

Abstract
Extracellular vesicles (EVs) have emerged as important mediators in cell–cell communication; however, their relevance in pulmonary hypertension (PH) secondary to human immunodeficiency virus (HIV) infection is yet to be explored. Considering that circulating monocytes are the source of the increased number of perivascular macrophages surrounding the remodeled vessels in PH, this study aimed to identify the role of circulating small EVs and EVs released by HIV-infected human monocyte–derived macrophages in the development of PH. We report significantly higher numbers of plasma-derived EVs carrying higher levels of TGF-β1 (transforming growth factor-β1) in HIV-positive individuals with PH compared with individuals without PH. Importantly, levels of these TGF-β1–loaded, plasma-derived EVs correlated with pulmonary arterial systolic pressures and CD4 counts but did not correlate with the Dl CO or viral load. Correspondingly, enhanced TGF-β1–dependent pulmonary endothelial injury and smooth muscle hyperplasia were observed. HIV-1 infection of monocyte-derived macrophages in the presence of cocaine resulted in an increased number of TGF-β1–high EVs, and intravenous injection of these EVs in rats led to increased right ventricle systolic pressure accompanied by myocardial injury and increased levels of serum ET-1 (endothelin-1), TNF-α, and cardiac troponin-I. Conversely, pretreatment of rats with TGF-β receptor 1 inhibitor prevented these EV-mediated changes. Findings define the ability of macrophage-derived small EVs to cause pulmonary vascular modeling and PH via modulation of TGF-β signaling and suggest clinical implications of circulating TGF-β–high EVs as a potential biomarker of HIV-associated PH.
Keywords: monocytes, smooth muscle dysfunction, endothelial injury, pulmonary hypertension, inflammation
Pulmonary arterial hypertension (PAH) is one of the most devastating noninfectious complications related to human immunodeficiency virus (HIV) infection, and echocardiographic evidence suggests a prevalence of increased pulmonary arterial systolic pressure (PASP) in 2.6–15.5% of people living with HIV (PLWH) (1). HIV-associated PAH (HIV-PAH) falls under group I pulmonary hypertension (PH), and the risk factors, such as cocaine, methamphetamine, and intravenous drug use in PLWH, can potentiate the development of HIV-PAH (2). Furthermore, with the increasing burden of cardiac and chronic respiratory complications in PLWH, the prevalence of secondary PH is suggested to increase as well in these individuals (3).
The presence of interstitial and perivascular infiltration of inflammatory cells has been linked to intimal and medial thickening of blood vessels associated with PH (4). Recently, we reported increased perivascular macrophages in the remodeled, thickened vessels in the lung sections from HIV-positive humans, simian immunodeficiency virus (SIV)–positive macaques, and HIV-transgenic (HIV-Tg) rats, particularly in the presence of exposure to illicit drugs (5, 6). However, the extent to which perivascular inflammation is involved in the thickening and occlusion of blood vessels is elusive. It is believed that inflammation precedes vascular remodeling in PAH and is more often an origin rather than a result of PAH (4, 7). Circulating monocytes contribute to increased interstitial macrophages observed around the remodeling pulmonary vessels in PH (8, 9).
Aside from directly releasing viral proteins, cytokines, and growth factors to the vascular cells, immune cells may also deliver their molecular cargo through the release of extracellular vesicles (EVs) (10, 11). EVs are small-membrane vesicles ranging from 30 to 1,000 nm in size and are involved in intercellular communication by delivering protein/nucleic acid cargo to surrounding cells. Studies have focused on how changes in the EV cargo contribute to the development of various diseases in animal models, including of PAH (12, 13). However, studies that have associated the levels of circulating membrane vesicles with pulmonary vascular resistance in patients with PAH have been limited (14, 15). These studies were focused on larger microparticles and lacked details on the type, size, and composition of EVs, leaving open the question of the role of small EVs.
Recent reports suggest the presence of viral proteins in BAL fluid– and plasma-derived EVs (PEVs) from PLWH (16–18). We demonstrated earlier that HIV infection and cocaine synergize in the activation of monocyte-derived macrophages (MDMs) (19) and that EVs released by these HIV-infected and cocaine-exposed (H + C) MDMs promote smooth muscle proliferation (18). Given the importance of TGF-β signaling in the development of PH (20–22), induction of TGF-β1 (transforming growth factor β1) in HIV-infected macrophages (23), and recent reports showing the presence of TGF-β1 on the surface of and within exosomes (24), we hypothesize that the increased levels of TGF-β–loaded EVs in the plasma from PLWH may be associated with the presence of PH and that EVs released by HIV-infected MDMs cause pulmonary vascular changes and right ventricle (RV) dysfunction in wild-type (WT) and noninfectious HIV-Tg rats. Some of the results of these studies have been previously reported in the form of a preprint (https://doi.org/10.1101/2020.09.09.20191338).
Methods
Human Samples and Data Collection
Plasma specimens used for this study were collected as part of the University of Pittsburgh HIV Cohort, and collection was approved by the institutional review board. Details on the inclusion/exclusion criteria have been published in previous reports (25, 26). A total of 73 deidentified human plasma samples from HIV-negative individuals without a history of cocaine use, HIV-negative individuals with a history of cocaine use, PLWH without a history of cocaine use, PLWH with a history of cocaine use (PLWH + Coc), and PLWH with PH (PLWH + PH) were analyzed. HIV-negative individuals with a history of cocaine use and PLWH + Coc were individuals who had self-reported regular use of cocaine (inhaled, crack, or intravenous use) within the last 6 months of their visit, with no history of consuming any other stimulants, opioids, or sedatives being reported. Individuals without a history of drug use reported that they had never used any type of illicit drug. Demographic data such as age, sex, and race and information on smoking history and use of antiretroviral therapy were collected from subjects by using a standardized questionnaire. Plasma CD4+ T-cell counts and viral load values closest to the current status within 6 months were used. Echocardiography, spirometry, and Dl CO measurements were performed as described (27, 28). The presence of PH was determined on the basis of echocardiography, was defined as having PASP greater than 40 mm Hg, and was not specific to the presence of group I PH. PASP data were also available for n = 18 PLWH without PH. All plasma samples were isolated from peripheral blood collected in EDTA tubes and stored at −80°C until use.
MDM Cultures
Mono-Mac-1 (a human monocytic cell line) cells were grown in RPMI-1640 and differentiated into MDMs, infected with HIVBal virus (5 ng/ml), and exposed to cocaine hydrochloride (1μM) for 4 days as described previously (18). The cell culture supernatants were collected and processed for EV isolation.
Animal Groups
Eight to 9-month old male HIV-Tg Fisher rats (n = 6–8/group) (ENVIGO) were intravenously administered 40 μg of EVs (four injections of 10 μg each at 5-d intervals) derived from uninfected MDMs (HIV-Tg control [Con] EV group) or H + C MDMs (HIV-Tg H + C EV group) together with cocaine (40 mg/kg, i.p.) once daily for 21 days. In parallel, the WT Fisher rats were administered uninfected MDM EVs (WT Con EV group) or H + C MDM EVs (WT H + C EV group). For TGF-β inhibitor studies, WT and HIV-Tg rats were either given TGF-β type I receptor inhibitor: GW788388 (1 mg/kg/day) (29) or vehicle orally for 21 days. The animals were housed at the University of Kansas Medical Center (KUMC) in strict accordance with the National Institutes of Health and Institutional Animal Care and Use Committee guidelines (protocol number 2018-2446). Animals administered EVs derived from HIV-infected cells were housed in a biohazard-contained room and killed in a biosafety cabinet.
Detailed descriptions of methods and statistical analyses are provided in the data supplement.
Results
PEVs Are Elevated in HIV-Positive Individuals with or without a History of Cocaine Use
Table 1 shows the demographic and clinical parameters of plasma samples from PLWH + Coc (n = 15) or PLWH without a history of cocaine use (n = 15) and HIV-negative individuals with (n = 15) or without (n = 15) a history of cocaine use. Sex, race, and the CD4+ T-cell count were the only variables observed to be significantly different among groups. Nanoparticle tracking analysis showed the highest total number of EVs in plasma from PLWH + Coc when isolated by using the exoEasy (QIAGEN) method and compared with all other groups (Figures 1A and 1B), with significantly higher numbers of EVs being observed in the size range of 100–250 nm (Figure 1C). The same trend in the increase of the EV number was also observed in EV samples isolated by using the ultracentrifugation method (see Figures E1A and E1B in the data supplement). Subdividing the HIV-positive subjects on the basis of their viral load (HIV-1 RNA copies/ml of plasma) showed higher levels of EVs in PLWH + Coc than in PLWH, regardless of a suppressed or higher (>50 copies/ml) (30) viral load (Figure 1D). These small EVs were further characterized by using transmission electron microscopy, which demonstrated classic, bilayered lipid structures (Figure 1E), and by using Western blotting, which showed the presence of exosomal markers (Figure 1F).
Table 1.
