

Abstract
The DNA damage response (DDR) is a coordinated cellular response to a variety of insults to the genome. DDR initiates the activation of cell cycle checkpoints preventing the propagation of damaged DNA followed by DNA repair, which are both critical in maintaining genome integrity. Several model systems have been developed to study the mechanisms and complexity of checkpoint function. Here we describe the application of cell-free extracts derived from Xenopus eggs as a model system to investigate signaling from DNA damage, modulation of DNA replication, checkpoint activation, and ultimately DNA repair. We outline the preparation of cell-free extracts, DNA substrates, and their subsequent use in assays aimed at understanding the cellular response to DNA damage. Cell-free extracts derived from the eggs of Xenopus laevis remain a robust and versatile system to decipher the biochemical steps underlying this essential characteristic of all cells, critical for genome stability.
Keywords: Xenopus cell-free extracts, DNA replication, DNA damage Response, Chromatin, S-phase, DNA repair
1. Introduction
Upon DNA damage, cells activate a DDR that causes a transient cell cycle arrest to facilitate DNA repair and/or programmed cell death. The DDR entails several steps: 1) recognition and signaling from the lesions, activation of cell cycle checkpoints, and fixing the lesions through a variety of DNA repair pathways. An operational DDR is critical for the maintenance of genome stability [1, 2].
Defects in sensing DNA damage, signaling from DNA damage, activating checkpoints, or repairing the lesions all lead to persistent DNA damage as well as improper propagation of genetic errors resulting in genomic instability and subsequent increased incidence in diseases, including cancer [3]. Genetic screens in yeast have been instrumental in identifying and characterizing components of the DDR, such as the identification of radiation-sensitive and checkpoint genes [4, 5]. These findings have been further validated in mammalian cells. However, the use of cellular systems has some limitations. For instance, standard genetic screens may be difficult to interpret for essential genes. Also, the DNA damage response is more complex in vertebrates than in yeast. Therefore, certain critical regulators of the damage response, such as p53 and BRCA1, can only be found in vertebrates [6]. Mammalian cell lines have been used to circumvent the above drawbacks. However, cell-based model systems do not allow the use of specific biochemical readouts because they are frequently based on cell growth, survival, or other phenotypes that derive from complex outputs [7]. Cell-free extracts from Xenopus provide an alternative that circumvents some of these limitations.
Xenopus cell-free extracts have been instrumental for the study of DNA replication, repair, and checkpoint signaling [8-16]. This system has key advantages [12, 17, 18]: First, the extracts contain cytoplasmic and nuclear proteins that can support up to 12 cell divisions in the absence of transcription. Second, the protein concentration in the extracts is high and sufficient to carry out DNA transactions, including complete rounds of semi-conservative, cell cycle regulated DNA replication. Third, the extracts allow the study of essential proteins following immunodepletion. Indeed, downregulation or deletion of essential genes using siRNA or CRISPR leads to complex phenotypes due to the gradual decrease of protein expression, which varies depending on their half-life. Extracts allow biochemical rescue experiments with recombinant proteins, wild-type, or engineered to contain mutations or deletions to further dissect its function. For example, depletion of endogenous proteins from the extract using specific antibodies has allowed to study extensively the role of essential proteins in DNA replication, repair, and checkpoint signaling [8, 17, 19-22]. Additionally, the absence of zygotic transcription has allowed the study of proteins whose activity would be interfered by gene expression, such as the role of the oncogene c-Myc in DNA replication [23] or the replication and transcription-independent mechanism of interstrand crosslink repair [19, 24].
DNA replication in eukaryotes requires the sequential loading of replication initiation factors ORC1-6, Cdc6 and Cdt1-MCM2-7 that assemble on chromatin to form the pre-replication complex. The subsequent initiation of DNA replication requires the activity of S-CDKs and DDK kinases and the recruitment of Cdc45 and GINS to form the active CMG helicase (Cdc45-MCM-GINS) as well as Treslin, MCM10, TopBP1, DNA Polymerase ϵ and other replication factors before active replication forks are established [10]. DNA replication experiments with Xenopus extracts involve the addition of sperm chromatin to egg extracts made from unfertilized Xenopus eggs by crushing the eggs by centrifugation at low speeds (≤ 20,000 × g) and recovering the fraction containing cytoplasmic and nucleoplasmic proteins along with lipids and small vesicles, with minimal dilution (Low-Speed Supernatant LSS, see Figure 1). The haploid maternal genomic DNA is pelleted during extract crushing. LSS can assemble nuclei from demembranated sperm heads that are capable of undergoing one complete round of semi-conservative DNA replication [8, 17]. By varying the method used to prepare the extract, different types of extracts mimicking specific phases of the cell cycle can be obtained (Figure 2). These extracts have been particularly useful in understanding the regulation of entry into S phase and mitosis [25-30].
Figure 1:

Preparation of LSS and HSS extracts. A: Freshly laid Xenopus eggs in their jelly coat. B: Dejellied egg following incubation in a L-cysteine solution. C: Packed eggs before low-speed centrifugation D: After low-speed centrifugation crude cytoplasm is recovered by piercing the side of the tube with a needle. The crude cytosol is then further fractionated by ultracentrifugation. D-E: clarified cytosol (transparent layer) and membranous layer are collected to form Low-Speed Supernatant Extract LSS. For HSS preparation, ultracentrifugation time is extended to separate the cytoplasm from lipid-rich layers better and only the clear layer (lacking membranes) is collected.
Fig. 2.
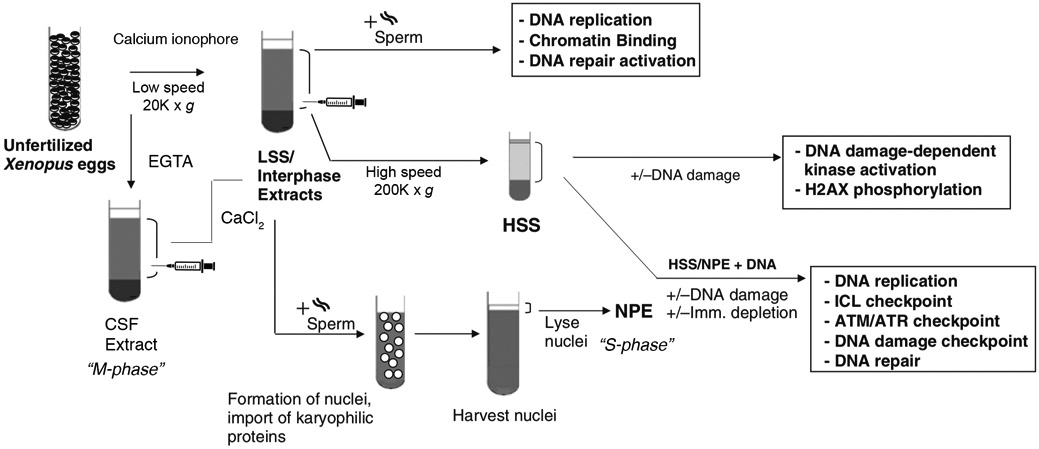
Schematic representation of Xenopus extracts used to the study the DNA Damage Response in the different cell-cycle phases. Following distinct centrifugation steps, extracts can be made replication-competent (LSS for chromatin templates, HSS/NPE for small plasmid templates) or replication-incompetent (HSS). See the text for details.
The initiation of DNA replication in extracts requires the assembly of a nuclear membrane surrounding the genomic DNA that locally imports and concentrates CDK and DDK kinase activities. Small DNA template, such as a circular plasmid, replicate poorly in crude cytosol (LSS), presumably because they do not promote nuclear membrane assembly efficiently. In contrast, extracts fractionated at high speeds (200,000 × g, High-Speed Supernatants, HSS) lack most lipids and vesicles and are unable to assemble nuclear envelopes, thus are replication-incompetent. HSS extracts are still capable of assembling chromatin and pre-Replication complexes on template DNA. Walter and Newport developed a “nucleus-free” system where the necessary kinase activity is supplemented in the form of an S-phase nucleoplasmic extract (NPE) derived from nuclei assembled in Low-Speed Supernatant extracts. Thus, artificially inducing the firing of pre-replication complexes [22]. This allows the study of plasmid replication and also alleviates other challenges faced in extracts involving nuclear assembly. Both types of Xenopus extracts (“nucleus-assembling” and “nucleus-free”) can be used to study checkpoint signaling by experimentally interfering with DNA replication or introducing damaged DNA.
In this chapter, we describe techniques that use the Xenopus model system to recapitulate several aspects of the DNA damage response, including cell cycle checkpoints and some aspects of DNA repair. The protocols used in the preparation of various extracts are described in Section 3.1. The intact and damaged DNA templates used in conjunction with the extracts are described in Section 3.2. The subsequent section (Section 3.3) covers the various assays that involve the use of the Xenopus system to investigate cell cycle checkpoints.
2. Materials
The animals and hormones described below are materials common to the protocols used for the preparation of extracts.
Animals: Xenopus laevis (the African clawed frog), females and males (Nasco). Females are used for obtaining eggs for extract preparation for at least three times, while males are used in the preparation of demembranated sperm chromatin.
Hormones to prime frogs: pregnant mare serum gonadotropin (PMSG, BioVendor) and pharmaceutical grade human chorionic gonadotropin HCG (Chorulon Injectable, Merk).
- General equipment:
- Refrigerated centrifuge (Sorvall RC-6 PLUS or similar) with HB-6 swinging bucket rotor and 14 mL round tube and Eppendorf tube adapters
- Refrigerated tabletop ultracentrifuge (Beckman OPTIMA MAX-XP or similar) equipped with TLS-55 swinging bucket rotor
- Fluorescent microscope (with a 385-400 nm excitation filter for DAPI-stained nuclei observation), plus glass slides and round 22mm coverslips and hemocytometer
- Tabletop centrifuges for 14 mL and Eppendorf-type tubes
- Water bath or temperature-controlled incubator (e.g., Eppendorf Thermomixer) capable of maintaining a temperature of 21-21°C
- Animal dissection kit
- Liquid nitrogen and cryogenic storage dewar
- Glass beakers
- 2.5 mL ultracentrifuge tubes (Beckman Coulter Ultra-Clear # 347356)
- 21-G needles and 3 mL disposable syringes
- 14 mL and 5 mL polypropylene Falcon tubes (Becton Dickinson # 352059 and #352063)
- Low retention 1.5-mL Eppendorf tubes (Fisher Scientific)
2.1. CSF Extract
MMR (Marc’s Modified Ringers) buffer: 5 mM HEPES (pH 7.7–7.8), 0.1 mM EDTA, 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 2 mM CaCl2
Dejellying buffer: 2% L-cysteine HCl monohydrate in MMR (pH 7.8 is critical)
XB Buffer: 10 mM HEPES (pH 7.7–7.8), 1 mM MgCl2, 0.1 mM CaCl2, 100 mM KCl, 50 mM sucrose
Cytostatic factor (CSF)-XB Buffer: 10 mM HEPES (pH 7.7–7.8), 2 mM MgCl2, 0.1 mM CaCl2, 100 mM KCl, 5 mM EGTA, 50 mM sucrose
Cytochalasin B. 10 mg/mL in ethanol. Store at −20°C
Energy Mix: 150 mM creatine phosphate, 20 mM ATP, 20 mM MgCl2 (Store at −20°C)
LPC protease inhibitors: leupeptin, pepstatin, chymostatin (Roche). Make a stock with 10 mg/mL each in DMSO (1000× stock) and store at −20°C
1 M CaCl2, for release into Interphase
2.2. Activated CSF Extract/Interphase Extract I
In addition to the materials required for CSF extract (see Section 2.1), the activated extract requires:
10 mg/mL cycloheximide
1M CaCl2
2.3. ELB Extract/LSS Extract
The materials required for the preparation of this extract are the same as those needed for CSF extract (see Section 2.1) except that ELB buffer is used instead of CSF buffer
Egg Lysis Buffer (ELB): 250 mM sucrose, 1× ELB Salts, 1 mM DTT, 50 μg/mL cycloheximide
10× ELB Salts (for ELB buffer): 25 mM MgCl2, 0.5 M KCl, 100 mM HEPES, pH 7.7–7.8, with KOH; filter sterilize. Store at 4°C
DAPI fix (for sperm decondensation and nuclei assembly observation): 3.7% formaldehyde, 2 μg/mL DAPI, 80 mM KCl, 15 mM NaCl, 15 mM PIPES–KOH (pH 7.2), 50% glycerol, stored at −20°C
2.4. Interphase Extract II
Dejellying buffer: 2% L-cysteine HCl monohydrate in 0.25× MMR, pH 7.8.
5× MMR: 100 mM HEPES–KOH, pH 7.5, 2 M NaCl, 10 mM KCl, 5 mM MgSO4, 10 mM CaCl2, 0.5 mM EDTA. Dilute to 1× or 0.25× as needed
5× S buffer: 250 mM HEPES–KOH, pH 7.5, 250 mM KCl, 12.5 mM MgCl2, 1.25 M sucrose. Keep at 4°C. Add β-mercaptoethanol to a final concentration of 2 mM; and LPC protease inhibitors (optional)
-
DAPI fix (for sperm decondensation observation): 3.7% formaldehyde, 2 μg/mL DAPI, 80 mM KCl, 15 mM NaCl, 15 mM PIPES–KOH (pH 7.2), 50% glycerol, stored at −20°C.
