential through altered ion influx. These transmembrane proteins, also known as gap junction proteins, play an important role in electrical coupling between cells by conducting ion currents (Fiori et al., 2012). Calcium is known to permeate through connexin hemichannels, and increased intracellular calcium may stimulate calcium-dependent proteolysis in response to denervation (Cisterna et al., 2020). In mice and in in vitro assays, repression of connexin-43 and connexin-45 (via acetylcholine receptor activation) prevented muscle atrophy induced by denervation, (Cisterna et al., 2020, 2016). It has thus been postulated that both enhanced ROS production and frequent excitation-contraction coupling mechanisms may work synergistically to regulate denervation/reinnervation during sarcopenia. Importantly, high levels of life-long physical activity can mitigate the loss of MUs and maintain the amount of excitable muscle mass, and this has been shown to be based on activation of the MN specific to the muscle action trained (Brocca et al., 2017a; Power et al., 2012).
Mitochondrial dysfunction has also emerged as a central factor in sarcopenia progression (Romanello and Sandri, 2010). There are several mechanisms that have been found to link dysfunctional mitochondria with muscle atrophy, which include increased mitochondrial ROS production, mitochondrial release of pro-apoptotic factors, and signaling events that are driven by diminished ATP production (Hyatt and Powers, 2021). For example, permeabilization of the outer mitochondrial membrane can result in cytochrome c release, which can activate caspase-3 (Hyatt and Powers, 2021). Interestingly, mitochondrial sensitivity to permeability transition is increased in both older humans as well as in a murine model of sporadic denervation (Gouspillou et al., 2014; Spendiff et al., 2016). Even in physically active older individuals showing no changes in mitochondrial ROS or maximal respiratory capacity, increased sensitization to mitochondrial permeability transition is still observed (Gouspillou et al., 2014; Spendiff et al., 2016). Furthermore, reduced ATP production by dysfunctional mitochondria can result in the activation of AMPK, which is associated with both MPS suppression (via mTOR inhibition) as well as enhanced MPD (via an AMPK-FOXO3-MuRF1/MAFbx axis) (Hyatt and Powers, 2021). With relevance to ageing specifically, several lines of evidence implicate mitochondrial dysfunction in sarcopenia. First, there are clear morphological alterations in aged muscles (Leduc-Gaudet et al., 2015). Additionally, ageing is characterized by perturbations in the expression of proteins that control mitochondrial dynamics (i.e., fusion and fission) (Tezze et al., 2017). Likewise, mitophagy regulators are also reduced in aged muscles, which could result in the accumulation of dysfunctional mitochondria (Gouspillou et al., 2014). Finally, further evidence for a relationship between mitochondrial dysfunction and sarcopenia is that in aged animals, treatment with compounds that improve mitochondrial function, such as SS-31 (elamipretide) (Campbell et al., 2019; Siegel et al., 2013) or sulforaphane (Bose et al., 2020) have been shown to prevent age-associated muscular deficits.
Notably, in sarcopenia there is also a loss of contractile function, even when normalized to muscle mass, which suggests that ageing may also be associated with impairments in cross-bridge force generation kinetics. For example, there is an evident loss of myosin content relative to cross-sectional area that results in less actomyosin interactions as well as decreased actin sliding speed (D’Antona et al., 2003). Functionally, there is a decline in muscle strength, shortening velocity, and power output during natural ageing (Frontera et al., 2000). The loss of strength may be attributed to structural changes in the muscle rather than an inability to activate the muscle. Older individuals tend to also be slower for voluntary angular contraction velocities of muscles, and the loss of strength and shortening velocity leads to alterations in the force-velocity relationship, impairing power production. These slower contractile properties may be a mechanism to achieve the same contraction at a lower firing rate (Raj et al., 2010). Older adults are also more fatigable for tasks relative to maximal force production (Dalton et al., 2010). Interestingly, eccentric contraction efficiency and strength are relatively maintained compared to isometric and concentric actions, which suggests that there may be reduced agonist and increased antagonist activation during concentric contractions, or that increases in muscle passive stiffness enhance lengthening actions (Hasson et al., 2011; Phillips et al., 1992). Finally, there is a reduction in the amount of calcium released from the SR and reduced calcium sensitivity associated with ageing (Ingalls et al., 1998).
The research on satellite cells in ageing have not led to consistent results. Some studies have shown that satellite cells decrease in number during ageing, but other studies have found no reduction (Renault et al., 2002; Roth et al., 2000). However, the overwhelming evidence remains that there is no reduction in myonuclei number with ageing; and despite a lower number of satellite cells, myonuclei number may actually be higher in aged muscle (Kadi et al., 2004). Therefore, satellite cells may have decreased functional ability regardless of the effect on their number, which can influence the ability of the satellite cells to proliferate and fuse, therefore potentially influencing changes seen in sarcopenia (Gallegly et al., 2004). An increased myonuclear number with decreased fiber size suggests decreased myonuclear efficiency in ageing, which contradicts the concept of a constant nuclear domain, since each myonucleus would instead have less cytoplasm to control (predicting a decrease in myonuclear domain) (Kadi et al., 2004). More recent evidence also supports a flexible myonuclear domain. For example, in studies in both rats and humans, muscle hypertrophy (especially in Type 2 fibers) has been demonstrated in the absence of myonuclear accretion (Murach et al., 2018a). This suggests satellite cell density can possibly increase in the absence of myonuclear accretion, which implies an expansion of the myonuclear domain.
Recent studies point to age-related differences in satellite cell requirements for skeletal muscle hypertrophy (Murach et al., 2018b). In a model of conditional satellite cell depletion, Murach et al. showed that adolescent mice (2–2.5 months), but not mature mice (>4 months), require satellite cells for overload-induced hypertrophy (Murach et al., 2017). In fact, hypertrophic growth still occurred in mature adult mice, even in the absence of satellite cells. In contrast, a previous study in aged mice (>24 months) found that overload-induced hypertrophy is impaired, regardless of satellite cell content (Lee et al., 2016). However, in this study, satellite cell depletion in aged muscle was also shown to result in increased skeletal muscle fibrosis (Lee et al., 2016). Interestingly, the breaking of satellite cell quiescence and proliferation initiation are driven by Notch, and Notch activation declines with ageing as a result of depressed mitogen-activated protein kinase (MAPK) activity, which may reduce satellite cell activation (Caldeira, 2009). Furthermore, transforming growth factor-beta (TGF-β) levels are also increased with ageing, which can lead to activation of cyclin-dependent kinase, which additionally inhibits satellite cells and prevents their regenerative response. Elevated levels of TGF-β may also play a role in the infiltration of non-contractile tissue into whole muscle with age (Ismaeel et al., 2019; Overend et al., 1992).
Despite the effects of satellite cells on acute muscle size, more recently, satellite cells have been shown to play an important role in physical activity-driven adaptations. In satellite cell-depleted adult mice assigned to wheel-running for 13 months (as a model of life-long physical activity), the adaptations to exercise, including balance, coordination, and running volume were all reduced (Englund et al., 2020). Thus, satellite cells may be critical for the maintenance of physical function and for preserving exercise adaptations during the ageing process. These adaptations may occur via recently discovered novel secretory and communication functions of satellite cells, which are independent from their better understood fusion roles (Murach et al., 2021).
Mechanical Unloading
In healthy individuals, muscle atrophy can occur independent of disease conditions due to mechanical unloading. In fact, one of the best human models of mechanical unloading is spaceflight. For example, one study showed that prolonged spaceflight (115–197 days) led to significant reductions in gastrocnemius, soleus, and quadricep muscle volume of 10–17% (LeBlanc et al., 2000). Other studies have also shown that this atrophy is greatest in type I fibers (Fitts et al., 2010). A method that is commonly used to model spaceflight is bed rest, especially prolonged bed rest with a 6° head down bed rest (HDBR). This model has consistently shown large reductions in muscle mass, which is also greater in type I fibers (Brocca et al., 2012). Other forms of immobilization, such as casting, have also been used to study mechanical unloading. Even as little as five days of casting can lead to significant declines in muscle cross-sectional area of ~4% (Wall et al., 2014). In addition, reducing daily step count for two weeks also has been shown to lead to ~3% losses in leg lean mass (Krogh-Madsen et al., 2010). Animal models used to study mechanical unloading include rodent hind limb unloading (HLU). For example, two weeks of HLU can lead to muscle mass losses of up to 34%, with the most rapid atrophy in slow muscles such as the soleus (Ohira et al., 1992). However, the mechanisms underlying unloading-induced muscle atrophy seem to differ between rodents and humans (Crossland et al., 2019). These differences are likely due to metabolic dissimilarities, including a higher relative MPS rate and basal metabolic rate in rodents. In addition, age differences between studied species may also play a role, as rodent models often use young animals that are still growing and in development (Crossland et al., 2019). This may explain the discrepancies noted above in the time-matched magnitude of muscle mass loss between animal and human unloading models. Thus, in this section, we begin by discussing the mechanisms of mechanical-unloading-induced muscle atrophy based on rodent data, followed by focusing on the impact of unloading in humans.
Animal Studies
A decrease in MPS is believed to be a major factor in muscle atrophy during unloading conditions (Gao et al., 2018). In a study by Baehr et al., MPS was shown to decrease in nearly all skeletal muscles in response to HLU, and changes in MPS were muscle-specific (Baehr et al., 2017). Notably, the Akt-mTOR pathway may play a role in HLU-mediated muscle atrophy, as there is evidence for reduced phosphorylation of Akt, S6K1, and 4E-BP1 in a model of HLU (Bodine et al., 2001b). HLU was also shown to increase binding of 4E-BP1 to eIF4e (inhibitory association) in just three days (Liu et al., 2012). HLU has also been shown to reduce the activation of mTORC1-independent MPS signaling pathways, as evidenced by reduced phosphorylation of GSK-3β (Mirzoev et al., 2016). More recently, mRNA levels of the mTOR signaling repressor, regulated in DNA damage and development 1 and 2 (REDD1/2), were significantly elevated in response to HLU (Kelleher et al., 2015, 2013). In addition to the regulation of translation initiation, mechanical unloading may also affect MPS at the level of elongation of mRNA translation and translational capacity. After two weeks of HLU phosphorylated (inactive form) levels of eukaryotic elongation factor 2 (eEF2), which plays a role in the elongation of translation, were doubled (Lomonosova et al., 2017). Furthermore, HLU has been shown to decrease total RNA and 28S rRNA content in rat muscle (markers of ribosome content, the main component of translational capacity) (Bajotto et al., 2011; Mirzoev et al., 2016).
With respect to MPD, all the MPD pathways seem to be involved in mechanical unloading-induced atrophy in rodents. For example, inhibition of either calpain or caspase-3 prevented immobilization-induced atrophy in rats (Talbert et al., 2013). However, the UPS is likely the most important. Gene expression of both MuRF1 and MAFbx have been shown to rapidly increase in mice in response to spaceflight (Allen et al., 2009; Gambara et al., 2017), rats in response to HLU (Baehr et al., 2017), and rats in response to both immobilization and HLU (Bodine et al., 2001a). Further evidence comes from the fact that mice deficient in MAFbx or MuRF1 are resistant to HLU-induced muscle atrophy (Bodine et al., 2001a; Labeit et al., 2010). Notably, MuRF-1 and MAFbx are regulated by upstream FOXO transcription factors, FOXO1 and FOXO3. FOXO are also controlled by Akt; phosphorylation of the transcription factors by Akt retains them in the cytosol, preventing nuclear translocation (Brunet et al., 1999b). However, when dephosphorylated, FOXO can translocate to the nucleus and upregulate the expression of MuRF-1 and MAFbx. Cast immobilization and spaceflight have been shown to increase FOXO3 and FOXO1 expression, respectively, in mice (Allen et al., 2009; Okamoto and Machida, 2017). Moreover, inhibition of FOXO3 during disuse in rats prevented increases in MuRF-1 and MAFbx (Senf et al., 2008).
In addition to the aforementioned effects on MPS and MPD, there is evidence that mechanical unloading leads to oxidative stress (Powers et al., 2012, 2007, 2005). The first evidence for this comes from a study in which rats subjected to immobilization demonstrated increased muscle lipid peroxidation, and vitamin E administration partially attenuated muscle atrophy (Kondo et al., 1991). Other studies, especially those focusing on mechanical ventilation-induced diaphragm muscle atrophy (a specific model of disuse) have demonstrated a link between increased oxidative stress and muscle loss. Powers and colleagues were able to show that in rats, mechanical ventilation led to increased mitochondrial ROS production and that using a mitochondrial-targeted antioxidant decreased ROS, with concomitant protection against diaphragm myofiber atrophy (Powers et al., 2011a). Furthermore, in mice, two weeks of hindlimb immobilization was shown to increase mitochondrial ROS production and muscle oxidative damage, measured as 4-HNE-conjugated proteins (Min et al., 2011). Notably, in this study, a mitochondrial-targeted antioxidant was also effective in preventing myofiber atrophy (Min et al., 2011).
Mechanical unloading is also associated with mitochondrial dysfunction and reduced mitochondrial quality. For example, expression of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), the transcriptional coactivator that regulates mitochondrial biogenesis, is reduced up to 40% following HLU (Rosa-Caldwell et al., 2020a). Earlier studies have shown that PGC-1α overexpression blunts decreases in muscle fiber diameter in mice induced by denervation and fasting (Sandri et al., 2006) and HLU (Cannavino et al., 2015, 2014) via reduced proteasomal degradation (MuRF1 and MAFbx induction) and reduced autophagy. In contrast to these studies, however, a recent study showed that PGC-1α overexpression does not protect against HLU-induced muscle atrophy (Rosa-Caldwell et al., 2020b). Although mice overexpressing PGC-1α had blunted expression of MuRF1 and MAFbx, these mice had an increase in MPD via autophagic mechanisms. In contrast, overexpression of mitochondrially targeted catalase (MCAT) did mitigate the response to HLU; however, this effect was shown to be sex specific, only protecting against muscle atrophy in female mice (Rosa-Caldwell et al., 2020b). Notably, this may be due to an augmented catabolic response reported in female mice (Rosa-Caldwell et al., 2021). The earlier studies (Cannavino et al., 2015, 2014; Sandri et al., 2006) all used male mice. However, the study by Rosa-Caldwell et al. included both males and females to investigate how interventions may impact males and females separately (Rosa-Caldwell et al., 2020a). Interestingly, recent data suggest a sex difference in the etiology of disuse muscle atrophy in rodents. Following 7 days of HLU, female mice had an average of 33% lower type IIB cross-sectional area, while male mice did not lose any significant type IIB fiber area (Rosa-Caldwell et al., 2021). Reductions in MPS were also more pronounced in female mice compared to male mice (40% reduction and 23% reduction, respectively) (Rosa-Caldwell et al., 2021). Finally, the differences could also be due to the differing ages of the mice. The studies by Cannavino and Sandri et al. (Cannavino et al., 2015, 2014; Sandri et al., 2006) used six-month old mice, while in Rosa-Caldwell et al.’s study, disuse atrophy was induced at ten weeks of age (Rosa-Caldwell et al., 2021). Overall, these data suggest that sex and age differences in muscle pathologies may exist and highlight the importance of investigating these mechanisms in both sexes and in different age groups.
Another final mediator that may be involved in unloading-induced muscle atrophy is calcium. An early study in 2001 by Ingalls, Wenke, and Armstrong showed that hindlimb suspending mice leads to significant increases in skeletal muscle intracellular calcium levels (up to 117% after just one week) (Ingalls et al., 2001). More recently, tetramethylpyrazine, which has been described as a calcium antagonist, was shown to suppress intracellular calcium and attenuate HLU-induced muscle atrophy in rats (Hu et al., 2017). Interestingly, calpains are calcium-dependent, and calpain1 and calpain2 have been shown to be activated by increased intracellular calcium following HLU (Matsumoto et al., 2014). Furthermore, an overload of intracellular calcium can activate caspase-12, which activates caspase-3 (Primeau et al., 2002b), and after HLU in rats, the Bax/Bcl-2 ratio has been shown to be significantly elevated (Hu et al., 2017).
It is important to note that despite the muscle mass losses observed during mechanical unloading, studies have also shown that reloading may restore these losses. For example, a study by Shimkus and colleagues showed that HLU-induced reductions in MPS, and the accompanying losses in muscle mass, are repeatable with serial reloading-unloading bouts (Shimkus et al., 2018). Specifically, after 28 days of HLU, a 56-day recovery period recovered the MPS rate and muscle size. However, exposure to a subsequent HLU period once more decreased MPS and muscle size, which again recovered after a final reloading period (Shimkus et al., 2018).
Human Studies
Although results in animal models have been consistent, the results of human models of mechanical unloading remain less clear. In a human model of disuse atrophy (lower leg suspension), rates of both myofibrillar and tendon protein synthesis rates progressively decreased over a three-week period (de Boer et al., 2007). More specifically, the reduction in MPS may involve attenuated activation of the Akt-mTOR pathway, as unloading is also associated with insulin resistance. For example, just five days of bed rest has been shown to lead to 67% increases in the insulin response to glucose (Hamburg et al., 2007). Likewise, seven days of immobilization also decreased insulin action on glucose uptake and MPS (Richter et al., 1989).
Still, the mechanistic drivers of possible MPS suppression in humans are unresolved. For example, following lower leg suspension for three weeks, although significant atrophy occurred and MPS rates decreased, there were no changes observed in the activation of any of the mTOR pathway components (de Boer et al., 2007). Similarly, following knee immobilization, although MPS rates were reduced, there were no differences in mTOR, GSK-3β, or eEF2 activation (Glover et al., 2008; Marimuthu et al., 2011). One pathway that may instead be involved in the MPS reduction in response to unloading in humans is focal adhesion kinase (FAK), a non-receptor protein tyrosine kinase that is sensitive to mechanical loading (Graham et al., 2015). For example, in de Boer et al.’s study, FAK activation was reduced by 30% after ten days of lower leg suspension (de Boer et al., 2007). Likewise, in another study unilateral immobilization for 14 days reduced FAK activation by 23% with a concomitant reduction in MPS in the restricted leg after amino acid infusion (Glover et al., 2008). While plasma insulin concentrations and fractional synthetic rates increased with higher amino acid doses, MPS rates were still lower in the muscles of the immobilized limb (Glover et al., 2008). Investigation of signaling proteins showed no differences between limbs in the phosphorylation of mTOR or downstream targets, suggesting this immobilization-induced suppression of MPS may be independent of mTOR activation (Glover et al., 2008). These data support prior in vitro studies demonstrating that FAK is co-immunoprecipitated with IRS-1 without catalytic activation, and FAK inhibition results in insulin resistance despite insulin administration (Bisht et al., 2007; Lebrun et al., 1998). There appears to be a degree of cross-talk regulation between the insulin signaling pathway and FAK, which could be a target for investigation in studies assessing inactivity-induced insulin resistance. Importantly, other etiological factors such as loss of muscle mass alone can also decrease the body’s ability to store and utilize glucose, which may contribute to insulin resistance from altered glucose homeostasis. However, this idea of altered glucose and protein metabolism in response to immobilization needs further investigation.
While increased MPD likely plays a major role in muscle mass loss in rodents in response to mechanical unloading, the involvement of MPD in human muscle atrophy is less clear. For example, gene expression of MuRF-1 and MAFbx were elevated in humans after three days of lower limb suspension (Gustafsson et al., 2010) and five days of immobilization (Dirks et al., 2014). In two other studies, however; one using a 20-day bed rest model, and the other two weeks of limb immobilization, only MAFbx increased, with no change in MuRF-1 (Jones et al., 2004; Ogawa et al., 2006). In contrast, in de Boer et al.’s study using lower leg suspension, after 10 days, there were significant increases in MuRF-1 mRNA expression, but no changes in MAFbx (de Boer et al., 2007). It is important to note that these differences between studies may be due to the varying timelines. Notably, other studies suggest that the effects of FOXO, MuRF-1, and MAFbx are muscle- and time course- specific and may explain the discrepancies reported between different studies (Atherton et al., 2016; Brocca et al., 2017b). Furthermore, in the context of the role of ROS in unloading, in humans, prolonged mechanical ventilation was shown to increase oxidative stress, measured as protein carbonyls and hydroxynonenal (HNE)-protein adducts (Hussain et al., 2010). However, results of other studies have been contradictory. For example, in one study in humans, two weeks of immobilization did not affect levels of the markers of oxidative stress, 4-HNE or protein carbonyls (Glover et al., 2010). Thus, whether unloading-induced ROS production does indeed play a role in muscle atrophy remains debated (Powers et al., 2012). Despite this, mechanistically, there is strong evidence that ROS can indeed lead to muscle atrophy. For example, exposure of myotubes to ROS leads to fiber atrophy (Gilliam et al., 2012). This is most likely due to allosteric regulation of proteases, via increased FOXO3 signaling, and thus expression of MuRF-1 and MAFbx (Min et al., 2011). Additionally, ROS may also activate calpain and caspase-3 (Powers et al., 2011a). Further, ROS can inhibit insulin action and may play a role in the development of insulin resistance (Di Meo et al., 2017), and oxidation of proteins is known to increase unfolding and proteolytic degradation (Powers et al., 2011b; Smuder et al., 2010).
