

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal interstitial lung disease with limited therapeutic options. Current evidence suggests that IPF may be initiated by repeated epithelial injuries in the distal lung, which are followed by abnormal wound healing responses that occur because of intrinsic and extrinsic factors. Mechanisms contributing to chronic damage of the alveolar epithelium in IPF include dysregulated cellular processes such as apoptosis, senescence, abnormal activation of the developmental pathways, aging, and genetic mutations. Therefore, targeting the regenerative capacity of the lung epithelium is an attractive approach in the development of novel therapies for IPF. Endogenous lung regeneration is a complex process involving coordinated cross-talk among multiple cell types and reestablishment of a normal extracellular matrix environment. This review will describe the current knowledge of reparative epithelial progenitor cells in the alveolar region of the lung and discuss potential novel therapeutic approaches for IPF, focusing on endogenous alveolar repair.
Keywords: epithelium, lung, regeneration, stem cells
Chronic respiratory diseases are currently the third leading cause of death globally (1). The only restorative treatment for end-stage chronic respiratory diseases is lung transplantation, with a median survival of 6 years being shown (2). The interstitial lung diseases (ILDs) are a heterogeneous group of chronic respiratory diseases characterized by varying degrees of inflammation and fibrosis in the parenchyma. Idiopathic pulmonary fibrosis (IPF) is one of the most common forms of ILD, with an estimated incidence of three to nine cases per population of 100,000 per year in North America and Europe, with a median survival of 2–3 years being shown (3, 4). However, a steep increase in the incidence has been observed during recent years in several studies from various regions (4, 5).
The treatment of patients with IPF presents a clinical challenge because of the unpredictability of the disease’s development and its rapid progression. Current care aims at slowing the disease’s progression, extending the life expectancy, and improving the quality of life. Numerous targets have been and are currently being assessed in clinical trials as potential treatments for IPF, including receptor antagonists for ligands of key pathways and cytokines (6) (Table 1). However, many of these clinical trials have failed, and only two drugs are currently approved by the U.S. Food and Drug Administration for the treatment of IPF: nintedanib and pirfenidone. Neither of these drugs are able to completely stop the progression of disease, and both are associated with significant side effects (7, 8).
Table 1.
Summary of Key Completed and Ongoing IPF Clinical Trials Spanning Multiple Drug Modalities
| Type | Drug | Latest Known Trial Identifier | Latest Known Stage | Outcome | Mechanism of Action | Reference: Latest Phase/Additional Relevant |
|---|---|---|---|---|---|---|
| Small molecule | Pirfenidone | NCT00287716 | Approved | Positive | Not established | 187–190 |
| NCT00287729 | ||||||
| NCT01366209 | ||||||
| NCT00662038 | ||||||
| Nintedanib (BIBF1120) | NCT00514683 | Approved | Positive | Tyrosine kinase receptor antagonist | 191, 192 | |
| NCT01335464 | ||||||
| NCT01335477 | ||||||
| Yifenidone | NCT03092102 | Phase I | Completed | Not established | N/A | |
| Bosentan | NCT00071461 | Phase II/III | Variable | Endothelin receptor antagonist | 193, 194 | |
| NCT00391443 | Phase III | Negative | ||||
| Losartan | NCT00981747 | Phase II/III | Terminated | Angiotensin II receptor antagonist | 195 | |
| TD139/GB0139 | NCT03832946 | Phase II | Ongoing* | Galectin-1 and galectin-3 antagonist | 141, 142 | |
| BMS-986020 | NCT01766817 | Phase II | Completed | Lysophosphatidic acid receptor antagonist | 196 | |
| BMS-986278 | NCT04308681 | Phase II | Ongoing | Lysophosphatidic acid receptor antagonist | 197 | |
| Omipalisib (GSK2126458) | NCT01725139 | Phase I | No safety signals | Phosphatidylinositol 3-kinase antagonist | 198 | |
| Tanzisertib (CC-930) | NCT01203943 | Phase II | Terminated | JUN N-terminal kinase antagonist | 199 | |
| Imatinib | NCT00131274 | Phase II/III | Negative | Tyrosine kinase antagonist | 200 | |
| Warfarin | NCT00957242 | Phase III | Terminated | Anticoagulant | 201 | |
| Ziritaxestat (GLPG1690) | NCT03711162 | Phase III | Terminated | Autotaxin antagonist | 143, 144 | |
| NCT03733444 | ||||||
| GLPG-1205 | NCT03725852 | Phase II | Ongoing | G-protein–coupled receptor 84 antagonist | N/A | |
| Vismodegib (with pirfenidone) | NCT02168530 | Phase II | Terminated | Hedgehog pathway antagonist | 202 | |
| Tipelukast (MN-001) | NCT02503657 | Phase II | Ongoing | Leukotriene antagonist | N/A | |
| Sirolimus | NCT01462006 | N/A | Ongoing | mTOR antagonist | N/A | |
| HEC-68498 | NCT03502902 | Phase I | Ongoing | Phosphoinositide 3-kinase antagonist/mTOR antagonist | N/A | |
| Nalbuphine hydrochloride ER, T-111 | NCT04030026 | Phase II | Ongoing | Opioid receptor agonist | N/A | |
| IDL-2965 | NCT03949530 | Phase I | Ongoing | Integrin antagonist | N/A | |
| PLN-74809 | NCT04072315 | Phase II | Ongoing | Integrin antagonist | 203 | |
| BBT-877 | NCT03830125 | Phase I | Completed | Autotaxin inhibitor | 204 | |
| BLD-2660 | NCT04244825 | Phase II | Pending | Cysteine protease inhibitor | N/A | |
| ND-L02-s0201 (BMS-986263) | NCT03538301 | Phase II | Ongoing | Heat shock protein 47 antagonist | 205 | |
| CC-539 (CC-90001) | NCT03142191 | Phase II | Ongoing | JUN N-terminal kinase antagonist | 206 | |
| GDC-3280 | NCT02471859 | Phase I | Ongoing | Growth factor antagonist | 207 | |
| LT-1002 | UMIN000009572 | Phase II | Completed | Superoxide dismutase stimulant | N/A | |
| KD025 (SLx-2119) | NCT02688647 | Phase II | Ongoing | Rho-associated kinase 2 inhibitor | N/A | |
| RVT-1601 (PA101B) | NCT03864328 | Phase II | Ongoing | Inhaled cromolyn sodium | N/A | |
| PBI-4050 + pirfenidone | NCT02538536 | Phase II | Positive | Not established | 208 | |
| ZSP1603 | NCT03619616 | Phase I | Completed | Tyrosine kinase inhibitor | N/A | |
| NAC | NCT00650091 | Phase III | Negative | Antioxidant | 209 | |
| Nucleic acid | TRK-250 | NCT03727802 | Phase I | Ongoing | Growth factor antagonist | N/A |
| Protein-based therapeutic | IFN-γ | NCT00047645 | Phase III | Negative | Antiinflammatory | 210, 211 |
| NCT00075998 | Phase III | Terminated | ||||
| Etanercept | NCT00063869 | Phase II | Negative | TNF-α receptor antagonist | 212 | |
| PRM-151 | NCT02550873 | Phase II | Positive | Recombinant human pentraxin 2 | 165–167 | |
| LTI-03 | NCT04233814 | Phase I | Ongoing | Not established | N/A | |
| Antibody | Pamrevlumab (FG-3019) | NCT03955146 | Phase II | Ongoing | Anti–connective tissue growth factor | 168–170 |
| NCT04419558 | ||||||
| Ianalumab (VAY736) | NCT03287414 | Phase II | Ongoing | Anti–B-cell activator | N/A | |
| Simtuzumab (GS6624) | NCT01769196 | Phase II | Terminated | Anti–lysyl oxidase-like protein 2 | 213 | |
| NCT01759511 | ||||||
| Dectrekumab (QAX576) | NCT01266135 | Phase II | Terminated | Anti–IL-13 | N/A | |
| Fresolimumab (GC1008) | NCT00125385 | Phase I | Completed | Transforming growth factor-β neutralizing | N/A | |
| STX100 | NCT01371305 | Phase II | Completed | Anti–αvβ6 integrin | 214 | |
| NCT03573505 | Phase II | Terminated | ||||
| Carlumab (CNTO888) | NCT00786201 | Phase II | Terminated | Anti–chemokine (C-C motif) ligand 2 | 215 | |
| Cell therapy | Placenta-derived MSCs | NCT01385644 | Phase I | No safety signals | Not established | 216 |
| Autologous bone marrow–derived MSCs | NCT01919827 | Phase I | Completed | Not established | N/A | |
| Allogeneic bone marrow–derived MSCs | NCT02013700 | Phase I | No safety signals | Not established | 180 | |
| Allogeneic bone marrow–derived MSCs | NCT02594839 | Phase I/II | No safety signals | Not established | 217 | |
| Adipose-derived MSCs | N/A | Phase I | No safety signals | Not established | 178, 218 | |
| Autologous bronchial basal-cell transplant | NCT03153800 | Phase I | Ongoing | Not established | N/A | |
| Autologous lung stem-cell transplant | NCT04262167 | Phase I | Pending | Not established | N/A | |
| Autologous lung stem-cell transplant | NCT02745184 | Phase I/II | Pending | Not established | N/A | |
| Autologous SVF, autologous adipose-derived MSCs | NCT02135380 | Phase I/II | Unknown | Not established | N/A | |
| Other | NAC + prednisone + azathioprine | NCT00639496 | Phase III | Positive | Antiinflammatory | 219, 220 |
| NCT00650091 | Phase III | Negative | ||||
| IK-7001 | NCT01457781 | Phase II | Positive | Vasodilator | 221, 222 | |
| GENOSYL | NCT01265888 | Phase II | Ongoing | Vasodilator | N/A | |
| BI-1015550 | NCT03422068 | Phase I | Completed | Not established | N/A |
Definition of abbreviations: ER = extended release; IPF = idiopathic pulmonary fibrosis; MSC = mesenchymal stem cell; N/A = not applicable; NAC = N-acetyl cysteine; SVF = stromal vascular fraction.
Study ongoing with GB0139 (3 mg) as a monotherapy only.
Although early trials focused on inhibiting inflammatory processes or on modulation of aberrant fibroblast behavior, recent work has indicated that the distal epithelium is the site of initial injury in pulmonary fibrosis and that modulation of aberrant distal epithelial-cell behavior is sufficient to slow or reverse fibrosis in animal models (9–11). In addition, an increase in epithelium-associated biomarkers identified in the serum of patients with IPF was shown to correlate with disease severity and to be predictive of mortality (12). Thus, targeting and promoting regeneration of the distal epithelium has emerged as a potential and promising new approach for IPF.
This review will summarize the current knowledge of lung epithelial repair, focusing on the alveolar compartment and how it is involved in IPF onset and progression. Moreover, potential therapeutic approaches targeting the lung epithelium will be discussed.
The Structure of the Lung Epithelium
The lung is a dynamic and complex organ composed of ∼40 different cell types located in specific regions of the lung. In this section, we will briefly cover the structural organization of the lung and its different cell types in the different regions (depicted in Figure 1). In particular, recent evidence suggests aberrant reepithelialization of the distal lung and remodeled alveolar regions in IPF, with cells expressing phenotypic markers of cells that are normally found in the airways (13–16). We will first briefly introduce potential progenitors that have been identified to date for the airway or alveolar epithelium (summarized in Table 2) and will further discuss these populations in more detail in the context of IPF in later sections of this review.
Figure 1.
