

Abstract
Purpose
Previous studies indicate that leukocytes, notably neutrophils, play a causal role in the capillary degeneration observed in diabetic retinopathy (DR), however, the mechanism by which they cause such degeneration is unknown. Neutrophil elastase (NE) is a protease released by neutrophils which participates in a variety of inflammatory diseases. In the present work, we investigated the potential involvement of NE in the development of early DR.
Methods
Experimental diabetes was induced in NE-deficient mice (Elane−/−), in mice treated daily with the NE inhibitor, sivelestat, and in mice overexpressing human alpha-1 antitrypsin (hAAT+). Mice were assessed for diabetes-induced retinal superoxide generation, inflammation, leukostasis, and capillary degeneration.
Results
In mice diabetic for 2 months, deletion of NE or selective inhibition of NE inhibited diabetes-induced retinal superoxide levels and inflammation, and inhibited leukocyte-mediated cytotoxicity of retinal endothelial cells. In mice diabetic for 8 months, genetic deletion of NE significantly inhibited diabetes-induced retinal capillary degeneration.
Conclusions
These results suggest that a protease released from neutrophils contributes to the development of DR, and that blocking NE activity could be a novel therapy to inhibit DR.
Keywords: diabetic retinopathy, neutrophils, microvasculature, neutrophil elastase, Elane
Diabetic retinopathy (DR) is the leading cause of vision loss in the working age population worldwide and remains as a major clinical challenge to overcome.1,2 Compelling evidence suggests that subclinical inflammation induced by hyperglycemia plays an important role in the pathogenesis of DR, including retinal capillary degeneration.3,4 Binding of neutrophils and perhaps other leukocytes to the endothelium plays a critical role in the degeneration of retinal capillaries in diabetes, because germline deletion of intercellular adhesion molecule-1 (ICAM-1) or CD18,3 or expression of neutrophil inhibitory factor5 significantly inhibit retinal capillary degeneration. Thus, vascular cell cytotoxicity caused by leukocytes, including neutrophils,6,7 apparently contributes to retinal capillary degeneration, however, the underlying mechanism remains elusive.
Neutrophil serine proteases (NSPs), including neutrophil elastase (NE), proteinase 3, and cathepsin G, are granule-associated enzymes that are secreted from activated neutrophils.8 In addition to their participation in destruction of bacteria and other pathogens, NSPs have been implicated in many inflammatory human diseases, including chronic obstructive pulmonary disease, cystic fibrosis, acute respiratory distress syndrome,9 chronic kidney disease, and inflammatory bowel disease.10,11 In all these conditions, activated neutrophils secrete activated NE, thus driving local inflammation and pathology.12–15 Some of the inflammatory changes induced by NE in other diseases are similar to what has been identified to play a role in the development of DR, including involvement of nuclear factor-κB (NF-κB), Toll-like receptor 4 (TLR4), myeloid differentiation primary response gene 88 (MyD88), and mitogen-activated protein kinases (MAPKs).16–18 Additionally, we have recently reported that NE, encoded by the Elane gene, contributes to the vascular leakage in the early stages of DR, potentially via PAR2, MyD88, and NF-κB signaling.19 We hypothesize that proteases released from neutrophils, most probably NE, might be the key factors in the pathogenesis of capillary degeneration and in the molecular abnormalities that contribute to the diabetes-associated retinopathy.
Research Design and Methods
Animals
This study was performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research, and with authorization of the Institutional Animal and Care Use Committees (IACUCs) at Case Western Reserve University (CWRU) and University of California-Irvine (UCI).
Wild type male control C57BL/6J and C57BL/6NJ and NE-deficient mice (Elanetm1Sds, abbreviated Elane−/−) were obtained from the Jackson Laboratory. Both the C57BL/6J and C57BL/6NJ are reported by the Jackson Laboratory to be acceptable controls for the Elane−/− mice, and we examined this again in nondiabetic and diabetic animals. Mice overexpressing hAAT+ in C57BL/6J background were obtained from Dr. Eli Lewis of the Ben-Gurion University of the Negev in Israel and were re-derived upon arrival to CWRU by the Transgenic and Targeting Facility.