Demographic and Clinical Characteristics of Participants by HIV Status and Cocaine Use
| Characteristic [n (%) or Median (IQR)] | HIV-Negative Individuals without a History of Cocaine Use (N = 15) | HIV-Negative Individuals with a History of Cocaine Use (N = 15) | PLWH (N = 15) | PLWH + Coc (N = 15) | PLWH + PH (N = 7) | PLWH + Coc with PH (N = 6) |
|---|---|---|---|---|---|---|
| Age, yr | 50.0 (43.5–56.5) | 51.0 (47.5–57.5) | 48.0 (39.0–53.5) | 50.0 (44.5–53.0) | 55.0 (46.5–58.0) | 48.5 (46.5–52.75) |
| Sex* | ||||||
| Male | 7 (46.6) | 6 (40.0) | 12 (80.0) | 12 (80.0) | 7 (100.0) | 2 (33.3) |
| Female | 8 (53.4) | 9 (60.0) | 3 (20.0) | 3 (20.0) | 0 (00.0) | 4 (66.7) |
| Race* | ||||||
| Black, non-Hispanic | 5 (33.3) | 13 (86.7) | 6 (40.0) | 11 (73.3) | 3 (42.9) | 5 (83.3) |
| White | 10 (66.7) | 1 (6.7) | 8 (53.3) | 4 (26.7) | 4 (57.1) | 1 (16.7) |
| Other | — | 1 (6.7) | 1 (6.7) | — | — | — |
| Smoking status | ||||||
| Current smoker | 4 (26.7) | 11 (73.3) | 7 (46.7) | 11 (73.3) | 3 (42.9) | 3 (50.0) |
| Former smoker | 3 (20.0) | 3 (20.0) | 3 (20.0) | 2 (13.3) | 2 (28.6) | 2 (33.3) |
| Never-smoker | 8 (53.3) | 1 (6.7) | 5 (33.3) | 2 (13.3) | 2 (28.6) | 1 (16.7) |
| Pack-years | 0 (0.0–1.3) | 8.8 (3.8–16.1) | 6.8 (0.0–20.0) | 17.0 (6.8–30.1) | 25.8 (0.5–35.3) | 19.4 (9.8–28.2) |
| COPD ever | 2 (13.3) | 2 (13.3) | 1 (6.7) | 2 (13.3) | 1 (14.3) | 2 (50.0) |
| Lung cancer ever | — | — | — | 1 (6.7) | — | — |
| Diabetes ever | 1 (6.7) | 2 (13.3) | 2 (13.3) | 2 (13.3) | 2 (28.6) | 0 |
| Hepatitis ever* | 0 | 5 (33.3) | 5 (33.3) | 8 (53.3) | 3 (42.9) | 2 (33.3) |
| HIV risk factor | ||||||
| Homosexual | — | — | 10 (66.7) | 8 (53.2) | 7 (100.0) | 2 (33.3) |
| Heterosexual | — | — | 2 (13.3) | 5 (33.3) | 0 (0) | 2 (33.3) |
| Other | — | — | 3 (20.0) | 2 (13.3) | 0 (0) | 2 (33.3) |
| CD4, cell count/μl* | — | — | 436.0 (307.0–657.0) | 551.5 (442.5–717.5) | 695.0 (691.0–860.0) | 234.0 (69.0–375.0) |
| Viral load, median (range) copies/ml | — | — | 39.0 (19.0–99,200.0) | 49.0 (19.0–1,250,000.0) | 39.0 (19.0–39.0) | 49.0 (9.0–336,000.0) |
| On ART | N/A | N/A | 14 (93.3) | 13(87) | 7 (100) | 3 (42.9) |
Definition of abbreviations: ART = antiretroviral therapy; CD4 = CD4+ T lymphocyte; COPD = chronic obstructive pulmonary disease; HIV = human immunodeficiency virus; IQR = interquartile range; N/A = not applicable; PH = pulmonary hypertension; PLWH = people living with HIV; PLWH + Coc = PLWH with a history of cocaine use; PLWH + PH = PLWH with PH.
Variables are statistically significant among the six groups. The differences among the groups were assessed by using a Fisher exact test for categorical variables and by using ANOVA or a Kruskal-Wallis test for continuous variables, depending on whether the data satisfied the normality assumption or not.
Figure 1.
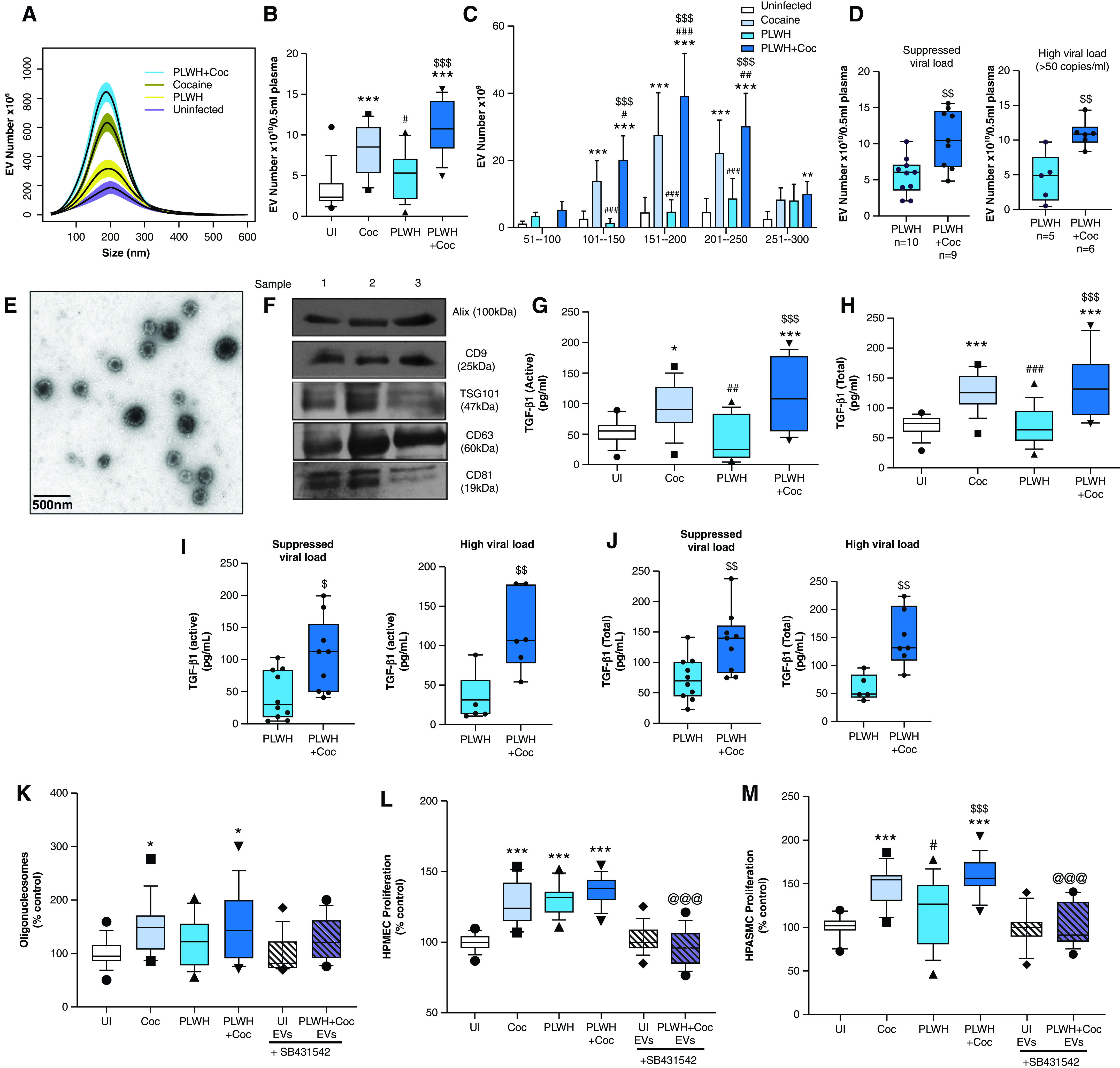
Analysis of plasma-derived extracellular vesicles (PEVs) from those with human immunodeficiency virus (HIV) and/or a history of cocaine use. Extracellular vesicles (EVs) were isolated from 0.5 milliliter (ml) of EDTA plasma from HIV-negative individuals without a history of cocaine use (UI), HIV-negative individuals with a history of cocaine use (Coc), people living with HIV (PLWH) without a history of cocaine use, and PLWH with a history of cocaine use (PLWH + Coc) (n = 15/group) by using an exoEasy kit. (A–D) Particles were counted and characterized for size distribution by using nanoparticle tracking analysis. Data represent the number of EVs/ml of a final EV suspension obtained from 0.5 ml of EDTA plasma by using an exoEasy kit. The high viral load group included PLWH with ⩾50 viral RNA copies of per ml of plasma. (E–F) Characterization of PEVs by using transmission electron microscopy (TEM) and Western blotting analysis. Representative TEM image of PEVs at 5,000× magnification (E) and Western blot of PEV protein extract from three different subjects for exosomal markers (F). Scale bar, 500 nm. (G–J) PEVs were analyzed for active (G and I) and total (H and J) TGF-β1 (transforming growth factor-β1) levels by using an ELISA. Analysis of (K) endothelial apoptosis and (L) proliferation on treatment with PEVs from HIV-negative individuals without a history of cocaine use, HIV-negative individuals with a history of cocaine use, PLWH, and PLWH + Coc (n = 15/group). Human pulmonary microvascular endothelial cells (HPMECs) were plated in a 96-well plate and were serum-starved after 24 hours in 0.5% serum–containing media, which was followed by the addition of 3 μg of PEVs/well. (K) Analysis of apoptosis by using a cell-death ELISA after 24 hours and (L) analysis of cell proliferation by using a cell proliferation assay after 48 hours. (M) Analysis of human pulmonary arterial smooth muscle cell (HPASMC) proliferation on treatment with PEVs. After 72 hours of plating in a 96-well plate, cells were serum starved for 48 hours, which followed by the addition of 3 μg of PEVs/well. A MTS cell proliferation assay was performed at 48 hours after treatment. Cell proliferation and apoptosis assays were also performed on cells treated with PLWH + Coc PEVs in the presence of 10μM SB431542 (TGFβ-R1 inhibitor [In.]). Boxes represent the median and interquartile range (IQR), and whiskers show the 10th to 90th percentiles. *P < 0.05, **P < 0.01, and ***P < 0.001 versus HIV-negative individuals without a history of cocaine use; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus HIV-negative individuals with a history of cocaine use; $P < 0.05, $$P < 0.01, and $$$P < 0.001 versus PLWH; and @@@P < 0.001 versus PLWH + Coc without TGFβ-R1 In.
PEVs from HIV-Positive Individuals with or without a History of Cocaine Use Promote TGF-β–Dependent Pulmonary Endothelial and Smooth Muscle Dysfunction
Analysis of TGF-β1 levels in PEVs showed significantly increased levels of active (Figure 1G) as well as total (active plus latent) (Figure 1H) TGF-β1 in PEVs from individuals in HIV-negative individuals with a history of cocaine use and PLWH + Coc when compared with PEVs from HIV-negative Con subjects. Importantly, levels of TGF-β1 in PEVs from the PLWH + Coc group were also significantly higher compared with levels from PLWH without a history of drug use (Figures 1G and 1H), and levels were high in both viremic and nonviremic patients (Figures 1I and 1J). Alterations in the levels of TGF-β1 in EVs isolated by using the exoEasy method from PLWH or HIV-negative individuals were also confirmed in a small set of EV samples isolated by using the ultracentrifugation method (Figure E1C).
EVs are readily taken up by human pulmonary arterial smooth muscle cells (HPASMCs) (18) as well as by human pulmonary microvascular endothelial cells (HPMECs) (Figure E2A). Elevated levels of TGF-β1 in the PEV cargo led us to explore the effect of PEVs on human endothelial cells (ECs) and smooth muscle cells (SMCs) in the presence and absence of the TGFβ-receptor 1 inhibitor SB431542 (10μM). The addition of PEVs from PLWH + Coc or HIV-negative individuals with a history of cocaine use to HPMECs for 24 hours resulted in significantly increased apoptosis when compared with treatment with an equal amount (micro-gram protein) of EVs from HIV-negative Con subjects (Figure 1K). Alternatively, significantly increased proliferation of HPMECs and HPASMCs was observed in response to 48-hour treatment with PEVs from the PLWH + Coc, PLWH, or HIV-negative individuals with a history cocaine use when compared with the cells loaded with uninfected PEVs (Figures 1L and 1M). This increased proliferation of both cell types on treatment with EVs from PLWH + Coc was higher when compared with EVs from PLWH without a history of cocaine use. Furthermore, a significantly higher number of EVs observed in plasma from the PLWH + Coc group (10.9 × 106 ± 3.2/0.5 ml) compared with plasma from the PLWH group (5.3 × 106 ± 2.8/0.5 ml) (Figure 1B) is expected to have a more cumulative effect on cellular injury in vivo. Treatment of cells in the presence of the TGFβ-receptor 1 inhibitor SB431542 (10μM) significantly rescued the PLWH + Coc PEV-mediated enhanced EC and SMC proliferation (Figures 1L and 1M) but did not show any significant effect on PEV-mediated EC apoptosis (Figure 1K).