While muscle mass loss is a known endpoint of immobilization in humans, there is still a lack of understanding of the mechanisms responsible for this, and a gap in the understanding of the pathophysiology of mechanical unloading exists. Due to important clinical ramifications (i.e., intensive care), future studies are necessary to clarify these mechanisms. These studies should be conducted in humans, include several time-points, and incorporate various assessments including functional deficit measures in addition to muscle size, muscle protein turnover and nutrient uptake measurements, and insulin sensitivity/resistance measures, in addition to molecular biology assays of signaling pathways. Furthermore, while muscle mass recovery has been demonstrated in humans after reloading as well, most studies have shown this in young, healthy individuals (Seene et al., 2012). Therefore, future work should assess the recovery potential of older and clinical populations.
Cancer Cachexia
Cachexia refers to the progressive atrophy of adipose tissue and skeletal muscle that leads to progressive loss of body weight (Tisdale, 2009). Weight loss is thought to occur in 30–80% of cancer patients, and 15% of these patients may lose more than 10% of their initial body weight (Dewys et al., 1980). The extent of weight loss varies between patients and between tumor types; pancreatic cancer and gastric cancer patients experience the most dramatic losses in body weight, reaching a median of 24.5% weight loss in pancreatic cancer patients before death (Dewys et al., 1980; Wigmore et al., 1997). Importantly, weight loss is an important prognostic factor in survival, as a greater amount of weight loss is associated with a reduced survival time (Dewys et al., 1980; Tisdale, 2009). In addition, loss of skeletal muscle leads to reductions in mobility and quality of life in cancer patients (Tisdale, 2009).
Anorexia is highly prevalent among cancer patients, even independent of chemotherapy and other treatments. In one study, 61% of cancer patients nearing end-of-life were shown to display anorexia (Tranmer et al., 2003). Several factors may lead to these findings, and possible causes of this include psychological depression, tumor encroachment on the gastrointestinal tract, and tumors affecting the mucosa (Knox et al., 1983). Furthermore, cytokines, including IL-1, IL-6, and TNF-α can suppress appetite (Banks, 2001). There is some evidence that there may also be an imbalance between orexigenic, or appetite-inducing, (i.e., neuropeptide Y (NPY)) and anorexigenic, or appetite-suppressing (i.e., proopiomelanocortin (POMC) signals in cancer (Jatoi et al., 2001; Wisse et al., 2001). Despite these links, anorexia does not fully explain body mass losses in cachexia; diminished caloric intake has been shown to not affect weight lost in patients (Bosaeus et al., 2001), and greater amounts of muscle mass are lost in cancer cachexia than can be explained by anorexia alone (Fearon, 1992).
In addition to decreased energy intake, cancer patients are also known to exhibit decreases in physical activity levels. Despite this, however, several types of cancers have been associated with an increased resting energy expenditure (REE), which may also contribute to cachexia. For example, in pancreatic, lung, and GI cancers, REE has been shown to be elevated in association with an elevated acute phase response (McMillan et al., 1998). One potential explanation for an elevated REE in cancer patients is increased thermogenesis in brown adipose tissue (BAT) and skeletal muscle due to uncoupling proteins (UCPs), which mediate proton leakage across the inner mitochondrial membrane, reducing the level of coupling of respiration to ATP synthesis. Interestingly, gene expression of UCPs have been shown to be elevated in tumor-bearing mice (Bing et al., 2000) as well as in cancer patients with weight loss compared to controls and cancer patients that did not lose weight (Collins et al., 2002). Additionally, futile cycles may play a role in increasing the energy expenditure of cancer patients. Notably, tumor hypoxia, which is induced by tumor growth beyond the vascular supply, activates the transcription factor hypoxia-inducible factor-1 (HIF-1). HIF-1 acts to increase glucose uptake via increased transcription of the glucose transporter GLUT1. Additionally, HIF-1 activates pyruvate dehydrogenase kinase (PDHK), which inactivates pyruvate dehydrogenase (PHD), the enzyme responsible for catalyzing the conversion of pyruvate into acetyl-CoA. This results in accumulation of pyruvate and subsequent conversion into lactate (Kim et al., 2006). Notably, this is an energy inefficient process compared to oxidative phosphorylation. Furthermore, the lactate is also inefficiently converted back to glucose in the liver via the Cori cycle. Overall, this process is thought to account for a loss of ~300 extra kcal/day in cancer patients (Edén et al., 1984).
The loss of muscle mass in cancer cachexia may also be attributed to a substantial decrease in MPS. Although a decrease in MPS may be attributed to anorexia, some animal studies suggest that cancer cachexia may be accompanied by depressed MPS independent of anorexia. For example, in mice with adenocarcinoma that lost 15–30% of their weight, MPS was reduced by 60% (Smith and Tisdale, 1993). Similarly, in a mouse model of colorectal cancer, MPS was reduced by 19% during cachexia initiation, with further reductions of up to 50% demonstrated with intermediate weight loss of 6–19% (White et al., 2011). Muscle mass was also significantly associated with MPS rates in these mice. Interestingly, in another murine model of colorectal cancer, although basal MPS was suppressed in cachectic mice, these mice still demonstrated a normal response of activated MPS after a single eccentric contraction bout, and the relative induction of MPS by eccentric contraction was not different from control mice (Hardee et al., 2018). Finally, in mice injected with Lewis lung carcinoma, MPS was reduced by 40% (Brown et al., 2018). However, results in humans have been less consistent. An early landmark study by Emery and colleagues showed that MPS was significantly reduced in patients with cancer (Emery et al., 1984). However, other more recent studies have shown no evidence of MPS suppression in patients with cancer cachexia (MacDonald et al., 2015). Several studies have also assessed markers of MPS to identify potential mechanisms affecting the protein synthetic machinery. In a murine model of cancer cachexia as well as in weight-losing cancer patients, there is evidence for elevated levels of phosphorylated, active double-stranded RNA-dependent protein kinase (PKR), which phosphorylates and inactivates eIF2 (which results in inhibition of translation initiation) (Helen L. Eley et al., 2007; H. L. Eley et al., 2007; Eley et al., 2008; Eley and Tisdale, 2007a). Additionally, mice with a cachexia-inducing tumor demonstrated an increase in eIF4E-4E-BP1 binding, a decrease in the active eIF4E-eIF4G complex, and a 5-fold increase in the phosphorylation of eEF2 (Helen L. Eley et al., 2007; H. L. Eley et al., 2007; Eley et al., 2008; Eley and Tisdale, 2007a). In the mouse models of colorectal cancer demonstrating reduced MPS, mechanistically, IGF-1 mRNA expression was shown to be reduced, and mTOR signaling was suppressed (reduced Akt, p70, and 4EBP1 phosphorylation) (Hardee et al., 2018; White et al., 2011). Despite this basal reduction, however, these mechano-sensitive signaling pathways were still induced following eccentric contractions (White et al., 2011).
With respect to MPD, the UPS seems to play a major role in cancer cachexia-induced muscle atrophy. In the common murine model of cancer cachexia of adenocarcinoma, proteasome proteolytic activity was increased in parallel with increasing weight loss (Chand et al., 2005). Furthermore, gene and protein expression of the proteasome subunits were shown to be elevated in cancer patients that lost >10% of their body weight (Khal et al., 2005). Expression of both MuRF-1 and MAFbx have also been shown to be elevated in several models of cancer cachexia (Bodine et al., 2001a; Gomes et al., 2001). In White et al.’s study of colorectal cancer in mice, MPD was increased by 45% in mice that lost ≤5% of their body weight. Further, mice that lost between 6–19% of their body weight, or ≥20% of their body weight, demonstrated elevations in MPD of 134% and 188%, respectively (White et al., 2011). These elevations in MPD were accompanied by significant and progressive (with weight loss severity) increases in MAFbx and MuRF1 expression (White et al., 2011). In mice injected with Lewis lung carcinoma, muscle protein ubiquitination was increased by around 50%, with concomitant increases in FOXO1 (50%) as well as MAFbx and MuRF1 (two to four-fold increase) (Brown et al., 2018). Likewise, apoptosis may also play a role in MPD. In the murine model of cancer cachexia, there were significant increases in the proteolytic activity of several of the caspases, including caspase-3 (by 151%) and caspase-9 (177%) (Belizário et al., 2001). Finally, in patients with upper GI cancer, skeletal muscle apoptosis was observed, measured as DNA fragmentation (Busquets et al., 2007).
Glucocorticoids are commonly used in the setting of cancer to aid with the symptoms; however, these compounds are known to induce skeletal muscle atrophy. Specifically, glucocorticoids attenuate activation of the PI3K/Akt pathway and augment the activity of forehead box (FOX)O transcription factors, which results in increased MAFbx expression (Hasselgren, 1999; Sandri et al., 2004). Another interesting link is the increase in intramuscular myostatin expression in response to glucocorticoids (Gilson et al., 2007a). Myostatin is a myokine and secreted growth differentiation factor that reduces myogenic gene expression, inhibits Akt, and increases FOXO1 expression (McFarlane et al., 2006). Interestingly, myostatin gene deletion actually prevents muscle atrophy in response to glucocorticoids (Gilson et al., 2007b). Furthermore, proteolysis-inducing factor (PIF) is a glycoprotein which has been identified in mice and humans with cancer cachexia, that has been speculated to also upregulate the UPS (Williams et al., 2004). In fact, a longitudinal study established a relationship between the presence of PIF in urine and weight loss in cancer patients (Williams et al., 2004). Interestingly, administration of PIF isolated from the urine of cancer cachexic patients to mice was shown to induce cachexia by reducing MPS, seemingly by PKR activation, and increasing MPD, seemingly by increasing UPS (Cariuk et al., 1997).
Several cancers are also associated with an excessive production of TNF-α, and in animal studies, there is ample evidence that TNF-α plays a role in the induction of cachexia. For example, in a murine Lewis lung carcinoma model, mice deficient in the TNF-α receptor exhibited reduced skeletal muscle wasting (Llovera et al., 1998). Notably, TNF-α treatment has been shown to decrease MPS and enhance MPD in rats (García-Martínez et al., 1993). The link between TNF-α and muscle wasting may also involve reactive oxygen species (ROS), as TNF-α has been shown to induce oxidative stress (Buck and Chojkier, 1996). Either by an ROS-dependent or independent mechanism, TNF-α leads to increased expression of MAFbx (Li et al., 2005). Moreover, there is evidence that at least in vitro, TNF-α can suppress MyoD mRNA at the posttranscriptional level (Guttridge et al., 2000). However, it is important to note that the involvement of TNF-α in human cancer cachexia is less clear, as some studies show no elevations of TNF-α in patients (Iwase et al., 2004). In contrast to TNF-α, there is much stronger evidence for the involvement of IL-6 in cancer cachexia in humans (Carson and Baltgalvis, 2010; White, 2017). IL-6, which can act as both a pro- or anti-inflammatory myokine, has been described as a predictor of weight loss in patients with cancer cachexia (Scott et al., 1996). In a model of murine colorectal cancer and cachexia, mice with the highest circulating IL-6 levels showed the most severe cachexia (Baltgalvis et al., 2008). Furthermore, IL-6−/− mice did not experience muscle wasting (Baltgalvis et al., 2008). Likewise, in a mouse model of colorectal cancer, administration of an IL-6 receptor antibody attenuated cachexia progression by suppressing MPD, without affecting MPS (White et al., 2011). Similarly, in murine colorectal cancer, NFκB was activated in response to cachexia and further induced by eccentric contractions (Hardee et al., 2018). Inhibition of NFκB improved both basal MPS and MPS in response to eccentric contractions (Hardee et al., 2018). These mechanism may involve MPD by both proteasomal and lysosomal pathways, as in vitro, IL-6 and NFκB have been shown to activate both (Cuervo et al., 1998; Ebisui et al., 1995). Finally, angiotensin II (Ang II) has been shown to be elevated in several cancers (Sanders et al., 2005). Ang II has been shown to both increase MPD by activation of PKR (Eley and Tisdale, 2007b; Russell et al., 2007) and the UPS (Sanders et al., 2005), as well as inhibit MPS by reducing levels of IGF-1 (Brink et al., 2001).
Abnormal mitochondrial morphology has been displayed in cancer cachexia, along with altered expression of mitochondrial fusion/fission proteins (de Castro et al., 2019; Marzetti et al., 2017). Furthermore, cachexia-associated muscle wasting is correlated with reductions in mitochondrial respiration and electron transport chain complex activity (Padrão et al., 2013). Interestingly, in murine models of lung and colorectal cancer, Brown et al. showed that mitochondrial degeneration actually precedes muscle atrophy (Brown et al., 2017). However, increasing mitochondrial biogenesis by transgenic PGC-1α overexpression was shown to have no effect on muscle atrophy in a mouse model of lung cancer (X. Wang et al., 2012).
Obesity-Related Chronic Inflammation
Obesity is defined as having a body mass index equal to or greater than 30 kg/m2 and is an epidemic primarily affecting developed countries. The CDC: National Center for Health Statistics reported that 39.8% of American adults and 18.5% of American children were obese in 2016 (Hales et al., 2017). Several health problems are related to obesity such as cardiovascular disease, cancer, insulin resistance, dyslipidemia, sleep apnea, and osteoarthritis. In addition, obesity is also a disease of low-grade chronic inflammation, which is likely due to the accumulation of excess adipose tissue (Lee et al., 2013). It is important to note that adipose tissue is not an inert energy storage depot; it is an endocrine organ that has been shown to have diverse roles within the body and has demonstrated communication with muscle (Coelho et al., 2013; Li et al., 2017). White adipose tissue secretes a variety of signaling molecules that are now commonly referred to as adipokines that function as hormones (Waki and Tontonoz, 2007).
As adipocytes hypertrophy and undergo hyperplasia, they secrete an increased amount of the hormone leptin, which upregulates monocyte chemoattractant protein‐1 expression, causing adipose tissue to become infiltrated with pro-inflammatory macrophages. These macrophages also release their own pro-inflammatory molecules, which exacerbates the inflammatory cascade (Burhans et al., 2018; Chawla et al., 2011; Haase et al., 2014; Lumeng et al., 2007; Monteiro et al., 2019; Weisberg et al., 2003). Therefore, adipose tissue expressing this inflammatory phenotype, can serve as a source of chronic low-grade inflammation, whereby inflammatory cytokines and chemokines are secreted into the blood to have negative effects on surrounding tissues. Once fat begins to display an inflammatory phenotype, it can stimulate an increased release of TNF-α, IL-6, leptin, visfatin, and resistin into systemic circulation, which may have negative influences on skeletal muscle (Makki et al., 2013). TNF-α is known to stimulate the UPS through NF-kB signaling and increased concentrations in circulations could stimulate muscle catabolism, but this direct mechanism has yet to be proven in the context of obesity in humans. Under normal conditions TNF-α and IL-6 can help promote muscle regeneration in an acute short-term inflammatory response such as during the post-exercise recovery; however, chronic exposure has been shown to induce muscle atrophy (Akhmedov and Berdeaux, 2013). This would seem to indicate that with obesity mediated low-grade chronic inflammation, muscle tissue could be chronically exposed to TNF-α and IL-6 and may have altered catabolic and anabolic activity.
The crosstalk between adipocytes and muscle cells has been investigated using an in-vitro co-culture model consisting of primary human satellite cells and mature adipocytes, isolated from subcutaneous adipose tissue from lean and obese individuals as well as visceral adipose tissue from obese subjects (Pellegrinelli et al., 2015). The researchers observed that the secretome of the obese visceral adipose tissue displayed a more pronounced inflammatory profile, while the lean, subcutaneous adipose tissue had the lowest inflammatory profile measured (Pellegrinelli et al., 2015). These findings support the idea that there is a spectrum of adipocyte cell size that may be related to inflammatory phenotype. When cocultured with muscle cells, the obese visceral adipose tissue decreased the expression of: MyoD, myogenin, titin, α-sarcoglycan, and caveolin-3, while disturbing the sarcomere structure (Pellegrinelli et al., 2015). The cell co-culture findings were supported by histological analysis of obese mice gastrocnemius muscle that showed abnormal muscle fiber sizes and adipocyte accumulation (Pellegrinelli et al., 2015). Additionally, the obese mice had increased expression of leptin and fatty acid binding protein-4 within the skeletal muscle, indicating a large amount of fat infiltration (Pellegrinelli et al., 2015). These results indicate a spectrum of inflammatory signaling can emerge from various adipose tissue locations and has the potential to influence muscle remodeling. In-vivo, leptin-deficient obese and leptin receptor-mutant diabetic mice have been found to have reduced muscle regenerative capacity several days after cardiotoxin injection (Nguyen et al., 2011). While the reduced regenerative capacity could potentially have been influenced by a dysregulated inflammatory response, the wild type mice fed a high fat diet to induce obesity and diabetes did not show any impairment in muscle regeneration or alterations in inflammatory cell accumulation as seen in the leptin-deficient or receptor-mutant mice (Nguyen et al., 2011). Notably, leptin has been previously found to stimulate muscle hypertrophy and can initiate the inflammatory process of macrophages to promote their M1 pro-inflammatory phenotype (Hamrick et al., 2010; Loffreda et al., 1998; Nguyen et al., 2011; Santos-Alvarez et al., 1999; Weisberg et al., 2003). These studies indicate that leptin may have a mediating role in muscle regeneration that could be a target of interest in obese populations.
Research in animal models of obesity and insulin resistance suggest varying effects on MPS at rest and in response to exercise (Nilsson et al., 2013, 2010). Basal mixed protein synthesis rates of sedentary obese Zucker rats were greater than lean rats in the quadriceps, red gastrocnemius, and white gastrocnemius muscles (Nilsson et al., 2013). Investigation of MPS-related signaling pathways showed that at rest, sedentary obese rats displayed decreased expression of both mTOR and DEP domain-containing mechanistic target of rapamycin (mTOR)-interacting protein (DEPTOR), a negative regulator of mTOR, coupled with significant elevations in p70S6k activation. In response to resistance exercise, there was an increase in mixed, myofibrillar, and cytosolic fractions of MPS in the lean rats that appeared to be blunted with obesity (Nilsson et al., 2013). This desensitization to the anabolic stimulus of resistance exercise was postulated to also be impacted by dysregulated mTOR signaling, since DEPTOR expression decreased after exercise in lean rats, but remained unchanged in obese rats (Nilsson et al., 2013). Additionally, despite the increased basal MPS seen in the obese rats, the muscle wet mass and total protein content of the of the quadriceps were significantly lower in the obese rats when compared to the lean rats, potentially indicating greater protein turnover with obesity.
Microscopy studies have found that intramyocellular lipid droplet content, size, and subsarcolemmal localization are directly associated with impaired insulin sensitivity in obesity. The impaired insulin sensitivity has been postulated to decrease fractional MPS rates through the dysregulation of mTOR signal transduction (Beals et al., 2019; Daemen et al., 2018; Nielsen et al., 2010; Smeuninx et al., 2017). However, there have been inconsistencies in findings related to mTOR activation in obesity, which may be due to variations between individuals (Beals et al., 2018; Tran et al., 2018). Despite this, the effects of obesity-induced anabolic resistance to exercise and dietary amino acids have also been displayed in humans (Beals et al., 2018; Smeuninx et al., 2017). Interestingly, the attenuated MPS response to exercise does not appear to be directly related to increased intramuscular NF-κB signaling (Beals et al., 2018). Instead, attenuated myofibrillar protein synthesis rate responses to nutrient ingestion are associated with large increases in serum insulin (Smeuninx et al., 2017). This association has been posited to suggest that obesity-induced insulin resistance may be a factor contributing to anabolic resistance (Smeuninx et al., 2017). While this mechanism of insulin resistance-induced anabolic resistance has been consistently linked to aberrant mTOR signaling in murine models, whether this mechanism involves dysregulated mTOR activation in humans is still unclear. Unlike animal models that give a more uniform presentation of obesity and insulin resistance, humans can display a range of obesity with or without metabolic disease (Jais et al., 2014; Stefan et al., 2013). This apparent difference in obesity severity or obesity-induced alterations in insulin sensitivity may be partially responsible for the inconsistent findings in human studies (Beals et al., 2019; Daemen et al., 2018; Nielsen et al., 2010; Smeuninx et al., 2017)(Beals et al., 2018)(Beals et al., 2018; Tran et al., 2018).
Overall, the collection of studies discussed indicate that obesity may influence skeletal muscle remodeling in a negative manner. Interestingly, while pro-inflammatory adipokines and signaling pathways may have the potential to alter muscle remodeling based on in vitro studies, the effects in vivo are less clear. Further, alterations in the regulatory mechanisms controlling mTOR (i.e., DEPTOR) due to obesity-induced insulin resistance may compromise skeletal muscle remodeling, leading to muscle atrophy. Still, a more concise characterization of obesity may be needed to distinguish between the divergent results of the different human studies. Further research is also needed to investigate how obesity alters adipocyte secretome, and the subsequent effects these signaling molecules have on regulating skeletal muscle mass in both animal models and humans.