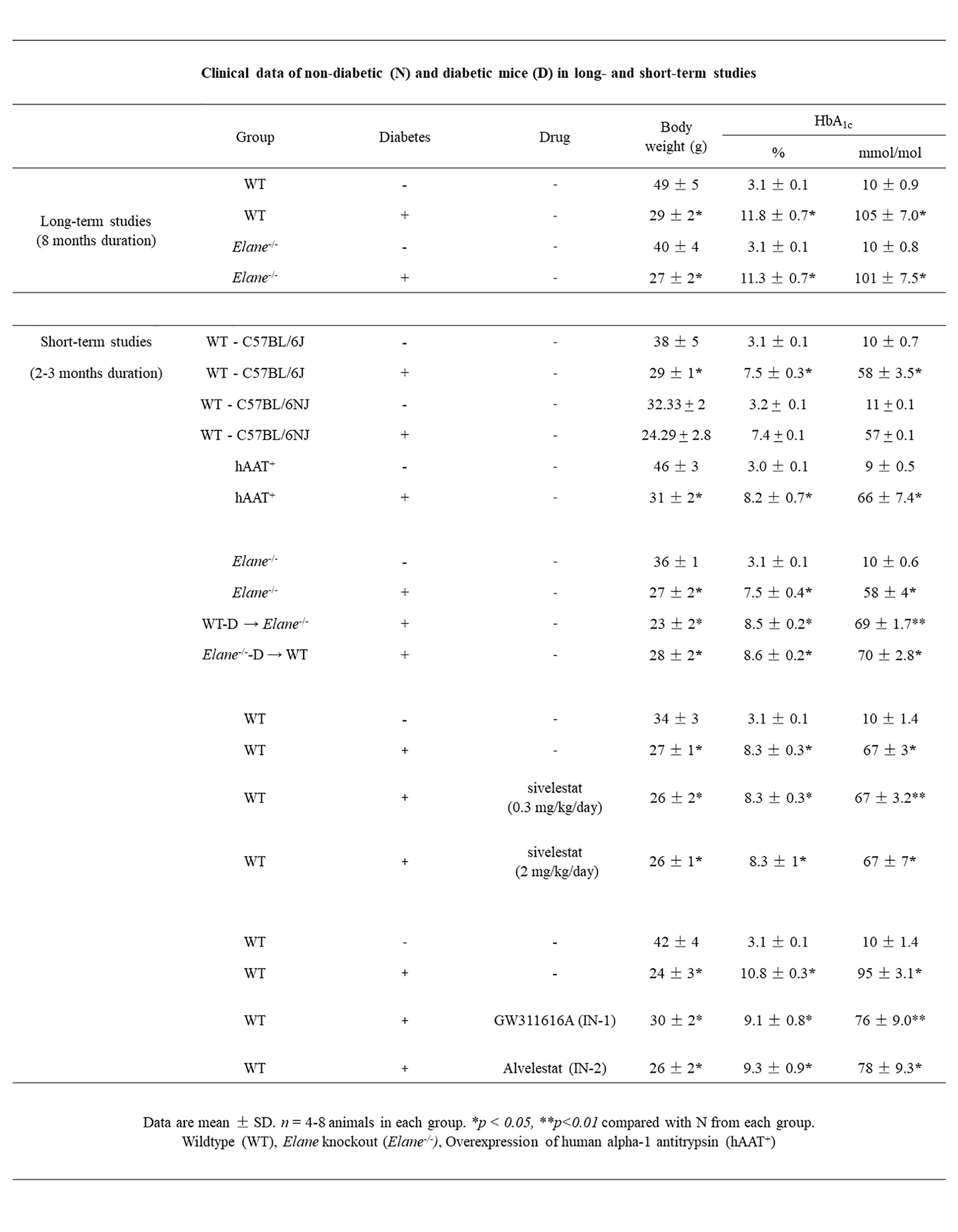
Diabetes was induced at 2 to 3 months of age by intraperitoneal injection (IP) of streptozotocin (60 mg/kg of body weight) for 5 consecutive days. The onset of diabetes was defined as 3 sequential measurements of blood glucose over 275 mg/dL. HbA1c was measured every 2 to 3 months throughout the experiment. In various drug-treated groups, diabetic mice were treated IP for 2 months with sivelestat (Abcam, Cambridge, MA, USA) at a dosage of 0.3 or 2.0 mg/kg/day, GW311616A (Axon Medchem, Reston, VA, USA) at a dosage of 4 mg/kg/injection twice a week, or alvelestat (Abmole Bioscience, Houston, TX, USA) at a dosage of 2 mg/kg/day. Other animals were treated with sivelestat eye-drops (5 µL per eye of a 3% solution in commonly available eye drops) once, twice, and 3 times per day for 2 months.
