

Abstract
Given the extent to which per- and polyfluoroalkyl substances (PFAS) are used in commercial and industrial applications, the need to evaluate treatment options that reduce environmental emissions and human and ecological exposures of PFAS is becoming more necessary. One specific chemical class of PFAS, fluorotelomer alcohols (FTOHs), have vapor pressures such that a significant fraction is expected to be present in the gas-phase even at ambient temperatures. FTOHs are used in a variety of PFAS applications, including synthesis and material coatings. Using two complementary mass spectrometric methods, the use of calcium oxide (CaO) was examined as a low temperature and potentially low-cost thermal treatment media for removal and destruction of four gas-phase FTOHs of varying molecular weights. This was accomplished by assessing the removal/destruction efficiency of the FTOHs and the formation of fluorinated byproducts as a function of treatment temperature (200 – 800 °C) in the presence of CaO compared to thermal-only destruction. During the treatment process, there is evidence that other PFAS compounds are produced at low temperatures (200 – 600 °C) as the primary FTOH partially degrades. At temperatures above 600 °C, thermal treatment with CaO prevented the formation or removed nearly all these secondary products.
Keywords: PFAS, FTOH, Thermal treatment, Gas-phase, Calcium oxide
Graphical Abstract
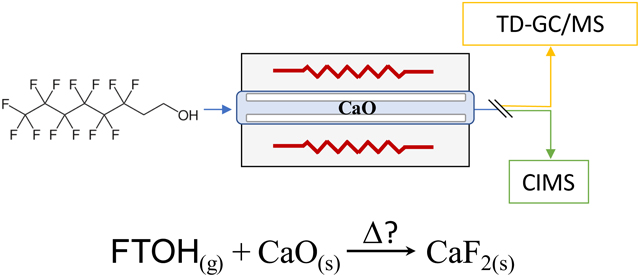
1. Introduction
Per- and polyfluoroalkyl substances (PFAS) are contaminants of emerging concern due to their relative resistance to chemical degradation and therefore potential for buildup in the environment as a persistent pollutant. PFAS are a family of thousands of anthropogenic molecules that have been widely used in numerous consumer, industrial, and military applications over the past 70 years (Buck et al., 2011). The chemical structure of these compounds (carbon-fluorine backbones coupled with a diverse number of other functional groups) lends a number of useful chemical and physical properties that have been leveraged for consumer products and industrial processes including fluoropolymer manufacturing and application of, protective, non-stick, water repellant, stain-resistant, and chemically inert coatings (Dinglasan-Panlilio and Mabury, 2006), robust surfactants, fire-fighting agents (DeYoung, 1994; Roth et al., 2020), and temperature resistant products to name a few (Lemal, 2004; Prevedouros et al., 2006). Many of the same properties that are uniquely useful to these products also create challenges in the disposal and destruction of PFAS. This has caused PFAS to become widespread persistent pollutants with few biological or environmental routes of destruction (Martin et al., 2002; Stock et al., 2004; Cordner et al., 2019; McCord and Strynar, 2019). PFAS are known to bioaccumulate in humans (Pan et al., 2017; Cordner et al., 2019), other animals (Haukås et al., 2007; Wan et al., 2020), and plants (Ghisi et al., 2019), and have been linked to adverse health effects (Hagen et al., 1981; Haukås et al., 2007; Cordner et al., 2019; Ghisi et al., 2019; Wan et al., 2020).
The progressive buildup of PFAS in various environmental media (such as groundwater and soils) requires that PFAS be disposed of by methods that do not further increase environmental levels and mitigate potential human and ecological exposures. Effective procedures for the removal and destruction of PFAS from a variety of contaminated media [stack emissions, consumer waste, wastewater, soil, ground water, landfill leachate, granular activated carbon (GAC), sewage sludge, and others] are needed to help alleviate environmental loadings. Due to the stability of these compounds, traditional waste disposal practices such as landfilling and water treatment often fail to adequately destroy or permanently contain PFAS to a point where they are no longer an environmental or exposure concern (Ahrens et al., 2011b; Lindstrom et al., 2011), and there is concern that incineration and other thermal treatment approaches may not fully destroy PFAS (Hogue, 2020). The carbon-fluorine bond is the strongest carbon single bond, with bond dissociation energies up to 544 kJ mol−1 in tetrafluoromethane, CF4 (Lemal, 2004). The energy required to break all of the carbon-fluorine bonds in PFAS can require temperatures in excess of 1,450 °C (Tsang et al., 1998), but incomplete oxidation can yield reactive species and other secondary PFAS products (Tsang et al., 1998; Han et al., 2011; Watanabe et al., 2018). While the primary PFAS compounds may have been destroyed, these secondarily formed products of incomplete destruction, sometimes referred to from an incineration perspective as products of incomplete combustion (PICs), may still contribute to the environmental PFAS burden and ecological and human health concerns. Often, these byproducts are more volatile than the parent compound allowing airborne dispersion in the environment (Cicerone, 1979; Ravishankara et al., 1993; Tsang et al., 1998; Krusic and Roe, 2004; Krusic et al., 2005; Prevedouros et al., 2006; Watanabe et al., 2018).
Moderately low temperature (<1000 °C) reaction of PFAS with various calcium species has recently shown promise as a potential destruction method that proceeds at temperatures significantly lower than those of conventional hazardous waste thermal destruction processes (>1000 °C) (Wang et al., 2011; Wang et al., 2015a). These calcium-based methods aim to mineralize the fluorine present in PFAS to produce calcium fluoride [CaF2, a largely inert and stable mineral (flourite)] and carbon dioxide (CO2). Klabunde et al. (1972) and Billups et al. (1984) demonstrated that calcium atoms can mineralize fluorinated organic molecules at low temperatures (<100 °C) to produce CaF2. Recently, calcium oxide (CaO), calcium hydroxide [Ca(OH)2], and calcium carbonate (CaCO3) have been used to facilitate the destruction of several PFAS at temperatures less than 600 °C (Wang et al., 2011; Wang et al., 2013a; Wang et al., 2015a; Wang et al., 2019). In these studies, the solid-phase calcium compounds were mixed with solid PFAS and heated to facilitate the solid-solid destruction interactions. Given that PFAS exhibit a variety of vapor pressures and can be effectively desorbed from soils and other solids at temperatures below 400 °C (Crownover et al., 2019), these solid-solid approaches can be inefficient if the PFAS desorbs before reaching an optimum temperature to react with the calcium compounds. Additionally, the comparatively low concentrations of many PFAS found in solid wastes would result in mass handling issues and exacerbate such inefficiencies. Further, PFAS are also present in liquid- and gas-phase waste streams, and phase-independent approaches that decouple mass transfer and kinetics are needed. Besides the potential to capture fluorine and prevent the emissions of extremely corrosive hydrofluoric acid (HF), a particular benefit to calcium-based treatment methods is the low energy/temperature requirements needed to facilitate the destruction reactions compared to incineration approaches involving a standing flame.
Fluorotelomer alcohols (FTOHs), shown in Figure S1, are an important class of PFAS with a number of uses including intermediates in the synthesis of other PFAS and fluoropolymers as well as components of aqueous film-forming foams (AFFF) and consumer products (Dinglasan-Panlilio and Mabury, 2006; Henderson and Smith, 2006; Kotthoff et al., 2015; Liu et al., 2015; Roth et al., 2020). FTOHs are volatile PFAS (Lei et al., 2004) and have been measured at elevated concentrations in air and condensed phase samples from wastewater treatment plants, landfills, textile plants, and remote regions as far as the Antarctic (Stock et al., 2004; Ahrens et al., 2011b; Wang et al., 2015b; Heydebreck et al., 2016). FTOHs can convert to bioaccumulative perfluorocarboxylic acids (PFCAs) (Hagen et al., 1981; Dinglasan et al., 2004; Ellis et al., 2004; Washington et al., 2020) which have traditionally been a major focus of environmental PFAS contamination and exposure in soils, groundwater, and surface water (Lau et al., 2007; Sun et al., 2016; Washington et al., 2020). Varying levels of FTOHs are commonly found in aqueous dispersions used to apply fluoropolymer coatings to textiles and other materials (Washington et al., 2014; Heydebreck et al., 2016). During commercial textile application, drying, and curing processes, many of the volatile components of the dispersions (including PFAS surfactants and processing aids) are vented to the atmosphere. Treatment of FTOHs and potentially other PFAS vapors at a point source such as stack emissions from fabric coating facilities represents one possible application of Ca-based control technologies. If successfully demonstrated, this technology offers the possibility of low temperature (<1000 °C) destruction of PFAS and simultaneous fluorine capture and conversion to CaF2, without the costs and complications of flame-based thermal approaches.