Muscular Dystrophy
Muscular dystrophy refers to a group of diseases that lead to progressive skeletal muscle degeneration, disability, and eventually death (Emery, 2002). Muscular dystrophies are caused by mutations in genes that encode for proteins including extracellular matrix (ECM) or nuclear matrix proteins (Blake et al., 2002). The most severe of muscular dystrophies result from mutations in the dystrophin-glycoprotein complex (DGC), the scaffold which localizes to the sarcolemma, providing mechanical stability to muscle (Hoffman et al., 1987). For example, one of the most common types of muscular dystrophy is Duchenne muscular dystrophy (DMD), which results in a lack of functional dystrophin protein, an important component of the DGC. Dystrophin plays a critical role in linking the cytoskeleton of muscle fibers to the ECM and transmembrane complexes (Prins et al., 2009). Without the presence of dystrophin, the mechanical stress from muscle contractions leads to damage of the sarcolemma and muscle fiber necrosis (Petrof et al., 1993). In addition to the direct effects of mechanical injury, the progression of muscular dystrophies also involves the activation of several pathological signaling pathways and cascades (Shin et al., 2013).
Studies have shown that cytosolic calcium concentrations are increased in mdx mice, the animal model of DMD, especially in the subsarcolemmal space (Mallouk et al., 2000). There is evidence that this may be associated with increased calcium uptake into the cells by both passive mechanisms, by increased leakage across channels, as well as active mechanisms, via physical openings of the sarcolemma (Mallouk et al., 2000; Wang et al., 2005). The role of calcium may be independent of membrane fragility, as studies have shown that increasing cytosolic influx by overexpressing transient receptor potential canonical 3 (TRPC3), an intracellular calcium release channel, results in a muscular dystrophy phenotype (Millay et al., 2009). Furthermore, inhibition of TRPC channels reduce dystrophic features of mdx mice (Millay et al., 2009). Notably, increased calpain activity has been shown in dystrophic muscle fibers, and inhibiting calpains reduces dystrophic pathology in mdx mice (Spencer and Mellgren, 2002).
Several studies analyzing muscle biopsies from humans with DMD as well as myofibers of mdx mice have demonstrated an enhanced inflammatory gene profile (Haslett et al., 2002; Porter et al., 2004). The source of the inflammatory molecules and cytokines is mostly attributed to immune cell infiltration by macrophages, T cells, and neutrophils (Evans et al., 2009). In fact, macrophage depletion has been shown to ameliorate muscle injury and muscle lesions in mdx mice (Wehling et al., 2001). Specifically, macrophages can exist either as M1, a proinflammatory state, or M2, which are generally anti-inflammatory (Tidball and Villalta, 2010). Activation of M1 macrophages is mediated by inflammatory cytokines such as TNF-α and interferon-γ (IFN-γ), and these cytokines also inhibit the transition to M2 macrophages (Tidball and Villalta, 2010). In contrast, anti-inflammatory cytokines such as IL-4 and IL-10 promote activation of M2 macrophages (Villalta et al., 2011). Furthermore, CD4+ T cells can produce inflammatory cytokines, and CD8+ T cells can trigger perforin-mediated muscle cell death; ablation of perforin or depletion of CD4+/CD8+ T cells resulted in reduced fiber necrosis and decreased pathology in mdx mice (Spencer et al., 2001, 1997). Finally, analysis of muscle biopsies from DMD patients have shown increases in these immune cells, in addition to neutrophils and mast cells (Evans et al., 2009). Finally, elevated levels of activated NF-κB and NF-κB-regulated inflammatory cytokines TNF-α and IL-1β have been shown in DMD patients as well as in different animal models of muscular dystrophy (Acharyya et al., 2007; Lammerding et al., 2004).
Increased oxidative stress, as assessed by lipid and protein oxidation, has also been reported in muscle of patients with DMD as well as in several animal models of muscular dystrophies (Haycock et al., 1996; Terrill et al., 2013). The major source of ROS in dystrophic muscle seems to be NADPH oxidase (NOX), which is upregulated in mdx muscle (Whitehead et al., 2010). Pharmacological inhibition of NOX protects against muscle damage in mdx mice (Whitehead et al., 2010). However, other sources of ROS exist, and include infiltrating neutrophils, which produce myeloperoxidase (MPO), mitochondria, and nitric oxide synthase (NOS) (Lawler, 2011). One of the mechanisms by which oxidative stress may be related to muscle loss in muscular dystrophy is via ROS-induced activation of NF-κB (Tidball and Wehling-Henricks, 2007). Interestingly, antioxidant treatments have been shown to reduce NF-κB activation and attenuate muscle wasting and myopathy in mdx mice (Hnia et al., 2007; Messina et al., 2006). Mitochondrial dysfunction is also a well-known feature of muscular dystrophies, resulting in up to 50% reductions in ATP production (Timpani et al., 2015). The connection between muscular dystrophy and mitochondrial dysfunction is thought to involve the increased calcium permeability, which can lead to calcium overload in the mitochondria, swelling, and enhanced opening of the mitochondrial permeability transition pore (Timpani et al., 2015). PGC-1α overexpression has been studied in murine models of muscular dystrophy as well, and has been shown to ameliorate muscle damage and improve muscular dystrophy (Chan et al., 2014).
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that are important for regulating ECM degradation, inflammation, fibrosis, and activation of cytokines and cell adhesion molecules (Page-McCaw et al., 2007). The most widely studied MMPs in skeletal muscle are MMP-2 and MMP-9. It has been suggested that increased levels of MMP-9 can target (proteolyze) β-dystroglycan in mdx mice, thus disrupting the link between the ECM and cell membrane (Matsumura et al., 2005; Roma et al., 2004). Serum levels of MMP-9 have been shown to be significantly higher in patients with DMD and to be linked to disease progression (Nadarajah et al., 2011). Several other MMPs have also been shown to be elevated in mdx muscle, and treatment with a broad spectrum inhibitor of MMPs reduced pathological features (Kumar et al., 2010). In addition to the effects of MMPs on breakdown of the cytoskeleton-ECM network causing sarcolemmal damage and fiber necrosis, MMPs can also activate latent inflammatory molecules and interfere with the process of muscle regeneration (Kumar et al., 2010; Li et al., 2009; Page-McCaw et al., 2007).
Furthermore, one of the major features of muscular dystrophies is fibrosis, or an excessive deposition of ECM proteins such as collagens and fibronectin, which impairs tissue function (Mann et al., 2011). Tissue fibrosis is also characterized by chronic inflammation and persistent production of pro-inflammatory cytokines (Mann et al., 2011). Endomysial fibrosis is the only pathological feature that significantly correlates with poor motor outcome (Desguerre et al., 2009). Cytokine members of the TGF-β superfamily play a role in the pathogenesis of fibrosis in most organs, including in skeletal muscle myopathies such as muscular dystrophy (Burks and Cohn, 2011). TGF-β1, for example, can promote fibrosis by enhancing collagen synthesis, promoting fibroblast growth and differentiation, and reducing expression of ECM degradation proteins (Biernacka et al., 2011). TGF-β1 can convert myoblasts into fibroblasts, suggesting that although satellite cells are activated, they can still differentiate into noncontractile tissue (Li et al., 2004). Several studies have shown that TGF-β levels are elevated in mdx mice and in several animal models of muscular dystrophies (Onofre-Oliveira et al., 2012), as well as in DMD patient muscle, in association with fibrosis, degree of pathology and clinical severity (Bernasconi et al., 1995; Song et al., 2017).
Apoptosis seems to be a relevant mechanism of muscle wasting in some muscular dystrophies, such as Bethlem myopathy and congenital muscular dystrophy; however, it does not appear to play a role in DMD pathogenesis (Miller and Girgenrath, 2006). For example, muscle from patients with merosin-deficient congenital muscular dystrophy type 1 A (MDC1A), caused by mutations in a laminin gene, exhibits signs of extensive apoptosis (Miller and Girgenrath, 2006). In a mouse model of laminin-deficient muscular dystrophy, overexpression of Bcl-2 (apoptosis inhibitor) or deletion of Bax (apoptosis inducer) reduced disease severity (Dominov et al., 2005; Girgenrath et al., 2004). In vitro, disruption of the binding of laminin-α2 to α-dystroglycan activates apoptosis via Akt inhibition (Langenbach and Rando, 2002). Likewise, in a mouse model of Bethlem myopathy and Ullrich congenital muscular dystrophy (Col6a1-knockout), there is an increased number of apoptotic nuclei. Treatment with cyclosporine A, an inhibitor of mitochondrial permeability transition pore opening, or inhibition of apoptosis with doxycycline attenuate disease progression (Davies et al., 2005; Irwin et al., 2003). However, in DMD, necrosis is primarily responsible for the death of myofibers (Tidball et al., 1995).
Interestingly, autophagy is impaired in muscle of mdx mice as well as DMD patients, which leads to accumulation of damaged organelles (De Palma et al., 2012). This attenuation of autophagy occurs along with activation of the Akt-mTOR pathway in mdx mice, and restoration of autophagy by chronic low-protein feeding improves pathological features (De Palma et al., 2012). Similarly, activation of AMP-activated protein kinase (AMPK) (which activates autophagy by activation of ULK1, a key initiator of autophagy, and inhibition of mTOR) by the agonist AICAR also improves pathology in mdx mice (Pauly et al., 2012). However, in other types of muscular dystrophy, such as in MDC1A, autophagy is actually highly upregulated, and inhibition of autophagy using 3-methyladenine has been shown to reduce dystrophic features in laminin-deficient mice (Carmignac et al., 2011).
Peripheral Artery Disease
Peripheral artery disease (PAD) refers to a group of diseases that affect arteries other than those supplying the brain or heart (Kullo and Rooke, 2016). PAD affects up to 27% of 45- to 74-year-olds, and with the expected increases in the elderly population, it is quickly becoming a major cause of morbidity (Wang et al., 2016). The most common cause of PAD is atherosclerosis, which most commonly affects the femoropopliteal or infrapopliteal arteries of the legs (Chen et al., 2013). Cycles of ischemia-reperfusion in PAD are thought to lead to significant muscle atrophy and degeneration. The earliest detailed report demonstrating that PAD is associated with abnormal skeletal muscle histology was published in 1977 (Mäkitie and Teräväinen, 1977). In this study, histochemical analysis of muscle biopsies obtained from the gastrocnemius and rectus femoris of PAD patients demonstrated myofiber atrophy, myofibrillar disorganization, and myofiber necrosis and phagocytosis (Mäkitie and Teräväinen, 1977). This was followed by a light- and electron microscopy study of gastrocnemius biopsies from PAD patients demonstrating significant morphological abnormalities characteristic of skeletal muscle myopathies, including angular fibers, fiber type grouping, central nuclei, and abundant connective tissue, or endomysial fibrosis (Hedberg et al., 1988). At the ultrastructural level, abnormalities in the contractile elements of myofibers have also been shown, such as Z-line disorganization and fragmentation as well as disintegration of the myofilaments (Hedberg et al., 1988). More recently, the use of quantitative fluorescence microscopy has allowed for more detailed studies of the changes that occur in PAD skeletal muscle. These studies have demonstrated that the average cross-sectional area of PAD myofibers is reduced ~30% compared to non-PAD controls (Weiss et al., 2013). There are also significant reductions in major and minor axes, equivalent diameter, perimeter, and density, and a significant increase in roundness of myofibers (Koutakis et al., 2015b). There is also evidence that there is selective degeneration of type II myofibers in PAD, as type II fibers demonstrate significantly greater reductions in CSA compared to type I and type I/II fibers (Koutakis et al., 2014). There is also a lower relative frequency of type II fibers compared to type I and type I/II fibers (Koutakis et al., 2014). In addition to the morphometric changes in skeletal muscle, PAD patients have also been shown to have significantly lower knee extension/flexion force-velocity parameters compared to non-PAD controls, assessed by isokinetic dynamometry, and lower limb muscle strength correlated with maximum walking distance (Dziubek et al., 2015). Likewise, PAD patients demonstrated reduced peak torque during isometric plantar flexion contractions compared to non-PAD controls, even at baseline and before the onset of ischemic pain (Schieber et al., 2017).
Interestingly, despite altered hemodynamics, elderly PAD patients have been shown to have similar basal MPS rates as healthy, non-PAD control elderly individuals. Furthermore, PAD patients were also shown to have a normal anabolic response to amino acid ingestion (Killewich et al., 2007). Thus, the skeletal muscle myopathy of PAD may not involve MPS-related mechanisms. Instead, the myopathy of PAD is believed to involve increases in ROS production and oxidative stress. Several studies have consistently demonstrated that PAD patients exhibit higher levels of oxidative stress markers in circulation compared to non-PAD controls at rest as well as in response to exercise. Likewise, there is clear evidence of oxidative stress in skeletal muscle as well (Koutakis et al., 2018). Following hindlimb ischemia (HLI) in mice, myofibers of ischemic skeletal muscle demonstrate increased levels of 4-HNE, a marker of lipid peroxidation, as well as carbonyl groups, a measure of protein oxidation (Pipinos et al., 2008c). Similarly, myofibers of gastrocnemius muscle from PAD patients also demonstrate greater labeling of carbonyl groups (30%) and HNE adducts (40%) compared to non-PAD control muscle (Weiss et al., 2013). Furthermore, the extent of oxidative damage is associated with significant atrophy of the myofibers, and oxidative stress markers are also correlated with walking distances of PAD patients (Dopheide et al., 2013; Weiss et al., 2013). PAD is also associated with mitochondrial dysfunction, and a number of studies in both human patients as well as murine models of hindlimb ischemia have shown impairments in skeletal muscle mitochondrial respiration and electron transport chain complex activity (Pipinos et al., 2008d, 2006, 2003). Importantly, skeletal muscle mitochondrial dysfunction in PAD is also associated with myofiber atrophy and abnormal myofiber morphology (Koutakis et al., 2015a).
Discussion
Muscle protein metabolism is an ever-changing dynamic system that involves both anabolic and catabolic mechanisms. When this equilibrium is altered to favor catabolic pathways, skeletal muscle atrophy occurs. This perturbation in protein balance can arise in response to various pathophysiological processes and conditions (Table 1). The proteolytic systems that govern MPD (i.e., the UPS, autophagy-lysosomal, caspase, calpains) appear to differentially upregulate in response to different atrophic conditions, as discussed in this review. In addition, skeletal muscle atrophy associated with certain catabolic states may also involve inhibition of MPS or resistance to anabolic stimuli (such as exercise and nutrient intake). Other factors that have also been implicated in muscle atrophy across certain pathological and physiological conditions include mitochondrial dysfunction, enhanced ROS production, and enhanced inflammatory processes.
Table 1:
The major mechanisms of skeletal muscle degradation are outlined for each condition visually in the table above.
| Overview of Skeletal Muscle Atrophy Mechanism by Condition | ||||||
|---|---|---|---|---|---|---|
| Cancer Cachexia | Obesity-Related Chronic Inflammation | Sarcopenia | Mechanical Unloading | Muscular Dystrophy | Peripheral Artery Disease | |
| UPS | X | X | ||||
| Reduced MPS | X | X | X | |||
| Inflammatory cytokines | X | X | X | X | X | |
| ROS | X | X | X | X | X | X |
| Mitochondrial dysfunction | X | X | X | X | X | X |
| Apoptosis | X | X | ||||
| Motor unit loss | X | |||||
| Reduced myonuclear efficiency | X | |||||
| Calpains | X | X | ||||
| Autophagy dysregulation | X | |||||
| Anabolic Resistance | X | X | X | |||
In 2001, Bodine and colleagues identified the mTOR pathway as a crucial regulator of muscle hypertrophy (Bodine et al., 2001b) as well as the involvement of the ubiquitin ligases, MuRF1 and MAFbx, in the muscle atrophy induced by different atrophic conditions (Bodine et al., 2001a). Since then, the primary focus during the past 20 years in the field of skeletal muscle atrophy has been to further identify and elucidate the molecular mechanisms that lead to muscle atrophy under different conditions. However, most researchers have focused on investigating the mechanisms of muscle atrophy in specific, isolated conditions. It is important to note, however, that there is significant overlap in many of the conditions associated with muscle atrophy. For example, PAD prevalence increases with age, and individuals with PAD may be at a greater risk for sarcopenia. Both sarcopenia and PAD are conditions that lead to significant losses of physical function (mechanical unloading), and notably, PAD patients with sarcopenia have also been shown to have decreased function compared to PAD patients without sarcopenia (Addison et al., 2018). Therefore, disuse atrophy is also strongly implicated in both PAD and sarcopenia development (Bell et al., 2016). Due to these overlapping etiologies, it is important to differentiate between the underlying factors that may be secondary to the condition of interest (Nishikawa et al., 2021). Additionally, most of the research has involved reductionist experiments, focused on specific proteins and pathway components. Despite new innovations in techniques and the identification of novel regulators of molecular signaling pathways, these factors are still understudied in humans. While mechanisms have been more clearly defined in vitro and in animal models, there have been inconsistencies related to the involvement of different factors in the muscle atrophy induced by different conditions in clinical populations. Due to the complexity of the signaling pathways involved and the plethora of factors that contribute to atrophy in response to varying catabolic stimuli, future studies are required to better define the involved pathogenesis. These studies should contain a human component and include larger sample sizes to account for large inter-personal variability in the responses to these stimuli.
Currently, physical exercise and nutritional strategies are considered the best approaches to attenuate muscle atrophy across various catabolic circumstances (Magne et al., 2013; Shen et al., 2018). Beyond this, considerable research has identified several novel drug targets to counter muscle atrophy induced by different conditions. However, no efficacious drug counteracting muscle atrophy has yet to reach the clinic. Although many in vitro and animal studies have demonstrated that various investigational drugs can protect against muscle atrophy, human clinical trials often result in important side effects or no significant improvements (Chen et al., 2021; Sartori et al., 2021). Only a limited number of therapeutics have been shown to be safe and effective in clinical trials. This highlights the importance of future development of novel therapies. In addition, new methods of combination therapy may be worthwhile since several intracellular degradation pathways may be upregulated simultaneously. Despite diverse etiology, muscles atrophying in response to different catabolic conditions share many common features. Thus, in the journey for the development of rational treatment strategies, targeting shared mechanisms and pathways associated with muscle wasting in a wide variety of atrophy conditions is logical when considering the potential impact.
Anabolic resistance seemingly afflicts individuals who are obese, sarcopenic, or are experiencing immobilization. One of the common lifestyle factors that is predominant within each of these populations is physical inactivity and mechanical unloading (muscular disuse). Anabolic resistance in each of these conditions appears to be partially mediated by the impaired integration of both mechanical load and nutrient signals, referred to as anabolic or inactivity-induced insulin resistance. It is possible that physical inactivity itself is one of the main contributors to anabolic resistance that is independent from the other factors such as age or body composition. Recently, researchers have indicated a necessity in separating the effects of ageing and disuse to specifically isolate the negative effects of age (Brocca et al., 2017a). It was found that the atrophic effects of age are exacerbated with disuse, indicating that physical inactivity is a major contributor to the loss of mass and anabolic resistance in this population. Like the age specific findings of Brocca et al., (2017a), the individual effects of obesity with and without chronic physical activity need to be elucidated to distinguish if separate mechanisms such as increased intramuscular ROS facilitate anabolic resistance in these populations as a separate mechanism.
Caspase-mediated apoptosis appears to be confined to cachexia and DMD and does not appear to play a major role in the other conditions discussed in this review. Additionally, increases in MPD via the UPS has been shown to occur with inflammatory signaling cascades induced by TNF-α, IL-6, and INF-γ in both cancer cachexia and DMD patients. These signaling molecules are primarily released from macrophages that infiltrate into the tissue to degrade cancer cells or the mutated dystrophin proteins. It has been hypothesized that macrophage-infiltrated, hypertrophied adipose tissue can secrete TNF-α and IL-6 into circulation and induce UPS-mediated MPD of skeletal muscle tissue. However, a definite link between adipocyte-associated inflammatory phenotypes and skeletal muscle atrophy has yet to be established in wild-type animals or in humans.
Increased ROS production and associated mitochondrial dysfunction appear to be the main mechanisms that are shared between all the reviewed conditions. However, despite this similar presentation of increased ROS, the mechanism for the increase and the sequential effects are somewhat distinct for various pathologies. For example, ROS production associated with cachexia and DMD has been shown to be modulated by TNF-α and the infiltration of immune cells, leading to UPS-mediated MPD. In sarcopenia, increased ROS production is mainly associated with mitochondrial dysfunction and has been postulated to be partially responsible for inducing denervation of MUs. In PAD, ROS production is thought to be driven by cycles of ischemia-reperfusion. It is important to note, however, that the majority of PAD patients are also elderly and may be sarcopenic; thus, large increases in ROS production in PAD could also increase the rate of ROS-driven denervation and MU loss. Therefore, the combination of distinct factors could mediate muscle atrophy via several mechanisms within the same condition, further highlighting the complexity of skeletal muscle atrophy research.