Chimeric mice were made as described previously.20 Chimeric mice in which only marrow-derived cells lacked Elane were generated by transplanting bone marrow from Elane−/− donors into irradiated wild type (WT) host mice (Elane−/− → WT). “Reverse” chimeric mice, where only marrow-derived cells have Elane (the rest of the animal is Elane-deficient), were made by transplanting bone marrow from WT donors into Elane−/− irradiated host mice (WT → Elane−/−). Transplantation of WT marrow cells back into irradiated nondiabetic and diabetic WT animals (WT N → WT N and WT D → WT D, respectively) served as controls for the irradiation process. Diabetes was induced in appropriate animals 2 to 3 weeks before irradiation to ensure that irradiation-induced destruction of immune cells did not affect diabetes severity. One week after the bone marrow transplantation was identified as the beginning of the experiment.
Two- and Eight-Month Studies
Diabetic mice and age-matched non-diabetic control mice were studied after 2 months or 8 months of diabetes (i.e. 4 and 10 months of age). For animals in the 8-month studies, one eye was used to analyze capillary degeneration (present manuscript), whereas the other eye was used to assess vascular permeability and was reported previously.19
Leukocyte-Mediated Cytotoxicity to Retinal Endothelial Cells
Leukocyte-mediated cytotoxicity toward mouse retinal endothelial cells (mRECs) was assayed as previously described.21,22 Transformed murine retinal endothelial cells, a kind gift of Dr. Nader Sheibani, were isolated from the retinas of Immorto-mice, as previously described.23 For some experiments, we compared the endothelial cytotoxicity of purified neutrophils from diabetic mice to that using the unfractionated leukocyte pool. Briefly, 100,000 white blood cells from peripheral blood or neutrophils isolated from bone marrow using the EasySep Mouse Neutrophil Enrichment Kit (Stemcell Technologies, Vancouver, Canada) from non-diabetic or diabetic mice were incubated with mRECs for 6 hours. In some experiments, a general protease inhibitor cocktail (Sigma, St. Louis, MO, USA) was added into the medium at a ratio of 1:200, or sivelestat (Abam, Cambridge, MA, USA) was used at a final concentration of 100 µM. The mRECs were then removed and labeled with an antibody against CD144 or CD31 (BD, San Diego, CA, USA), and viability was measured by flow cytometry after immunostaining of mRECs with 7-aminoactinomycin D (7-AAD; BD Biosciences, San Diego, CA, USA). A total of 10,000 events were counted for each sample. Results were analyzed by Flow Jo 7.6 and/or NovoExpress.
NE-induced Apoptosis of Retinal Endothelial Cells
Primary mouse retinal endothelial cells (Cell Biologics, Chicago, IL, USA) were set up at a density of 100,000 per well in a 6-well plate with culture medium per vendor instructions. The medium was changed every other day for 3 days. When cells reached 80% to 90% confluency, human NE (Innovative Research, Novi, MI, USA) was added to the medium at 50 nmol/L concentration and incubated for 1, 6, and 12 hours. After the indicated times, endothelial cells were removed and labeled with an antibody against CD31 (BD Biosciences, San Diego, CA, USA). An Annexin V apoptosis detection kit (Stem Cell Technologies, Seattle, WA, USA) was used to evaluate early apoptosis. Cells were stained also with 7-AAD to identify late-stage apoptosis and necrosis. Viability was measured by flow cytometry as described in the previous section. A total of 10,000 events were counted for each sample. Results were analyzed by NovoExpress and graphed as reported in the literature.24
Retinal Superoxide
Retinas from animals were analyzed for superoxide production as previously described.21,22 Briefly, retinas were placed in 0.2 mL of Krebs/HEPES buffer and allowed to equilibrate in the dark at 37°C under 95% O2/5% CO2 conditions for 20 minutes. To each tube, 0.5 mM lucigenin was added and incubated at 37°C for an additional 10 minutes before having the photon emission detected by a luminometer (Analytical Luminescence Laboratory, San Diego, CA, USA). Retinal protein was quantified, and the luminescence expressed per mg of protein.
