

Abstract
Molecular patterns and pathways in idiopathic pulmonary fibrosis (IPF) have been extensively investigated, but few studies have assimilated multiomic platforms to provide an integrative understanding of molecular patterns that are relevant in IPF. Herein, we combine the coding and noncoding transcriptomes, DNA methylomes, and proteomes from IPF and healthy lung tissue to identify molecules and pathways associated with this disease. RNA sequencing, Illumina MethylationEPIC array, and liquid chromatography–mass spectrometry proteomic data were collected on lung tissue from 24 subjects with IPF and 14 control subjects. Significant differential features were identified by using linear models adjusting for age and sex, inflation, and bias when appropriate. Data Integration Analysis for Biomarker Discovery Using a Latent Component Method for Omics Studies was used for integrative multiomic analysis. We identified 4,643 differentially expressed transcripts aligning to 3,439 genes, 998 differentially abundant proteins, 2,500 differentially methylated regions, and 1,269 differentially expressed long noncoding RNAs (lncRNAs) that were significant after correcting for multiple tests (false discovery rate < 0.05). Unsupervised hierarchical clustering using 20 coding mRNA, protein, methylation, and lncRNA features with the highest loadings on the top latent variable from the four data sets demonstrates perfect separation of IPF and control lungs. Our analysis confirmed previously validated molecules and pathways known to be dysregulated in disease and implicated novel molecular features as potential drivers and modifiers of disease. For example, 4 proteins, 18 differentially methylated regions, and 10 lncRNAs were found to have strong correlations (|r| > 0.8) with MMP7 (matrix metalloproteinase 7). Therefore, by using a system biology approach, we have identified novel molecular relationships in IPF.
Keywords: systems biology, transcriptome, methylome, proteome, multiomics
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal disease of the aging lung (1, 2). Its prevalence is increasing (3), and it is likely underdiagnosed (4, 5). Although cigarette smoke remains the most significant environmental risk factor for this complex disease (6), the gain-of-function MUC5B promoter variant is the strongest risk factor, genetic or otherwise, for the development of IPF. However, 13 other common variants and several rare variants including telomerase pathway genes also contribute to the risk of developing IPF (7–9). Although pirfenidone (10) and nintedanib (11) slow IPF progression, no treatment short of lung transplantation impacts survival. IPF is characterized by dysplastic bronchiolar metaplasia, alveolar epithelial injury and repair, proliferation of resident fibroblasts, formation of myofibroblastic foci, accumulation of extracellular matrix (ECM), and lung remodeling (12).
Genomic approaches have been used to characterize the molecular landscape of IPF. Gene expression studies have identified several thousand genes that are differentially regulated in the IPF lung (13–20), consistently reporting common genes and pathways (ECM organization and regulation, TGF-β signaling, endoplasmic reticulum stress, epithelial–mesenchymal transition, mitochondrial homeostasis, bronchial epithelial genes, fibroblast genes, smooth muscle markers, cytokines and chemokines, growth factors, and receptors) that are differentially expressed in fibrotic lungs. A recent deep proteome profiling study has confirmed that many of these genomic differences result in differential protein abundance in the IPF lung, with key genes such as MMP7 (matrix metalloproteinase 7) and MUC5B showing increased abundance (21). At the regulatory level, DNA methylation changes have been associated with many of the key transcriptional changes in IPF lung tissue (22–24), and hypermethylation of genes such as CXCL10 (25), PTGER2 (26), and THY1 (27) has been shown to contribute to IPF pathogenesis. Genomic microRNA (miRNA) profiles have revealed several miRNAs that are known to affect fibroproliferation, epithelial–mesenchymal transition, and the TGF-β1 signaling pathway (28–32). Although studies of long noncoding RNAs (lncRNAs) in pulmonary fibrosis are limited, there appears to be an antifibrotic role for FENDRR (33) and a profibrotic role for DNM3OS (34). In addition, studies in peripheral blood have identified biomarkers of disease (35) and disease outcomes (36, 37).
Despite the successful application of multiple single platform “omic” technologies to characterize the molecular landscape of IPF, integrative approaches using system biology have not yet been applied to the IPF lung. Stimulated by a recent application of multiomics to a small-sample, big-data study in newborns (38), we obtained DNA methylome, coding and noncoding transcriptome, and proteome results from 24 IPF lungs and 14 control lungs. Leveraging supervised (39) and unsupervised (40) machine learning methods allowed us to identify integrated molecules and pathways across the multiple omic platforms to more comprehensively characterize the complex molecular features of IPF.
Methods
Ethics Statement
Human tissue was collected after appropriate ethical review for the protection of human subjects through the National Heart, Lung, and Blood Institute–sponsored LTRC (Lung Tissue Research Consortium) and lung donor program at the University of Pittsburgh. Deidentified data and samples were approved for use in this study by the University of Colorado (Colorado Multiple Institutional Review Board number 15-1147).
Study Population
We selected 24 subjects with IPF from the LTRC and 14 control subjects from the University of Pittsburgh Lung Core, all of whom were non-Hispanic white individuals. Details of the study population are provided in the Methods of the data supplement.
Sample Processing
DNA and RNA were isolated from the same sample of lung tissue using the AllPrep kit (Qiagen). Samples with an RNA integrity number and DNA integrity number >5 were used. Genotyping for the MUC5B rs35705950 variant was performed by using a TaqMan assay (Thermo Fisher Scientific). Sample preparation for proteomic analysis is described in the Methods of the data supplement.
Omic Data Collection
mRNA libraries were prepared from 500 ng of total RNA by using TruSeq stranded mRNA library preparation kits (Illumina) and were sequenced at the average depth of 80 million reads on the Illumina NovaSeq 6000 system. A total of 4,011 unique proteins were detected by using published mass spectrometry methods (41) and are described in the Methods of the data supplement. One microgram of DNA was bisulfite treated by using the Zymo EZ DNA Methylation Kit and was labeled and hybridized to an Illumina Infinium Human MethylationEPIC BeadChip by using standard protocols. Ribosomal RNA–depleted libraries were prepared from 1 μg of RNA by using the Epidemiology Ribo-Zero Gold rRNA Removal Kit (Illumina) and were sequenced at the average depth of 80 million reads on the Illumina NovaSeq 6000 system. RNA sequencing (RNA-seq) count-level data and Infinium Human MethylationEPIC BeadChip methylation data have been deposited to the Gene Expression Omnibus under accession number GSE173357.