Significantly Increased TGF-β1 Levels in PEVs from PLWH + PH Compared with Those without PH
Next, to assess alterations in EV numbers and associated TGF-β1 levels in PLWH + PH, we compared findings from PLWH + PH and PLWH + Coc with PH (n = 13) with findings from PLWH without PH (n = 30) and HIV-negative individuals without PH (both with and without a history of cocaine use) (n = 30) (Table 2). PEVs isolated from PLWH + PH by using the exoEasy method were significantly higher in number than those from PLWH without PH (Figure 2A), even after adjusting for sex and race (Table 2). Correspondingly, the presence of PH among PLWH was also associated with significantly higher levels of both active and total TGFβ-1 in PEVs (Figure 2C) before and after adjusting for covariates (Table 2). Likewise, PEVs isolated by using the ultracentrifugation method also showed higher numbers of EVs (Figure E1D) that were linked with higher levels of TGFβ-1 (Figure E1E) in PLWH + PH compared with PLWH without PH. Although EV numbers and TGF-β1 levels in PLWH + PH were also significantly higher than those of HIV-negative Con subjects, not much difference was observed between the PLWH without PH and the HIV-negative individuals without a history of cocaine use (Table 2). Even though only two subjects from the PLWH + PH group had very high viremia, subanalysis of PLWH based on the viral load showed higher levels of EVs in PH subjects than in non-PH subjects in both the viremic and nonviremic groups (Figure 2B). However, a clear distinction in EV-linked TGF-β1 levels between PLWH + PH and PLWH without PH was observed only in patients with a suppressed viral load (Figures 2D and 2E). Spearman correlation analyses showed no significant correlation of viral load or CD4+ T-cell count with the EV number in HIV-positive individuals (Table 3). However, a significant association of TGF-β1 levels in EVs was observed with the CD4+ T-cell count but not with the viral load (Figure 2F and Table 3). Importantly, the levels of both the active and total isoforms of TGF-β in EVs showed a significant positive correlation with PASP, whereas the negative correlation with Dl CO in PLWH was not significant (Figure 2F and Table 3).
Table 2.
Comparison of Lung Function, PASP, EV Numbers, and TGF-β Levels in PLWH with or without PH and HIV-Negative Control Subjects
| Characteristic [Median (IQR)] | HIV-Negative Individuals (N = 30) | PLWH without PH (N = 30) | PLWH with PH (N = 13) | All PLWH with and without PH (N = 43) | Unadjusted P Value (P Value Adjusted for Sex and Race) |
|||
|---|---|---|---|---|---|---|---|---|
| Difference between HIV-Negative Individuals and PLWH without PH | Difference between HIV-Negative Individuals and PLWH with PH | Difference between HIV-Negative Individuals and All PLWH | Difference between PLWH with PH and PLWH without PH | |||||
| CD4, cell count/μl | — | 531.0 (327.5–687.0) | 633.5 (269.2–708.5) | 543.0 (307.0–0.695) | — | — | — | 0.8123 (0.9388) |
| Viral load, median (range) copies/ml | — | 49 (19–1,250,000) | 39 (9–336,000) | 39 (9–1,250,000) | — | — | — | 0.8549 (0.6420) |
| FVC% predicted after BD | 94.83 (0.87.31–103.92) | 84.00 (79.51–95.95) | 77.90 (71.90–96.40) | 83.88 (77.04–96.29) | 0.2683 (0.5629) | 0.0268 (0.0537) | 0.0181 (0.0792) | 0.3397 (0.2817) |
| FEV1% predicted after BD | 93.87 (83.50–106.80) | 88.04 (81.51–94.29) | 77.00 (56.70–95.50) | 87.21 (77.32–95.20) | 0.5639 (0.6221) | 0.0210 (0.0263) | 0.0532 (0.0721) | 0.1386 (0.1447) |
| FEV1/FVC after BD | 80.90 (75.55–84.75) | 80.60 (74.60–83.30) | 75.30 (65.70–84.70) | 80.00 (74.05–83.97) | 0.9431 (0.9977) | 0.3889 (0.3284) | 0.7519 (0.5569) | 0.2664 (0.2952) |
| DlCO% predicted | 86.83 (75.71–96.33) | 78.59 (64.98–82.60) | 70.85 (52.52–80.45) | 77.10 (62.56–82.44) | 0.0146 (0.1149) | 0.0012 (0.0015) | <0.001 (<0.001) | 0.2998 (0.1058) |
| PASP, mm Hg | — | 30.00 (26.00–33.00) | 44.00 (39.00–46.00) | 35.00 (27.50–41.00) | — | — | — | <0.001 (<0.001) |
| EV number × 1010 | 4.60 (2.39–9.13) | 7.65 (5.01–10.23) | 12.20 (9.21–13.10) | 8.80 (6.08–11.80) | 0.1014 (0.1171) | <0.001 (<0.001) | <0.001 (<0.001) | <0.001 (<0.001) |
| TGF-β1 active, pg/ml | 67.52 (48.87–90.43) | 73.42 (24.76–107.01) | 169.62 (126.86–250.50) | 85.49 (44.23–161.22) | 0.4141 (0.6112) | <0.001 (<0.001) | 0.5675 (0.3970) | <0.001 (<0.001) |
| TGF-β1 total, pg/ml | 90.55 (74.05–125.55) | 91.75 (66.08–137.90) | 258.70 (230.30–360.00) | 122.20 (75.75–226.90) | 0.9599 (0.9909) | <0.001 (<0.001) | 0.0335 (0.0220) | <0.001 (<0.001) |
Definition of abbreviations: BD = bronchodilator test; EV = extracellular vesicle; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; IQR = interquartile range; PASP = pulmonary arterial systolic pressure; TGF = transforming growth factor.
Differences among groups were assessed by using ANOVA followed by post hoc pairwise tests with multiple comparison adjustments. For the covariate-adjusted analyses, linear models were used and followed by post hoc pairwise tests. For variables present in only two groups (CD4, viral load, and PASP), t tests were used.
Figure 2.
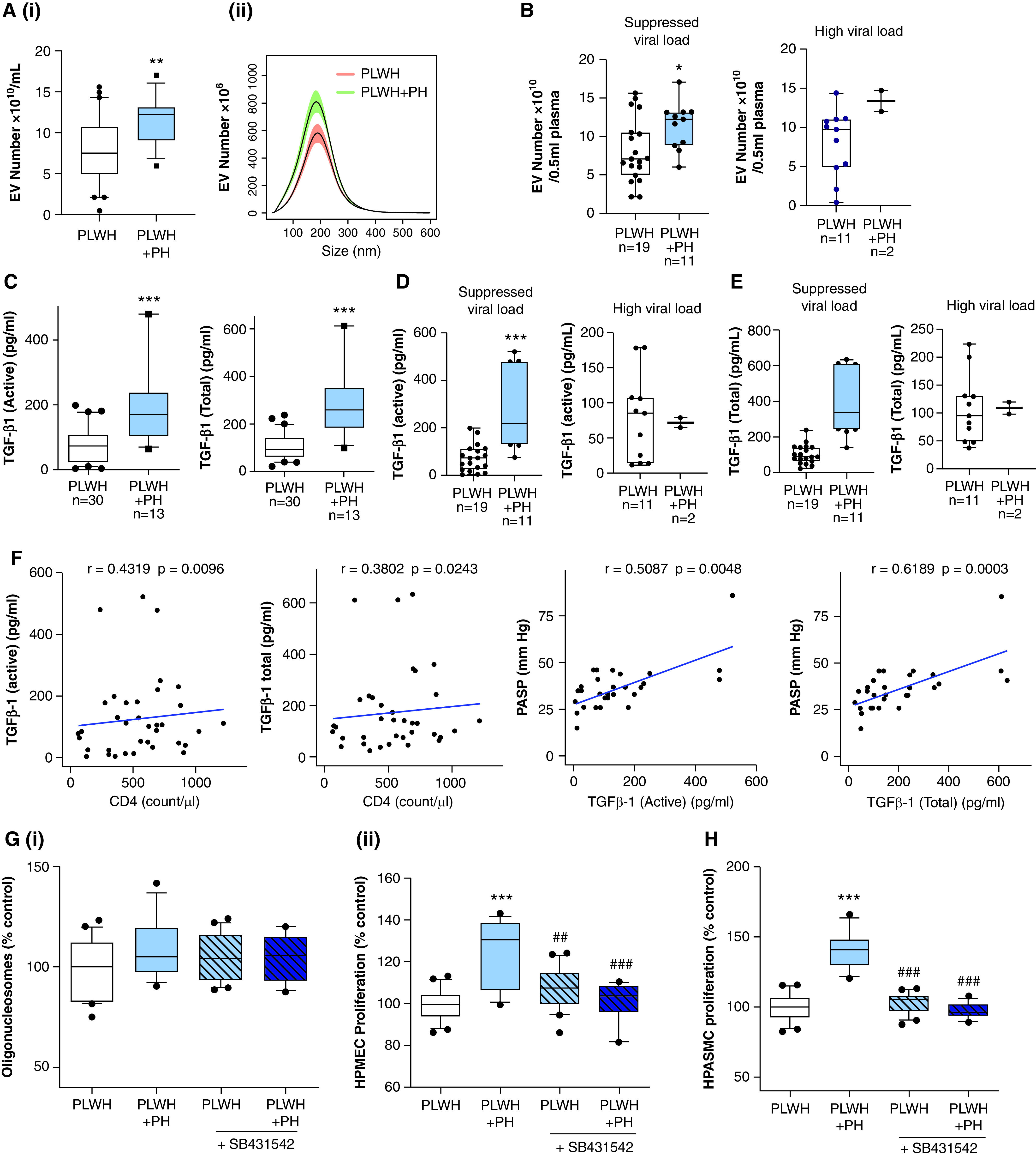
TGF-β1 levels in PEVs correlate with the presence of pulmonary hypertension (PH) in HIV-positive individuals. (A) The size distribution (i) and total number (ii) of PEVs were compared between PLWH without PH (n = 30) and PLWH with PH (PLWH + PH) (n = 13) by using NanoSight. For this, PEVs from PLWH without a history of drug use (n = 15) and PLWH + Coc (n = 15) without PH were grouped as the PLWH group (n = 30). For the PLWH + PH group (n = 13), plasma samples from PLWH + PH without a history of drug use (n = 8) and PLWH + Coc with PH (n = 7) were included (Table 1). (B) EV numbers in PLWH and PLWH + PH when segregated on the basis of the viral load (high viral load, ⩾50 copies/ml plasma). (C–E) Levels of active and total TGF-β1 in PEVs as analyzed by using an ELISA. (F) Correlation analysis of EV-linked TGFβ1 (active and total) with PASP and CD4 cell count. (G) To compare the effect of PEVs from PLWH + PH and PLWH without PH on the survival and proliferation of endothelial cells, HPMECs were treated with PEVs in the presence and absence of 10μM SB431542 (TGFβ-R1 In.) (n = 13–15/group), which was followed by performance of a cell-death ELISA (i) and an MTS assay (ii) at 24 hours and 48 hours after treatment. (H) Cell proliferation analysis of quiescent HPASMCs treated with PEVs from PLWH + PH and PLWH without PH at 48 hours after treatment in the presence and absence of 10μM SB431542 (TGFβ-R1 In.). For box and whisker plots, boxes represent the median and IQR, and whiskers show the 10th to 90th percentiles. *P < 0.05, **P < 0.01 and ***P < 0.001 versus PLWH; and ##P < 0.01 and ###P < 0.001 versus PLWH + PH without TGFβ-R1 In. PASP = pulmonary arterial systolic pressure.