Western Blot Analysis
Retinal homogenates were subjected to Western blot analysis as reported by us previously.25 Briefly, murine retinas were isolated and sonicated in RIPA buffer (Santa Cruz Biotechnology, Dallas, TX, USA). Lysates were collected by centrifugation at 12,000 rpm for 20 minutes at 4°C. Protein concentration was estimated using Bradford assay. All samples were mixed with Laemmli sample buffer and boiled for 5 minutes. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes for immuno-blotting. Antibodies against inducible nitric oxide synthase (iNOS, 1:500 dilution; Transduction Laboratories, Lexington, KY, USA), intercellular adhesion molecule 1 (ICAM-1, 1:1000 dilution; Proteintech Group, Rosemont, IL, USA), phospho-IκB and IκB (1:500 and 1:1000 dilution respectively; Cell Signaling, Dallas, TX, USA) were used.
Quantitative Real-Time PCR
Total RNA was isolated from mouse retina using PureLink RNA Mini kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. A total of 400 ng RNA was reverse transcribed into cDNA using High-Capacity cDNA Reverse Transcription kit. Quantitative real-time PCR was performed using FastStart Universal SYBR Green Master Mix (Roche, Mannheim, Germany) on an ABI Step OnePlus Real-Time PCR System (Applied Biosystems; Foster City, CA, USA). Experiments were performed in duplicate. PCR conditions were 400 nmol primer for 10 minutes at 95°C, 44 cycles of 15 seconds at 95°C, and at 57°C for 30 seconds. The fold-change expression of target genes was calculated using the 2−ΔΔCt fold-change formula normalized to β-actin. The following primer sets were used: ICAM-1 forward (5' → 3')ATCACCGTGTATTCGTTTCC and ICAM-1 reverse (5' → 3')CAGCACCGTGAATGTGATCT, iNOS forward (5' → 3')TCTTTGACGCTCGGA ACTGTAGCA, and iNOS reverse (5' → 3')TAGGTCGATGCACAACTGGGTGAA, β-actin forward (5' → 3')GATTCCATACCCAAGAAGGAAGGCTG and β-actin reverse (5' → 3')AGCTGAGAGGGAAATCGTGCGT.
Protease Activity Assay
Leukocytes or plasma isolated from 2 months diabetic animals or from nondiabetic controls were incubated in 96 well plates with elastase substrate following the EnzChek Elastase Assay Kit protocol (Molecular Probes, Eugene, OR, USA). Samples were incubated with or without NE inhibitor (sivelestat; 100 µM) for 20, 40, 60, and 80 minutes at room temperature protected from light. Fluorescence intensity was measured in a fluorescence microplate reader equipped with standard fluorescence filters. For each time point, correction for background fluorescence was performed by subtracting the value derived from the no-cell control.
Enzyme-Linked Immunosorbent Assay
Mouse alpha-1 antitrypsin enzyme-linked immunosorbent assay (ELISA) kit (Innovative Research, Novi, MI, USA; IRKTAH1147) was used for detecting and quantifying mouse AAT in the circulating plasma. Following the kit instructions, samples were prepared at a dilution of 1/1000, and the amount of AAT in the test samples was determined by absorbance at 450 nm, with the amount interpolated from a standard curve constructed standards after correction for sample dilution.