Conclusion
In each of the conditions listed throughout the review there is an overall shift in the protein regulation within skeletal muscle that favors MPD. Despite the shared atrophic effect these conditions have on muscle, the mechanisms in which they operate are somewhat distinct. Regardless of their apparent uniqueness, these situational conditions and signaling events are not mutually exclusive and multiple overlapping circumstances can augment protein breakdown in a synergistic manner. This review highlights the major mechanisms involved to better summarize the current understanding of the role the individual complex proteolytic events play across disease conditions. Future interventions that target the specific mechanisms will be critical in the development of novel treatments for patients suffering from these conditions.
Highlights.
The ubiquitin-proteasome is involved in muscle degradation in cachexia and unloading.
Co-morbid conditions such as obesity and PAD increase ROS and muscle loss with ageing.
Without co-morbidities, anabolic resistance leads to muscle loss in ageing.
Acknowledgements:
Graphical Abstract and Figure 1 were created with BioRender.com.
Figure 1:
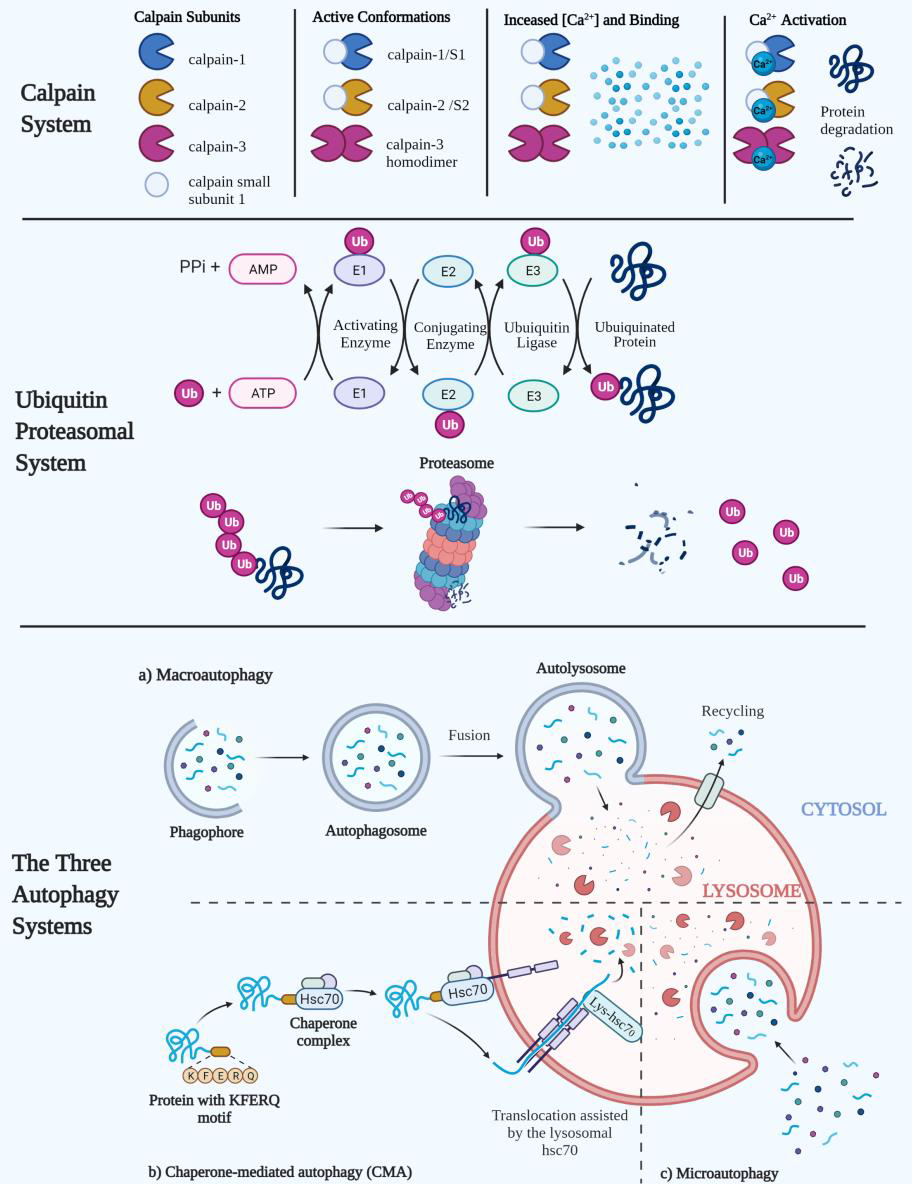
Above indicates the three major proteolytic systems that regulate skeletal muscle atrophy. Calpain activation can induce protein degradation by calcium dependent binding. The ubiquitin proteasomal system can degrade proteins through post-translational polyubiquitination through a series of E1, E2, and E3 relaying mechanisms. The polyubiquinated protein is then subsequently shuttled into the 26S proteasome for degradation. The three autophagy pathways degrade polypeptides, proteins, and organelles by integrating substrates into the lysosome. Macro-autophagy and micro-autophagy are non-specific in their degradation targets, but chaperone-mediated autophagy uses the specific pentapeptide sequence (KFERQ) to selectively bind, shuttle, and translocate proteins into the lysosome with the help of cytosolic and lysosomal heat shock cognate 71 kDa protein (Hsc70).
Funding:
This work was supported by the National Institute on Aging at the National Institutes of Health under [grant number R01AG064420] to [PK]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PML, Carathers M, Li Z-W, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC, 2007. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest 117, 889–901. 10.1172/JCI30556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addison O, Prior SJ, Kundi R, Serra MC, Katzel LI, Gardner AW, Ryan AS, 2018. Sarcopenia in Peripheral Arterial Disease: Prevalence and Effect on Functional Status. Arch. Phys. Med. Rehabil 99, 623–628. 10.1016/j.apmr.2017.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmedov D, Berdeaux R, 2013. The effects of obesity on skeletal muscle regeneration. Front. Physiol 4, 371. 10.3389/fphys.2013.00371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DL, Bandstra ER, Harrison BC, Thorng S, Stodieck LS, Kostenuik PJ, Morony S, Lacey DL, Hammond TG, Leinwand LL, Argraves WS, Bateman TA, Barth JL, 2009. Effects of spaceflight on murine skeletal muscle gene expression. J. Appl. Physiol. Bethesda Md 1985 106, 582–595. 10.1152/japplphysiol.90780.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, Goldberg AL, Ulfhake B, 2010. Muscle Wasting in Aged, Sarcopenic Rats Is Associated with Enhanced Activity of the Ubiquitin Proteasome Pathway. J. Biol. Chem 285, 39597–39608. 10.1074/jbc.M110.129718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton PJ, Greenhaff PL, Phillips SM, Bodine SC, Adams CM, Lang CH, 2016. Control of skeletal muscle atrophy in response to disuse: clinical/preclinical contentions and fallacies of evidence. Am. J. Physiol. Endocrinol. Metab 311, E594–604. 10.1152/ajpendo.00257.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, Furlow JD, Bodine SC, 2011. Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J. Physiol 589, 4759–4776. 10.1113/jphysiol.2011.212845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, West DWD, Marshall AG, Marcotte GR, Baar K, Bodine SC, 2017. Muscle-specific and age-related changes in protein synthesis and protein degradation in response to hindlimb unloading in rats. J. Appl. Physiol. Bethesda Md 1985 122, 1336–1350. 10.1152/japplphysiol.00703.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajotto G, Sato Y, Kitaura Y, Shimomura Y, 2011. Effect of branched-chain amino acid supplementation during unloading on regulatory components of protein synthesis in atrophied soleus muscles. Eur. J. Appl. Physiol 111, 1815–1828. 10.1007/s00421-010-1825-8 [DOI] [PubMed] [Google Scholar]
- Baltgalvis KA, Berger FG, Pena MMO, Davis JM, Muga SJ, Carson JA, 2008. Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol.-Regul. Integr. Comp. Physiol 294, R393–R401. 10.1152/ajpregu.00716.2007 [DOI] [PubMed] [Google Scholar]
- Beals JW, Burd NA, Moore DR, van Vliet S, 2019. Obesity Alters the Muscle Protein Synthetic Response to Nutrition and Exercise. Front. Nutr 6. 10.3389/fnut.2019.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals JW, Skinner SK, McKenna CF, Poozhikunnel EG, Farooqi SA, Vliet S. van, Martinez IG, Ulanov AV, Li Z, Paluska SA, Burd NA, 2018. Altered anabolic signalling and reduced stimulation of myofibrillar protein synthesis after feeding and resistance exercise in people with obesity. J. Physiol 596, 5119–5133. 10.1113/JP276210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcastro AN, 1993. Skeletal muscle calcium-activated neutral protease (calpain) with exercise. J. Appl. Physiol. Bethesda Md 1985 74, 1381–1386. 10.1152/jappl.1993.74.3.1381 [DOI] [PubMed] [Google Scholar]
- Belcastro AN, Shewchuk LD, Raj DA, 1998. Exercise-induced muscle injury: a calpain hypothesis. Mol. Cell. Biochem 179, 135–145. 10.1023/a:1006816123601 [DOI] [PubMed] [Google Scholar]
- Belizário JE, Lorite MJ, Tisdale MJ, 2001. Cleavage of caspases-1, −3, −6, −8 and −9 substrates by proteases in skeletal muscles from mice undergoing cancer cachexia. Br. J. Cancer 84, 1135–1140. 10.1054/bjoc.2001.1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KE, von Allmen MT, Devries MC, Phillips SM, 2016. Muscle Disuse as a Pivotal Problem in Sarcopenia-related Muscle Loss and Dysfunction. J. Frailty Aging 5, 33–41. 10.14283/jfa.2016.78 [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G, Sharon M, 2014. Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 4, 862–884. 10.3390/biom4030862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, Cornelio F, Morandi L, Mantegazza R, 1995. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Invest. 96, 1137–1144. 10.1172/JCI118101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagetti B, Simó R, 2021. GH/IGF-1 Abnormalities and Muscle Impairment: From Basic Research to Clinical Practice. Int. J. Mol. Sci 22, E415. 10.3390/ijms22010415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A, Dobaczewski M, Frangogiannis NG, 2011. TGF-β signaling in fibrosis. Growth Factors Chur Switz. 29, 196–202. 10.3109/08977194.2011.595714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing C, Brown M, King P, Collins P, Tisdale MJ, Williams G, 2000. Increased Gene Expression of Brown Fat Uncoupling Protein (UCP)1 and Skeletal Muscle UCP2 and UCP3 in MAC16-induced Cancer Cachexia. Cancer Res. 60, 2405–2410. [PubMed] [Google Scholar]
- Bisht B, Goel HL, Dey CS, 2007. Focal adhesion kinase regulates insulin resistance in skeletal muscle. Diabetologia 50, 1058–1069. 10.1007/s00125-007-0591-6 [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE, 2002. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev 82, 291–329. 10.1152/physrev.00028.2001 [DOI] [PubMed] [Google Scholar]
- Bodine SC, Baehr LM, 2014. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab 307, E469–484. 10.1152/ajpendo.00204.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ, 2001a. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. 10.1126/science.1065874 [DOI] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD, 2001b. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol 3, 1014–1019. 10.1038/ncb1101-1014 [DOI] [PubMed] [Google Scholar]
- Bosaeus I, Daneryd P, Svanberg E, Lundholm K, 2001. Dietary intake and resting energy expenditure in relation to weight loss in unselected cancer patients. Int. J. Cancer 93, 380–383. 10.1002/ijc.1332 [DOI] [PubMed] [Google Scholar]
- Bose C, Alves I, Singh P, Palade PT, Carvalho E, Børsheim E, Jun S-R, Cheema A, Boerma M, Awasthi S, Singh SP, 2020. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell 19, e13261. 10.1111/acel.13261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink M, Price SR, Chrast J, Bailey JL, Anwar A, Mitch WE, Delafontaine P, 2001. Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology 142, 1489–1496. 10.1210/endo.142.4.8082 [DOI] [PubMed] [Google Scholar]
- Brocca L, Cannavino J, Coletto L, Biolo G, Sandri M, Bottinelli R, Pellegrino MA, 2012. The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J. Physiol 590, 5211–5230. 10.1113/jphysiol.2012.240267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca L, McPhee JS, Longa E, Canepari M, Seynnes O, De Vito G, Pellegrino MA, Narici M, Bottinelli R, 2017a. Structure and function of human muscle fibres and muscle proteome in physically active older men. J. Physiol 595, 4823–4844. 10.1113/JP274148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca L, Toniolo L, Reggiani C, Bottinelli R, Sandri M, Pellegrino MA, 2017b. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 595, 1143–1158. 10.1113/JP273097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook Matthew S., Wilkinson DJ, Mitchell WK, Lund JN, Phillips BE, Szewczyk NJ, Greenhaff PL, Smith K, Atherton PJ, 2016. Synchronous deficits in cumulative muscle protein synthesis and ribosomal biogenesis underlie age‐related anabolic resistance to exercise in humans. J. Physiol 594, 7399–7417. 10.1113/JP272857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook MS, Wilkinson DJ, Phillips BE, Perez‐Schindler J, Philp A, Smith K, Atherton PJ, 2016. Skeletal muscle homeostasis and plasticity in youth and ageing: impact of nutrition and exercise. Acta Physiol. Oxf. Engl 216, 15–41. 10.1111/apha.12532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Lee DE, Rosa-Caldwell ME, Brown LA, Perry RA, Haynie WS, Huseman K, Sataranatarajan K, Remmen HV, Washington TA, Wiggs MP, Greene NP, 2018. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 987–1002. 10.1002/jcsm.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP, 2017. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 8, 926–938. 10.1002/jcsm.12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME, 1999a. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. 10.1016/s0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME, 1999b. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. 10.1016/s0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- Buck M, Chojkier M, 1996. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J. 15, 1753–1765. [PMC free article] [PubMed] [Google Scholar]
- Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, Kratz M, 2018. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol 9, 1–58. 10.1002/cphy.c170040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burks TN, Cohn RD, 2011. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 1, 19. 10.1186/2044-5040-1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch WA, Stromer MH, Goll DE, Suzuki A, 1972. Ca2+-SPECIFIC REMOVAL OF Z LINES FROM RABBIT SKELETAL MUSCLE. J. Cell Biol 52, 367–381. 10.1083/jcb.52.2.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquets S, Deans C, Figueras M, Moore-Carrasco R, López-Soriano FJ, Fearon KCH, Argilés JM, 2007. Apoptosis is present in skeletal muscle of cachectic gastro-intestinal cancer patients. Clin. Nutr. Edinb. Scotl 26, 614–618. 10.1016/j.clnu.2007.06.005 [DOI] [PubMed] [Google Scholar]
- Byrd SK, 1992. Alterations in the sarcoplasmic reticulum: a possible link to exercise-induced muscle damage. Med. Sci. Sports Exerc 24, 531–536. [PubMed] [Google Scholar]
- Caldeira S, 2009. Welcome to EMBO Molecular Medicine! EMBO Mol. Med 1, 1. 10.1002/emmm.200900010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MD, Duan J, Samuelson AT, Gaffrey MJ, Merrihew GE, Egertson JD, Wang L, Bammler TK, Moore RJ, White CC, Kavanagh TJ, Voss JG, Szeto HH, Rabinovitch PS, MacCoss MJ, Qian W-J, Marcinek DJ, 2019. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med 134, 268–281. 10.1016/j.freeradbiomed.2018.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, McComas AJ, Petito F, 1973. Physiological changes in ageing muscles. J. Neurol. Neurosurg. Psychiatry 36, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavino J, Brocca L, Sandri M, Bottinelli R, Pellegrino MA, 2014. PGC1-α over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J. Physiol 592, 4575–4589. 10.1113/jphysiol.2014.275545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA, 2015. The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. J. Physiol 593, 1981–1995. 10.1113/jphysiol.2014.286740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariuk P, Lorite MJ, Todorov PT, Field WN, Wigmore SJ, Tisdale MJ, 1997. Induction of cachexia in mice by a product isolated from the urine of cachectic cancer patients. Br. J. Cancer 76, 606–613. 10.1038/bjc.1997.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignac V, Svensson M, Körner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, Durbeej M, 2011. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet 20, 4891–4902. 10.1093/hmg/ddr427 [DOI] [PubMed] [Google Scholar]
- Carson JA, Baltgalvis KA, 2010. Interleukin-6 as a Key Regulator of Muscle Mass during Cachexia. Exerc. Sport Sci. Rev 38, 168–176. 10.1097/JES.0b013e3181f44f11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang M-L, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S, 2001. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain11Edited by J. Karn. J. Mol. Biol 306, 717–726. 10.1006/jmbi.2001.4448 [DOI] [PubMed] [Google Scholar]
- Chan MC, Rowe GC, Raghuram S, Patten IS, Farrell C, Arany Z, 2014. Post-natal induction of PGC-1α protects against severe muscle dystrophy independently of utrophin. Skelet. Muscle 4, 2. 10.1186/2044-5040-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand A, Wyke SM, Tisdale MJ, 2005. Effect of cancer cachexia on the activity of tripeptidyl-peptidase II in skeletal muscle. Cancer Lett. 218, 215–222. 10.1016/j.canlet.2004.07.047 [DOI] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, Goh YPS, 2011. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol 11, 738–749. 10.1038/nri3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhang H, Chi M, Yang Q, Guo C, 2021. Drugs for the Treatment of Muscle Atrophy, Background and Management of Muscular Atrophy. IntechOpen. 10.5772/intechopen.93503 [DOI] [Google Scholar]
- Chen Q, Smith CY, Bailey KR, Wennberg PW, Kullo IJ, 2013. Disease Location Is Associated With Survival in Patients With Peripheral Arterial Disease. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis 2. 10.1161/JAHA.113.000304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG T, PARK JS, KIM S, MONTES N, WALSTON J, HÖKE A, 2017. EVIDENCE FOR DYING-BACK AXONAL DEGENERATION IN AGE-ASSOCIATED SKELETAL MUSCLE DECLINE. Muscle Nerve 55, 894–901. 10.1002/mus.25267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisterna BA, Vargas AA, Puebla C, Fernández P, Escamilla R, Lagos CF, Matus MF, Vilos C, Cea LA, Barnafi E, Gaete H, Escobar DF, Cardozo CP, Sáez JC, 2020. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat. Commun 11, 1073. 10.1038/s41467-019-14063-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisterna BA, Vargas AA, Puebla C, Sáez JC, 2016. Connexin hemichannels explain the ionic imbalance and lead to atrophy in denervated skeletal muscles. Biochim. Biophys. Acta BBA - Mol. Basis Dis 1862, 2168–2176. 10.1016/j.bbadis.2016.08.020 [DOI] [PubMed] [Google Scholar]
- Coelho M, Oliveira T, Fernandes R, 2013. Biochemistry of adipose tissue: an endocrine organ. Arch. Med. Sci. AMS 9, 191–200. 10.5114/aoms.2013.33181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KH, Herzog W, MacDonald GZ, Reimer RA, Rios JL, Smith IC, Zernicke RF, Hart DA, 2018. Obesity, Metabolic Syndrome, and Musculoskeletal Disease: Common Inflammatory Pathways Suggest a Central Role for Loss of Muscle Integrity. Front. Physiol 9, 112. 10.3389/fphys.2018.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins P, Bing C, McCulloch P, Williams G, 2002. Muscle UCP-3 mRNA levels are elevated in weight loss associated with gastrointestinal adenocarcinoma in humans. Br. J. Cancer 86, 372–375. 10.1038/sj.bjc.6600074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossland H, Skirrow S, Puthucheary ZA, Constantin-Teodosiu D, Greenhaff PL, 2019. The impact of immobilisation and inflammation on the regulation of muscle mass and insulin resistance: different routes to similar end-points. J. Physiol 597, 1259–1270. 10.1113/JP275444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, Martin FC, Michel J-P, Rolland Y, Schneider SM, Topinková E, Vandewoude M, Zamboni M, European Working Group on Sarcopenia in Older People, 2010. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 39, 412–423. 10.1093/ageing/afq034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Hu W, Lim B, Dice JF, 1998. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 9, 1995–2010. 10.1091/mbc.9.8.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ, 2005. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. 19, 1–22. 10.1096/fj.04-2640fje [DOI] [PubMed] [Google Scholar]
- Cuthbertson DJ, Babraj J, Smith K, Wilkes E, Fedele MJ, Esser K, Rennie M, 2006. Anabolic signaling and protein synthesis in human skeletal muscle after dynamic shortening or lengthening exercise. Am. J. Physiol. Endocrinol. Metab 290, E731–738. 10.1152/ajpendo.00415.2005 [DOI] [PubMed] [Google Scholar]
- Daemen S, Gemmink A, Brouwers B, Meex RCR, Huntjens PR, Schaart G, Moonen-Kornips E, Jörgensen J, Hoeks J, Schrauwen P, Hesselink MKC, 2018. Distinct lipid droplet characteristics and distribution unmask the apparent contradiction of the athlete’s paradox. Mol. Metab 17, 71–81. 10.1016/j.molmet.2018.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton BH, Power GA, Vandervoort AA, Rice CL, 2010. Power loss is greater in old men than young men during fast plantar flexion contractions. J. Appl. Physiol 109, 1441–1447. 10.1152/japplphysiol.00335.2010 [DOI] [PubMed] [Google Scholar]
- D’Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Canepari M, Saltin B, Bottinelli R, 2003. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J. Physiol 552, 499–511. 10.1113/jphysiol.2003.046276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antona G, Pellegrino MA, Carlizzi CN, Bottinelli R, 2007. Deterioration of contractile properties of muscle fibres in elderly subjects is modulated by the level of physical activity. Eur. J. Appl. Physiol 100, 603–611. 10.1007/s00421-007-0402-2 [DOI] [PubMed] [Google Scholar]