Table 3.
Correlation Analyses of Clinical Characteristics with EV Number and TGF-β Levels (Total and Active) in PLWH and HIV-Negative Individuals
| EV Number [Correlation (P Value)] |
TGF-β, Active [Correlation (P Value)] |
TGF-β, Total [Correlation (P Value)] |
||||
|---|---|---|---|---|---|---|
| HIV-Negative Individuals (N = 30) | PLWH (N = 43) | HIV-Negative Individuals (N = 30) | PLWH (N = 43) | HIV-Negative Individuals (N = 30) | PLWH (N = 43) | |
| Age, yr | 0.0154 (0.9357) | 0.2284 (0.1407) | −0.0036 (0.9851) | 0.3061 (0.0459) | −0.0188 (0.9213) | 0.3241 (0.0340) |
| White | −0.5874 (0.0006) | −0.0978 (0.5328) | −0.5155 (0.0036) | −0.1303 (0.4049) | −0.5115 (0.0039) | −0.1380 (0.3776) |
| Sex | −0.0350 (0.8544) | −0.1043 (0.5058) | −0.2448 (0.1923) | −0.2041 (0.1894) | −0.2565 (0.1713) | −0.1508 (0.3343) |
| Pack-years | 0.2855 (0.1489) | 0.3166 (0.0386) | 0.1580 (0.4313) | 0.3532 (0.0201) | 0.2563 (0.1970) | 0.3346 (0.0283) |
| Cocaine usage | 0.7279 (5.2 × 10−6) | 0.1495 (0.3388) | 0.6123 (3.2 × 10−4) | 0.1416 (0.3652) | 0.7742 (5.2 × 10−7) | 0.0747 (0.6340) |
| CD4, cell count/μl* | — | 0.0868 (0.6200) | — | 0.4319 (0.0096) | — | 0.3802 (0.0243) |
| Viral load, copies/ml* | — | 0.1507 (0.3666) | — | −0.1056 (0.5281) | — | −0.1514 (0.3641) |
| FVC% after BD* | 0.2546 (0.1911) | 0.2420 (0.1325) | 0.1701 (0.3869) | 0.1032 (0.5264) | 0.0714 (0.7182) | 0.0790 (0.6280) |
| FEV1% predicted after BD* | 0.3930 (0.0386) | 0.0203 (0.9009) | 0.1276 (0.0.5177) | 0.0110 (0.9462) | 0.0959 (0.6271) | −0.0010 (0.9950) |
| FEV1/FVC after BD* | 0.3039 (0.1159) | −0.4951 (0.0012) | −0.1771 (0.3674) | −0.3723 (0.0180) | −0.0767 (0.6981) | −0.3421 (0.0307) |
| DlCO% predicted* | −0.0099 (0.9608) | −0.3515 (0.0305) | 0.3636 (0.0623) | −0.2978 (0.0694) | 0.2883 (0.1447) | −0.2881 (0.0794) |
| PASP, mm Hg* | — | 0.3456 (0.0663) | — | 0.5087 (0.0048) | — | 0.6189 (0.0003) |
| EV number × 1010* | — | — | 0.2948 (0.1277) | 0.5068 (0.0007) | 0.3637 (0.0571) | 0.5307 (0.0004) |
Spearman rank correlation analysis was adjusted for sex and race.
Although PLWH + PH PEVs showed no significant difference in terms of the apoptosis of HPMECs when compared with EVs from PLWH without PH (Figure 2Gi), augmented proliferation of ECs and SMCs was observed with PLWH + PH PEVs (Figures 2Gii and 2H). This increased EC and SMC proliferation by EVs from PLWH + PH was also found to be TGFβ dependent (Figures 2Gii and 2E).
Increased TGF-β1 Levels in EVs Released by HIV-infected MDMs on Exposure to Cocaine Correspond to Augmented Endothelial and Smooth Muscle Dysfunction
In light of recent findings demonstrating that circulating blood monocytes are a source of perivascular macrophages surrounding the remodeled vessels in PH (4, 7, 8), we next examined the TGF-β1 levels in CD14+ PEVs from a small set of PLWH (Figure E1F). As shown in Figure E1G, the levels of total TGF-β1 were found to be elevated in CD14+ PEVs from the PLWH + PH group when compared with PLWH without PH or with HIV-negative Con subjects. Furthermore, we examined the levels of TGF-β1 in EVs released by HIV-infected MDMs in the presence and absence of cocaine, and in findings similar to those for PEVs, we observed a significant increase in both the active and total forms of TGF-β1 in the EVs from supernatants of H + C MDMs (Figure 3A).
Figure 3.
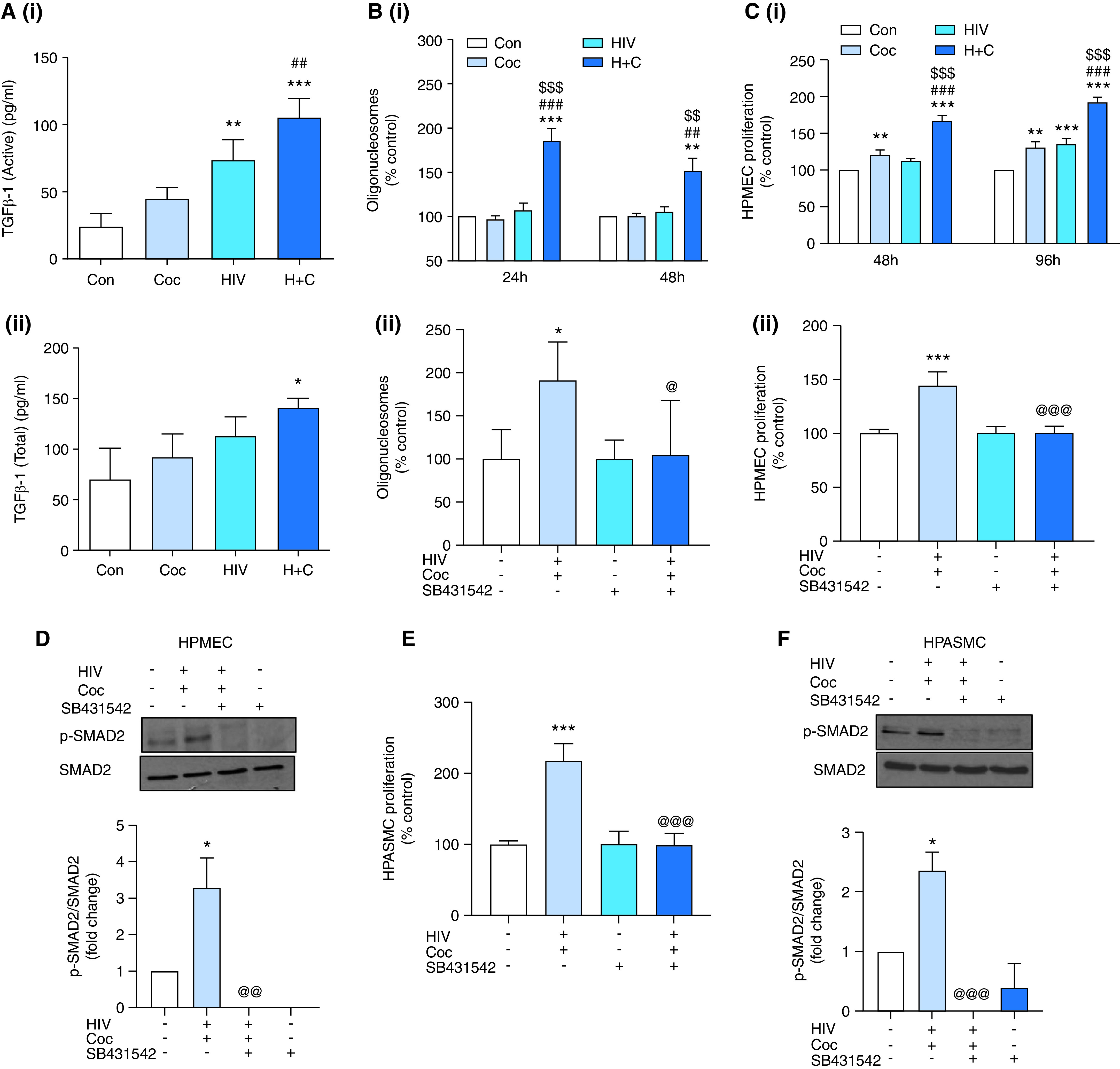
TGFβ-1 levels are upregulated in EVs from HIV-infected monocyte–derived macrophages (MDMs) on treatment with cocaine, resulting in enhanced MDM EV–mediated endothelial dysfunction and smooth muscle hyperplasia. (A) EVs isolated from supernatants of Mono-Mac-1–derived macrophages infected with HIVBal for 4 days in the presence of cocaine (HIV-infected and Coc [H + C] macrophages) and absence of cocaine (HIV only) or UI MDMs exposed (Coc) or unexposed to cocaine (Con) were analyzed for active (i) and total (ii) TGFβ-1 levels by using an ELISA. (B) HPMECs were treated with 4 μg of MDM EVs, which was followed by performance of a cell-death ELISA at 24 and 48 hours after EV addition (i) and at 24 hours EV addition in the presence or absence of TGFβ-R1 In. (SB431542) (ii). (C) An MTS assay was performed on HPMECs at 48 or 96 hours after treatment with MDM EVs in the absence of SB431542 (i) or at 48 hours after EV treatment in the presence of SB431542 (ii). (D) HPMECs treated with H + C MDM EVs in the presence and absence of SB431542 were lysed by using radio-immunoprecipitation assay buffer at 16 hours after treatment, which was followed by Western blot analysis for phosphorylated and total SMAD2. The graphs represent the densitometry analysis of three independent experiments (mean ± SEM). (E) An MTS assay was performed on HPASMCs treated with H + C MDM EVs in the presence and absence of SB431542 at 48 hours after treatment. (F) Western blot analysis of HPASMCs for phosphorylated and total SMAD2 at 1 hour after treatment. For all In. studies, cells were pretreated with SB431542 (10μM) for 30 minutes before the addition of MDM EVs to cells. Values of experiments using MDM EVs are the average of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus Con EVs; ##P < 0.01 and ###P < 0.001 versus Coc EVs; $$P < 0.01 and $$$P < 0.001 versus HIV EVs; and @P < 0.05, @@P < 0.01, and @@@P < 0.001 versus H + C EVs without TGFβ-R1 In. Con = control.