The objective of this study is to demonstrate the utility of CaO-based thermal treatment of gas-phase PFAS waste streams. To do so, the temperature dependent gas-solid reaction of four gas-phase FTOHs (4:2, 6:2, 8:2 and 10:2 FTOH – structures provided in Figure S1) with CaO were examined. To provide a more comprehensive assessment of the potential emissions from the treatment of FTOHs by CaO, products of incomplete thermal destruction were monitored in addition to the removal efficiency of the primary FTOHs using two independent mass spectrometric techniques: iodide-adduct chemical ionization mass spectrometry and thermal-desorption gas chromatography/mass spectrometry. The chemical ionization mass spectrometer provided rapid real-time data allowing for destruction measurements with fine scale temperature resolution while the gas chromatograph/mass spectrometer detected a large array of gas-phase PFAS. The low temperature range examined (200 – 800 °C) does not represent combustion or incineration, and the testing apparatus was entirely flameless.
2. Materials and methods
2.1. Experimental setup
The experimental setup is depicted in Figure S2. A flameless tube furnace (Model TF55030A-1, Lindberg/Blue M) was used to provide a stable and controlled temperature environment for the potential FTOH destruction/removal reactions. Temperatures within the furnace were set by the furnace temperature controller and monitored at 1 Hz by a Type E thermocouple located at the center of the quartz glass reaction tube (45 cm length; 2.54 cm outer diameter; 2.2 cm inner diameter). Approximately 1 g of CaO (Sigma-Aldrich, 99.9% purity) powder was distributed along the central 40 cm of the quartz glass reactor tube. The tube was then rolled on a benchtop to coat the reactor walls with a layer of CaO ~1 mm in thickness, and over the course of an experimental run, the CaO coating remained intact. Initially, a packed bed of CaO within the quartz reactor was planned to be used. However, it was determined that the supporting materials needed to hold a packed bed in place were either reactive with or provided substantial surface area for the adsorption of gas-phase analytes. These surface area effects are discussed further in section 2.4. Thermal treatment below. Additionally, there was insufficient pore space in the packed CaO bed to facilitate the flow rate used in the experiments, which resulted in pressure build up, and the CaO plug was often pushed out of the reactor. In order to remove these confounding effects, the coated wall approach was instead used, providing a clean system containing only CaO and the quartz glass reactor that relied on gas-phase diffusion to foster mass transfer of the FTOH to the CaO. An analysis of the relative time scales for diffusion and advection is presented in section 3.1. Assessing diffusion vs. advection in the reactor.
Gas-phase FTOHs were delivered through the tube furnace system by a 0.5 L min−1 flow of carrier air (~20% relative humidity) from a laboratory zero air generator set by a mass flow controller. The volumetric residence time in the heated portion of the reactor tube was ~20 s. Apart from the quartz glass reaction tube, polytetrafluoroethylene (PTFE) fittings and tubing (3.175 mm inner diameter) were used for all transfer lines. Glassware/tubing transitions were sufficiently long and narrow so that PTFE fittings and tubing never experienced gases with temperatures >30 °C. 4:2, 6:2, 8:2, and 10:2 FTOH standards were purchased from Synquest Laboratories (www.synquestlabs.com). To deliver a stable concentration of gas-phase 4:2 and 6:2 FTOH through the treatment system and to the analytical instrumentation, a 75 μL aliquot of the liquid standard was placed in a room temperature (~23 °C) diffusion cell equipped with a 1.5 cm long, 1.57 mm inner diameter capillary over which the carrier air was passed. For the solid-phase 8:2 and 10:2 FTOH standards, ~300 mg of the solid standard was placed in a room temperature 200 mL vessel that was continually purged with the carrier air. These two delivery approaches entrained a sufficient amount [approximately 50 parts per billion by volume (ppbv) or less] of each FTOH for reliable detection by the instrumentation used.
2.2. Chemical Ionization Mass Spectrometry
An iodide-adduct high-resolution time-of-flight chemical ionization mass spectrometer (CIMS, Aerodyne Research Inc/TOFWERK AG) was used to monitor gas-phase levels of the primary FTOH compounds and several other compounds of interest [including hydrofluoric acid (HF)] during thermal treatment (Lee et al., 2014; Riedel et al., 2019). Gas-phase analytes were detected as ion-molecular clusters with the iodide reagent ion. This was a soft ionization process, so parent ions were typically preserved with little or no fragmentation. Additional details regarding instrumental setup, iodide reagent, ionization, and the detection of different PFAS chemical classes (including FTOHs) have been described in a previous publication (Riedel et al., 2019).
During operation, the ion-molecule reaction region (IMR) and the first focusing quadrupole region (SSQ) of the instrument were maintained at pressures of 100 and 2 mbar, respectively (Bertram et al., 2011; Aljawhary et al., 2013). This resulted in an inlet draw of 2.0 L min−1. During CIMS sampling, the 0.5 L min−1 sample air flow from the tube furnace was transferred directly to the inlet of the instrument through a 15 cm length of PTFE tubing. A humidified stream of nitrogen teed in immediately upstream of the inlet accounted for the remaining 1.5 L min−1 of inlet draw (see Figure S2). This also served to maintain the ratio of the iodide-water cluster to iodide [I(H2O)/I−] over the course of sampling. Full mass spectra were recorded at an acquisition rate of 0.5 or 0.4 Hz, depending on the mass-to-charge ratio (m/z) window being scanned, and all reported m/z signals were normalized to the total reagent ion signal [I− + I(H2O)−].
Primary FTOHs were identified at the expected m/z (molecular weight + I−). In addition to the FTOHs, additional compounds, including HF, that lacked standards were identified throughout the thermal treatment process and assigned probable molecular compositions using the high-resolution m/z peak location (Riedel et al., 2019). System blanks, operating the CIMS with carrier air, the furnace operating at temperatures between 200 – 800 °C (see section 2.4. Thermal treatment), but without FTOHs being introduced, did not indicate the presence of measurable fluorinated compounds.
2.3. Thermal Desorption-Gas Chromatography/Mass Spectrometry
As an additional measurement of the FTOHs and other gas-phase PFAS that were not detectable by iodide-CIMS, samples were collected onto three-bed Universal sorbent tubes (C3-AXXX-5266 Tenax TA/Carbograph 1TD/Carboxen 1003, Markes International) at a variety of treatment temperatures and analyzed by thermal desorption-gas chromatography/mass spectrometry (TD-GC/MS, Markes International TD-100xR coupled to an Agilent Technologies 7890B GC/5977B MSD). During sorbent tube sampling, a 0.1 L min−1 sample flow was drawn through each sorbent tube from the 0.5 L min−1 carrier flow by a personal sampling pump (AirCheck Touch, SKC Inc.). Samples were collected 5 cm from the exit of the tube furnace for ten minutes. Throughout sample collection, despite the furnace being at temperatures as high as 800 °C, the air passing through the sorbent tubes had cooled in the glassware to <30 °C for all reactor temperatures tested. Prior to introduction of FTOHs to the tube furnace, blank sorbent tube samples were collected at temperatures from 250 – 800 °C in the presence of CaO. The blank samples were analyzed by TD-GC/MS to ensure that no fluorinated compounds were present as a result of tube background or potential PTFE contamination. All blank samples were found to be absent of fluorinated compounds.
For thermal desorption, a standby split of 20 mL min−1 and a flow path temperature of 120 °C were utilized. Sorbent tubes were dry purged with 50 mL min−1 of research grade helium (99.9999% purity) for 2 min prior to desorption. Sorbent tubes were heated at 315 °C for 10 min with a trap flow rate of 50 mL min−1 to desorb analyte compounds which were then captured onto a secondary Air Toxics cold trap (Markes International, U-T15ATA-2S). This trap was then purged for 2 min at 50 mL min−1 before being heated from 25 to 315 °C at a rate of 40 °C s−1 and held for 3 min. An outlet split of 2 mL min−1 was used. Analytes were chromatographically separated on a 30 m × 0.25 mm × 1.4 μm Restek Rtx-VMS GC column with a helium flow rate of 2 mL min−1. The column temperature was held at 30 °C for 3 min then increased at a rate of 8 °C min−1 to 150 °C followed by a 36 °C min−1 ramp to 240 °C and held for 5 min for a total run time of 25.5 min. The mass selective detector (MSD) transfer line temperature was set to 290 °C, and the quadrupole and source were set to 150 and 230 °C, respectively. The electron ionization (EI) ion source was operated at 70 eV. The mass spectrometer was operated in selected ion monitoring (SIM)/scan mode, and unit mass ions were scanned from 30 – 300 m/z. All TD-GC/MS data were analyzed using Agilent’s ChemStation software.
Ions indicative of FTOHs and other PFAS compounds [m/z 31 (CF+), m/z 69 (CF3+), and m/z 95 (CF2C2H5O+)] were included in the SIM method with dwell times of 100 ms, the peak areas of which were used as analyte signals (Henderson et al., 2007; Roth et al., 2020). The peak area for m/z 69 was used as the signal for the FTOHs. Chromatographic retention times were confirmed with sorbent tubes directly loaded with the FTOH standards, and the unique ratio of m/z 69 to 95 for each FTOH was used as additional confirmation. The chromatographic peaks and retention times for the FTOH standards along with representative mass spectra are provided in Figure S3–S6. As shown in those figures, the FTOHs eluted in a predictable order according to molecular weight: 10.74, 13.24, 15.63, and 17.50 min for 4:2, 6:2, 8:2, and 10:2 FTOH, respectively. The areas of additional ions characteristic of PFAS compounds (m/z 50, 75, 77, 113, and 131) were extracted from the scan chromatograms of each FTOH to assess the relative amount of non-targeted degradation products present at each temperature.