- Davies JE, Wang L, Garcia-Oroz L, Cook LJ, Vacher C, O’Donovan DG, Rubinsztein DC, 2005. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med 11, 672–677. 10.1038/nm1242 [DOI] [PubMed] [Google Scholar]
- de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ, 2007. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J. Physiol 585, 241–251. 10.1113/jphysiol.2007.142828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro GS, Simoes E, Lima JDCC, Ortiz-Silva M, Festuccia WT, Tokeshi F, Alcântara PS, Otoch JP, Coletti D, Seelaender M, 2019. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 11, 1264. 10.3390/cancers11091264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M, Sandri M, Clementi E, 2012. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 3, e418. 10.1038/cddis.2012.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desguerre I, Mayer M, Leturcq F, Barbet J-P, Gherardi RK, Christov C, 2009. Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol 68, 762–773. 10.1097/NEN.0b013e3181aa31c2 [DOI] [PubMed] [Google Scholar]
- Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC, 1980. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 69, 491–497. 10.1016/s0149-2918(05)80001-3 [DOI] [PubMed] [Google Scholar]
- Di Meo S, Iossa S, Venditti P, 2017. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J. Endocrinol 233, R15–R42. 10.1530/JOE-16-0598 [DOI] [PubMed] [Google Scholar]
- Dirks ML, Wall BT, Snijders T, Ottenbros CLP, Verdijk LB, van Loon LJC, 2014. Neuromuscular electrical stimulation prevents muscle disuse atrophy during leg immobilization in humans. Acta Physiol. Oxf. Engl 210, 628–641. 10.1111/apha.12200 [DOI] [PubMed] [Google Scholar]
- Dirks ML, Wall BT, van de Valk B, Holloway TM, Holloway GP, Chabowski A, Goossens GH, van Loon LJC, 2016. One Week of Bed Rest Leads to Substantial Muscle Atrophy and Induces Whole-Body Insulin Resistance in the Absence of Skeletal Muscle Lipid Accumulation. Diabetes 65, 2862–2875. 10.2337/db15-1661 [DOI] [PubMed] [Google Scholar]
- Doherty TJ, Brown WF, 1997. Age-related changes in the twitch contractile properties of human thenar motor units. J. Appl. Physiol 82, 93–101. 10.1152/jappl.1997.82.1.93 [DOI] [PubMed] [Google Scholar]
- Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Beermann ML, Miller JB, 2005. Muscle-specific BCL2 expression ameliorates muscle disease in laminin {alpha}2-deficient, but not in dystrophin-deficient, mice. Hum. Mol. Genet 14, 1029–1040. 10.1093/hmg/ddi095 [DOI] [PubMed] [Google Scholar]
- Dopheide JF, Doppler C, Scheer M, Obst V, Radmacher M-C, Radsak MP, Gori T, Warnholtz A, Fottner C, Münzel T, Daiber A, Espinola-Klein C, 2013. Critical limb ischaemia is characterised by an increased production of whole blood reactive oxygen species and expression of TREM-1 on neutrophils. Atherosclerosis 229, 396–403. 10.1016/j.atherosclerosis.2013.05.029 [DOI] [PubMed] [Google Scholar]
- Dupont-Versteegden EE, 2006. Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol. WJG 12, 7463–7466. 10.3748/wjg.v12.i46.7463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziubek W, Bulioska K, Stefaoska M, Woźniewski M, Kropielnicka K, Jasioski T, Jasioski R, Pilch U, Dąbrowska G, Skórkowska-Telichowska K, Wojcieszczyk-Latos J, Kałka D, Janus A, Zywar K, Paszkowski R, Szuba A, 2015. Peripheral arterial disease decreases muscle torque and functional walking capacity in elderly. Maturitas 81, 480–486. 10.1016/j.maturitas.2015.06.001 [DOI] [PubMed] [Google Scholar]
- Ebisui C, Tsujinaka T, Morimoto T, Kan K, Iijima S, Yano M, Kominami E, Tanaka K, Monden M, 1995. Interleukin-6 induces proteolysis by activating intracellular proteases (cathepsins B and L, proteasome) in C2C12 myotubes. Clin. Sci. Lond. Engl. 1979 89, 431–439. 10.1042/cs0890431 [DOI] [PubMed] [Google Scholar]
- Edén E, Edström S, Bennegård K, Scherstén T, Lundholm K, 1984. Glucose Flux in Relation to Energy Expenditure in Malnourished Patients with and without Cancer during Periods of Fasting and Feeding. Cancer Res. 44, 1718–1724. [PubMed] [Google Scholar]
- Eley Helen L., Russell ST, Baxter JH, Mukerji P, Tisdale MJ, 2007. Signaling pathways initiated by beta-hydroxy-beta-methylbutyrate to attenuate the depression of protein synthesis in skeletal muscle in response to cachectic stimuli. Am. J. Physiol. Endocrinol. Metab 293, E923–931. 10.1152/ajpendo.00314.2007 [DOI] [PubMed] [Google Scholar]
- Eley HL, Russell ST, Tisdale MJ, 2007. Attenuation of muscle atrophy in a murine model of cachexia by inhibition of the dsRNA-dependent protein kinase. Br. J. Cancer 96, 1216–1222. 10.1038/sj.bjc.6603704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley HL, Skipworth RJE, Deans D. a. C., Fearon KCH, Tisdale MJ, 2008. Increased expression of phosphorylated forms of RNA-dependent protein kinase and eukaryotic initiation factor 2alpha may signal skeletal muscle atrophy in weight-losing cancer patients. Br. J. Cancer 98, 443–449. 10.1038/sj.bjc.6604150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley HL, Tisdale MJ, 2007a. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J. Biol. Chem 282, 7087–7097. 10.1074/jbc.M610378200 [DOI] [PubMed] [Google Scholar]
- Eley HL, Tisdale MJ, 2007b. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J. Biol. Chem 282, 7087–7097. 10.1074/jbc.M610378200 [DOI] [PubMed] [Google Scholar]
- Elsasser S, Chandler-Militello D, Müller B, Hanna J, Finley D, 2004. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem 279, 26817–26822. 10.1074/jbc.M404020200 [DOI] [PubMed] [Google Scholar]
- Emery AEH, 2002. The muscular dystrophies. Lancet Lond. Engl 359, 687–695. 10.1016/S0140-6736(02)07815-7 [DOI] [PubMed] [Google Scholar]
- Emery PW, Edwards RH, Rennie MJ, Souhami RL, Halliday D, 1984. Protein synthesis in muscle measured in vivo in cachectic patients with cancer. Br. Med. J. Clin. Res. Ed 289, 584–586. 10.1136/bmj.289.6445.584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- English KL, Paddon-Jones D, 2010. Protecting muscle mass and function in older adults during bed rest. Curr. Opin. Clin. Nutr. Metab. Care 13, 34–39. 10.1097/MCO.0b013e328333aa66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund DA, Murach KA, Dungan CM, Figueiredo VC, Vechetti IJ, Dupont-Versteegden EE, McCarthy JJ, Peterson CA, 2020. Depletion of resident muscle stem cells negatively impacts running volume, physical function, and muscle fiber hypertrophy in response to lifelong physical activity. Am. J. Physiol. Cell Physiol 318, C1178–C1188. 10.1152/ajpcell.00090.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Grange RW, 2009. Immune-mediated mechanisms potentially regulate the disease time-course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R 1, 755–768. 10.1016/j.pmrj.2009.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K, 1992. Fearon KCHThe mechanisms and treatment of weight loss in cancer. Proc Nut Soc 51: 251–265. Proc. Nutr. Soc. 51, 251–265. 10.1079/PNS19920036 [DOI] [PubMed] [Google Scholar]
- Fernando R, Drescher C, Nowotny K, Grune T, Castro JP, 2019. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med 132, 58–66. 10.1016/j.freeradbiomed.2018.08.037 [DOI] [PubMed] [Google Scholar]
- Fiori MC, Figueroa V, Zoghbi ME, Saéz JC, Reuss L, Altenberg GA, 2012. Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem 287, 40826–40834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Igoudjil A, Magrané J, Li Y, Hansen JM, Manfredi G, Glass JD, 2011. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 134, 196–209. 10.1093/brain/awq314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Li Y, Asress SA, Jones DP, Glass JD, 2012. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol 233, 163–171. 10.1016/j.expneurol.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts RH, Trappe SW, Costill DL, Gallagher PM, Creer AC, Colloton PA, Peters JR, Romatowski JG, Bain JL, Riley DA, 2010. Prolonged space flight-induced alterations in the structure and function of human skeletal muscle fibres. J. Physiol 588, 3567–3592. 10.1113/jphysiol.2010.188508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontera WR, Hughes VA, Fielding RA, Fiatarone MA, Evans WJ, Roubenoff R, 2000. Aging of skeletal muscle: a 12-yr longitudinal study. J. Appl. Physiol. Bethesda Md 1985 88, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L, Fanò G, 2004. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp. Gerontol 39, 17–24. 10.1016/j.exger.2003.09.012 [DOI] [PubMed] [Google Scholar]
- Gallegly JC, Turesky NA, Strotman BA, Gurley CM, Peterson CA, Dupont-Versteegden EE, 2004. Satellite cell regulation of muscle mass is altered at old age. J. Appl. Physiol. Bethesda Md 1985 97, 1082–1090. 10.1152/japplphysiol.00006.2004 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G, 2017. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov 16, 487–511. 10.1038/nrd.2017.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambara G, Salanova M, Ciciliot S, Furlan S, Gutsmann M, Schiffl G, Ungethuem U, Volpe P, Gunga H-C, Blottner D, 2017. Microgravity-Induced Transcriptome Adaptation in Mouse Paraspinal longissimus dorsi Muscle Highlights Insulin Resistance-Linked Genes. Front. Physiol 8, 279. 10.3389/fphys.2017.00279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Arfat Y, Wang H, Goswami N, 2018. Muscle Atrophy Induced by Mechanical Unloading: Mechanisms and Potential Countermeasures. Front. Physiol 9. 10.3389/fphys.2018.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez C, López-Soriano FJ, Argilés JM, 1993. Acute treatment with tumour necrosis factor-α induces changes in protein metabolism in rat skeletal muscle. Mol. Cell. Biochem 125, 11–18. 10.1007/BF00926829 [DOI] [PubMed] [Google Scholar]
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G, 2006. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 13, 1423–1433. 10.1038/sj.cdd.4401950 [DOI] [PubMed] [Google Scholar]
- Gilliam LAA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, Reid MB, 2012. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol 302, C195–202. 10.1152/ajpcell.00217.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP, 2007a. Myostatin Gene Deletion Prevents Glucocorticoid-Induced Muscle Atrophy. Endocrinology 148, 452–460. 10.1210/en.2006-0539 [DOI] [PubMed] [Google Scholar]
- Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP, 2007b. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 148, 452–460. 10.1210/en.2006-0539 [DOI] [PubMed] [Google Scholar]
- Girgenrath M, Dominov JA, Kostek CA, Miller JB, 2004. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J. Clin. Invest 114, 1635–1639. 10.1172/JCI22928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ, 2005. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol 37, 1974–1984. 10.1016/j.biocel.2005.04.018 [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S, Macleod KF, 2010. Autophagy: cellular and molecular mechanisms. J. Pathol 221, 3–12. 10.1002/path.2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, Smith K, Rennie MJ, 2008. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J. Physiol 586, 6049–6061. 10.1113/jphysiol.2008.160333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EI, Yasuda N, Tarnopolsky MA, Abadi A, Phillips SM, 2010. Little change in markers of protein breakdown and oxidative stress in humans in immobilization-induced skeletal muscle atrophy. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab 35, 125–133. 10.1139/H09-137 [DOI] [PubMed] [Google Scholar]
- Goll DE, Neti G, Mares SW, Thompson VF, 2008. Myofibrillar protein turnover: the proteasome and the calpains. J. Anim. Sci 86, E19–35. 10.2527/jas.2007-0395 [DOI] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL, 2001. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U. S. A 98, 14440–14445. 10.1073/pnas.251541198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Miu MH, Frey JW, Mabrey DM, Lincoln HC, Ge Y, Chen J, Hornberger TA, 2010. A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Mol. Biol. Cell 21, 3258–3268. 10.1091/mbc.E10-05-0454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, 2017. Reinnervated muscle fiber type-grouping-inevitable? Oncotarget 8, 17410–17411. 10.18632/oncotarget.15757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, Hegedus J, Tam SL, 2004. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease. Neurol. Res 26, 174–185. 10.1179/016164104225013806 [DOI] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves-Smith F, Norris B, Pion CH, Barbat-Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin-Leheudre M, Hepple RT, 2014. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 28, 1621–1633. 10.1096/fj.13-242750 [DOI] [PubMed] [Google Scholar]
- Graham ZA, Gallagher PM, Cardozo CP, 2015. Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil 36, 305–315. 10.1007/s10974-015-9415-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhaff PL, Karagounis LG, Peirce N, Simpson EJ, Hazell M, Layfield R, Wackerhage H, Smith K, Atherton P, Selby A, Rennie MJ, 2008. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am. J. Physiol. Endocrinol. Metab 295, E595–604. 10.1152/ajpendo.90411.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson T, Osterlund T, Flanagan JN, von Waldén F, Trappe TA, Linnehan RM, Tesch PA, 2010. Effects of 3 days unloading on molecular regulators of muscle size in humans. J. Appl. Physiol. Bethesda Md 1985 109, 721–727. 10.1152/japplphysiol.00110.2009 [DOI] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS, 2000. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366. 10.1126/science.289.5488.2363 [DOI] [PubMed] [Google Scholar]
- Haase J, Weyer U, Immig K, Klöting N, Blüher M, Eilers J, Bechmann I, Gericke M, 2014. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 57, 562–571. 10.1007/s00125-013-3139-y [DOI] [PubMed] [Google Scholar]
- Haghighat A, Mader S, Pause A, Sonenberg N, 1995. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14, 5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakuno F, Takahashi S-I, 2018. IGF1 receptor signaling pathways. J. Mol. Endocrinol 61, T69–T86. 10.1530/JME-17-0311 [DOI] [PubMed] [Google Scholar]
- Hales CM, Carroll MD, Fryar CD, Ogden CL, 2017. Prevalence of Obesity Among Adults and Youth: United States, 2015–2016. NCHS Data Brief 1–8. [PubMed] [Google Scholar]
- Hamburg NM, McMackin CJ, Huang AL, Shenouda SM, Widlansky ME, Schulz E, Gokce N, Ruderman NB, Keaney JF, Vita JA, 2007. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler. Thromb. Vasc. Biol 27, 2650–2656. 10.1161/ATVBAHA.107.153288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrick MW, Herberg S, Arounleut P, He H-Z, Shiver A, Qi R-Q, Zhou L, Isales CM, Mi Q-S, 2010. The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem. Biophys. Res. Commun 400, 379–383. 10.1016/j.bbrc.2010.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann ZS, Ji C, Olsen SK, Lu X, Lux MC, Tan DS, Lima CD, 2019. Structural basis for adenylation and thioester bond formation in the ubiquitin E1. Proc. Natl. Acad. Sci 116, 15475–15484. 10.1073/pnas.1905488116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S-I, Natsume T, Guan J-L, Mizushima N, 2008. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol 181, 497–510. 10.1083/jcb.200712064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardee JP, Counts BR, Gao S, VanderVeen BN, Fix DK, Koh H, Carson JA, 2018. Inflammatory signalling regulates eccentric contraction‐induced protein synthesis in cachectic skeletal muscle. J. Cachexia Sarcopenia Muscle 9, 369–383. 10.1002/jcsm.12271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslett JN, Sanoudou D, Kho AT, Bennett RR, Greenberg SA, Kohane IS, Beggs AH, Kunkel LM, 2002. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl. Acad. Sci. U. S. A 99, 15000–15005. 10.1073/pnas.192571199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselgren PO, 1999. Glucocorticoids and muscle catabolism. Curr. Opin. Clin. Nutr. Metab. Care 2, 201–205. 10.1097/00075197-199905000-00002 [DOI] [PubMed] [Google Scholar]
- Hasson CJ, Miller RH, Caldwell GE, 2011. Contractile and elastic ankle joint muscular properties in young and older adults. PloS One 6, e15953. 10.1371/journal.pone.0015953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi C, Ono Y, Doi N, Kitamura F, Tagami R, Mineki R, Arai T, Taguchi H, Yanagida M, Hirner S, Labeit D, Labeit S, Sorimachi H, 2008. Multiple molecular interactions implicate the connectin/titin N2A region as a modulating scaffold for p94/calpain 3 activity in skeletal muscle. J. Biol. Chem 283, 14801–14814. 10.1074/jbc.m708262200 [DOI] [PubMed] [Google Scholar]
- Haycock JW, MacNeil S, Jones P, Harris JB, Mantle D, 1996. Oxidative damage to muscle protein in Duchenne muscular dystrophy. Neuroreport 8, 357–361. 10.1097/00001756-199612200-00070 [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ, 2009. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet 43, 67–93. 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg B, Angquist KA, Sjöström M, 1988. Peripheral arterial insufficiency and the fine structure of the gastrocnemius muscle. Int. Angiol. J. Int. Union Angiol 7, 50–59. [PubMed] [Google Scholar]
- Hepple RT, Rice CL, 2016. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol 594, 1965–1978. 10.1113/JP270561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnia K, Hugon G, Rivier F, Masmoudi A, Mercier J, Mornet D, 2007. Modulation of p38 mitogen-activated protein kinase cascade and metalloproteinase activity in diaphragm muscle in response to free radical scavenger administration in dystrophin-deficient Mdx mice. Am. J. Pathol 170, 633–643. 10.2353/ajpath.2007.060344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Kunkel LM, 1987. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. 10.1016/0092-8674(87)90579-4 [DOI] [PubMed] [Google Scholar]
- Höhn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B, Kehm R, König J, Grune T, Castro JP, 2016. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 11, 482–501. 10.1016/j.redox.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Sasaki T, Iemura SI, Natsume T, Hara T, Mizushima N, 2009. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979. 10.4161/auto.5.7.9296 [DOI] [PubMed] [Google Scholar]
- Hsieh Y-Y, Chang C-C, Hsu K-H, Tsai F-J, Chen C-P, Tsai H-D, 2008. Effect of exercise training on calpain systems in lean and obese Zucker rats. Int. J. Biol. Sci 300–308. 10.7150/ijbs.4.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu N-F, Chang H, Du B, Zhang Q-W, Arfat Y, Dang K, Gao Y-F, 2017. Tetramethylpyrazine ameliorated disuse-induced gastrocnemius muscle atrophy in hindlimb unloading rats through suppression of Ca2+/ROS-mediated apoptosis. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab 42, 117–127. 10.1139/apnm-2016-0363 [DOI] [PubMed] [Google Scholar]
- Huang J, Forsberg NE, 1998. Role of calpain in skeletal-muscle protein degradation. Proc. Natl. Acad. Sci. U. S. A 95, 12100–12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SNA, Mofarrahi M, Sigala I, Kim HC, Vassilakopoulos T, Maltais F, Bellenis I, Chaturvedi R, Gottfried SB, Metrakos P, Danialou G, Matecki S, Jaber S, Petrof BJ, Goldberg P, 2010. Mechanical Ventilation–induced Diaphragm Disuse in Humans Triggers Autophagy. Am. J. Respir. Crit. Care Med 182, 1377–1386. 10.1164/rccm.201002-0234OC [DOI] [PubMed] [Google Scholar]