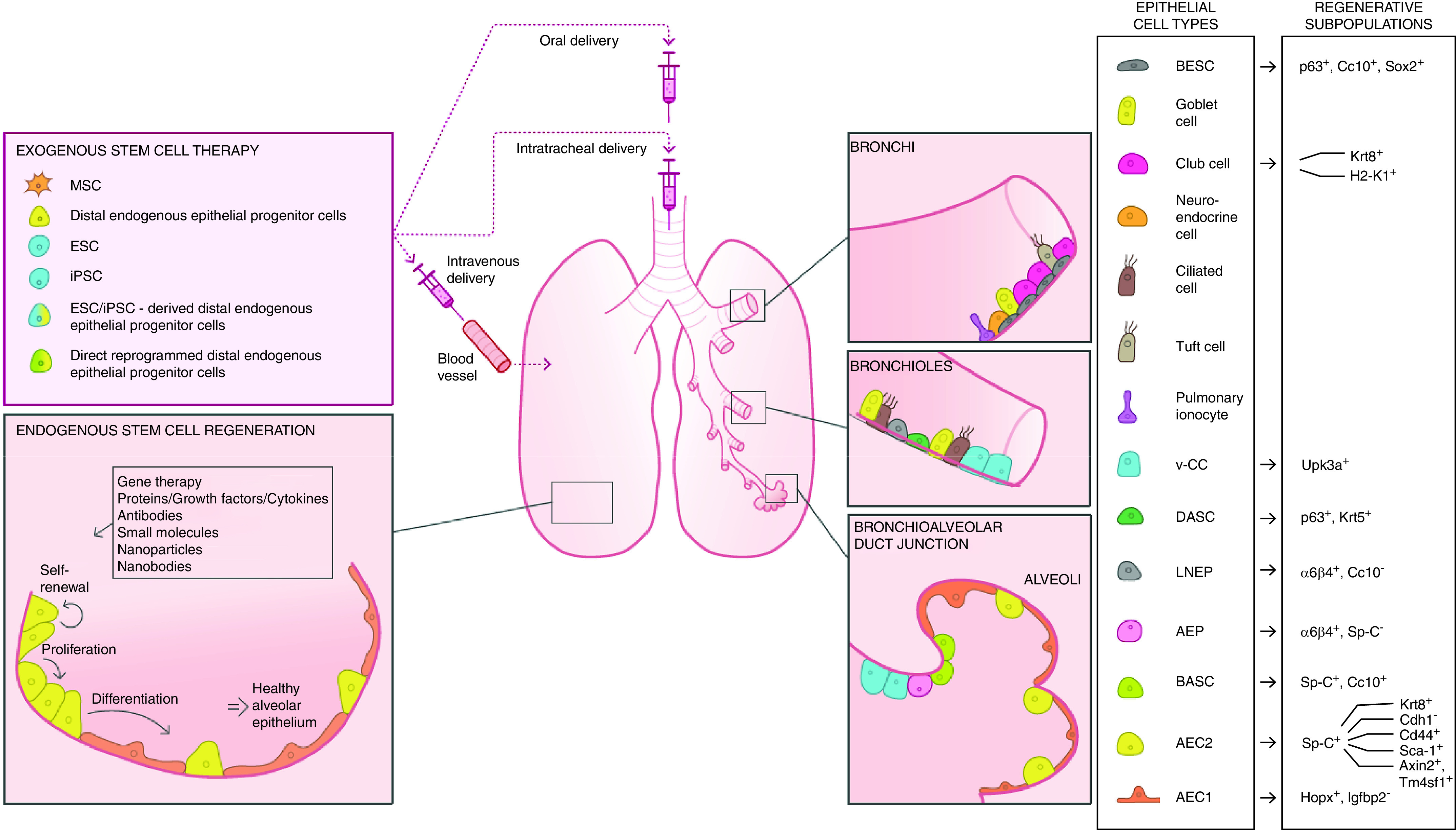
Strategies to promote therapeutic lung regeneration. Regenerative subpopulations have been found to reside in the bronchi (BESCs and subpopulations of club cells), bronchiolar epithelium (DASCs, LNEPs, and v-CCs), and alveoli (BASCs, AEPs, AEC2s, and subpopulations of AEC1s) in lung injury models as described in Table 2. Strategies to promote therapeutic lung regeneration include the exogenous administration of stem cells, including primary distal endogenous epithelial progenitor cells, multi- or pluripotent stem cells (MSCs, ESCs, iPSCs), stem cell–derived distal epithelial progenitor cells, and directly reprogrammed distal epithelial progenitor cells. Endogenous activation of distal epithelial progenitor cells to promote self-renewal, proliferation, and differentiation of the alveolar epithelium can be induced through modulation of the molecular environment (signaling pathways, extracellular matrix) or by directly targeting individual cell types. Potential future therapeutics include gene therapy (administration of oligonucleotides and gene editing); administration of peptides such as growth factors, cytokines, and proteins; inhibition of receptors and targets with antibodies; and administration of small molecules, nanoparticles, and nanobodies designed for specific targets of interest. AEC1 = alveolar epithelial type 1 cell; AEC2 = alveolar epithelial type 2 cell; AEP = alveolar epithelial progenitor cell; BASC = bronchioalveolar stem cell; BESC = bronchial epithelial stem cell; DASC = distal airway stem cell; ESC = embryonic stem cell; iPSC = induced pluripotent stem cell; LNEP = lineage-negative epithelial progenitor cell; MSC = mesenchymal stem cell; Sp-C = SFTPC; v-CC = variant club cell.
Table 2.
Putative Epithelial Stem-Cell and Progenitor-Cell Populations Contributing to Injury Repair in the Alveolar Region of the Lung
| Stem-Cell Type | Species | In Vivo Injury Model | Marker | Daughter Cell Type | Reference |
|---|---|---|---|---|---|
| AEC2 | Mouse, Human | Bleomycin (mouse) | Sp-C+ | AEC1, AEC2 | 41 |
| AEC2 | Rat | Hyperoxia | Sp-C+, Cdh1− | AEC2 (assessed proliferation but not differentiation potential) | 99 |
| AEC2 | Mouse | Homeostatic conditions | Sp-C+, Cd44+ | AEC1, AEC2 | 100 |
| AEC2 | Mouse | P. aeruginosa–induced pneumonia | Sp-C+, Sca-1+ | AEC1 | 101 |
| Wnt-responsive AEC2 | Mouse, Human | Influenza virus infection (mouse) | Sp-C+, Axin2+, Tm4sf1+ | AEC1, AEC2 | 102, 103 |
| AEC1 | Mouse | Pneumonectomy | Hopx+, Igfbp2− | AEC1, AEC2 | 104, 105 |
| AEP | Mouse | Bleomycin | α6β4+, Sp-C−, | AEP, AEC2, secretory cells | 106 |
| Alveolar/club cell–derived epithelial progenitor cell | Mouse | Bleomycin, influenza virus infection, LPS | Krt8+ | AEC1 | 32, 33 |
| BASC | Mouse | Naphthalene, bleomycin | Sp-C+, Cc10+ | BASC, AEC1, AEC2, secretory cells, ciliated cells | 35, 36 |
| BESC | Mouse | Bleomycin | p63+ Cc10+ Sox2+ | AEC1, AEC2 | 11 |
| DASC | Mouse | Influenza virus infection | p63+, Krt5+ | DASC, AEC1, AEC2, secretory cells | 24, 25 |
| LNEP | Mouse | Influenza virus infection | α6β4+, Cc10− | LNEP, AEC2, secretory cells | 107 |
| v-CC | Mouse | Naphthalene, bleomycin | Upk3a+ | AEC1, AEC2, secretory cells | 30 |
| Club cell–derived epithelial progenitor cell | Mouse | Bleomycin | H2-K1+ | AEC1, AEC2, airway epithelial cells | 31 |
Definition of abbreviations: AEC1 = alveolar epithelial type 1 cell; AEC2 = alveolar epithelial type 2 cell; AEP = alveolar epithelial progenitor cell; BASC = bronchioalveolar stem cell; BESC = bronchial epithelial stem cell; DASC = distal airway stem cell; Krt5 = cytokeratin 5; LNEP = lineage-negative epithelial progenitor cell; P. aeruginosa = Pseudomonas aeruginosa; Sp-C = SFTPC; v-CC = variant club cell.
The Airway Epithelium
Structurally, the proximal part of the lung is composed of the bronchi, which branch out into smaller bronchioles along the distal axis. The trachea and bronchi are lined by a pseudostratified epithelium, which is mainly composed of basal cells, secretory cells, and ciliated cells (17) (Figure 1). Although murine models are often used to study lung diseases, major differences in the cellular composition and architecture exist along the epithelium between humans and mice. Human airways are composed of a pseudostratified epithelium with basal cells extending from the main bronchi down to the distal bronchioles. In mice, the trachea has a pseudostratified epithelial organization similar to that of the large airways in humans, but the main bronchi in murine lungs are instead lined by a simple, cuboidal epithelium without basal cells (18).
Basal cells.
The basal cells are the progenitor cells of the proximal epithelium and are characterized by their expression of the transcription factor Trp-63 and Krt5 (cytokeratin 5) (19–21), although it has also been recognized that Krt14 is expressed in a subset of basal cells in mice and humans (22). Basal cells have an important role in maintaining bronchial epithelial homeostasis and serve as a critical pool of progenitor cells for repair of the injured epithelium (21, 22). In humans, evidence of basal cells as progenitors in the proximal lung came from the observation of similar mitochondrial DNA mutations in clones of differentiated basal, secretory, and ciliated cells in the bronchial epithelium (23). In mice, recent studies have demonstrated more extensive plasticity of the basal cell population, in which subsets of cells can generate distal epithelial cells upon injury (11, 24, 25) (Table 2). In addition to the rare neuroendocrine cells and tuft cells also present in the proximal lung, recent single-cell transcriptomic analyses have identified a novel proximal cell type termed the “pulmonary ionocyte,” which can be derived from tracheal murine basal cells and from human bronchial epithelial cells (26, 27).
Secretory cells.
Secretory cells are characterized by the expression of Scgb1a1 (secretoglobin family 1A member 1) (28). This group of cells is mainly composed of goblet cells and club cells. In humans, goblet cells are relatively abundant in the airways, whereas club cells increase in proportion in the distal bronchioles. Mice have a smaller proportion of goblet cells but have more club cells in the tracheobronchial region (18, 29). Club cells produce secretoglobin and provide protection to the airway epithelium against infections and oxidative stress (29). In mice, several recent studies have identified that subsets of club cells have the capacity to repair the airway and alveolar epithelium upon injury with naphthalene (30), bleomycin (30–33), influenza infection (32), or LPS (32) (Table 2). The goblet cells are responsible for producing mucus and play a vital role in the mucociliary clearance of the airways (18). The ciliated cells, characterized by the expression of the transcription factor Foxj1 (forkhead box J1), transport the mucus along the distal to proximal airway axis to be expectorated or swallowed through coordinated ciliary beating on the apical side (28, 34).
The Alveolar Epithelium
Distally, the bronchioles branch out into the alveoli. Human alveoli are formed via respiratory bronchioles, which can compose multiple alveolar ducts, whereas mouse airways open out into the alveoli through bronchioalveolar duct junctions. In mice, a specific reparative cell population termed “bronchioalveolar stem cells” (BASCs; Figure 1 and Table 2) has been identified in this region, and these cells can contribute to the repair of both the airway and alveolar epithelium upon injury with naphthalene and bleomycin (35, 36). In humans, however, BASCs or a similar cell subpopulation have not yet been identified.
The alveolar epithelium consists of alveolar epithelial type 1 cells (AEC1s) and alveolar epithelial type 2 cells (AEC2s). AEC1s facilitate the gas exchange process with their thin morphology, interfacing with the vascular endothelium (37). AEC2s are structurally cuboidal in shape and contain secretory vesicles (lamellar bodies) containing surfactant material such as SP-C (SFTPC). The secreted surfactant reduces the tension of the alveoli, preventing alveolar collapse upon expiration (38). Early studies of the rat distal epithelium in which AEC2s were labeled with [‘H]thymidine in development (39) and upon injury due to nitrogen oxide (40) were among the first to demonstrate that AEC2s act as progenitors and are able to self-renew and transdifferentiate into AEC1s. Later studies using lineage-tracing experiments in transgenic Sftpc-CreER;Rosa-Tm reporter mice, labeling AEC2s in vivo, confirmed that AEC2s act as progenitors that are able to self-renew and transdifferentiate into AEC1s, thereby maintaining the alveolar epithelium during homeostasis and injury (41, 42).
Dysregulation of the Alveolar Epithelium in IPF
Increasing evidence suggests that IPF arises as a consequence of repeated injury and failed repair of the alveolar epithelium, which is followed by an abnormal wound healing response. Histopathologically, typical abnormalities in the IPF lung include thickening of the alveolar walls, fibroblastic foci composed of myofibroblasts, and excessive extracellular matrix (ECM) deposition (43). Epithelial abnormalities include the presence of areas of honeycombing in which cells aberrantly express bronchiolar markers (e.g., bronchiolization), the presence of hyperplastic AEC2s, and the loss of AEC1s (44, 45). Functional reepithelialization of the fibrotic areas is significantly impaired, leading to a progressively damaged alveolar epithelium and loss in the surface area available for gas exchange. In this section, we will describe some of the processes associated with a dysregulated alveolar epithelium that are commonly observed in IPF. Several factors contributing to the derangement of the epithelium in IPF have been studied, including dysregulated cellular processes such as apoptosis, senescence, stress of the endoplasmic reticulum (ER) and genetic predispositions.