General Statistical Methods
All analyses were performed in R (version 3.6.2, R Foundation for Statistical Computing). Principal component analysis was used for quality control, and no samples had to be excluded on the basis of this criterion. Principal component regression analysis was used to identify variables associated with top principal components, and strong batch effects were regressed out by using ComBat software (42). Differentially abundant features in each data set were identified by using linear models that adjusted for age and sex. In the mRNA sequencing, lncRNA sequencing, and DNA methylation data sets, P values were adjusted for inflation and bias by using Bacon software (43). To control for multiple comparisons, P values were adjusted to a 5% false discovery rate (FDR) by using the Benjamini-Hochberg procedure (44) in all data sets. Detailed methods for data processing and statistical analysis of individual data sets are provided in the Methods of the data supplement.
Data Integration Analysis for Biomarker Discovery Using Latent Variable Approaches for Omics Studies
The Data Integration Analysis for Biomarker Discovery Using a Latent Component Method for Omics Studies (DIABLO) framework (39) was used to determine correlated omic features associated with diagnosis. DIABLO is a supervised learning approach that builds on Regularized and Sparse Generalized Canonical Correlation Analysis, maximizing correlations among multiple data sets containing the same individuals and a classifier (diagnosis). DIABLO seeks common information across different data types through the selection of a subset of molecular features while discriminating between IPF and control lung tissue. By using simulations in DIABLO, we determined that a single latent variable sufficiently captures most of the variation associated with diagnosis. For input into DIABLO, we used the four lists of differential features at an FDR <0.05, with the mRNA set limited to 1,109 transcripts with a |fold change| >4 to have a number of features similar to those of the remaining three data sets.
Results
We selected subjects with IPF and control subjects to have similar demographic (age and sex) characteristics, the same race and ethnicity (non-Hispanic white), and similar smoking histories (all ever-smokers or former smokers) (Table 1). All control subjects have the GG MUC5B genotype, and among the subjects with IPF, 50% have the GG MUC5B genotype and 50% have the GT MUC5B genotype.
Table 1.
Clinical Characteristics of the Subjects Included in the Multiomic Profiling
| Subjects with IPF (n = 24) | Control Subjects (n = 14) | P | |
|---|---|---|---|
| Age, yr, mean ± SD | 62 ± 5.9 | 64 ± 5.7 | 0.323* |
| Sex, M, n (%) | 20 (83.3) | 10 (71.4) | 0.433† |
| Race, W, n (%) | 24 (100) | 14 (100) | 1† |
| Ethnicity, NH, n (%) | 24 (100) | 14 (100) | 1† |
| Smoking status, ever | 17 (70.1) | 9 (64.3) | 0.521† |
| MUC5B genotype, GG | 13 (50) | 14 (100) | N/A‡ |
Definition of abbreviations: IPF = idiopathic pulmonary fibrosis; M = male; N/A = not applicable; NH = non-Hispanic; W = white.
Assessed by using a Student’s t test.
Assessed by using a Fisher’s exact Test.
By design.
Coding RNA
A total of 116,503 transcripts were detected through polyA-enriched RNA-seq, 75,382 of which are annotated as protein-coding or retained-intron transcripts (alternatively spliced transcripts). A total of 4,643 transcripts (protein-coding and retained-intron transcripts) aligning to 3,439 genes are differentially expressed in IPF lung tissue compared with normal control lung tissue at an FDR <0.05 after stringent adjustment for inflation and bias (see Figure E1A in the data supplement). As an alternative to adjustment for inflation and bias, we performed cell deconvolution analysis by using the xCell software (45) and adjusted for cell proportions in the statistical model, but this method did not perform as well (Figure E2). Of the 4,634 differentially expressed transcripts, 1,425 transcripts are upregulated in IPF lung tissue, whereas 3,218 RNAs are more abundant in control lung tissue (Table E1A). The majority of differentially expressed transcripts are protein-coding transcripts (74%; Figure 1A), and the remaining 26% (Figure 1B) are alternatively spliced transcripts. Upregulated mRNAs are strongly enriched for protein products localizing to the mitochondria as well as transcripts involved in oxidation. Downregulated mRNAs are enriched for focal adhesion and immune signaling pathways. Differentially expressed genes previously reported to be associated with IPF include MMP7, a gene that is the most established biomarker for IPF (17, 46, 47), and EGF (epidermal growth factor). Our recent analysis of transcriptional profiles of airway epithelial cells grown at the air–liquid interface at different time points identified an interaction of the receptor EGFR (EGF receptor) and the inducible transcriptional coactivator (YAP) as being critical to the migratory phenotype of IPF cells (48). In addition, we observed genes involved in other fibrotic lung diseases, such as CUX1, a transcription factor that regulates COL1 expression and is upregulated in systemic sclerosis (49). CUX1 isoforms are localized within ACTA2+ cells in systemic sclerosis skin sections and IPF lung tissue sections, suggesting an important role for CUX1 in regulation of COL1 expression in fibrosis in multiple organs (50).
Figure 1.

Volcano plots depicting features that are statistically significant in idiopathic pulmonary fibrosis (IPF) lung tissue compared with control lung tissue at a false discovery rate <0.05 (blue dots). (A) Protein-coding and (B) alternatively spliced RNA were captured by using polyA or mRNA sequencing, whereas (E) long noncoding RNAs (lncRNAs) were captured by using ribosomal RNA–depleted sequencing. Alternative splicing includes retained exon annotations from GENCODE. Noncoding RNA includes long intergenic noncoding RNA, antisense RNA, miscellaneous RNA, sense intronic RNA, small nuclear RNA, microRNA, small nucleolar RNA, sense overlapping RNA, bidirectional promoter lncRNA, 3′ overlapping noncoding RNA, small Cajal body–specific RNA, ribozymes, noncoding macro lncRNA, small conditional RNA, and vault RNA GENCODE annotations. All data other than the proteome data set were adjusted for bias and inflation (43). (C) Protein data were not adjusted for bias and inflation because of an inherent bias in the proteomic assay focusing on proteins/peptides known to be involved in IPF; therefore, inflation is expected in this data set. (D) DNA methylome data were collected and analyzed using standard methods for Illumina Infinium Human MethylationEPIC BeadChip. EGF = epidermal growth factor; LINC-PINT = long intergenic non–protein-coding RNA p53–induced transcript; MMP7 = matrix metalloproteinase; PRKRA = protein activator of IFN induced protein kinase EIF2AK2.