2.4. Thermal treatment
During an experimental run with CIMS sampling, the tube furnace was held at ~800 °C for 5 min, then turned off and allowed to cool to ~200 °C with measurements taking place continuously throughout the cooling cycle, which usually was about 15 min in duration. This was repeated at least two more times (a minimum of three replicate experiments per FTOH) in order to capture any experimental variability. An average cooling profile for all performed CIMS runs is provided in Figure S7. It was found to be preferable to analyze only the cooling portion of a temperature cycle because at temperatures below 200 °C, appreciable amounts of the FTOHs began to deposit onto the surfaces of the quartz reactor, CaO coating, and sampling manifold. Then during the early portions of a temperature ramp, the FTOHs desorbed, rendering the data from ~200 to 275 °C mostly unusable. By examining only the cooling portion, any adsorbed FTOH was already sufficiently removed during reactor heating to 800 °C, and adsorption effects were avoided until 200 °C when analysis was stopped. CIMS signals from each of the 3+ runs were averaged into 10 °C temperature bins. In addition to CaO thermal treatment assessments, similar measurements were also taken in the absence of CaO in order to assess the effectiveness of the treatment compared to purely thermal degradation. The real-time and fast measurements by CIMS provided an efficient method to calculate removal efficiencies with high temperature resolution.
TD-GC/MS runs were conducted separately from the CIMS runs in order to make appropriate use of the fast acquisition rate afforded by the CIMS and to accommodate the longer sorbent tube sampling times needed for reliable analyte collection and detection at each temperature step. Sorbent tube samples were collected during tube furnace cooling at constant temperature steps of approximately 800, 700, 600, 500, 400, 350, 300, and 250 °C for the 10 min sampling duration.
Figure S2 illustrates the setup used for the two sampling conditions (CIMS and TD-GC/MS sampling) including flow paths and flow rates.
2.5. Calculation of removal efficiency
Temperature dependent destruction/removal efficiencies were determined for both CaO thermal treatment (CIMS and TD-GC/MS) and thermal-only degradation (CIMS only) of the FTOHs using Equation 1. These were calculated for individual temperature points as 1 minus the ratio of the FTOH signal at that temperature to the maximum observed FTOH signal, which, as expected, were the signals measured at the lowest temperatures.
| (1) |
Destruction and removal are used interchangeably throughout the manuscript to describe either the thermal breakdown of gas-phase compounds or the irreversible reaction of said compounds with the CaO treatment media.
3. Results and discussion
3.1. Assessing diffusion vs. advection in the reactor
Entering the heated reactor, the FTOH was thoroughly mixed with carrier air and begins to diffuse to and react with the CaO coated reactor walls creating a radial concentration gradient. Flow was laminar at all treatment temperatures with Reynolds numbers (Re) of approximately 10. A ratio of the characteristic times for advection (tadv) and diffusion (tdiff), was used to determine whether mass transfer of the FTOHs was primarily dominated by advection or diffusion. The characteristic time for diffusion in the radial direction can be defined by Equation 2,
| (2) |
where r is the inner radius of the reactor, and D is the gas-phase mass transfer coefficient or diffusion coefficient. The characteristic time for advection, tadv, is given simply by Equation 3,
| (3) |
where U is the speed of advection, and L is the length of heated portion of the reactor. The dimensionless ratio of these characteristic times is then given by Equation 4, which provides a relative magnitude of the importance of advection and diffusion.
| (4) |
Values ~1 indicate a mixture of advection and diffusion, values >>1 indicate advection dominated mass transfer, and <<1 indicate diffusion dominated mass transfer with time scales for diffusion to the walls being much shorter than the reactor residence time.
10:2 FTOH, the largest FTOH sampled, and 4:2 FTOH, the smallest FTOH sampled, were used to bracket the range in possible sampling conditions. A mass transfer coefficient at 298 K (D298) of 0.054 cm2 s−1 and 0.07 cm2 s−1 was used for 10:2 FTOH and 4:2 FTOH, respectively (Ahrens et al., 2011a). The mass transfer coefficients were then adjusted for the elevated temperatures using Equation 5 (Tang et al., 2014).
| (5) |
Figure 1 shows the estimated range in tdiff/tadv for all the experimental conditions examined. Under the flow conditions and temperatures used, there is sufficient residence time for mass transfer by diffusion. The system is estimated to be slightly diffusion dominated at lower temperatures and more so at the higher sampling temperatures, with tdiff/tadv<1 for all cases.
Fig. 1.
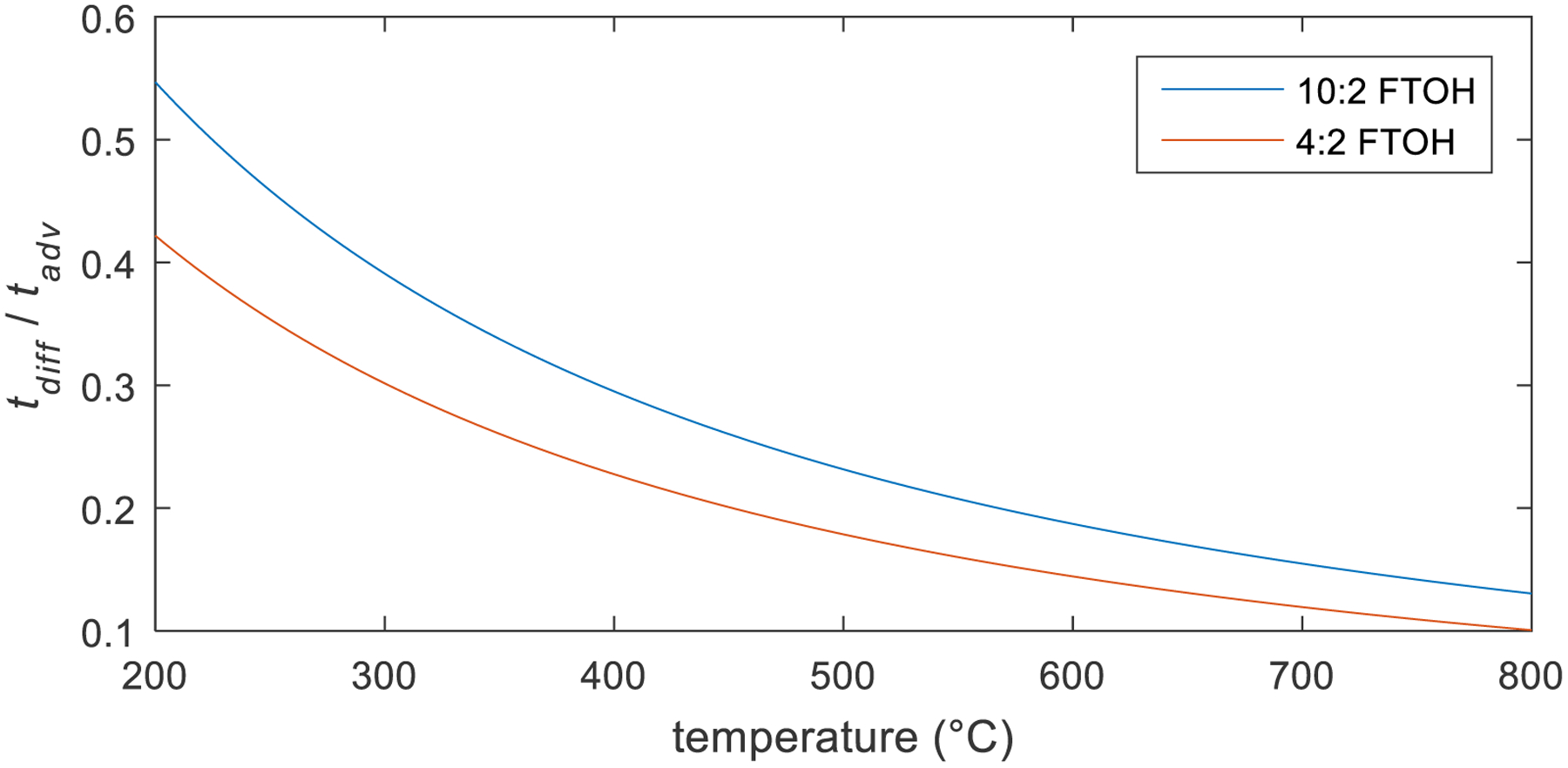
Ratio of the characteristic times for advection and diffusion (tdiff/tadv) in the reactor for 10:2 and 4:2 FTOH over the treatment temperature range.
3.2. Removal Efficiency of FTOHs by CaO
The amount of CaO in the reactor (1 g) was far in excess of the maximum amount of fluorine provided by the FTOHs, so a reduction in the yield of the FTOH + CaO reaction due to saturation of CaO was not a concern. For example, even if the setup was allowed to run for 24 hours (the longest TD-GC/MS runs lasted only ~3 hours) at 800 °C (the maximum reaction efficiency for the temperatures studied), <0.002% of the CaO is predicted to be consumed.