Senescence and Apoptosis of AEC2s
Senescence plays important roles in processes such as embryonic development (46, 47) and wound healing (48, 49). Senescent cells also naturally accumulate in tissues with age (50). Recent work has found that both fibroblasts and distal lung epithelial cells express increased amounts of senescent markers in animal models of fibrosis and in patients with IPF (16, 51). The distal epithelium in IPF lungs has an increased expression of well-known senescence-associated markers, including the tumor suppressor genes p53 and p21 (52). In particular, studies examining transcriptomic differences in human AEC2s derived from IPF lungs and healthy control lungs have predicted p53 to be one of the main transcription factors driving aberrant changes in fibrotic AEC2s (53). Several different potential mechanisms have been shown as factors contributing to the onset of the senescent cell phenotype, including DNA damage–induced senescence and replicative senescence. Furthermore, chronic activation of pathways such as the Wnt/β-catenin pathway have been shown to induce cellular senescence in AEC2s (51). Other soluble factors have also been associated with the induction of the senescent phenotype in AEC2s and may represent promising therapeutic targets. PAI-1 (plasminogen activation inhibitor 1; SERPINE1) is known to be upregulated in IPF, and its inhibition has been observed to reduce the expression of p53 and p21 in rat AEC2s in vitro upon bleomycin treatment. Furthermore, AEC2-specific deletion of PAI-1 in mice reduced bleomycin-induced fibrosis and the senescence of AEC2 (9). PAI-1 itself has also been implicated to be a major component of the senescence-associated secretory phenotype, which is also postulated to play a role in the development of fibrotic diseases. Senolytic compounds, drugs that selectively clear senescent cells, have been shown to be capable of inhibiting pulmonary fibrosis ex vivo and in vivo (16, 54); however, no clinical trials have yet been conducted to test this concept in patients with IPF. These compounds currently clear both alveolar and fibroblastic senescent cells and mediate their effects by both clearing senescent cells and reducing a variety of senescence-associated secretory phenotype components. Taken together, these studies highlight the significant involvement of senescence in IPF and suggest that targeting senescence by using senolytic drugs might be an attractive clinical approach.
In addition to senescence, aberrant activation of apoptotic pathways, including incomplete apoptosis, has been shown to play a major role in the pathogenesis of IPF. Early studies examining the epithelium in IPF lungs found greater proportions of apoptotic cells and increased expression of well-known apoptosis-associated markers, including bax (BCL2-associated X protein) and caspase-3 (55). A correlation between apoptosis and disease pathogenesis is supported by the observation that mice that are administered antibodies against the apoptotic ligand Fas and its receptor showed a decreased number of apoptotic cells and that these animals were resistant to pulmonary fibrosis induced by bleomycin (56). More recently, GSTP (glutathione-S-transferase π) was shown to be upregulated in AEC2s in IPF and to mediate S-glutathionylation of FAS (leading to increased apoptosis) (57). Gstp−/− animals did not develop pulmonary fibrosis after bleomycin administration, and animals receiving GSTP inhibitors after fibrosis established via bleomycin or adenovirus-driven TGF-β (transforming growth factor β) overexpression showed a reduction of the fibrotic burden as compared with control animals (57). Taken together, these studies highlight the potential of targeting induction of aberrant apoptotic pathways, including those prominently upregulated in AEC2s.
Genetic Predispositions
Genetic predispositions have been suggested to contribute to the dysfunction of epithelial cells and the development of both familial and sporadic cases of pulmonary fibrosis. One such predisposition is the SNP rs35705950 in the MUC5B (mucin 5B) promoter, which is responsible for regulating mucus secretion in the airways (58, 59). The polymorphism drives chronic hypersecretion of mucus, causing impaired mucociliary clearance and increased cough severity, as reported by patients with IPF (60, 61). Mouse models overexpressing Muc5b in the distal lung under the SFTPC promoter had a marked reduction in survival after administration of bleomycin, something that was intriguingly not observed in mice expressing Muc5b in the proximal lung under the Scgb1a1 promoter or in Muc5b−/− mice (62). Both SFTPC-Muc5b and Scgb1a1-Muc5b mice produced higher amounts of collagen and lung injury upon bleomycin challenge than the Muc5b−/− mice, suggesting that overexpression of Muc5b enhances the fibrotic response (62). However, the fact that the mice overexpressing Muc5b had no spontaneous phenotype (62) indicates that overexpression of the Muc5b gene contributes as a modifier rather than as a driver of lung fibrosis. In contrast to the observations in the animal models, although the rs35705950 polymorphism is a strong risk factor for the development of human IPF, the carriers appear to have a survival rate that is improved by twofold in comparison with that of noncarriers (63), suggesting protective effects of the overexpressed mucins in human IPF.
Genetic predispositions intimately associated with aging of cells and abnormal tissue repair have also been observed in epithelial cells of fibrotic lungs. These include mutations in hTERT (human telomerase), the enzyme responsible for maintaining the length of the telomeres, which cause accelerated shortening of the telomeres (64, 65). Short telomeres have been found both in cases of familial IPF (65) and in cases of sporadic IPF (66). Studies assessing the mechanisms by which hTERT mutations may contribute to cases of familial IPF identified two variants, V791I and V867M, that caused defects in telomere repeat addition processivity in vitro (67). Supporting evidence on the contribution of telomere dysfunction to pulmonary fibrosis has also been provided in SPC-Cre TRF1fl/fl mice modeling AEC2-localized deletion of the telomere-capping shelterin component Trf1 (telomeric repeat binding factor 1), causing spontaneous development of pulmonary fibrosis, or in Tert−/− mice, leading to development of pulmonary fibrosis after a low dose of bleomycin, which did not induce fibrosis in wild-type mice (68, 69). Notably, another study found that young Tert−/− mice showed loss of AEC2s and club cells, fibroblast activation, DNA damage, senescence, and apoptosis at levels comparable with changes observed in aged versus young wild-type animals (70). Furthermore, deletion of Trf2 in mice induced spontaneous senescence of AEC2s through signaling of the p53 pathway and induction of immune signaling (71). These mice also showed higher mortality upon challenge with bleomycin (71), supporting the role of telomere shortening in lung fibrosis.
Several studies have shown that restoration of telomerase activity may be a promising therapeutic target for pulmonary fibrosis. Administration of viral vectors containing telomerase (adeno-associated virus 9 [AAV9]-Tert) to bleomycin-injured mice deficient in Tert ameliorated the inflammation and contributed to the resolution of fibrosis (72), and the same type of treatment could also prevent the onset of profibrotic pathologies due to aging in both Tert-deficient and wild-type mice (70). These studies suggest that treatments that prevent the shortening of telomeres could be a potential approach in the context of human IPF.
ER Stress
Another abnormality intimately linked to aging is the accumulation of misfolded proteins. This accumulation leads to induction of ER stress, which is another process linked to IPF (73). In a mouse model with an inducible AEC2-specific knockout of the ER chaperone Grp78 (Grp78SCE), ER stress and the unfolded protein response were triggered, which in turn led to development of spontaneous lung fibrosis (74). Moreover, the AEC2s from the Grp78 knockout had elevated cleavage of caspase-3 and induced markers of senescence, including p53, γ-H2A.X, and p21 (74), demonstrating the link between ER stress and apoptosis and senescence. Interestingly, mice deficient in CHOP (C/EBP homologous protein), a transcriptional factor regulated by ER stress, were protected against alveolar epithelial-cell apoptosis after bleomycin exposure and hypoxic conditions, simultaneously suggesting a potential role of localized hypoxia in IPF (75). Markers indicating activation of the unfolded protein response due to ER stress have also been observed in AEC2s of patients with IPF, but whether this is causative of or a consequence of IPF has not yet been defined (76).
ER stress due to mutations in surfactant proteins.
Mutations in the surfactant proteins produced by AEC2s, including SP-C (77–79), SP-A1 (80), and SP-A2 (81), have been linked to the development of familial pulmonary fibrosis. Some of these mutations, such as the L188Q mutation of the SFTPC gene, are suggested to prevent the correct folding of the proteins in the ER, thereby causing ER stress and the triggering of the unfolded protein response (82–84). Although ER stress was observed in mice expressing the mutant form L188Q of the SFTPC gene, this was insufficient to cause the development of pulmonary fibrosis on its own (85). However, administration of bleomycin to these mice caused pulmonary fibrosis that was exaggerated compared with that of wild-type control animals, as assessed by fibrosis scores, collagen expression, the number of myofibroblasts, and measurements of lung compliance (85). In contrast to the L188Q mutation, the C121G Sftpc mutation causes both spontaneous lung fibrosis and retention of immaturely processed proteins in the ER of AEC2s, resulting in subsequent ER stress (86).
In addition to SP-C mutations that result in misfolded proteins and ER stress, other Sftpc mutants have been associated with spontaneous development of pulmonary fibrosis (87). Expression of Sftpc with a different IPF-linked mutation (SP-CI73T) in mice has been shown to cause excess accumulation of mutant SP-C in the plasma membrane and within early and late endosomes of AEC2s, which spontaneously drives rapid parenchymal injury associated with infiltration of immune cells and expression of inflammatory cytokines (88). This was followed by remodeling manifested by AEC2 hyperplasia, collagen deposition, and the appearance of α-SMA+ (ACTA2+) cells (88). A different study conducted by the same group demonstrated that the initial inflammatory phase induced by mutant SP-CI73T is characterized by the influx of immature CCR2+Ly6Chi macrophages (89). Taken together, these studies suggest potential genetic factors in IPF that might aid in determining which individuals are at risk for developing IPF at an early stage and furthermore highlights the surfactant proteins as possible therapeutic targets.
ER stress due to autophagy and mitochondrial dysfunction.
ER stress in AEC2s can also be triggered by defective autophagy. Autophagy is a cellular mechanism through which aberrantly processed proteins and organelles are delivered to lysosomes for degradation. This process is needed under conditions of an insufficient supply of energy to the cells; however, excessive activation of autophagy may initiate apoptosis (90). Inhibition of autophagy in cultures of human alveolar epithelial cells induced reprogramming associated with a mesenchymal phenotype (discussed more in detail in a later section) (91). In particular, impaired autophagy of mitochondrial content (mitophagy) and subsequent accumulation of dysfunctional mitochondria in AEC2s has been linked to the development of IPF (92). AEC2s from IPF lungs exhibit accumulation of dysfunctional mitochondria, a common characteristic of age-related diseases (93). The contribution of age-related mitochondrial dysfunction in AEC2s to pulmonary fibrosis is further supported by experimental evidence in vivo that demonstrates increased susceptibility to pulmonary fibrosis in mice deficient in the Pink1 (PTEN-induced kinase 1) gene, which is associated with development and accumulation of dysfunctional mitochondria (93). Boosting autophagy to restore the clearance of misprocessed organelles in AEC2s might thus be a potential approach for future therapeutics for IPF.
Epithelial-to-Mesenchymal Transition
Epithelial-to-mesenchymal transition (EMT) is a cellular process that has been well described in a variety of biological processes, such as embryonic development, which has also been discussed in the context of IPF pathogenesis. Seminal in vitro studies demonstrated that TGF-β promotes an EMT phenotype in cultured rat primary AEC2s by upregulating the expression of the mesenchymal markers α-SMA, type I collagen, and vimentin (94). Similar observations were also made in mouse primary AEC2s in vitro, and a mechanistic link to the SNAI transcription factors was demonstrated (95). However, the coexpression of EMT markers in AEC2s in IPF and in preclinical mouse models is controversial (42, 96, 97), making this process less accepted as being a driving fibrotic mechanism in human IPF. Nonetheless, recent single-cell data, as discussed further below, have identified aberrant distal epithelial cells in IPF (marked by their KRT5/KRT17 status), which coexpress classical epithelial markers but also have increased expression of mesenchymal genes, such as vimentin and tenascin C, as well as collagens, indicating an altered or EMT cellular phenotype as compared with that of the normal epithelium (13, 14). Potentially, future therapeutics may be developed to prevent the induction of these aberrant cell phenotypes in IPF or restoration to a more normal epithelial state; however, the functional contribution of these deranged cell types to human disease still needs further study.
Epithelial Reprogramming
Accumulating evidence indicates that the lung epithelium consists of diverse cell types along the airway epithelium and that individual cell subpopulations have differing functional characteristics depending on not only their physical location but also their disease or transitory state (98). The development of next-generation sequencing techniques has permitted transcriptomic analyses of bulk material and analysis at the single-cell level to provide detailed insight into the behavior of different cell types and subpopulations in the lung. Many studies have aimed to identify potential reparative subpopulations of epithelial progenitor cells contributing to alveolar epithelial repair by using experimental techniques such as lineage tracing of individual cell types and high-throughput sequencing techniques in combination with various experimental lung injury models (11, 24, 25, 30–33, 35, 36, 41, 42, 99–107). A summary of the described putative reparative subpopulations with the defining markers is given in Table 2, and their proposed location in the lung is given in Figure 1. Single-cell RNA sequencing analysis of human lung epithelial cells enriched for the 7AAD– (7-amino actinomycin D–negative), CD31–, CD45–, and CD326+ surface markers (EPCAM [epithelial-cell adhesion molecule]) first demonstrated that the distal lung tissue in patients with IPF harbors cell populations with a mixed molecular phenotype (i.e., an “indeterminate” phenotype) that coexpress markers of AEC1s, AEC2s, and conductive airway epithelial cells, indicating dysregulated repair of the epithelium in IPF tissue (53). Since then, and with improved sequencing depth, sorting, and throughput of cell numbers, several groups have described more detailed analyses at the single-cell level in lung tissue from IPF and other ILDs (e.g., the IPF Lung Cell Atlas [http://ipfcellatlas.com/]), which are available online for use in explorative analyses (13, 14, 108–110). These studies have further identified changes in the lung epithelial subpopulations in patients with IPF or ILD tissue as compared with healthy tissue.