Exposure of HPMECs to H + C MDM EVs led to a significant increase in apoptosis at 24 hours and 48 hours after treatment as compared with cells treated with EVs derived from uninfected Con, HIV-infected, or cocaine-exposed (Coc) MDMs (Figure 3Bi and Figure E2B). In addition, a significant endothelial proliferation of apoptosis-resistant HPMECs was observed on treatment with H + C MDM EVs when compared with all other groups at 48 hours and 96 hours (Figure 3Ci and Figure E2C). However, the addition of MDM EVs in the presence of the TGFβ-R1 inhibitor resulted in significant abrogation of H + C MDM EV–mediated induction of endothelial apoptosis (Figure 3Bii) and proliferation (Figure 3Cii), together with the prevention of SMAD2 activation (Figure 3D).
Similarly, H + C MDM EV–mediated hyperproliferation of SMCs was observed to be TGF-β dependent (Figure 3E). In parallel, increased expression of phosphorylated SMAD2 at treatment with H + C MDM EVs was attenuated at pretreatment of cells with the TGFβ-R1 inhibitor (Figure 3F).
RV Dysfunction in Rats Treated with H + C MDM EVs
To see the in vivo effect of HIV-infected monocyte/macrophage–derived EVs in the development of PH, we first confirmed the cellular uptake of PKH67-labeled human MDM EVs by rat cells (Figure E3A) and in vivo biodistribution of intravenously injected DiR-labeled human MDM EVs in rat lungs (Figure E3B). Next, we investigated the effect of H + C MDM EVs on the hemodynamics in WT rats and whether they can further potentiate the RV dysfunction seen in our previously established noninfectious HIV-Tg rat PAH model (6). We observed a significant increase in the RV systolic pressure (RVSP) (P = 0.003, unpaired t test) in WT rats given H + C EVs compared with rats given uninfected MDM EVs (Con EVs). However, comparison among multiple groups (Figure 4B) revealed a significant increase in the RVSP in only HIV-Tg rats treated with either Con EVs or H + C EVs when compared with the WT Con EV group. Interestingly, the RVSP in HIV-Tg rats given H + C EVs exhibited a higher trend than HIV-Tg rats given Con EVs and showed a significant increase compared with WT rats administered H + C EVs. Assessment of the Fulton index (RV/left ventricle [LV] + septum), however, showed significant RV hypertrophy only in HIV-Tg rats that were given Con EVs and showed an increased trend only in HIV-Tg rats given H + C EVs when compared with the WT groups (Figure 4C). The trichrome staining of RVs exhibited maximum fibrosis and collagen deposition in HIV-Tg rats administered H + C EVs (Figure 4E), and no significant differences were observed in the mean arterial pressure among all four groups (Figure 4D). Furthermore, HIV-Tg rat treatment with H + C EVs resulted in significantly increased levels of serum ET-1 (endothelin-1) and TNF-α, together with an increased trend in CRP (C-reactive protein), compared with WT and HIV-Tg rats treated with Con EVs (Figure 4F). However, no differences were observed in IL-6 levels (Figure 4F). Both ET-1 and TNF-α levels had a significantly positive correlation with the RVSP, as shown in Figure 4G.
Figure 4.
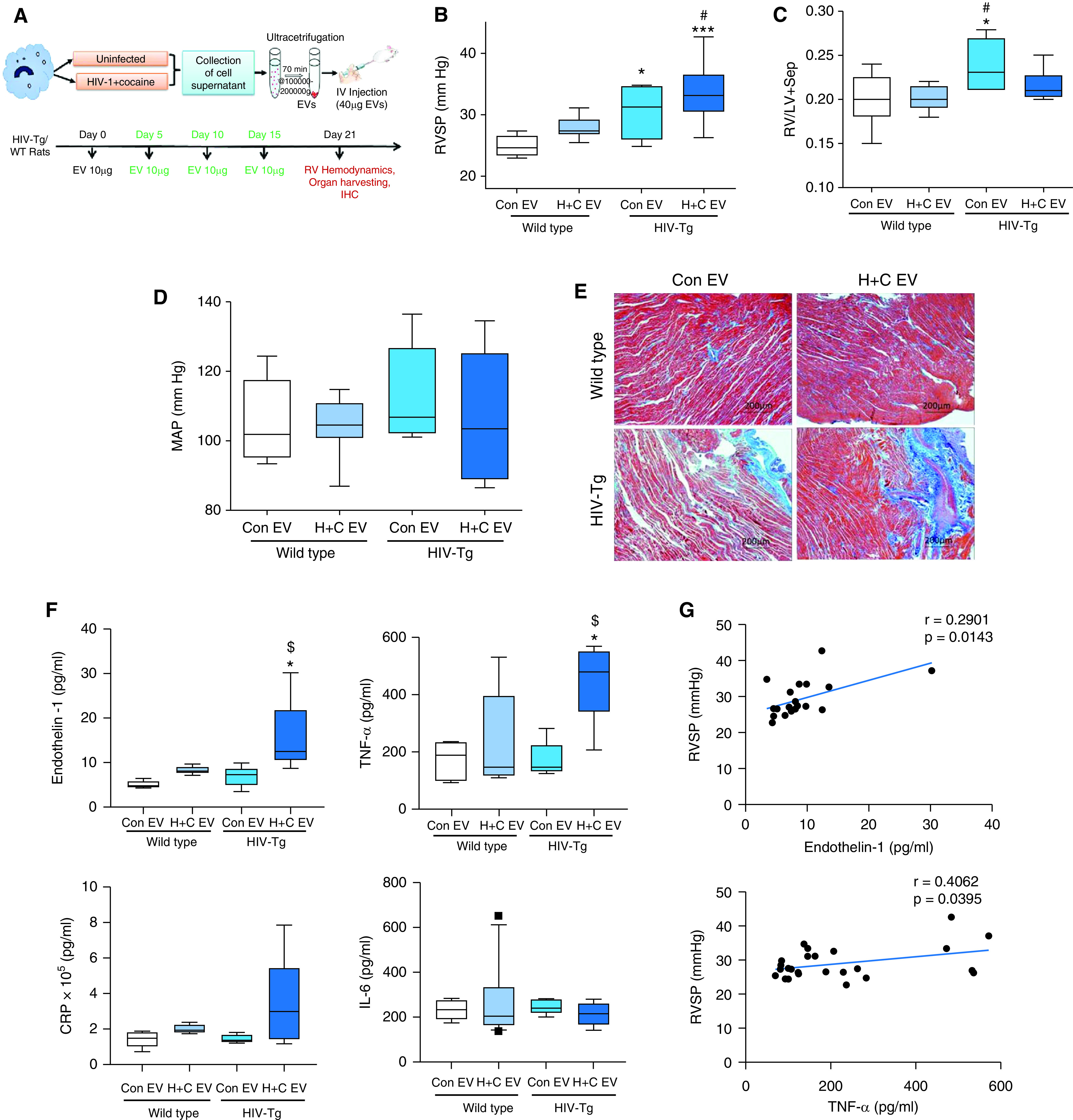
Hemodynamic measurements, right ventricle (RV) fibrosis, and biomarkers of PH in rats injected with H + C MDM EVs. (A) Figure showing the timeline for the administration of H + C MDM EVs (H + C EVs) in wild-type (WT)/HIV-transgenic (HIV-Tg) rats. HIV-Tg/WT rats injected with EVs from UI MDMs (Con EVs) were used for comparison. Rats were IV injected with four doses of 10 μg of EVs each at 5-day intervals. (B–D) Box and whisker plots showing RV systolic pressure (RVSP) (B), Fulton index (C), and MAP measurements (D). (E) Representative micrographs show RV fibrosis after Masson's trichrome staining on formaldehyde-fixed, paraffin-embedded RV sections. Scale bars, 200 mm. (F) ELISAs were performed to assess serum levels of endothelin-1, TNF-α, CRP (C-reactive protein), and IL-6. (G) Graphs showing correlation of RVSP with endothelin-1 and TNF-α serum levels in rats. n = 6–8 rats per group. Box and whisker plots show the median value and 10th to 90th percentiles. *P < 0.05 and ***P < 0.001 versus WT Con EV; #P < 0.05 versus WT H + C EV; and $P < 0.05 versus HIV-Tg Con EV. IHC = immunohistochemistry; IV = intravenously; MAP = mean arterial pressure.
Myocardial Injury in Rats Treated with H + C MDM EVs
Given that we observed no further increase in the Fulton index in HIV-Tg rats on administration of H + C EVs compared with Con EVs, we separately compared the RV and LV + septum mass with body weight. Although body weight remained constant in WT rats and HIV-Tg rats injected with Con EVs or H + C EVs (Figure E4A), both WT and HIV-Tg rats that were administered H + C EVs had decreased RV and LV + septum mass (Figures 5A and 5B). However, the effect on RV mass was far smaller than the effect on the LV + septum mass (22–30% RV vs. 60–70% LV) among H + C EV–treated rats. This was associated with significantly increased caspase-3 expression in whole-LV lysates (Figure 5D) but not in RV lysates (Figure 5C) from HIV-Tg rats treated with H + C EVs when compared with rats treated with Con EVs. Interestingly, a significant increase in caspase-3 expression was observed in RVs from WT rats given H + C EVs compared with WT rats treated with Con EVs (Figure 5C). A decrease in both RV and LV cardiomyocyte size in HIV-Tg rats treated with H + C EVs compared with HIV-Tg rats given Con EVs (Figures 5E and 5F) further suggests cardiac atrophy in HIV-Tg rats in response to treatment with H + C MDM EVs. Finally, serum levels of cardiac troponin-I, an indicator of cardiomyocyte damage, were increased in HIV-Tg animals given H + C EVs compared with Con EV–treated animals (Figure 5G) and were found to positively correlate with RVSP (Figure 5H).