Store the following reagents in small aliquots at −20°C:- Calcium Ionophore A23187 (Sigma). Stock: 10 mg/mL in DMSO
- 10 mg/mL Cytochalasin B (Sigma) in DMSO or ethanol
- 1 M creatine phosphate (Sigma) in Milli-Q water
- 10 mg/mL creatine phosphokinase (CPK, Sigma) in Milli-Q water
- 15 mg/mL leupeptin (Roche) in DMSO
- 10 mg/mL cycloheximide (Sigma) in Milli-Q water
- Other materials: Styrofoam box or wooden platform, liquid nitrogen, cryogenic vials (1.5- or 2-mL)
2.5. High-Speed Supernatant (HSS)
Marc’s Modified Ringer’s Buffer (MMR): 100 mM NaCl, 2 mM KCl, 0.5 mM MgSO4, 2.5 mM CaCl2, 0.1 mM EDTA, 5 mM HEPES–KOH, pH 7.7–7.8
Dejellying solution: 2% L-cysteine HCl monohydrate in MMR, pH 7.8.
-
Egg Lysis Buffer (ELB): 1× ELB salts, 1 mM DTT, 50 μg/mL cycloheximide, 0.25 M sucrose
The 10× salts (stock) for Egg Lysis Buffer: 25 mM MgCl2, 500 mM KCl, 100 mM HEPES–KOH, pH 7.8, filter sterilized and stored at 4°C
Cytochalasin B (Sigma) 5 mg/mL in ethanol; stored at −20°C
Cycloheximide (Sigma) 10 mg/mL in water and stored at −20°C
Dithiothreitol (DTT) 1M in water; stored at −20°C
LPC protease inhibitors: leupeptin, pepstatin, chymostatin. Make a stock with 10 mg/mL each in DMSO (1000× stock) and store at −20°C
Energy Mix: 10 mM Creatine phosphate, 10 μg/mL Creatine kinase, 2 mM ATP, 2 mM MgCl2, 5 mM HEPES, pH 7.5, 1 mM DTT
2.6. Nucleoplasmic Extract (NPE)
Marc’s Modified Ringer’s Buffer (MMR): 100 mM NaCl, 2 mM KCl, 0.5 mM MgSO4, 2.5 mM CaCl2, 0.1 mM EDTA, 5 mM HEPES–KOH, pH 7.7–7.8
Dejellying solution: 2% L-cysteine HCl monohydrate in MMR, pH7.8.
Egg lysis buffer: 250 mM sucrose, 2.5 mM MgCl2, 50 mM KCl, 10 mM HEPES, pH 7.8, 50 μg/mL cycloheximide, 1 mM DTT
LPC protease inhibitor cocktail: leupeptin, pepstatin, chymostatin. Make a stock with 10 mg/mL each in DMSO (1000× stock) and store at −20°C
Cytochalasin B (Sigma) 5 mg/mL in ethanol; stored at −20°C
Cycloheximide (Sigma) 10 mg/mL in water and stored at −20°C
Nocodazole (Sigma) 5 mg/mL in DMSO; Sigma
Dithiothreitol (DTT) 1M in water; stored at −20°C
NPE–ATP regeneration system: Prepare stocks of 0.2M ATP (pH 7.0), 1M phosphocreatine disodium salt (pH 7.0) and 5 mg/mL creatine phosphokinase (CPK) in in 50 mM NaCl, 50% glycerol, 10 mM HEPES- KOH (pH 7.5). Make small aliquots and store at −20°C for up to six months. Make fresh before use. Mix on ice 10 μL of phosphocreatine, 5 μL of 0.2 M ATP, and 0.5 μL of 5 mg/mL CPK. Use 1μL per 30μL of extract. Discard leftovers after use.
Demembranated Sperm Chromatin (See Sections 2.8 and 3.2.1)
DAPI fix (for nuclear assembly observation): 3.7% formaldehyde, 2 μg/mL DAPI, 80 mM KCl, 15 mM NaCl, 15 mM PIPES–KOH (pH 7.2), 50% glycerol, stored at −20°C
Falcon Tube (Becton Dickinson # 352063)
Becton Dickinson tube # 352059
Beckman tube # P60720
2.7. Immuno- depletion of Extracts to Study Protein Function
Protein-A Sepharose CL-4B agarose beads (GE Healthcare)
Spin Columns (Snap Cap, Pierce #69725 or equivalent)
Phosphate-buffered saline (PBS buffer), pH 7–7.5
2.8. Demembranated Sperm Chromatin
Benzocaine. Dissolve 1 g in 10 mL of 100% ethanol. Dispense drop-wise into 2 L of water with continuous stirring. This gives a 0.05% solution of benzocaine. Make fresh. IMPORTANT: always follow your Institutional Animal Care and Use Committee guidelines on animal euthanasia
Marc’s Modified Ringer’s Buffer (MMR): 100 mM NaCl, 2 mM KCl, 0.5 mM MgSO4, 2.5 mM CaCl2, 0.1 mM EDTA, 5 mM HEPES–KOH, pH 7.7–7.8
Nuclei Preparation Buffer (NPB): 250 mM sucrose, 15 mM HEPES–KOH, pH 7.8, 1 mM EDTA, 0.5 mM Spermidine, 0.2 mM Spermine, 1 mM DTT. Prepare as a 2× solution
Triton X-100, 10% v/v in water; store at room temperature.
Bovine Serum Albumin. 10% w/v in water. Store at −20°C or make fresh
Glycerol
PMSF 0.3 M made in 100% ethanol. Make fresh before use
LPC protease inhibitors: leupeptin, pepstatin, chymostatin. Make a stock with 10 mg/mL each in DMSO (1000× stock) and store at −20°C
Spermidine 10 mM
Spermine 10 mM
Sucrose 1.5 M; store at 4°C
2.9. Generation of Double-Strand Breaks (DSB) in Chromatin
LSS or HSS Xenopus egg extract.
Demembranated sperm nuclei (see Section 3.2.1)
PflMI or equivalent restriction endonuclease
Chromatin isolation buffer: 50 mM Hepes-KOH pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 0.125% Triton X-100
Sucrose cushion: 30% sucrose in Chromatin Isolation buffer without Triton X-100
2.10. Preparation of Plasmid with an interstrand Crosslinks (ICL)
pBS plasmid
SJG-136 (NCI/Spriogen LTD)
Triethanolamine, 50 mM in EDTA, 2mM
Ethanol for DNA precipitation
2.11. Preparation of DNA with Double-Strand Breaks
pBR322 plasmid
HaeIII restriction endonuclease (NEB)
Phenol/chloroform to extract digested DNA
2.12. Preparation of Biotinylated Substrates
DNA fragment of interest
Gel purification kit
T4 polymerase
dATP, dGTP, dTTP, biotin–dCTP
EDTA, 0.5 M pH 8.0
PCR purification kit (Qiagen)
2.13. DNA Damage Checkpoint Induced by Single-Strand DNA Gaps in Chromatin
LSS or HSS Xenopus egg extract
Demembranated sperm nuclei
Etoposide or Exonuclease III
Tris–HCl, 1M pH 8.0
MgCl2, 1M
Nuclei Preparation Buffer (NPB): 250 mM sucrose, 15 mM HEPES–KOH, pH 7.8, 1 mM EDTA, 0.5 mM Spermidine, 0.2 mM Spermine, 1 mM DTT, LPC protease inhibitors 10 μg/mL each, 0.3 mM PMSF
Caffeine 100 mM in PIPES 10 mM, pH 8.0. Prepare fresh before use
Specific PIKK inhibitors (KU55933 for ATM and VE-821 for ATR, Selleck Chemicals Inc.)
α-32P–dCTP (Pelkin Elmer)
Stop Solution: 0.5% SDS, 80 mM Tris (pH 8.0), 8 mM EDTA
Proteinase K solution (Roche)
Phenol/chloroform to extract DNA
Trichloroacetic acid, 50% w/v in water
Whatman 3 MM blotting paper
2.14. DNA Damage Checkpoint Induced by DSB in Chromatin
LSS or HSS Xenopus egg extract
Circular control plasmid or plasmids containing double-strand breaks (DSB)
Caffeine, 100 mM in PIPES, pH 8.0. Prepare fresh before use.
Specific PIKK inhibitors (KU55933 for ATM and VE-821 for ATR, Selleck Chemicals Inc).
Sodium Acetate, 3M
Stop Solution: 0.5% SDS, 80 mM Tris (pH 8.0), 8 mM EDTA
Proteinase K solution (Roche)
Phenol/chloroform to extract DNA
Trichloroacetic acid
Whatman 3 MM blotting paper
2.15. Assay to Study Checkpoint Induced by ICL
Xenopus extracts (HSS/NPE, Sections 3.1.5 and 3.1.6)
Control and ICL DNA (Section 3.2.3)
NPE–ATP regeneration system: Prepare stocks of 0.2M ATP (pH 7.0), 1M phosphocreatine disodium salt (pH 7.0) and 5 mg/mL creatine phosphokinase (CPK) in in 50 mM NaCl, 50% glycerol, 10 mM HEPES- KOH (pH 7.5). Make small aliquots and store at −20°C for up to six months. Make fresh before use. Mix on ice 10 μL of phosphocreatine, 5 μL of 0.2 M ATP, and 0.5 μL of 5 mg/mL CPK. Use 1μL per 30μL of extract. Discard leftovers after use.
Caffeine, 100 mM in PIPES, pH 8.0. Prepare fresh before use.
Specific PIKK inhibitors (KU55933 for ATM and VE-821 for ATR, Selleck Chemicals Inc).
α-32P–dCTP (Pelkin Elmer)
Stop Solution: 0.5% SDS, 80 mM Tris (pH 8.0), 8 mM EDTA
Proteinase K solution (Roche)
Phenol: chloroform: isoamyl alcohol (25:24:1, Sigma)
Sodium acetate, 3M
Ethanol
DNA loading buffer with bromophenol blue and cyan blue dyes
Trichloroacetic acid
2.16. Study of the Checkpoint Induced by DNA Damage In Trans on DNA Replication
Xenopus Extracts (HSS/NPE; see Sections 3.1.5 and 3.1.6)
NPE–ATP regeneration system: Prepare stocks of 0.2M ATP (pH 7.0), 1M phosphocreatine disodium salt (pH 7.0) and 5 mg/mL creatine phosphokinase (CPK) in in 50 mM NaCl, 50% glycerol, 10 mM HEPES- KOH (pH 7.5). Make small aliquots and store at −20°C for up to six months. Make fresh before use. Mix on ice 10 μL of phosphocreatine, 5 μL of 0.2 M ATP, and 0.5 μL of 5 mg/mL CPK. Use 1μL per 30μL of extract. Discard leftovers after use.
Control or ICL DNA (see Section 3.2.3)
α-32P–dCTP (Pelkin Elmer)
2.17. Phosphorylated Histone H2AX Detection (Endogenous)
Activated CSF extract or Interphase Extract I (see Section 3.1.2)
Demembranated sperm chromatin (see Section 3.2.1)
Chromatin isolation buffer: 50 mM Hepes-KOH pH 7.5, 50 mM KCl, 2.5 mM MgCl2, LPC protease inhibitors, 0.125% Triton X-100 (made fresh before use); this buffer is supplemented with 1 mM NaF, 1 mM Na vanadate, and 0.125% Triton X-100
Sucrose cushion: Chromatin isolation buffer (without Triton X-100) supplemented with 30% sucrose
2.18. Histone H2AX Phosphorylation Assay (Exogenous Substrate)
Xenopus extracts (See Section 3.1)
Biotinylated DNA fragments to induce DNA damage
Streptavidin magnetic beads (Dynabeads, Thermo Fisher)
ELB Buffer: 250 mM sucrose, 1× ELB Salts, 1 mM DTT, 50 μg/mL cycloheximide (10× ELB Salts – for ELB buffer: 25 mM MgCl2, 0.5 M KCl, 100 mM HEPES, pH 7.7–7.8 with KOH); filter sterilize. Store at 4°C
Wild-type H2AX peptide: AVGKKASQASQEY
Mutant H2AX peptide: AVGKKAAQAAQEY
EB Buffer: 20 mM HEPES, pH 7.5, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT
ATP
γ-32P–ATP (Pelkin Elmer)
EDTA, 0,5M pH 8.0
p81 phospho-cellulose filter papers (Upstate Biotechnology)
Acetic acid
Caffeine, 100 mM in PIPES, pH 8.0. Prepare fresh before use.
Specific PIKK inhibitors (KU55933 for ATM and VE-821 for ATR, Selleck Chemicals Inc).