Protein
The liquid chromatography–mass spectrometry platform we used detected 22,198 peptides associated with 4,011 unique proteins/genes. A total of 1,040 proteins were differentially abundant in IPF tissue compared with control tissue at an FDR <0.05 (Figure 1C; Table E1B). A total of 522 proteins (including 27 core matrisome–associated proteins and 19 matrisome-associated proteins) are increased in IPF, and 518 (including 24 core matrisome–associated proteins and 22 matrisome-associated proteins) are decreased. Differentially abundant proteins are significantly enriched for core matrisome and matrisome-associated proteins (Fisher’s P = 0.001). We also detected multiple upregulated thioredoxin-related genes (P4HB, QSOX1, TXN2, TXNDC5, TXNL1) in IPF tissue. Thioredoxins are upregulated by reactive oxygen species and reduce oxidative stress. TMEM231 shows the greatest increase in IPF. TMEM231 is a transmembrane protein present in the transition zone of cilia that prevents protein mislocalization by blocking protein diffusion across the ciliary membrane and is necessary for proper ciliogenesis. Our group has previously implicated ciliary dysfunction in IPF pathogenesis through patient clustering of gene expression microarray data (19).
Methylation
After stringent control for bias and inflation (Figure E1C), we identified 2,500 differentially methylated regions (DMRs) overlapping or within 10 kb of 1,840 genes (Figure 1D; Table E1C). As an alternative to adjustment for inflation and bias, we performed cell deconvolution analysis by using the RefFreeEWAS package in R (51) and adjusted for cell confounding, but this method did not perform as well (Figure E3). Of the 2,568 DMR–gene relationships for the DMRs, 31% of the DMRs are intronic to genes, 24% overlap an exonic region, and 11% are in promoters (defined as within 2 kb upstream of the transcription start site). On average, significant DMRs contained four Illumina probes and spanned 335 bp. The absolute average difference in the percentage of methylation of CpGs within significant DMRs in IPF lung tissue versus control lung tissue is 9.6%. The greatest hypomethylated DMR shows a 30% decrease in methylation relative to control tissue. This 709-bp region contains six probes and overlaps the 3' untranslated region of VMP1 as well as most of the transcribed region of miR-21, a microRNA shown to promote fibrogenesis through upregulation of TGF-β signaling, for which differential methylation has not been previously reported (29). Additional DMRs overlap genes shown to be involved in lung development and fibrosis. We observe hypomethylation in FOXP1, a transcription factor involved in secretory epithelial cell fate determination in the lung (52), as well as differential methylation of genes previously shown to be involved in IPF, such as CCL2 (53).
lncRNA
We identified 1,269 differentially expressed transcripts associated with 1,067 unique genes (FDR < 0.05) (Figure 1E; Table E1D) after controlling for bias and inflation (Figure E1D). The majority of differentially expressed noncoding RNAs are lncRNAs (39%) and antisense RNAs (43%). As expected, most of the lncRNAs are those of unknown function. Among most differentially expressed lncRNAs with known function that are upregulated in IPF are MUC5B-AS1, a noncoding RNA antisense to MUC5B, and LINP2, which has multiple roles in cancer (54). lncRNAs of known function downregulated in IPF include LINC-PINT (long intergenic non–protein-coding RNA p53–induced transcript), which reduces lung cancer progression via sponging of miR-543 to induce the tumor suppressor PTEN (phosphatase and tensin homolog) (55).
Integrative Analyses
Protein-coding transcriptome and proteome interactions.
To begin to integrate data sets, we first performed pairwise comparisons of coding mRNA and protein data, initially focusing on transcripts and proteins with significant changes in both data sets. Comparing the fold change of protein to mRNA, we demonstrated that most changes with large effect sizes (fourfold change in IPF vs. control) are consistent directionally (Figure 2A). PRKRA (protein activator of IFN induced protein kinase EIF2AK2) is especially highly upregulated at the mRNA and protein level. This protein kinase is activated by double-stranded RNA and mediates the effects of IFN in response to viral infections, which are known risk factors of disease (56, 57). PRKRA promoter hypomethylation has been previously reported in IPF lung tissue (23), and our novel observation of mRNA and protein upregulation further suggests a role for this gene in disease.
Figure 2.
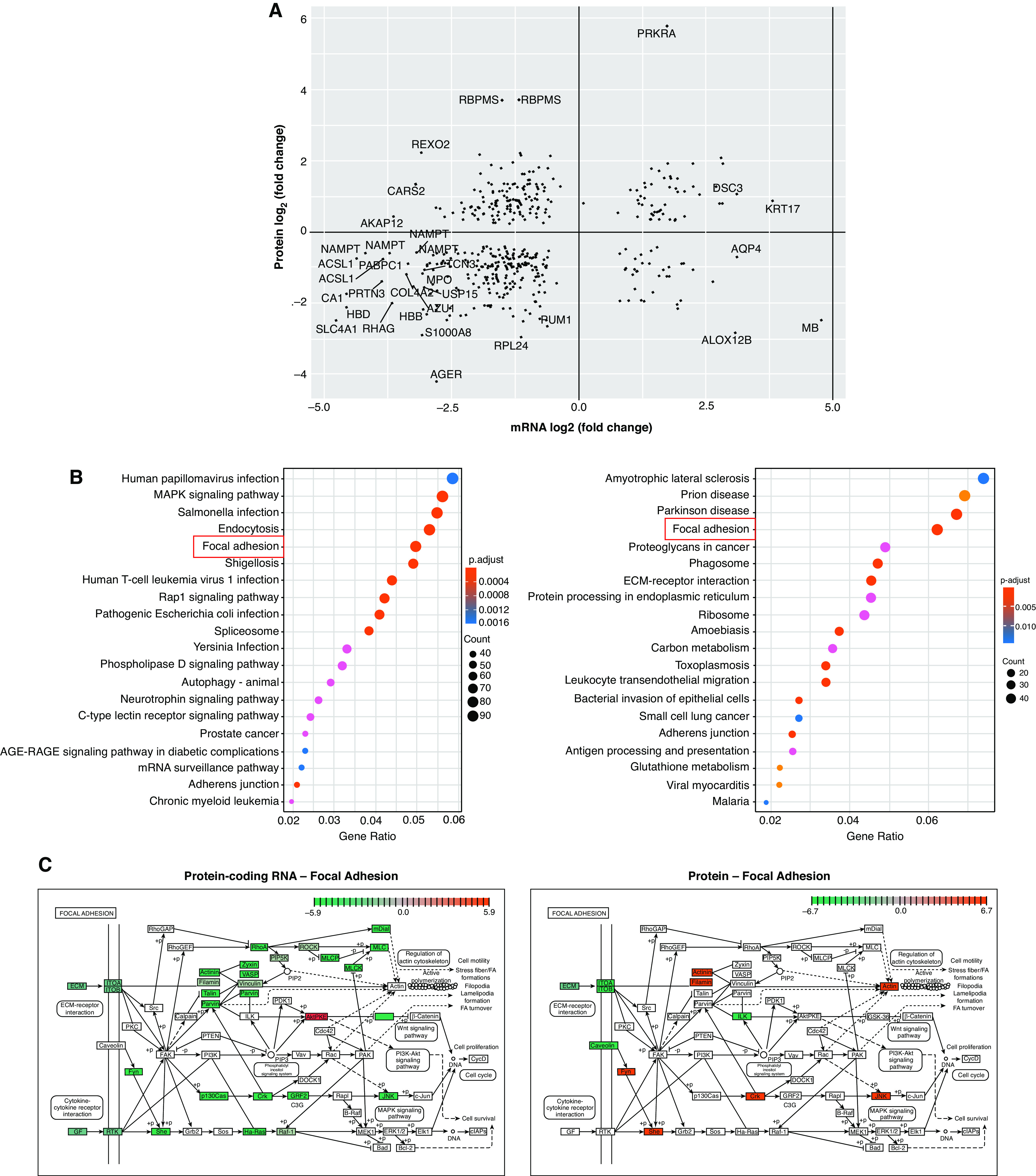
Comparison of protein-coding mRNA and protein data sets. (A) The protein fold change plotted against the fold change for the corresponding protein-coding mRNA. Transcripts/proteins with an absolute fold change >4 (2 on the log2 scale) are highlighted. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment in the mRNA (left) and protein (right) data sets. Boxes highlight pathways of interest in common to the two data sets. (C) mRNAs/proteins in the focal adhesion KEGG pathway are highly dysregulated in IPF lung tissue. Red represents upregulation and green represents downregulation. ECM = extracellular matrix; PTEN = phosphatase and tensin homolog.
Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis revealed common pathways, specifically focal adhesion and adherens junctions, in the mRNA and protein data sets (Figure 2B). Focal adhesion, adhesive contact between the cell and ECM through the interaction of integrin transmembrane proteins with their extracellular ligand, is strongly enriched in both the mRNA and the protein data sets. Although the majority of transcripts in the focal adhesion pathway are downregulated in IPF, protein-level data demonstrate a mix of upregulation and downregulation, highlighting the importance of studying disease-related genes across omic data sets. We observed downregulation of integrin α1 and β5 subunits at the RNA level (ITGA1A and ITGA5B) and integrin α1 and β1 subunits at the protein level (ITGA1A and ITGA1B). Published findings have established a profibrotic role of αvβ1 and αvβ6 integrins at the protein level by activation of TGF-β (58); more work is needed to understand the roles of integrins we identified in lung fibrosis.
Effect of DNA methylation on gene expression.
We next assessed the effect of DNA methylation on expression of the nearest protein-coding RNA (Figure 3A), alternatively spliced RNA (Figure 3B), and protein (Figure 3C). We observed only a few relationships within 10 kb among significantly differentially expressed genes, acknowledging that methylation marks do not always regulate the nearest gene (59). Inversely correlated methylation and expression were observed for genes of interest in IPF, such as AGER (60), α catenin 2 (61), KRT17 (62), and CASZ1 (a gene we previously reported as regulated by methylation in IPF [24]). Increasing the distance of overlap to 100 kb reveals many more potential cis relationships between DNA methylation and gene expression. A potentially interesting novel finding in these data is regulation by methylation of COL17A1, a transmembrane protein that is a structural component of hemidesmosomes and has been reported to be regulated by promoter methylation in epithelial cancers (63). Even with the limitation of only focusing on relationships with the nearest genes, DNA methylation appears to be an important feature of gene regulation in IPF.
Figure 3.
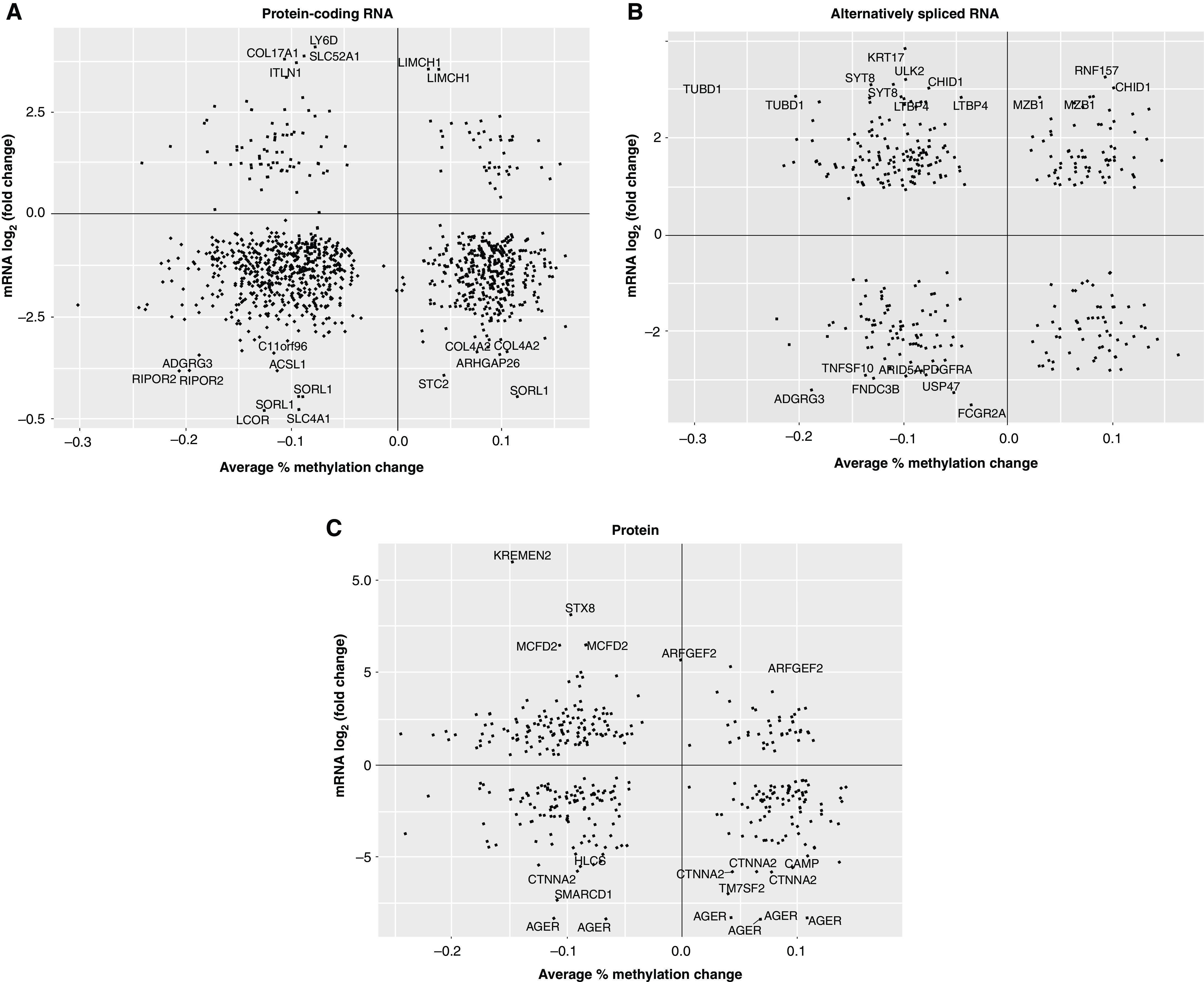
The effect of DNA methylation on dysregulated gene expression in IPF lung tissue. (A) Protein-coding mRNA, (B) alternatively spliced mRNA (retained intron), and (C) the protein fold change plotted against the percent change in DNA methylation in DMRs assigned to the same genes. All fold changes are presented on the log2 scale. DNA methylation changes are presented as percent methylation changes (on the scale 0–1). DMR = differentially methylated region.
