

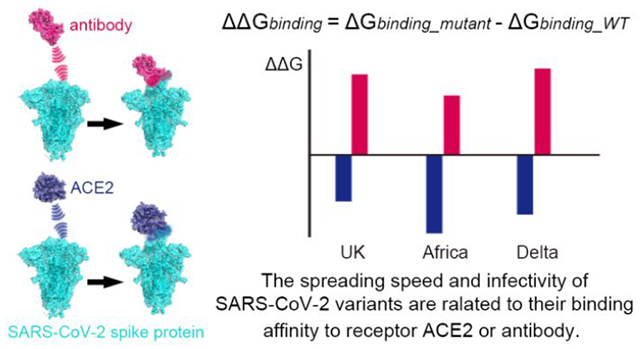
Abstract
The pandemic caused by SARS-CoV-2 has cost millions of lives and tremendous social/financial loss. The virus continues to evolve and mutate. In particular, the recent emerged of the “UK”, “South Africa” and Delta variants show higher infectivity and spreading speed. Thus the relationship between the mutations of certain amino acids and the spreading speed of the virus is a problem of great importance. In this respect, understanding the mutational mechanism is crucial for surveillance and prediction of future mutations as well as antibody/vaccine development. In this work, we used a coarse-grained model ( that was used previously in predicting the importance of mutations of N501) in calculating the free energy change of various types of single site or combined sites mutations. This was done for the UK mutant, the South African mutant and the Delta mutant. We investigated the underlying mechanisms of the binding affinity changes for mutations at different spike protein domains of SARS-CoV-2 and provided the energy basis for the resistance of the E484 mutant to the antibody m396. Other potential mutation sites were also predicted. Furthermore, the in-silico predictions were assessed by functional experiments. The results establish that the faster spreading of recently observed mutants is strongly correlated with the binding affinity enhancement between virus and human receptor as well as with the reduction of the binding to the m396 antibody. Significantly, the current approach offers a way for predicting new variants and to assess the effectiveness of different antibodies towards such variants.
Graphical Abstract
I. Introduction
The spreading of the novel coronavirus (SARS-CoV-2) outbreak has cost million lives and immeasurable financial loss up to July 16th 2021, according to World Health Organization (WHO)1. To fight the pandemic, extensive efforts have been put into the structure determination, mechanism study, and drug/antibody/vaccine design2–12. However, the SARS-CoV-2 continues to evolve and mutate, bringing essential impact on the spreading speed, the fatality rate, and the relative population of mutants13–19. Moreover, such mutations would in turn reduce the recognition of virus by human antibody-mediated vaccines. This would lead to ineffectiveness of vaccines or suppress diagnostic detection20–26. It is crucial to understand the structural/energy basis of the mechanism of the mutational effects and to provide reliable predictions in order to facilitate the development of cures such as antibodies and vaccines.
Previous works have suggested that the affinity of the binding the spike protein (in the following text “spike” is referred to the spike domain of SARS-CoV-2 virus) to the ACE2 receptor correlates with the spreading speed27, the infectivity28, and the population of the mutants17. Our work in June 2020 predicted that certain mutations of the spike protein at N501 might lead to stronger binding of ACE2, ahead of the appearance on December of the “UK” mutant (SARS-CoV-2 VOC 202012/01) that was found to contains the N501Y substitution29. This mutant has spread across the United Kingdom with increased transmissibility30 . Another “South Africa” variant (501Y.V2) that was reported by national authorities in South Africa on 18 December contains three substitutions: K417N, E484K, and N501Y31. This variant spread extremely fast and displaced other lineages circulating in South Africa. During early 2021, the Delta variant (B.1.617.2) exploded in India and rapidly spread to other continents of the world. It was proposed that the dominance of the Delta variant in India is a result of evasion of antibodies, increased activity32, and increased transmissibility33.
The spread of the variants challenges the scientific community to find ways to predict the effects of different mutations on the virus binding to the receptor and the binding of the antibodies to the virus. Arguably one of the earliest attempt to address this issue computationally has been our work that predicted some residues with significant mutational effects and in particular the N501 residue whose mutations appeared in several of the new variants29. Subsequent experimental alanine scanning plus computational study led to interesting retinal enhancement in the binding of the spike to the receptor34. The work provided (despite the limitation of the energy calculations used) an important prove of principle, but was not used in exploring the evolution of variants.
Another interesting computational study was reported by Zimmerman et al. who run a very massive simulations observing a large conformational change landscape35. However this study has not provided a way to obtain mutational free energies. Another long simulation was reported by Sztain et al. who simulated the glycan gate controls opening of the spike36. Furthermore, this approach did not provide binding free energies. It is also obvious that the two studies mentioned above cannot provide fast estimates of mutational effects.
Another interesting approach was reported by Hie et al. and Maher et al. used machine learning to assess mutational effects37–38. However, the free energy changes of new variants were not assessed. It is also important to mention directed evolution study by Higuchi and his coworkers that engineered an ACE2 construct to bind and neutralize SARS-Cov-2 at an EC50 of 100-fold below wild-type ACE2, overcoming mutational escape39.
Of particular interest are recent studies that analyzed the structural effects of mutations contributed to the understanding of the corresponding biological implications and provided clear information on some mutational induced structural variations40–42. However, despite the clear importance of these works they do not provide the crucial estimates of the changes in binding energies.
It is also important to mention experimental studies that detected large conformational changes with several different structures of the free spike region43. It was also concluded that the binding process involves at least one step. The observation of conformational changes is clearly important and should be taken into account in some way. However, starting from the structure of the bound complex reduces the uncertainties associated with conformational changes including those induced by mutations (see below). Basically, studies of binding energies should use mutated structure if it is available, but use the structure if the mutated structure is not available (see below).
Regardless of the advances discussed above, none of the mentioned works analyzed energetics of the key new variants considering both the spike binding to the receptor and the binding to antibodies.
The challenge that we like to address in this work is to explore the energetics of the different variants by an approach that can be used in relatively fast screening.
To better explore the energy/structural basis of the three variants we used here the same coarse-grained (CG) model44–46 as in the previous work29 to systemically investigate the binding free energy changes upon mutations of the individual or combination of amino acid substitutions for the ACE2-spike complex or for the m396(antibody)-spike complex. The overlapped structures of ACE2-Spike complex and m396-Spike complex are shown in Fig. 1, only part of ACE2-Spike binding interface is covered by m396 antibody.
Fig. 1. The overlap of ACE2-Spike complex and m396-Spike complex.
