

Abstract
The neuropathological hallmarks of Alzheimer’s Disease are plaques and neurofibrillary tangles. Yet, Alzheimer’s is a complex disease with many contributing factors, such as energy-metabolic changes, which have been documented in autopsy brains from individuals with Alzheimer’s and animal disease models alike. One conceivable explanation is that the interplay of age-related extracellular and intracellular alterations pertaining to Alzheimer’s, such as cerebrovascular changes, protein aggregates and inflammation, evoke a mitochondrial response. However, it is not clear if and how mitochondria can contribute to Alzheimer’s pathophysiology. This study focuses on one particular aspect of this question by investigating the functional interaction between the microtubule-associated protein tau and the mitochondrial inner membrane fusion machinery, which shows alterations in Alzheimer’s brains. OPA1 is an essential inner membrane-fusion protein regulated by the two membrane proteases OMA1 and YME1L1. Assessment of OPA1 proteolysis—usually found in dividing mitochondria—and posttranslational tau modifications in mouse and human neuroblastoma cells under different experimental conditions clarified the relationship between these two pathways: OPA1 hydrolysis and phosphorylation or dephosphorylation of tau may coincide, but are not causally related. OPA1 cleavage did not alter tau’s phosphorylation pattern. Conversely, tau’s phosphorylation state did not induce nor correlate with OPA1 proteolysis. These results irrefutably demonstrate that there is no direct functional interaction between posttranslational tau modifications and the regulation of the OMA1-OPA1 pathway, which implies a common root cause modulating both pathways in Alzheimer’s.
Keywords: Mitochondria, Alzheimer’s, OMA1, tau hyperphosphorylation, fusion
1. Introduction
The neuropathological hallmarks of Alzheimer’s Disease are plaques and neurofibrillary tangles—abnormally folded amyloid-β deposits and tau protein aggregates in the atrophic brain, which are believed to be causally related to neurodegenerative processes [1–9]. Apolipoprotein (APO) E4 on the other hand is a major risk factor for Alzheimer’s [10, 11]. Also mitochondria may contribute to Alzheimer’s pathophysiology [12–16]. For example, damaged and dysfunctional mitochondria were recognized in electron micrographs of brain specimens from deceased individuals with Alzheimer’s [17, 18]. Metabolic deficits are also evident in the brains from individuals with Alzheimer’s [19–25].
Mitochondria form dynamic networks of interconnected tubules, whose morphology is determined by balanced fission and fusion events. These events are intimately linked to the energy metabolism as well as quality control measures, such as mitochondrial autophagy (aka mitophagy) and apoptotic signaling. The dynamin-related GTPases DNM1L (DRP1), MFN1, MFN2, and OPA1 coordinate mitochondrial fission and fusion [26–28]. A number of studies described changes in these proteins alongside with mitochondrial fragmentation and other functional deficits evoked by amyloid-β and tau [29–33]. Likewise, APOE4 was associated with mitochondrial dysfunction [34–36]. Yet it is still not clear, whether mitochondrial alterations are a cause for, the consequence of, or coincide with neurodegeneration in Alzheimer’s.
The present study focuses on one particular aspect of the interrelationship between mitochondria and Alzheimer’s by asking, is there a functional interaction between tau (MAPT) and mitochondrial fusion? More specifically, OPA1-mediated mitochondrial inner membrane fusion? OPA1 is an essential structural protein organizing the inner membranes [37–39]. The protein is proteolytically deactivated when mitochondria divide. For this reason, OPA1 exists in two forms, membrane-anchored L-OPA1 and truncated S-OPA1. Mitochondria fusion strictly depends on L-OPA1 [40, 41], which is hydrolyzed for example in damaged organelles destined for mitophagy [42]. The i-AAA protease YME1L1 maintains stoichiometric ratios of the OPA1 isoforms contingent on energy levels [43, 44]. OMA1 in contrast releases L-OPA1 under stress conditions into the intermembrane space. OMA1’s activation and L-OPA1 proteolysis, mitochondrial fragmentation, outer membrane permeabilization, cytochrome c release, and cell death are all highly correlated events [45–49].
Here, I addressed the question whether the regulation of the OMA1 protease and proteolysis of OPA1 show any direct functional cross-communication with posttranslational tau modifications. This question was prompted by five observations. (1) Two independent studies of the mitochondrial fission and fusion proteins in Alzheimer’s autopsy brains reported reduced OPA1 protein levels, which implies increased proteolytic activity in these specimens [30, 31]. (2) Cortical neurons from tau-knockout mice transfected with truncated or pseudophosphorylated tau-GFP fusion proteins displayed fragmented mitochondria and reduced OPA1 levels [29]. (3) PHB2-deficient mice displayed an OMA1-dependent tauopathy phenotype [50, 51]. The prohibitins PHB1 and PHB2 are scaffolding proteins found in the mitochondrial inner membrane, where they are involved, among other things, in the coordination of OMA1 [52, 53]. (4) Mutations in OPA1 cause dominant optic atrophy, a form of neurodegeneration that affects mainly the optic nerve in humans and in animal disease models [54]. Intriguingly, optic nerve degeneration is also evident in individuals with Alzheimer’s [55]. And (5) patient-derived induced pluripotent stem cells further revealed an OPA1-dependent deficiency to differentiate specifically into GABAergic interneurons [56]. GABAergic dysfunction and abnormalities also pertain to Alzheimer’s [57–61]. These observations provided the rational for examining the relationship between tau and the OMA1-OPA1 pathway by interrogating neuroblastoma cells with numerous stimuli and assessing the impact on OPA1 and tau.
2. Material and methods
2.1. Chemicals
Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) and okadaic acid were from Sigma (Sigma-Aldrich, St. Louis, MO, USA). AZD1080, CHIR98014, cyclosporin A, GNE7915, LRRK2in1, oligomycin, rotenone, SB216763, sorafenib, staurosporine, and valinomycin were from Cayman Chemical Company (Ann Arbor, MI, USA). Tipranavir (#11285) was obtained through the NIH’s HIV Reagent Program, which is supported by National Institute of Allergy and Infectious Diseases. 10 mM stock solutions in dimethyl sulfoxide (DMSO) were prepared for all compounds with the exception of staurosporine (1mg/ml in ethyl acetate) and stored at −20°C until further use. Working dilutions were prepared fresh for each experiment.
2.2. Cell culture
All cell lines were obtained from the cell culture core facility at UC Berkeley (CA, USA). All cell culture reagents were from GIBCO (Invitrogen, Carlsbad, CA, USA). Neuro2A mouse neuroblastoma cells and SH-SY5Y human neuroblastoma cells were grown under standard conditions in DMEM/F12 supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 units/ml) and streptomycin (100 μg/ml) at 37°C in a humidified atmosphere with 5% CO2. Hek293T human embryonic kidney cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/ml) and streptomycin (100 μg/ml). Unless stated otherwise, cells were treated with the denoted compounds diluted in serum-free DMEM/F12 for 3 hours at 37°C in a humidified incubator with 5% CO2.
2.3. Gene silencing
Protein knockdown was accomplished by silencing gene expression with pooled siRNAs targeting mouse Oma1 or mouse Opa1. 50-60% confluent Neuro2A cells were transfected with 300 ng Oma1 siRNA (#EMU088111, Sigma and #SC-151297, Santa Cruz Biotechnology, Dallas, TX, USA), Opa1 siRNA (#EMU010511, Sigma) or fluorescent control siRNA (#SIC007, Sigma) using X-tremeGENE 360 transfection reagent (Roche Diagnostics, Mannheim, Germany) at an siRNA-to-transfection reagent ratio of 1:5 following the manufacturer’s protocol. All cells were investigated 36 hours post transfection.
2.4. Protein expression
Human OMA1-FLAG under the control of a CMV-promotor in a pcDNA3.1-vector was from GenScript (Pissatthem, NJ, USA; clone ID: OHu74904). Hek293T cells and Neuro2A cells were transfected with X-tremeGENE 360 with a plasmid-to-transfection reagent ratio of 1:3 and 1:2, respectively. All cells were studied 24 hours post transfection.
2.5. Cell assays
OMA1 protease activity was measured with a luciferase-based reporter protein stably expressed in Neuro2A cells. The reporter is targeted to the mitochondrial inner membrane, where it can be hydrolyzed by the OMA1 protease thereby eliminating its luciferase activity [62]. About 50,000 reporter cells per well of a 96-well half-area plate (Costar #3688, Corning, New York, USA) were exposed for 30 minutes to serial dilutions of denoted compounds in DMEM/F12, after which luciferase substrate was added and bioluminescence measured with a Fluoroskan FL plate reader (ThermoFisher Scientific, Waltham, MA, USA). Δψ was measured with tetramethylrhodamine ethyl ester perchlorate (TMRE, #87917, Sigma). 10,000 Neuro2A cells per well of a 96-well half-area plate were incubated for 3 hours with 4 μM TMRE in DMEM/F12 without or with 30 μM of the denoted compounds. Cells were washed twice in PBS and fluorescence was measured with the Fluoroskan FL plate reader (543 nm excitation filter and a 580 nm emission filter). ATP levels and cell viability were measured with the CellTiter-Glo assay (#G7570, Promega, Madison, WI, USA) and the CellTiter-Fluor assay (#G6080, Promega), respectively. Briefly, 2,000 Neuro2A cells per well of a 96-well half-area plate were incubated for 3 hours with 30 μM of the denoted compounds in DMEM/F12. Cell viability was determined first by adding the CellTiter-Fluor assay reagents according to the manufactures protocol and measuring fluorescence with aforementioned plate reader (405 nm excitation filter and 490 nm emission filter). ATP levels were measured thereafter by adding the CellTiter-Glo assay reagents as specified by the manufacturer and measuring bioluminescence with the same instrument.