Multiomic modeling.
To fully integrate all four data sets (protein-coding RNA, protein, DNA methylation, and noncoding RNA), we used the DIABLO multiomics integrative method. The DIABLO model differentiates IPF and control lungs by using one latent variable (Figure E4A), demonstrates strong correlations of individual features with the top latent variable (Figure E4B), and demonstrates strong correlations across features from different data sets (Figure E4C). Contributions of individual data set features on the top latent variable are shown in Figure E4D and in Tables E2A–E2D. Unsupervised hierarchical clustering using 20 coding mRNA, protein, methylation, and lncRNA features with highest loadings on the top latent variable from the four data sets (80 features total; Figure 4A) demonstrates perfect separation of IPF and control lungs (Figure 4B). Among the top protein-coding mRNA features are MMP7, a key biomarker of pulmonary fibrosis (13, 47); PROM2, a gene expressed in basal cells that differentiates airway from the alveolar transcriptional subtype of IPF (19); COL17A1, discussed above; and LAMC3, a focal adhesion gene. Among the top protein features are periostin, a protein that promotes myofibroblast differentiation and type 1 collagen production (64); palladin, a protein involved in cell adhesion; AGER, a gene polymorphic protein in IPF that encodes soluble RAGE decoy receptor (60); focal adhesion proteins LAMC2 and ITGA3; and PECAM1, a protein involved in leukocyte migration, angiogenesis, and integrin activation. Among the top DNA methylation features (all hypomethylated) are DMRs 5' to miR-21, a key profibrotic miRNA upregulated in IPF (29); the promoter of CCL2, a T cell–recruiting chemokine with an established role in IPF (65); the promoter of TNXB, a gene that has been reported to be hypomethylated and upregulated in IPF fibroblasts (66); and an intron of the LTBP1 gene that is upregulated in IPF, especially in honeycomb cysts, and regulates the effects of TGF-β1 (67). lncRNA data are more difficult to dissect because of currently unknown functions of many of the lncRNAs. Of the top 20 lncRNAs, RARA-AS1 is promising as a potential regulator of RARA, a gene that has been shown to be downregulated in IPF fibroblasts (68). LINC01565 or GR6 is another potential candidate on the basis of its expression patterns (highest in lung and bone marrow), but no studies have shown its role in fibrosis at this time. MIR34AHG is the host gene for miR-34, which has been shown to regulate cellular senescence in IPF type II alveolar epithelial cells (69). FENDRR, a lncRNA previously associated with IPF (33) is 28th on the extended list of features ranked by the strength of association with the top latent variable (Table E2D), and we observed hypermethylation of a FENDRR enhancer predicted by GeneHancer.
Figure 4.
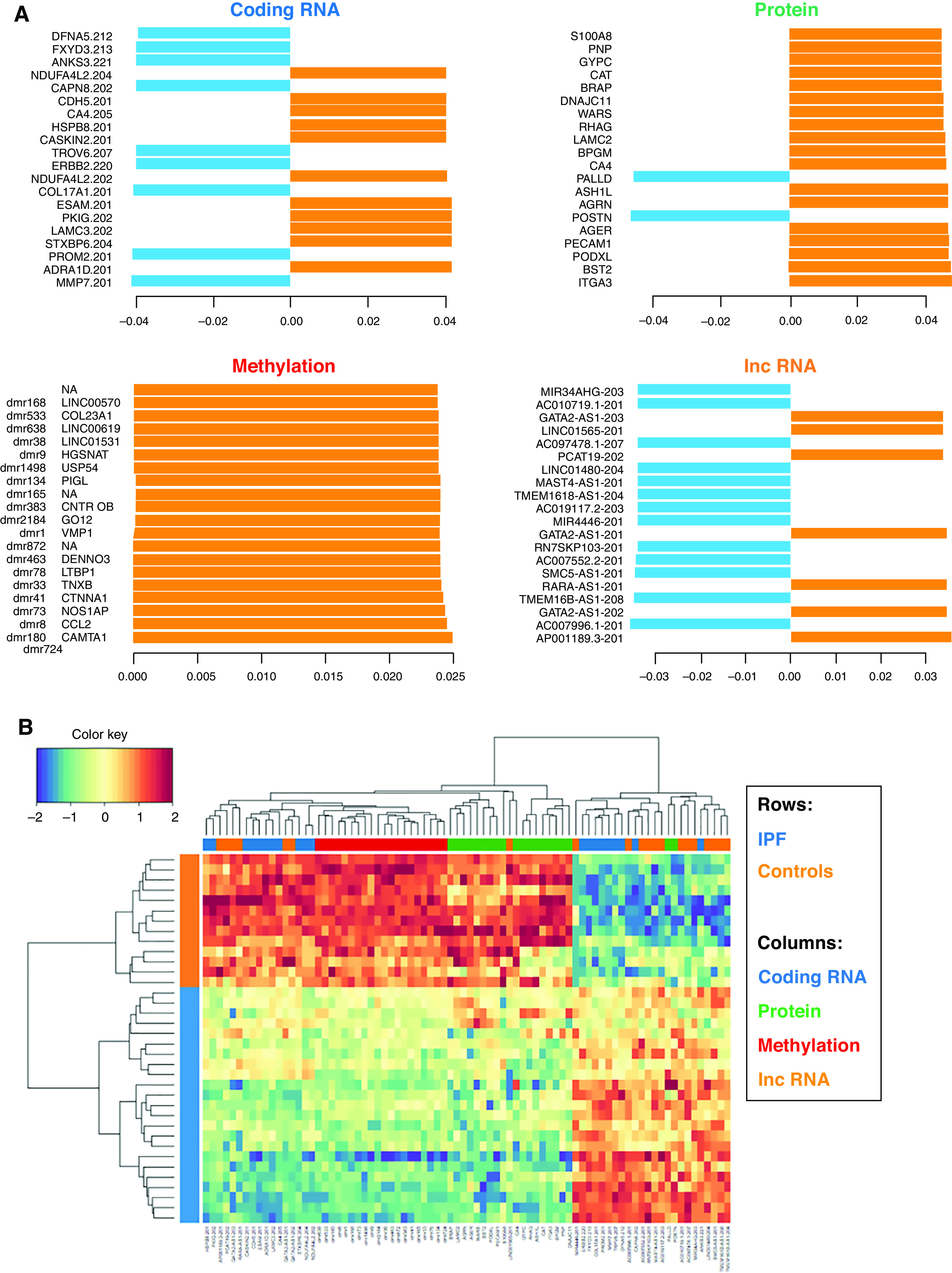

Data Integration Analysis for Biomarker Discovery Using a Latent Component Method for Omics Studies multiomic model results for the top 20 features in each data set (blue indicates coding RNA, green indicates protein, red indicates methylation, and orange indicates noncoding RNA). IPF lung tissue is represented in blue, and control lung tissue is represented in orange. (A) Top 20 features from each data set contributing to the top latent component. (B) Clustering of subjects with IPF versus clustering of control subjects on the basis of top 20 features from each data set. (C) Circos plot of correlations (|r| > 0.8) for all features contributing to the top latent component. Red lines represent positive correlations, and blue lines represent negative correlations. (D) An interactome network of top features from each of the individual data sets. Red lines represent positive correlations, and blue lines represent negative correlations.