Temperature dependent destruction/removal efficiencies for 4:2, 6:2, 8:2, and 10:2 FTOH by CaO are provided in Figure 2 along with the thermal decomposition temperature profiles. To our knowledge, these are the first and only investigations on the thermal stability of gas-phase FTOHs in the presence and absence of CaO. Results for the two measurement approaches (CIMS and TD-GC/MS) agreed well, showing good overlap for the CaO treatment temperature profiles (Figure 2). Error bars on the CIMS-reported measurements (both CaO treatment and thermal degradation only) represent the standard deviation for each temperature bin for all experimental runs. As the temperature in the tube furnace was increased above 250 °C, removal of each FTOH due to treatment started to become apparent. As the carbon chain length increased across the FTOHs sampled, there was a clear decrease in treatment efficiency presumably due to steric hinderances in the reactivity with the CaO solid brought on by the longer and likely less linear chains. At 400 °C, 4:2 FTOH was removed with >95% efficiency, but as the size of each FTOH increased, the efficiency dropped: <87% removal efficiency for 6:2 FTOH, <78% for 8:2 FTOH, and <39% for 10:2 FTOH at 400 °C. This decrease in removal efficiency across the FTOHs appeared to approach the thermal decomposition only profile. This is most apparent for the 10:2 FTOH, the largest FTOH of the set, where treatment with CaO may offer minimal advantages over thermal destruction for primary compound removal. We note though that there is a tendency in modern PFAS applications to employ shorter chain molecules over the longer chain homologues (Wang et al., 2013b). For the shorter chain 4:2 and 6:2 FTOHs, there are significant advantages to CaO treatment. Greater than 90% of the 4:2 and 6:2 FTOHs were removed by 500 °C, a two-fold improvement over just thermal treatment. Above 750 °C, the four FTOHs tested had essentially aligned with the purely thermal decomposition profile, indicating that CaO treatment beyond such temperature was no longer required to remove the primary FTOH. That said, despite primary FTOH removal, there was evidence of formation of additional fluorinated compounds throughout the treatment process that originate from the degradation of the primary FTOHs. Reaction of the FTOHs with CaO did not seem consistent with the hydrodefluorination removal mechanism proposed by Wang et al. (2015a) for reaction of perfluorooctanesulfonate (PFOS) with Ca(OH)2. There was no evidence of significant m/z 51 (CHF2+) signals in the GC/MS spectra, and at temperatures above 500 °C, any formed Ca(OH)2 would decompose to CaO and water vapor.
Fig. 2.
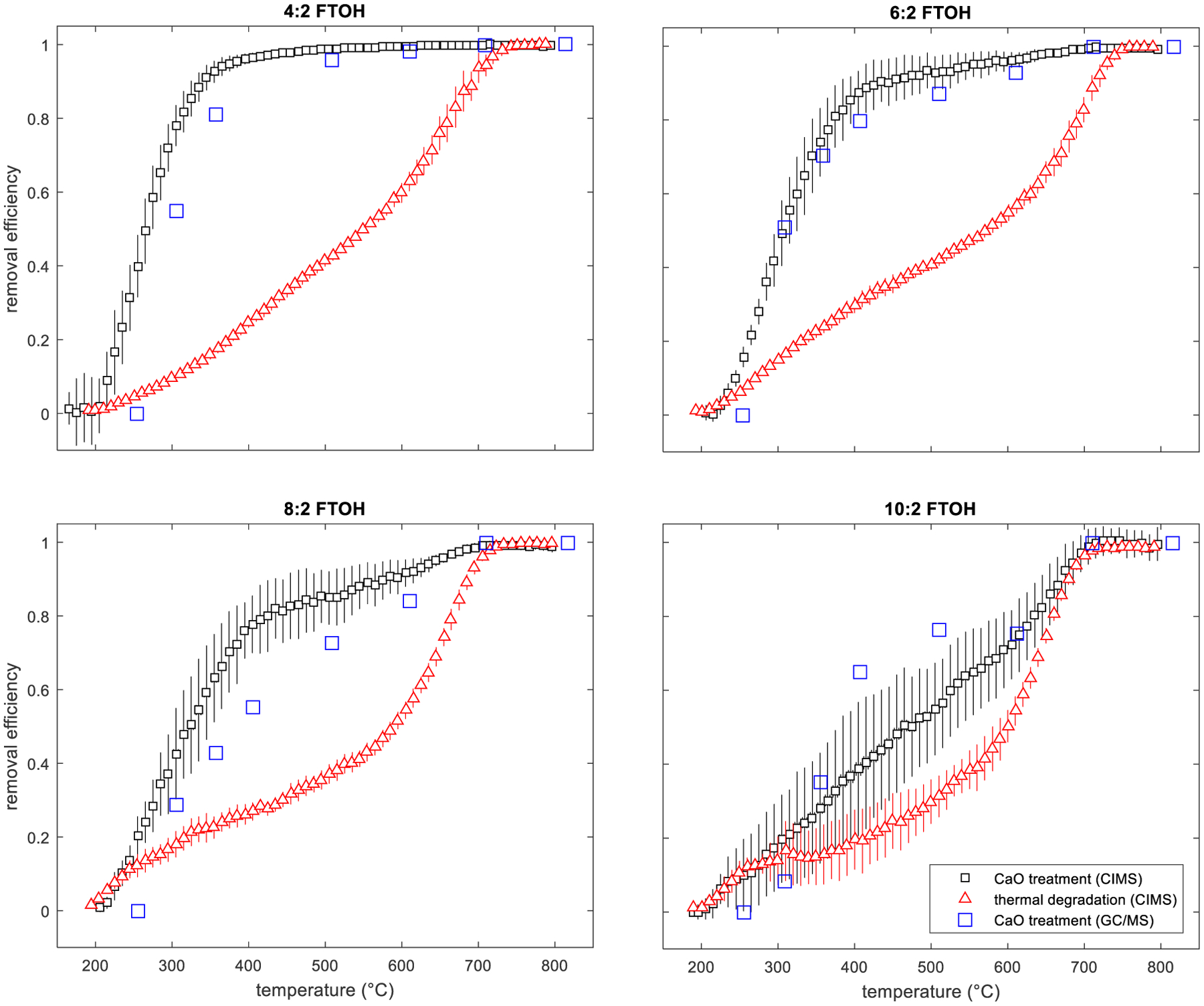
Temperature dependent removal efficiencies by CaO treatment and thermal decomposition for 4:2, 6:2, 8:2, and 10:2 FTOH as measured by CIMS and TD-GC/MS.
3.3. Products of incomplete destruction
Beyond their use to determine the destruction/removal efficiency of the FTOHs, the two measurement approaches can assess, to varying degrees, the extent to which other PFAS compounds (referred to here as products of incomplete destruction or removal) are formed. Depending on whether polar functional groups remained on secondarily formed compounds, the CIMS was able to detect a small selection of such compounds and report an accurate mass and thus a probable molecular composition/formula was determined. The TD-GC/MS sorbent tube approach, while less chemically specific due to quadrupole mass selection and the comparatively harder ionization employed, was used as a qualitative measurement of any gas-phase PFAS exiting the treatment apparatus, regardless of any functionality concerns. Thus, sorbent tubes sampling captured compounds that were not observed by the CIMS. Any PFAS signals observed using TD-GC/MS unrelated to the primary FTOH were indicative of products of incomplete destruction. Given the ionization scheme, unit mass resolution, and lack of chemical standards, identifying possible molecular formula and structure of these secondary species was outside of the scope of this work.
For each of the FTOHs tested, the CIMS was able to identify two degradation product molecular compositions that likely retained the hydroxyl functional group. These compositions were indicative of loss of HF or CH2. This was most obvious for 4:2 FTOH, which is shown in Figure 3 where the error bars (Figure 3A) again represent the standard deviation for each temperature bin. As the FTOH (C6F9H5O) started to degrade at the lower temperatures, C6F8H4O (loss of HF) and C5F9H3O (loss of CH2) signals rose from their respective <200 °C values. These compounds persisted to slightly higher temperatures with ~50 °C offset in peak concentration compared to the primary FTOH before they ceased to be formed or started to undergo significant losses by reaction with the CaO treatment media. To show this trend, Figure 3A presents CIMS measurements for the original 4:2 FTOH as well as C6F8H4O and C5F9H3O products of incomplete destruction as a function of temperature with each signal normalized to the maximum signal for each species. The relative signal of each species is shown in Figure 3B by normalizing the same data to the maximum 4:2 FTOH signal. Figure 3B indicates that relative signals of both products of incomplete destructions are very small compared to that of 4:2 FTOH, are formed at low temperatures, peak, and decay as temperatures increase. Similar plots to Figure 3A for 6:2, 8:2, and 10:2 FTOH are provided in Figure S8–S10. These products of incomplete destruction were also observed during thermal only treatment but peaked at substantially higher temperatures (~400 °C higher), indicating effective low temperature destruction/removal by CaO. Compared to the fluorocarbon backbone, the hydroxyl group is likely the most labile portion of the FTOH molecules and would therefore be expected to be eliminated most easily or first with treatment (thermal-only or CaO thermal treatment). As such, the CIMS likely only captures a small subset of degradation products, and another technique capable of additional non-polar or weakly polar PFAS detection is needed, which was the benefit of sorbent tube sampling and analysis by TD-GC/MS.