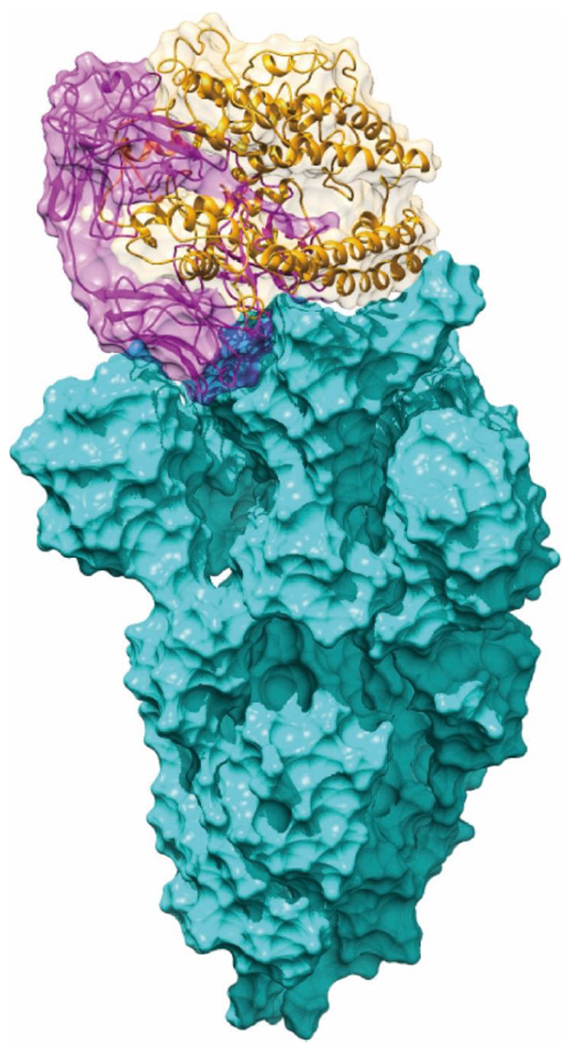
The ACE2 of the ACE2-Spike complex is shown in orange. The m396 of the m396-Spike complex is shown in magenta. The Spike protein is shown in cyan.
The binding free energy change for the ACE2-spike complex is defined here as follows:
| Eq. 1 |
where indicates how the ACE2-spike complex is stabilized after mutation while shows how spike protein is destabilized, respectively. For the human antibody m396 and the spike protein complex we use the same definition of and (see methods).
At this point it might be useful to comment about mutational induced structural changes. Actually we have considered the effects of large structural changes on binding very early (e.g. Refs47, 48. In these works, we pointed out that while it is possible to include the structural reorganization in the calculations at great expense it is likely to add to the instability of the calculations and many times we do not have information of the relevant structural changes. Thus it is very useful to incorporate the effect of the structural change implicitly be increasing the effective dielectric constant. This philosophy is used here. In fact, when we and other who are dealing with fast screening of mutational effects believe that the best option is to use the original wild-type structure as the first order approximation in predicting mutational effects.
II. Results and Discussion
Our main results for the binding free energy change following different types of mutations are shown in Fig. 2, considering individual substitutions and combinations. The “UK” variant contains 23 substitutions30 and a part of them is considered in this work. Each mutant with a single substituted site from the “UK” variant shows an enhancement of the binding affinity (d144 to D1118H, see Fig. 2). In particular, D614G (Fig. 2A) and the “South Africa” (Fig. 2C) mutants demonstrated the largest binding affinity increase. On the other hand, some of the mutants show a weakening of binding energy (positive red bar in Fig. 2) for the m396-spike complex, indicating potential reduction in the effectiveness of the antibody. For single substitution, the E484K mutant shows the largest weakening of binding between and spike protein. The trend is consistent with the recent observations of antibody/vaccine resistance on E484K20–22. These calculations validate that the binding energy change is an effective approach for predicting mutational effects. From the UK and Delta results (Fig. 2A and 2B) we noticed that the individual substitutions lead to mixed binding effects of m396-spike complex, but when mutating all sites together the destabilization effect of the m396-spike complex is apparent (the red bar with star sign). This indicates that the targeting of antibodies on the SARS-CoV-2 variants is highly specific; multiple substituted residues would intensively break the “look and key” interactions between the old antibodies and spike protein.
Fig. 2. The calculated CG binding energy differences between the ACE2-spike/m396-spike mutants and the wild-type.
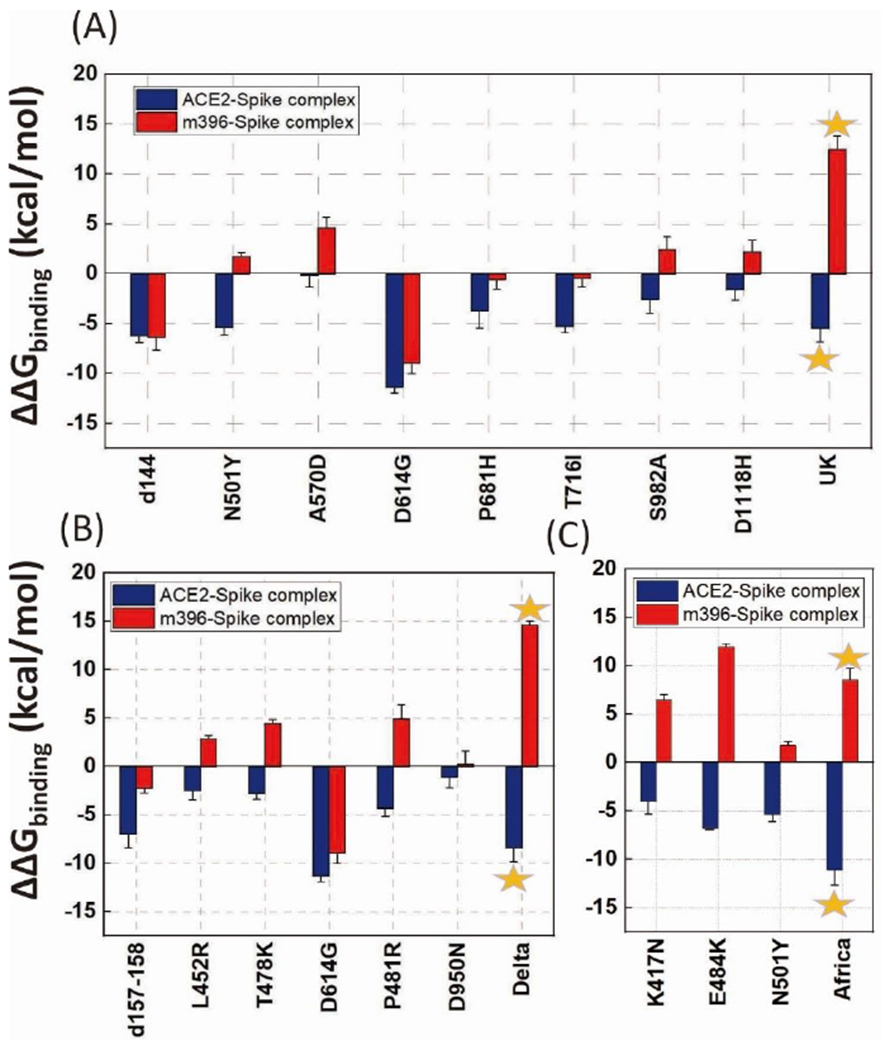
(A) part of the amino acid substitutions of the UK variant, (d144, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H), the star designates a mutation of all the 8 sites together. (B) amino acid substitutions of the Delta variant (d157-158, L452R, T478K, D614G, P481R, D950N), the star designates a mutation of all the 6 sites together. (C) amino acid substitutions of the Africa variant (K417N, E484K, N501Y), the star designates a mutation of all the 3 sites together. Error bars represent standard error.