Aberrant proximal patterning of alveolar epithelium.
Recent evidence suggests that the alveolar regions undergo aberrant reepithelialization and remodeling with cells of a proximal phenotype in IPF (13, 14). Adams and colleagues (13) observed an increased proportion of airway epithelial cells in IPF lung tissue and also observed a decline in AEC1s and AEC2s. Moreover, they identified “aberrant basaloid cells” that are exclusively present in IPF lung tissue, a subpopulation that coexpresses basal markers (e.g., TP63 and KRT17), mesenchymal markers (VIM, CDH2, FN1, etc.), and senescence markers (e.g., CDKN1A and CDKN2A). The study by Habermann and colleagues (14) also identified a previously unrecognized epithelial subpopulation capable of producing collagen and other ECM components, which was highly enriched in fibrotic lungs (not restricted to IPF) and was characterized by KRT5−/KRT17+ expression. Intriguingly, the aberrant basaloid cells identified by Adams and colleagues (13) also showed lack of KRT5 expression, suggesting that those two studies might have independently identified similar cell subpopulations. Similar to what Adams and colleagues (13) described, Morse and colleagues (110) observed an increased presence of airway epithelial cells in their patients with IPF as compared with healthy control subjects and in particular observed increased proliferation of TP63+/KRT5+ basal cells. Together, these studies suggest that epithelial cells with phenotypes normally observed in the airways contribute to the reepithelialization of the alveolar epithelium in IPF and that these aberrant cell subpopulations might be contributing to the tissue remodeling through production of ECM components and through senescence. However, the functional link among these subpopulations has not yet been established in human IPF, and the origin of these subpopulations has still not been fully elucidated. A potential source of these aberrant epithelial subpopulations includes airway epithelial cells that migrate to the scarred alveolar parts of the lung, as has been suggested to be the case in mice upon lung injury caused by bleomycin (11) and influenza infection (24, 25). Alternatively, alveolar epithelial cells could undergo reprogramming to aberrantly express mesenchymal markers and markers associated with the airway epithelium and downregulate alveolar markers as a consequence of the profibrotic environment, as has been suggested in earlier studies that used the bleomycin mouse model (111). Further studies focusing more closely on the alveolar epithelium in relevant model systems are needed to delineate the mechanisms of the aberrant proximal patterning of alveolar epithelial cells.
Transitional AEC2–AEC1 states.
Reyfman and colleagues (108) sequenced lung tissue from patients with ILD but stratified their data on the basis of the specific diagnosis. This study identified a subpopulation of AEC2s that is exclusively present in fibrotic lungs and has a characteristic differential molecular signature of the genes DMBT1, SERPINA1, and CHI3L1, which are markers that have previously been associated with pulmonary fibrosis and regulation of the immune response (108). The expression of these genes was, however, variable among the patients, and the contribution of this cell population is similar to that of the aberrant basaloid cell population in that it has not yet been validated functionally. A subsequent study by a different group further assessed the molecular phenotype of this unique fibrotic epithelial cluster and identified high expression of KRT8 (32). High expression of KRT8 has been linked to a transitional AEC2–AEC1 state and has been observed in subpopulations appearing in both in vivo lung injury models and IPF (32, 33). The KRT8+ subpopulation has been computationally inferred (i.e., via RNA velocity) to arise from both AEC2s and MHC-II+ airway club cells, demonstrating the contribution of both alveolar and airway cells to parenchymal regeneration in fibrosis (33). Moreover, this subpopulation is transcriptionally similar to the KRT5−/KRT17+ aberrant basaloid cells described by Adams and colleagues (13) (and likely also by Habermann and colleagues [14]) and to another dysfunctional, profibrotic AEC2 cell state identified in a mouse model of pneumonectomy-induced regeneration (112). Taken together, these studies suggest that there are similarities among the recently discovered dysfunctional subpopulations in IPF. Potentially, similar subpopulations may contribute to the pathogenesis of IPF through numerous mechanisms such as halted alveolar repair, ECM production, and senescence. Even though the similarities in the expressed markers and observed functions of these subpopulations are appealing and potentially suggest that observations in mice are translatable to human disease, there are caveats of such studies to be considered when making such comparisons. Identified subpopulations might be similar but are certainly not the same, as they depend on differences in the diagnosis (e.g., ILDs or IPF only); furthermore, isolation methods used to increase the number of cells used for sequencing and differences in sequencing and analysis strategies may lead to bias within or between studies. Nevertheless, the concept that the epithelium is aberrantly reprogrammed in human IPF and ILDs is strongly supported across multiple, independent analysis techniques and across cohorts.
Putative Reparative Subpopulations of Alveolar Epithelial Cells
The information obtained from patients with diagnosed IPF has mainly been obtained through transbronchial biopsies or through end-stage biopsies and has furthered our understanding of late-stage disease, but the factors contributing to the early development of IPF have remained challenging to elucidate by using patient samples. Understanding cell subpopulations that are activated or reprogrammed early and the molecular mechanisms driving these changes might contribute to earlier detection and potentially prevention of IPF and the development of potential reparative therapies. Compounds or therapies that promote normal reepithelialization or restoration of a normal epithelial state may be viable new approaches for the treatment of IPF if they can be started during earlier stages of the disease, at which time it is believed that the lung will be more amenable to treatment. The following section will discuss subpopulations of distal alveolar epithelial cells that are candidate targets to promote restoration or regeneration of normal lung tissue.
Subpopulations of AEC2s
Early studies identified differences in the reparative potential of AEC2s in rats exposed to hyperoxia on the basis of their expression of the epithelial marker E-cadherin (99). E-cadherin–negative AEC2s from animals exposed to hyperoxia showed greater proliferative potential and higher telomerase activity than the E-cadherin–positive subpopulation, suggesting that the E-cadherin–negative AEC2s might act as reparative progenitors upon lung injury (99). Furthermore, studies of the alveolar epithelium under homeostatic conditions in mice demonstrated the presence of a subpopulation of AEC2s expressing high levels of the marker CD44, which expressed higher proliferative capacity and augmented differentiation properties (100). Interestingly, this subpopulation increases in abundance in mice above 18 months of age but showed decreased progenitor properties in relation to younger mice, potentially explaining the impaired regenerative capacity of aged mice after lung injury in experimental models (100, 113, 114). Moreover, AEC2 populations showing particular responsiveness to the Wnt/β-catenin signaling pathway, a pathway that has been described to be implicated in the pathogenesis of IPF (115), have been identified in infection models of Pseudomonas aeruginosa–induced pneumonia and influenza virus in mice (101–103). Together, there is increasing evidence suggesting that the AEC2 population consists of several subpopulations with potentially different reparative capacities, which might differ between different types of lung injury.
Subpopulations of AEC1s
Interestingly, subpopulations contributing to alveolar epithelial repair after lung injury have also been identified in the AEC1 population (104, 105). In a mouse model of postnatal lung regeneration through pneumonectomy, two distinct subpopulations of AEC1s with variable expression of Hopx and Igfbp2 were identified and were found to differentially contribute to the increase in AEC2s after compensatory lung growth (105). The background for this particular study came from prior lineage-tracing experiments of Hopx+ AEC1s in mice, which demonstrated the ability of Hopx+ AEC1s to transdifferentiate into AEC2s after pneumonectomy, although this mechanism was rather rare (104). However, because the expression of Hopx has also been observed in transitional or intermediate cells (116), the true plasticity of the mature AEC1 population remains uncertain and needs to be further investigated.
Subpopulations of Airway Epithelial Cells
Several studies have also examined the contribution of additional epithelial-cell sources, particularly airway cells, for alveolar epithelial regeneration after lung injury (11, 24, 25, 30, 31, 33, 35, 36, 106, 107). As described earlier, mice are known to harbor a specific reparative subpopulation of BASCs that have been demonstrated to contribute to alveolar repair upon naphthalene- and bleomycin-induced lung injury (35, 36). In addition, secreted factors such as FGF10 (fibroblast growth factor 10) have been shown to mediate alveolar epithelial regeneration from bronchial epithelial stem cells (BESCs) in bleomycin-induced pulmonary fibrosis in mice and in ex vivo cultures of human IPF tissue (11). The contribution of airway epithelial cells contributing to alveolar repair upon injury in mice is further strengthened by single-cell RNA sequencing of β4+CD200+ lung epithelial cells at Day 9 after bleomycin injury (31). Cells with high expression of the marker H2-K1 clustered with the club-like cell population and were found to expand upon injury. Furthermore, RNA velocity analysis of this population predicted it to contribute to the formation of both AEC1s and AEC2s (31). Other studies have also identified the potential contribution of club cells to alveolar repair upon bleomycin and naphthalene injury in mice (30). Interestingly, the population of H2-K1 club-like cells was highly enriched for markers also present in the Krt8+ epithelial subpopulation identified by Struntz and colleagues (33), indicating they could be analyzing a similar subpopulation. Thus, multiple studies in mice have identified the potential of a more proximal population of airway cells contributing to repair of the alveolar epithelium in the context of murine pulmonary fibrosis. However, there is currently a limited amount of evidence in human tissue as to the contribution of airway cells to the repair of the alveolar epithelium. Humanized in vitro and ex vivo models of pulmonary fibrosis may offer an opportunity to study the potential contribution of different epithelial-cell populations to alveolar repair, but in the absence of methods that allow direct tracing or visualization of repair in humans in vivo, such analyses will always have some degree of limitation.
The Importance of Functional Repair in the Lung
Although several of the injurious stimuli described above promote expansion of suggested reparative subpopulations in the lung during the recovery phase, this is not always associated with functional repair. One such example was demonstrated in the mouse influenza virus infection model, in which local lung hypoxia through HIF1α (hypoxia-inducible factor 1α) promoted the differentiation of a KRT5+ basal-like cell population from p63+ lineage–negative progenitors that displayed upregulated core pathways associated with migration and squamous metaplasia that were suggestive of aberrant epithelial repair (117). The contribution of KRT5+/p63+ cells to alveolar repair upon influenza infection in mice has also been documented in earlier studies (24, 107). Intriguingly, this was in agreement with another observation in the same study, which was obtained by using single-cell analysis of fibrotic AEC2s that revealed a hypoxic subpopulation of KRT5+ basal-like cells (117). Deletion of HIF1α in mice promoted functional alveolar repair after influenza viral injury and was associated with more rapid increases in arterial oxygen saturation and improvement in restoration of the alveolar barrier (117), highlighting the importance of functional regeneration. Boosting the regenerative capacity of progenitor subpopulations is an attractive potential therapeutic strategy for IPF and other chronic lung diseases. However, before such approaches can be developed clinically, the presence of these cell types needs to be validated in human lung tissue, and their functional contribution to proper repair in human IPF needs to be confirmed. This requires further development of relevant experimental models of epithelial injury and repair that can be applied early in the drug discovery process to aid in the understanding of relevant mechanisms implicated in human IPF and in the early evaluation of potential novel therapeutic compounds.
Dysregulation of ECM
A central part of the pathogenesis of IPF is the increased deposition of ECM in the lung parenchyma, which leads to thickening of the basement membrane, fibrotic foci, and the appearance of honeycombing structures. Transcriptomic studies of bulk tissue relating observations from fibrotic mouse lungs to human IPF have identified differential expression of genes associated with ECM formation as well as degradation, such as MMP7 (matrix metalloprotease 7) expression. In particular, distinct populations of activated/matrix-producing fibroblasts and myofibroblasts have been recently identified in both murine and human pulmonary fibrosis (118–120). Activated fibroblasts and myofibroblasts are known to contribute to the excessive production and deposition of ECM components, leading to scarring and stiffening of the lung parenchyma (121). Furthermore, in addition to changes in the ECM composition in IPF, increased cross-linking of ECM proteins makes the matrix less susceptible to degradation, despite the increased presence of MMPs in IPF (121). Proteomic analysis of IPF and normal human and murine lungs from the bleomycin model of pulmonary fibrosis has indicated a change in the amount and type of proteins present in the insoluble fraction of the lung (i.e., ECM), indicating potentially irreversible changes during the development of pulmonary fibrosis (122–125).