Fig. 3.
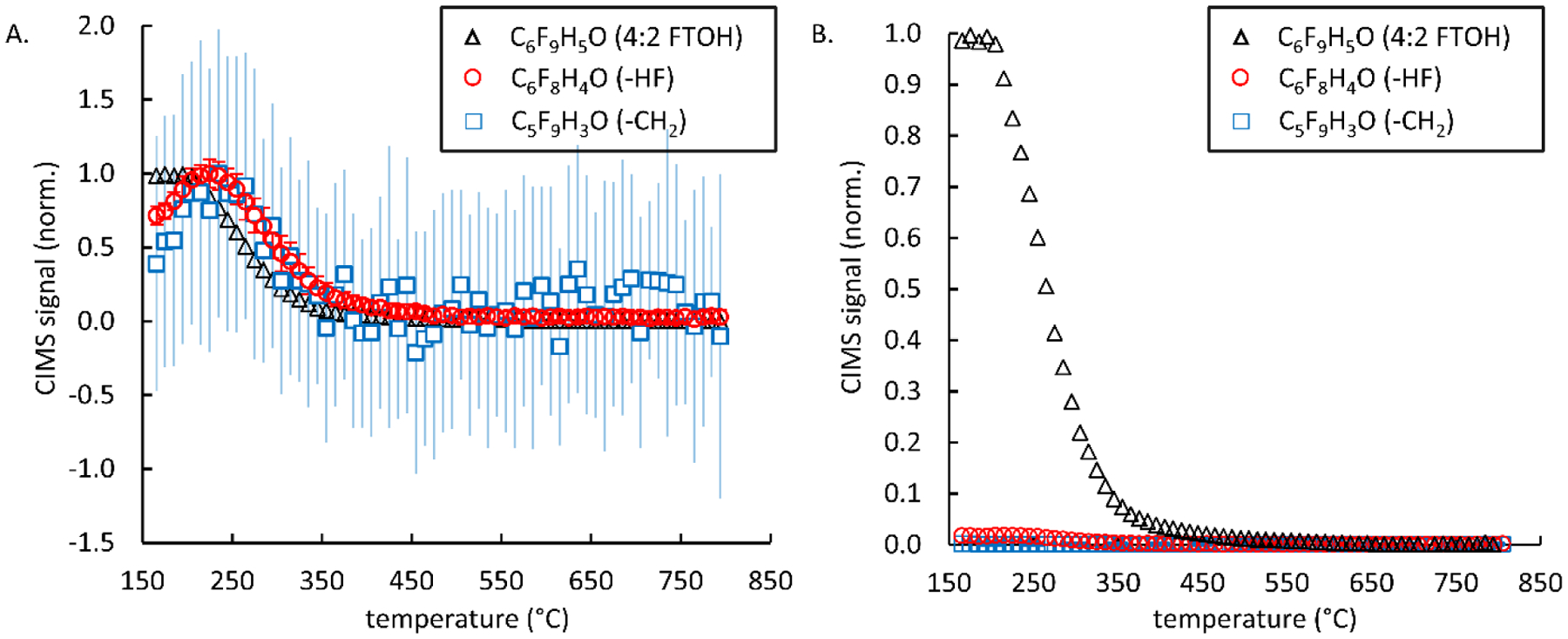
Temperature dependent destruction/removal and formation of products of incomplete destruction resulting from CaO thermal treatment of 4:2 FTOH. 3A) Presents CIMS measurements of the 4:2 FTOH and two products of incomplete destruction each normalized to the maximum CIMS value of each species. 3B) Presents CIMS measurement of the 4:2 FTOH and two products of incomplete destruction each normalized to the maximum CIMS value of the 4:2 FTOH.
In addition to measuring FTOH levels, the TD-GC/MS was used to assess gas-phase GC-elutable fluorinated products of incomplete destruction present in the emissions from the treatment process. The presence of PFAS indicative ions in the GC/MS mass spectra at retention times different from the primary FTOH compounds were assumed to represent these products. In addition to m/z 31, 69, and 95, m/z 50 (CF2+), m/z 75 (C2HCF2+), m/z 77 (C2H3CF2+), m/z 113 (CF2CF2CH+), and m/z 131 (CF3CF2C+) were also considered as potential PFAS ions (Roth et al., 2020). Extracted ion chromatogram (EIC) peak areas were compared for this set of ions at various retention times as a measure of breakdown product levels across treatment temperatures. A selection of representative temperature dependent EIC areas with their associated retention times from each of the four FTOH treatment experiments are shown in Figure 4, where the areas were normalized by their maximum values for ease of comparison. As in Figure 3A, this normalization approach presents relative concentrations of all compounds on a scale from 0 – 1, regardless of their actual concentrations, but allows the effect of temperature to be seen. Similar to the CIMS-measured products of incomplete destruction described above (Figure 3), an initial rise was typical as the primary FTOH was destroyed/removed, followed by destruction/removal of the breakdown products by the treatment media at elevated temperatures. It is likely that products identified by the CIMS are contributors to these GC/MS signals. At temperatures above 600 °C, the majority of EIC areas were reduced to below 90% of their maximum values, indicating that most of the products of incomplete destruction were being destroyed/removed by CaO treatment or no longer being formed. A larger list of the retention times, ions, and EIC areas, including the data used in Figure 4, is provided in Table S1, where the analysis was limited to retention times <10 minutes. Table S1 is not intended to be an exhaustive list or a quantitative measure of the amount of non-primary PFAS exiting the treatment system but more of an assessment of the presence or absence of fluorinated products of incomplete destruction being formed during the treatment process. In general, it was clear that more chromatographic peaks corresponding to fluorocarbon ions were present as the size of the FTOHs increased (see Table S1).
Fig. 4.
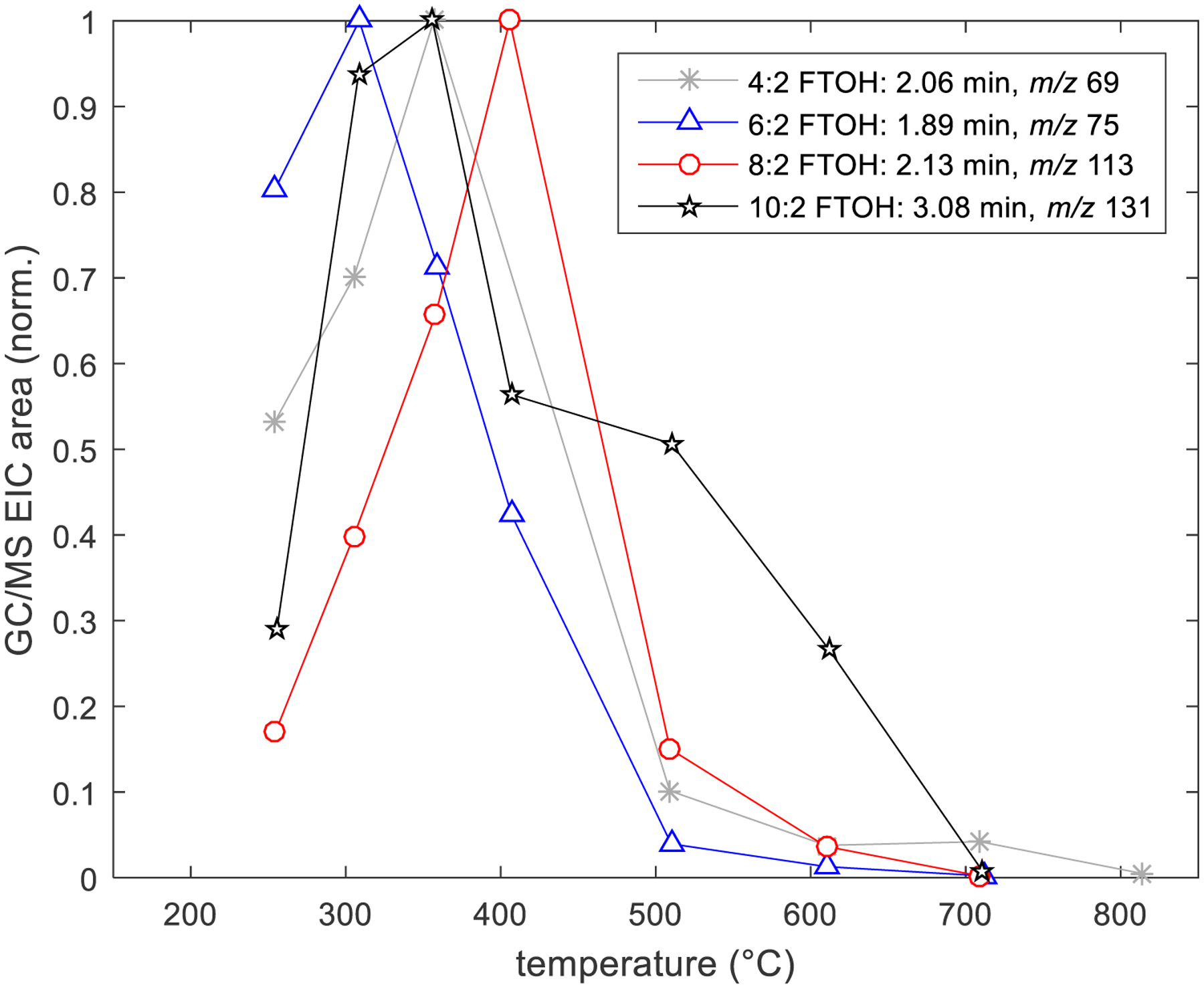
Extracted ion chromatogram (EIC) peak areas, normalized to the species’ largest area counts observed, at various CaO treatment temperatures for select non-FTOH PFAS ions measured by TD-GC/MS. Each trace is identified by the precursor FTOH experiment, the retention time, and the nominal ion mass.