Although the trend in the calculated binding free energies is nicely correlated with the overall observed mutational effects, it is important to examine the effects of structural variations. Here we focus on the three substituted sites of the “South Africa” variant (Fig. 3A–3C) to explore the structural/energetic basis.
Fig. 3. The CG representation of the position and environment of key residues of “South Africa” variant.
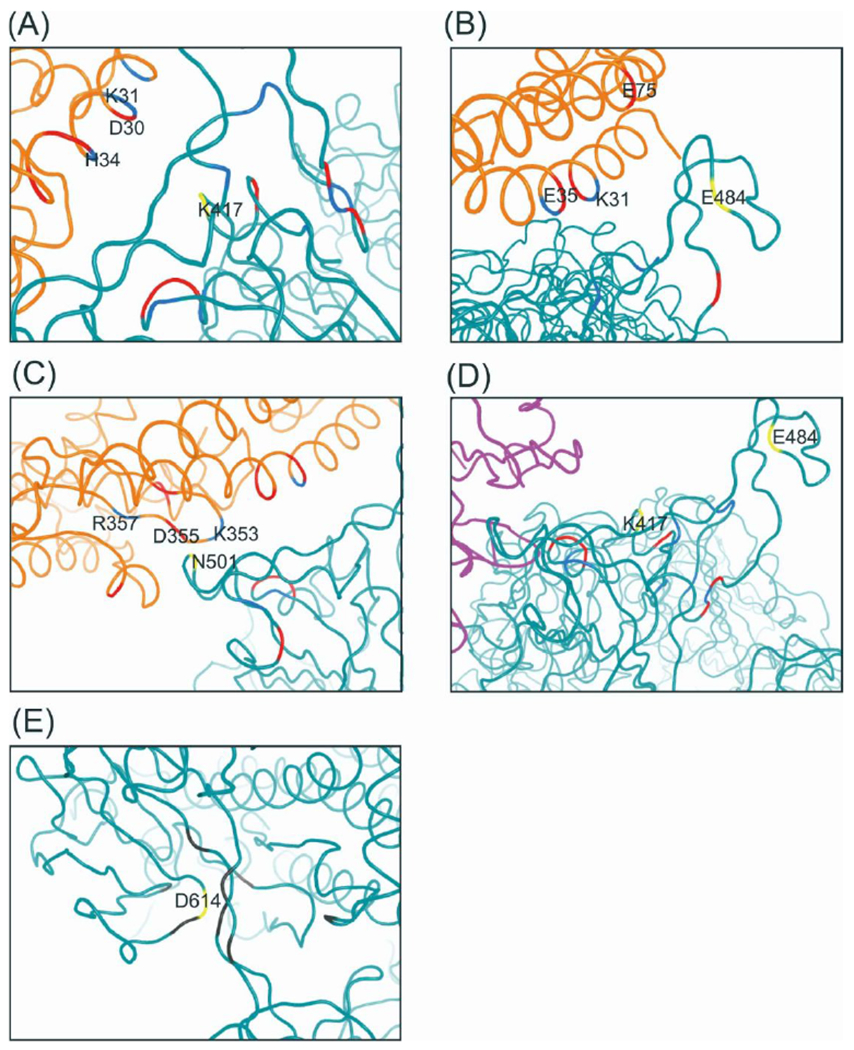
(A) K417 in ACE2-spike complex. (B) E484 in ACE2-spike complex, (C) N501 in ACE2-spike complex, (D) K417 and E484 in m396-spike complex, (E) D614 in SD2 domain. Yellow: “South Arica” relevant residues; Blue: positively charged residues; Red: negatively charged residues; Gray: hydrophobic residues; Orange: ACE2 receptor; Magenta: m396 antibody; Cyan: SARS-CoV-2 spike protein.
The mutant has been found to have a of , which can be further decomposed into and are defined in Eq. 1 and shown in Table S1). The result indicates that both the ACE2-spike complex stabilization and the spike protein destabilization contribute to the overall mutational effect. We further decomposed the CG energy into individual terms (Table S1) and found that the electrostatic term dominates for both and . This finding is consistent with the idea that the electrostatic interaction plays dominant role in determining the free energy profile of biophysical systems29, 44, 49–53. On the other hand, it is important to note that we should take into account the change of the overall electrostatic energy, in contrast of just considering several “key interactions” between the substituted site and nearby residues as has been assumed by some researchers54–57. This point is supported by the results reported in Table S2 and Fig. S2 which show the distance changes of the nearest charged residues for K417N in either the ACE2-spike, separate spike protein, or the m396-spike systems. The change in the distances between the nearest charged residues is mainly a relaxation in response to the change of charge and cannot account for the sign and the magnitude of and (Table S1), which is determined by the electrostatic energy change. This conclusion is also true for other mutants.
The E484K mutant shows capability of escaping antibodies produced by vaccination 21, 26. This mutation was originally observed in the “South Africa” variant but has also been recently identified in the United Kingdom. Remarkably, this antibody resistance effect is also reproduced by our calculations. The for the m396-spike after mutation is , which indicates a strong resistance to the antibody binding and hence neutralization. Further decomposition of the energy shows a and . Although this is clearly an overestimate it indicates that a major contribution comes from the destabilization of the m396-spike complex. Similarly, the electrostatic term plays a dominating role (Table S1). Structurally, the binding interface of the m396-spike complex contains uncovered E484 (Fig. 3D), in contrast to the ACE2-spike interface where the two residues are fully covered. The differences in binding patters between the ACE2-spike and the m396-spike systems has led to opposite sign of the and for the E484K mutants.