- Hwee DT, Bodine SC, 2009. Age-related deficit in load-induced skeletal muscle growth. J. Gerontol. A. Biol. Sci. Med. Sci 64, 618–628. 10.1093/gerona/glp026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt HW, Powers SK, 2021. Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants 10, 588. 10.3390/antiox10040588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt HW, Powers SK, 2020. The Role of Calpains in Skeletal Muscle Remodeling with Exercise and Inactivity-induced Atrophy. Int. J. Sports Med 41, 994–1008. 10.1055/a-1199-7662 [DOI] [PubMed] [Google Scholar]
- Ingalls CP, Warren GL, Williams JH, Ward CW, Armstrong RB, 1998. E-C coupling failure in mouse EDL muscle after in vivo eccentric contractions. J. Appl. Physiol. Bethesda Md 1985 85, 58–67. [DOI] [PubMed] [Google Scholar]
- Ingalls CP, Wenke JC, Armstrong RB, 2001. Time course changes in [Ca2+]i, force, and protein content in hindlimb-suspended mouse soleus muscles. Aviat. Space Environ. Med 72, 471–476. [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P, 2003. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet 35, 367–371. 10.1038/ng1270 [DOI] [PubMed] [Google Scholar]
- Ismaeel A, Kim J-S, Kirk JS, Smith RS, Bohannon WT, Koutakis P, 2019. Role of Transforming Growth Factor-β in Skeletal Muscle Fibrosis: A Review. Int. J. Mol. Sci 20, 2446. 10.3390/ijms20102446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivannikov MV, Van Remmen H, 2015. Sod1 gene ablation in adult mice leads to physiological changes at the neuromuscular junction similar to changes that occur in old wild-type mice. Free Radic. Biol. Med 84, 254–262. 10.1016/j.freeradbiomed.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Murakami T, Saito Y, Nakagawa K, 2004. Steep elevation of blood interleukin-6 (IL-6) associated only with late stages of cachexia in cancer patients. Eur. Cytokine Netw 15, 312–316. [PubMed] [Google Scholar]
- Jackman RW, Kandarian SC, 2004. The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol 287, C834–843. 10.1152/ajpcell.00579.2003 [DOI] [PubMed] [Google Scholar]
- Jackson MJ, McArdle A, 2011. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J. Physiol 589, 2139–2145. 10.1113/jphysiol.2011.206623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jais A, Einwallner E, Sharif O, Gossens K, Lu TT-H, Soyal SM, Medgyesi D, Neureiter D, Paier-Pourani J, Dalgaard K, Duvigneau JC, Lindroos-Christensen J, Zapf T-C, Amann S, Saluzzo S, Jantscher F, Stiedl P, Todoric J, Martins R, Oberkofler H, Müller S, Hauser-Kronberger C, Kenner L, Casanova E, Sutterlüty-Fall H, Bilban M, Miller K, Kozlov AV, Krempler F, Knapp S, Lumeng CN, Patsch W, Wagner O, Pospisilik JA, Esterbauer H, 2014. Heme Oxygenase-1 Drives Metaflammation and Insulin Resistance in Mouse and Man. Cell 158, 25–40. 10.1016/j.cell.2014.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen I, Heymsfield SB, Wang ZM, Ross R, 2000. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. Bethesda Md 1985 89, 81–88. 10.1152/jappl.2000.89.1.81 [DOI] [PubMed] [Google Scholar]
- Jatoi A, Loprinzi CL, Sloan JA, Klee GG, Windschitl HE, 2001. Neuropeptide Y, leptin, and cholecystokinin 8 in patients with advanced cancer and anorexia: a North Central Cancer Treatment Group exploratory investigation. Cancer 92, 629–633. [DOI] [PubMed] [Google Scholar]
- Jones SW, Hill RJ, Krasney PA, O’Conner B, Peirce N, Greenhaff PL, 2004. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 18, 1025–1027. 10.1096/fj.03-1228fje [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y, 2005. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 16, 2544–2553. 10.1091/mbc.e04-08-0669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadi F, Charifi N, Denis C, Lexell J, 2004. Satellite cells and myonuclei in young and elderly women and men. Muscle Nerve 29, 120–127. 10.1002/mus.10510 [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM, 2018. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol 19, 365–381. 10.1038/s41580-018-0001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher AR, Kimball SR, Dennis MD, Schilder RJ, Jefferson LS, 2013. The mTORC1 signaling repressors REDD1/2 are rapidly induced and activation of p70S6K1 by leucine is defective in skeletal muscle of an immobilized rat hindlimb. Am. J. Physiol. Endocrinol. Metab 304, E229–236. 10.1152/ajpendo.00409.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher AR, Pereira SL, Jefferson LS, Kimball SR, 2015. REDD2 expression in rat skeletal muscle correlates with nutrient-induced activation of mTORC1: responses to aging, immobilization, and remobilization. Am. J. Physiol. Endocrinol. Metab 308, E122–129. 10.1152/ajpendo.00341.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khal J, Hine AV, Fearon KCH, Dejong CHC, Tisdale MJ, 2005. Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight loss. Int. J. Biochem. Cell Biol 37, 2196–2206. 10.1016/j.biocel.2004.10.017 [DOI] [PubMed] [Google Scholar]
- Killewich LA, Tuvdendorj D, Bahadorani J, Hunter GC, Wolfe RR, 2007. Amino acids stimulate leg muscle protein synthesis in peripheral arterial disease. J. Vasc. Surg 45, 554–559; discussion 559–560. 10.1016/j.jvs.2006.11.033 [DOI] [PubMed] [Google Scholar]
- Kim J, Tchernyshyov I, Semenza GL, Dang CV, 2006. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. 10.1016/j.cmet.2006.02.002 [DOI] [PubMed] [Google Scholar]
- Kleiger G, Mayor T, 2014. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 24, 352–359. 10.1016/j.tcb.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CS, Marsh GD, Petrella RJ, Rice CL, 2003. Muscle fiber number in the biceps brachii muscle of young and old men. Muscle Nerve 28, 62–68. 10.1002/mus.10386 [DOI] [PubMed] [Google Scholar]
- Knecht E, Salvador N, 2013. Chaperone-Mediated Autophagy, Madame Curie Bioscience Database [Internet]. Landes Bioscience. [Google Scholar]
- Knox LS, Crosby LO, Feurer ID, Buzby GP, Miller CL, Mullen JL, 1983. Energy expenditure in malnourished cancer patients. Ann. Surg 197, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Miura M, Itokawa Y, 1991. Oxidative stress in skeletal muscle atrophied by immobiIization. Acta Physiol. Scand 142, 527–528. 10.1111/j.1748-1716.1991.tb09191.x [DOI] [PubMed] [Google Scholar]
- Koutakis P, Ismaeel A, Farmer P, Purcell S, Smith RS, Eidson JL, Bohannon WT, 2018. Oxidative stress and antioxidant treatment in patients with peripheral artery disease. Physiol. Rep 6, e13650. 10.14814/phy2.13650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutakis P, Miserlis D, Myers SA, Kim JK-S, Zhu Z, Papoutsi E, Swanson SA, Haynatzki G, Ha DM, Carpenter LA, McComb RD, Johanning JM, Casale GP, Pipinos II, 2015a. Abnormal accumulation of desmin in gastrocnemius myofibers of patients with peripheral artery disease: associations with altered myofiber morphology and density, mitochondrial dysfunction and impaired limb function. J. Histochem. Cytochem. Off. J. Histochem. Soc 63, 256–269. 10.1369/0022155415569348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutakis P, Myers SA, Cluff K, Ha DM, Haynatzki G, McComb RD, Uchida K, Miserlis D, Papoutsi E, Johanning JM, Casale GP, Pipinos II, 2015b. Abnormal myofiber morphology and limb dysfunction in claudication. J. Surg. Res 196, 172–179. 10.1016/j.jss.2015.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutakis P, Weiss DJ, Miserlis D, Shostrom VK, Papoutsi E, Ha DM, Carpenter LA, McComb RD, Casale GP, Pipinos II, 2014. Oxidative damage in the gastrocnemius of patients with peripheral artery disease is myofiber type selective. Redox Biol. 2, 921–928. 10.1016/j.redox.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh-Madsen R, Thyfault JP, Broholm C, Mortensen OH, Olsen RH, Mounier R, Plomgaard P, van Hall G, Booth FW, Pedersen BK, 2010. A 2-wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J. Appl. Physiol. Bethesda Md 1985 108, 1034–1040. 10.1152/japplphysiol.00977.2009 [DOI] [PubMed] [Google Scholar]
- Kullo IJ, Rooke TW, 2016. CLINICAL PRACTICE. Peripheral Artery Disease. N. Engl. J. Med 374, 861–871. 10.1056/NEJMcp1507631 [DOI] [PubMed] [Google Scholar]
- Kumar Akhilesh, Bhatnagar S, Kumar Ashok, 2010. Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am. J. Pathol 177, 248–260. 10.2353/ajpath.2010.091176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Selby A, Rankin D, Patel R, Atherton P, Hildebrandt W, Williams J, Smith K, Seynnes O, Hiscock N, Rennie MJ, 2009. Age-related differences in the dose–response relationship of muscle protein synthesis to resistance exercise in young and old men. J. Physiol 587, 211–217. 10.1113/jphysiol.2008.164483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S, Kohl CH, Witt CC, Labeit D, Jung J, Granzier H, 2010. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J. Biomed. Biotechnol 2010, 693741. 10.1155/2010/693741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT, 2004. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest 113, 370–378. 10.1172/JCI19670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbach KJ, Rando TA, 2002. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve 26, 644–653. 10.1002/mus.10258 [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM, 2012. mTOR signaling in growth control and disease. Cell 149, 274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson L, 1978. Morphological and functional characteristics of the ageing skeletal muscle in man. A cross-sectional study. Acta Physiol. Scand. Suppl 457, 1–36. [PubMed] [Google Scholar]
- Lawler JM, 2011. Exacerbation of pathology by oxidative stress in respiratory and locomotor muscles with Duchenne muscular dystrophy. J. Physiol. 589, 2161–2170. 10.1113/jphysiol.2011.207456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc A, Lin C, Shackelford L, Sinitsyn V, Evans H, Belichenko O, Schenkman B, Kozlovskaya I, Oganov V, Bakulin A, Hedrick T, Feeback D, 2000. Muscle volume, MRI relaxation times (T2), and body composition after spaceflight. J. Appl. Physiol. Bethesda Md 1985 89, 2158–2164. 10.1152/jappl.2000.89.6.2158 [DOI] [PubMed] [Google Scholar]
- Lebrun P, Mothe-Satney I, Delahaye L, Van Obberghen E, Baron V, 1998. Insulin Receptor Substrate-1 as a Signaling Molecule for Focal Adhesion Kinase pp125FAK and pp60src*. J. Biol. Chem 273, 32244–32253. 10.1074/jbc.273.48.32244 [DOI] [PubMed] [Google Scholar]
- Lecker SH, Goldberg AL, Mitch WE, 2006. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol 17, 1807–1819. 10.1681/ASN.2006010083 [DOI] [PubMed] [Google Scholar]
- Leduc-Gaudet J-P, Picard M, St-Jean Pelletier F, Sgarioto N, Auger M-J, Vallée J, Robitaille R, St-Pierre DH, Gouspillou G, 2015. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937. 10.18632/oncotarget.4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lee IS, Choue R, 2013. Obesity, Inflammation and Diet. Pediatr. Gastroenterol. Hepatol. Nutr 16, 143–152. 10.5223/pghn.2013.16.3.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Fry CS, Mula J, Kirby TJ, Jackson JR, Liu F, Yang L, Dupont-Versteegden EE, McCarthy JJ, Peterson CA, 2016. Aged Muscle Demonstrates Fiber-Type Adaptations in Response to Mechanical Overload, in the Absence of Myofiber Hypertrophy, Independent of Satellite Cell Abundance. J. Gerontol. Ser. A 71, 461–467. 10.1093/gerona/glv033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Li Y, Duan Y, Hu C-AA, Tang Y, Yin Y, 2017. Myokines and adipokines: Involvement in the crosstalk between skeletal muscle and adipose tissue. Cytokine Growth Factor Rev 33, 73–82. 10.1016/j.cytogfr.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A, 2009. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet 18, 2584–2598. 10.1093/hmg/ddp191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J, Huard J, 2004. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am. J. Pathol 164, 1007–1019. 10.1016/s0002-9440(10)63188-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-P, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB, 2005. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 19, 362–370. 10.1096/fj.04-2364com [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Blough ER, Arvapalli R, Wang Y, Reiser PJ, Paturi S, Katta A, Harris R, Nepal N, Wu M, 2012. Regulation of contractile proteins and protein translational signaling in disused muscle. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol 30, 1202–1214. 10.1159/000343310 [DOI] [PubMed] [Google Scholar]
- Llovera M, García-Martínez C, López-Soriano J, Carbó N, Agell N, López-Soriano FJ, Argiles JM, 1998. Role of TNF receptor 1 in protein turnover during cancer cachexia using gene knockout mice. Mol. Cell. Endocrinol 142, 183–189. 10.1016/s0303-7207(98)00105-1 [DOI] [PubMed] [Google Scholar]
- Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM, 1998. Leptin regulates proinflammatory immune responses. FASEB J. 12, 57–65. 10.1096/fsb2fasebj.12.1.57 [DOI] [PubMed] [Google Scholar]
- Lomonosova YN, Belova SP, Mirzoev TM, Kozlovskaya IB, Shenkman BS, 2017. Eukaryotic elongation factor 2 kinase activation in M. soleus under 14-day hindlimb unloading of rats. Dokl. Biochem. Biophys 474, 165–167. 10.1134/S1607672917030048 [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR, 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest 117, 175–184. 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald AJ, Johns N, Stephens N, Greig C, Ross JA, Small AC, Husi H, Fearon KCH, Preston T, 2015. Habitual Myofibrillar Protein Synthesis Is Normal in Patients with Upper GI Cancer Cachexia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 21, 1734–1740. 10.1158/1078-0432.CCR-14-2004 [DOI] [PubMed] [Google Scholar]
- Magne H, Savary-Auzeloux I, Rémond D, Dardevet D, 2013. Nutritional strategies to counteract muscle atrophy caused by disuse and to improve recovery. Nutr. Res. Rev 26, 149–165. 10.1017/S0954422413000115 [DOI] [PubMed] [Google Scholar]
- Mäkitie J, Teräväinen H, 1977. Histochemical changes in striated muscle in patients with intermittent claudication. Arch. Pathol. Lab. Med 101, 658–663. [PubMed] [Google Scholar]
- Makki K, Froguel P, Wolowczuk I, 2013. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, e139239. 10.1155/2013/139239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallouk N, Jacquemond V, Allard B, 2000. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. U. S. A 97, 4950–4955. 10.1073/pnas.97.9.4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Muñoz-Cánoves P, 2011. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 1, 21. 10.1186/2044-5040-1-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Klionsky DJ, 2011. AMPK Activates Autophagy by Phosphorylating ULK1. Circ. Res 108, 787–788. 10.1161/RES.0b013e3182194c29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marimuthu K, Murton AJ, Greenhaff PL, 2011. Mechanisms regulating muscle mass during disuse atrophy and rehabilitation in humans. J. Appl. Physiol. Bethesda Md 1985 110, 555–560. 10.1152/japplphysiol.00962.2010 [DOI] [PubMed] [Google Scholar]
- Marx JO, Kraemer WJ, Nindl BC, Larsson L, 2002. Effects of Aging on Human Skeletal Muscle Myosin Heavy-Chain mRNA Content and Protein Isoform Expression. J. Gerontol. Ser. A 57, B232–B238. 10.1093/gerona/57.6.B232 [DOI] [PubMed] [Google Scholar]
- Marzetti E, Lorenzi M, Landi F, Picca A, Rosa F, Tanganelli F, Galli M, Doglietto GB, Pacelli F, Cesari M, Bernabei R, Calvani R, Bossola M, 2017. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp. Gerontol 87, 92–99. 10.1016/j.exger.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Fujita N, Arakawa T, Fujino H, Miki A, 2014. Influence of electrical stimulation on calpain and ubiquitin-proteasome systems in the denervated and unloaded rat tibialis anterior muscles. Acta Histochem. 116, 936–942. 10.1016/j.acthis.2014.03.006 [DOI] [PubMed] [Google Scholar]
- Matsumura K, Zhong D, Saito F, Arai K, Adachi K, Kawai H, Higuchi I, Nishino I, Shimizu T, 2005. Proteolysis of beta-dystroglycan in muscular diseases. Neuromuscul. Disord. NMD 15, 336–341. 10.1016/j.nmd.2005.01.007 [DOI] [PubMed] [Google Scholar]
- Mayhew AJ, Amog K, Phillips S, Parise G, McNicholas PD, de Souza RJ, Thabane L, Raina P, 2019. The prevalence of sarcopenia in community-dwelling older adults, an exploration of differences between studies and within definitions: a systematic review and meta-analyses. Age Ageing 48, 48–56. 10.1093/ageing/afy106 [DOI] [PubMed] [Google Scholar]
- McCarthy JJ, Esser KA, 2010. Anabolic and catabolic pathways regulating skeletal muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 13, 230–235. 10.1097/MCO.0b013e32833781b5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComas AJ, 1977. Chapter 20 - WALLERIAN DEGENERATION AND FIBRE REGENERATION, in: McComas AJ (Ed.), Neuromuscular Function and Disorders. Butterworth-Heinemann, pp. 221–232. 10.1016/B978-0-407-00058-2.50029-2 [DOI] [Google Scholar]
- McCormick R, Vasilaki A, 2018. Age-related changes in skeletal muscle: changes to life-style as a therapy. Biogerontology 19, 519–536. 10.1007/s10522-018-9775-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC, 2002. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J. Cell Biol 157, 125–136. 10.1083/jcb.200108089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Dikic I, 2010. Not all autophagy membranes are created equal. Cell 141, 564–566. 10.1016/j.cell.2010.04.030 [DOI] [PubMed] [Google Scholar]
- McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R, 2006. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J. Cell. Physiol 209, 501–514. 10.1002/jcp.20757 [DOI] [PubMed] [Google Scholar]
- Mercer CA, Kaliappan A, Dennis PB, 2009. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5, 649–662. 10.4161/auto.5.5.8249 [DOI] [PubMed] [Google Scholar]
- Messina S, Bitto A, Aguennouz M, Minutoli L, Monici MC, Altavilla D, Squadrito F, Vita G, 2006. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp. Neurol 198, 234–241. 10.1016/j.expneurol.2005.11.021 [DOI] [PubMed] [Google Scholar]
- Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD, 2009. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A 106, 19023–19028. 10.1073/pnas.0906591106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JB, Girgenrath M, 2006. The role of apoptosis in neuromuscular diseases and prospects for anti-apoptosis therapy. Trends Mol. Med 12, 279–286. 10.1016/j.molmed.2006.04.003 [DOI] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon O, Kavazis AN, Szeto HH, Powers SK, 2011. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol 111, 1459–1466. 10.1152/japplphysiol.00591.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoev T, Tyganov S, Vilchinskaya N, Lomonosova Y, Shenkman B, 2016. Key Markers of mTORC1-Dependent and mTORC1-Independent Signaling Pathways Regulating Protein Synthesis in Rat Soleus Muscle During Early Stages of Hindlimb Unloading. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol 39, 1011–1020. 10.1159/000447808 [DOI] [PubMed] [Google Scholar]