EB Kinase buffer: 20 mM HEPES, pH 7.5, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 1 mM NaF, 1 mM Na3VO4, 10 mM MnCl2
2.19. Phosphorylation of ATM/ATR Target Proteins
Xenopus Extracts (see Section 3.1)
Energy Mix: 10 mM Creatine phosphate, 10 μg/mL Creatine kinase, 2 mM ATP, 2 mM MgCl2, 5 mM HEPES, pH 7.5, 1 mM DTT
Control and damaged DNA (either chromatin or small DNA templates can be used; see Section 3.2)
Caffeine, 100 mM in PIPES, pH 8.0. Prepare fresh before use
Specific PIKK inhibitors (KU55933 for ATM and VE-821 for ATR, Selleck Chemicals Inc).
Phospho-Chk1 antibody (Cell Signaling)
Phospho-ATM antibody (Rockland Immunochemicals, PA)
Geminin, roscovitine or aphidicolin for DNA replication inhibition
Curcumin dissolved in ethanol
2.20. Chromatin Binding Assay
Xenopus extracts (see Section 3.1).
Demembranated sperm chromatin (see Section 3.2.1)
Chromatin isolation buffer: 50 mM Hepes-KOH pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 0.125% Triton X-100 (made fresh before use)
Chromatin isolation buffer + Sucrose: 50 mM Hepes-KOH pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 30% sucrose
Micrococcal Nuclease (NEB)
2.21. Binding Assay Using HSS/NPE and ICL Plasmid
Xenopus extracts (HSS/NPE see Sections 3.1.5 and 3.1.6)
NPE–ATP regeneration system: Prepare stocks of 0.2M ATP (pH 7.0), 1M phosphocreatine disodium salt (pH 7.0) and 5 mg/mL creatine phosphokinase (CPK) in in 50 mM NaCl, 50% glycerol, 10 mM HEPES- KOH (pH 7.5). Make small aliquots and store at −20°C for up to six months. Make fresh before use. Mix on ice 10 μL of phosphocreatine, 5 μL of 0.2 M ATP, and 0.5 μL of 5 mg/mL CPK. Use 1μL per 30μL of extract. Discard leftovers after use.
Protein-A Sepharose CL-4B beads
Spin Columns (Snap Cap, Pierce #69705 or equivalent)
Phosphate-buffered saline (PBS buffer), pH 7–7.5
Control or ICL plasmid DNA (see Section 3.2.3)
Chromatin isolation buffer (high KCL): 50 mM Hepes-KOH pH 7.5, 100 mM KCl, 2.5 mM MgCl2, supplement with 0.125% Triton X-100
Sucrose cushion: 30% sucrose in chromatin isolation buffer (without Triton X-100)
3. Methods
The methods described below include (a) the preparation of different types of extracts (Section 3.1), (b) the preparation of DNA substrates – both chromosomal (Sections 3.2.1 and 3.2.2) and small template DNA (Sections 3.2.3, 3.2.4 and 3.2.5) and (c) assays used to study checkpoints – (i) DNA replication assays (Sections 3.3.1 and 3.3.2), (ii) assays to study protein modifications such as phosphorylation to detect checkpoint activation (Sections 3.3.5, 3.3.6, 3.3.7 and 3.3.8), and (iii) Chromatin/DNA binding assays (Sections 3.3.,9 and 3.3.10).
3.1. Preparation of Extracts
Studies of the DDR using Xenopus involve at least four different types of extracts. Three of the extracts (Cytostatic Factor Extract, Low-Speed Interphase Extract, and High-Speed Extract) are derived from Xenopus eggs, whereas the fourth type (Nucleoplasmic Extract) requires Xenopus eggs and sperm chromatin to assemble and isolate nuclei from which a nucleoplasm fraction is extracted. The addition of sperm chromatin to activated CSF or LSS extracts leads to nuclear assembly and chromosomal replication. HSS lacks lipid vesicles and is unable to form nuclear envelopes; therefore, it is not replication competent, although DNA templates incubated in HSS form pre-replication complexes. DNA replication can be initiated subsequentially in HSS by supplementing with NPE (Figure 2) [16, 22].
Below, we describe the preparation of Cytostatic-arrested (CSF) extracts arrested in M phase and activated CSF extracts. Since CSF and activated CSF extracts are obtained by relatively low-speed centrifugation, they are also referred to as Low-Speed Supernatant (LSS, Figure 1). These extracts can be further fractionated for checkpoint studies to obtain fractionated membrane-free cytosol. Of note, a modification of the protocol to prepare LSS allows storing the extract, without appreciable loss of activity. We are referring to such extract as Interphase Extract II, which is also described in this chapter. Furthermore, we describe the preparation of High-Speed Supernatant (HSS), which is a membrane-free extract that supports the assembly of the pre-replication complexes on chromatin [16, 22]. HSS extract is also used with Nucleoplasmic Extract (NPE) to bypass the nuclear envelop formation step, thus allowing normal DNA replication of chromatin and plasmid [16, 22, 31]. The NPE system is particularly useful since it allows for the modification of the nuclear environment and supports plasmid replication very efficiently [22, 32]. The preparation of demembranated sperm chromatin and NPE are also outlined in this chapter. Energy Mix is added to extracts before use. Figure 2 summarizes the various types of extracts and their applications.
3.1.1. CSF Extract
Mature unfertilized eggs laid by Xenopus are arrested in meiosis by a calcium-sensitive activity (Cyclo-Static Factor, CSF) that inhibits the release from metaphase II into anaphase and meiotic exit into interphase. In Xenopus CSF extracts, the mitotic state stabilized by CSF is preserved by chelating free calcium during extract preparation, thus maintaining the characteristics of an M phase of the cell cycle. The addition of calcium allows controlled arrest release into interphase. CSF extract has been used extensively over the years for reconstitution of cell cycle transitions, studies of chromosome condensation, cohesion and microtubule spindle assembly [33]. Extracts are typically prepared in the presence of cytochalasin B or D to prevent actin polymerization, preventing local gelation that impairs extract homogeneity and might affect other processes. Protocols for the preparation of extracts in the absence of actin polymerization inhibitors are available [34]. The preparation of fresh CSF-arrested extracts by our group has been previously described in [7, 35].
Prime 4–6 female frogs with 500 μL/frog of PMSG (PMSG stock: 100 IU/μL). This step is generally performed 3–7 days before the frogs are induced with HCG.
Induce females to lay eggs with 800 IU of HCG/frog injected the evening before egg collection. Place each frog in a container with at least 1 L of 1× MMR (pH 7.7–7.8) after HCG injection. Eggs are laid overnight and collected the next morning (Figure 1A, see Notes 1 and 2).
Wash eggs in 1× MMR. Discard bad eggs with a plastic Pasteur pipette. Remove as much buffer as possible by decanting. Transfer eggs to a glass beaker.
Dejelly the eggs in 2% L-cysteine pH 7.8 (Figure 1B, see Note 3).
Remove the cysteine solution by pouring off the excess liquid.
Rapidly wash eggs three times with XB and then remove all XB.
Wash eggs three times in CSF-XB and remove as much buffer as possible.
Wash eggs two times in XB containing protease inhibitors (LPC, leupeptin, pepstatin, chymostatin, 1,000× stock: 10 mg/mL each in DMSO).
Transfer eggs into 1 mL of CSF-XB with LPC protease inhibitors and 100 μg/mL cytochalasin B [36] in 1.5 mL Eppendorf tubes (see Note 4). Alternatively, if there is a large volume of eggs, 14-mL Falcon tubes can be used.
Pack the eggs by spinning for 1 min at 800 rpm and remove excess buffer (Figure 1C, see Note 5).
Combine eggs from different tubes and fill each Eppendorf tube as much as possible. Crush the eggs at 4–8°C for 15 min at 10,000 rpm.
Collect the extract with an 18-gauge needle by puncturing the side of the tube and gently aspirate out the cloudy intermediate cytoplasmic layer. This layer is located between the top opaque lipid layer (yellow) and the solid pellet of pigments and egg debris (dark brown).
Supplement the cytosolic extract with 1/1,000 volume of cytochalasin B (10 mg/mL stock), 1/15 volume of energy mix, LPC protease inhibitors, and 1/40 volume of 2 M sucrose.
Clarify the cytosolic extract by centrifugation for 30 min at 13,000×g in an Eppendorf centrifuge at 4°C. The transparent cytoplasmic layer is collected for immediate use (see Notes 6 and 7).
3.1.2. Activated CSF Extract/Interphase Extract I
Upon activation by calcium, CSF extracts exit M phase and enter interphase. Such activated extracts are used in DNA replication, chromatin binding, and checkpoint assays.
Supplement freshly prepared CSF extracts (see Section 3.1.1) with 50 μg/mL of cycloheximide and 0.4 mM CaCl2.
Incubate for 15 min at 21°C. This treatment mimics the Calcium flux that usually takes place after fertilization of eggs and triggers degradation of mitotic cyclins, inactivation of Cdc2/Cyclin B, and exit from mitosis (see Note 7).
3.1.3. ELB Extract/LSS Extract
A variation of the activated extract is to prepare a “crude” S-phase extract/Low-Speed Supernatant Extract with MMR buffer that does not contain EGTA. Thus, eggs are already activated, and the buffer does not need to be supplemented with CaCl2. The protocol, in this case, is the same as for CSF extract except that the eggs are washed with ELB (Egg Lysis Buffer) instead of CSF-XB buffer. Since the ELB buffer already contains sucrose, it is also not necessary to supplement the cytosolic extract with 1/40 volume of 2 M sucrose (during Step 13 of CSF extract Section 3.1.1). Supplement extract with energy mix before use.
3.1.4. Interphase Extract II
The major advantage of this type of extract is that it can be stored as frozen drops at −80°C and thawed before use [37], allowing multiple experiments or technical replicates to be performed with a batch of extracts on different days. This particularly useful when working with extracts depleted with precious antibodies. Frozen extracts can be kept for about six months without appreciable loss of quality.
Induce 4–6 frogs to lay eggs in 0.25× Interphase Extract II MMR (See MMR recipe, the frogs are induced to lay eggs as described in Section 3.1.1 above.
Collect and pool good eggs by aspirating them out with a plastic transfer pipette.
Decant as much buffer as you can and then add dejelly buffer (prepare 100 mL buffer per frog), for 5–10 min. If there are more eggs, change de-jelly buffer every 2–3 min, but extending the time of dejellying may be detrimental to the quality of the extract (see Note 3).
Wash three times with 0.25× MMR buffer.
Add 1/10,000 of 10 mg/mL Calcium ionophore A23187 and incubate for 5 min to activate eggs. You might see how egg hemispheres (poles) become more apparent.
Wash three times with 0.25× MMR buffer.
Wash three times with S-buffer. Prepare 50 mL per frog, add + 7μL β-mercaptoethanol per 50 mL, and in the last wash, add 1/1,000 leupeptin.
Transfer eggs to a 14-mL Falcon tube.
Spin in Sorvall swing-bucket HB-6 rotor for 1 min at 800 rpm. This is the packing spin. Remove excess buffer.
Spin at 10,000 rpm in Sorvall for 30 min at 4°C. This is the crushing spin (Figure 1D).
Pool and transfer the cytoplasm layer to a 5 ml Falcon tube using a 21-G needle/syringe. Care should be taken not to aspirate the dark layer immediately below containing mitochondria.
Add cytochalasin B (1/500).
Seal tubes tightly with parafilm and rotate in the cold room for 5 min to completely homogenize the extract.
Centrifuge in Beckman tabletop centrifuge with TLS-55 swing-bucket rotor for 55,000 rpm for 15 min at 4°C.
Take only the cytoplasmic and membrane layers. Leave out the mitochondrial layer, the oily layer and the debris (typically, the debris accumulates at the bottom-most part of the tube; immediately above the debris is the oily layer above which is the mitochondrial layer). Use a cut pipette tip (P1000 or P200) to recover the desired fractions (Figure 1E).
-
Supplement with the following:
1/30 30 mM Creatine phosphate
1/60 150 μg/mL Phosphocreatine kinase
1/500 20 μg/mL Cycloheximide
Invert tube several times to make sure all components are adequately mixed.
-
Use extract within 3 hours or freeze as follows:
Freezing the Interphase Extract II:- (i) Add glycerol (3% final concentration) to extract and mix thoroughly by inverting the tube.
- (ii) On a Styrofoam platform, place P60 plastic petri dishes filled with liquid nitrogen. Fill labeled cryotubes with liquid nitrogen and place on Styrofoam rack.
- (iii) With a cut P200 tip, drop beads of 25 μL of extract into a P60 dish containing liquid nitrogen. Allow extract to flash-freeze in the liquid nitrogen (see Note 8).
- (iv) Store the beads of frozen extract in cryogenic vials at −80°C.
3.1.5. High-Speed Supernatant (HSS)
Prime 4–6 female frogs with 500 μL/frog of PMSG (PMSG stock: 100 IU/μL) within a week of extract preparation.
Induce females to lay eggs with 800 IU of HCG/frog injected the night before egg collection. Place each frog in a container with 1 L of 1× MMR (pH 7.7–7.8) after HCG injection (Note 9). Eggs are laid overnight and collected the next morning.