2.6. Protein analysis
Whole cell extracts were prepared by harvesting cells in lysis buffer A (25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.1% NP-40, 0.1% sodium deoxycholate, 0.01% SDS) supplemented with 1 × HALT protease inhibitor cocktail (ThermoFisher) and 1 × phosphatase inhibitor cocktail 3 (Sigma). Whole cell lysates were cleared from debris by centrifugation at 20,000 × g for 5 minutes at 4°C. The supernatant (about 30 μg protein per lane) was separated under reducing conditions on 8% tris-glycine mini gels (Invitrogen) following the manufacturer’s protocol, blotted onto PVDF membranes, and labeled with antibodies following standard procedures. For a list of primary antibodies, antibody concentrations and blocking conditions see Table 1. All tau phosphorylation sites throughout the manuscript, in Table 1, and in the figures are referred to by their amino acid position in the human 2N4R tau protein. Please see Figure S1A in the supplementary material for the corresponding sites in the mouse protein. Alkaline-phosphatase-conjugated secondary antibodies diluted 1:5,000 in 5% (w/v) dry milk powder in TBST (tris buffered saline with 0.1% Tween20) and 1-step NBT/BCIP substrate (ThermoFisher) were used for protein detection.
Table 1:
Primary antibodies.
| Antibody | Supplier (Catalog No.) | Antigen | Host (clone) | Dilution (diluent) |
|---|---|---|---|---|
| β-Actin | Proteintech (#66009-1-Ig) | human ACTB | mouse (2D4H5) | 1:1,000 (5% milk powder) |
| DELE1 | Novus Bio. (#NBP2-84765) | human DELE1 (aa 164-213) | rabbit (polyclonal) | 1:500 (5% milk powder) |
| OMA1 | Santa Cruz Biotech. (#sc-515788) | human OMA1 (aa 230-399) | mouse (H-11) | 1:250 (5% milk powder) |
| OPA1 | BD Biosciences (#612606) | human OPA1 (aa 708-830) | mouse (18/OPA-1) | 1:1,000 (5% milk powder) |
| P-Tau S199/S202 | ThermoFisher (#44-768G) | human TAU phosphopeptide around aa 199-202 | rabbit (polyclonal) | 1:1,000 (3% BSA) |
| P-Tau S396 | Novus Bio. (#NB100-82243) | human TAU phosphopeptide around aa 396 | rabbit (polyclonal) | 1:1,000 (3% BSA) |
| P-Tau S396 (PHF13) | Santa Cruz Biotech. (#sc-32275) | human PHF-Tau | mouse (PHF13) | 1:250 (3% BSA) |
| P-Tau S404 | Cell Signaling Tech. (#20194) | human tau phosphorylated at S404; S400 & S404; T403 & S404; S400, T403 & S404 | rabbit (D2Z4G) | 1:500 (3% BSA) |
| P-Tau T231 | Novus Bio. (#NB100-82249) | human tau phosphopeptide around aa 231 | rabbit (polyclonal) | 1:1,000 (3% BSA) |
| PGAM5 | Proteintech (#28445-1-AP) | human PGAM5 isoform 2 | rabbit (polyclonal) | 1:1,000 (5% milk powder) |
| Phosphoserine | Novus Bio. (#NB100-1953) | phosphorylated serine residues | rabbit (polyclonal) | 1:100 (3% BSA) |
| Phosphothreonine | Novus Bio. (#NB110-96896) | phosphorylated threonine residues | rabbit (polyclonal) | 1:125 (3% BSA) |
| PINK1 | Novus Bio. (#BC100-494) | human PINK1 (aa 175-250) | rabbit (polyclonal) | 1:1,000 (5% milk powder) |
| Tau | EMD Millipore (#MABN2472 ) | phosphorylation-independent N-terminal region | mouse (2A1-2E1) | 1:1,000 (5% milk powder) |
2.7. Data analysis
Western blots were digitalized and images adjusted for brightness and contrast. Regions of interest were defined manually and quantified by densitometry using NIH’s imageJ software [63]. Statistical analyses were performed with GraphPad Prism 5.0c (GraphPad Software Inc., San Diego, CA, USA). All results are represented as mean ± standard deviation (SD). Groups from at least 3 independent experiments were compared by Student’s t-test or using a 1-way or 2-way analysis of variance (ANOVA) with Bonferroni’s multiple-group comparison posttest as applicable. Differences between groups were considered to be significant at p values of ≤ 0.05. Dose-response relationships were plotted as semi-logarithmic graphs and half maximal effective concentrations (EC50) were calculated using a non-linear inhibitor regression model with variable Hill-slope.
3. Results
3.1. Establishing the experimental paradigm.
The first step was to establish an experimental framework for the analysis of the functional interaction of the OMA1-OPA1 pathway and tau. The OMA1 protease shows only little activity under physiological conditions but is readily activated by cell stress and upon apoptotic stimuli. Active OMA1 rapidly cleaves L-OPA1 whereby S-OPA1 is generated. OMA1 can be activated by dissipation of the mitochondrial membrane potential Δψ with the protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) [64, 65]. As illustrated in Figure 1A, treatment of Neuro2A neuroblastoma cells with 1 μM CCCP for 30 minutes led to loss of L-OPA1 isoforms in Western blots. Valinomycin is an alternative ionophore, which activated OMA1 in Neuro2A cells at concentrations above 0.1 μM (Figure 1B). CCCP’s half maximal effective concentration (EC50) was determined in cellular OMA1 protease assays at about 0.2 μM (Figure 1C). Valinomycin’s EC50 in the OMA1 protease assay was 22 nM (Figure 1D).
Figure 1: CCCP induced OPA1 proteolysis and tau dephosphorylation.
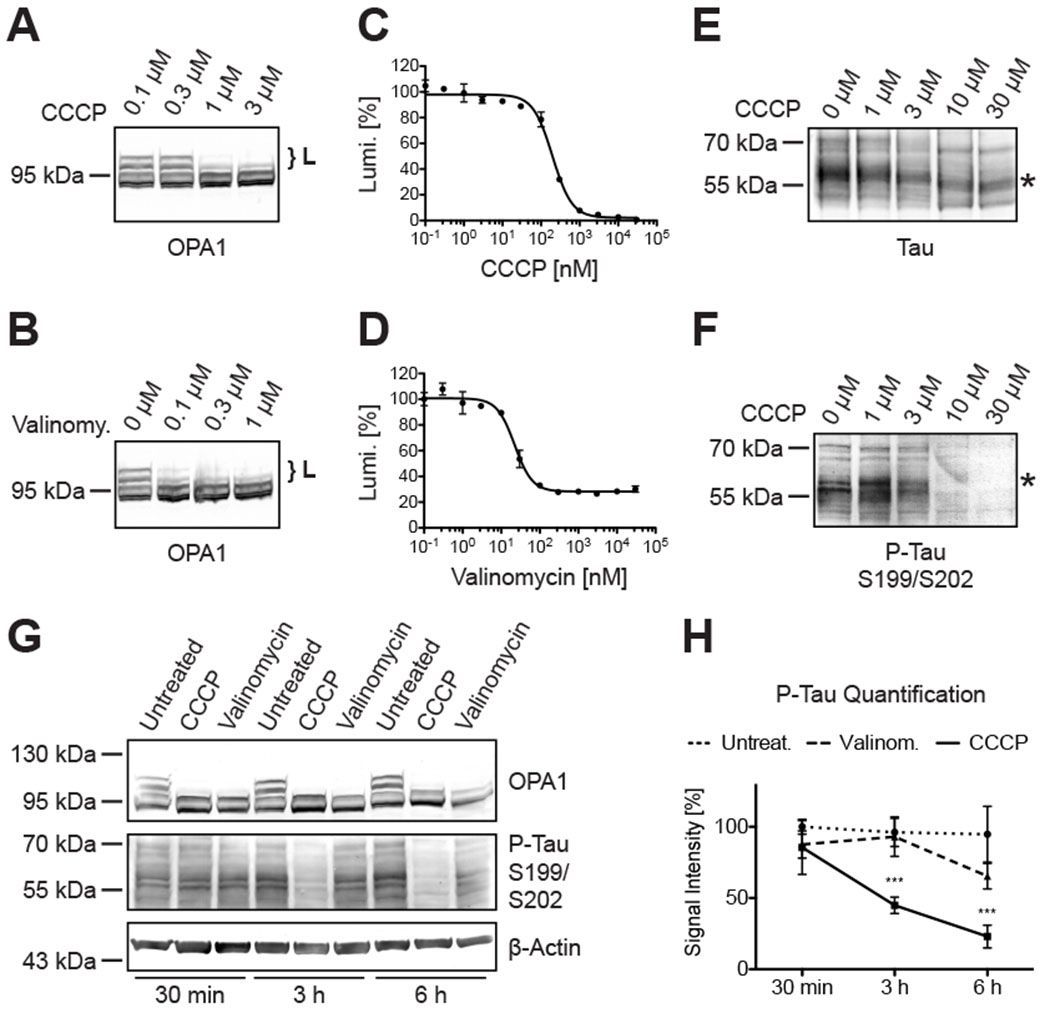
L-OPA1 isoforms (accolades) were hydrolyzed in a dose-dependent manner in Western blots of Neuro2A cells treated with CCCP (A) and valinomycin (B). An engineered and mitochondria-targeted luciferase functioning as artificial OMA1 substrate was expressed in Neuro2A cells to determine dose-response relationships of CCCP (C; EC50: 198.0 nM) and valinomycin (D; EC50: 22.2 nM). Tau showed a CCCP-dependent electrophoretic mobility shift in Western blots (E; asterisk), consistent with tau dephosphorylation with increasing CCCP concentrations (F). OPA1 cleavage and tau dephosphorylation had different kinetics in CCCP-, and in valinomycin-treated Neuro2A cells (G), whereby CCCP, but not valinomycin, significantly reduced tau phosphorylation after 3 of treatment (H). Scatter plot shows mean ± SD; n(30 min) = 3; n(3 h) = 6; n(6 h) = 3; 2-way ANOVA: p ≤ 0.001.