In the case of the N501Y mutant, the substitution of Asparagine by Tyrosine has introduced hydrophobicity and led to intrinsic changes to the spike protein structure. As shown in Table S3, the N501Y mutation resulted in large RMSD change, of non-RBD (RBD: the receptor binding domain) domain after mutation, in contrast to for and for . This indicates that the interactions near the N501 site plays a key role in determining the local structural stability. With and for N501Y we find that the negative mainly comes from the stabilization of the ACE2-spike complex. We note that Table S1 shows a major contribution to , which is again the electrostatic term.
Another important mutant is D614G. This variant has showed higher infectivity, spreading, and fatality rate14. It was first detected at a significant level in March 2020 and then spread globally in April 2020. The mechanism behind the larger of is different compared to the other mutants discussed above since D614 is not located in the RBD but at the SD2 domain (Fig. 3E). Recent work58 suggests that D614 might be essential for the interaction with and stabilization of fusion peptide (FP), and mutation of D614 to Glycine could disrupt the interaction and potentially lead to activation of FP and decrease in the barrier of structural change from pre- to fusion prone-open state. However, our result does not support such a hypothesis. The deeply increased of D614G comes from both ACE2-spike stabilization and spike protein destabilization . On one hand, our result shows that the D614G mutation reduces the nearby interactions but it does not always destabilize the structure. The free energy change is positive for the separate spike protein, but negative for the ACE2-spike complex. Overall, the large negative suggests the faster spreading, infectivity, and enhanced furin cleavage efficiency of D614G mutant could still be a result of the increased binding affinity but not of the local interaction disruption. Very recently, the enhanced ACE2-spike binding affinity and increased entry efficiency of the D614G mutant is validated by functional experiments59. We propose that once the spike protein binds the receptor, it will eventually undergo a structural change that will lead to spike protein unpacking. The barrier of the structure transformation process after binding may not be a dominant factor for the whole infection process (not the rate-determining step). In contrast, the receptor binding process and the binding affinity might play a much more essential role. However, this hypothesis needs to be validated by construction of the complete free energy landscape of the activation and binding process, which will be conducted in a subsequent study. The current results show: 1. mutational stabilization/destabilization effects for ACE2-spike complex or separate spike protein should be discussed individually; 2. The underlying mechanism behind mutational effects should not be accounted just by distance analysis of several nearby interacting residues.
We noticed that mutations of the three residues (K417N, E484K, N501Y) of the “South Africa” variant result in (Fig. 2) is not simply a linear addition of the individual ones. It is reasonable to deduce that there is a synergistic effect between the three substitutions. It is worth noting here that for the of the South Africa mutant and the of the D614G mutant, the hydrophobic energy terms became the largest contribution.
III. Experimental Assessment of previous predictions
The RBD domain is an important drug/vaccine/antibody target. Semiquantitative prediction of potential mutation sites at RBD domain would provide tremendous help to the mutation surveillance and antibody design process. An example is provided by our early predicted of the importance of mutations at the N501 site29. Other than N501, we calculated the for another 4 sites at RBD: N439, F486, Q493, and Q498. These are the important substitutions for SARS-CoV-2 spike compared to SARS-CoV at the RBD29, 60. The results show that the binding energy of F486L, Q493N, and Q498Y has been enhanced by −1.7, −4.4, and , respectively. This suggests they should be potential mutation sites for the future variants. Another issue is the possibility of large conformational changes that would be reflected in a reduction of the predicted changes in binding free energy. To explore the above issues, it is important to have experimentally determined mutational effects our in silico predictions, we performed mutational and functional experiments to examine our binding free energy calculation. All plasmids used for protein expression were confirmed by Sanger sequencing. The S protein RBD WT and mutants were expressed in the Sf9 insect cells using the Bac-to-Bac baculovirus expression system. As shown in Fig. S4A, B and C, there has been some protein impurity (higher than ) in S RBD WT and mutants (around ) after NI-NTA resin purification. Therefore, size exclusion chromatography was used for protein purification. The size exclusion chromatography results also indicated that protein impurity (higher than ) exist in samples (Fig. S5.A, C–E). The peak of S protein WT and mutants’ elution (16 mL-21 mL) from size exclusion chromatography were collected for SDS-PAGE. The result shows that most of the protein impurity was removed by size exclusion chromatography (Fig. S4D). Then Biolayer interferometry assay was used to detect the binding affinity of S protein to ACE2 61. As shown in Fig. 4, Fig. 5 and Table S4, the order of the binding affinity of S protein to ACE2 is Q493N . The magnitude of differences between the values are relatively small and it is more appropriate to understand them qualitatively. The experimental results show an increase of binding affinity of these residues and has a similar trend to that found in the calculations except for Q498Y. However, the calculations drastically overestimated the observed magnitudes. A scatter plot of experimental vs. computed values are given in Fig. S6. The data points are colored based on the data source (black for simulated results and red for experimental ones). This indicates that the dielectric compensation effect is underestimated in our calculations. A simple example is the E484K mutant. As we found very early62 that mutations to Lys results in major compensation of the expected electrostatic effect since the Lys residues finds way to increase its solvation by going to water . To compare the influence of different experimental structures in modeling, we calculated the mutational effects of N501Y mutant with two experimental structures (Fig. S3). It is clear the discrepancy of crystal structure does not alter the sign of .
Fig. 4. protein WT and mutants bind to ACE2.