- Mitchell WK, Williams J, Atherton P, Larvin M, Lund J, Narici M, 2012. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol 3, 260. 10.3389/fphys.2012.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Noguchi M, Takemasa T, 2008. Intermittent reloading attenuates muscle atrophy through modulating Akt/mTOR pathway. Med. Sci. Sports Exerc 40, 848–855. 10.1249/MSS.0b013e318163275f [DOI] [PubMed] [Google Scholar]
- Monteiro L, Pereira JA, da S, Palhinha L, Moraes-Vieira PMM, 2019. Leptin in the regulation of the immunometabolism of adipose tissue-macrophages. J. Leukoc. Biol 106, 703–716. 10.1002/JLB.MR1218-478R [DOI] [PubMed] [Google Scholar]
- Mourkioti F, Kratsios P, Luedde T, Song Y-H, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N, 2006. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest 116, 2945–2954. 10.1172/JCI28721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murach KA, Englund DA, Dupont-Versteegden EE, McCarthy JJ, Peterson CA, 2018a. Myonuclear Domain Flexibility Challenges Rigid Assumptions on Satellite Cell Contribution to Skeletal Muscle Fiber Hypertrophy. Front. Physiol 9, 635. 10.3389/fphys.2018.00635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murach KA, Fry CS, Kirby TJ, Jackson JR, Lee JD, White SH, Dupont-Versteegden EE, McCarthy JJ, Peterson CA, 2018b. Starring or Supporting Role? Satellite Cells and Skeletal Muscle Fiber Size Regulation. Physiol. Bethesda Md 33, 26–38. 10.1152/physiol.00019.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murach KA, Peck BD, Policastro RA, Vechetti IJ, Van Pelt DW, Dungan CM, Denes LT, Fu X, Brightwell CR, Zentner GE, Dupont-Versteegden EE, Richards CI, Smith JJ, Fry CS, McCarthy JJ, Peterson CA, 2021. Early satellite cell communication creates a permissive environment for long-term muscle growth. iScience 24, 102372. 10.1016/j.isci.2021.102372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murach KA, White SH, Wen Y, Ho A, Dupont-Versteegden EE, McCarthy JJ, Peterson CA, 2017. Differential requirement for satellite cells during overload-induced muscle hypertrophy in growing versus mature mice. Skelet. Muscle 7, 14. 10.1186/s13395-017-0132-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musaro A, 2010. Comments on Point:Counterpoint: IGF is/is not the major physiological regulator of muscle mass. The strange case of IGF-1. J. Appl. Physiol. Bethesda Md 1985 108, 1826. 10.1152/japplphysiol.00312.2010 [DOI] [PubMed] [Google Scholar]
- Nadarajah VD, van Putten M, Chaouch A, Garrood P, Straub V, Lochmüller H, Ginjaar HB, Aartsma-Rus AM, van Ommen GJB, den Dunnen JT, t Hoen P. a. C., 2011. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in Duchenne muscular dystrophy (DMD). Neuromuscul. Disord. NMD 21, 569–578. 10.1016/j.nmd.2011.05.011 [DOI] [PubMed] [Google Scholar]
- Neti G, Novak SM, Thompson VF, Goll DE, 2009. Properties of easily releasable myofilaments: are they the first step in myofibrillar protein turnover? Am. J. Physiol.-Cell Physiol 296, C1383–C1390. 10.1152/ajpcell.00022.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M-H, Cheng M, Koh TJ, 2011. Impaired muscle regeneration in ob/ob and db/db mice. ScientificWorldJournal 11, 1525–1535. 10.1100/tsw.2011.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Mogensen M, Vind BF, Sahlin K, Højlund K, Schrøder HD, Ørtenblad N, 2010. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab 298, E706–E713. 10.1152/ajpendo.00692.2009 [DOI] [PubMed] [Google Scholar]
- Nilsson MI, Dobson JP, Greene NP, Wiggs MP, Shimkus KL, Wudeck EV, Davis AR, Laureano ML, Fluckey JD, 2013. Abnormal protein turnover and anabolic resistance to exercise in sarcopenic obesity. FASEB J. 27, 3905–3916. 10.1096/fj.12-224006 [DOI] [PubMed] [Google Scholar]
- Nilsson MI, Greene NP, Dobson JP, Wiggs MP, Gasier HG, Macias BR, Shimkus KL, Fluckey JD, 2010. Insulin resistance syndrome blunts the mitochondrial anabolic response following resistance exercise. Am. J. Physiol.-Endocrinol. Metab 299, E466–E474. 10.1152/ajpendo.00118.2010 [DOI] [PubMed] [Google Scholar]
- Nishikawa H, Fukunishi S, Asai A, Yokohama K, Nishiguchi S, Higuchi K, 2021. Pathophysiology and mechanisms of primary sarcopenia (Review). Int. J. Mol. Med 48, 1–8. 10.3892/ijmm.2021.4989 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Furochi H, Mameoka M, Hirasaka K, Onishi Y, Suzue N, Oarada M, Akamatsu M, Akima H, Fukunaga T, Kishi K, Yasui N, Ishidoh K, Fukuoka H, Nikawa T, 2006. Ubiquitin ligase gene expression in healthy volunteers with 20-day bedrest. Muscle Nerve 34, 463–469. 10.1002/mus.20611 [DOI] [PubMed] [Google Scholar]
- Ohira Y, Jiang B, Roy RR, Oganov V, Ilyina-Kakueva E, Marini JF, Edgerton VR, 1992. Rat soleus muscle fiber responses to 14 days of spaceflight and hindlimb suspension. J. Appl. Physiol. Bethesda Md 1985 73, 51S–57S. 10.1152/jappl.1992.73.2.S51 [DOI] [PubMed] [Google Scholar]
- Okamoto T, Machida S, 2017. Changes in FOXO and proinflammatory cytokines in the late stage of immobilized fast and slow muscle atrophy. Biomed. Res. Tokyo Jpn 38, 331–342. 10.2220/biomedres.38.331 [DOI] [PubMed] [Google Scholar]
- Oku M, Sakai Y, 2018. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays News Rev. Mol. Cell. Dev. Biol 40, e1800008. 10.1002/bies.201800008 [DOI] [PubMed] [Google Scholar]
- Ono Y, Sorimachi H, 2012. Calpains — An elaborate proteolytic system. Biochim. Biophys. Acta BBA - Proteins Proteomics, Proteolysis 50 years after the discovery of lysosome 1824, 224–236. 10.1016/j.bbapap.2011.08.005 [DOI] [PubMed] [Google Scholar]
- Onofre-Oliveira PCG, Santos ALF, Martins PM, Ayub-Guerrieri D, Vainzof M, 2012. Differential expression of genes involved in the degeneration and regeneration pathways in mouse models for muscular dystrophies. Neuromolecular Med. 14, 74–83. 10.1007/s12017-012-8172-3 [DOI] [PubMed] [Google Scholar]
- Overend TJ, Cunningham DA, Paterson DH, Lefcoe MS, 1992. Thigh composition in young and elderly men determined by computed tomography. Clin. Physiol. Oxf. Engl 12, 629–640. [DOI] [PubMed] [Google Scholar]
- Padrão AI, Oliveira P, Vitorino R, Colaço B, Pires MJ, Márquez M, Castellanos E, Neuparth MJ, Teixeira C, Costa C, Moreira-Gonçalves D, Cabral S, Duarte JA, Santos LL, Amado F, Ferreira R, 2013. Bladder cancer-induced skeletal muscle wasting: disclosing the role of mitochondria plasticity. Int. J. Biochem. Cell Biol 45, 1399–1409. 10.1016/j.biocel.2013.04.014 [DOI] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, Werb Z, 2007. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol 8, 221–233. 10.1038/nrm2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-M, Jung CH, Seo M, Otto NM, Grunwald D, Kim KH, Moriarity B, Kim Y-M, Starker C, Nho RS, Voytas D, Kim D-H, 2016. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 12, 547–564. 10.1080/15548627.2016.1140293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, Liang F, Hussain S, Matecki S, Petrof BJ, 2012. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol 181, 583–592. 10.1016/j.ajpath.2012.04.004 [DOI] [PubMed] [Google Scholar]
- Pellegrinelli V, Rouault C, Rodriguez-Cuenca S, Albert V, Edom-Vovard F, Vidal-Puig A, Clément K, Butler-Browne GS, Lacasa D, 2015. Human Adipocytes Induce Inflammation and Atrophy in Muscle Cells During Obesity. Diabetes 64, 3121–3134. 10.2337/db14-0796 [DOI] [PubMed] [Google Scholar]
- Penet M-F, Bhujwalla ZM, 2015. Cancer Cachexia, Recent Advances, and Future Directions. Cancer J. Sudbury Mass 21, 117–122. 10.1097/PPO.0000000000000100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL, 1993. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A. 90, 3710–3714. 10.1073/pnas.90.8.3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SK, Bruce SA, Newton D, Woledge RC, 1992. The weakness of old age is not due to failure of muscle activation. J. Gerontol. 47, M45–49. [DOI] [PubMed] [Google Scholar]
- Pipinos II, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, Dodd SL, 2008a. The myopathy of peripheral arterial occlusive disease: Part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc. Endovascular Surg. 42, 101–112. 10.1177/1538574408315995 [DOI] [PubMed] [Google Scholar]
- Pipinos II, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, Dodd SL, 2008b. The Myopathy of Peripheral Arterial Occlusive Disease: Part 1. Functional and Histomorphological Changes and Evidence for Mitochondrial Dysfunction. Vasc. Endovascular Surg 41, 481–489. 10.1177/1538574407311106 [DOI] [PubMed] [Google Scholar]
- Pipinos II, Judge AR, Zhu Z, Selsby JT, Swanson SA, Johanning JM, Baxter BT, Lynch TG, Dodd SL, 2006. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med 41, 262–269. 10.1016/j.freeradbiomed.2006.04.003 [DOI] [PubMed] [Google Scholar]
- Pipinos II, Sharov VG, Shepard AD, Anagnostopoulos PV, Katsamouris A, Todor A, Filis KA, Sabbah HN, 2003. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J. Vasc. Surg 38, 827–832. 10.1016/s0741-5214(03)00602-5 [DOI] [PubMed] [Google Scholar]
- Pipinos II, Swanson SA, Zhu Z, Nella AA, Weiss DJ, Gutti TL, McComb RD, Baxter BT, Lynch TG, Casale GP, 2008c. Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am. J. Physiol. Regul. Integr. Comp. Physiol 295, R290–296. 10.1152/ajpregu.90374.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipinos II, Swanson SA, Zhu Z, Nella AA, Weiss DJ, Gutti TL, McComb RD, Baxter BT, Lynch TG, Casale GP, 2008d. Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am. J. Physiol. Regul. Integr. Comp. Physiol 295, R290–296. 10.1152/ajpregu.90374.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JD, Merriam AP, Leahy P, Gong B, Feuerman J, Cheng G, Khanna S, 2004. Temporal gene expression profiling of dystrophin-deficient (mdx) mouse diaphragm identifies conserved and muscle group-specific mechanisms in the pathogenesis of muscular dystrophy. Hum. Mol. Genet 13, 257–269. 10.1093/hmg/ddh033 [DOI] [PubMed] [Google Scholar]
- Power GA, Dalton BH, Behm DG, Doherty TJ, Vandervoort AA, Rice CL, 2012. Motor unit survival in lifelong runners is muscle dependent. Med. Sci. Sports Exerc 44, 1235–1242. 10.1249/MSS.0b013e318249953c [DOI] [PubMed] [Google Scholar]
- Powers S, Smuder A, Judge A, 2012. Oxidative stress and disuse muscle atrophy: cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 15, 240–245. 10.1097/MCO.0b013e328352b4c2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ, 2011a. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med 39, 1749–1759. 10.1097/CCM.0b013e3182190b62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, DeRuisseau KC, 2005. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol 288, R337–344. 10.1152/ajpregu.00469.2004 [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, McClung JM, 2007. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. Bethesda Md 1985 102, 2389–2397. 10.1152/japplphysiol.01202.2006 [DOI] [PubMed] [Google Scholar]
- Powers SK, Smuder AJ, Criswell DS, 2011b. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox Signal 15, 2519–2528. 10.1089/ars.2011.3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primeau AJ, Adhihetty PJ, Hood DA, 2002a. Apoptosis in heart and skeletal muscle. Can. J. Appl. Physiol. Rev. Can. Physiol. Appl 27, 349–395. 10.1139/h02-020 [DOI] [PubMed] [Google Scholar]
- Primeau AJ, Adhihetty PJ, Hood DA, 2002b. Apoptosis in heart and skeletal muscle. Can. J. Appl. Physiol. Rev. Can. Physiol. Appl 27, 349–395. 10.1139/h02-020 [DOI] [PubMed] [Google Scholar]
- Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM, 2009. Dystrophin is a microtubule-associated protein. J. Cell Biol 186, 363–369. 10.1083/jcb.200905048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E, 2010. Autophagy and metabolism. Science 330, 1344–1348. 10.1126/science.1193497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj IS, Bird SR, Shield AJ, 2010. Aging and the force-velocity relationship of muscles. Exp. Gerontol 45, 81–90. 10.1016/j.exger.2009.10.013 [DOI] [PubMed] [Google Scholar]
- Raught B, Peiretti F, Gingras A-C, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW, 2004. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23, 1761–1769. 10.1038/sj.emboj.7600193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault V, Thornell L-E, Eriksson P-O, Butler-Browne G, Mouly V, Thorne L-E, 2002. Regenerative potential of human skeletal muscle during aging. Aging Cell 1, 132–139. [DOI] [PubMed] [Google Scholar]
- Richter EA, Kiens B, Mizuno M, Strange S, 1989. Insulin action in human thighs after one-legged immobilization. J. Appl. Physiol. Bethesda Md 1985 67, 19–23. 10.1152/jappl.1989.67.1.19 [DOI] [PubMed] [Google Scholar]
- Rolland Y, Czerwinski S, Abellan Van Kan G, Morley JE, Cesari M, Onder G, Woo J, Baumgartner R, Pillard F, Boirie Y, Chumlea WMC, Vellas B, 2008. Sarcopenia: its assessment, etiology, pathogenesis, consequences and future perspectives. J. Nutr. Health Aging 12, 433–450. 10.1007/BF02982704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roma J, Munell F, Fargas A, Roig M, 2004. Evolution of pathological changes in the gastrocnemius of the mdx mice correlate with utrophin and beta-dystroglycan expression. Acta Neuropathol. (Berl.) 108, 443–452. 10.1007/s00401-004-0908-1 [DOI] [PubMed] [Google Scholar]
- Romanello V, Sandri M, 2010. Mitochondrial Biogenesis and Fragmentation as Regulators of Muscle Protein Degradation. Curr. Hypertens. Rep 12, 433–439. 10.1007/s11906-010-0157-8 [DOI] [PubMed] [Google Scholar]
- Rosa-Caldwell ME, Brown JL, Perry RA, Shimkus KL, Shirazi-Fard Y, Brown LA, Hogan HA, Fluckey JD, Washington TA, Wiggs MP, Greene NP, 2020a. Regulation of mitochondrial quality following repeated bouts of hindlimb unloading. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab 45, 264–274. 10.1139/apnm-2019-0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa-Caldwell ME, Lim S, Haynie WA, Brown JL, Deaver JW, Morena Da Silva F, Jansen LT, Lee DE, Wiggs MP, Washington TA, Greene NP, 2021. Female mice may have exacerbated catabolic signalling response compared to male mice during development and progression of disuse atrophy. J. Cachexia Sarcopenia Muscle 12, 717–730. 10.1002/jcsm.12693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa-Caldwell ME, Lim S, Haynie WS, Jansen LT, Westervelt LC, Amos MG, Washington TA, Greene NP, 2020b. Altering aspects of mitochondrial quality to improve musculoskeletal outcomes in disuse atrophy. J. Appl. Physiol. Bethesda Md 1985 129, 1290–1303. 10.1152/japplphysiol.00407.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SM, Martel GF, Ivey FM, Lemmer JT, Metter EJ, Hurley BF, Rogers MA, 2000. Skeletal muscle satellite cell populations in healthy young and older men and women. Anat. Rec 260, 351–358. [DOI] [PubMed] [Google Scholar]
- Rowan SL, Rygiel K, Purves-Smith FM, Solbak NM, Turnbull DM, Hepple RT, 2012. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PloS One 7, e29082. 10.1371/journal.pone.0029082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell ST, Eley H, Tisdale MJ, 2007. Role of reactive oxygen species in protein degradation in murine myotubes induced by proteolysis-inducing factor and angiotensin II. Cell. Signal 19, 1797–1806. 10.1016/j.cellsig.2007.04.003 [DOI] [PubMed] [Google Scholar]
- Saido TC, Sorimachi H, Suzuki K, 1994. Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J. 8, 814–822. 10.1096/fasebj.8.11.8070630 [DOI] [PubMed] [Google Scholar]
- Sanders PM, Russell ST, Tisdale MJ, 2005. Angiotensin II directly induces muscle protein catabolism through the ubiquitin–proteasome proteolytic pathway and may play a role in cancer cachexia. Br. J. Cancer 93, 425–434. 10.1038/sj.bjc.6602725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM, 2006. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci 103, 16260–16265. 10.1073/pnas.0607795103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL, 2004. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412. 10.1016/s0092-8674(04)00400-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Alvarez J, Goberna R, Sánchez-Margalet V, 1999. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell. Immunol 194, 6–11. 10.1006/cimm.1999.1490 [DOI] [PubMed] [Google Scholar]
- Sartori R, Romanello V, Sandri M, 2021. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat. Commun 12, 330. 10.1038/s41467-020-20123-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber MN, Hasenkamp RM, Pipinos II, Johanning JM, Stergiou N, DeSpiegelaere HK, Chien JH, Myers SA, 2017. Muscle strength and control characteristics are altered by peripheral artery disease. J. Vasc. Surg 66, 178–186.e12. 10.1016/j.jvs.2017.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HR, McMillan DC, Crilly A, McArdle CS, Milroy R, 1996. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br. J. Cancer 73, 1560–1562. 10.1038/bjc.1996.294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seene T, Kaasik P, Riso E-M, 2012. Review on aging, unloading and reloading: Changes in skeletal muscle quantity and quality. Arch. Gerontol. Geriatr. 54, 374–380. 10.1016/j.archger.2011.05.002 [DOI] [PubMed] [Google Scholar]
- Senf SM, Dodd SL, McClung JM, Judge AR, 2008. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 22, 3836–3845. 10.1096/fj.08-110163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Sabatini DM, 2010. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40, 310–322. 10.1016/j.molcel.2010.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Meng X, Zhang Z, Wang T, 2018. Physical Exercise for Muscle Atrophy. Adv. Exp. Med. Biol 1088, 529–545. 10.1007/978-981-13-1435-3_24 [DOI] [PubMed] [Google Scholar]
- Shimkus KL, Shirazi-Fard Y, Wiggs MP, Ullah ST, Pohlenz C, Gatlin DM, Carroll CC, Hogan HA, Fluckey JD, 2018. Responses of skeletal muscle size and anabolism are reproducible with multiple periods of unloading/reloading. J. Appl. Physiol. Bethesda Md 1985 125, 1456–1467. 10.1152/japplphysiol.00736.2017 [DOI] [PubMed] [Google Scholar]
- Shin J, Tajrishi MM, Ogura Y, Kumar A, 2013. Wasting Mechanisms in Muscular Dystrophy. Int. J. Biochem. Cell Biol 45, 2266–2279. 10.1016/j.biocel.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS, Marcinek DJ, 2013. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12, 763–771. 10.1111/acel.12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeuninx B, Mckendry J, Wilson D, Martin U, Breen L, 2017. Age-Related Anabolic Resistance of Myofibrillar Protein Synthesis Is Exacerbated in Obese Inactive Individuals. J. Clin. Endocrinol. Metab 102, 3535–3545. 10.1210/jc.2017-00869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KL, Tisdale MJ, 1993. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br. J. Cancer 67, 680–685. 10.1038/bjc.1993.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smuder AJ, Kavazis AN, Hudson MB, Nelson WB, Powers SK, 2010. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med 49, 1152–1160. 10.1016/j.freeradbiomed.2010.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Yao S, Liu Y, Long L, Yang H, Li Q, Liang J, Li X, Lu Y, Zhu H, Zhang N, 2017. Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med 13, 1209–1214. 10.3892/etm.2017.4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, Suzuki K, 1989. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem 264, 20106–20111. [PubMed] [Google Scholar]