Earlier studies have demonstrated dephosphorylation of tau proteins in rat brain cortical slides treated with CCCP and in PC12 cells treated with the structurally related protonophore FCCP [66, 67]. This could be replicated in Neuro2A cells incubated with 10 μM CCCP for 3 hours. CCCP thereby accelerated migration of tau in Western blots, which led to a dose-dependent shift in the size of the detected tau species (Figure 1E, asterisk). This antibody recognized tau isoforms of approximately 45-70 kDA and 90-130 kDa irrespective of the phosphorylation state (Table 1; supplementary material, Figure S1). The higher migrating proteins, however, were only detected in mouse Neuro2A neuroblastoma cells but not in human SH-SY5Y neuroblastoma cells (supplementary material, Figure S2). Increased electrophoretic mobility indicates the loss of posttranslational modifications, which could be confirmed with a phospho-tau specific antibody (Figure 1F, asterisk). This particular antibody is specific for tau phosphorylated at serine 199 and serine 202 (corresponding to mouse tau serine residues 188 and 191, see supplementary material, Figure S1A).
A time series to better understand the dynamics revealed significant tau dephosphorylation after 3 hours of CCCP exposure (Figure 1G & H). Valinomycin had much slower kinetics and tau alterations became only apparent after 6 hours of treatment. Discernable OPA1 cleavage on the other hand was already detectable within 30 minutes of CCCP or valinomycin exposure. Note, 30 μM CCCP and 30 μM valinomycin in these experiments were 30 to 300-times above the critical concentrations for OPA1 proteolysis in Western blots. Still, 3 hours of treatment with 30 μM CCCP or 30 μM valinomycin provided practically a binary readout for tau alterations and was therefore defined as standard paradigm for the following experiments.
3.2. CCCP but not valinomycin stimulated abundant tau dephosphorylation.
CCCP-induced tau dephosphorylation was not restricted to serines 199 and 202. Probing the samples with different phospho-tau specific antibodies demonstrated loss of phosphates also at threonine 231, serine 396 and serine 404 (Figure 2A; see also supplementary material, Figure S2). Valinomycin, however, did not show such an effect. Dephosphorylation of tau appeared to be somewhat specific because a pan-phosphoserine antibody did not display any changes in the CCCP-treated samples (supplementary material, Figure S3). A pan-phosphothreonine specific antibody at the same time indicated dephosphorylation of an unidentified protein migrating just above 34 kDa. Valinomycin again had also no effect on this unidentified protein. On the other hand, both CCCP and valinomycin induced OPA1 proteolysis as well as (autocatalytic) OMA1 degradation (Figure 2B). OPA1 is the main substrate of the OMA1 protease, which was reported to also cleave PGAM5 [68], misrouted PINK1 [69], and DELE1 [70, 71]. With the available antibodies, however, and under the herein defined experimental conditions no alterations in the other OMA1 substrates were noted (Figure 2B).
Figure 2: Abundant CCCP-dependent tau dephosphorylation.
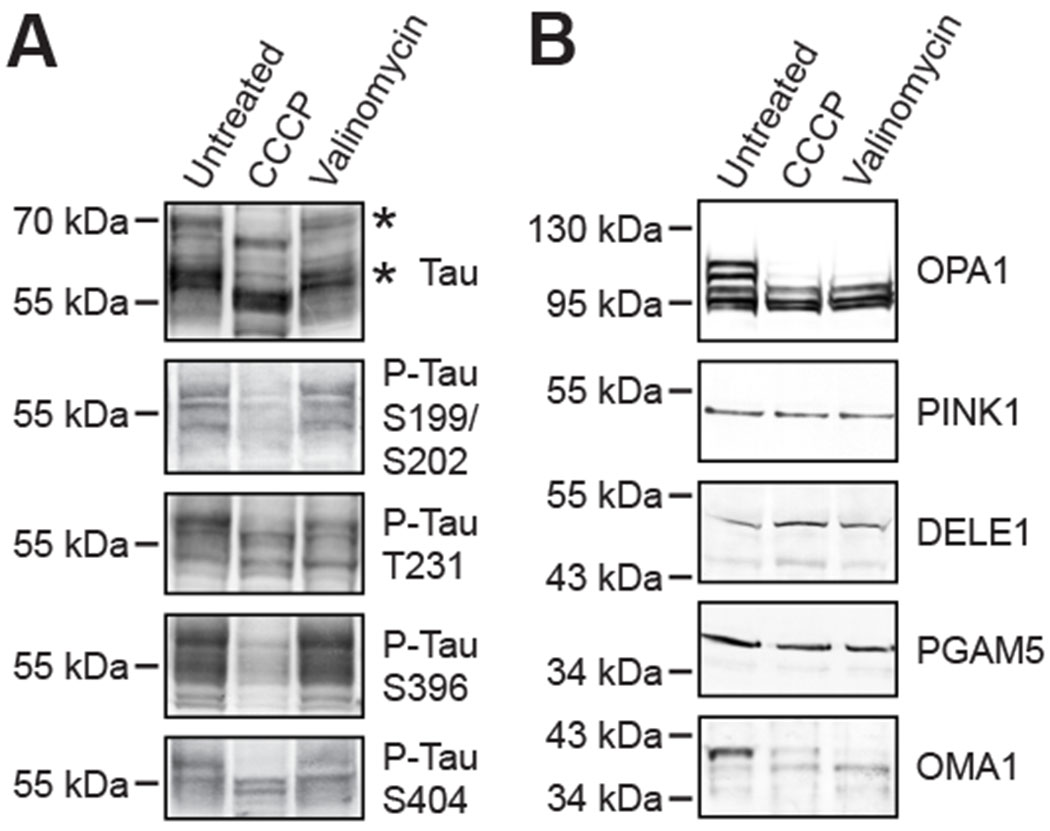
Western blots with 4 different phosphorylation site-specific antibodies consistently showed tau dephosphorylation at all sites in CCCP-treated Neuro2A cells, which coincided with tau’s increased electrophoretic mobility in these samples (A; asterisk); valinomycin had no effects on tau’s posttranslational modifications or its migration (A). Both CCCP and valinomycin stimulated proteolytic OPA1 hydrolysis and autocatalytic OMA1 cleavage in Neuro2A cells, but did not alter PINK1, DELE1 or PGAM5 (B).
3.3. No functional interaction between OMA1/OPA1 and tau dephosphorylation.
Discordance between CCCP and valinomycin, inconsistencies in the timing, and differences in the dosing implied proteolytic OPA1 regulation and tau dephosphorylation are unrelated events. Previous studies suggested that CCCP-dependent tau dephosphorylation was mediated by calcium signaling, because the chelating agent EGTA as well as cyclosporin A, which can inhibit protein phosphatase 2B (calcineurin), mitigated CCCP’s effects on tau [66]. Neuro2A cells cultured for 3 hours in the presence of 10 mM EGTA showed about 50% reduction in tau phosphorylation even in the absence of CCCP (Figure 3). The presence of CCCP in this experiment did not significantly change levels of phosphorylated tau when compared to cells treated with EGTA alone. 100 μM cyclosporin A for 3 hours appeared to increase the levels of phosphorylated tau (though without reaching statistical significance), while cyclosporin A could not prevent CCCP-dependent tau dephosphorylation in these experiments (Figure 3B). EGTA and cyclosporin A did not alter the ratios of the OPA1 isoforms in the absence of CCCP nor prevented OPA1 cleavage in the presence of CCCP (see Figure 3A). Together, these results separated tau dephosphorylation from OPA1 hydrolysis in Neuro2A cells.