Figure 5.
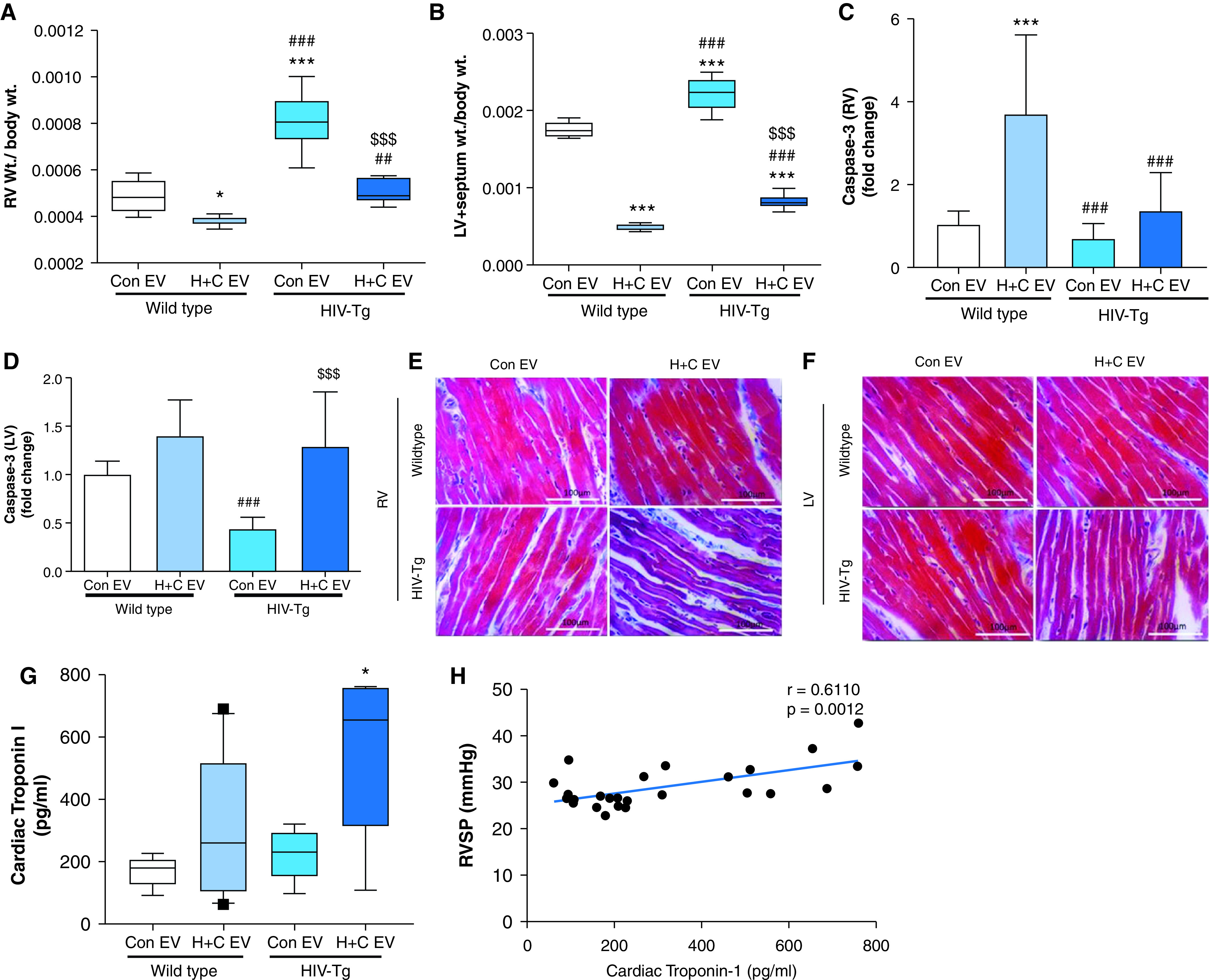
Analysis of cardiac atrophy in rats injected with EVs from H + C macrophages. (A and B) Ratio of RV weight to total body weight (A) and left ventricle (LV) + septum weight to body weight (B) for WT and HIV-Tg rats injected with EVs from UI MDMs (Con EVs) or EVs from H + C MDMs (H + C EVs). Values are from n = 7–8 rats per group. (C and D) Quantitative RT-PCR analysis of caspase-3 in the RV (C) and LV (D) tissues from rats (n = 6/group). The values are expressed as the fold change. (E and F) Representative micrographs showing RV (E) and LV (F) cardiomyocytes from all four groups after Masson's trichrome staining. Scale bars, 100 mm. (G) ELISA was performed to check the levels of cardiac troponin-1 in rat serum from all groups. Values are from n = 7–8 rats per group. (H) Graph showing correlation of RVSP with cardiac troponin-1 levels in rats. For box and whisker plots, boxes represent the median and IQR, and whiskers show the 10th to 90th percentiles. *P < 0.05 and ***P < 0.001 versus WT Con EVs; ##P < 0.01 and ###P < 0.001 versus WT H + C EVs; and $$$P < 0.001 versus HIV-Tg Con EVs.
Enhanced Pulmonary Endothelial Injury and SMC Hyperplasia in Rats Administered H + C MDM EVs
Staining of lung sections for vWF (von Willebrand factor) and α-SMA (ACTA2) indicated enhanced smooth muscle positivity (red fluorescence) in the pulmonary vessels of HIV-Tg rats injected with Con EVs or H + C EVs as compared with both groups of WT rats. Increased SMA+ pulmonary vessels were also found in WT rats injected with H + C EVs when compared with WT rats given Con EVs (Figure 6A). Concordantly, rat pulmonary arterial SMCs (RPASMCs) from HIV-Tg rats injected with H + C EVs exhibited significant hyperproliferation as early as Day 4 of serum-stressed conditions compared with HIV-Tg or WT rats given Con EVs (Figure 6B). On Day 6 of serum starvation, RPASMCs from both HIV-Tg rats and WT rats given H + C EVs exhibited hyperproliferation when compared with WT Con EV–treated rats. Simultaneously, RPASMCs from HIV-Tg rats showed less apoptotic cell death than those from WT rats at 48 hours of serum starvation (Figure E4B). These findings corresponded with increased TGFβ-R1 activation in RPASMCs (Figure 6C) and downstream p-SMAD2 expression (Figure E4C) from H + C EV–treated rats compared with Con EV–treated rats.
Figure 6.

Pulmonary smooth muscle proliferation and endothelial injury in lungs from rats injected with H + C MDM EVs. (A) Representative micrographs showing immunofluorescence staining of vWF (von Willebrand factor) (green) and α-SMA (ACTA2) (red) in pulmonary vessels of sizes >100 μm, 50–70 μm, and <30 μm in all four groups. Scale bars, 50 mm. (B) Graph showing the percentage of proliferation of rat pulmonary arterial smooth muscle cells (RPASMCs) isolated from all four groups. RPASMCs were cultured in 96-well plates, and MTS assays were performed on Days 2, 4, and 6 to assess the cell proliferation. (C) The total extract of RPASMCs (n = 3/group) underwent IP, which was conducted by using a TGFβ-R1 antibody, Western blotting (IB), and the detection of phosphorylated and total levels of TGFβ-R1. Graphs represent densitometric analysis of n = 3 animals per group. (D) A cell-death ELISA was performed on rat pulmonary microvascular endothelial cells (RPMECs) isolated from all four groups. RPMECs isolated from the left lung lobe were cultured in 96-well plates, and the oligonucleosome ELISA was performed at 24 and 48 hours to assess the cell apoptosis. (E) IP–Western blot and densitometric analysis showing phosphorylated and total levels of TGFβ-R1 in RPMECs. Values are the mean ± SEM of n = 3/group. *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT Con EVs; #P < 0.05 versus WT H + C EVs; and $P < 0.05 versus HIV-Tg Con EVs.
In contrast to RPASMCs, rat pulmonary microvascular ECs from HIV-Tg rats given H + C EVs demonstrated significantly higher apoptosis in response to serum starvation when compared with the other groups (Figure 6D) and exhibited high activated caspase-3 levels (Figure E4D). In addition, these ECs exhibited less survivability in response to serum starvation (Figure E4E) and exhibited increased TGFβ-R1 (Figure 6E) and SMAD2 activation (Figure E4F).
Pharmacologic Inhibition of TGFβ-R1 Resulted in Attenuation of H + C MDM EV–mediated Cardiopulmonary Dysfunction
For in vivo inhibition of TGF-β downstream signaling, we treated rats with GW788388, which is considered to be a more potent TGFβ-R1 (Alk-5) inhibitor than SB431542 without having any nonspecific effects on BMPR-2 (bone morphogenetic protein receptor 2) signaling. Animals from both the WT and HIV-Tg groups pretreated with GW788388 showed improvement in the H + C EV–mediated increase in the RVSP (Figure 7A). Echocardiography performed on this set of rats revealed an increase in the RV end-diastolic area after 21 days of treatment with H + C EVs, whereas blocking of TGF-βRI prevented this increase (Figure 7B), as also shown by a significant drop in the differences between RV/LV end-diastolic area as assessed at Day 21 and at Day 0 (Figure 7C). Correspondingly, a significant increase in the difference between the RV/LV ejection fraction before and after treatment was observed in WT and HIV-Tg rats treated with H + C EVs in the presence of the inhibitor compared with rats treated with H + C EVs and the vehicle (Figure 7D). A parallel mitigation of the H + C EV–mediated increase of the levels of ET-1, TNF-α, and cardiac troponin I was also observed on treatment with GW7883888 (Figure 7E).
Figure 7.

Attenuation of H + C MDM EVs mediated cardiopulmonary dysfunction in the presence of TGFβ-RI In. HIV-Tg or WT rats were IV injected with four doses of H + C MDM EVs (10 μg each at a 5-d interval) in the presence of treatment with TGFβ receptor In.: GW788388 (1 mg/kg/d) or vehicle, once daily orally for 21 days. (A) RVSP in WT and HIV-Tg rats given H + C EVs with and without In. in comparison with WT rats treated with Con EVs (n = 5/group). (B) RVEDA at Day 0 and Day 21 for WT and HIV-Tg rats given H + C EVs in the presence or absence of GW788388. (C and D) Percent change in RV/LV EDA (C) and EF (D) in rats on treatment with or without GW788388. (E) ELISAs were performed to assess serum levels of endothelin-1, TNF-α, and cardiac troponin-I. Box and whisker plots show the median value and 10th to 90th percentiles. @@P < 0.01 and @@@P < 0.001 versus WT Con EVs; *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT H + C EVs; and $$P < 0.01 and $$$P < 0.001 versus HIV-Tg H + C EVs. EDA = end-diastolic area; EF = ejection fraction; RVEDA = EDA of RV.