Wash eggs with 1× MMR. Remove dead eggs/floaters/debris with a transfer pipette.
Combine eggs in a glass beaker and dejelly the eggs using a 2% L-cysteine solution (pH 7.8) for about 5–10 min (see Note 3).
Wash eggs three times with 0.5× MMR, pH 7.8 (see Note 10).
Wash eggs three times with ELB, pH 7.8. At the last wash, add 1 mL of 10 mg/mL cycloheximide per 100 mL ELB.
Transfer eggs into 14-mL polypropylene tubes (Falcon tube #352059) without caps and centrifuge them at 800 rpm for 1 min at 4°C (Sorvall HB-6 rotor, use adapters to enable centrifugation of the tubes). This is the packing spin. Remove excess buffer after this spin and add cytochalasin B on top to a final concentration of 5 μg/mL (stock 10 mg/mL).
Centrifuge eggs at 10,000 rpm for 20 min at 4°C. This is the crushing spin and will result in the formation of different layers.
Transfer the cytoplasmic layer (central layer) to a 50-mL tube. Remove the layer by using a 5-mL syringe with a 25-gauge needle. Add 100 μg/mL cycloheximide, 5 μg/mL cytochalasin B, 1 mM DTT, and LPC protease inhibitors.
Pipet cytoplasmic layer in ultracentrifuge tubes (make sure to have at least 1.75 mL per tube or the tube might be crushed onto itself). Centrifuge tubes at 55,000 rpm for 2h at 4°C (TLS-55, see Note 11).
Collect clear cytoplasm with a pipette with a cut-tip, avoiding the membrane rich layer below. You might re-spin again for 15 minutes at 55,000 rpm if required.
Aliquot into 0.5mL tubes (100 μL/tube), freeze with liquid nitrogen and store at −80°C. HSS is stable for several years.
3.1.6. Nucleoplasmic Extract (NPE)
The preparation of nucleoplasmic extracts was initially reported by Walter et al. [22]. Nucleoplasmic extracts, when used in combination with HSS, have several advantages. In particular, they support plasmid replication [16, 21, 22, 38]. Therefore, this type of extract allows to study modified plasmid DNA templates to study the DNA damage response [19, 24].
Inject 20 female frogs with PMSG (500 μL/frog). Leave them for 3–5 days.
Induce egg-laying by injecting each frog with 1 mL of HCG and placing them in individual containers with 1 L 1× MMR. It must be noted that the final yield of NPE will be a small fraction of the crude S extract obtained from the eggs (see Notes 1 and 2).
Combine good eggs, remove and discard buffer. Wash eggs with 1× MMR.
Dejelly the eggs with 2% L-cysteine pH 7.8 (see Note 3).
Wash the eggs rapidly three times with 0.5× MMR and three times with 1× ELB, pH 7.8. Transfer eggs to 14-mL Falcon Tube (# 352059).
Pack eggs by centrifugation for 1 minute at 1,000 rpm. Remove excess buffer, add LPC protease inhibitor cocktail, Aprotinin 5 μg/mL (1:2,000), and 2.5 μg/mL cytochalasin B.
Crush eggs by centrifugation at 10,000 rpm in a Sorvall Centrifuge with an HB6 rotor for 15 min at 4°C.
The cytoplasmic layer is withdrawn by puncturing the side of the tube with a 21-gauge needle. This is the crude S extract (the middle layer), and is supplemented with LPC protease inhibitor cocktail, 10 μg/mL Aprotinin, 5 μg/mL Cytochalasin B, 50 μg/mL cycloheximide, 3.3 μg/mL nocodazole (stock 1,000× in DMSO), and 1 mM DTT.
The crude S-phase extract thus obtained is re-centrifuged at 10,000 rpm for 10 min at 4°C in a Falcon tube of appropriate size.
All of the residual lipids and the viscous dark brown material located just below it are entirely aspirated off. About 1 mL of the extract could be lost at this step, but all of the upper layers must be removed. The remaining cytoplasm is decanted into a fresh tube and supplemented with NPE–ATP regeneration system (Section 2.6, prepare fresh before use).
Add memembranated sperm chromatin (see Section 3.2.1). This nuclear assembly reaction is carried out in volumes of 4.5 mL per tube. The assembly reaction is made by thoroughly mixing 1 mL of the extract with 90 μL of 200,000/μL sperm. Pipetting several times with a cut blue tip is recommended before transferring the mix into a 5-mL Falcon tube (Becton Dickinson # 352063). To this mix, 3.5 mL of the extract is added and mixed by inverting multiple times. It is essential to have at least 4 mL of extract in each tube to ascertain that the layer of nuclei formed on the top after centrifugation will be thick enough to harvest.
The nuclear assembly reaction is incubated at room temperature and mixed by inversion every 10 min. A few microliters of the reaction are observed under the microscope. It is recommended to wait approximately 30 min after the time when round nuclei first become visible. The total time after sperm addition is around 70–80 min. Large nuclei are essential to ensure a good layer of nuclei in the subsequent step.
To collect the assembled nuclei, the reaction is centrifuged for 2 min at 10,000 rpm at 4°C in an HB-4 rotor in a Sorvall centrifuge. The Falcon tube (Becton Dickinson # 352063) with the extract is placed in a tube (Becton Dickinson # 352059) containing 2 mL of water to help support it during the spin. After this step, the nuclei should be visible as a clear layer about 2 mm thick on the top of the extract. If the layer is thinner than expected, let the nuclei grow more next time and make sure that the sperm concentration used is correct. Nocodazole must be added at this step since it prevents microtubule assembly allowing nuclei to float to the top of the tube during centrifugation.
Hold the tube to the light to better distinguish the clear nuclear layer from the underlying cytoplasm and remove the nuclei with a cut-off 200 μL pipette tip. Transfer all of the nuclei to a Beckman 5 × 21 mm ultracentrifuge tube (# P60720).
Supplement the nuclei with ELB to a final volume of 225 μL (assuming a 4.5-mL nuclear assembly reaction); mix thoroughly with a cut-off pipette tip and centrifuge for 30 min at 55,000 rpm at 2°C. Centrifugation is carried out in a Beckman TL100 tabletop ultracentrifuge using a TL55 swinging bucket rotor furnished with Teflon adapters (Beckman) to accommodate the 5 × 21 mm tubes. If there is more than 750 μL of the NPE+ELB mixture, a thick-walled 11 × 34 mm tube (Beckman # 343778) can be used with a centrifugation time of 40 min.
Carefully aspirate any lipids at the top of the sample after the spin and harvest the clear nucleoplasm taking care to avoid the pellet consisting of nuclear envelopes and chromatin (see Notes 12 and 13).
The expected volume of NPE from each 4.5 mL nuclear assembly reaction is around 180 μL. NPE is snap-frozen (in 10-20 μL aliquots) in liquid nitrogen, stored at −80°C and is stable for several years if freeze-thawing is prevented.
3.1.7. Immunodepletion of Extracts to Study Protein Function
Quantitative immunodepletion allows the complete removal of an endogenous protein from the extract by using antibodies against the protein of interest. The removal of proteins or protein complexes from the extract is a valuable tool to investigate protein function; however, care should be taken to retain the overall activity and functionality of the extract. Therefore, the general protocol may need modification based on the antibody used and the subsequent assay that employs the immunodepleted extract. The following protocol is for rabbit polyclonal antibodies that bind avidly to Protein A agarose beads.
Wash Protein-A Sepharose CL-4B beads at least three times with large volumes of 1× PBS (beads to buffer ratio is about 1:10) to remove the sodium-azide present in the storage buffer. After each wash, the beads are pelleted by spinning at 800 × g for 1 min on a tabletop centrifuge.
Add 1–4 volumes of sera to one volume of beads. If affinity-purified antibodies are used, the volume of antibody added can be lower. The bead-antibody suspension is placed in spin columns and mixed thoroughly by incubating overnight at 4°C on a rotator.
Wash the bead-antibody mixture in the same buffer that was used to make the extract. If the beads are washed in the spin column itself, then at least five washes are recommended. After each wash, the columns are placed in Eppendorf tubes and spun at 800 × g for 1 min to remove residual liquid. Care should be taken to prevent drying of the beads during these spins. If necessary, the time/speed of the spin should be reduced.
Resuspend the beads with the extract that needs to be depleted. Typically, for one volume of beads, 3–5 volumes of the extract are used, but this can vary depending on the protein being depleted. The column containing the bead/extract mixture is incubated at 4°C on a rotator for 30–40 min.
To collect the extract, place the column in a fresh Eppendorf tube and spin at 800 × g for 1 min. The beads are retained in the column and the extract flows through into the Eppendorf tube. For additional rounds of depletion, the depleted extract is incubated in a new column containing a second batch of (antibody-treated) beads and Steps 4 and 5 are repeated. Complete depletion of most proteins requires two to three rounds of depletion.
The depleted extract must be compared to a control extract by Western Blotting to monitor the efficiency of depletion. The control extract is one which is subjected to the same procedure with control IgG instead of antibodies. Testing protein depletion levels are strongly recommended before using the depleted extract in assays designed to investigate protein function.
Alternatively, Protein A non-aggregating iron oxide magnetic beads can also be used, allowing for the depletion of small quantities of extract (≤20μL) with minimal loss of volume through magnetic separation.
3.2. Preparation of DNA Templates
To study DNA replication and the DDR, Xenopus extracts are used in conjunction with a variety of DNA templates, including sperm chromatin and plasmid DNA. In this section, we describe the preparation of both chromosomal as well as small DNA templates typically used to investigate the DDR. Whereas intact templates are widely used to understand normal DNA replication, the DNA is sometimes subjected to damage in order to investigate damage-related physiological processes such as checkpoint and repair assays. Therefore, the preparation of damaged DNA templates is also discussed.
Chromatin templates are used in several assays to monitor DNA replication and the binding of DNA-associated proteins. In some instances, the DNA is appropriately damaged to investigate DNA damage-related phenomena. The protocols to make both intact and damaged chromatin are described below (Sections 3.2.1 and 3.2.2). Moreover, defined human chromosome segments using purified bacterial artificial chromosomes [39] have also been used in LSS extracts. This opens the possibility of performing biochemical and proteomics analyses using specific chromatin contexts.
The use of small DNA templates has specific advantages, notably the possibility of engineering site-specific alterations on the DNA that can be easily accomplished using synthetic oligonucleotides and plasmids. Furthermore, the introduced lesions can be designed to mimic the damage caused by UV radiation or chemotherapeutic agents such as mitomycin C and cisplatin. These templates, in combination with Xenopus extracts, notably the HSS/NPE system, proved to be extremely useful to study specific checkpoint signaling pathways and the repair of Protein-DNA adducts [11].
The protocols to obtain two types of small DNA molecules are described in Sections 3.2.3 and 3.2.4 below.
3.2.1. Demembranated Sperm Chromatin
Demembranated sperm chromatin described below are used to study chromosome replication and as starting material to prepare NPE, a nucleus-free system used in replication, repair and checkpoint assays.
Prime male frogs with 25U PMSG (per frog) 3–5 days before use and inject with 125U HCG (per frog) the day before Sperm Chromatin Preparation (see Note 1). It is suggested that 5–6 male frogs be used to obtain adequate sperm of high concentration (200,000 nuclei/μL).
Put frogs in 0.05% benzocaine in water for 30 min.
Dissect frogs (incision on the ventral side), cut out testes and immediately place in cold 1× MMR in a petri dish on ice.
With a clean razor blade, remove fat tissue and blood vessels and rinse three times in MMR. The final testes should be as free from fat and blood tissues as possible. If necessary, use a dissecting microscope to dissect out unwanted tissue.
Wash three times in 1× NPB.
Remove buffer and macerate extensively with a clean, sharp razor blade. Thorough maceration is critical to obtain maximum recovery. Place the testes in a 60-mm plastic petri dish on ice and macerate for about 20 min.
Add 10 mL cold NPB and aspirate a few times with a plastic Pasteur pipette. Filter the sperm suspension through two layers of cheesecloth into a 50-mL Falcon tube. Squeeze excess liquid and any remaining sperm from the cheesecloth. Wash cheesecloth in 2–4 mL NPB and squeeze out any remaining sperm into the tube. Split the contents of the 50-mL tube into two 14-mL Falcon tubes (Becton Dickinson).
Centrifuge using an HB-6 rotor at 6,000 rpm (~6,000 × g) for 15 min at 4°C to recover sperm and remove debris.
Remove supernatant and wash pellet with 10 mL NPB, trying to remove any blood vessels and debris. Repeat the wash process 2–3 times and spin for 10 min between washes. If blood vessels or other debris are still present, centrifuge at low speed (800 rpm) and transfer the liquid containing the sperm to a new tube. The debris should remain at the bottom of the tube. Re-centrifuge at 6,000 rpm for 15 min.
Remove supernatant from the last wash and resuspend pellet in 1 mL NPB + 0.2% Triton X-100. Incubate for 15 min at room temperature with gentle mixing.
Add 10 mL of cold NPB + 3% BSA and mix gently to obtain a homogenous suspension. Centrifuge for 10 min at 6,000 rpm.