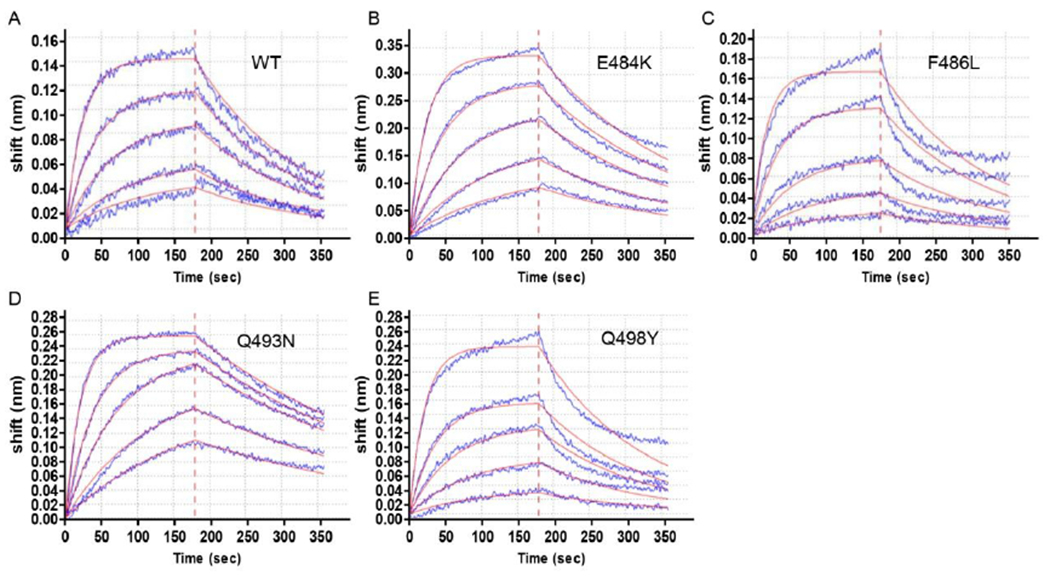
A Bio-Layer Interferometry (BLI) sensorgram is shown, displaying the binding between ACE2 and S protein RBD (WT and different mutants). The data are shown as blue lines and the best fit of the data to a 1:1 binding model is shown in red.
Fig. 5. values of protein and mutants.
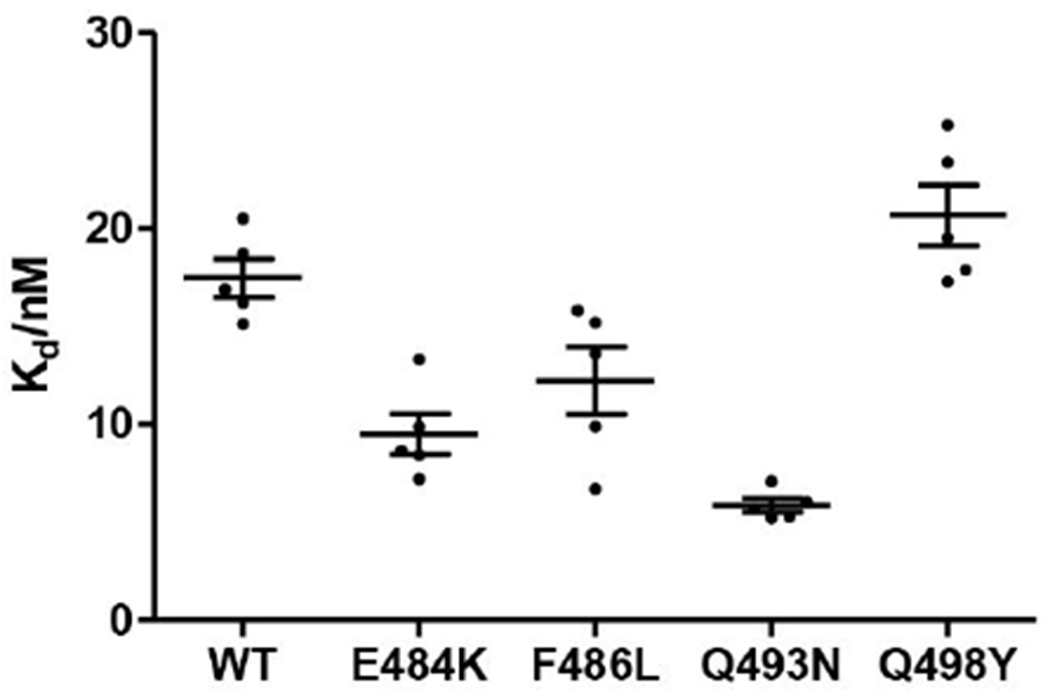
Equilibrium dissociation constants were obtained from (Table S4). The data are shown as mean ± S.E.M, based on five separate experiments.
IV. Concluding remarks
Following our early predictions of sites that can lead to mutations of SARS-CoV-2 (that include the N501 mutation) we now use the same approach for a more systematic analysis of the energetics of the binding of the spike protein of recently emerged mutants to the receptor. We also explored the change in binding of the spike protein to the m396 antibody. Encouragingly, our results appeared to be consistent with the observed behavior in terms of the binding to the receptor and the effectiveness of the antibody. Although the calculated results are correlated with the observed results, they clearly overestimate these results. This discrepancy is likely to reflect the neglect of significant structural changes that should be represented implicitly by increasing the effective dielectric constant (see above). More accurate approaches such as the Protein Dipole Langevin Dipole method in the linear response approximation, with a scaled non electrostatic term (PDLD/S-LRA/β)63 can be used for further improvements. . At any rate, the calculated trend is quite reasonable, showing that the newly appeared key variants have stronger binding to the receptor and weaker binding to the m396 antibody. Considering the speed of our CG method we can use it for screening of possible new mutation sites and for exploring the effectiveness of available new antibodies.
V. Method
Modeling the complex structures
By using Modeler64, we utilized recently published high resolution cryo-EM structures to perform homology modeling to get the atomic coordinates for ACE2, spike protein, m396, ACE2-spike, and m396-spike complexes. The SARS-CoV-2 crystal structure is from PDB data bank with PDB ID 6VSB27. However, this structure lacks part of the loop in the receptor binding domain (RBD). In this case, we used RBD structure (PDB ID 6M0J)65 to compensate the missing part. For m396-spike protein structure, we used the m396-SARS-CoV (PDB ID: 2DD8)66 as the template. After getting the complex structures, we trim them into single ACE2, m396, and spike proteins.
The coarse-grained (CG) potential surface
Next we converted the all-atom (AA) structures into CG representation and perform extensive relaxation on the structures before energy evaluation. The potential surface was evaluated by our constantly developing CG model. The CG model focuses on the precise treatment of the electrostatic interactions. The total CG energy has the following terms45:
| Eq. 2 |
where the terms on the right are the side chain van der Waals energy, main chain solvation energy, main chain hydrogen bond energy, side chain electrostatic energy, side chain polar energy, side chain hydrophobic energy, main chain/side chain electrostatic energy, and main chain/side chain van der Waals energy, respectively. The c1, c2, and c3 are scaling coefficients, same as in previous work they taking values of 0.10, 0.25, and 0.15.
Before energy evaluation we used a Monte Carlo Proton Transfer (MCPT) method45 to determine the charge states of residues in each system. During MCPT, protons are “jumped” between ionizable residues and a standard Metropolis criterion is utilized to calculate the acceptance probability. After obtaining the optimized charge distribution we evaluated the free energy of each structure and hence the binding free energies. All relative calculations were performed using the Molaris-XG package67–68.