In general, we observed strong correlations among features from different omic platforms that were prioritized by the DIABLO model, as would be expected. Many of the DNA methylation marks are negatively correlated with protein-coding RNAs as well as lncRNAs (Figure 4C). This led us to construct a network of the top 20 features from each of the individual data sets (Figure 4D). MMP7 RNA, for example, has strong correlations (|r| > 0.8) with four proteins (ASH1L, BRAP, RHAG; all negative) and 18 DMRs (all negative), negative correlations with three lncRNAs (AP001189.3, GATA2-AS1, and RARA-AS1), and positive correlations with seven lncRNAs (AC007552.2, AC007996.1, AC097478.1, LINC01480, MAST4-AS1, SMC5-AS1, and TMEM161B-AS1). MMP7 illustrates how this multiomic approach may uncover novel relationships that will require additional computational (replication) and experimental (functional) validation.
Validation of the multiomic model.
We used the unsupervised approach Multi-Omics Factor Analysis to independently identify the principal sources of variation in our multiomic data sets. Results of this confirmatory analysis are discussed and summarized in the data supplement (Figure E5 and Table E4). Overall, a number of the same transcriptome and proteome features emerge as prioritized by both the DIABLO and Multi-Omics Factor Analysis multiomic methods, whereas more work will be needed to further assess the reproducibility of regulatory features of the transcriptome (DNA methylation and lncRNAs).
Discussion
We present the first application of multiomic integration modalities to IPF lung tissue, leveraging coding and noncoding RNA expression, proteomic, and DNA methylation data to construct a multiomic network to gain insights into relevant pathogenic molecules and pathways in disease. Our analyses confirm previously validated molecules and pathways known to be dysregulated in disease and implicate novel molecular features as potential drivers and modifiers of disease.
The multiomic model provides a more complete characterization of molecular pathways in IPF and provides insight into the complex biology of IPF. It also provides better power for identification of such pathways by using analysis of multiple sets of data on the same tissue, as has been previously shown in other settings (38). Multiomic analysis may be used to identify pathways that are dysregulated in IPF at both the transcript and the protein level, such as focal adhesion or regulators of genes/proteins already identified as important in IPF. For example, our multiomic model indicated that 18 regions of differential methylation and 10 lncRNAs may be important in regulation of MMP7, a gene known with a known role in IPF. Interestingly, RARA-AS1, a potential regulator of RARA, a gene that has been shown to be downregulated in IPF fibroblasts (68), is highly negatively correlated with MMP7, suggesting that cross-talk between RARA and MMP7 may provide novel targets for IPF therapeutics. However, additional computational (replication) and experimental (functional) validation is needed.
Given that IPF is a complex disease biologically (2), it is not surprising that narrowly targeted drugs have generally failed in IPF and that currently approved drugs target multiple pathways, such as inhibition of several growth factors with nintedanib (70). Analyzing proteomic, transcriptomic, and regulatory (methylome and noncoding transcriptome) molecules at the same time provides a more complete picture of IPF pathophysiology and will raise the interest for new compounds in IPF. Data mining has become an important research direction in drug discovery that should take advantage of multiomic analysis (71). Future work in IPF will likely apply these multiomic methods to longitudinally collected data in peripheral blood, capturing earlier or preclinical stages of disease for the development of treatments that can be used before the fibrotic process involves large portions of the lung and becomes irreversible (72).
By study design, cases were evenly distributed between patients homozygous for the major allele and patients heterozygous for the MUC5B promoter polymorphism rs3570950 to allow for examination of the multiomic signal in relation to the MUC5B variant. However, differential testing within data sets based on the variant did not yield statistically significant results; much larger sample sizes will likely be needed to detect the effect of the variant in mixed tissue, given that the small number of distal airway epithelial cells in which the variant exhibits the strongest effect (73, 74). Furthermore, although MUC5B transcript and protein are upregulated in IPF (7.7-fold at the transcript level and 2-fold at the protein level in our samples), the MUC5B transcript and protein are not differentially abundant at an FDR <0.05, which is due to the heterogeneity in abundance among cases in whole-lung tissue. This could also explain why MUC5B-AS1, one of the top differentially abundant (increased) antisense RNAs in our RNA-seq data, is not a top-weighted feature in the DIABLO model. These results are not surprising because of our limited sample size. Further studies examining MUC5B genotype contributions to molecular signaling will have increased power if cell types of interest can be isolated through microdissection or cell selection or enrichment methodologies.
We applied stringent corrections for bias and inflation by using methodology specifically developed for transcriptome-wide association studies (TWAS) and epigenome-wide association studies (EWAS). Methods developed for genome-wide association studies assume a Gaussian distribution of test statistics; this is a valid assumption in genome-wide association studies, as the vast majority of (generally binary) variants are not expected to be associated with the trait of interest. In TWAS and EWAS of complex traits such as IPF, it is common to identify changes in hundreds to thousands of features, most of which are likely to be true associations but some of which may be spurious because of inherent inflation that has been documented in TWAS and EWAS (43). Because of this, we applied stringent corrections within our data for bias and inflation by using Bacon software (43) to empirically derive a null testing distribution from our data, which takes into account the nonnormal mean and variance of the data. This greatly reduced the number of differential features meeting significance within our data sets, compared with previous publications (19). However, some residual inflation remains in the DNA methylation data set, an issue that is common in the field (75).