Figure 3: Tau phosphorylation potentially calcium-dependent but independent from cyclosporin A.
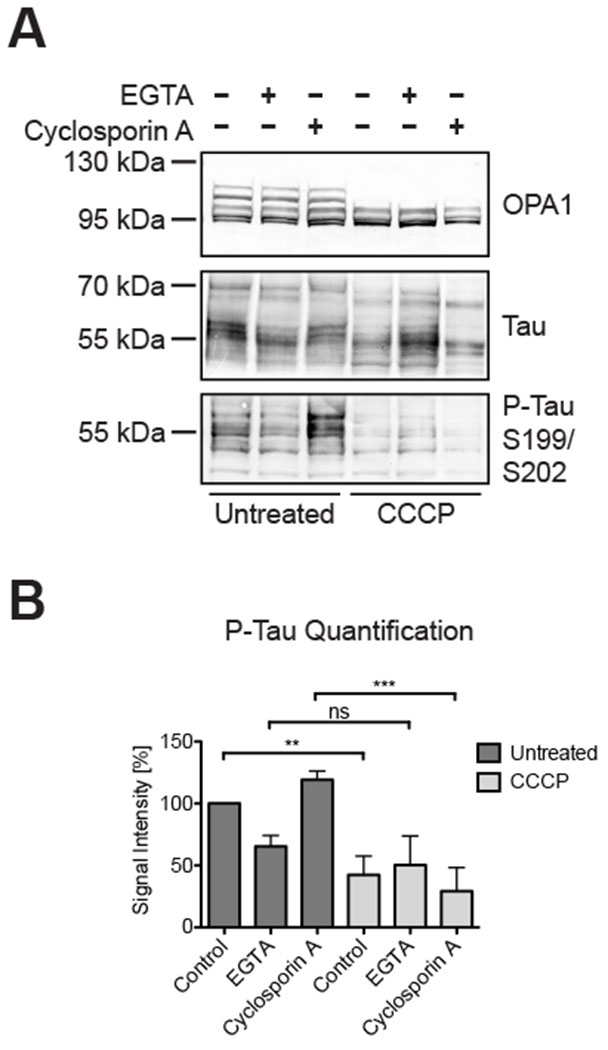
EGTA and cyclosporin A treatment of Neuro2A cells had no influence on CCCP-dependent OPA1 hydrolysis in Western blots (A). EGTA reduced tau phosphorylation irrespective of the presence of CCCP, whereas CCCP significantly reduced tau phosphorylation in untreated and cyclosporin A-treated cells, but not in EGTA-treated cells (B). Bar graph shows mean ± SD; n=3; 1-way ANOVA: p ≤ 0.01 (**); p ≤ 0.001 (***).
This conclusion could be confirmed with additional bioactive compounds also in human SH-SY5Y cells. Neuro2A cells (Figure 4A) and SH-SY5Y cells (Figure 4B) treated with 30 μM oligomycin A for 3 hours showed some reduction in L-OPA1 and no significant changes in tau phosphorylation. 30 μM staurosporine and rotenone instead significantly reduced tau phosphorylation but did not alter OPA1. Staurosporine most likely blocked GSK3B and other kinases which phosphorylate tau [72, 73]. Follow-up experiments with 3 different GSK3B inhibitors demonstrated tau dephosphorylation after 3 hours of exposure again without signs of OPA1 hydrolysis (Figure 4C). And 30 μM tipranavir as well as sorafenib induced OPA1 cleavage, while no effects on tau were noted. The HIV protease inhibitor tipranavir had an EC50 in the OMA1 protease assay of 9.7 μM (Figure 4D) and the kinase inhibitor sorafenib had an EC50 of 11.3 μM (Figure 4E). No differences were noted between Neuro2A and SH-SY5Y cells in their response to the different chemicals, which further substantiated that tau and OMA1-OPA1 are unrelated pathways.
Figure 4: Inconsistencies between OPA1 hydrolysis and tau dephosphorylation under different experimental conditions.
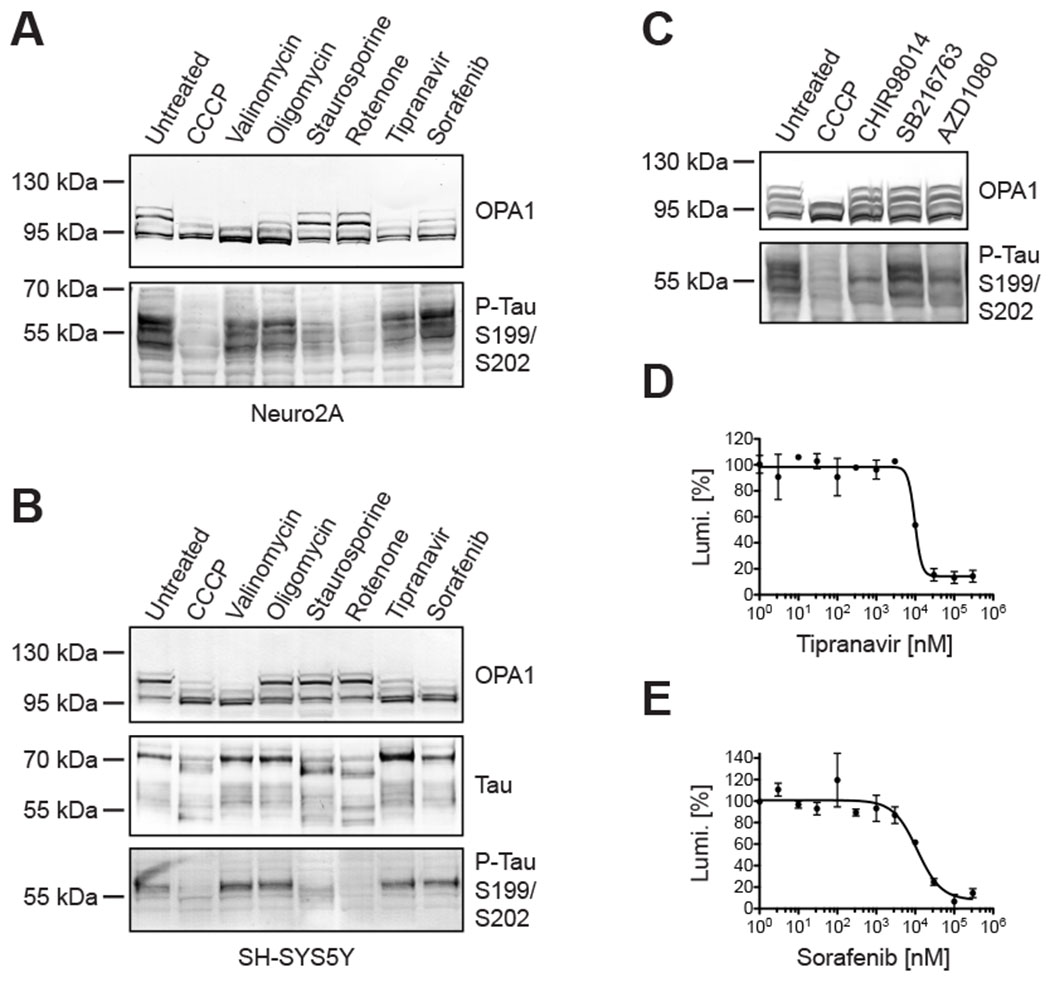
Tau dephosphorylation could be experimentally uncoupled from proteolytic OPA1 hydrolysis by exposing Neuro2A cells (A) and SH-SY5Y cells (B) to different molecules, whereby CCCP led to OPA1 hydrolysis and tau dephosphorylation in Western blots, while staurosporine and rotenone only evoked tau dephosphorylation, and tipranavir and sorafenib only induced OPA1 hydrolysis. The denoted GSK3B inhibitors impacted tau phosphorylation in Neuro2A cells without changing OPA1’s cleavage pattern in Western blots (C). The dose-response relationship of tipranavir (D; EC50: 9.7 μM) and sorafenib (E; EC50: 11.3 μM) in Neuro2A cells expressing an engineered luciferase as artificial OMA1 substrate.
3.4. Mitochondrial membrane potential Δψ not correlated with tau nor OPA1.
The protonophore CCCP disrupts the mitochondrial inner membrane potential Δψ, which powers the ATP synthase [74, 75]. To better understand the relationship of the inner membrane potential with OPA1 and tau, Δψ was determined in Neuro2A cells under the aforementioned experimental conditions. Relative Δψ levels were measured with the cationic fluorescent dye TMRE, which readily accumulates in active mitochondria [76, 77]. TMRE fluorescence was significantly reduced in Neuro2A cells treated with CCCP, rotenone, and tipranavir (Figure 5A). Again, tau was dephosphorylated in cells treated with CCCP, rotenone, and staurosporine (Figure 5B). And L-OPA1 was reduced in cells treated with CCCP, valinomycin, tipranavir, and sorafenib, but not in cells treated with rotenone or staurosporine (Figure 5C). Oligomycin showed some reduction of all three parameters without reaching statistical significance. All bioactives, however, reduced cellular ATP levels by 31.2% to 72.9% (Figure 5D), whereby cell viability was largely unaffected (supplementary material, Figure S4).