Wash 2 times with 10 mL NPB + 0.3% BSA.
Resuspend in 0.5 mL NPB + 0.3% BSA and 30% glycerol.
Take a 2 μL aliquot of sperm. Mix with 2μL of DAPI fix and then dilute in 96 μL. Calculate sperm concentration with a hemocytometer. Sperm tend to sediment over time and sometimes to clump together, leading to inaccurate counts. Tap the tube before mixing or aliquoting but avoid vertexing or hard pipetting. Adjust sperm concentration to 200,000/μL with NPB + 0.3% BSA 30% glycerol.
Freeze nuclei using liquid nitrogen and store at −80°C in aliquots (see Note 14).
3.2.2. Generation of Double-Strand Breaks (DSBs) in Chromatin
Xenopus extracts can be used to assess the physiological consequences of DNA damage and identify the protein complexes that assemble at the site of DNA damage. The use of S-phase and M-phase extracts (Section 3.1) further enables the identification of cell cycle “phase-dependent” composition of the DNA repair complexes. To introduce DNA damage in chromatin, we use PflMI restriction endonuclease [40, 41], although other suitable enzymes can also be used.
To the extract, add demembranated sperm nuclei/chromatin (2,500-5,000 nuclei/μL). Incubate for 10 min at 21°C.
Remove 15 μL of extract into a separate Eppendorf tube. This is the “No-Damage” control.
To the remaining extract–sperm mixture, add 0.05 U/μL of PflMI restriction endonuclease. Incubate at 21°C.
At desired time intervals, remove 15 μL of extract and stop the reaction by mixing with 800 μL of ice-cold chromatin isolation buffer (supplemented with 0.125% Triton X-100). Keep on ice for 5 min.
This protocol is used to perform both the chromatin binding assay to detect proteins involved in various stages of the cell cycle and in other checkpoint assays. HSS can also be used for these types of studies. Alternatively, DSBs can be generated using the radiomimetic drug neocarzinostatin (200ng/μl), which generates breaks with heterogenous ends or etoposide (20 μM), which stabilizes Topoisomerase 2 covalent protein complexes at the DNA ends.
3.2.3. Preparation of Plasmid with a Single Interstrand Crosslink (ICL)
DNA interstrand crosslinks (ICLs) can be induced by chemotherapeutic, environmental or endogenous agents. They represent a class of damaged DNA where the strands are covalently linked, usually leading to a distorted DNA helix. Interstrand crosslinks are highly toxic since they can block DNA replication and transcription. Their removal or bypass is accomplished by the concerted effort of multiple repair pathways and repair failure can result in mutations, rearrangements, tumors, or cell death.
Checkpoint signaling induced by ICLs and their repair mechanisms have been intensively studied over the last decade using Xenopus cell-free extracts. The advantages of the Xenopus egg extract system mentioned above have been key for unraveling the molecular details that repair this class of genotoxic lesions.
Interstrand crosslinks can be generated on plasmid DNA with SJG-136 (NCI/Spriogen LTD), a rationally designed crosslinking chemical, that creates ICLs between opposing guanine residues [42].
Incubate pBS vector with 3μM SJG-136 (NCI/Spriogen LTD) in 50 mM triethanolamine and 2 mM EDTA, overnight at 37°C
Purify DNA by ethanol precipitation and resuspended in water
Asses the quality and quantity of plasmids recovered by agarose gel electrophoresis with ethidium bromide.
Alternatively, is it possible to obtain a plasmid with a single interstrand crosslink by incubating specific oligonucleotides with a single pair of opposing guanines with SJG-136 and then cloning the resulting crosslinked duplex product onto a plasmid backbone [42].
3.2.4. Preparation of DNA with Double-Strand Breaks
DNA molecules with DSBs have been generated in our laboratory by using either restriction enzymes or PCR. To obtain DNA fragments with DSBs, we use the circular pBR322 plasmid as a template and digest it to completion with restriction endonucleases. We tested different enzymes generating different types of DNA ends (blunt, 3′ overhang, or 5′ overhang) and did not observe differences in the DNA-damage response [43]. Such DNA can be used in assays that investigate the effect of damage on DNA replication and also in chromatin binding experiments.
Digest 0.5 mg of pBR322 with HaeIII (NEB). HaeIII cuts pBR322 plasmid 25 times, thus generating 26 fragments containing 2 DSBs each.
Digest DNA and extract twice in phenol/chloroform, then precipitate in ethanol and sodium acetate.
Resuspend DBS-containing DNA in water at a concentration of 1 mg/mL.
Dilute the DSBs stock solution into the extracts to the desired concentration.
Alternatively, we have used λ-DNA that was digested with a series of restriction enzymes giving rise to different numbers of restriction fragments. λ-DNA is digested with XbeI, NcoI, HindIII, and BstEI enzymes that generate 2, 5, 7, and 14 fragments, respectively. This approach enables us to increase the concentration of DSBs in the extracts while keeping the mass of added DNA constant.
To obtain 1 kb DNA fragments by PCR, we use the M13 ssDNA template using 22nt primers complementary to positions 5,570 and 6,584, as described before [44].
3.2.5. Preparation of Biotinylated Substrates
Biotinylated substrates are extensively used in replication, checkpoint and repair assays. We have established a system in which we use biotinylated DNA substrates to understand the initiation of DNA replication and DNA damage checkpoint signaling [45].
One microgram of the gel-purified DNA fragment is end-labeled with 1-unit T4 polymerase in the presence of 33 μM each of dATP, dGTP, dTTP, and biotin–dCTP for 15 min at 12°C.
Stop reaction by addition of 50 mM EDTA and incubate at 76°C.
Purify the labeled DNA using the PCR purification kit as per the manufacturers’ instructions.
Quantify the purified DNA by UV absorbance at 260 nm.
In our experience, we observed that DNA damage response and checkpoint activation were induced by the relatively short substrates [45], thus increasing the potential use of such substrates in checkpoint studies.
3.3. Use of the Xenopus System to Study Cell Cycle Checkpoints/ Checkpoint Assays
The maintenance of genome integrity in cells is constantly challenged by DNA damaging agents. The response to such DNA lesions involves the prompt detection, signaling and repair of the damaged DNA. Two proteins with established roles in damage signaling are the ATM and ATR protein kinases [6].
The ATM- and ATR-dependent checkpoints can be classified based on whether or not they depend on active DNA replication and fork progression. During G1 or S phases, double-strand breaks (DSB) and ssDNA can induce replication-independent checkpoints, namely the G1/S checkpoint and the Intra-S checkpoint. However, there also exist replication-dependent checkpoints such as the Replication checkpoint and the S/M checkpoint [46].
Replication-independent checkpoints:
The G1/S checkpoint can result from IR or radiomimetic agents and functions to prevent origin firing until the damage is repaired. This pathway can be either p53 dependent or independent. The two p53-independent pathways involve ATM and ATR: Firstly, the presence of DSBs activates an ATM-dependent checkpoint resulting in the phosphorylation and inhibition of Cdk2 activity. This inhibition of Cdk2 prevents the loading of Cdc45 onto chromatin and blocks origin firing [46]. The second p53-independent G1/S checkpoint ensues upon the generation of aberrant DNA structures comprising of ssDNA-RPA intermediates generated in G1. Such intermediates can be generated experimentally by treatment with exonuclease III or by addition of etoposide, an inhibitor of DNA topoisomerase II [41, 47]. This RPA-dependent signaling results in ATR activation, which in turn inhibits origin firing [48].
The Intra-S checkpoint inhibits late origin firing. The proper installation of this checkpoint requires the phosphorylation of serine residues 278 and 343 of Nbs1 by ATM. After sensing the damage, ATM and ATR activation are followed by phosphorylation of their targets- Chk2 and Chk1, respectively. The ultimate result of the ensuing cascade is to downregulate Cdk2 and Cdc7 protein kinases, which in turn prevent cdc45 loading and origin activation.
S-phase checkpoint:
The S-phase checkpoint is activated in response to UV, IR and HU treatments and usually requires that active DNA replication be elicited. This checkpoint results in ATR activation and phosphorylation of its target Chk1 on serine residues 317 and 345 [49]. The phosphorylation of Chk1, in turn, culminates in the phosphorylation-dependent degradation of Cdc25A and inhibition of Cdk2-Cyclin E [50]. Other regulators of the S-phase checkpoints are Claspin/Mrc1, MRN complex, BRCA1 and FANC proteins. The phosphorylation of Nbs1 by ATM is another event crucial for the S-phase checkpoint [51].
Replication-dependent checkpoints:
The Replication checkpoint and the S/M checkpoint are other replication-dependent checkpoints that are active during the S phase. The Replication checkpoint is initiated by stalled forks in response to genotoxic stresses, aberrant DNA structures or DNA damage and inhibits origins through the activity of ATR and Chk1. The S/M checkpoint ensures that DNA replication is complete before the cell enters mitosis. This checkpoint prevents mitotic entry by enabling Chk2 phosphorylation and activation, which in turn inhibits Cdc25 phosphatase activity and prevents the activation of the Cyclin B-cdk1 complexes. The S/M checkpoint also involves ATM substrates such as Nbs1 and BRCA1 [52, 53].
Replication stress, which is defined as the slowing and stalling of replication forks during DNA replication, is a potent inducer of ATR mediated DNA damage response. Defects in ATR pathway signaling are associated with replication fork collapse resulting in double-strand breaks that can lead to chromosomal rearrangements and genome instability [54]. Replication stress is induced in cellular systems with hydroxyurea, a ribonucleotide reductase inhibitor that quickly leads to dNTP pools exhaustion. Since the egg stockpile of dNTP pools in the extracts is remarkably high, hydroxyurea is ineffective in fork slowdown in extracts. However, aphidicolin, an inhibitor of replicative polymerases, is a potent activator of ATR in extracts replicating chromatin templates. Additionally, fork collapse can be mimicked in the extracts by treatment with nicking endonucleases such as S1 or mung bean nucleases that result in one-ended double-strand breaks after fork passage [55].
G2/M checkpoint:
The G2/M checkpoint targets Cdk1-Cyclin B kinase and prevents mitotic entry in response to DNA damage or incomplete S phase. Cell cycle arrest, in this case, involves both the ATM and the ATR pathways [56]. The G2 checkpoint is abrogated by caffeine and is sometimes used as an assay to test the involvement of the ATM pathway.
In addition to the above functions, the ATM/ATR kinases also assist in DNA repair by inducing repair proteins and activating them by post-translational modifications such as phosphorylation. The activation of ATM is, in turn, an indicator of checkpoint activation and can be assayed by assessing the phosphorylation of its substrates such as Smc1, Chk2, or H2AX.
The above paragraphs (in Section 3.3) are a few examples of the proteins involved in and the mechanisms that drive various cell cycle checkpoints activated as a result of DNA damage. To assess whether a checkpoint has been activated or to investigate checkpoint-related phenomena further, the proteins involved, their levels, phosphorylation status, and other modifications are frequently used as readouts. For instance, phosphorylation of Chk1, Chk2, or H2AX indicates that the ATR, ATM, or the ATM/ATR pathways have (respectively) been activated.
The Xenopus cell-free system can be used to investigate cell cycle checkpoints in a variety of ways such as monitoring DNA replication, studying the phosphorylation or activation of target proteins, and the assembly or localization of repair proteins on the site of DNA damage. The following sections describe the assays used to study checkpoints.
DNA replication assay to study checkpoint activation:
Extracts derived from Xenopus eggs can support semi-conservative DNA replication of genomic DNA upon the addition of DNA templates [17]. However, in the presence of DNA damage, checkpoints are activated. These checkpoints prevent the initiation or progression of DNA replication upon DNA damage [57, 58] and coordinate DNA replication, recombination and repair processes [6, 59]. This inhibition occurs both when the damage is present on the template during replication in “cis” as well as when DNA damage signaling is induced in “trans” by DNA containing exogenous DSBs. Both types of DNA damage can activate a checkpoint. The use of the Xenopus system to investigate such checkpoints is described below.
3.3.1. DNA Damage Response Induced by Single-Strand DNA Gaps in Chromatin
We have developed a cell-free system that recapitulates the inhibition of DNA replication in the presence of single-strand DNA gaps [60, 61].
Single-strand DNA gaps are generated by incubating chromatin in cell-free extracts in the presence of etoposide, an inhibitor of topoisomerase II, or by in vitro treatment of chromatin by DNA exonuclease III. Etoposide generates lesions in the chromatin templates that are undergoing DNA replication [47] by blocking the activity of DNA topoisomerase covalently linked to DNA 5′ termini [41, 62].
In addition to studying checkpoints induced by ssDNA gaps in chromatin, the protocol described below (the portion that describes the monitoring of DNA replication) can also be used to assess the replication of chromatin and/or small DNA templates in various extracts.
Incubate 20 μL of activated extract with 5,000 demembranated sperm nuclei/μL in the presence of etoposide at 21°C for 90 min (see Note 15).
Concentrations of etoposide ranging from 10 to 50 μM are effective in inducing a checkpoint response, as seen by the inhibition of genomic DNA replication (see Note 16).