Changes in ECM composition have been shown to affect lung epithelial repair in ex vivo models of both murine and human IPF (126). Interestingly, ECMs derived from patients with IPF have been shown to have a more significant impact on fibroblast behavior than the origin of the cells themselves, indicating that changes in the ECM are an important driver of disease pathogenesis (127, 128). In addition to changes in ECM composition, increased mechanical stimuli due to both increased tissue stiffness and changes in mechanical stretch have been shown to play roles in the activation of pathways known to be deranged in fibrosis. The IPF lung is known to have increased stiffness in the parenchyma as compared with normal lung tissue, which has been shown to result in increased fibroblast proliferation and matrix production (128, 129). The mechanotransducers YAP (yes-associated protein 1) and TAZ (transcriptional coactivator with PDZ-binding motif) have also been identified to be increased in fibroblasts of IPF lungs and to have a pathogenic role in disease progression (130). Although the effect of the matrix and stiffness on fibroblasts has been well described in IPF, less is known about their effects on alveolar epithelial cells. Mechanical stretch of fibrotic lung tissue ex vivo has been observed to trigger the release and activation of latent TGF-β1. Interestingly, this was not observed in healthy lung tissue, supporting the contribution of fibrosis-related stiffness to this mechanism (131). The role of the mechanical tension in the lung has also been studied in the context of alveolar regeneration upon injury, demonstrating a role of YAP/TAZ activation in this process (132), which has also been observed in the lung epithelium of patients with IPF (133). Further understanding of the role of the dysregulated extracellular environment in IPF, including more specifically understanding its role in promoting aberrant reepithelialization and fibrosis progression, may help to identify new potential therapeutic targets.
Repairing the Damaged Lung Epithelium: The Future of IPF Therapeutics?
Currently, a large number of ongoing clinical trials for IPF are including drugs developed for a multitude of different cellular targets or signaling pathways implicated in IPF. Examples of these targets are the lysophosphatidic acid, Wnt, hedgehog, and TNF-α and TGF-β pathways; integrins; galectin-3; matrix processing enzymes (e.g., LOX and LOXL); and several more (drug candidates targeting these and other pathways are listed in Table 1) (6, 134, 135). A large majority of the preclinical and clinical investigations for IPF therapies have focused on a pathological fibroblast behavior inhibition approach or an antiinflammatory approach, and the two currently approved therapies (i.e., pirfenidone and nintedanib) have been classified as belonging to the former approach. The latter approach (e.g., treatment with IFN-γ, etanercept, and triple treatment with N-acetyl cysteine, prednisone, and azathioprine) has so far not been a viable treatment strategy for IPF, as a vast majority of the clinical trials have reported negative results (with NCT00639496 being one exception).
The increased understanding of the significant involvement of lung epithelial cells in IPF has prompted the investigation of new approaches to target repair of the lung epithelium. This includes modulation of endogenous lung epithelial progenitor cells and their environment with small and large molecules, as well as exogenous delivery of stromal cells or lung epithelial progenitor cells or their secreted products to promote regeneration (136). These approaches have shown promise in vitro and in preclinical in vivo models (10, 137–140), and there are clinical trials of compounds targeting proteins expressed by epithelial cells that have reached phase II (NCT03832946, galectin-1 and galectin-3 inhibitor GB0139) or phase III (NCT03711162 and NCT03733444, autotaxin antagonist GLPG1690), albeit targeting of the epithelium was not part of the initial rationale for the implementation of these compounds (141–144). Autotaxin is known to convert lysophosphatidylcholine to lysophosphatidic acid, which can then elicit pleiotropic effects on numerous cell types, including fibroblasts, endothelial cells, and epithelial cells (145). GB0139 (previously referred to in the literature as TD139 and 33DFTG) is known to inhibit the activity of galectin-1 and galectin-3, which are expressed and secreted notably by macrophages but are also expressed by many other cell types, including epithelial cells (146, 147). GB0139 has also been shown to have direct effects on several cell types, including immune, mesenchymal, and epithelial cells (138, 148), but the exact role of galectin-1 and galectin-3 remains incompletely understood in IPF. However, safety concerns were recently raised at interim analyses for two IPF clinical trials, which has resulted in the termination of one clinical trial and in the termination of two arms of another trial. The ziritaxestat (GLPG1690) trial (NCT03711162 and NCT03733444) was terminated because of an unfavorable benefit–risk profile (149), whereas two treatment arms of the ongoing GB0139 trial (NCT03832946) were terminated because of an imbalance in the serious adverse experiences across the study groups identified in one arm using a high dose of GB0139 alone (10 mg) and in one arm using a low dose of GB0139 arm (3 mg), which included standard-of-care treatment with nintedanib or pirfenidone in combination with GB0139 (150). However, no major concerns were raised with the lower dose of GB0139 (3 mg) as a monotherapy, and this arm has continued. It will be important to more completely understand the exact reasons for the recommendation to terminate these clinical trials (or to terminate subgroups) and to identify whether the compound or target itself caused the unfavorable outcome or whether the fact that both compounds simultaneously target multiple cell types led to serious adverse events and to unfavorable risk–benefit profiles. Furthermore, as the low-dose arm of the GB0139 trial has not been terminated after this interim analysis, it will be important to watch how this trial continues to develop.
Currently, there are very few drugs in clinical trials for therapies that are aimed at targeting the epithelium. This is, in part, due to a lack of appropriate models that permit the study of the effects of potential therapeutic compounds on epithelial cells in lung fibrosis. Development of new models that allow for understanding epithelial-cell injury across different stages of IPF are urgently needed for the discovery of potential new compounds.
Targeting Endogenous Epithelial Regeneration Processes in the Lung by Using Exogenous Molecules or Peptides
An increasingly attractive perspective for emerging therapies for IPF is the pharmacological manipulation of pathways that restore endogenous lung repair or targeting of pathways that inhibit dysregulated regeneration. The reparative properties of endogenous progenitor cells rely heavily on appropriate interactions with their environment, including interactions with the mesenchyme, ECM, and molecular signaling. Many of the signaling pathways normally implicated in lung development are also activated during and after lung damage, thereby promoting tissue repair. Chronic aberrant activation of several of these pathways has, however, been associated with the development of chronic lung diseases. Although we now have extensive knowledge about the aberrantly activated signaling pathways and how they contribute to the pathogenesis of IPF, several of these pathways intersect and influence each other (151). This makes the differential targeting of these pathways a significant challenge, and we may need combinations of therapeutics to overcome this hurdle. Likely, modulation of the profibrotic environment in combination with stimulation of reparative endogenous progenitor cells will be necessary to promote therapeutic lung regeneration in IPF (Figure 1 and Table 1 provide an overview of approaches explored to date).
Even though pharmacological targeting of the distal epithelium is a relatively new concept, some of the compounds originally designed to target nonepithelial cells have been shown to be able to reverse pathological effects on AEC2s and phenotypic markers of AEC2s (152). Both pirfenidone and nintedanib have recently been shown to have an effect on epithelial-cell phenotypes when evaluated in an ex vivo precision-cut lung slice (PCLS) model with induction of early fibrotic remodeling via a cocktail of growth factors (152, 153); however, how similar this injury model is to IPF is not yet known. Alternatively, other work has evaluated potential compounds in explanted IPF tissue, representing end-stage disease (154, 155). How relevant either the PCLS approach or even the use of TGF-β stimulation alone on PCLSs might be in predicting clinical efficacy remains unknown. However, several omics-based approaches have recently been optimized for PCLSs and will most likely accelerate the characterization of this ex vivo model (156–159). In addition, the use of other small molecules in phase I and phase II trials (e.g., GB0139) and the use of direct modulation of targets expressed in epithelial cells (e.g., JNK) have been shown to be capable of regulating epithelial-cell behavior (126, 138, 160). This highlights the need for further characterizing both the molecular and cellular targets of drugs in development for IPF, which has not been done for the majority of drugs listed in Table 1. Determining the cell-specific responses for IPF treatment strategies may increase the success rate of translating promising drugs into the clinic. The rapid development of novel in vitro/ex vivo models for IPF, including those in which human cell types can be selectively built into the model (e.g., de- and recellularization models, organoid models, and fibrosis on a chip) will likely shed more light on this matter (161–163).
Although industry has significant experience with developing small molecules such as nintedanib and pirfenidone for therapeutic purposes, other strategies are needed to modify targets such as genes or to reach a target specific to a certain cell type. In this regard, emerging approaches such as nanoparticle- or nanobody-based therapies may aid in directing pharmacological agents to their intended cell type through the use of surface receptors (164). This technology is particularly well suited for delivery of molecules such as small molecules or nucleic acids (164). Other strategies that are currently explored are depicted in Figure 1 and Table 1 and include the delivery of protein-based molecules such as recombinant human pentraxin 2 (NCT02550873) (165–167) and antibodies such as Pamrevlumab (FG-3019, NCT03955146, and NCT04419558), an anti–connective tissue growth factor antibody (168–170). Further emergence of novel therapeutic strategies and refinement of targets as well as potential delivery directly to the lung are needed for the development of successful therapeutics for IPF.
Exogenous Stem Cell–based Therapies for Lung Regeneration and IPF
Exogenous stem-cell therapies have shown promise in hematological disorders (171, 172), and clinical applications for the lung is an area of active research (173, 174). In particular, investigation has been done on the use of mesenchymal stem cells (MSCs), which are multipotent cells with remarkable immunomodulatory and migratory properties that can be isolated from multiple tissues, including bone marrow, peripheral blood, adipose tissue, skeletal muscle, amniotic fluid, and the lung (175, 176). In IPF, MSCs have been explored with the aim of modifying fibrosis development and progression (177). In preclinical animal models, the administration of MSCs resulted in improved histopathology, reduced lung collagen levels, and decreased neutrophil counts in bronchoalveolar lavage fluid (175). Administration of MSCs isolated from adipose tissue (178), the placenta (179), or bone marrow (180) in clinical trials proved to be safe and well tolerated by patients, with no worsening fibrosis being observed (Table 1). However, future clinical studies that examine efficacy are needed. A number of studies have also investigated the approach of delivering distal airway stem cells (10), differentiated AEC2s from embryonic stem cells (181), and induced pluripotent stem cells (182) or undifferentiated induced pluripotent stem cells (183, 184). These approaches have been evaluated preclinically and have shown promising effects in terms of tolerability and reduced collagen deposition and inflammation (177). Intratracheal, allogeneic AEC2 transplantation combined with immunosuppression and prophylactic drug treatment has been performed in a small cohort of patients with IPF (n = 16) with moderate and progressive disease. The AEC2 transplantation was determined to be safe, as there were no adverse side effects observed (185). Although the observations from the preclinical studies and clinical studies seem promising for further therapeutic exploration and development, at present, no approved cell-based therapies for chronic lung diseases are available (174).
Conclusions
To prevent chronic lung diseases from climbing even higher on the list of the leading causes of death globally, novel innovative therapeutic approaches are needed. Although in utero targeting of the genetic mutations leading to IPF may be a possibility (186), not all patients with IPF or other ILDs have identifiable genetic mutations. Therefore, promoting endogenous lung regeneration is a promising strategy with potential wide-scale applicability to reversing the disease progression of IPF and other chronic lung diseases. By targeting endogenous repair, processes that normally support homeostatic maintenance and repair could be stimulated directly in patients, thereby avoiding the potential issues associated with exogenous cell delivery. However, endogenous progenitor-cell stimulation might cause safety issues such as neoplasia if not carefully balanced, and pharmacological interventions aimed at endogenous epithelial progenitor cells may have detrimental effects on other cell types if delivered without specifically targeting deranged epithelial cells. Another potential issue may lie in ensuring that the mechanisms induced by the pharmacological interventions are sustained, especially when targeting subpopulations of cells that might be present only in the injured lung. To avoid these safety concerns about an otherwise exciting therapeutic approach, further research is needed to better understand homeostatic mechanisms and how proliferation is regulated in the healthy lung. Developing innovative models of epithelial injury and repair will aid in better understanding of the molecular mechanisms implicated in chronic lung diseases such as IPF. Furthermore, the characterization of reparative progenitor cells in chronic lung diseases will help further our understanding of the complex process of lung repair and will aid in the design of novel therapies. In addition, development of new approaches and refinement of clinical delivery strategies will increase the accessibility by novel therapeutics of their targets. We are heading toward an exciting era of addressing the unmet clinical need of patients with IPF with novel and successful therapeutic approaches, and curing patients with chronic lung diseases in the future is the aim.
Acknowledgments
Acknowledgment
The authors thank Dr. Petra Hazon (AstraZeneca) for editorial support and the graphic designers from Xerox Sverige AB for the graphic realization of Figure 1.
Footnotes
Supported by AstraZeneca; the Knut and Alice Wallenberg Foundation (PA2016-1522 [D.E.W.]); the Medical Faculty at Lund University, Region Skåne (D.E.W.); and the Swedish Research Council (registration number 2018-02352 [D.E.W.]).