Binding free energy change calculation
The binding energy for the ACE2-spike protein and m396-spike protein are defined as follows:
For the ACE2-virus complexes:
| Eq. 3 |
For the m396-virus complexes:
| Eq. 4 |
For the mutated binding free energy change of ACE2-spike protein:
| Eq. 5 |
For the mutated binding free energy change of m396-spike protein:
| Eq. 6 |
S protein RBD expression and purification
The mutant genes coding for S protein RBD were amplified by overlap Polymerase Chain Reaction (PCR) from pFastBac S protein RBD Wild Type (S protein RBD WT) using specific primers (Genewiz, Suzhou, China) (Table S5). The coding sequence of S protein RBD mutants (E484K, F486L, Q493N, Q498Y) were cloned into pFastBac with an N-terminal GP67 signal peptide and a C-terminal 8x His tag.
protein and mutants were expressed in insect cells using the Bac-to-Bac baculovirus expression system. Cell cultures were grown in SIM SF Expression Medium (Sino Biological Inc., MSF1, Beijing, China) to a density of cells and then infected with baculovirus by Cellfectin™ II Reagent (Life Technologies, Gaithersburg, MD, USA). Sixty hours after infection, the supernatant was collected by centrifugation at for . For the purification of WT S protein RBD and its mutants, the supernatant was transferred to large beaker and treated with Tris (, final concentration (final concentration (final concentration ) and stirred at room temperature for . Supernatant was collected by centrifugation at g for and then incubated for at with pre-equilibrated Nickel-NTA resin. After incubation, the Nickel-NTA resin was spun down at for , poured into a glass column, and washed with wash buffer (20 mM HEPES pH 7.5, 500 imidazole). The S protein RBD WT and mutants were eluted with elution buffer (20 mM HEPES pH 7.5, imidazole) and further applied to Superdex-200 (10/300 GL) column with AKTA FPLC. The proteins were concentrated using a 10 molecular weight cutoff Millipore concentrator and fast-frozen by liquid nitrogen and stored at until further use.
S protein RBD /ACE2 binding
The binding of protein and ACE2 was detected by biolayer interferometry. Biotinylation labeling of ACE2 protein (biotinylation ratio of Biotin:ACE2=2:1). Biotin-tagged ACE2 (15 ) was immobilized to a Streptavidin (SA) sensortip (Gator Bio) using an GatorPrime (Gator Bio) . The sensortip was then dipped into WT, E484K, F486L, Q493N, Q498Y (100 nM, 50 to measure association before being dipped into the well containing only running buffer composed of PBS ( PH 7.4) , 0.02% Tween bovine serum albumin to measure dissociation. Data were reference subtracted and fit to a 1:1 binding model using Gator Data Analysis Software v1.7.2.0129 (Gator Bio). Equilibrium dissociation constants were obtained from .
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Health R35 GM122472 and the National Science Foundation Grant MCB 1707167. This work was also supported by the Special Fund for COVID-19 Research from the Longgang District (LGKCXGZX2020013). We thank Dr. Veselin Kolev for discussion and manuscript preparation. We thank the University of Southern California High-Performance Computing and Communication Center for computational resources.
Footnotes
Supporting Information
The Supporting Information is available free of charge at
Additional computational details and results (PDF)
References
- 1.Coronavirus Death Toll. https://www.worldometers.info/coronavirus/coronavirus-death-toll/ (accessed 2021-07-16).
- 2.Corbett KS; Edwards DK; Leist SR; Abiona OM; Boyoglu-Barnum S; Gillespie RA; Himansu S; Schäfer A; Ziwawo CT; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature, 2020, 586 (7830), 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 2020, 368 (6489), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henderson R; Edwards RJ; Mansouri K; Janowska K; Stalls V; Gobeil SMC; Kopp M; Li D; Parks R; Hsu AL; Borgnia MJ; Haynes BF; Acharya P Controlling the SARS-CoV-2 spike glycoprotein conformation. Nature Structural & Molecular Biology, 2020, 27 (10), 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amanat F; Krammer F SARS-CoV-2 Vaccines: Status Report. Immunity, 2020, 52 (4), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao L; Goreshnik I; Coventry B; Case JB; Miller L; Kozodoy L; Chen RE; Carter L; Walls AC; Park Y-J; Strauch E-M; Stewart L; Diamond MS; Veesler D; Baker D De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science, 2020, 370 (6515), 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai W; Zhang B; Jiang X-M; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; Li C; Li Y; Bai F; Wang H; Cheng X; Cen X; Hu S; Yang X; Wang J; Liu X; Xiao G; Jiang H; Rao Z; Zhang L-K; Xu Y; Yang H; Liu H Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science, 2020, 368 (6497), 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Widge AT; Rouphael NG; Jackson LA; Anderson EJ; Roberts PC; Makhene M; Chappell JD; Denison MR; Stevens LJ; Pruijssers AJ; et al. Durability of Responses after SARS-CoV-2 mRNA-1273 Vaccination. New England Journal of Medicine, 2020, 384 (1), 80–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thoguluva Chandrasekar V; Venkatesalu B; Patel HK; Spadaccini M; Manteuffel J; Ramesh M Systematic review and meta-analysis of effectiveness of treatment options against SARS-CoV-2 infection. Journal of Medical Virology, 2021, 93 (2), 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voysey M; Clemens SAC; Madhi SA; Weckx LY; Folegatti PM; Aley PK; Angus B; Baillie VL; Barnabas SL; Bhorat QE; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. The Lancet, 2021, 397 (10269), 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ku Z; Xie X; Davidson E; Ye X; Su H; Menachery VD; Li Y; Yuan Z; Zhang X; Muruato AE; i Escuer AG; Tyrell B; Doolan K; Doranz BJ; Wrapp D; Bates PF; McLellan JS; Weiss SR; Zhang N; Shi P-Y; An Z Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nature Communications, 2021, 12 (1), 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaebler C; Wang Z; Lorenzi JCC; Muecksch F; Finkin S; Tokuyama M; Cho A; Jankovic M; Schaefer-Babajew D; Oliveira TY; et al. Evolution of antibody immunity to SARS-CoV-2. Nature, 2021, 591, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becerra-Flores M; Cardozo T SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. International journal of clinical practice, 2020, 74 (8), e13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung K; Pei Y; Leung GM; et al. Empirical transmission advantage of the D614G mutant strain of SARS-CoV-2. 2020.09.22. medRxiv. 10.1101/2020.09.22.2019981Q (accessed 2020-09-23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos JC; Passos GA The high infectivity of SARS-CoV-2 B. 1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. 2020.12.29. bioRxiv. 10.1101/2020.12.29.4247Q8 (accessed 2021-01-01). [DOI] [Google Scholar]
- 16.Lauring AS; Hodcroft EB Genetic Variants of SARS-CoV-2-What Do They Mean? Jama, 2021, 325 (6), 529–531. [DOI] [PubMed] [Google Scholar]
- 17.Zahradník J; Marciano S; Shemesh M; et al. SARS-CoV-2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. 2021.01.06. bioRxiv. 10.1101/2021.01.06.425392 (accessed 2021-01-29). [DOI] [Google Scholar]
- 18.Leung K; Shum MH; Leung GM; Lam TT; Wu JT Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance, 2021, 26 (1), 2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.New COVID mutation may have higher mortality rate. https://thelincolnite.co.uk/2021/01/new-covid-mutation-may-have-higher-mortality-rate/ (accessed 2021-01-22).