Etoposide-induced inhibition of DNA replication is rescued by the addition of 5 mM caffeine, an inhibitor of checkpoint signaling kinases, including ATM and ATR [63].
Monitor DNA replication by incorporation of α-32P–dCTP into the chromatin. Add 0.2 μCi of α-32P–dCTP to each replication reaction.
Stop DNA replication reactions by diluting the samples in 200 μL of stop solution.
Incubate the samples with 1 mg/mL of proteinase K for 30 min at 37°C.
Extract DNA with 1 volume of phenol/chloroform.
Centrifuge the samples for 10 min at room temperature.
Recover the aqueous phase and precipitate with 2 volumes of ethanol and 1/10 volume of sodium acetate (3 M stock).
Resuspend the pellet in DNA loading buffer and run on a 0.8% agarose gel in TBE.
Fix the gel in 7% TCA (some protocols require a higher percentage of TCA). Position the gel between two layers of Whatman 3 MM paper and stacks of filter paper. Dry the gel overnight on the bench.
Expose the dried gel for autoradiography.
3.3.2. DNA Damage Responses Induced by DSB in Chromatin
To investigate the cell cycle response to DNA damage at the onset of S phase, we use the Xenopus cell-free system designed to study the initiation of DNA replication [64]. Activated extracts are treated with either circular plasmid DNA, plasmid DNA containing DSBs, or λ-DNA containing DSBs. The treatment of the cytosolic extracts with DSBs-containing DNA activates a checkpoint in trans. In this protocol, the damaged DNA is only used to trigger the checkpoint and is subsequently removed, following which the extract is tested for its ability to replicate intact DNA templates. The damaged template is removed to avoid any interference with genomic DNA replication, such as titration of essential factors required in the elongation step of genomic DNA replication.
Incubate 100 μL of activated LSS extract at 21°C in the presence of 50 ng/μL of circular plasmid DNA or digested plasmid (DSB) for 15 min to activate the checkpoint (see Note 15).
For rescue experiments, extracts are pre-treated with 5 mM caffeine to inhibit PIKK kinases non-specifically, 100 μM KU55933 to specifically inhibit ATM, or 50 μM VE821 to inhibit ATR specifically. Our lab has also been successful in inhibiting ATM activity by the addition of purified anti-ATM antibodies for 15 min at 21°C, before incubation with the damaged DNA.
For replication assays, add 0.2 μL α-32P–dCTP and sperm chromatin (2,000/μL) to 10 μL of each extract treated with different types of DNA molecules, caffeine, or specific ATR/ATR inhibitors.
Incubate the reactions for 30 min at 21°C.
Stop reaction by adding 200 μL of Stop Solution. Mix well.
Add 1/20 volumes (10 μL) of Proteinase K and incubate at 55°C for 1–2 h.
Cool samples to room temperature and spin down for 10 s.
Add 200 μL of phenol: chloroform: isoamyl alcohol (25:24:1) and mix thoroughly by inverting several times.
Centrifuge samples at 13,000 rpm for 10 min. Transfer the aqueous phase to a fresh tube. Add 1/10 volumes (20 μL) of sodium acetate (3 M Stock) and 2.5 volumes (500 μL) of cold 100% ethanol. Incubate at −20°C overnight.
Spin samples for 10 min at 13,000 rpm.
Discard supernatant, briefly dry pellet and dissolve it in 15-μL DNA loading buffer with bromophenol blue and cyan blue dyes.
Load samples and electrophorese them on a 0.8% agarose gel.
Treat gel with trichloroacetic acid (Sigma) 50% made up in water for 20–30 min.
Dry gel overnight between 3 M blotting paper and several layers of absorbent paper towels. The next day, the gel may be dried for an additional hour at 70°C using a vacuum dryer.
Subject dried gel to autoradiography; multiple exposure times may be required, ranging from 15 min to overnight.
3.3.3. Assay to Study Checkpoint Induced by ICL
To investigate whether the damaged DNA activates an ATM/ATR checkpoint, replication assays are conducted in the presence of caffeine, which is an inhibitor of PIKK kinases (ATR, DNA-PK and ATM) or specific inhibitors (see Note 16). The experiment described below uses DNA containing an interstrand crosslink; however, the protocol can be modified to investigate whether other damaged DNA templates similarly activate the ATM/ATR checkpoints.
The protocol for the ICL induced damage assay is described below:
Xenopus extracts (HSS and NPE) are prepared as described above (Sections 3.1.5 and 3.1.6). Experiments typically use a ratio of HSS to NPE at 1:2.
Take two sets of two tubes. To each set of tubes, add 3 μL of HSS, 0.1 μL of NPE–ATP regeneration system (Section 2.6), and 5 ng control DNA (tube 1) or 5 ng ICL DNA (tube 2) (Control and ICL DNA as described in Section 3.2.3). To the first set of tubes, 5 mM caffeine is also added. The second set serves as a “no-caffeine control,” and only buffer is added instead. Mix. Incubate for 30 min at 21°C.
Add 6 μL NPE and 0.2 μL α-32P–dCTP, mix by pipetting and incubate for 2 h at 21°C.
Monitor DNA synthesis by the incorporation of 32P for 90 min at 21°C, followed by agarose gel electrophoresis (as described in Section 3.3.1; Steps 4 to 12).
3.3.4. Study of the Checkpoint Induced by DNA Damage In-Trans on DNA Replication
Checkpoint activation can be assessed by monitoring the effect of the ICL DNA (or other forms of damaged DNA) on the replication of an intact plasmid. The experiment described below uses ICL DNA; however, a similar protocol can be used to study the impact of other types of damaged DNA on the replication of intact DNA templates.
Prepare Xenopus extracts (HSS and NPE) as described (Sections 3.1.5 and 3.1.6).
Add 3 μL of HSS, 0.1 μL of NPE–ATP regeneration system (Section 2.6), and 5 ng Control or ICL DNA. Mix. Incubate for 30 min at 21°C.
Add 6 μL NPE; mix by pipetting and incubate for 1 h at 21°C (Try to keep HSS to NPE ratio to 1:2).
Add the plasmid that is to be replicated to each of the tubes and also add 0.2 μL α-32P–dCTP. Mix well and incubate at 21°C for 20 min.
Analyze the replication of the plasmid DNA by agarose gel electrophoresis as described earlier (Section 3.3.1).
Since the two plasmids (control /ICL plasmid and the plasmid to be replicated) are of different sizes, they can be distinguished by gel electrophoresis.
3.3.5. Study of Phosphorylation to Assess Checkpoint Activation
It is known that DNA damage blocks DNA polymerases, allows DNA unwinding by MCM helicases and triggers an ATM or ATR-dependent checkpoint [65, 66]. The ATR checkpoint pathway is activated during S phase to sense and coordinate cellular responses to DNA damage. Several checkpoint proteins are recruited to the site of damaged DNA and play a role in the assembly of the ATR kinase signaling complex [67, 68]. The activated ATR phosphorylates its target proteins, including Chk1, to initiate the cellular response to the DNA damage. This phosphorylation of Chk1 is commonly used as a readout for ATR activation. In Xenopus, Chk1 phosphorylation at the S344 site is observed in response to DNA damage induced by UV rays, methyl methanesulfonate (MMS), 4-nitroquinoline 1-oxide (4-NQO), interstrand crosslinks (ICL), and aphidicolin [19, 69, 70]. This is best detected by a phospho-specific antibody capable of detecting Xenopus Chk1 phosphorylated at S344.
The phosphorylation of Histone H2AX at the Serine 139 residue is another frequently used indicator of DNA damage. Histone H2AX is a well-characterized substrate for activated protein kinases and is phosphorylated by both ATM (in response to IR) and ATR kinases (in response to UV) or DNA replication blocks [71, 72] at Serine 139 [73, 74]. In combination with 53BP1, histone H2AX is also thought to play a role in G2-M checkpoint activation [75]. In our laboratory, we have monitored the phosphorylation of both endogenous histone H2AX as well as exogenous H2AX peptide [20, 76].
Several other proteins get phosphorylated upon DNA damage. For instance, MRE11, a protein that is involved in the repair of double-strand chromosome breaks [77] and Nbs1 that forms a complex with MRE11 and Rad50 are both phosphorylated in response to DNA damage [78]. The above examples collectively suggest that the phosphorylation of various proteins can be used to assess the presence of DNA damage as well as, in some instances, determine the activation of cell cycle checkpoints. The use of phosphorylation assays and Xenopus cell-free extracts in checkpoint studies are outlined in the following sections (Sections 3.3.5, 3.3.6, 3.3.7 and 3.3.8).
3.3.6. Phosphorylated Histone H2AX Detection (Endogenous Substrate)
The detection of phosphorylated Histone H2AX (γH2AX) is a sensitive assay to monitor DNA damage-induced checkpoint signaling.
Incubate 50 μL of interphase extract (or extract in which the occurrence of DNA damage will be assessed) with 10,000 nuclei/μL for 90 min at 21°C.
Isolate post replicative chromatin by diluting the extracts in chromatin isolation buffer containing 1 mM NaF, 1 mM sodium vanadate, and 0.125% Triton X-100.
Layer samples onto sucrose cushions (chromatin isolation buffer containing 30% sucrose and lacking Triton X-100), then spin at 6,000×g for 20 min at 4°C.
Prepare a positive control by incubating sperm nuclei for 30 min in an interphase extract to decondense chromatin.
Isolate the chromatin and digest for 4 h with NotI restriction endonuclease.
Re-isolate the digested chromatin through a sucrose cushion and incubate in interphase extract for 60 min.
Boil chromatin in Laemmli buffer and process for SDS-PAGE electrophoresis.
Use anti-γH2AX antibody for Western blotting at 1/2,000 dilution.
3.3.7. Histone H2AX Phosphorylation Assay (Exogenous Substrate)
Another assay to monitor checkpoint signaling is the measurement of H2AX peptide phosphorylation [76]. Histone H2AX is a well-characterized substrate for protein kinases that are activated by DNA damage and is phosphorylated in vivo at serine 139 by ATM and ATR. We have reported earlier the use of the C-terminal peptide of mouse H2AX as a reporter substrate to monitor the response to damage [43]. The actual assay involves incubation of interphase extracts (LSS/Interphase extracts I/II or Activated CSF) with fragmented DNA and either wild-type (AVGKKASQASQEY) or control/mutant (AVGKKAAQAAQEY) H2AX peptides. The presence of fragmented/damaged DNA results in rapid phosphorylation of the exogenous peptide.
The extract (LSS or Activated CSF extracts) is incubated with DNA fragments to elicit a DSB response. The DNA fragments we use are biotinylated at one end and immobilized on Streptavidin magnetic beads (Dynabeads, Thermo Fisher) at concentrations of 20 and 60 ng/μL (corresponds to 1.2×1011 and 3.6×1011 ends per microliter, which in turn simulate irradiation doses of 70 Gy and 210 Gy respectively for a human lymphocyte).
Separate the DNA-bound beads from the extracts (according to the manufacturer’s instructions) and wash DNA six times with ELB buffer supplemented with 0.1% (v/v) Triton. The resulting DNA-bound and soluble fractions of the extracts can be evaluated for various parameters.
Mix 2 μL of the above extract with 20 μL of EB buffer supplemented with 50 μM ATP, 0.4 μL γ32P–ATP (stock – 10 mCi/mL), and 1 mg/mL of either wild-type or mutant H2AX peptide (Sigma).
Incubate samples for 15 min at 30°C.
Stop reactions by adding 2 μL of 0.5 M EDTA.
Spot reactions on p81 phosphocellulose filter papers (Upstate Biotechnology).
Wash filters three times in 5% (v/v) acetic acid and twice with water.
Air dry filters and quantify radioactivity in a scintillation counter.
It should be noted that the c.p.m. incorporated into the control mutant H2AX peptide is subtracted from the c.p.m. of the wild-type H2AX peptide and the values are normalized to those from control extracts treated with streptavidin beads in the absence of DNA (no DSB). Second, the extracts may be depleted of a specific protein by using antibodies (see Section 3.1.7), and the depleted extract can be analyzed as above to assess the function of a particular protein. Third, the extract can be pre-incubated with 5 mM caffeine to serve as a control in the H2AX peptide phosphorylation assay.
An alternate protocol for H2AX peptide phosphorylation is as follows:
Take 2 μL of LSS Extract (that has been preincubated with undamaged or damaged DNA as above) and mix with 20 μL of EB Kinase Buffer supplemented with 0.5 mg/mL histone H2AX peptide (wild-type or mutant), 50 μM ATP and 1 μL of γ32P–ATP (10 mCi/μL, greater than 3,000 Ci/ mM).
Incubate samples at 30°C for 20 min.
Stop reaction by adding 20 μL of 50% acetic acid.
Spot samples on p81 phospho-cellulose filter papers.
Wash filters three times with 10% (v/v) acetic acid and twice with water. Air dry filters.
Quantify radioactivity using a scintillation counter.
Subtract the number of c.p.m. incorporated into the control mutant H2AX peptide from the number incorporated into the wild-type H2AX peptide; normalize the values to those from control extracts treated with streptavidin beads in the absence of DNA (negative control).