Author Contributions: V.A.P., J.S., M.G.B., D.E.W., and L.A.M. contributed to the development of the manuscript, including direction of content, review, revision of all drafts, and approval of the final draft.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0476TR on June 15, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.The top 10 causes of death. Geneva, Switzerland: World Health Organization; 2018https://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death [Google Scholar]
- 2. Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, et al. The registry of the International Society for Heart and Lung Transplantation. Thirty-second official adult lung and heart-lung transplantation report: 2015; focus theme: early graft failure. J Heart Lung Transplant. 2015;34:1264–1277. doi: 10.1016/j.healun.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 3. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795–806. doi: 10.1183/09031936.00185114. [DOI] [PubMed] [Google Scholar]
- 4. Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther. 2018;35:724–736. doi: 10.1007/s12325-018-0693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 6. Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov. 2017;16:755–772. doi: 10.1038/nrd.2017.170. [DOI] [PubMed] [Google Scholar]
- 7. Galli JA, Pandya A, Vega-Olivo M, Dass C, Zhao H, Criner GJ. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology. 2017;22:1171–1178. doi: 10.1111/resp.13024. [DOI] [PubMed] [Google Scholar]
- 8. Ogura T, Taniguchi H, Azuma A, Inoue Y, Kondoh Y, Hasegawa Y, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382–1392. doi: 10.1183/09031936.00198013. [DOI] [PubMed] [Google Scholar]
- 9. Jiang C, Liu G, Luckhardt T, Antony V, Zhou Y, Carter AB, et al. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell. 2017;16:1114–1124. doi: 10.1111/acel.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shi Y, Dong M, Zhou Y, Li W, Gao Y, Han L, et al. Distal airway stem cells ameliorate bleomycin-induced pulmonary fibrosis in mice. Stem Cell Res Ther. 2019;10:161. doi: 10.1186/s13287-019-1257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan T, Volckaert T, Redente EF, Hopkins S, Klinkhammer K, Wasnick R, et al. FGF10-FGFR2B signaling generates basal cells and drives alveolar epithelial regeneration by bronchial epithelial stem cells after lung injury. Stem Cell Reports. 2019;12:1041–1055. doi: 10.1016/j.stemcr.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 2017;5:946–955. doi: 10.1016/S2213-2600(17)30430-7. [DOI] [PubMed] [Google Scholar]
- 13. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Korfei M, Schmitt S, Ruppert C, Henneke I, Markart P, Loeh B, et al. Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J Proteome Res. 2011;10:2185–2205. doi: 10.1021/pr1009355. [DOI] [PubMed] [Google Scholar]
- 16. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J. 2017;50:1602367. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mercer RR, Russell ML, Roggli VL, Crapo JD. Cell number and distribution in human and rat airways. Am J Respir Cell Mol Biol. 1994;10:613–624. doi: 10.1165/ajrcmb.10.6.8003339. [DOI] [PubMed] [Google Scholar]
- 18. Rock JR, Randell SH, Hogan BLM. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3:545–556. doi: 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Daniely Y, Liao G, Dixon D, Linnoila RI, Lori A, Randell SH, et al. Critical role of p63 in the development of a normal esophageal and tracheobronchial epithelium. Am J Physiol Cell Physiol. 2004;287:C171–C181. doi: 10.1152/ajpcell.00226.2003. [DOI] [PubMed] [Google Scholar]
- 20. Schoch KG, Lori A, Burns KA, Eldred T, Olsen JC, Randell SH. A subset of mouse tracheal epithelial basal cells generates large colonies in vitro. Am J Physiol Lung Cell Mol Physiol. 2004;286:L631–L642. doi: 10.1152/ajplung.00112.2003. [DOI] [PubMed] [Google Scholar]
- 21. Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. In vivo differentiation potential of tracheal basal cells: evidence for multipotent and unipotent subpopulations. Am J Physiol Lung Cell Mol Physiol. 2004;286:L643–L649. doi: 10.1152/ajplung.00155.2003. [DOI] [PubMed] [Google Scholar]
- 22. Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teixeira VH, Nadarajan P, Graham TA, Pipinikas CP, Brown JM, Falzon M, et al. Stochastic homeostasis in human airway epithelium is achieved by neutral competition of basal cell progenitors. eLife. 2013;2:e00966. doi: 10.7554/eLife.00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147:525–538. doi: 10.1016/j.cell.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zuo W, Zhang T, Wu DZA, Guan SP, Liew A-A, Yamamoto Y, et al. p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nature. 2015;517:616–620. doi: 10.1038/nature13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560:377–381. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560:319–324. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rawlins EL, Hogan BLM. Epithelial stem cells of the lung: privileged few or opportunities for many? Development. 2006;133:2455–2465. doi: 10.1242/dev.02407. [DOI] [PubMed] [Google Scholar]
- 29.Harkema JR, Nikula KJ, Haschek WM. In: 3rd. ed. Wallig MA, Haschek WM, Rousseaux CG, Bolon B, editors. Cambridge, MA: Academic Press; 2018. Respiratory system; pp. 351–393. [Google Scholar]
- 30. Guha A, Deshpande A, Jain A, Sebastiani P, Cardoso WV. Uroplakin 3a+ cells are a distinctive population of epithelial progenitors that contribute to airway maintenance and post-injury repair. Cell Rep. 2017;19:246–254. doi: 10.1016/j.celrep.2017.03.051. [DOI] [PubMed] [Google Scholar]
- 31. Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell. 2020;26:346–358.e4. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, et al. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med. 2020;201:1443–1447. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun. 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 35. Kim CFB, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 36. Liu Q, Liu K, Cui G, Huang X, Yao S, Guo W, et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat Genet. 2019;51:728–738. doi: 10.1038/s41588-019-0346-6. [DOI] [PubMed] [Google Scholar]
- 37. Lee J-H, Rawlins EL. Developmental mechanisms and adult stem cells for therapeutic lung regeneration. Dev Biol. 2018;433:166–176. doi: 10.1016/j.ydbio.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 38. Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med. 2014;20:822–832. doi: 10.1038/nm.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adamson IY, Bowden DH. Derivation of type 1 epithelium from type 2 cells in the developing rat lung. Lab Invest. 1975;32:736–745. [PubMed] [Google Scholar]
- 40. Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol. 1973;70:175–198. [PMC free article] [PubMed] [Google Scholar]
- 41. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tiitto L, Bloigu R, Heiskanen U, Pääkkö P, Kinnula VL, Kaarteenaho-Wiik R. Relationship between histopathological features and the course of idiopathic pulmonary fibrosis/usual interstitial pneumonia. Thorax. 2006;61:1091–1095. doi: 10.1136/thx.2005.055814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8:e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, et al. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax. 2011;66:651–657. doi: 10.1136/thx.2010.151555. [DOI] [PubMed] [Google Scholar]
- 46. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 47. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 48. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, Von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–323. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 51. Lehmann M, Hu Q, Hu Y, Hafner K, Costa R, van den Berg A, et al. Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell Signal. 2020;70:109588. doi: 10.1016/j.cellsig.2020.109588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, et al. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996;154:477–483. doi: 10.1164/ajrccm.154.2.8756825. [DOI] [PubMed] [Google Scholar]
- 53. Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 56. Kuwano K, Hagimoto N, Kawasaki M, Yatomi T, Nakamura N, Nagata S, et al. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J Clin Invest. 1999;104:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McMillan DH, van der Velden JLJ, Lahue KG, Qian X, Schneider RW, Iberg MS, et al. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase π. JCI Insight. 2016;1:e85717. doi: 10.1172/jci.insight.85717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y, Noth I, Garcia JGN, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–1577. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scholand MB, Wolff R, Crossno PF, Sundar K, Winegar M, Whipple S, et al. Severity of cough in idiopathic pulmonary fibrosis is associated with MUC5 B genotype. Cough. 2014;10:3. doi: 10.1186/1745-9974-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang IV, Fingerlin TE, Evans CM, Schwarz MI, Schwartz DA. MUC5B and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2015;12:S193–S199. doi: 10.1513/AnnalsATS.201503-110AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9:5363. doi: 10.1038/s41467-018-07768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Peljto AL, Zhang Y, Fingerlin TE, Ma S-F, Garcia JGN, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–2239. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 66. Alder JK, Chen JJL, Lancaster L, Danoff S, Su SC, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Alder JK, Cogan JD, Brown AF, Anderson CJ, Lawson WE, Lansdorp PM, et al. Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet. 2011;7:e1001352. doi: 10.1371/journal.pgen.1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015;12:286–299. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 69. Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1:e86704. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Piñeiro-Hermida S, Autilio C, Martínez P, Bosch F, Pérez-Gil J, Blasco MA. Telomerase treatment prevents lung profibrotic pathologies associated with physiological aging. J Cell Biol. 2020;219:e202002120. doi: 10.1083/jcb.202002120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci USA. 2015;112:5099–5104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Povedano JM, Martinez P, Serrano R, Tejera Á, Gómez-López G, Bobadilla M, et al. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife. 2018;7:e31299. doi: 10.7554/eLife.31299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest. 2018;128:64–73. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Borok Z, Horie M, Flodby P, Wang H, Liu Y, Ganesh S, et al. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med. 2020;201:198–211. doi: 10.1164/rccm.201902-0451OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Burman A, Kropski JA, Calvi CL, Serezani AP, Pascoalino BD, Han W, et al. Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight. 2018;3:e99543. doi: 10.1172/jci.insight.99543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng D-S, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 77. van Moorsel CHM, van Oosterhout MFM, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJT, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med. 2010;182:1419–1425. doi: 10.1164/rccm.200906-0953OC. [DOI] [PubMed] [Google Scholar]
- 78. Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 79. Thomas AQ, Lane K, John Phillips I, Prince M, Markin C, Speer M, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 80. Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet. 2016;25:1457–1467. doi: 10.1093/hmg/ddw014. [DOI] [PubMed] [Google Scholar]
- 81. Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol. 2007;293:L720–L729. doi: 10.1152/ajplung.00025.2007. [DOI] [PubMed] [Google Scholar]
- 84. Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. 2005;32:521–530. doi: 10.1165/rcmb.2005-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA. 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, et al. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight. 2019;4:e126125. doi: 10.1172/jci.insight.126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Thurm T, Kaltenborn E, Kern S, Griese M, Zarbock R. SFTPC mutations cause SP-C degradation and aggregate formation without increasing ER stress. Eur J Clin Invest. 2013;43:791–800. doi: 10.1111/eci.12107. [DOI] [PubMed] [Google Scholar]
- 88. Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest. 2018;128:4008–4024. doi: 10.1172/JCI99287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Venosa A, Katzen J, Tomer Y, Kopp M, Jamil S, Russo SJ, et al. Epithelial expression of an interstitial lung disease-associated mutation in surfactant protein-C modulates recruitment and activation of key myeloid cell populations in mice. J Immunol. 2019;202:2760–2771. doi: 10.4049/jimmunol.1900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 91. Hill C, Li J, Liu D, Conforti F, Brereton CJ, Yao L, et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 2019;10:591. doi: 10.1038/s41419-019-1820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One. 2015;10:e0121246. doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bueno M, Lai Y-C, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-β1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jayachandran A, Königshoff M, Yu H, Rupniewska E, Hecker M, Klepetko W, et al. SNAI transcription factors mediate epithelial-mesenchymal transition in lung fibrosis. Thorax. 2009;64:1053–1061. doi: 10.1136/thx.2009.121798. [DOI] [PubMed] [Google Scholar]