It is worth noting that use of a standard reference library (NIST Chemistry WebBook) to interpret the GC/MS mass spectra did not prove useful for compound identification of the CaO treatment byproducts. This was unsurprising considering 8:2 FTOH was the only one of the primary FTOHs tested that had available reference spectra in the NIST library utilized for compound identification. For the most part, there were relatively few PFAS standard spectra for comparison purposes. However, when referencing the library, the compound identification suggestions with the highest confidence were those that contained fluorine, so despite not having a conclusive assignment, the library was still useful as a confirmatory check that the compounds were likely fluorocarbons.
3.4. PFAS disposal and destruction considerations
Since most thermal disposal methods involving combustion and incineration operate well above 800 °C, it may seem suitable to simply rely on these thermal approaches to decompose PFAS for disposal. This raises an important point regarding the destruction of PFAS. PFAS are often composed of a fluorinated alkyl carbon backbone and a non-fluorinated functional group. There are concerns that partial destruction might be accomplished by dissociation of the functional group that also results in incomplete defluorination of the stable fluorocarbon backbone. Such a situation would result in seemingly high destruction of the original compound but potential emission of fluorocarbon products of incomplete destruction, especially if validation focusses on analysis only of targeted compounds without analysis of additional non-targeted species. These concerns apply to both low temperature reactions and high temperature incineration approaches. However, one benefit of heterogeneous reactions of gas-phase PFAS with compounds like CaO is that they occur at easily achieved, relatively low temperatures without the need for a standing flame and large quantities of auxiliary energy.
In addition to PFAS-type products of incomplete destruction, hydrofluoric acid can also be released during thermal treatment of PFAS. HF is classified as a hazardous air pollutant (HAP) which requires additional post-thermal treatment to reduce emissions (U.S. EPA, 2015). Highly corrosive HF can cause extensive damage to silica-based refractories and metal ductwork used in many thermal treatment devices, a significant operational and maintenance issue (Che et al., 2002). Compared to the thermal-only experiments, the reduction in HF produced in the presence of the CaO treatment media was significant. Figure 5 illustrates this for the 8:2 FTOH case. At the maximum sampled temperature, HF is reduced by a factor of 66 with the inclusion of CaO. Reductions in HF with CaO treatment were observed for all FTOHs sampled. At 800 °C, CaO treatment reduced the amount of HF formed by a factor of 3.4–66, depending on which FTOH was treated.
Figure 5.
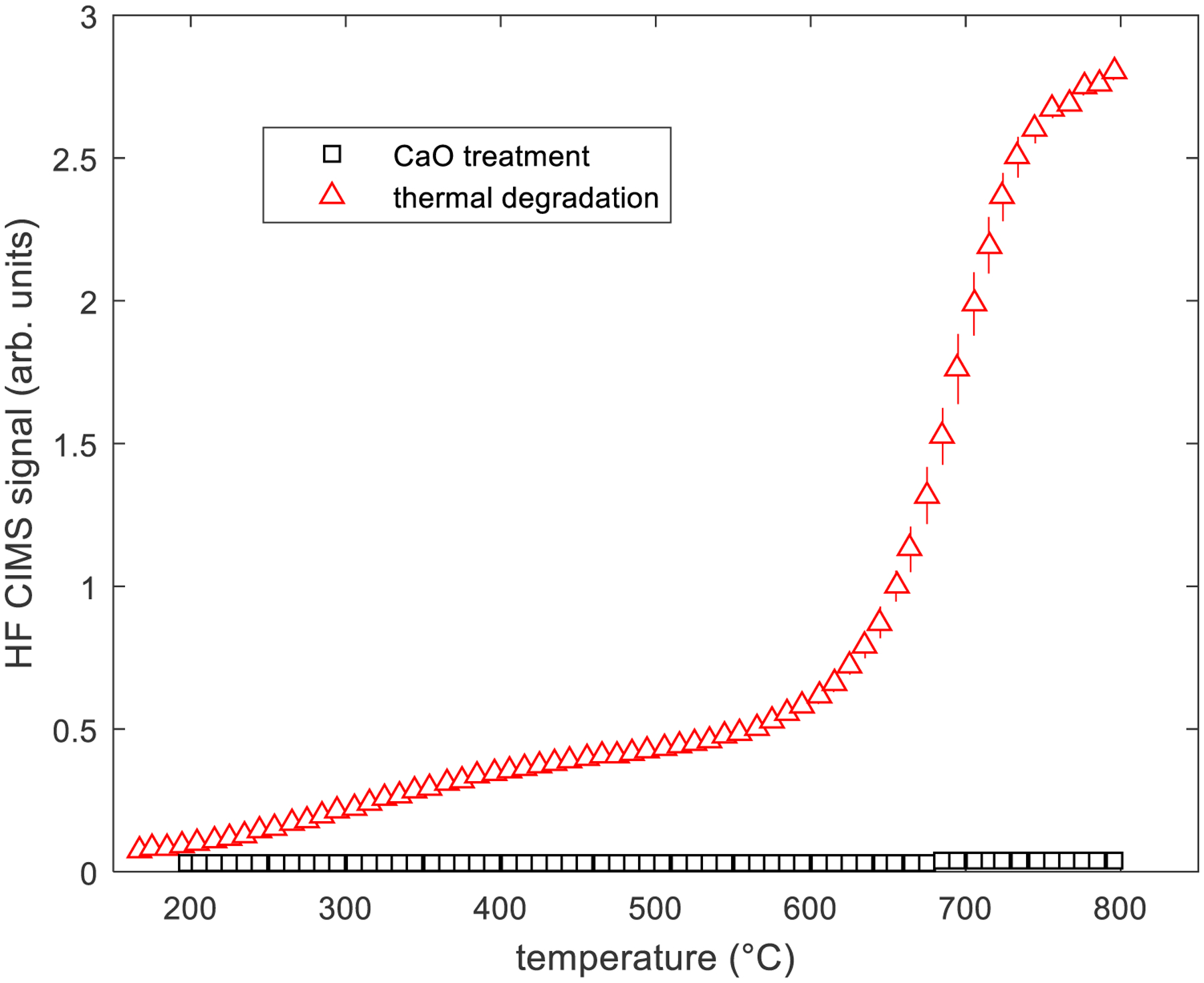
Hydrofluoric acid (HF) signals during CaO thermal treatment and thermal decomposition of 8:2 FTOH.
The absence of significant HF emissions suggests the formation of CaF2 when CaO is present. CaF2 was not measured in our work, but it has been reported (Wang et al., 2015a) that calcium compounds such as CaO and Ca(OH)2 are very reactive defluorination agents which convert fluorine in PFAS into CaF2 in thermal treatment of PFAS at low temperatures (300–900 °C) similar to those in our work. Wang et al. (2015a) characterized CaF2 and reported over 95% of the fluorine in a PFAS compound, perfluorooctanesulfonate (PFOS), was converted into CaF2 when the compound was treated with CaO at 700 °C. CaF2 is a most stable species in nature, and most fluorine used to produce PFAS originates from the pyrolysis of the mineral form of CaF2 (fluorite). The results for thermal treatment with CaO are in contrast to the thermal only case (without CaO) where a significant HF signal is seen beginning at low temperatures and increasing notably at temperatures (~650 °C) corresponding to increased rates of thermal destruction (see Figure 2). The absence of significant amounts of HF in the air emissions also represents an important advantage of reacting PFAS with calcium compounds, namely inherent control and neutralization of this extremely corrosive hazardous air pollutant.
The large reduction in HF emissions with CaO thermal treatment compared to thermal treatment only is independent evidence that relative time scales calculated for mass transfer by diffusion and advection made in section 3.1. Assessing diffusion vs. advection in the reactor are valid. Ample interaction and reaction of the FTOHs with the CaO coating is needed for such a reduction in HF, giving confidence that the treatment was not limited by diffusion of the FTOHs to the CaO coated walls of the reactor.