Future multiomic research in IPF should attempt to increase the power as well as the genetic context of these molecular patterns. Larger cohort studies will provide the power to derive and then test and validate sparse multiomic signatures for replication in independent samples. Larger numbers will also allow for clustering of IPF cases into potentially meaningful subgroups. The inclusion of genetic data, which explains a significant proportion of disease variability, will aid in patient clustering and recognition of distinct molecular subtypes. These improved integrative models hold promise to focus our attention on key molecules and pathways involved in the complex biology of lung fibrosis and will potentially enable us to identify critical checkpoints that can manipulated pharmacologically.
Acknowledgments
Acknowledgment
The authors thank the University of Colorado Genomics and Microarray Core Facility for collection of Illumina BeadChip and RNA-seq data.
Footnotes
Supported by the National Heart, Lung, and Blood Institute grant P01-HL092870. I.R.K. was supported by the University of Colorado Team Oriented Training across the Translational Sciences Spectrum program (National Center for Advancing Translational Sciences grant NCATS TL1-TR002533). R.B. was supported by the European Respiratory Society Long Term Research Fellowship.
Author Contributions: T.E.F., I.V.Y., and D.A.S. conceived and designed the study. R.B., A.D.W., F.M., and S.M.H. collected the data. I.R.K., R.B., J.C., and F.M. analyzed the data. M.R. performed clinical phenotyping of the subjects. I.R.K., R.B., I.V.Y., and D.A.S. wrote the manuscript. All authors edited and approved the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0546OC on May 24, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–284. doi: 10.1164/rccm.200701-044OC. [DOI] [PubMed] [Google Scholar]
- 2. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 3. Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, Fogarty A, et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011;66:462–467. doi: 10.1136/thx.2010.148031. [DOI] [PubMed] [Google Scholar]
- 4. Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. 2013;368:2192–2200. doi: 10.1056/NEJMoa1216076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators; COPDGene Investigators. Association between interstitial lung abnormalities and all-cause mortality. JAMA. 2016;315:672–681. doi: 10.1001/jama.2016.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 7. Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2020;201:564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moore C, Blumhagen RZ, Yang IV, Walts A, Powers J, Walker T, et al. Resequencing study confirms that host defense and cell senescence gene variants contribute to the risk of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:199–208. doi: 10.1164/rccm.201810-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathai SK, Newton CA, Schwartz DA, Garcia CK. Pulmonary fibrosis in the era of stratified medicine. Thorax. 2016;71:1154–1160. doi: 10.1136/thoraxjnl-2016-209172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 11. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 12. King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 13. Kaminski N. Microarray analysis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29(3)(Suppl):S32–S36. [PubMed] [Google Scholar]
- 14. Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R, et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;180:167–175. doi: 10.1164/rccm.200810-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Selman M, Carrillo G, Estrada A, Mejia M, Becerril C, Cisneros J, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One. 2007;2:e482. doi: 10.1371/journal.pone.0000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2006;173:188–198. doi: 10.1164/rccm.200504-644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002;99:6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, et al. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One. 2009;4:e5134. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68:1114–1121. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang IV, Burch LH, Steele MP, Savov JD, Hollingsworth JW, McElvania-Tekippe E, et al. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med. 2007;175:45–54. doi: 10.1164/rccm.200601-062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiller HB, Mayr CH, Leuschner G, Strunz M, Staab-Weijnitz C, Preisendörfer S, et al. Deep proteome profiling reveals common prevalence of MZB1-positive plasma B cells in human lung and skin fibrosis. Am J Respir Crit Care Med. 2017;196:1298–1310. doi: 10.1164/rccm.201611-2263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:525–535. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang IV, Pedersen BS, Rabinovich E, Hennessy CE, Davidson EJ, Murphy E, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:1263–1272. doi: 10.1164/rccm.201408-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP, et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- 26. Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC, et al. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol. 2010;177:2245–2255. doi: 10.2353/ajpath.2010.100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:610–618. doi: 10.1165/rcmb.2007-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, et al. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45:287–294. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oak SR, Murray L, Herath A, Sleeman M, Anderson I, Joshi AD, et al. A micro RNA processing defect in rapidly progressing idiopathic pulmonary fibrosis. PLoS One. 2011;6:e21253. doi: 10.1371/journal.pone.0021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pottier N, Maurin T, Chevalier B, Puisségur MP, Lebrigand K, Robbe-Sermesant K, et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS One. 2009;4:e6718. doi: 10.1371/journal.pone.0006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang C, Liang Y, Zeng X, Yang X, Xu D, Gou X, et al. Long noncoding RNA FENDRR exhibits antifibrotic activity in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2020;62:440–453. doi: 10.1165/rcmb.2018-0293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Savary G, Dewaeles E, Diazzi S, Buscot M, Nottet N, Fassy J, et al. The long noncoding RNA DNM3OS is a reservoir of fibromiRs with major functions in lung fibroblast response to TGF-β and pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:184–198. doi: 10.1164/rccm.201807-1237OC. [DOI] [PubMed] [Google Scholar]
- 35. Yang IV, Luna LG, Cotter J, Talbert J, Leach SM, Kidd R, et al. The peripheral blood transcriptome identifies the presence and extent of disease in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e37708. doi: 10.1371/journal.pone.0037708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herazo-Maya JD, Sun J, Molyneaux PL, Li Q, Villalba JA, Tzouvelekis A, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med. 2017;5:857–868. doi: 10.1016/S2213-2600(17)30349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma SF, Tseng GC, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5:205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee AH, Shannon CP, Amenyogbe N, Bennike TB, Diray-Arce J, Idoko OT, et al. EPIC Consortium. Dynamic molecular changes during the first week of human life follow a robust developmental trajectory. Nat Commun. 2019;10:1092. doi: 10.1038/s41467-019-08794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh A, Shannon CP, Gautier B, Rohart F, Vacher M, Tebbutt SJ, et al. DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics. 2019;35:3055–3062. doi: 10.1093/bioinformatics/bty1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, et al. Multi-omics factor analysis: a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. 2018;14:e8124. doi: 10.15252/msb.20178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lepper MF, Ohmayer U, von Toerne C, Maison N, Ziegler AG, Hauck SM. Proteomic landscape of patient-derived CD4+ T cells in recent-onset type 1 diabetes. J Proteome Res. 2018;17:618–634. doi: 10.1021/acs.jproteome.7b00712. [DOI] [PubMed] [Google Scholar]