- 20.Vasques Nonaka CK; Miranda. Franco. M; Gräf T; et al. Genomic Evidence of a Sars-Cov-2 Reinfection Case With E484K Spike Mutation, Brazil. Emerg Infect Dis, 2021, 27(5), 1522–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jangra S; Ye C; Rathnasinghe R; et al. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. 2021.01.26. medRxiv. 10.1101/2021.01.26.21250543 (accessed 2021-01-29). [DOI] [Google Scholar]
- 22.Why E484K mutation is worrying vaccine developers. https://www.msn.com/en-au/news/australia/why-e484k-mutation-is-worrying-vaccine-developers/ar-BB1dvZXy (accessed 2021-02-09).
- 23.Greaney AJ; Starr TN; Gilchuk P; Zost SJ; Binshtein E; Loes AN; Hilton SK; Huddleston J; Eguia R; Crawford KHD; Dingens AS; Nargi RS; Sutton RE; Suryadevara N; Rothlauf PW; Liu Z; Whelan SPJ; Carnahan RH; Crowe JE; Bloom JD Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host & Microbe, 2021, 29 (1), 44–57.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z; VanBlargan LA; Bloyet L-M; Rothlauf PW; Chen RE; Stumpf S; Zhao H; Errico JM; Theel ES; Liebeskind MJ; Alford B; Buchser WJ; Ellebedy AH; Fremont DH; Diamond MS; Whelan SPJ Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host & Microbe, 2021, 29(3), 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomson EC; Rosen LE; Shepherd JG; Spreafico R; da Silva Filipe A; Wojcechowskyj JA; Davis C; Piccoli L; Pascall DJ; Dillen J; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell, 2021, 184(5), 1171–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ascoli CA Could mutations of SARS-CoV-2 suppress diagnostic detection? Nature Biotechnology, 2021, 39, 274–275. [DOI] [PubMed] [Google Scholar]
- 27.Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh C-L; Abiona O; Graham BS; McLellan JS Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 2020, 367 (6483), 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starr TN; Greaney AJ; Hilton SK; Ellis D; Crawford KHD; Dingens AS; Navarro MJ; Bowen JE; Tortorici MA; Walls AC; King NP; Veesler D; Bloom JD Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell, 2020, 182 (5), 1295–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai C; Warshel A Critical Differences between the Binding Features of the Spike Proteins of SARS-CoV-2 and SARS-CoV. The Journal of Physical Chemistry B, 2020, 124 (28), 5907–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.SARS-CoV-2 Variants: COVID-19 - Global. https://www.who.int/emergencies/disease-outbreak-news/item/2020-DON305 (accessed 2020-12-31).
- 31.Tegally H; Wilkinson E; Giovanetti M; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. 2020.12.21. medRxiv. 10.1101/2020.12.21.20248640 (accessed 2020-12-22). [DOI] [Google Scholar]
- 32.Mlcochova P; Kemp S; Dhar MS; et al. SARS-CoV-2 B.1.617.2 Delta variant emergence and vaccine breakthrough. 2021.05.08. BioRxiv. 10.1101/2021.05.08.443253 (accessed 2021-06-28). [DOI] [Google Scholar]
- 33.Salvatore M; Bhattacharyya R; Purkayastha S; et al. Resurgence of SARS-CoV-2 in India: Potential role of the B.1.617.2 (Delta) variant and delayed interventions. 2021.06.23. medRxiv. 10.1101/2021.06.23.21259405 (accessed 2021-06-30). [DOI] [Google Scholar]
- 34.Glasgow A; Glasgow J; Limonta D; Solomon P; Lui I; Zhang Y; Nix MA; Rettko NJ; Zha S; Yamin R; Kao K; Rosenberg OS; Ravetch JV; Wiita AP; Leung KK; Lim SA; Zhou XX; Hobman TC; Kortemme T; Wells JA Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci, 2020, 117 (45), 28046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zimmerman MI; Porter JR; Ward MD; Singh S; Vithani N; Meller A; Mallimadugula UL; Kuhn CE; Borowsky JH; Wiewiora RP; Hurley MFD; Harbison AM; Fogarty CA; Coffland JE; Fadda E; Voelz VA; Chodera JD; Bowman GR SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nature Chemistry, 2021, 13 (7), 651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sztain T; Ahn S-H; Bogetti AT; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. 2021.02.15. bioRxiv. 10.1101/2021.02.15.431212 (accessed 2021-05-17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hie B; Zhong ED; Berger B; Bryson B Learning the language of viral evolution and escape. Science, 2021, 371 (6526), 284. [DOI] [PubMed] [Google Scholar]
- 38.Maher MC; Bartha I; Weaver S; et al. Predicting the mutational drivers of future SARS-CoV-2 variants of concern. 2021.06.21. medRxiv. 10.1101/2021.06.21.21259286 (accessed 2021-06-22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higuchi Y; Suzuki T; Arimori T; Ikemura N; Mihara E; Kirita Y; Ohgitani E; Mazda O; Motooka D; Nakamura S; Sakai Y; Itoh Y; Sugihara F; Matsuura Y; Matoba S; Okamoto T; Takagi J; Hoshino A Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nature Communications, 2021, 12 (1), 3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gobeil SMC; Janowska K; McDowell S; Mansouri K; Parks R; Stalls V; Kopp MF; Manne K; Li D; Wiehe K; Saunders KO; Edwards RJ; Korber B; Haynes BF; Henderson R; Acharya P Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science, 2021, 373 (6555), eabi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai Y; Zhang J; Xiao T; Lavine CL; Rawson S; Peng H; Zhu H; Anand K; Tong P; Gautam A; Lu S; Sterling SM; Walsh RM; Rits-Volloch S; Lu J; Wesemann DR; Yang W; Seaman MS; Chen B Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science, 2021, 373 (6555), 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan M; Huang D; Lee C-CD; Wu NC; Jackson AM; Zhu X; Liu H; Peng L; van Gils MJ; Sanders RW; Burton DR; Reincke SM; Prüss H; Kreye J; Nemazee D; Ward AB; Wilson IA Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science, 2021, 373 (6556), 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu M; Uchil PD; Li W; Zheng D; Terry DS; Gorman J; Shi W; Zhang B; Zhou T; Ding S; Gasser R; Prévost J; Beaudoin-Bussières G; Anand SP; Laumaea A; Grover JR; Liu L; Ho DD; Mascola JR; Finzi A; Kwong PD; Blanchard SC; Mothes W Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host & Microbe, 2020, 28 (6), 880–891.