Discussion
In this study, we report a higher number of TGF-β–loaded EVs in the plasma of individuals with HIV-associated PH (HIV-PH) compared with plasma from individuals without HIV-PH, suggesting its potential as a biomarker for HIV-PH. We showed that the EVs from the plasma of PLWH + Coc and from H + C MDMs augment pulmonary endothelial injury and smooth muscle hyperplasia via activation of TGF-β1 signaling. We show for the first time that the EVs derived from H + C MDMs can cause pulmonary vascular remodeling and promote the development of PH in vivo.
PAH has been well known as a secondary complication of HIV infection. The link between HIV infection leading to pulmonary vascular remodeling (5, 31) and the further contribution of intravenous drug use in exacerbating the development of PAH has been well established (32–34). Viral proteins show an additive effect with illicit drugs such as cocaine and opioids in potentiating pulmonary vascular remodeling and development of PH in infectious nonhuman primate or noninfectious HIV-Tg rat models (5, 6, 35). Recently, EVs have been recognized as novel communicators between cells, thereby contributing to disease pathogenesis. Importantly, we and others have shown that the level and content of EVs released by immune cells change with virus replication (12, 16). Many studies have reported increased levels of PEVs or platelet-derived EVs in patients with idiopathic, heritable, or connective tissue disease–associated PAH (12, 18, 36). Here, we show an increased number of small-sized PEVs in individuals with HIV-PH not only compared with HIV-negative individuals but also compared with HIV-positive individuals without PH. Furthermore, consistent with our earlier findings of augmented pulmonary vascular damage in response to the dual hit of drug use and HIV infection, we observed a higher number of PEVs in PLWH + Coc compared with PLWH without a history of cocaine use and HIV-negative individuals with a history cocaine use.
Inflammation is the hallmark feature of all forms of PAH, with increased infiltration of inflammatory cells, such as macrophages and T cells surrounding the pulmonary vessels, being shown (4, 7, 9). We previously demonstrated significant perivascular inflammation in morphine-treated simian human immunodeficiency virus (SHIV)-infected macaques as well as in HIV-Tg rats exposed to cocaine (5). Similarly, increased numbers of proinflammatory and profibrotic monocytes have been observed in the peripheral blood (37) of SHIV Nef– or SIV-infected macaques. Although these studies suggest the association of inflammation with HIV-PAH, studies defining the direct role of inflammatory cells in the development of the disease are limited. Our current study demonstrates the direct effect of EVs derived from HIV-infected MDMs in causing cardiopulmonary dysfunction.
Macrophage depletion has been reported to prevent the development of PH in animals (38). A recent study by Florentin and colleagues (39) reported that an increased number of blood monocytes in PAH get recruited to lung perivascular spaces and differentiate into inflammatory macrophages responsible for pulmonary vascular remodeling. Our pivotal findings suggest that the altered cargo within the circulating EVs derived from H + C MDMs are able to cause EC injury and SMC proliferation, consequently leading to the development of PH in WT rats. In addition, these H + C MDM EVs potentiated the vascular damage in the Coc HIV-Tg rat model of PAH (6) and increased serum levels of TNF-α and ET-1, which are known to affect vascular-cell function and facilitate the development of PH (40, 41). Previous studies on PEVs from mice with monocrotaline-induced PAH have been shown to cause PAH when injected into healthy mice (12, 13). However, EVs are known to transfer their contents not only to different cell types but also across different species (42, 43). Our data show that injecting HIV-infected human cell–derived EVs into HIV-Tg rats can be used as a tool to delineate the relationship between immune activation/inflammation and non–acquired immunodeficiency syndrome comorbidities in this noninfectious HIV-Tg rat model.
Although administration of H + C EVs in WT and HIV-Tg rats resulted in increased RVSP, analysis of the RVs and LVs indicated a significant decrease in LV + septum mass. LV dysfunction with a decreased LV mass index has been reported earlier in HIV-positive individuals (44). Changes in rat LV mass could be explained by interventricular dependence as a result of RV overload associated with high pulmonary vascular resistance (45–47). Another possibility could be the direct effect of H + C EVs on cardiomyocytes, leading to increased apoptosis and atrophy. We observed increased levels of circulating cardiac troponin I, CRP, and TNF-α, indicative of cardiac injury and inflammation in response to H + C EV treatment, but these occurred without an increase in proinflammatory IL-6.
TGF-β1, a growth factor and proinflammatory cytokine, is known to play an important role in pulmonary vascular remodeling and PH (20, 48, 49). We found that cocaine exposure not only augmented the levels of TGF-β in PEVs from PLWH and EVs from HIV-infected MDMs but also increased the levels of EVs independently of infection. Pretreatment of cells with a TGF-β receptor inhibitor resulted in attenuation of H + C PEV/MDM EV–mediated pulmonary vascular dysfunction. Recent reports demonstrated that TGFβ-1–associated exosomes promote more potent and sustained activation of downstream signaling than free TGF-β1 (24). Our findings showing TGF-β–dependent escalation of cardiopulmonary dysfunction in H + C EV–treated rats further emphasize its significance in EV-mediated pathologic changes. However, TGF-β1 may not be the only molecule driving these changes; additional cargo molecules, including other proteins, viral proteins, and microRNAs (miRNAs), in these EVs may also contribute to pulmonary vascular remodeling in vivo. We earlier reported that increased levels of miRNA 130 in H + C MDM EVs mediates prosurvival signaling in HPASMCs (18). However, we were unable to detect miRNA 130 in PEVs from any of the groups tested. Still, it could be that the prevention of TGF-β1–coated EVs binding to TGF receptors on the surface of cells in the presence of a TGF-β type I receptor inhibitor may have prevented the uptake of EVs, thereby completely inhibiting the effect of EVs and downstream signaling in response to other cargo molecules as well. Furthermore, as in our previously published findings, HIV-Tg rat PAH models treated with Con EVs are also expected to have increased levels of phosphorylated TGFβR1 when compared with WT rats, even though the levels of activation further increased on treatment of these rats with H + C EVs. Therefore, there is a possibility that the observed robust effect of GW78838 in preventing the cardiopulmonary dysfunction in HIV-Tg rats treated with H + C EVs could be due to the additive effect of the blockage of the H + C EV response as well as to the inhibition of endogenous TGF activation in the Coc HIV-Tg rat model. Nonetheless, the effect of the TGF-receptor 1 inhibitor in WT rats given H + C EVs is expected to be mainly due to the inhibition of TGF EV uptake and/or downstream signaling.
The exoEasy method that we used to isolate PEVs has recently been reported to be more practical, faster, and more reproducible than the traditional approaches used to isolate good-quality EVs from small-volume serum or plasma samples for downstream applications (50). We opted to validate our exoEasy findings by using the ultracentrifugation method as well. Nevertheless, our study includes the limitations of having a small sample size and a lack of confirmation of PH through right heart catheterization in PLWH. Further investigations using a larger cohort are needed to better characterize the EVs and to confirm the correlation of TGFβ-loaded EVs with the clinical parameters of PH to use TGFβ as a circulatory biomarker of HIV-PH.
In summary, we report for the first time the direct effect of HIV-infected macrophage–derived small EVs on pulmonary vascular modeling and PH development via modulation of TGF-β signaling in pulmonary SMCs and ECs. Most importantly, this study correlates an increased number of TGF-β1–linked PEVs with the presence of PH in HIV-positive individuals, highlighting the importance of TGF-β–rich EVs as mediators and markers of HIV-PH. Considering that TGF-β signaling also plays a pivotal role in the development of other forms of PH, it will be interesting to see whether these TGF-β–rich plasma EVs are also high in other forms of PH. Further investigation of these EVs to elucidate their potential as biomarkers of and therapeutic targets in PH are warranted.
Acknowledgments
Acknowledgment
The authors thank Dr. Ghazwan Burrous, University of Kent, Canterbury, United Kingdom, for providing invaluable feedback and critical evaluation of this manuscript. They thank Julie Allen, Department of Molecular and Integrative Physiology, KUMC, for helping in animal surgery; Michael Wulser, Department of Internal Medicine, KUMC, for morphometric analysis; and Pranjali Dalvi and Himanshu Sharma, Department of Internal Medicine, KUMC, for initial standardization of the in vitro EV uptake and proliferation/apoptosis experiments. They also thank the Electron Microscope Research Laboratory for transmission electron microscopy analysis and the Flow Cytometry Core for EV analyses.
Footnotes
Supported by the National Institutes of Health (NIH) grants R01DA042715 (N.K.D.), R01DA034542 (N.K.D.), R01HL129875 (N.K.D.), R01HL125049 (A. Morris), R01HL120398 (A. Morris) and P01HL103455 (A. Morris). The work of the Electron Microscope Research Laboratory was supported by NIH/National Institute of General Medical Sciences Centers of Biomedical Research Excellence grant P20GM104936 and NIH grant 1S10RR027564. The Flow Cytometry Core Laboratory, is sponsored, in part, by the NIH/NIGMS COBRE grant P30GM103326 and the NIH/ NCI Cancer Center grant P30 CA168524.