- 42. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Iterson M, van Zwet EW, Heijmans BT. BIOS Consortium. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18:19. doi: 10.1186/s13059-016-1131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 45. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185:67–76. doi: 10.1164/rccm.201101-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stancil IT, Michalski JE, Davis-Hall D, Magin CM, Yang IV, Dobrinskikh E, et al. Aberrant airway epithelial function and signaling promote lung fibrosis. Nat Commun. 2021 doi: 10.1038/s41467-021-24853-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ikeda T, Fragiadaki M, Shi-Wen X, Ponticos M, Khan K, Denton C, et al. Transforming growth factor-β-induced CUX1 isoforms are associated with fibrosis in systemic sclerosis lung fibroblasts. Biochem Biophys Rep. 2016;7:246–252. doi: 10.1016/j.bbrep.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ikeda T, Fragiadaki M, Shi-Wen X, Ponticos M, Khan K, Denton C, et al. Data on CUX1 isoforms in idiopathic pulmonary fibrosis lung and systemic sclerosis skin tissue sections. Data Brief. 2016;8:1377–1380. doi: 10.1016/j.dib.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics. 2016;17:259. doi: 10.1186/s12859-016-1140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li S, Wang Y, Zhang Y, Lu MM, DeMayo FJ, Dekker JD, et al. Foxp1/4 control epithelial cell fate during lung development and regeneration through regulation of anterior gradient 2. Development. 2012;139:2500–2509. doi: 10.1242/dev.079699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Osafo-Addo AD, Herzog EL. CCL2 and T cells in pulmonary fibrosis: an old player gets a new role. Thorax. 2017;72:967–968. doi: 10.1136/thoraxjnl-2017-210517. [DOI] [PubMed] [Google Scholar]
- 54. Zhang Y, He Q, Hu Z, Feng Y, Fan L, Tang Z, et al. Long noncoding RNA LINP1 regulates repair of DNA double-strand breaks in triple-negative breast cancer. Nat Struct Mol Biol. 2016;23:522–530. doi: 10.1038/nsmb.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang S, Jiang W, Zhang X, Lu Z, Geng Q, Wang W, et al. LINC-PINT alleviates lung cancer progression via sponging miR-543 and inducing PTEN. Cancer Med. 2020;9:1999–2009. doi: 10.1002/cam4.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moore BB, Moore TA. Viruses in idiopathic pulmonary fibrosis: etiology and exacerbation. Ann Am Thorac Soc. 2015;12:S186–S192. doi: 10.1513/AnnalsATS.201502-088AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sheng G, Chen P, Wei Y, Yue H, Chu J, Zhao J, et al. Viral infection increases the risk of idiopathic pulmonary fibrosis: a meta-analysis. Chest. 2020;157:1175–1187. doi: 10.1016/j.chest.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 59. Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet. 2019;20:437–455. doi: 10.1038/s41576-019-0128-0. [DOI] [PubMed] [Google Scholar]
- 60. Yamaguchi K, Iwamoto H, Horimasu Y, Ohshimo S, Fujitaka K, Hamada H, et al. AGER gene polymorphisms and soluble receptor for advanced glycation end product in patients with idiopathic pulmonary fibrosis. Respirology. 2017;22:965–971. doi: 10.1111/resp.12995. [DOI] [PubMed] [Google Scholar]
- 61. Lovgren AK, Kovacs JJ, Xie T, Potts EN, Li Y, Foster WM, et al. β-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci Transl Med. 2011;3:74ra23. doi: 10.1126/scitranslmed.3001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis Science Advances 20206eaba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thangavelu PU, Krenács T, Dray E, Duijf PH. In epithelial cancers, aberrant COL17A1 promoter methylation predicts its misexpression and increased invasion. Clin Epigenetics. 2016;8:120. doi: 10.1186/s13148-016-0290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. O’Dwyer DN, Moore BB. The role of periostin in lung fibrosis and airway remodeling. Cell Mol Life Sci. 2017;74:4305–4314. doi: 10.1007/s00018-017-2649-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Milger K, Yu Y, Brudy E, Irmler M, Skapenko A, Mayinger M, et al. Pulmonary CCR2+CD4+ T cells are immune regulatory and attenuate lung fibrosis development. Thorax. 2017;72:1007–1020. doi: 10.1136/thoraxjnl-2016-208423. [DOI] [PubMed] [Google Scholar]
- 66. Garner IM, Evans IC, Barnes JL, Maher TM, Renzoni EA, Denton CP, et al. Hypomethylation of the tnxb gene contributes to increased expression and deposition of tenascin-x in idiopathic pulmonary fibrosis [abstract] Am J Respir Crit Care Med. 2014;189:A3378. [Google Scholar]
- 67. Khalil N, Parekh TV, O’Connor R, Antman N, Kepron W, Yehaulaeshet T, et al. Regulation of the effects of TGF-β1 by activation of latent TGF-β1 and differential expression of TGF-β receptors (TβR-I and TβR-II) in idiopathic pulmonary fibrosis. Thorax. 2001;56:907–915. doi: 10.1136/thorax.56.12.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Emblom-Callahan MC, Chhina MK, Shlobin OA, Ahmad S, Reese ES, Iyer EP, et al. Genomic phenotype of non-cultured pulmonary fibroblasts in idiopathic pulmonary fibrosis. Genomics. 2010;96:134–145. doi: 10.1016/j.ygeno.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 69. Disayabutr S, Kim EK, Cha SI, Green G, Naikawadi RP, Jones KD, et al. Mir-34 miRNAs regulate cellular senescence in type ii alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Spagnolo P, Tzouvelekis A, Bonella F. The management of patients with idiopathic pulmonary fibrosis. Front Med (Lausanne) 2018;5:148. doi: 10.3389/fmed.2018.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agatonovic-Kustrin S, Morton D. In: Puri M, Pathak Y, Sutariya VK, Tipparaju S, Moreno W, editors. Boston, MA: Academic Press; 2016. Data mining in drug discovery and design; pp. 181–193. [Google Scholar]
- 72. Salisbury ML, Hewlett JC, Ding G, Markin CR, Douglas K, Mason W, et al. Development and progression of radiologic abnormalities in individuals at risk for familial interstitial lung disease. Am J Respir Crit Care Med. 2020;201:1230–1239. doi: 10.1164/rccm.201909-1834OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nakano Y, Yang IV, Walts AD, Watson AM, Helling BA, Fletcher AA, et al. MUC5B promoter variant rs35705950 affects MUC5B expression in the distal airways in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193:464–466. doi: 10.1164/rccm.201509-1872LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2017;57:91–99. doi: 10.1165/rcmb.2017-0046OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mansell G, Gorrie-Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 2019;20:366. doi: 10.1186/s12864-019-5761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]