4. Conclusions
FTOHs were chosen as model PFAS given their importance in industrial PFAS applications and because CIMS was capable of providing fast real-time measurements of these compounds. Similar CaO facilitated thermal destruction is expected with other gas-phase PFAS classes, but future studies are needed to confirm this. Thermal treatment with CaO offered several advantages over thermal only treatment and was most effective for the shorter chain FTOHs sampled. At the higher temperatures examined (~800 °C), upwards of 99% of the primary compound was destroyed/removed, but even at the more moderate treatment temperatures (~400 °C), removal efficiencies were >85%. As the molecular length of the compounds increased, the effectiveness of treatment was reduced, but inclusion of the CaO treatment media still significantly reduced the formation of secondary fluorinated products of incomplete destruction as well as HF compared to strictly thermal destruction, thus minimizing significant environmental and operational issues. The formation and release of these secondary PFAS products may result from several different treatment and/or disposal processes but will likely be most relevant to stack emissions from elevated temperature applications such as material coating, annealing, and sintering of products that incorporate PFAS. Secondary products that do not contain alcohol or other oxidized moieties almost certainly have higher vapor pressures than the primary compounds, and therefore, likely result in air emissions. Under atmospheric conditions, they are likely to be oxidized to lower volatility compounds (like PFCAs) that ultimately will be deposited through typical atmospheric wet and dry deposition processes (Ellis et al., 2004; Wallington et al., 2006; Young et al., 2007).
Extension of CaO treatment could represent an economical solution to waste gas streams containing PFAS like FTOHs. Removal of FTOH vapors and subsequent products of incomplete destruction through CaO thermal treatment requires moderately low temperatures (<800 °C), thus reducing the energy needed to achieve thermal destruction. Stacks that traditionally vent PFAS vapors directly to the atmosphere from drying and sintering processes could efficiently remove such vapors at lower temperatures by incorporating CaO treatment prior to emission. Additionally, use of CaO as a pretreatment media before traditional gas-phase control processes like GAC packed beds could also extend the life of GAC and reduce the frequency of needed reactivation or disposal of PFAS-laden GAC. In the case of PFAS-laden GAC, the resulting reactivation waste gas stream would contain volatilized PFAS and treatment of these vapors prior to emission would prevent potential environmental concerns.
Supplementary Material
Highlights:
FTOHs, a volatile subset of PFAS, were destroyed more efficiently from heated gas streams interacting with calcium oxide than from heating only.
As primary FTOHs were heated, secondary gas-phase PFAS compounds were formed as the FTOHs partially thermally decompose.
Calcium oxide efficiently removed hydrofluoric acid and effectively destroyed FTOHs and secondarily formed gas-phase PFAS compared to heating alone.
Acknowledgements
The authors thank Stephen Jackson (U.S. EPA) for his assistance in sampling setup. The U.S. EPA through its Office of Research and Development funded and managed the research described here. This paper has been subjected to the Agency’s administrative review and approved for publication. The U.S. EPA does not endorse the purchase of any commercial products or services mentioned in the publication.
Footnotes
Appendix A. Supplementary data
Supplementary information and data related to this article can be found at [insert web address here].
References
- Ahrens L, Shoeib M, Harner T, Lane DA, Guo R, Reiner EJ, 2011a. Comparison of Annular Diffusion Denuder and High Volume Air Samplers for Measuring Per- and Polyfluoroalkyl Substances in the Atmosphere. Analytical Chemistry 83, 9622–9628. [DOI] [PubMed] [Google Scholar]
- Ahrens L, Shoeib M, Harner T, Lee SC, Guo R, Reiner EJ, 2011b. Wastewater Treatment Plant and Landfills as Sources of Polyfluoroalkyl Compounds to the Atmosphere. Environ. Sci. Technol 45, 8098–8105. [DOI] [PubMed] [Google Scholar]
- Aljawhary D, Lee AKY, Abbatt JPD, 2013. High-resolution chemical ionization mass spectrometry (ToF-CIMS): application to study SOA composition and processing. Atmos. Meas. Tech 6, 3211–3224. [Google Scholar]
- Bertram TH, Kimmel JR, Crisp TA, Ryder OS, Yatavelli RLN, Thornton JA, Cubison MJ, Gonin M, Worsnop DR, 2011. A field-deployable, chemical ionization time-of-flight mass spectrometer. Atmos. Meas. Tech 4, 1471–1479. [Google Scholar]
- Billups WE, Bell JP, Margrave JL, Hauge RH, 1984. Activation of hexafluoroethane by calcium atoms. Journal of Fluorine Chemistry 26, 165–167. [Google Scholar]
- Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, Jensen AA, Kannan K, Mabury SA, van Leeuwen SP, 2011. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess. Manage 7, 513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che SC, Iaquaniello G, Olivieri L, 2002. Selection of refractory for thermal oxidizers on gas streams containing fluorine. Environmental Progress 21, 116–120. [Google Scholar]
- Cicerone RJ, 1979. Atmospheric Carbon Tetrafluoride: A Nearly Inert Gas. Science 206, 59. [DOI] [PubMed] [Google Scholar]
- Cordner A, De La Rosa VY, Schaider LA, Rudel RA, Richter L, Brown P, 2019. Guideline levels for PFOA and PFOS in drinking water: the role of scientific uncertainty, risk assessment decisions, and social factors. Journal of Exposure Science & Environmental Epidemiology 29, 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crownover E, Oberle D, Kluger M, Heron G, 2019. Perfluoroalkyl and polyfluoroalkyl substances thermal desorption evaluation. Remediation Journal 29, 77–81. [Google Scholar]
- DeYoung DJ, 1994. Seventy Years of Science for the Navy and the Nation (1923–1993). Naval Research Laboratory, Washington, DC. [Google Scholar]
- Dinglasan-Panlilio MJA, Mabury SA, 2006. Significant Residual Fluorinated Alcohols Present in Various Fluorinated Materials. Environ. Sci. Technol 40, 1447–1453. [DOI] [PubMed] [Google Scholar]
- Dinglasan MJA, Ye Y, Edwards EA, Mabury SA, 2004. Fluorotelomer Alcohol Biodegradation Yields Poly- and Perfluorinated Acids. Environ. Sci. Technol 38, 2857–2864. [DOI] [PubMed] [Google Scholar]
- Ellis DA, Martin JW, De Silva AO, Mabury SA, Hurley MD, Sulbaek Andersen MP, Wallington TJ, 2004. Degradation of Fluorotelomer Alcohols: A Likely Atmospheric Source of Perfluorinated Carboxylic Acids. Environ. Sci. Technol 38, 3316–3321. [DOI] [PubMed] [Google Scholar]
- Ghisi R, Vamerali T, Manzetti S, 2019. Accumulation of perfluorinated alkyl substances (PFAS) in agricultural plants: A review. Environmental Research 169, 326–341. [DOI] [PubMed] [Google Scholar]
- Hagen DF, Belisle J, Johnson JD, Venkateswarlu P, 1981. Characterization of fluorinated metabolites by a gas chromatographic-helium microwave plasma detector—The biotransformation of 1H,1H,2H,2H-perfluorodecanol to perfluorooctanoate. Analytical Biochemistry 118, 336–343. [DOI] [PubMed] [Google Scholar]
- Han S-H, Park H-W, Kim T-H, Park D-W, 2011. Large Scale Treatment of Perfluorocompounds Using a Thermal Plasma Scrubber. Clean Technology 17, 250–258. [Google Scholar]
- Haukås M, Berger U, Hop H, Gulliksen B, Gabrielsen GW, 2007. Bioaccumulation of per- and polyfluorinated alkyl substances (PFAS) in selected species from the Barents Sea food web. Environmental Pollution 148, 360–371. [DOI] [PubMed] [Google Scholar]
- Henderson WM, Smith MA, 2006. Perfluorooctanoic Acid and Perfluorononanoic Acid in Fetal and Neonatal Mice Following In Utero Exposure to 8–2 Fluorotelomer Alcohol. Toxicol. Sci 95, 452–461. [DOI] [PubMed] [Google Scholar]
- Henderson WM, Weber EJ, Duirk SE, Washington JW, Smith MA, 2007. Quantification of fluorotelomer-based chemicals in mammalian matrices by monitoring perfluoroalkyl chain fragments with GC/MS. Journal of Chromatography B 846, 155–161. [DOI] [PubMed] [Google Scholar]