3.3.8. Phosphorylation of ATM/ATR Target Proteins
Response to DNA damage involves the sensing of the damage, transduction of the signal and activating the response. Each component of the damage response is carried out by specific proteins. The ATM and ATR proteins are master regulators of the pathway. The levels and phosphorylation status of these proteins and their downstream targets are commonly used readouts to investigate the activation of a checkpoint. For instance, to distinguish between ATR and ATM activation, phosphorylation of Chk1 protein at S344 and phosphorylation of ATM at S1981 can be investigated as follows:
Prepare Xenopus extracts (HSS and NPE) as described earlier (Sections 3.1.5 and 3.1.6).
Take two sets of two tubes. To each set of tubes, add 3 μL of HSS, 0.1 NPE–ATP regeneration system, and 5 ng control DNA (tube 1) or damaged DNA (tube 2). To the first set of tubes, 5 mM caffeine (made up in buffer) is also added. The second set serves as a “no-caffeine control,” and only buffer is added instead. Mix. Incubate for 30 min at 21°C.
Add 6 μL NPE and incubate for 90 min at 21°C.
Analyze the soluble extracts by Western blotting with phospho-Chk1 antibody (Cell Signaling) and phospho-ATM antibody (Rockland Immunochemicals, PA).
To differentiate between ICL and canonical DNA replication checkpoints, the above experiment can be carried out with purified recombinant geminin (50 ng/mL stock, added at 1/50 final concentration) and roscovitine (500 μM in DMSO) instead of caffeine. We have observed that ICL induced Chk1 phosphorylation is resistant to geminin/roscovitine treatment indicating the existence of a replication-independent pathway [19]. To evaluate the role of the Fanconi Anemia (FA) pathway in the ICL-induced checkpoint, the above experiment is carried out with curcumin – an inhibitor of the FA pathway (100 μM in ethanol; Sigma).
Binding assays to study the localization of damage-dependent proteins: The association or binding of proteins on to chromatin or small DNA templates is frequently used to understand the composition of complexes that bind DNA and to investigate the function of a specific protein. Furthermore, understanding the binding patterns of proteins onto intact or damaged DNA can provide insights into DNA damage, signaling, and repair. The protocols to identify the proteins bound to either intact or damaged DNA (either chromatin or plasmid DNA) are described below.
3.3.9. Chromatin Binding Assay
A critical aspect of the DNA-damage response is the damage-dependent localization of proteins to the DNA. This is exemplified by the formation of damage-induced foci within the nuclei of mammalian cells. Cell-free systems allow the rapid mixing and subsequent separation of chromatin, nuclear, and cytoplasmic fractions. In chromatin binding assays, depleted extracts allow to investigate the function of a specific protein in coordinating the binding specificities of other proteins. To analyze the status of the proteins that are recruited to the chromatin in replication or checkpoint-dependent manner, chromatin binding assays can be performed.
Thaw the appropriate amount of LSS or HSS extracts.
Assemble chromatin in 15–50 μL of interphase extracts in which a checkpoint has or has not been activated.
Incubate 2,500-5,000 nuclei/μL for 30–120 min.
Following incubation, dilute each reaction in 800 μL of chromatin isolation buffer supplemented with 0.125% Triton X-100.
Lay diluted extracts over 350 μL sucrose cushions (chromatin isolation buffer containing 30% sucrose, omit the Triton X-100 in this sucrose-containing buffer).
Centrifuge the chromatin at 6,000×g for 20 min at 4°C.
Carefully aspirate supernatant leaving a few μL at the bottom of the tube.
Add 0.5 μL (50 U) micrococcal nuclease and incubate min at RT. Avoid pipetting on the sidewalls of the tube. Transfer samples to clean tubes.
Add 2× Laemmli sample loading buffer and run the samples on SDS-PAGE.
Analyze by Western blotting with specific antibodies.
Either intact or damaged chromosomal DNA can serve as templates in the above assay. Optimal sperm concentration should be optimized for each experiment. In chromatin binding assays, there is the possibility of contaminating the chromatin-bound proteins with cytosolic proteins. Therefore, it required to include a “no DNA” control (extract processed exactly as other samples except that the DNA template or sperm nuclei are omitted from the reaction tube).
The same protocol can also be conducted with HSS. For HSS/NPE, use a 1:2 ratio and demembranated sperm chromatin as the DNA template on which proteins are bound.
3.3.10. Binding Assay Using HSS/NPE and ICL Plasmid
To investigate the role of specific proteins in checkpoint signaling, we deplete the specific protein and determine the activation of downstream pathways and the phosphorylation of target proteins. Although the assay described below involves ICL plasmid, it must be noted that the protocol can easily be adapted to use chromatin or other small DNA templates instead of the ICL plasmid.
This assay is extensively applied to study the levels, phosphorylation status and identity of the proteins that are bound to DNA under intact or damaged conditions. We have used this assay in our laboratory to investigate checkpoint signaling from and the repair of a single, site-specific interstrand crosslink [19, 24, 42]. Alternatively, this assay can also be modified to carry out rescue experiments where recombinant wild-type or mutant protein is added back to the extract.
Xenopus HSS, NPE extracts prepared as described above ([79]).
Take 3 μL of HSS, 0.1 μL of NPE–ATP regeneration system, and 5 ng plasmid DNA (control DNA or the ICL DNA) in an Eppendorf tube. Mix. Incubate for 30 min at 21°C. Add 6 μL NPE, mix well, and incubate for 1 h at 21°C.
Dilute the reaction with 800 μL of Chromatin isolation buffer supplemented with Triton X 100.
Layer the entire mixture on 350 μL of 30% sucrose cushions made with chromatin isolation buffer (without Triton X-100). Low retention 1.5-mL Eppendorf tubes are used for this step.
Centrifuge at 6,000 rpm (8,000 rpm for plasmid DNA template) in HB-6 rotor for 30 min at 4°C.
Carefully remove most of the liquid from the tubes after the spin but leave about 10 μL in the tube. This should contain the DNA/chromatin-bound protein. Freeze the tube in liquid nitrogen and store at −20°C overnight.
Thaw out tubes with DNA bound protein and resuspend the pellets with 10 μL of 2× Laemmli sample buffer. Denature samples by boiling for 3 min and analyze by SDS-PAGE and Western Blotting.
A control sample without DNA must also be analyzed simultaneously to ascertain that there is no contamination from non-chromatin bound proteins. A mock- depleted extract is also included as a control in these assays.
4. Notes
Depending on age and size, a female Xenopus will lay close to 50 mL of eggs, which results in between 5 and 10 mL of fully dejellied eggs. This yields between 2 and 4 mL of egg cytosol. Frogs are generally not fed after this initial priming in order to reduce the possibility of fecal matter contaminating the eggs. However, the experiments must be scheduled so that the initial priming and egg collection are done within a week so that the frogs are not deprived of eating for extended periods. Always follow your Institutional Animal Care and Use Committee guidelines.
Healthy eggs have a dark and a light hemisphere, almost symmetrical, with even and dense coloration (Figure 1A). Throughout extract preparation, pale gray eggs, with no sharp hemispheres, floating or broken eggs must be discarded using a transfer pipette. Depending on age and size, a female Xenopus will lay close to 50 mL of eggs, which results in between 5 and 10 mL of fully dejellied eggs (Figure 1B). This yields between 2 and 4 mL of clarified egg cytosol.
Gently swirl the eggs with a plastic pipette to facilitate the dejellying. Once completed, the total volume of eggs will be reduced by more than half (Figure 1B), and you might notice transparent shells floating in the cysteine solution. Eggs are extremely fragile once the protective jelly coats are removed, handle with care. Over-dejellying of eggs while preparing any of the extracts is detrimental and will cause eggs to burst/pop and render them unusable. Typically, dejellying takes about 5-8 min. However, each batch of eggs is unique and therefore, dejellying times must be adjusted accordingly. Proper pH adjustment of the cysteine solution is critical, as slightly acidic solutions will not work. Quickly remove and discard any floating or white egg/shells with a plastic Pasteur pipette.
If Falcon tubes and HB-6 rotors are used, then the caps of the tubes should be removed before centrifugation to allow proper closing of the rotor’s lid. We routinely prepare our extracts at 4°C. Other groups have reported successful extract preparation at room temperature.
The cytoplasmic layer can also be pipetted by directly sliding a pipette tip against the wall of the tube through the top lipid layer. Alternatively, the lipid layer can be removed first using a cotton swab.
Cytosolic extracts (CSF or activated) must be used immediately after preparation. Freezing and thawing the extract triggers apoptosis.
While freezing the Interphase extracts II, to avoid clumping of drops, it must be ascertained that the previous drop is completely frozen before adding the next drop. Frozen drops generally sink to the bottom of the P60 dish containing liquid nitrogen. The frozen drops can be picked with clean forceps and dropped into a frozen cryotube. Several drops can be placed in a single cryogenic vial. Do not fully close the vials unless all the liquid nitrogen in it has evaporated to avoid pressure rupture. Loosely place the cap and gently shake before fully screwing.
For the preparation of HSS, the MMR buffer can be made as a 10× stock, stored at 4°C and diluted to 1× MMR. It is recommended to check that the pH of the diluted MMR is 7.7–7.8 before use.
For the ultracentrifugation step, if thin tubes are used, the catalog number is Beckman 326819.
If the NPE is contaminated by insoluble material that appears cloudy or opaque, the extract is re-centrifuged in the TL55 rotor with Teflon adapters for 15 min.
It is recommended that NPE is flash-frozen in liquid nitrogen and stored in −80°C as small aliquots. Repeated freeze-thaw cycles can result in loss of activity and must be avoided.
Demembranated sperm chromatin is stored at −80°C in small aliquots (10 μL/tube) for replication assays and in larger aliquots (100 μL/tube) for making NPE. We routinely test before use sperm preparations to avoid background DNA damage and poor replication dynamics. Discard leftover sperm after thawing.
Pipetting the extracts gently but thoroughly is critical to the success of all procedures to achieve a homogenous extract. The formation of aggregates or incomplete mixing would lead to variable or irreproducible experimental results. Always avoid vortexing the extracts or introducing bubbles when working with these extracts, as this would result in protein oxidation and denaturation.
Alternatively, supplement activated extract with ExoIII chromatin at the same concentration of nuclei. ExoIII chromatin preparation: Incubate 106 sperm nuclei with 100 U DNA exonuclease III (Roche) for 10 min at 37°C (in 60 mM Tris–HCl, pH 8.0, and 0.6 mM MgCl2 buffer); stop the reaction by addition of 1 mL NPB buffer and
We routinely use 100 μM KU55933[80] for ATM inhibition, 50 μM VE-821[81] for ATR inhibition or 100 μM NU7441[82] for DNAPKcs.
5. Acknowledgments
This work was supported by grants from the National Institutes of Health P01 CA174653 and R35 CA197606 to J.G. and R50 CA233182 to T.A.