Figure 5: Δψ not correlated with tau phosphorylation nor OPA1 hydrolysis.
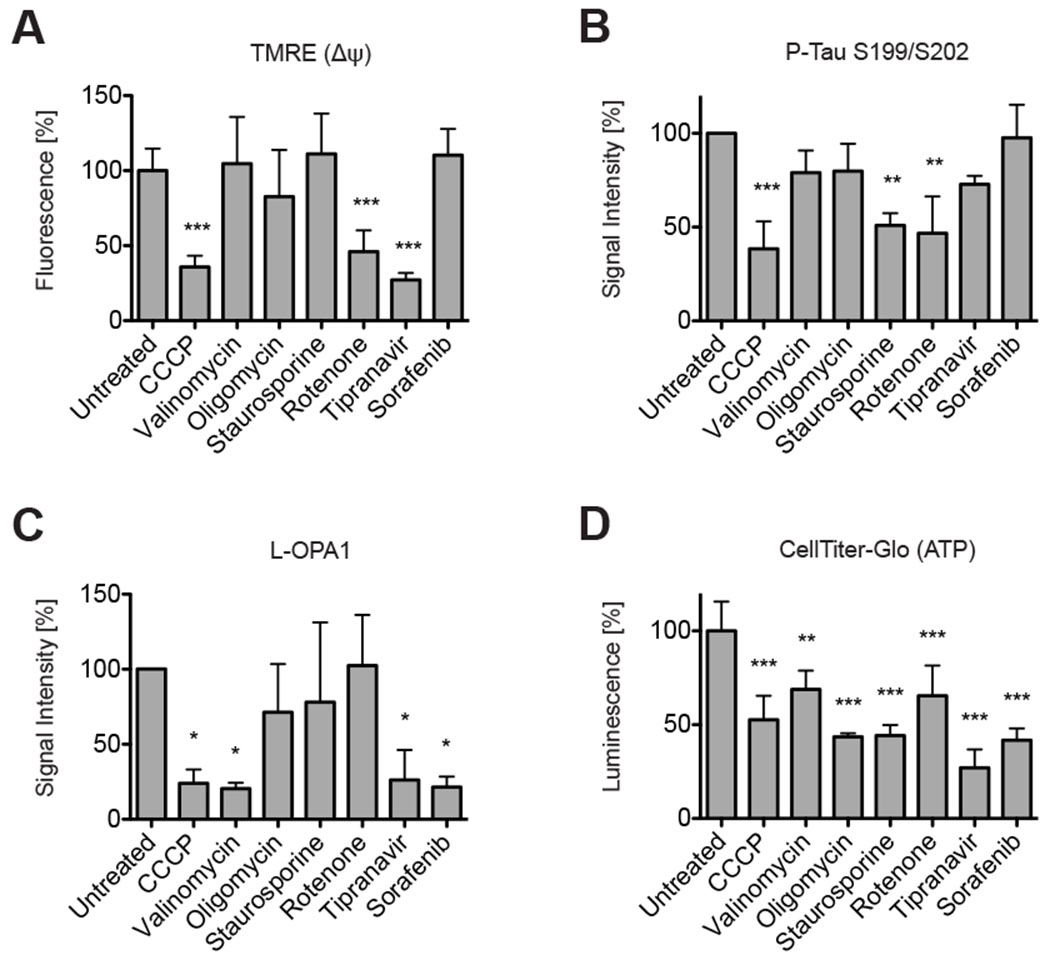
CCCP, rotenone, and tipranavir significantly reduced TMRE fluorescence, which can serve as proxy for the mitochondrial inner membrane potential Δψ (A). Densitometry of Western blots revealed significant phospho-tau reduction in Neuro2A cells treated with CCCP, staurosporine, and rotenone (B). L-OPA1 was significantly reduced in Western blots in Neuro2A cells treated with CCCP, valinomycin, tipranavir, and sorafenib (C). All bioactives significantly reduced ATP levels in the CellTiter-Glo assay, which measures ATP levels (D). All bar graphs show means of 3 independent experiments ± SD; 1-way ANOVA: p ≤ 0.05 (*); p ≤ 0.01 (**); p ≤ 0.001 (***).
Overall, there was no correlation between Δψ and the phosphorylation state of tau. Notably, there was also no correlation between Δψ and OMA1 activation. Rotenone lowered the membrane potential without stimulating L-OPA1 hydrolysis, while valinomycin had no effects on Δψ under the outlined experimental conditions, but evoked OPA1 cleavage. This shows OMA1’s activity is not Δψ-dependent.
3.5. No functional interaction between tau hyperphosphorylation and OMA1/OPA1.
The experiments so far demonstrated that OPA1 proteolysis, tau dephosphorylation and the membrane potential Δψ are not interrelated. Moreover, neither knockdown of OMA1 or OPA1 altered tau in Neuro2A cells regardless of CCCP treatment (Figure 6). OMA1 overexpression in Hek293T and Neuro2A cells had also no influence on tau (supplementary material, Figure S5).
Figure 6: OMA1 and OPA1 knockdown did not alter tau’s phosphorylation and tau hyperphosphorylation had no impact on OPA1 hydrolysis.
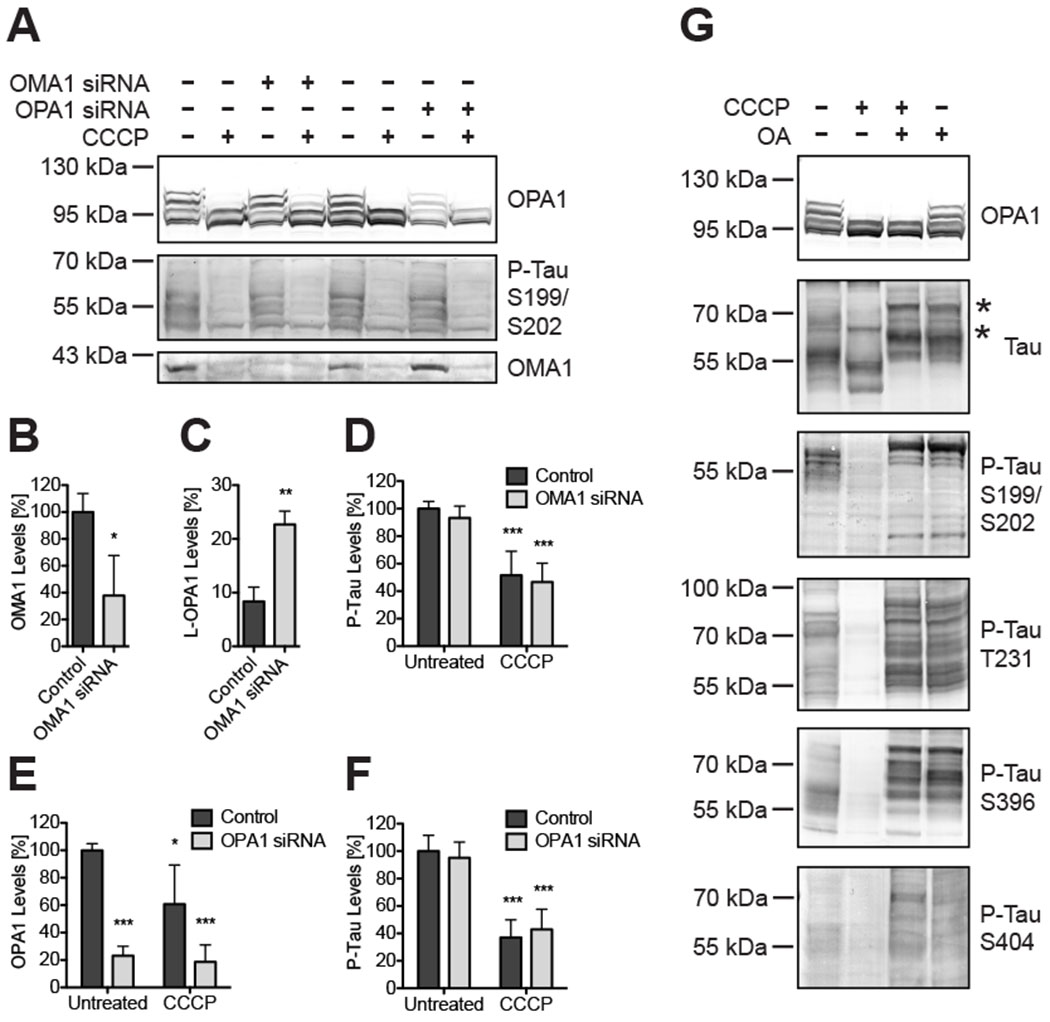
Western blots of Neuro2A cells indicated siRNA-mediated OMA1 knockdown or OPA1 knockdown does not change tau’s phosphorylation (A). Densitometry of Western blots showed significant OMA1 protein reduction in OMA1 knockdown cells to 37.8% ± 29.9 SD (B; t-test: p = 0.03; n=3), which rescued L-OPA1 in CCCP-treated cells (C; control: 8.4% ± 2.7 SD; OMA1 siRNA: 22.7% ± 2.5 SD; t-test: p ≤ 0.01; n=3). Only CCCP, but not OMA1 knockdown, resulted in significant changes in tau phosphorylation levels (D). OPA1 siRNA significantly reduced OPA1 protein levels by 76.7% ± 8.5 SD (E) without changing phospho-tau levels (F). Inhibiting phosphatases with okadaic acid (OA) resulted in tau hyperphosphorylation and electrophoretic mobility shifts (G; asterisk). CCCP-induced OPA1 proteolysis still occurred in OA-treated cells, but CCCP-dependent tau dephosphorylation was blocked by OA. All bar graphs show means of 3 independent experiments ± SD; 2-way ANOVA: p ≤ 0.05 (*); p ≤ 0.01 (**); p ≤ 0.001 (***).