- Heydebreck F, Tang J, Xie Z, Ebinghaus R, 2016. Emissions of Per- and Polyfluoroalkyl Substances in a Textile Manufacturing Plant in China and Their Relevance for Workers’ Exposure. Environ. Sci. Technol 50, 10386–10396. [DOI] [PubMed] [Google Scholar]
- Hogue C, 2020. Incineration may spread, not break down PFAS. Chem. Eng. News https://cen.acs.org/environment/persistent-pollutants/Incincerators-spread-break-down-https://cen.acs.org/environment/persistent-pollutants/Incincerators-spread-break-down-PFAS/98/web/2020/04PFAS/98/web/2020/04. [Google Scholar]
- Klabunde KJ, Low JYF, Scott Key M, 1972. Metal atom reactions with fluorocarbons. II. Defluorination by calcium atoms1, 2. Journal of Fluorine Chemistry 2, 207–209. [Google Scholar]
- Kotthoff M, Müller J, Jürling H, Schlummer M, Fiedler D, 2015. Perfluoroalkyl and polyfluoroalkyl substances in consumer products. Environ. Sci. Pollut. Res 22, 14546–14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusic PJ, Marchione AA, Roe DC, 2005. Gas-phase NMR studies of the thermolysis of perfluorooctanoic acid. Journal of Fluorine Chemistry 126, 1510–1516. [Google Scholar]
- Krusic PJ, Roe DC, 2004. Gas-Phase NMR Technique for Studying the Thermolysis of Materials: Thermal Decomposition of Ammonium Perfluorooctanoate. Analytical Chemistry 76, 3800–3803. [DOI] [PubMed] [Google Scholar]
- Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J, 2007. Perfluoroalkyl Acids: A Review of Monitoring and Toxicological Findings. Toxicol. Sci 99, 366–394. [DOI] [PubMed] [Google Scholar]
- Lee BH, Lopez-Hilfiker FD, Mohr C, Kurtén T, Worsnop DR, Thornton JA, 2014. An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds. Environ. Sci. Technol 48, 6309–6317. [DOI] [PubMed] [Google Scholar]
- Lei YD, Wania F, Mathers D, Mabury SA, 2004. Determination of Vapor Pressures, Octanol–Air, and Water–Air Partition Coefficients for Polyfluorinated Sulfonamide, Sulfonamidoethanols, and Telomer Alcohols. J. Chem. Eng. Data 49, 1013–1022. [Google Scholar]
- Lemal DM, 2004. Perspective on Fluorocarbon Chemistry. The Journal of Organic Chemistry 69, 1–11. [DOI] [PubMed] [Google Scholar]
- Lindstrom AB, Strynar MJ, Delinsky AD, Nakayama SF, McMillan L, Libelo EL, Neill M, Thomas L, 2011. Application of WWTP Biosolids and Resulting Perfluorinated Compound Contamination of Surface and Well Water in Decatur, Alabama, USA. Environ. Sci. Technol 45, 8015–8021. [DOI] [PubMed] [Google Scholar]
- Liu X, Guo Z, Folk EE, Roache NF, 2015. Determination of fluorotelomer alcohols in selected consumer products and preliminary investigation of their fate in the indoor environment. Chemosphere 129, 81–86. [DOI] [PubMed] [Google Scholar]
- Martin JW, Muir DCG, Moody CA, Ellis DA, Kwan WC, Solomon KR, Mabury SA, 2002. Collection of Airborne Fluorinated Organics and Analysis by Gas Chromatography/Chemical Ionization Mass Spectrometry. Analytical Chemistry 74, 584–590. [DOI] [PubMed] [Google Scholar]
- McCord J, Strynar M, 2019. Identification of Per- and Polyfluoroalkyl Substances in the Cape Fear River by High Resolution Mass Spectrometry and Nontargeted Screening. Environ. Sci. Technol 53, 4717–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Zhang H, Cui Q, Sheng N, Yeung LWY, Guo Y, Sun Y, Dai J, 2017. First Report on the Occurrence and Bioaccumulation of Hexafluoropropylene Oxide Trimer Acid: An Emerging Concern. Environ. Sci. Technol 51, 9553–9560. [DOI] [PubMed] [Google Scholar]
- Prevedouros K, Cousins IT, Buck RC, Korzeniowski SH, 2006. Sources, Fate and Transport of Perfluorocarboxylates. Environ. Sci. Technol 40, 32–44. [DOI] [PubMed] [Google Scholar]
- Ravishankara AR, Solomon S, Turnipseed AA, Warren RF, 1993. Atmospheric Lifetimes of Long-Lived Halogenated Species. Science 259, 194. [DOI] [PubMed] [Google Scholar]
- Riedel TP, Lang JR, Strynar MJ, Lindstrom AB, Offenberg JH, 2019. Gas-Phase Detection of Fluorotelomer Alcohols and Other Oxygenated Per- and Polyfluoroalkyl Substances by Chemical Ionization Mass Spectrometry. Environ. Sci. Technol. Lett 6, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Abusallout I, Hill T, Holton C, Thapa U, Hanigan D, 2020. Release of Volatile Per- and Polyfluoroalkyl Substances from Aqueous Film-Forming Foam. Environ. Sci. Technol. Lett 7, 164–170. [Google Scholar]
- Stock NL, Lau FK, Ellis DA, Martin JW, Muir DCG, Mabury SA, 2004. Polyfluorinated Telomer Alcohols and Sulfonamides in the North American Troposphere. Environ. Sci. Technol 38, 991–996. [DOI] [PubMed] [Google Scholar]
- Sun M, Arevalo E, Strynar M, Lindstrom A, Richardson M, Kearns B, Pickett A, Smith C, Knappe DRU, 2016. Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environ. Sci. Technol. Lett 3, 415–419. [Google Scholar]
- Tang MJ, Cox RA, Kalberer M, 2014. Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: volume 1. Inorganic compounds. Atmos. Chem. Phys 14, 9233–9247. [Google Scholar]
- Tsang W, Burgess DR, Babushok V, 1998. On the Incinerability of Highly Fluorinated Organic Compounds. Combustion Science and Technology 139, 385–402. [Google Scholar]
- U.S. EPA, 2015. Initial List of Hazardous Air Pollutants with Modifications. https://www.epa.gov/haps/initial-list-hazardous-air-pollutants-modifications.
- Wallington TJ, Hurley MD, Xia J, Wuebbles DJ, Sillman S, Ito A, Penner JE, Ellis DA, Martin J, Mabury SA, Nielsen OJ, Sulbaek Andersen MP, 2006. Formation of C7F15COOH (PFOA) and Other Perfluorocarboxylic Acids during the Atmospheric Oxidation of 8:2 Fluorotelomer Alcohol. Environ. Sci. Technol 40, 924–930. [DOI] [PubMed] [Google Scholar]
- Wan HT, Lai KP, Wong CKC, 2020. Comparative Analysis of PFOS and PFOA Toxicity on Sertoli Cells. Environ. Sci. Technol 54, 3465–3475. [DOI] [PubMed] [Google Scholar]
- Wang F, Lu X, Li X. y., Shih K, 2015a. Effectiveness and Mechanisms of Defluorination of Perfluorinated Alkyl Substances by Calcium Compounds during Waste Thermal Treatment. Environ. Sci. Technol 49, 5672–5680. [DOI] [PubMed] [Google Scholar]
- Wang F, Lu X, Shih K, Liu C, 2011. Influence of calcium hydroxide on the fate of perfluorooctanesulfonate under thermal conditions. Journal of Hazardous Materials 192, 1067–1071. [DOI] [PubMed] [Google Scholar]
- Wang F, Shih K, Lu X, Liu C, 2013a. Mineralization Behavior of Fluorine in Perfluorooctanesulfonate (PFOS) during Thermal Treatment of Lime-Conditioned Sludge. Environ. Sci. Technol 47, 2621–2627. [DOI] [PubMed] [Google Scholar]
- Wang M, Tan Q, Liu L, Li J, 2019. A Facile, Environmentally Friendly, and Low-Temperature Approach for Decomposition of Polyvinylidene Fluoride from the Cathode Electrode of Spent Lithium-ion Batteries. ACS Sustainable Chemistry & Engineering 7, 12799–12806. [Google Scholar]
- Wang Z, Cousins IT, Scheringer M, Hungerbühler K, 2013b. Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors. Environment International 60, 242–248. [DOI] [PubMed] [Google Scholar]
- Wang Z, Xie Z, Mi W, Möller A, Wolschke H, Ebinghaus R, 2015b. Neutral Poly/Per-Fluoroalkyl Substances in Air from the Atlantic to the Southern Ocean and in Antarctic Snow. Environ. Sci. Technol 49, 7770–7775. [DOI] [PubMed] [Google Scholar]
- Washington JW, Naile JE, Jenkins TM, Lynch DG, 2014. Characterizing Fluorotelomer and Polyfluoroalkyl Substances in New and Aged Fluorotelomer-Based Polymers for Degradation Studies with GC/MS and LC/MS/MS. Environ. Sci. Technol 48, 5762–5769. [DOI] [PubMed] [Google Scholar]
- Washington JW, Rosal CG, McCord JP, Strynar MJ, Lindstrom AB, Bergman EL, Goodrow SM, Tadesse HK, Pilant AN, Washington BJ, Davis MJ, Stuart BG, Jenkins TM, 2020. Nontargeted mass-spectral detection of chloroperfluoropolyether carboxylates in New Jersey soils. Science 368, 1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Takata M, Takemine S, Yamamoto K, 2018. Thermal mineralization behavior of PFOA, PFHxA, and PFOS during reactivation of granular activated carbon (GAC) in nitrogen atmosphere. Environ. Sci. Pollut. Res 25, 7200–7205. [DOI] [PubMed] [Google Scholar]
- Young CJ, Furdui VI, Franklin J, Koerner RM, Muir DCG, Mabury SA, 2007. Perfluorinated Acids in Arctic Snow: New Evidence for Atmospheric Formation. Environ. Sci. Technol 41, 3455–3461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.