- 96. Yamada M, Kuwano K, Maeyama T, Hamada N, Yoshimi M, Nakanishi Y, et al. Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol. 2008;129:453–462. doi: 10.1007/s00418-008-0388-9. [DOI] [PubMed] [Google Scholar]
- 97. Marmai C, Sutherland RE, Kim KK, Dolganov GM, Fang X, Kim SS, et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;301:L71–L78. doi: 10.1152/ajplung.00212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Reddy R, Buckley S, Doerken M, Barsky L, Weinberg K, Anderson KD, et al. Isolation of a putative progenitor subpopulation of alveolar epithelial type 2 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L658–L667. doi: 10.1152/ajplung.00159.2003. [DOI] [PubMed] [Google Scholar]
- 100. Chen Q, Suresh Kumar V, Finn J, Jiang D, Liang J, Zhao YY, et al. CD44high alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am J Physiol Lung Cell Mol Physiol. 2017;313:L41–L51. doi: 10.1152/ajplung.00564.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liu Y, Kumar VS, Zhang W, Rehman J, Malik AB. Activation of type II cells into regenerative stem cell antigen-1+ cells during alveolar repair. Am J Respir Cell Mol Biol. 2015;53:113–124. doi: 10.1165/rcmb.2013-0497OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jain R, Barkauskas CE, Takeda N, Bowie EJ, Aghajanian H, Wang Q, et al. Plasticity of Hopx+ type I alveolar cells to regenerate type II cells in the lung. Nat Commun. 2015;6:6727. doi: 10.1038/ncomms7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang Y, Tang Z, Huang H, Li J, Wang Z, Yu Y, et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc Natl Acad Sci USA. 2018;115:2407–2412. doi: 10.1073/pnas.1719474115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, et al. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest. 2011;121:2855–2862. doi: 10.1172/JCI57673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. McDonough JE, Ahangari F, Li Q, Jain S, Verleden SE, Herazo-Maya J, et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight. 2019;4:131597. doi: 10.1172/jci.insight.131597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. 2019;54:1802441. doi: 10.1183/13993003.02441-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell. 2020;180:107–121, e17. doi: 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 113. Redente EF, Jacobsen KM, Solomon JJ, Lara AR, Faubel S, Keith RC, et al. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;301:L510–L518. doi: 10.1152/ajplung.00122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sueblinvong V, Neujahr DC, Mills ST, Roser-Page S, Ritzenthaler JD, Guidot D, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. 2012;344:41–51. doi: 10.1097/MAJ.0b013e318234c132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chilosi M, Poletti V, Zamò A, Lestani M, Montagna L, Piccoli P, et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162:1495–1502. doi: 10.1016/s0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Liebler JM, Marconett CN, Juul N, Wang H, Liu Y, Flodby P, et al. Combinations of differentiation markers distinguish subpopulations of alveolar epithelial cells in adult lung. Am J Physiol Lung Cell Mol Physiol. 2016;310:L114–L120. doi: 10.1152/ajplung.00337.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol. 2017;19:904–914. doi: 10.1038/ncb3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep. 2018;22:3625–3640. doi: 10.1016/j.celrep.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, et al. Defining the activated fibroblast population in lung fibrosis using single-cell sequencing. Am J Respir Cell Mol Biol. 2019;61:74–85. doi: 10.1165/rcmb.2018-0313OC. [DOI] [PubMed] [Google Scholar]
- 120. Tsukui T, Sun K-H, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun. 2020;11:1920. doi: 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Upagupta C, Shimbori C, Alsilmi R, Kolb M. Matrix abnormalities in pulmonary fibrosis. Eur Respir Rev. 2018;27:180033. doi: 10.1183/16000617.0033-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Schiller HB, Fernandez IE, Burgstaller G, Schaab C, Scheltema RA, Schwarzmayr T, et al. Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair. Mol Syst Biol. 2015;11:819. doi: 10.15252/msb.20156123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Schiller HB, Mayr CH, Leuschner G, Strunz M, Staab-Weijnitz C, Preisendörfer S, et al. Deep proteome profiling reveals common prevalence of MZB1-positive plasma B cells in human lung and skin fibrosis. Am J Respir Crit Care Med. 2017;196:1298–1310. doi: 10.1164/rccm.201611-2263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sokocevic D, Bonenfant NR, Wagner DE, Borg ZD, Lathrop MJ, Lam YW, et al. The effect of age and emphysematous and fibrotic injury on the re-cellularization of de-cellularized lungs. Biomaterials. 2013;34:3256–3269. doi: 10.1016/j.biomaterials.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Åhrman E, Hallgren O, Malmström L, Hedström U, Malmström A, Bjermer L, et al. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J Proteomics. 2018;189:23–33. doi: 10.1016/j.jprot.2018.02.027. [DOI] [PubMed] [Google Scholar]
- 126. van der Velden JL, Wagner DE, Lahue KG, Abdalla ST, Lam Y-W, Weiss DJ, et al. TGF-β1-induced deposition of provisional extracellular matrix by tracheal basal cells promotes epithelial-to-mesenchymal transition in a c-Jun NH2-terminal kinase-1-dependent manner. Am J Physiol Lung Cell Mol Physiol. 2018;314:L984–L997. doi: 10.1152/ajplung.00053.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–876. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–L357. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Froese AR, Shimbori C, Bellaye P-S, Inman M, Obex S, Fatima S, et al. Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 132. Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, et al. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep. 2016;16:1810–1819. doi: 10.1016/j.celrep.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 133. Gokey JJ, Sridharan A, Xu Y, Green J, Carraro G, Stripp BR, et al. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight. 2018;3:e98738. doi: 10.1172/jci.insight.98738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kolb M, Bonella F, Wollin L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir Med. 2017;131:49–57. doi: 10.1016/j.rmed.2017.07.062. [DOI] [PubMed] [Google Scholar]
- 135. Aryal S, Nathan SD. An update on emerging drugs for the treatment of idiopathic pulmonary fibrosis. Expert Opin Emerg Drugs. 2018;23:159–172. doi: 10.1080/14728214.2018.1471465. [DOI] [PubMed] [Google Scholar]
- 136. De Santis MM, Bölükbas DA, Lindstedt S, Wagner DE. How to build a lung: latest advances and emerging themes in lung bioengineering. Eur Respir J. 2018;52:1601355. doi: 10.1183/13993003.01355-2016. [DOI] [PubMed] [Google Scholar]
- 137. Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. 2018;24:39–49. doi: 10.1038/nm.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, et al. Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012;185:537–546. doi: 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ninou I, Kaffe E, Müller S, Budd DC, Stevenson CS, Ullmer C, et al. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm Pharmacol Ther. 2018;52:32–40. doi: 10.1016/j.pupt.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 140. Oikonomou N, Mouratis M-A, Tzouvelekis A, Kaffe E, Valavanis C, Vilaras G, et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47:566–574. doi: 10.1165/rcmb.2012-0004OC. [DOI] [PubMed] [Google Scholar]
- 141. Hirani N, Nicol L, MacKinnon AC, Ford P, Schambye H, Pedersen A, et al. TD139, a novel inhaled galectin-3 inhibitor for the treatment of idiopathic pulmonary fibrosis (IPF): results from the first in (IPF) patients study. QJM. 2016;109:S16. [Google Scholar]
- 142. Hirani N, MacKinnon AC, Nicol L, Ford P, Schambye H, Pedersen A, et al. Target-inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2021;57:2002559. doi: 10.1183/13993003.02559-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Maher TM, Kreuter M, Lederer DJ, Brown KK, Wuyts W, Verbruggen N, et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2) BMJ Open Respir Res. 2019;6:e000422. doi: 10.1136/bmjresp-2019-000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. 2018;6:627–635. doi: 10.1016/S2213-2600(18)30181-4. [DOI] [PubMed] [Google Scholar]
- 145. Zulfikar S, Mulholland S, Adamali H, Barratt SL. Inhibitors of the autotaxin-lysophosphatidic acid axis and their potential in the treatment of interstitial lung disease: current perspectives. Clin Pharmacol. 2020;12:97–108. doi: 10.2147/CPAA.S228362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Delaine T, Collins P, MacKinnon A, Sharma G, Stegmayr J, Rajput VK, et al. Galectin-3-binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. ChemBioChem. 2016;17:1759–1770. doi: 10.1002/cbic.201600285. [DOI] [PubMed] [Google Scholar]
- 147. Johannes L, Jacob R, Leffler H. Galectins at a glance. J Cell Sci. 2018;131:jcs208884. doi: 10.1242/jcs.208884. [DOI] [PubMed] [Google Scholar]
- 148. Lepur A, Carlsson MC, Novak R, Dumić J, Nilsson UJ, Leffler H. Galectin-3 endocytosis by carbohydrate independent and dependent pathways in different macrophage like cell types. Biochim Biophys Acta. 2012;1820:804–818. doi: 10.1016/j.bbagen.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 149.Gilead Sciences. Foster City, CA: Gilead Sciences; 2021. https://www.gilead.com/news-and-press/press-room/press-releases/2021/2/galapagos-and-gilead-discontinue-isabela-phase-3-trials-in-ipf [Google Scholar]
- 150.Galecto Copenhagen, Denmark: Galecto; 2021https://ir.galecto.com/news-releases/news-release-details/galecto-announces-outcome-data-safety-monitoring-board-interim. [Google Scholar]
- 151. Rackow AR, Nagel DJ, McCarthy C, Judge J, Lacy S, Freeberg MAT, et al. The self-fulfilling prophecy of pulmonary fibrosis: a selective inspection of pathological signalling loops. Eur Respir J. 2020;56:2000075. doi: 10.1183/13993003.00075-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lehmann M, Buhl L, Alsafadi HN, Klee S, Hermann S, Mutze K, et al. Differential effects of nintedanib and pirfenidone on lung alveolar epithelial cell function in ex vivo murine and human lung tissue cultures of pulmonary fibrosis. Respir Res. 2018;19:175. doi: 10.1186/s12931-018-0876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Alsafadi HN, Staab-Weijnitz CA, Lehmann M, Lindner M, Peschel B, Königshoff M, et al. An ex vivo model to induce early fibrosis-like changes in human precision-cut lung slices. Am J Physiol Lung Cell Mol Physiol. 2017;312:L896–L902. doi: 10.1152/ajplung.00084.2017. [DOI] [PubMed] [Google Scholar]
- 154. Wei Y, Dong W, Jackson J, Ho T-C, Le Saux CJ, Brumwell A, et al. Blocking LOXL2 and TGFβ1 signalling induces collagen I turnover in precision-cut lung slices derived from patients with idiopathic pulmonary fibrosis. Thorax. 2021;76:729–732. doi: 10.1136/thoraxjnl-2020-215745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Mercer PF, Woodcock HV, Eley JD, Platé M, Sulikowski MG, Durrenberger PF, et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax. 2016;71:701–711. doi: 10.1136/thoraxjnl-2015-207429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Khan MM, Poeckel D, Halavatyi A, Zukowska-Kasprzyk J, Stein F, Vappiani J, et al. An integrated multiomic and quantitative label-free microscopy-based approach to study pro-fibrotic signalling in ex vivo human precision-cut lung slices. Eur Respir J. 2020;58:2000221. doi: 10.1183/13993003.00221-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Roach KM, Sutcliffe A, Matthews L, Elliott G, Newby C, Amrani Y, et al. A model of human lung fibrogenesis for the assessment of anti-fibrotic strategies in idiopathic pulmonary fibrosis. Sci Rep. 2018;8:342. doi: 10.1038/s41598-017-18555-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Stegmayr J, Alsafadi HN, Langwinski W, Niroomand A, Lindstedt S, Leigh ND, et al. Isolation of high yield and quality RNA from human precision-cut lung slices for RNA-sequencing and computational integration with larger patient cohorts. Am J Physiol Lung Cell Mol Physiol. 2021;320:L232–L240. doi: 10.1152/ajplung.00401.2020. [DOI] [PubMed] [Google Scholar]
- 159. Stegmayr J, Wagner DE. The dawn of the omics era in human precision-cut lung slices. Eur Respir J. 2021;58:2100203. doi: 10.1183/13993003.00203-2021. [DOI] [PubMed] [Google Scholar]
- 160. van der Velden JL, Alcorn JF, Chapman DG, Lundblad LKA, Irvin CG, Davis RJ, et al. Airway epithelial specific deletion of Jun-N-terminal kinase 1 attenuates pulmonary fibrosis in two independent mouse models. PLoS One. 2020;15:e0226904. doi: 10.1371/journal.pone.0226904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Gilpin SE, Wagner DE. Acellular human lung scaffolds to model lung disease and tissue regeneration. Eur Respir Rev. 2018;27:180021. doi: 10.1183/16000617.0021-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Asmani M, Velumani S, Li Y, Wawrzyniak N, Hsia I, Chen Z, et al. Fibrotic microtissue array to predict anti-fibrosis drug efficacy. Nat Commun. 2018;9:2066. doi: 10.1038/s41467-018-04336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Oglesby IK, Schweikert A, Fox B, Redmond C, Donnelly SC, Hurley K. Lung organoids and other preclinical models of pulmonary fibrosis. QJM. 2021;114:167–173. doi: 10.1093/qjmed/hcaa281. [DOI] [PubMed] [Google Scholar]