A still open question was whether tau hyperphosphorylation would impact OMA1 and OPA1. Tau is mainly dephosphorylated by the protein phosphatase 2A [78], which can be inhibited by okadaic acid [79]. Okadaic acid hence can enhance phosphorylation of tau and other proteins [80–83]. 1 μM okadaic acid induced tau hyperphosphorylation also in Neuro2a cells, which was distinguishable by a marked shift upwards in tau’s electrophoretic mobility (Figure 6G; see also supplementary material, Figure S6). Tau hyperphosphorylation was consistent between all tested antibodies. CCCP had no impact on tau’s phosphorylation pattern in cells simultaneously treated with okadaic acid and CCCP. Conversely, okadaic acid did not change the ratios of the OPA1 isoforms nor inhibit L-OPA1 proteolysis in CCCP-treated samples. These results clearly demonstrate hyperphosphorylated tau and OPA1 are not functionally connected. In conclusion, there is no direct functional interaction between posttranslational tau modifications and regulation of the OMA1-OPA1 pathway.
4. Discussion
The present study focused on a potential functional interaction of the mitochondrial fusion protein OPA1 and the microtubule-associated protein tau. Neuroblastoma cells were cultured under defined conditions that either triggered OPA1 proteolysis, tau phosphorylation/dephosphorylation, or both. These experiments clearly demonstrated that posttranslational tau modifications and the OMA1-OPA1 pathway are two independent mechanisms, which are not causally connected. The 3-hour duration of the various interventions herein was purposefully set fairly short to mitigate secondary effects due to slower processes, such as mitophagy or apoptosis. The experiments were also designed in a straightforward manner, assessing OPA1 and tau directly, thus limiting the impact of confounding factors and secondary effects. For example, the mitochondrial network morphology changes over the course of the cell-cycle and with energy-metabolic demands [84], which makes its evaluation as a marker rather complicated–even more so in malignant neuroblasts in the context of neurodegeneration. (The cells of this study were not differentiated, for instance by retinoic acid, to exhibit a more neuronal phenotype.) The dynamic mitochondrial network can also adapt to various factors and numerous stressors by reorganizing its morphology. 18% of all approved cancer drugs activated OMA1 in a high-throughput drug screening campaign [62]. And almost 10% of a panel of genes encoding the mitochondrial proteome altered the mitochondrial network morphology in an siRNA-screen [85]. Tau may be modulated by an equally diverse set of factors and variables [86–88]. The plain and simple experimental approach of this study thus might help tame at least some of the complexity.
On the other hand, the different modes of action and pleiotropic effects of the herein investigated compounds do not lend themselves to an easy interpretation of the results. For instance, GSK3B is among the tau-phosphorylating kinases and also involved in the regulation of mitochondria [89–93]. Interestingly, OMA1’s proteasomal degradation appears to be GSK3B-dependent, because the protease was stabilized (and OPA1 cleavage restored) in mesenchymal stem cells treated for 24 hours with 10 μM of the GSK3B-inhibitor SB216763 [94]. Yet, three different GSK3B inhibitors showed no effects on OPA1 under the experimental paradigm of this study (i.e. 30 μM for 3 hours), while leading to tau-dephosphorylation. Another example is CCCP, which disrupts the inner membrane potential by carrying protons across the membrane with a pH-independent maximum conductivity at a concentration of about 10 μM [95]. Yet, 1 μM sufficed to convert all L-OPA1 into S-OPA1 within short time. Also the potassium-ionophore valinomycin activated OMA1 without changing Δψ [96]. (Again, OMA1 is unlikely to be regulated by the membrane potential Δψ.) However, 10 μM CCCP over an extended period of time were necessary to observe an effect on tau, suggesting Δψ could indeed be connected with tau. Such a connection could involve calcium ions, which are stored in electronegative mitochondria and which are released upon loss of the membrane potential [97]. This would explain why valinomycin had little effect on tau as well as the results obtained with EGTA. Yet, the calcineurin inhibitor cyclosporin A failed in preventing CCCP-induced tau dephosphorylation, which is at odds with previous reports [98, 99]. Alternative models for CCCP’s mechanism of action may involve microtubules. For example, 60 μM FCCP (but not 1.8 μM valinomycin) disassembled microtubules within 4 hours in HeLa and BHK21 cells [100]. And also rotenone, a complex I inhibitor [101, 102], was found to destabilize microtubules [103–105]. To conclude, this study is not suited to elucidate the means that regulate tau or OMA1 (nor was it designed to). Still, the data conclusively demonstrated that tau and the OMA1-OPA1 pathway are two independent mechanisms, which are not causally linked in neuroblasts.
A limitation of the present study is the focus on tau’s posttranslational modifications, because proteotoxicity is another aspect likely tied to OMA1’s regulation. For instance, OMA1 and YME1L1 double-knockout yeast cells grew only under reduced temperatures [106]. Mammalian cells challenged by heat stress showed OMA1-dependent OPA1 hydrolysis [107]. And knockdown of the m-AAA protease in fibroblasts resulted in OMA1 activation, which was rescued by chloramphenicol [108]. Chloramphenicol can inhibit ribosomal translation, which together with the aforementioned experiments suggests involvement of proteotoxicity in OMA1’s regulation. Certain tau species or conformers can propagate from one cell to another and seed endogenous tau in a prion-like mechanism [109]. There is the possibility that these tau aggregates and neurofibrillary tangles influence OMA1’s activity, which is an area of ongoing research. An alternative possibility is that the factors and/or conditions that lead to the formation of tau aggregates and neurofibrillary tangles also impact the OMA1-OPA1 pathway.
Supplementary Material
Highlights.
Damaged mitochondria a cause for, the consequence of, or coincide with Alzheimer’s?
OMA1-dependent OPA1 proteolysis is a signature event of mitochondrial damage.
This study demonstrates OPA1 cleavage does not cause tau hyperphosphorylation.
Furthermore, tau hyperphosphorylation does not activate OMA1.
Concluding, tau and OPA1/OMA1 may act in parallel but are functionally independent.
Acknowledgments
Thank you for the generous support of my research by the National Institute on Aging (NIA) of the National Institutes of Health (NIH) under Award Number R43AG063642.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Dr. Marcel V. Alavi is shareholder of 712 North Inc., a California-based pharmaceutical company.