Author Contributions: In vitro experiments were performed by B.K., A. Mahajan, and S.A. A.K. and L.C. performed animal experiments. Human samples were provided by A. Morris and processed for extracellular vesicle isolation by B.K., A. Mohan, and A.K. B.K., A. Mahajan, A.K., S.A., and N.K.D. analyzed and interpreted the data. Statistical analysis was performed by B.K., A. Mahajan, and P.C. B.K., A. Mahajan, S.A., and N.K.D. contributed to writing the manuscript. P.Y.H. and A. Morris critically read and edited the manuscript. N.K.D. designed, conceptualized, and supervised the research. All authors read the manuscript and approved the study.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0010OC on May 18, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Triplette M, Crothers K, Attia EF. Non-infectious pulmonary diseases and HIV. Curr HIV/AIDS Rep. 2016;13:140–148. doi: 10.1007/s11904-016-0313-0. [DOI] [PubMed] [Google Scholar]
- 2. Harter ZJ, Agarwal S, Dalvi P, Voelkel NF, Dhillon NK. Drug abuse and HIV-related pulmonary hypertension: double hit injury. AIDS. 2018;32:2651–2667. doi: 10.1097/QAD.0000000000002030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Almodovar S. The complexity of HIV persistence and pathogenesis in the lung under antiretroviral therapy: challenges beyond AIDS. Viral Immunol. 2014;27:186–199. doi: 10.1089/vim.2013.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spikes L, Dalvi P, Tawfik O, Gu H, Voelkel NF, Cheney P, et al. Enhanced pulmonary arteriopathy in simian immunodeficiency virus-infected macaques exposed to morphine. Am J Respir Crit Care Med. 2012;185:1235–1243. doi: 10.1164/rccm.201110-1909OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dalvi P, Spikes L, Allen J, Gupta VG, Sharma H, Gillcrist M, et al. Effect of cocaine on pulmonary vascular remodeling and hemodynamics in human immunodeficiency virus-transgenic rats. Am J Respir Cell Mol Biol. 2016;55:201–212. doi: 10.1165/rcmb.2015-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 8. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 9. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 10. Osada-Oka M, Shiota M, Izumi Y, Nishiyama M, Tanaka M, Yamaguchi T, et al. Macrophage-derived exosomes induce inflammatory factors in endothelial cells under hypertensive conditions. Hypertens Res. 2017;40:353–360. doi: 10.1038/hr.2016.163. [DOI] [PubMed] [Google Scholar]
- 11. Tang N, Sun B, Gupta A, Rempel H, Pulliam L. Monocyte exosomes induce adhesion molecules and cytokines via activation of NF-κB in endothelial cells. FASEB J. 2016;30:3097–3106. doi: 10.1096/fj.201600368RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aliotta JM, Pereira M, Wen S, Dooner MS, Del Tatto M, Papa E, et al. Exosomes induce and reverse monocrotaline-induced pulmonary hypertension in mice. Cardiovasc Res. 2016;110:319–330. doi: 10.1093/cvr/cvw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aliotta JM, Pereira M, Amaral A, Sorokina A, Igbinoba Z, Hasslinger A, et al. Induction of pulmonary hypertensive changes by extracellular vesicles from monocrotaline-treated mice. Cardiovasc Res. 2013;100:354–362. doi: 10.1093/cvr/cvt184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amabile N, Heiss C, Real WM, Minasi P, McGlothlin D, Rame EJ, et al. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med. 2008;177:1268–1275. doi: 10.1164/rccm.200710-1458OC. [DOI] [PubMed] [Google Scholar]
- 15. Bakouboula B, Morel O, Faure A, Zobairi F, Jesel L, Trinh A, et al. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:536–543. doi: 10.1164/rccm.200706-840OC. [DOI] [PubMed] [Google Scholar]
- 16. Chelvanambi S, Bogatcheva NV, Bednorz M, Agarwal S, Maier B, Alves NJ, et al. HIV-Nef protein persists in the lungs of aviremic patients with HIV and induces endothelial cell death. Am J Respir Cell Mol Biol. 2019;60:357–366. doi: 10.1165/rcmb.2018-0089OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chelvanambi S, Gupta SK, Chen X, Ellis BW, Maier BF, Colbert TM, et al. HIV-Nef protein transfer to endothelial cells requires Rac1 activation and leads to endothelial dysfunction implications for statin treatment in HIV patients. Circ Res. 2019;125:805–820. doi: 10.1161/CIRCRESAHA.119.315082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sharma H, Chinnappan M, Agarwal S, Dalvi P, Gunewardena S, O’Brien-Ladner A, et al. Macrophage-derived extracellular vesicles mediate smooth muscle hyperplasia: role of altered miRNA cargo in response to HIV infection and substance abuse. FASEB J. 2018;32:5174–5185. doi: 10.1096/fj.201701558R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dhillon NK, Williams R, Peng F, Tsai YJ, Dhillon S, Nicolay B, et al. Cocaine-mediated enhancement of virus replication in macrophages: implications for human immunodeficiency virus-associated dementia. J Neurovirol. 2007;13:483–495. doi: 10.1080/13550280701528684. [DOI] [PubMed] [Google Scholar]
- 20. Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat Commun. 2017;8:15494. doi: 10.1038/ncomms15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalvi P, Sharma H, Konstantinova T, Sanderson M, Brien-Ladner AO, Dhillon NK. Hyperactive TGF-β signaling in smooth muscle cells exposed to HIV-protein(s) and cocaine: role in pulmonary vasculopathy. Sci Rep. 2017;7:10433. doi: 10.1038/s41598-017-10438-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hemnes AR, Humbert M. Pathobiology of pulmonary arterial hypertension: understanding the roads less travelled. Eur Respir Rev. 2017;26:170093. doi: 10.1183/16000617.0093-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lima RG, Van Weyenbergh J, Saraiva EM, Barral-Netto M, Galvão-Castro B, Bou-Habib DC. The replication of human immunodeficiency virus type 1 in macrophages is enhanced after phagocytosis of apoptotic cells. J Infect Dis. 2002;185:1561–1566. doi: 10.1086/340412. [DOI] [PubMed] [Google Scholar]
- 24. Shelke GV, Yin Y, Jang SC, Lässer C, Wennmalm S, Hoffmann HJ, et al. Endosomal signalling via exosome surface TGFβ-1. J Extracell Vesicles. 2019;8:1650458. doi: 10.1080/20013078.2019.1650458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gingo MR, George MP, Kessinger CJ, Lucht L, Rissler B, Weinman R, et al. Pulmonary function abnormalities in HIV-infected patients during the current antiretroviral therapy era. Am J Respir Crit Care Med. 2010;182:790–796. doi: 10.1164/rccm.200912-1858OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morris A, Gingo MR, George MP, Lucht L, Kessinger C, Singh V, et al. Cardiopulmonary function in individuals with HIV infection in the antiretroviral therapy era. AIDS. 2012;26:731–740. doi: 10.1097/QAD.0b013e32835099ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu WY, Meijer K, Delbressine JM, Willems PJ, Franssen FM, Wouters EF, et al. Reproducibility and validity of the 6-minute walk test using the gait real-time analysis interactive lab in patients with COPD and healthy elderly. PLoS One. 2016;11:e0162444. doi: 10.1371/journal.pone.0162444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simon MA, Lacomis CD, George MP, Kessinger C, Weinman R, McMahon D, et al. Isolated right ventricular dysfunction in patients with human immunodeficiency virus. J Card Fail. 2014;20:414–421. doi: 10.1016/j.cardfail.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F, et al. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008;73:705–715. doi: 10.1038/sj.ki.5002717. [DOI] [PubMed] [Google Scholar]
- 30. Doyle T, Smith C, Vitiello P, Cambiano V, Johnson M, Owen A, et al. Plasma HIV-1 RNA detection below 50 copies/ml and risk of virologic rebound in patients receiving highly active antiretroviral therapy. Clin Infect Dis. 2012;54:724–732. doi: 10.1093/cid/cir936. [DOI] [PubMed] [Google Scholar]
- 31. Fitzpatrick M, Crothers K, Morris A. Future directions: lung aging, inflammation, and human immunodeficiency virus. Clin Chest Med. 2013;34:325–331. doi: 10.1016/j.ccm.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Porter KM, Walp ER, Elms SC, Raynor R, Mitchell PO, Guidot DM, et al. Human immunodeficiency virus-1 transgene expression increases pulmonary vascular resistance and exacerbates hypoxia-induced pulmonary hypertension development. Pulm Circ. 2013;3:58–67. doi: 10.4103/2045-8932.109915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. George MP, Champion HC, Gladwin MT, Norris KA, Morris A. Injection drug use as a “second hit” in the pathogenesis of HIV-associated pulmonary hypertension. Am J Respir Crit Care Med. 2012;185:1144–1146. doi: 10.1164/rccm.201204-0609ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Janda S, Quon BS, Swiston J. HIV and pulmonary arterial hypertension: a systematic review. HIV Med. 2010;11:620–634. doi: 10.1111/j.1468-1293.2010.00829.x. [DOI] [PubMed] [Google Scholar]
- 35. Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, et al. Effect of cocaine on human immunodeficiency virus-mediated pulmonary endothelial and smooth muscle dysfunction. Am J Respir Cell Mol Biol. 2011;45:40–52. doi: 10.1165/rcmb.2010-0097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao L, Luo H, Li X, Li T, He J, Qi Q, et al. Exosomes derived from human pulmonary artery endothelial cells shift the balance between proliferation and apoptosis of smooth muscle cells. Cardiology. 2017;137:43–53. doi: 10.1159/000453544. [DOI] [PubMed] [Google Scholar]
- 37. Schweitzer F, Tarantelli R, Rayens E, Kling HM, Mattila JT, Norris KA. Monocyte and alveolar macrophage skewing is associated with the development of pulmonary arterial hypertension in a primate model of HIV infection. AIDS Res Hum Retroviruses. 2019;35:63–74. doi: 10.1089/aid.2018.0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, et al. Blocking macrophage leukotriene B4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. 2013;5:200ra117. doi: 10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J Immunol. 2018;200:3612–3625. doi: 10.4049/jimmunol.1701287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, et al. Krüppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res. 2011;12:128. doi: 10.1186/1465-9921-12-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Selimovic N, Bergh CH, Andersson B, Sakiniene E, Carlsten H, Rundqvist B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur Respir J. 2009;34:662–668. doi: 10.1183/09031936.00174908. [DOI] [PubMed] [Google Scholar]
- 42. Dorronsoro A, Robbins PD. Regenerating the injured kidney with human umbilical cord mesenchymal stem cell-derived exosomes. Stem Cell Res Ther. 2013;4:39. doi: 10.1186/scrt187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou Y, Xu H, Xu W, Wang B, Wu H, Tao Y, et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res Ther. 2013;4:34. doi: 10.1186/scrt194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martínez-García T, Sobrino JM, Pujol E, Galvez J, Benítez E, Girón-González JA. Ventricular mass and diastolic function in patients infected by the human immunodeficiency virus. Heart. 2000;84:620–624. doi: 10.1136/heart.84.6.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Homsi R, Luetkens JA, Skowasch D, Pizarro C, Sprinkart AM, Gieseke J, et al. Left ventricular myocardial fibrosis, atrophy, and impaired contractility in patients with pulmonary arterial hypertension and a preserved left ventricular function: a cardiac magnetic resonance study. J Thorac Imaging. 2017;32:36–42. doi: 10.1097/RTI.0000000000000248. [DOI] [PubMed] [Google Scholar]
- 46. Manders E, Rain S, Bogaard HJ, Handoko ML, Stienen GJ, Vonk-Noordegraaf A, et al. The striated muscles in pulmonary arterial hypertension: adaptations beyond the right ventricle. Eur Respir J. 2015;46:832–842. doi: 10.1183/13993003.02052-2014. [DOI] [PubMed] [Google Scholar]
- 47. Vizza CD, Lynch JP, Ochoa LL, Richardson G, Trulock EP. Right and left ventricular dysfunction in patients with severe pulmonary disease. Chest. 1998;113:576–583. doi: 10.1378/chest.113.3.576. [DOI] [PubMed] [Google Scholar]
- 48. Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, et al. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang S, Banerjee S, Freitas Ad, Cui H, Xie N, Abraham E, et al. miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2012;302:L521–L529. doi: 10.1152/ajplung.00316.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stranska R, Gysbrechts L, Wouters J, Vermeersch P, Bloch K, Dierickx D, et al. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J Transl Med. 2018;16:1. doi: 10.1186/s12967-017-1374-6. [DOI] [PMC free article] [PubMed] [Google Scholar]