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee M; Kolev V; Warshel A Validating a coarse-grained voltage activation model by comparing its performance to the results of monte carlo simulations. The Journal of Physical Chemistry B, 2017, 121 (50), 11284–11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vorobyov I; Kim I; Chu ZT; Warshel A Refining the treatment of membrane proteins by coarse-grained models. Proteins: Structure, Function, and Bioinformatics, 2016, 84 (1), 92–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vicatos S; Rychkova A; Mukherjee S; Warshel A An effective coarse-grained model for biological simulations: recent refinements and validations. Proteins, 2014, 82 (7), 1168–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muegge I; Schweins T; Warshel A Electrostatic contributions to protein-protein binding affinities: Application to Rap/Raf interaction. Proteins, 1998, 30 (4), 407–423. [DOI] [PubMed] [Google Scholar]
- 48.Sham YY; Muegge I; Warshel A The Effect of Protein Relaxation on Charge-Charge Interactions and Dielectric Constants of Proteins. Biophysical Journal, 1998, 74 (4), 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bai C; Asadi M; Warshel A The catalytic dwell in ATPases is not crucial for movement against applied torque. Nature Chemistry, 2020, 12(12):1187–1192. [DOI] [PubMed] [Google Scholar]
- 50.Alhadeff R; Warshel A A free-energy landscape for the glucagon-like peptide 1 receptor GLP1R. Proteins: Structure, Function, and Bioinformatics, 2020, 88 (1), 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai C; Warshel A Revisiting the protomotive vectorial motion of F0-ATPase. Proceedings of the National Academy of Sciences, 2019, 116 (39), 19484–19489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee M; Bai C; Feliks M; Alhadeff R; Warshel A On the control of the proton current in the voltage-gated proton channel Hv1. Proceedings of the National Academy of Sciences, 2018, 115 (41), 10321–10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim I; Warshel A Analyzing the electrogenicity of cytochrome c oxidase. Proceedings of the National Academy of Sciences, 2016, 113 (28), 7810–7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou W; Xu C; Wang P; Luo M; Xu Z; Cheng R; Jin X; Guo Y; Xue G; Juan L; Anashkina AA; Nie H; Jiang Q N439K Variant in Spike Protein Alter the Infection Efficiency and Antigenicity of SARS-CoV-2 Based on Molecular Dynamics Simulation. Frontiers in cell and developmental biology, 2021, 9, 697035–697035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han Y; Wang Z; Wei Z; Schapiro I; Li J Binding affinity and mechanisms of SARS-CoV-2 variants. Computational and Structural Biotechnology Journal, 2021, 19, 4184–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gan HH; Twaddle A; Marchand B; Gunsalus KC Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface can Identify High-Affinity Variants Associated with Increased Transmissibility. Journal of Molecular Biology, 2021, 433 (15), 167051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang WB; Liang Y; Jin YQ; et al. E484K mutation in SARS-CoV-2 RBD enhances binding affinity with hACE2 but reduces interactions with neutralizing antibodies and nanobodies: Binding free energy calculation studies. 2021.02.17. bioRxiv. 10.1101/2021.02.17.431566 (accessed 2021-02-17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gobeil SMC; Janowska K; McDowell S; Mansouri K; Parks R; Manne K; Stalls V; Kopp MF; Henderson R; Edwards RJ; Haynes BF; Acharya P D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Reports, 2021, 34 (2), 108630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ozono S; Zhang Y; Ode H; Sano K; Tan TS; Imai K; Miyoshi K; Kishigami S; Ueno T; Iwatani Y; Suzuki T; Tokunaga K SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nature Communications, 2021, 12 (1), 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walls AC; Park Y-J; Tortorici MA; Wall A; McGuire AT; Veesler D Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell, 2020, 181 (2), 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong J; Huang B; Wang B; Titong A; Gallolu Kankanamalage S; Jia Z; Wright M; Parthasarathy P; Liu Y Development of humanized tri-specific nanobodies with potent neutralization for SARS-CoV-2. Scientific reports, 2020, 10 (1), 17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cutler RL; Davies AM; Creighton S; Warshel A; Moore GR; Smith M; Mauk AG Role of arginine-38 in regulation of the cytochrome c oxidation-reduction equilibrium. Biochemistry, 1989, 28 (8), 3188–3197. [DOI] [PubMed] [Google Scholar]
- 63.Singh N; Warshel A Absolute binding free energy calculations: on the accuracy of computational scoring of protein-ligand interactions. Proteins, 2010, 78 (7), 1705–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Webb B; Sali A Comparative Protein Structure Modeling Using MODELLER. Current protocols in bioinformatics, 2016, 54, 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lan J; Ge J; Yu J; Shan S; Zhou H; Fan S; Zhang Q; Shi X; Wang Q; Zhang L; Wang X Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature, 2020, 581,215–220. [DOI] [PubMed] [Google Scholar]
- 66.Prabakaran P; Gan J; Feng Y; Zhu Z; Choudhry V; Xiao X; Ji X; Dimitrov DS Structure of Severe Acute Respiratory Syndrome Coronavirus Receptor-binding Domain Complexed with Neutralizing Antibody Journal of Biological Chemistry, 2006, 281 (23), 15829–15836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamerlin SCL; Vicatos S; Dryga A; Warshel A Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annual Review of Physical Chemistry, 2011, 62 (1), 41–64. [DOI] [PubMed] [Google Scholar]
- 68.Lee FS; Chu ZT; Warshel A Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. Journal of Computational Chemistry, 1993, 14 (2), 161–185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.