CRediT author statement
Marcel V. Alavi: Conceptualization, Investigation, Formal analysis, Visualization, Writing – Original Draft, Review & Editing
References
- [1].Arnsten AFT, Datta D, Tredici KD, Braak H, Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease, Alzheimers Dement 17 (2021) 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I, Tau pathology in Alzheimer disease and other tauopathies, Biochim Biophys Acta 1739 (2005) 198–210. [DOI] [PubMed] [Google Scholar]
- [3].Karran E, Mercken M, De Strooper B, The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics, Nature reviews. Drug discovery 10 (2011) 698–712. [DOI] [PubMed] [Google Scholar]
- [4].Taylor JP, Hardy J, Fischbeck KH, Toxic proteins in neurodegenerative disease, Science 296 (2002) 1991–1995. [DOI] [PubMed] [Google Scholar]
- [5].De Strooper B, Karran E, The Cellular Phase of Alzheimer’s Disease, Cell 164 (2016) 603–615. [DOI] [PubMed] [Google Scholar]
- [6].Chang CW, Shao E, Mucke L, Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies, Science 371 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hardy JA, Higgins GA, Alzheimer’s disease: the amyloid cascade hypothesis, Science 256 (1992) 184–185. [DOI] [PubMed] [Google Scholar]
- [8].Wang Y, Mandelkow E, Tau in physiology and pathology, Nat Rev Neurosci 17 (2016) 5–21. [DOI] [PubMed] [Google Scholar]
- [9].Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW, Tau, tangles, and Alzheimer’s disease, Biochim Biophys Acta 1739 (2005) 216–223. [DOI] [PubMed] [Google Scholar]
- [10].Suidan GL, Ramaswamy G, Targeting Apolipoprotein E for Alzheimer’s Disease: An Industry Perspective, Int J Mol Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Serrano-Pozo A, Das S, Hyman BT, APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches, Lancet Neurol 20 (2021) 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bell SM, Barnes K, De Marco M, Shaw PJ, Ferraiuolo L, Blackburn DJ, Venneri A, Mortiboys H, Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future?, Biomedicines 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, Wallace DC, A mitochondrial etiology of Alzheimer and Parkinson disease, Biochim Biophys Acta 1820 (2012) 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Weidling IW, Swerdlow RH, Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis, Exp Neurol 330 (2020) 113321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Flannery PJ, Trushina E, Mitochondrial dynamics and transport in Alzheimer’s disease, Mol Cell Neurosci 98 (2019) 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Albensi BC, Dysfunction of mitochondria: Implications for Alzheimer’s disease, Int Rev Neurobiol 145 (2019) 13–27. [DOI] [PubMed] [Google Scholar]
- [17].Baloyannis SJ, Costa V, Michmizos D, Mitochondrial alterations in Alzheimer’s disease, Am J Alzheimers Dis Other Demen 19 (2004) 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA, Mitochondrial abnormalities in Alzheimer’s disease, The Journal of neuroscience : the official journal of the Society for Neuroscience 21 (2001) 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Butterfield DA, Drake J, Pocernich C, Castegna A, Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide, Trends in molecular medicine 7 (2001) 548–554. [DOI] [PubMed] [Google Scholar]
- [20].Gibson GE, Sheu KF, Blass JP, Abnormalities of mitochondrial enzymes in Alzheimer disease, Journal of neural transmission 105 (1998) 855–870. [DOI] [PubMed] [Google Scholar]
- [21].Maurer I, Zierz S, Moller HJ, A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients, Neurobiology of aging 21 (2000) 455–462. [DOI] [PubMed] [Google Scholar]
- [22].Parker WD Jr., Filley CM, Parks JK, Cytochrome oxidase deficiency in Alzheimer’s disease, Neurology 40 (1990) 1302–1303. [DOI] [PubMed] [Google Scholar]
- [23].Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N, Oxidative damage in Alzheimer’s, Nature 382 (1996) 120–121. [DOI] [PubMed] [Google Scholar]
- [24].Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X, Abnormal mitochondrial dynamics and neurodegenerative diseases, Biochim Biophys Acta 1802 (2010) 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bonilla E, Tanji K, Hirano M, Vu TH, DiMauro S, Schon EA, Mitochondrial involvement in Alzheimer’s disease, Biochim Biophys Acta 1410 (1999) 171–182. [DOI] [PubMed] [Google Scholar]
- [26].Sabouny R, Shutt TE, Reciprocal Regulation of Mitochondrial Fission and Fusion, Trends Biochem Sci 45 (2020) 564–577. [DOI] [PubMed] [Google Scholar]
- [27].Chan DC, Mitochondrial Dynamics and Its Involvement in Disease, Annu Rev Pathol 15 (2020) 235–259. [DOI] [PubMed] [Google Scholar]
- [28].Youle RJ, van der Bliek AM, Mitochondrial fission, fusion, and stress, Science 337 (2012) 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Perez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA, Caspase-Cleaved Tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease, Mol Neurobiol 55 (2018) 1004–1018. [DOI] [PubMed] [Google Scholar]
- [30].Manczak M, Calkins MJ, Reddy PH, Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage, Hum Mol Genet 20 (2011) 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X, Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease, The Journal of neuroscience : the official journal of the Society for Neuroscience 29 (2009) 9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Birnbaum JH, Wanner D, Gietl AF, Saake A, Kundig TM, Hock C, Nitsch RM, Tackenberg C, Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-beta and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients, Stem Cell Res 27 (2018) 121–130. [DOI] [PubMed] [Google Scholar]
- [33].Oliver D, Reddy PH, Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases, Cells 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Orr AL, Kim C, Jimenez-Morales D, Newton BW, Johnson JR, Krogan NJ, Swaney DL, Mahley RW, Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function, J Alzheimers Dis 68 (2019) 991–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW, Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease, J Biol Chem 286 (2011) 5215–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schmukler E, Solomon S, Simonovitch S, Goldshmit Y, Wolfson E, Michaelson DM, Pinkas-Kramarski R, Altered mitochondrial dynamics and function in APOE4-expressing astrocytes, Cell death & disease 11 (2020) 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Faelber K, Dietrich L, Noel JK, Wollweber F, Pfitzner AK, Muhleip A, Sanchez R, Kudryashev M, Chiaruttini N, Lilie H, Schlegel J, Rosenbaum E, Hessenberger M, Matthaeus C, Kunz S, von der Malsburg A, Noe F, Roux A, van der Laan M, Kuhlbrandt W, Daumke O, Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1, Nature 571 (2019) 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang D, Zhang Y, Ma J, Zhu C, Niu T, Chen W, Pang X, Zhai Y, Sun F, Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding, Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yan L, Qi Y, Ricketson D, Li L, Subramanian K, Zhao J, Yu C, Wu L, Sarsam R, Wong M, Lou Z, Rao Z, Nunnari J, Hu J, Structural analysis of a trimeric assembly of the mitochondrial dynamin-like GTPase Mgm1, Proceedings of the National Academy of Sciences of the United States of America 117 (2020) 4061–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH, Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane, Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Akepati VR, Muller EC, Otto A, Strauss HM, Portwich M, Alexander C, Characterization of OPA1 isoforms isolated from mouse tissues, J Neurochem 106 (2008) 372–383. [DOI] [PubMed] [Google Scholar]
- [42].Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS, Fission and selective fusion govern mitochondrial segregation and elimination by autophagy, EMBO J 27 (2008) 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T, The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission, J Cell Biol 204 (2014) 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rainbolt TK, Saunders JM, Wiseman RL, YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress, EMBO Rep 16 (2015) 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L, D’Hooge R, Scorrano L, De Strooper B, Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling, Cell 126 (2006) 163–175. [DOI] [PubMed] [Google Scholar]
- [46].Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G, Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis, J Biol Chem 278 (2003) 7743–7746. [DOI] [PubMed] [Google Scholar]
- [47].Song Z, Chen H, Fiket M, Alexander C, Chan DC, OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L, J Cell Biol 178 (2007) 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS, Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology, J Biol Chem 281 (2006) 37972–37979. [DOI] [PubMed] [Google Scholar]
- [49].Ishihara N, Fujita Y, Oka T, Mihara K, Regulation of mitochondrial morphology through proteolytic cleavage of OPA1, EMBO J 25 (2006) 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, Rugarli EI, Langer T, Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration, PLoS Genet 8 (2012) e1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Korwitz A, Merkwirth C, Richter-Dennerlein R, Troder SE, Sprenger HG, Quiros PM, Lopez-Otin C, Rugarli EI, Langer T, Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria, J Cell Biol 212 (2016) 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li L, Martin-Levilain J, Jimenez-Sanchez C, Karaca M, Foti M, Martinou JC, Maechler P, In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way, J Biol Chem 294 (2019) 12581–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Anderson CJ, Kahl A, Fruitman H, Qian L, Zhou P, Manfredi G, Iadecola C, Prohibitin levels regulate OMA1 activity and turnover in neurons, Cell Death Differ 27 (2020) 1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Alavi MV, Fuhrmann N, Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics, Mol Neurodegener 8 (2013) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hinton DR, Sadun AA, Blanks JC, Miller CA, Optic-nerve degeneration in Alzheimer’s disease, N Engl J Med 315 (1986) 485–487. [DOI] [PubMed] [Google Scholar]