- 164. Bölükbas DA, Datz S, Meyer-Schwickerath C, Morrone C, Doryab A, Gößl D, et al. Organ-restricted vascular delivery of nanoparticles for lung cancer therapy. Adv Ther (Weinh) 2020;3:2000017. doi: 10.1002/adtp.202000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. van den Blink B, Dillingh MR, Ginns LC, Morrison LD, Moerland M, Wijsenbeek M, et al. Recombinant human pentraxin-2 therapy in patients with idiopathic pulmonary fibrosis: safety, pharmacokinetics and exploratory efficacy. Eur Respir J. 2016;47:889–897. doi: 10.1183/13993003.00850-2015. [DOI] [PubMed] [Google Scholar]
- 166. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA. 2018;319:2299–2307. doi: 10.1001/jama.2018.6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: an open-label extension study. Lancet Respir Med. 2019;7:657–664. doi: 10.1016/S2213-2600(19)30172-9. [DOI] [PubMed] [Google Scholar]
- 168. Raghu G, Scholand MB, de Andrade J, Lancaster L, Mageto Y, Goldin J, et al. FG-3019 anti-connective tissue growth factor monoclonal antibody: results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur Respir J. 2016;47:1481–1491. doi: 10.1183/13993003.01030-2015. [DOI] [PubMed] [Google Scholar]
- 169. Mageto Y, Flaherty K, Brown K, Fong A, Raghu G. Safety and tolerability of human monoclonal antibody fg-3019, anti-connective tissue growth factor, in patients with idiopathic pulmonary fibrosis. Chest. 2004;126:773S. [Google Scholar]
- 170. Richeldi L, Fernández Pérez ER, Costabel U, Albera C, Lederer DJ, Flaherty KR, et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2020;8:25–33. doi: 10.1016/S2213-2600(19)30262-0. [DOI] [PubMed] [Google Scholar]
- 171. DeFilipp Z, Advani AS, Bachanova V, Cassaday RD, Deangelo DJ, Kebriaei P, et al. hematopoietic cell transplantation in the treatment of adult acute lymphoblastic leukemia: updated 2019 evidence-based review from the American Society for Transplantation and Cellular Therapy. Biol Blood Marrow Transplant. 2019;25:2113–2123. doi: 10.1016/j.bbmt.2019.08.014. [DOI] [PubMed] [Google Scholar]
- 172. Kassim AA, Sharma D. Hematopoietic stem cell transplantation for sickle cell disease: the changing landscape. Hematol Oncol Stem Cell Ther. 2017;10:259–266. doi: 10.1016/j.hemonc.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 173. Saeedi P, Halabian R, Imani Fooladi AA. A revealing review of mesenchymal stem cells therapy, clinical perspectives and modification strategies. Stem Cell Investig. 2019;6:34. doi: 10.21037/sci.2019.08.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ikonomou L, Wagner DE, Turner L, Weiss DJ. Translating basic research into safe and effective cell-based treatments for respiratory diseases. Ann Am Thorac Soc. 2019;16:657–668. doi: 10.1513/AnnalsATS.201812-890CME. [DOI] [PubMed] [Google Scholar]
- 175. Tzouvelekis A, Toonkel R, Karampitsakos T, Medapalli K, Ninou I, Aidinis V, et al. Mesenchymal stem cells for the treatment of idiopathic pulmonary fibrosis. Front Med (Lausanne) 2018;5:142. doi: 10.3389/fmed.2018.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Wecht S, Rojas M. Mesenchymal stem cells in the treatment of chronic lung disease. Respirology. 2016;21:1366–1375. doi: 10.1111/resp.12911. [DOI] [PubMed] [Google Scholar]
- 177. Serrano-Mollar A. Cell therapy in idiopathic pulmonary fibrosis. Med Sci (Basel) 2018;6:64. doi: 10.3390/medsci6030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Tzouvelekis A, Paspaliaris V, Koliakos G, Ntolios P, Bouros E, Oikonomou A, et al. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J Transl Med. 2013;11:171. doi: 10.1186/1479-5876-11-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Chambers DC, Enever D, Ilic N, Sparks L, Whitelaw K, Ayres J, et al. A phase 1b study of placenta-derived mesenchymal stromal cells in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19:1013–1018. doi: 10.1111/resp.12343. [DOI] [PubMed] [Google Scholar]
- 180. Glassberg MK, Minkiewicz J, Toonkel RL, Simonet ES, Rubio GA, DiFede D, et al. Allogeneic human mesenchymal stem cells in patients with idiopathic pulmonary fibrosis via intravenous delivery (AETHER): a phase I safety clinical trial. Chest. 2017;151:971–981. doi: 10.1016/j.chest.2016.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Banerjee ER, Laflamme MA, Papayannopoulou T, Kahn M, Murry CE, Henderson WR., Jr Human embryonic stem cells differentiated to lung lineage-specific cells ameliorate pulmonary fibrosis in a xenograft transplant mouse model. PLoS One. 2012;7:e33165. doi: 10.1371/journal.pone.0033165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zhou Q, Ye X, Sun R, Matsumoto Y, Moriyama M, Asano Y, et al. Differentiation of mouse induced pluripotent stem cells into alveolar epithelial cells in vitro for use in vivo. Stem Cells Transl Med. 2014;3:675–685. doi: 10.5966/sctm.2013-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. How C-K, Chien Y, Yang K-Y, Shih H-C, Juan C-C, Yang Y-P, et al. Induced pluripotent stem cells mediate the release of interferon gamma–induced protein 10 and alleviate bleomycin-induced lung inflammation and fibrosis. Shock. 2013;39:261–270. doi: 10.1097/SHK.0b013e318285f2e2. [DOI] [PubMed] [Google Scholar]
- 184. Zhou Y, He Z, Gao Y, Zheng R, Zhang X, Zhao L, et al. Induced pluripotent stem cells inhibit bleomycin-induced pulmonary fibrosis in mice through suppressing TGF-β1/Smad-mediated epithelial to mesenchymal transition. Front Pharmacol. 2016;7:430. doi: 10.3389/fphar.2016.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Serrano-Mollar A, Gay-Jordi G, Guillamat-Prats R, Closa D, Hernandez-Gonzalez F, Marin P, et al. Pneumocyte Study Group. Safety and tolerability of alveolar type II cell transplantation in idiopathic pulmonary fibrosis. Chest. 2016;150:533–543. doi: 10.1016/j.chest.2016.03.021. [DOI] [PubMed] [Google Scholar]
- 186. Alapati D, Zacharias WJ, Hartman HA, Rossidis AC, Stratigis JD, Ahn NJ, et al. In utero gene editing for monogenic lung disease. Sci Transl Med. 2019;11:eaav8375. doi: 10.1126/scitranslmed.aav8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 188. Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone Clinical Study Group in Japan. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- 189. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 190. Costabel U, Albera C, Lancaster LH, Lin CY, Hormel P, Hulter HN, et al. An open-label study of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis (RECAP) Respiration. 2017;94:408–415. doi: 10.1159/000479976. [DOI] [PubMed] [Google Scholar]
- 191. Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 192. Kolb M, Richeldi L, Behr J, Maher TM, Tang W, Stowasser S, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72:340–346. doi: 10.1136/thoraxjnl-2016-208710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. King TE, Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:92–99. doi: 10.1164/rccm.201011-1874OC. [DOI] [PubMed] [Google Scholar]
- 194. King TE, Jr, Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:75–81. doi: 10.1164/rccm.200705-732OC. [DOI] [PubMed] [Google Scholar]
- 195. Couluris M, Kinder BW, Xu P, Gross-King M, Krischer J, Panos RJ. Treatment of idiopathic pulmonary fibrosis with losartan: a pilot project. Lung. 2012;190:523–527. doi: 10.1007/s00408-012-9410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Palmer SM, Snyder L, Todd JL, Soule B, Christian R, Anstrom K, et al. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest. 2018;154:1061–1069. doi: 10.1016/j.chest.2018.08.1058. [DOI] [PubMed] [Google Scholar]
- 197. Tirucherai G, Yu D, Revankar R, Klinger G, Van Lier JJ, Taubel J, et al. BMS-986278, a lysophosphatidic acid 1 (LPA1) receptor antagonist, in healthy participants: a single/multiple ascending dose (SAD/MAD) phase 1 study. Eur Respir J. 2019;54:PA1398. [Google Scholar]
- 198. Lukey PT, Harrison SA, Yang S, Man Y, Holman BF, Rashidnasab A, et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur Respir J. 2019;53:1801992. doi: 10.1183/13993003.01992-2018. [DOI] [PubMed] [Google Scholar]
- 199. van der Velden JL, Ye Y, Nolin JD, Hoffman SM, Chapman DG, Lahue KG, et al. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin Transl Med. 2016;5:36. doi: 10.1186/s40169-016-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR. Imatinib-IPF Study Investigators. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010;181:604–610. doi: 10.1164/rccm.200906-0964OC. [DOI] [PubMed] [Google Scholar]
- 201. Noth I, Anstrom KJ, Calvert SB, Andrade JD, Flaherty KR, Glazer C, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:88–95. doi: 10.1164/rccm.201202-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Prasse A, Ramaswamy M, Mohan S, Pan L, Kenwright A, Neighbors M, et al. A phase 1b study of vismodegib with pirfenidone in patients with idiopathic pulmonary fibrosis. Pulm Ther. 2019;5:151–163. doi: 10.1007/s41030-019-0096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Decaris M, Turner S, Lepist I, Wong S, Park E, Cha J, et al. PLN-74809, a dual αV/β6/αVβ1, oral, selective integrin inhibitor, is well tolerated and reduces lung TGF-β activity in healthy volunteers [abstract] Am J Respir Crit Care Med. 2020;201:A4554. [Google Scholar]
- 204. Kang S-U, Ryou J-H, Lim J-J, Lee D-Y, Kwon H-J, Ha G-H, et al. BBT-877, a potent autotaxin inhibitor in clinical development to treat idiopathic pulmonary fibrosis [abstract] Am J Respir Crit Care Med. 2019;199:A2577. [Google Scholar]
- 205. Soule B, Tirucherai G, Kavita U, Kundu S, Christian R. Safety, tolerability, and pharmacokinetics of BMS-986263/ND-L02-s0201, a novel targeted lipid nanoparticle delivering HSP47 siRNA, in healthy participants: a randomised, placebo-controlled, double-blind, phase 1 study [abstract] J Hepatol. 2018;68:S112. [Google Scholar]
- 206. Blease K, Ye Y, Azaryan A, Ramirez-Valle F, Ceres R, Horan G, et al. CC-90001, a second generation Jun N-terminal kinase (JNK) inhibitor for the treatment of idiopathic pulmonary fibrosis [abstract] Am J Respir Crit Care Med. 2017;195:A5409. [Google Scholar]
- 207. Wang J, Van Parys M, Pan L, Chen Y, Cheung D, McKnight J, et al. Determination of a novel antifibrotic small molecule GDC-3280 in human plasma and urine by liquid chromatography tandem mass spectrometry to support its first-in-human clinical trial. Biomed Chromatogr. 2019;33:e4482. doi: 10.1002/bmc.4482. [DOI] [PubMed] [Google Scholar]
- 208. Khalil N, Manganas H, Ryerson CJ, Shapera S, Cantin AM, Hernandez P, et al. Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2019;53:800663. doi: 10.1183/13993003.00663-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Jr, Raghu G. Idiopathic Pulmonary Fibrosis Clinical Research Network. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2093–2101. doi: 10.1056/NEJMoa1401739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Raghu G, Brown KK, Bradford WZ, Starko K, Noble PW, Schwartz DA, et al. Idiopathic Pulmonary Fibrosis Study Group. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350:125–133. doi: 10.1056/NEJMoa030511. [DOI] [PubMed] [Google Scholar]
- 211. King TE, Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. INSPIRE Study Group. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374:222–228. doi: 10.1016/S0140-6736(09)60551-1. [DOI] [PubMed] [Google Scholar]
- 212. Raghu G, Brown KK, Costabel U, Cottin V, du Bois RM, Lasky JA, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178:948–955. doi: 10.1164/rccm.200709-1446OC. [DOI] [PubMed] [Google Scholar]
- 213. Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ, et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. Lancet Respir Med. 2017;5:22–32. doi: 10.1016/S2213-2600(16)30421-0. [DOI] [PubMed] [Google Scholar]
- 214. Mouded M, Culver DA, Hamblin MJ, Golden JA, Veeraraghavan S, Enelow RI, et al. Randomized, double-blind, placebo-controlled, multiple dose, dose-escalation study of BG00011 (formerly STX-100) in patients with idiopathic pulmonary fibrosis (IPF) [abstract] Am J Respir Crit Care Med. 2018;197:A7785. [Google Scholar]
- 215. Raghu G, Martinez FJ, Brown KK, Costabel U, Cottin V, Wells AU, et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur Respir J. 2015;46:1740–1750. doi: 10.1183/13993003.01558-2014. [DOI] [PubMed] [Google Scholar]
- 216. Chambers DC, Enever D, Ilic N, Sparks L, Whitelaw K, Ayres J, et al. A phase 1b study of placenta-derived mesenchymal stromal cells in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19:1013–1018. doi: 10.1111/resp.12343. [DOI] [PubMed] [Google Scholar]
- 217. Averyanov A, Koroleva I, Konoplyannikov M, Revkova V, Lesnyak V, Kalsin V, et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl Med. 2020;9:6–16. doi: 10.1002/sctm.19-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Ntolios P, Manoloudi E, Tzouvelekis A, Bouros E, Steiropoulos P, Anevlavis S, et al. Longitudinal outcomes of patients enrolled in a phase Ib clinical trial of the adipose-derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. Clin Respir J. 2018;12:2084–2089. doi: 10.1111/crj.12777. [DOI] [PubMed] [Google Scholar]
- 219.Raghu G, Anstrom KJ, King TE, Jr, Lasky JA, Martinez FJIdiopathic Pulmonary Fibrosis Clinical Research Network Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis N Engl J Med 20123661968–1977.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, et al. IFIGENIA Study Group. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 221. Yoshida M, Taguchi O, Gabazza EC, Yasui H, Kobayashi T, Kobayashi H, et al. The effect of low-dose inhalation of nitric oxide in patients with pulmonary fibrosis. Eur Respir J. 1997;10:2051–2054. doi: 10.1183/09031936.97.10092051. [DOI] [PubMed] [Google Scholar]
- 222. Blanco I, Ribas J, Xaubet A, Gómez FP, Roca J, Rodriguez-Roisin R, et al. Effects of inhaled nitric oxide at rest and during exercise in idiopathic pulmonary fibrosis. J Appl Physiol (1985) 2011;110:638–645. doi: 10.1152/japplphysiol.01104.2010. [DOI] [PubMed] [Google Scholar]