- Sorimachi H, Kinbara K, Kimura S, Takahashi M, Ishiura S, Sasagawa N, Sorimachi N, Shimada H, Tagawa K, Maruyama K, 1995. Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J. Biol. Chem 270, 31158–31162. 10.1074/jbc.270.52.31158 [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Mellgren RL, 2002. Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum. Mol. Genet 11, 2645–2655. 10.1093/hmg/11.21.2645 [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG, 2001. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. Orlando Fla 98, 235–243. 10.1006/clim.2000.4966 [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Walsh CM, Dorshkind KA, Rodriguez EM, Tidball JG, 1997. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J. Clin. Invest 99, 2745–2751. 10.1172/JCI119464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spendiff S, Vuda M, Gouspillou G, Aare S, Perez A, Morais JA, Jagoe RT, Filion M-E, Glicksman R, Kapchinsky S, MacMillan NJ, Pion CH, Aubertin-Leheudre M, Hettwer S, Correa JA, Taivassalo T, Hepple RT, 2016. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol 594, 7361–7379. 10.1113/JP272487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan N, Häring H-U, Hu FB, Schulze MB, 2013. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol 1, 152–162. 10.1016/S2213-8587(13)70062-7 [DOI] [PubMed] [Google Scholar]
- Strucksberg K-H, Tangavelou K, Schröder R, Clemen CS, 2010. Proteasomal activity in skeletal muscle: a matter of assay design, muscle type, and age. Anal. Biochem 399, 225–229. 10.1016/j.ab.2009.12.026 [DOI] [PubMed] [Google Scholar]
- Takekura H, Fujinami N, Nishizawa T, Ogasawara H, Kasuga N, 2001. Eccentric exercise-induced morphological changes in the membrane systems involved in excitation-contraction coupling in rat skeletal muscle. J. Physiol 533, 571–583. 10.1111/j.1469-7793.2001.0571a.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert EE, Smuder AJ, Min K, Kwon OS, Powers SK, 2013. Calpain and caspase-3 play required roles in immobilization-induced limb muscle atrophy. J. Appl. Physiol. Bethesda Md 1985 114, 1482–1489. 10.1152/japplphysiol.00925.2012 [DOI] [PubMed] [Google Scholar]
- Tando T, Hirayama A, Furukawa M, Sato Y, Kobayashi T, Funayama A, Kanaji A, Hao W, Watanabe R, Morita M, Oike T, Miyamoto K, Soga T, Nomura M, Yoshimura A, Tomita M, Matsumoto M, Nakamura M, Toyama Y, Miyamoto T, 2016. Smad2/3 Proteins Are Required for Immobilization-induced Skeletal Muscle Atrophy. J. Biol. Chem 291, 12184–12194. 10.1074/jbc.M115.680579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrill JR, Radley-Crabb HG, Iwasaki T, Lemckert FA, Arthur PG, Grounds MD, 2013. Oxidative stress and pathology in muscular dystrophies: focus on protein thiol oxidation and dysferlinopathies. FEBS J. 280, 4149–4164. 10.1111/febs.12142 [DOI] [PubMed] [Google Scholar]
- Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C, Loefler S, Kern H, Franceschi C, Salvioli S, Conte M, Blaauw B, Zampieri S, Salviati L, Scorrano L, Sandri M, 2017. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 25, 1374–1389.e6. 10.1016/j.cmet.2017.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJ, 1995. Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci 108 ( Pt 6), 2197–2204. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Villalta SA, 2010. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol 298, R1173–1187. 10.1152/ajpregu.00735.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M, 2007. The role of free radicals in the pathophysiology of muscular dystrophy. J. Appl. Physiol. Bethesda Md 1985 102, 1677–1686. 10.1152/japplphysiol.01145.2006 [DOI] [PubMed] [Google Scholar]
- Timpani CA, Hayes A, Rybalka E, 2015. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 85, 1021–1033. 10.1016/j.mehy.2015.08.015 [DOI] [PubMed] [Google Scholar]
- Tipton KD, Elliott TA, Cree MG, Wolf SE, Sanford AP, Wolfe RR, 2004. Ingestion of Casein and Whey Proteins Result in Muscle Anabolism after Resistance Exercise. Med. Sci. Sports Exerc 36, 2073–2081. 10.1249/01.MSS.0000147582.99810.C5 [DOI] [PubMed] [Google Scholar]
- Tisdale MJ, 2009. Mechanisms of cancer cachexia. Physiol. Rev 89, 381–410. 10.1152/physrev.00016.2008 [DOI] [PubMed] [Google Scholar]
- Tomlinson BE, Irving D, 1977. The numbers of limb motor neurons in the human lumbosacral cord throughout life. J. Neurol. Sci 34, 213–219. [DOI] [PubMed] [Google Scholar]
- Tran L, Kras KA, Hoffman N, Ravichandran J, Dickinson JM, D’Lugos A, Carroll CC, Patel SH, Mandarino LJ, Roust L, Katsanos CS, 2018. Lower Fasted-State but Greater Increase in Muscle Protein Synthesis in Response to Elevated Plasma Amino Acids in Obesity. Obesity 26, 1179–1187. 10.1002/oby.22213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranmer JE, Heyland D, Dudgeon D, Groll D, Squires-Graham M, Coulson K, 2003. Measuring the symptom experience of seriously ill cancer and noncancer hospitalized patients near the end of life with the memorial symptom assessment scale. J. Pain Symptom Manage 25, 420–429. 10.1016/s0885-3924(03)00074-5 [DOI] [PubMed] [Google Scholar]
- Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG, 2011. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet 20, 790–805. 10.1093/hmg/ddq523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi E, Rasmussen BB, 2000. Nutrition and muscle protein metabolism in the elderly. Diabetes Nutr. Metab 13, 99–107. [PubMed] [Google Scholar]
- Volpi E, Sheffield-Moore M, Rasmussen BB, Wolfe RR, 2001. Basal muscle amino acid kinetics and protein synthesis in healthy young and older men. JAMA 286, 1206–1212. 10.1001/jama.286.10.1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki H, Tontonoz P, 2007. Endocrine Functions of Adipose Tissue. Annu. Rev. Pathol. Mech. Dis 2, 31–56. 10.1146/annurev.pathol.2.010506.091859 [DOI] [PubMed] [Google Scholar]
- Wall BT, Dirks ML, Snijders T, Senden JMG, Dolmans J, van Loon LJC, 2014. Substantial skeletal muscle loss occurs during only 5 days of disuse. Acta Physiol. Oxf. Engl 210, 600–611. 10.1111/apha.12190 [DOI] [PubMed] [Google Scholar]
- Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A, Casey DC, Charlson FJ, Chen AZ, Coates MM, Coggeshall M, Dandona L, Dicker DJ, Erskine HE, Ferrari AJ, Fitzmaurice C, Foreman K, Forouzanfar MH, Fraser MS, Fullman N, Gething PW, Goldberg EM, Graetz N, Haagsma JA, Hay SI, Huynh C, Johnson CO, Kassebaum NJ, Kinfu Y, Kulikoff XR, Kutz M, Kyu HH, Larson HJ, Leung J, Liang X, Lim SS, Lind M, Lozano R, Marquez N, Mensah GA, Mikesell J, Mokdad AH, Mooney MD, Nguyen G, Nsoesie E, Pigott DM, Pinho C, Roth GA, Salomon JA, Sandar L, Silpakit N, Sligar A, Sorensen RJD, Stanaway J, Steiner C, Teeple S, Thomas BA, Troeger C, VanderZanden A, Vollset SE, Wanga V, Whiteford HA, Wolock T, Zoeckler L, Abate KH, Abbafati C, Abbas KM, Abd-Allah F, Abera SF, Abreu DMX, Abu-Raddad LJ, Abyu GY, Achoki T, Adelekan AL, Ademi Z, Adou AK, Adsuar JC, Afanvi KA, Afshin A, Agardh EE, Agarwal A, Agrawal A, Kiadaliri AA, Ajala ON, Akanda AS, Akinyemi RO, Akinyemiju TF, Akseer N, Lami FHA, Alabed S, Al-Aly Z, Alam K, Alam NKM, Alasfoor D, Aldhahri SF, Aldridge RW, Alegretti MA, Aleman AV, Alemu ZA, Alexander LT, Alhabib S, Ali R, Alkerwi A, Alla F, Allebeck P, Al-Raddadi R, Alsharif U, Altirkawi KA, Martin EA, Alvis-Guzman N, Amare AT, Amegah AK, Ameh EA, Amini H, Ammar W, Amrock SM, Andersen HH, Anderson BO, Anderson GM, Antonio CAT, Aregay AF, Ärnlöv J, Arsenijevic VSA, Artaman A, Asayesh H, Asghar RJ, Atique S, Avokpaho EFGA, Awasthi A, Azzopardi P, Bacha U, Badawi A, Bahit MC, Balakrishnan K, Banerjee A, Barac A, Barker-Collo SL, Bärnighausen T, Barregard L, Barrero LH, Basu A, Basu S, Bayou YT, Bazargan-Hejazi S, Beardsley J, Bedi N, Beghi E, Belay HA, Bell B, Bell ML, Bello AK, Bennett DA, Bensenor IM, Berhane A, Bernabé E, Betsu BD, Beyene AS, Bhala N, Bhalla A, Biadgilign S, Bikbov B, Abdulhak AAB, Biroscak BJ, Biryukov S, Bjertness E, Blore JD, Blosser CD, Bohensky MA, Borschmann R, Bose D, Bourne RRA, Brainin M, Brayne CEG, Brazinova A, Breitborde NJK, Brenner H, Brewer JD, Brown A, Brown J, Brugha TS, Buckle GC, Butt ZA, Calabria B, Campos-Nonato IR, Campuzano JC, Carapetis JR, Cárdenas R, Carpenter DO, Carrero JJ, Castañeda-Orjuela CA, Rivas JC, Catalá-López F, Cavalleri F, Cercy K, Cerda J, Chen W, Chew A, Chiang PP-C, Chibalabala M, Chibueze CE, Chimed-Ochir O, Chisumpa VH, Choi J-YJ, Chowdhury R, Christensen H, Christopher DJ, Ciobanu LG, Cirillo M, Cohen AJ, Colistro V, Colomar M, Colquhoun SM, Cooper C, Cooper LT, Cortinovis M, Cowie BC, Crump JA, DamsereDerry J, Danawi H, Dandona R, Daoud F, Darby SC, Dargan PI, Neves J. das, Davey G, Davis AC, Davitoiu DV, Castro E.F. de, Jager P. de, Leo DD, Degenhardt L, Dellavalle RP, Deribe K, Deribew A, Dharmaratne SD, Dhillon PK, Diaz-Torné C, Ding EL, Santos K.P.B. dos, Dossou E, Driscoll TR, Duan L, Dubey M, Duncan BB, Ellenbogen RG, Ellingsen CL, Elyazar I, Endries AY, Ermakov SP, Eshrati B, Esteghamati A, Estep K, Faghmous IDA, Fahimi S, Faraon EJA, Farid TA, Farinha C.S. e S., Faro A, Farvid MS, Farzadfar F, Feigin VL, Fereshtehnejad S-M, Fernandes JG, Fernandes JC, Fischer F, Fitchett JRA, Flaxman A, Foigt N, Fowkes FGR, Franca EB, Franklin RC, Friedman J, Frostad J, Fürst T, Futran ND, Gall SL, Gambashidze K, Gamkrelidze A, Ganguly P, Gankpé FG, Gebre T, Gebrehiwot TT, Gebremedhin AT, Gebru AA, Geleijnse JM, Gessner BD, Ghoshal AG, Gibney KB, Gillum RF, Gilmour S, Giref AZ, Giroud M, Gishu MD, Giussani G, Glaser E, Godwin WW, Gomez-Dantes H, Gona P, Goodridge A, Gopalani SV, Gosselin RA, Gotay CC, Goto A, Gouda HN, Greaves F, Gugnani HC, Gupta Rahul, Gupta Rajeev, Gupta V, Gutiérrez RA, Hafezi-Nejad N, Haile D, Hailu AD, Hailu GB, Halasa YA, Hamadeh RR, Hamidi S, Hancock J, Handal AJ, Hankey GJ, Hao Y, Harb HL, Harikrishnan S, Haro JM, Havmoeller R, Heckbert SR, Heredia-Pi IB, Heydarpour P, Hilderink HBM, Hoek HW, Hogg RS, Horino M, Horita N, Hosgood HD, Hotez PJ, Hoy DG, Hsairi M, Htet AS, Htike MMT, Hu G, Huang C, Huang H, Huiart L, Husseini A, Huybrechts I, Huynh G, Iburg KM, Innos K, Inoue M, Iyer VJ, Jacobs TA, Jacobsen KH, Jahanmehr N, Jakovljevic MB, James P, Javanbakht M, Jayaraman SP, Jayatilleke AU, Jeemon P, Jensen PN, Jha V, Jiang G, Jiang Y, Jibat T, Jimenez-Corona A, Jonas JB, Joshi TK, Kabir Z, Kamal R, Kan H, Kant S, Karch A, Karema CK, Karimkhani C, Karletsos D, Karthikeyan G, Kasaeian A, Katibeh M, Kaul A, Kawakami N, Kayibanda JF, Keiyoro PN, Kemmer L, Kemp AH, Kengne AP, Keren A, Kereselidze M, Kesavachandran CN, Khader YS, Khalil IA, Khan AR, Khan EA, Khang Y-H, Khera S, Khoja TAM, Kieling C, Kim D, Kim YJ, Kissela BM, Kissoon N, Knibbs LD, Knudsen AK, Kokubo Y, Kolte D, Kopec JA, Kosen S, Koul PA, Koyanagi A, Krog NH, Defo BK, Bicer BK, Kudom AA, Kuipers EJ, Kulkarni VS, Kumar GA, Kwan GF, Lal A, Lal DK, Lalloo R, Lallukka T, Lam H, Lam JO, Langan SM, Lansingh VC, Larsson A, Laryea DO, Latif AA, Lawrynowicz AEB, Leigh J, Levi M, Li Y, Lindsay MP, Lipshultz SE, Liu PY, Liu S, Liu Y, Lo L-T, Logroscino G, Lotufo PA, Lucas RM, Lunevicius R, Lyons RA, Ma S, Machado VMP, Mackay MT, MacLachlan JH, Razek HMAE, Magdy M, Razek AE, Majdan M, Majeed A, Malekzadeh R, Manamo WAA, Mandisarisa J, Mangalam S, Mapoma CC, Marcenes W, Margolis DJ, Martin GR, Martinez-Raga J, Marzan MB, Masiye F, Mason-Jones AJ, Massano J, Matzopoulos R, Mayosi BM, McGarvey ST, McGrath JJ, McKee M, McMahon BJ, Meaney PA, Mehari A, Mehndiratta MM, Mejia-Rodriguez F, Mekonnen AB, Melaku YA, Memiah P, Memish ZA, Mendoza W, Meretoja A, Meretoja TJ, Mhimbira FA, Micha R, Millear A, Miller TR, Mirarefin M, Misganaw A, Mock CN, Mohammad KA, Mohammadi A, Mohammed S, Mohan V, Mola GLD, Monasta L, Hernandez JCM, Montero P, Montico M, Montine TJ, Moradi-Lakeh M, Morawska L, Morgan K, Mori R, Mozaffarian D, Mueller UO, Murthy GVS, Murthy S, Musa KI, Nachega JB, Nagel G, Naidoo KS, Naik N, Naldi L, Nangia V, Nash D, Nejjari C, Neupane S, Newton CR, Newton JN, Ng M, Ngalesoni FN, Ngirabega J. de D., Nguyen QL, Nisar MI, Pete PMN, Nomura M, Norheim OF, Norman PE, Norrving B, Nyakarahuka L, Ogbo FA, Ohkubo T, Ojelabi FA, Olivares PR, Olusanya BO, Olusanya JO, Opio JN, Oren E, Ortiz A, Osman M, Ota E, Ozdemir R, Pa M, Pain A, Pandian JD, Pant PR, Papachristou C, Park E-K, Park J-H, Parry CD, Parsaeian M, Caicedo AJP, Patten SB, Patton GC, Paul VK, Pearce N, Pedro JM, Stokic LP, Pereira DM, Perico N, Pesudovs K, Petzold M, Phillips MR, Piel FB, Pillay JD, Plass D, Platts-Mills JA, Polinder S, Pope CA, Popova S, Poulton RG, Pourmalek F, Prabhakaran D, Qorbani M, Quame-Amaglo J, Quistberg DA, Rafay A, Rahimi K, Rahimi-Movaghar V, Rahman M, Rahman MHU, Rahman SU, Rai RK, Rajavi Z, Rajsic S, Raju M, Rakovac I, Rana SM, Ranabhat CL, Rangaswamy T, Rao P, Rao SR, Refaat AH, Rehm J, Reitsma MB, Remuzzi G, Resnikoff S, Ribeiro AL, Ricci S, Blancas MJR, Roberts B, Roca A, Rojas-Rueda D, Ronfani L, Roshandel G, Rothenbacher D, Roy A, Roy NK, Ruhago GM, Sagar R, Saha S, Sahathevan R, Saleh MM, Sanabria JR, Sanchez-Niño MD, Sanchez-Riera L, Santos IS, Sarmiento-Suarez R, Sartorius B, Satpathy M, Savic M, Sawhney M, Schaub MP, Schmidt MI, Schneider IJC, Schöttker B, Schutte AE, Schwebel DC, Seedat S, Sepanlou SG, Servan-Mori EE, Shackelford KA, Shaddick G, Shaheen A, Shahraz S, Shaikh MA, Shakh-Nazarova M, Sharma R, She J, Sheikhbahaei S, Shen J, Shen Z, Shepard DS, Sheth KN, Shetty BP, Shi P, Shibuya K, Shin M-J, Shiri R, Shiue I, Shrime MG, Sigfusdottir ID, Silberberg DH, Silva DAS, Silveira DGA, Silverberg JI, Simard EP, Singh A, Singh GM, Singh JA, Singh OP, Singh PK, Singh V, Soneji S, Søreide K, Soriano JB, Sposato LA, Sreeramareddy CT, Stathopoulou V, Stein DJ, Stein MB, Stranges S, Stroumpoulis K, Sunguya BF, Sur P, Swaminathan S, Sykes BL, Szoeke CEI, Tabarés-Seisdedos R, Tabb KM, Takahashi K, Takala JS, Talongwa RT, Tandon N, Tavakkoli M, Taye B, Taylor HR, Ao BJT, Tedla BA, Tefera WM, Have MT, Terkawi AS, Tesfay FH, Tessema GA, Thomson AJ, Thorne-Lyman AL, Thrift AG, Thurston GD, Tillmann T, Tirschwell DL, Tonelli M, Topor-Madry R, Topouzis F, Towbin JA, Traebert J, Tran BX, Truelsen T, Trujillo U, Tura AK, Tuzcu EM, Uchendu US, Ukwaja KN, Undurraga EA, Uthman OA, Dingenen RV, Donkelaar A. van, Vasankari T, Vasconcelos AMN, Venketasubramanian N, Vidavalur R, Vijayakumar L, Villalpando S, Violante FS, Vlassov VV, Wagner JA, Wagner GR, Wallin MT, Wang L, Watkins DA, Weichenthal S, Weiderpass E, Weintraub RG, Werdecker A, Westerman R, White RA, Wijeratne T, Wilkinson JD, Williams HC, Wiysonge CS, Woldeyohannes SM, Wolfe CDA, Won S, Wong JQ, Woolf AD, Xavier D, Xiao Q, Xu G, Yakob B, Yalew AZ, Yan LL, Yano Y, Yaseri M, Ye P, Yebyo HG, Yip P, Yirsaw BD, Yonemoto N, Yonga G, Younis MZ, Yu S, Zaidi Z, Zaki MES, Zannad F, Zavala DE, Zeeb H, Zeleke BM, Zhang H, Zodpey S, Zonies D, Zuhlke LJ, Vos T, Lopez AD, Murray CJL, 2016. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. The Lancet 388, 1459–1544. 10.1016/S0140-6736(16)31012-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B, 2012. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338, 956–959. 10.1126/science.1225967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pickrell AM, Zimmers TA, Moraes CT, 2012. Increase in Muscle Mitochondrial Biogenesis Does Not Prevent Muscle Loss but Increased Tumor Size in a Mouse Model of Acute Cancer-Induced Cachexia. PLOS ONE 7, e33426. 10.1371/journal.pone.0033426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Weisleder N, Collet C, Zhou J, Chu Y, Hirata Y, Zhao X, Pan Z, Brotto M, Cheng H, Ma J, 2005. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat. Cell Biol 7, 525–530. 10.1038/ncb1254 [DOI] [PubMed] [Google Scholar]
- Weber J, Polo S, Maspero E, 2019. HECT E3 Ligases: A Tale With Multiple Facets. Front. Physiol 10. 10.3389/fphys.2019.00370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG, 2001. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol 155, 123–131. 10.1083/jcb.200105110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, 2003. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest 112, 1796–1808. 10.1172/JCI19246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DJ, Casale GP, Koutakis P, Nella AA, Swanson SA, Zhu Z, Miserlis D, Johanning JM, Pipinos II, 2013. Oxidative damage and myofiber degeneration in the gastrocnemius of patients with peripheral arterial disease. J. Transl. Med 11, 230. 10.1186/1479-5876-11-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG, 1998. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 421, 125–130. 10.1016/s0014-5793(97)01548-2 [DOI] [PubMed] [Google Scholar]
- White JP, 2017. IL-6, cancer and cachexia: metabolic dysfunction creates the perfect storm. Transl. Cancer Res 6, S280–S285. 10.21037/tcr.2017.03.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA, 2011. The Regulation of Skeletal Muscle Protein Turnover during the Progression of Cancer Cachexia in the ApcMin/+ Mouse. PLOS ONE 6, e24650. 10.1371/journal.pone.0024650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW, Froehner SC, Allen DG, 2010. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PloS One 5, e15354. 10.1371/journal.pone.0015354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC, 1997. Changes in nutritional status associated with unresectable pancreatic cancer. Br. J. Cancer 75, 106–109. 10.1038/bjc.1997.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes EA, Selby AL, Atherton PJ, Patel R, Rankin D, Smith K, Rennie MJ, 2009. Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age-related sarcopenia. Am. J. Clin. Nutr 90, 1343–1350. 10.3945/ajcn.2009.27543 [DOI] [PubMed] [Google Scholar]
- Wilkinson DJ, Piasecki M, Atherton PJ, 2018. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res. Rev 47, 123–132. 10.1016/j.arr.2018.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisse BE, Frayo RS, Schwartz MW, Cummings DE, 2001. Reversal of cancer anorexia by blockade of central melanocortin receptors in rats. Endocrinology 142, 3292–3301. 10.1210/endo.142.8.8324 [DOI] [PubMed] [Google Scholar]
- Wu M, Fannin J, Rice KM, Wang B, Blough ER, 2011. Effect of aging on cellular mechanotransduction. Ageing Res. Rev 10, 1–15. 10.1016/j.arr.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Katta A, Gadde MK, Liu H, Kakarla SK, Fannin J, Paturi S, Arvapalli RK, Rice KM, Wang Y, Blough ER, 2009. Aging-Associated Dysfunction of Akt/Protein Kinase B: S-Nitrosylation and Acetaminophen Intervention. PLOS ONE 4, e6430. 10.1371/journal.pone.0006430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Rape M, 2009. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol 10, 755–764. 10.1038/nrm2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Chen Y, Tooze SA, 2018. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14, 207–215. 10.1080/15548627.2017.1378838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Ktistakis NT, 2020. Mammalian Mitophagosome Formation: A Focus on the Early Signals and Steps. Front. Cell Dev. Biol 8. 10.3389/fcell.2020.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]