- [56].Caglayan S, Hashim A, Cieslar-Pobuda A, Jensen V, Behringer S, Talug B, Chu DT, Pecquet C, Rogne M, Brech A, Brorson SH, Nagelhus EA, Hannibal L, Boschi A, Tasken K, Staerk J, Optic Atrophy 1 Controls Human Neuronal Development by Preventing Aberrant Nuclear DNA Methylation, iScience 23 (2020) 101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ambrad Giovannetti E, Fuhrmann M, Unsupervised excitation: GABAergic dysfunctions in Alzheimer’s disease, Brain Res 1707 (2019) 216–226. [DOI] [PubMed] [Google Scholar]
- [58].Govindpani K, Calvo-Flores Guzman B, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A, Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease, Int J Mol Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Solas M, Puerta E, Ramirez MJ, Treatment Options in Alzheimer s Disease: The GABA Story, Curr Pharm Des 21 (2015) 4960–4971. [DOI] [PubMed] [Google Scholar]
- [60].Marczynski TJ, GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease revisited, Brain Res Bull 45 (1998) 341–379. [DOI] [PubMed] [Google Scholar]
- [61].Xu Y, Zhao M, Han Y, Zhang H, GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment, Front Neurosci 14 (2020) 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Alavi MV, OMA1 High-throughput Screen Reveals Protease Activation by Kinase Inhibitors, ACS Chem Biol submitted (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis, Nat Methods 9 (2012) 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T, Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1, J Cell Biol 187 (2009) 1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM, Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells, J Cell Biol 187 (2009) 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Norman SG, Johnson GV, Compromised mitochondrial function results in dephosphorylation of tau through a calcium-dependent process in rat brain cerebral cortical slices, Neurochem Res 19 (1994) 1151–1158. [DOI] [PubMed] [Google Scholar]
- [67].Luo Y, Ingram VM, Uncoupling of mitochondria activates protein phosphatases and inactivates MBP protein kinases, J Alzheimers Dis 3 (2001) 593–598. [DOI] [PubMed] [Google Scholar]
- [68].Wai T, Saita S, Nolte H, Muller S, Konig T, Richter-Dennerlein R, Sprenger HG, Madrenas J, Muhlmeister M, Brandt U, Kruger M, Langer T, The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L, EMBO Rep 17 (2016) 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, Youle RJ, Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1, Mol Cell 73 (2019) 1028–1043 e1025. [DOI] [PubMed] [Google Scholar]
- [70].Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, Jae LT, A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol, Nature 579 (2020) 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M, Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway, Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, Eisenbrand G, Meijer L, Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors?, J Biol Chem 276 (2001) 251–260. [DOI] [PubMed] [Google Scholar]
- [73].Dillon GM, Henderson JL, Bao C, Joyce JA, Calhoun M, Amaral B, King KW, Bajrami B, Rabah D, Acute inhibition of the CNS-specific kinase TTBK1 significantly lowers tau phosphorylation at several disease relevant sites, PLoS One 15 (2020) e0228771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Heytler PG, Uncoupling of oxidative phosphorylation by carbonyl cyanide phenylhydrazones. I. Some characteristics of m-Cl-CCP action on mitochondria and chloroplasts, Biochemistry 2 (1963) 357–361. [DOI] [PubMed] [Google Scholar]
- [75].Heytler PG, Prichard WW, A new class of uncoupling agents--carbonyl cyanide phenylhydrazones, Biochem Biophys Res Commun 7 (1962) 272–275. [DOI] [PubMed] [Google Scholar]
- [76].Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM, Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes, Biophys J 53 (1988) 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Scaduto RC Jr., Grotyohann LW, Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives, Biophys J 76 (1999) 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K, Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease, J Biol Chem 275 (2000) 5535–5544. [DOI] [PubMed] [Google Scholar]
- [79].Bialojan C, Takai A, Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics, Biochem J 256 (1988) 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Furiya Y, Sahara N, Mori H, Okadaic acid enhances abnormal phosphorylation on tau proteins, Neurosci Lett 156 (1993) 67–69. [DOI] [PubMed] [Google Scholar]
- [81].Pei JJ, Gong CX, An WL, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K, Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer’s disease, Am J Pathol 163 (2003) 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Harris KA, Oyler GA, Doolittle GM, Vincent I, Lehman RA, Kincaid RL, Billingsley ML, Okadaic acid induces hyperphosphorylated forms of tau protein in human brain slices, Ann Neurol 33 (1993) 77–87. [DOI] [PubMed] [Google Scholar]
- [83].Rametti A, Esclaire F, Yardin C, Terro F, Linking alterations in tau phosphorylation and cleavage during neuronal apoptosis, J Biol Chem 279 (2004) 54518–54528. [DOI] [PubMed] [Google Scholar]
- [84].Alavi MV, Targeted OMA1 therapies for cancer, Int J Cancer 145 (2019) 2330–2341. [DOI] [PubMed] [Google Scholar]
- [85].Cretin E, Lopes P, Vimont E, Tatsuta T, Langer T, Gazi A, Sachse M, Yu-Wai-Man P, Reynier P, Wai T, High-throughput screening identifies suppressors of mitochondrial fragmentation in OPA1 fibroblasts, EMBO Mol Med (2021) e13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Song L, Wells EA, Robinson AS, Critical Molecular and Cellular Contributors to Tau Pathology, Biomedicines 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wegmann S, Biernat J, Mandelkow E, A current view on Tau protein phosphorylation in Alzheimer’s disease, Curr Opin Neurobiol 69 (2021) 131–138. [DOI] [PubMed] [Google Scholar]
- [88].Alquezar C, Arya S, Kao AW, Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation, Front Neurol 11 (2020) 595532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yang K, Chen Z, Gao J, Shi W, Li L, Jiang S, Hu H, Liu Z, Xu D, Wu L, The Key Roles of GSK-3beta in Regulating Mitochondrial Activity, Cell Physiol Biochem 44 (2017) 1445–1459. [DOI] [PubMed] [Google Scholar]
- [90].Lauretti E, Dincer O, Pratico D, Glycogen synthase kinase-3 signaling in Alzheimer’s disease, Biochim Biophys Acta Mol Cell Res 1867 (2020) 118664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hooper C, Killick R, Lovestone S, The GSK3 hypothesis of Alzheimer’s disease, Journal of neurochemistry 104 (2008) 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Reddy PH, Amyloid beta-induced glycogen synthase kinase 3beta phosphorylated VDAC1 in Alzheimer’s disease: implications for synaptic dysfunction and neuronal damage, Biochim Biophys Acta 1832 (2013) 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sayas CL, Avila J, GSK-3 and Tau: A Key Duet in Alzheimer’s Disease, Cells 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yang F, Wu R, Jiang Z, Chen J, Nan J, Su S, Zhang N, Wang C, Zhao J, Ni C, Wang Y, Hu W, Zeng Z, Zhu K, Liu X, Hu X, Zhu W, Yu H, Huang J, Wang J, Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation, Cell death & disease 9 (2018) 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Leblanc OH Jr., The effect of uncouplers of oxidative phosphorylation on lipid bilayer membranes: Carbonylcyanidem-chlorophenylhydrazone, J Membr Biol 4 (1971) 227–251. [DOI] [PubMed] [Google Scholar]
- [96].Moore C, Pressman BC, Mechanism of Action of Valinomycin on Mitochondria, Biochemical and Biophysical Research Communications 16 (1964) 562-&.5871847 [Google Scholar]
- [97].Beatrice MC, Palmer JW, Pfeiffer DR, The relationship between mitochondrial membrane permeability, membrane potential, and the retention of Ca2+ by mitochondria, J Biol Chem 255 (1980) 8663–8671. [PubMed] [Google Scholar]
- [98].Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K, Alzheimer’s disease abnormally phosphorylated tau is dephosphorylated by protein phosphatase-2B (calcineurin), Journal of neurochemistry 62 (1994) 803–806. [DOI] [PubMed] [Google Scholar]
- [99].Drewes G, Mandelkow EM, Baumann K, Goris J, Merlevede W, Mandelkow E, Dephosphorylation of tau protein and Alzheimer paired helical filaments by calcineurin and phosphatase-2A, FEBS Lett 336 (1993) 425–432. [DOI] [PubMed] [Google Scholar]
- [100].Maro B, Marty MC, Bornens M, In vivo and in vitro effects of the mitochondrial uncoupler FCCP on microtubules, EMBO J 1 (1982) 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Okun JG, Lummen P, Brandt U, Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase), J Biol Chem 274 (1999) 2625–2630. [DOI] [PubMed] [Google Scholar]
- [102].Heinz S, Freyberger A, Lawrenz B, Schladt L, Schmuck G, Ellinger-Ziegelbauer H, Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation, Sci Rep 7 (2017) 45465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Brinkley BR, Barham SS, Barranco SC, Fuller GM, Rotenone inhibition of spindle microtubule assembly in mammalian cells, Exp Cell Res 85 (1974) 41–46. [DOI] [PubMed] [Google Scholar]
- [104].Marshall LE, Himes RH, Rotenone inhibition of tubulin self-assembly, Biochim Biophys Acta 543 (1978) 590–594. [DOI] [PubMed] [Google Scholar]
- [105].Srivastava P, Panda D, Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding, FEBS J 274 (2007) 4788–4801. [DOI] [PubMed] [Google Scholar]
- [106].Bohovych I, Donaldson G, Christianson S, Zahayko N, Khalimonchuk O, Stress-triggered activation of the metalloprotease Oma1 involves its C-terminal region and is important for mitochondrial stress protection in yeast, J Biol Chem 289 (2014) 13259–13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T, Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics, EMBO J 33 (2014) 578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Richter U, Ng KY, Suomi F, Marttinen P, Turunen T, Jackson C, Suomalainen A, Vihinen H, Jokitalo E, Nyman TA, Isokallio MA, Stewart JB, Mancini C, Brusco A, Seneca S, Lombes A, Taylor RW, Battersby BJ, Mitochondrial stress response triggered by defects in protein synthesis quality control, Life Sci Alliance 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ayers JI, Paras NA, Prusiner SB, Expanding spectrum of prion diseases, Emerg Top Life Sci 4 (2020) 155–167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.