6. References
- [1].Jackson SP, Bartek J, The DNA-damage response in human biology and disease, Nature 461(7267) (2009) 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lanz MC, Dibitetto D, Smolka MB, DNA damage kinase signaling: checkpoint and repair at 30 years, EMBO J 38(18) (2019) e101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goldstein M, Kastan MB, The DNA damage response: implications for tumor responses to radiation and chemotherapy, Annu Rev Med 66 (2015) 129–43. [DOI] [PubMed] [Google Scholar]
- [4].Murray AW, The genetics of cell cycle checkpoints, Curr Opin Genet Dev 5(1) (1995) 5–11. [DOI] [PubMed] [Google Scholar]
- [5].Thompson SL, Bakhoum SF, Compton DA, Mechanisms of chromosomal instability, Curr Biol 20(6) (2010) R285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhou BB, Elledge SJ, The DNA damage response: putting checkpoints in perspective, Nature 408(6811) (2000) 433–9. [DOI] [PubMed] [Google Scholar]
- [7].Costanzo V, Gautier J, Xenopus cell-free extracts to study DNA damage checkpoints, Methods Mol Biol 241 (2004) 255–67. [DOI] [PubMed] [Google Scholar]
- [8].Blow JJ, Laskey RA, Xenopus cell-free extracts and their contribution to the study of DNA replication and other complex biological processes, Int J Dev Biol 60(7-8-9) (2016) 201–207. [DOI] [PubMed] [Google Scholar]
- [9].Sannino V, Pezzimenti F, Bertora S, Costanzo V, Xenopus laevis as Model System to Study DNA Damage Response and Replication Fork Stability, Methods Enzymol 591 (2017) 211–232. [DOI] [PubMed] [Google Scholar]
- [10].Parker MW, Botchan MR, Berger JM, Mechanisms and regulation of DNA replication initiation in eukaryotes, Crit Rev Biochem Mol Biol 52(2) (2017) 107–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hoogenboom WS, Klein Douwel D, Knipscheer P, Xenopus egg extract: A powerful tool to study genome maintenance mechanisms, Dev Biol 428(2) (2017) 300–309. [DOI] [PubMed] [Google Scholar]
- [12].Sannino V, Kolinjivadi AM, Baldi G, Costanzo V, Studying essential DNA metabolism proteins in Xenopus egg extract, Int J Dev Biol 60(7-8-9) (2016) 221–227. [DOI] [PubMed] [Google Scholar]
- [13].Cupello S, Richardson C, Yan S, Cell-free Xenopus egg extracts for studying DNA damage response pathways, Int J Dev Biol 60(7-8-9) (2016) 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Willis J, DeStephanis D, Patel Y, Gowda V, Yan S, Study of the DNA damage checkpoint using Xenopus egg extracts, J Vis Exp (69) (2012) e4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hashimoto Y, Costanzo V, Studying DNA replication fork stability in Xenopus egg extract, Methods Mol Biol 745 (2011) 437–45. [DOI] [PubMed] [Google Scholar]
- [16].Lebofsky R, Takahashi T, Walter JC, DNA replication in nucleus-free Xenopus egg extracts, Methods Mol Biol 521 (2009) 229–52. [DOI] [PubMed] [Google Scholar]
- [17].Blow JJ, Laskey RA, Initiation of DNA replication in nuclei and purified DNA by a cell-free extract of Xenopus eggs, Cell 47(4) (1986) 577–87. [DOI] [PubMed] [Google Scholar]
- [18].Smythe C, Newport JW, Systems for the study of nuclear assembly, DNA replication, and nuclear breakdown in Xenopus laevis egg extracts, Methods Cell Biol 35 (1991) 449–68. [DOI] [PubMed] [Google Scholar]
- [19].Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J, Checkpoint signaling from a single DNA interstrand crosslink, Mol Cell 35(5) (2009) 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M, Carroll D, Gautier J, Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication, Mol Cell 8(1) (2001) 137–47. [DOI] [PubMed] [Google Scholar]
- [21].Walter J, Newport J, Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha, Mol Cell 5(4) (2000) 617–27. [DOI] [PubMed] [Google Scholar]
- [22].Walter J, Sun L, Newport J, Regulated chromosomal DNA replication in the absence of a nucleus, Mol Cell 1(4) (1998) 519–29. [DOI] [PubMed] [Google Scholar]
- [23].Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, Dalla-Favera R, Non-transcriptional control of DNA replication by c-Myc, Nature 448(7152) (2007) 445–51. [DOI] [PubMed] [Google Scholar]
- [24].Williams HL, Gottesman ME, Gautier J, Replication-independent repair of DNA interstrand crosslinks, Mol Cell 47(1) (2012) 140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gautier J, Minshull J, Lohka M, Glotzer M, Hunt T, Maller JL, Cyclin is a component of maturation-promoting factor from Xenopus, Cell 60(3) (1990) 487–94. [DOI] [PubMed] [Google Scholar]
- [26].Gautier J, Norbury C, Lohka M, Nurse P, Maller J, Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+, Cell 54(3) (1988) 433–9. [DOI] [PubMed] [Google Scholar]
- [27].Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW, cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2, Cell 67(1) (1991) 197–211. [DOI] [PubMed] [Google Scholar]
- [28].Lohka MJ, Masui Y, Formation in vitro of sperm pronuclei and mitotic chromosomes induced by amphibian ooplasmic components, Science 220(4598) (1983) 719–21. [DOI] [PubMed] [Google Scholar]
- [29].Murray AW, Kirschner MW, Cyclin synthesis drives the early embryonic cell cycle, Nature 339(6222) (1989) 275–80. [DOI] [PubMed] [Google Scholar]
- [30].Murray AW, Solomon MJ, Kirschner MW, The role of cyclin synthesis and degradation in the control of maturation promoting factor activity, Nature 339(6222) (1989) 280–6. [DOI] [PubMed] [Google Scholar]
- [31].Lupardus PJ, Van C, Cimprich KA, Analyzing the ATR-mediated checkpoint using Xenopus egg extracts, Methods 41(2) (2007) 222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tutter AV, Walter JC, Chromosomal DNA replication in a soluble cell-free system derived from Xenopus eggs, Methods Mol Biol 322 (2006) 121–37. [DOI] [PubMed] [Google Scholar]
- [33].Maresca TJ, Heald R, Methods for studying spindle assembly and chromosome condensation in Xenopus egg extracts, Methods Mol Biol 322 (2006) 459–74. [DOI] [PubMed] [Google Scholar]
- [34].Field CM, Nguyen PA, Ishihara K, Groen AC, Mitchison TJ, Xenopus egg cytoplasm with intact actin, Methods Enzymol 540 (2014) 399–415. [DOI] [PubMed] [Google Scholar]
- [35].Costanzo V, Robertson K, Gautier J, Xenopus cell-free extracts to study the DNA damage response, Methods Mol Biol 280 (2004) 213–27. [DOI] [PubMed] [Google Scholar]
- [36].test note.
- [37].Trenz K, Errico A, Costanzo V, Plx1 is required for chromosomal DNA replication under stressful conditions, EMBO J 27(6) (2008) 876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sparks J, Walter JC, Extracts for Analysis of DNA Replication in a Nucleus-Free System, Cold Spring Harb Protoc (2018). [DOI] [PubMed] [Google Scholar]
- [39].Aze A, Sannino V, Soffientini P, Bachi A, Costanzo V, Centromeric DNA replication reconstitution reveals DNA loops and ATR checkpoint suppression, Nat Cell Biol 18(6) (2016) 684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Peterson SE, Li Y, Chait BT, Gottesman ME, Baer R, Gautier J, Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair, J Cell Biol 194(5) (2011) 705–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Aparicio T, Baer R, Gottesman M, Gautier J, MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts, J Cell Biol 212(4) (2016) 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kato N, Kawasoe Y, Williams H, Coates E, Roy U, Shi Y, Beese LS, Scharer OD, Yan H, Gottesman ME, Takahashi TS, Gautier J, Sensing and Processing of DNA Interstrand Crosslinks by the Mismatch Repair Pathway, Cell Rep 21(5) (2017) 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Costanzo V, Paull T, Gottesman M, Gautier J, Mre11 assembles linear DNA fragments into DNA damage signaling complexes, PLoS Biol 2(5) (2004) E110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C, Human Rad50/Mre11 is a flexible complex that can tether DNA ends, Mol Cell 8(5) (2001) 1129–35. [DOI] [PubMed] [Google Scholar]
- [45].Kurth I, Gautier J, Origin-dependent initiation of DNA replication within telomeric sequences, Nucleic Acids Res 38(2) (2010) 467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kastan MB, Bartek J, Cell-cycle checkpoints and cancer, Nature 432(7015) (2004) 316–23. [DOI] [PubMed] [Google Scholar]
- [47].Costanzo V, Gautier J, Single-strand DNA gaps trigger an ATR- and Cdc7-dependent checkpoint, Cell Cycle 2(1) (2003) 17. [DOI] [PubMed] [Google Scholar]
- [48].Kim JM, Yamada M, Masai H, Functions of mammalian Cdc7 kinase in initiation/monitoring of DNA replication and development, Mutat Res 532(1-2) (2003) 29–40. [DOI] [PubMed] [Google Scholar]
- [49].Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ, Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint, Genes Dev 14(12) (2000) 1448–59. [PMC free article] [PubMed] [Google Scholar]
- [50].Busino L, Chiesa M, Draetta GF, Donzelli M, Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis, Oncogene 23(11) (2004) 2050–6. [DOI] [PubMed] [Google Scholar]
- [51].Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, Shay JW, Ziv Y, Shiloh Y, Lee EY, Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products, Nature 405(6785) (2000) 473–7. [DOI] [PubMed] [Google Scholar]
- [52].Buscemi G, Savio C, Zannini L, Micciche F, Masnada D, Nakanishi M, Tauchi H, Komatsu K, Mizutani S, Khanna K, Chen P, Concannon P, Chessa L, Delia D, Chk2 activation dependence on Nbs1 after DNA damage, Mol Cell Biol 21(15) (2001) 5214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC, BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage, Nat Genet 30(3) (2002) 285–9. [DOI] [PubMed] [Google Scholar]
- [54].Zeman MK, Cimprich KA, Causes and consequences of replication stress, Nat Cell Biol 16(1) (2014) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hashimoto Y, Puddu F, Costanzo V, RAD51-and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks, Nat Struct Mol Biol 19(1) (2012) 17–U30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].O'Connell MJ, Walworth NC, Carr AM, The G2-phase DNA-damage checkpoint, Trends Cell Biol 10(7) (2000) 296–303. [DOI] [PubMed] [Google Scholar]
- [57].Garner E, Costanzo V, Studying the DNA damage response using in vitro model systems, DNA Repair (Amst) 8(9) (2009) 1025–37. [DOI] [PubMed] [Google Scholar]
- [58].Smith E, Costanzo V, Responding to chromosomal breakage during M-phase: insights from a cell-free system, Cell Div 4 (2009) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cimprich KA, Fragile sites: breaking up over a slowdown, Curr Biol 13(6) (2003) R231–3. [DOI] [PubMed] [Google Scholar]
- [60].Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J, An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication, Mol Cell 11(1) (2003) 203–13. [DOI] [PubMed] [Google Scholar]
- [61].MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA, The structural determinants of checkpoint activation, Genes Dev 21(8) (2007) 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Burden DA, Osheroff N, Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme, Biochim Biophys Acta 1400(1-3) (1998) 139–54. [DOI] [PubMed] [Google Scholar]
- [63].Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT, Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine, Cancer Res 59(17) (1999) 4375–82. [PubMed] [Google Scholar]
- [64].Chong JP, Mahbubani HM, Khoo CY, Blow JJ, Purification of an MCM-containing complex as a component of the DNA replication licensing system, Nature 375(6530) (1995) 418–21. [DOI] [PubMed] [Google Scholar]
- [65].Pichierri P, Rosselli F, Fanconi anemia proteins and the s phase checkpoint, Cell Cycle 3(6) (2004) 698–700. [PubMed] [Google Scholar]
- [66].Pichierri P, Rosselli F, The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways, EMBO J 23(5) (2004) 1178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Melo J, Toczyski D, A unified view of the DNA-damage checkpoint, Curr Opin Cell Biol 14(2) (2002) 237–45. [DOI] [PubMed] [Google Scholar]
- [68].Cook JG, Replication licensing and the DNA damage checkpoint, Front Biosci (Landmark Ed) 14 (2009) 5013–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kumagai A, Guo Z, Emami KH, Wang SX, Dunphy WG, The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts, J Cell Biol 142(6) (1998) 1559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, Cimprich KA, A requirement for replication in activation of the ATR-dependent DNA damage checkpoint, Genes Dev 16(18) (2002) 2327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Fernandez-Capetillo O, Celeste A, Nussenzweig A, Focusing on foci: H2AX and the recruitment of DNA-damage response factors, Cell Cycle 2(5) (2003) 426–7. [PubMed] [Google Scholar]
- [72].Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM, A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage, Curr Biol 10(15) (2000) 886–95. [DOI] [PubMed] [Google Scholar]
- [73].Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM, DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139, J Biol Chem 273(10) (1998) 5858–68. [DOI] [PubMed] [Google Scholar]
- [74].Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ, ATM phosphorylates histone H2AX in response to DNA double-strand breaks, J Biol Chem 276(45) (2001) 42462–7. [DOI] [PubMed] [Google Scholar]
- [75].Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A, DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1, Nat Cell Biol 4(12) (2002) 993–7. [DOI] [PubMed] [Google Scholar]
- [76].Dupre A, Boyer-Chatenet L, Gautier J, Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex, Nat Struct Mol Biol 13(5) (2006) 451–7. [DOI] [PubMed] [Google Scholar]
- [77].Ogawa H, Johzuka K, Nakagawa T, Leem SH, Hagihara AH, Functions of the yeast meiotic recombination genes, MRE11 and MRE2, Adv Biophys 31 (1995) 67–76. [DOI] [PubMed] [Google Scholar]
- [78].Haber JE, The many interfaces of Mre11, Cell 95(5) (1998) 583–6. [DOI] [PubMed] [Google Scholar]
- [79].Shechter D, Costanzo V, Gautier J, ATR and ATM regulate the timing of DNA replication origin firing, Nat Cell Biol 6(7) (2004) 648–55. [DOI] [PubMed] [Google Scholar]
- [80].Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC, Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM, Cancer Res 64(24) (2004) 9152–9. [DOI] [PubMed] [Google Scholar]
- [81].Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR, Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR, Nat Chem Biol 7(7) (2011) 428–30. [DOI] [PubMed] [Google Scholar]
- [82].Cano C, Barbeau OR, Bailey C, Cockcroft XL, Curtin NJ, Duggan H, Frigerio M, Golding BT, Hardcastle IR, Hummersone MG, Knights C, Menear KA, Newell DR, Richardson CJ, Smith GC, Spittle B, Griffin RJ, DNA-dependent protein kinase (DNA-PK) inhibitors. Synthesis and biological activity of quinolin-4-one and pyridopyrimidin-4-one surrogates for the chromen-4-one chemotype, J Med Chem 53(24) (2010) 8498–507. [DOI] [PubMed] [Google Scholar]