Quantification of Neutrophil Population by Flow Cytometry
Blood was drawn by cardiac puncture and leukocytes were immunostained for flow cytometry. Antibodies including PE-CF594 Rat Anti-Mouse CD45R, BV650 Rat Anti-Mouse CD11b, PE-Cy 7 Mouse Anti-Mouse NK-1.1, BV786 Rat Anti-Mouse CD4, BV786 Rat Anti-Mouse CD8a, BV421 Rat Anti-Mouse Ly-6C, and APC Rat Anti-Mouse Ly-6G were purchased from Becton Dickinson. Samples were incubated with BD Fc block for 15 minutes and then with antibodies in brilliant stain buffer for 30 minutes at 4°C. Cell populations were analyzed using a LSRII flow cytometer. CD11b positive, Ly6G positive, and Ly6C intermediate (CD11b+ Ly6G+ Ly6Cinterm) cells were identified as neutrophils.
Diabetes-Induced Retinal Vascular Histopathology
Retinal capillary degeneration was assessed at 8 months of diabetes (10 months of age) as described previously.21 Briefly, eyes were removed and fixed in 10% formalin for at least 10 days. Next, retinas were isolated and digested in elastase pH 6.5 for 2 hours at 37°C followed by incubation of samples in 100 mM Tris buffer pH 8.5 at room temperature overnight. Retinal neurons were brushed away from the vasculature, and the cleaned vasculature was laid out on a glass microscope slide to dry. Samples were stained with hematoxylin and periodic acid-Schiff reagent. Capillary-sized vessel tubes having no nuclei along their length were counted as acellular capillaries in six-seven field areas around the mid-retina.
Quantitative Measurement of Leukostasis
The number of leukocytes adherent to the microvasculature was determined at 3 months of diabetes. After cardiac catheterization, anesthetized mice (100 mg/mL ketaset to 100 mg/mL xylazine ratio of 5:1) were exsanguinated by perfusion with PBS. Fluorescein-coupled concanavalin A lectin (20 µg/mL in PBS; Vector Laboratories, Burlingame, CA, USA) was then infused as previously described.26 Flat-mounted retinas were viewed via fluorescence microscopy, and brightly fluorescent leukocytes were counted in the entire retina.
Statistical Analysis
Data between groups were analyzed by ANOVA followed by the Fisher post hoc tests. The P values < 0.05 were considered statistically significant. Superoxide and co-culture data were normalized to controls because these assays can show day-to-day variations in absolute values, but relative differences between groups are maintained.
Results
Animals
Based on HbA1c and blood glucose levels, diabetes was successfully induced in mice by serial injections of streptozotocin (STZ). Clinical data of nondiabetic (N), diabetic mice (D) with or without NE inhibitor treatment, and genetically modified mice are reported in Supplementary Table S1. There were significant differences between STZ-treated mice and nondiabetic control mice in HbA1c and body weight (BW). Neither genetic modifications nor pharmacologic treatments had an effect on HbA1c or BW compared with the WT diabetic control group. The Jackson Laboratory reports that C57BL/6J and C57BL/6NJ mice are acceptable controls for the Elane−/- mice, so we compared both strains with respect to the effect of diabetes on development of molecular abnormalities that have been implicated in the pathogenesis of the retinopathy. We did not find differences with regard to BW and blood glucose levels (Supplementary Table S1) between both strains. Similarly, diabetes-induced increase in retinal superoxide and expression of inflammatory proteins and leukocyte-mediated cytotoxicity of retinal endothelial cells were comparable between the two strains (Supplementary Figs. S1A–D), suggesting that both strains respond to diabetes similarly. Because the C57BL/6NJ mice carries the rd8 mutation,27 we subsequently used C57BL/6J mice as the control for Elane−/− studies. Although the Elane−/− strain has a mixed background of C57BL/6J and C57BL/6NJ according to the Jackson Laboratory, and thus carries the rd8 mutation, we did not observe any significant effect of the C57BL/6NJ strain or NE deficiency on visual function, retinal thickness, or photoreceptors survival in Elane−/− mice compared to C57BL/6J mice.19
Enzymatic Activity of NE is Significantly Higher in Circulating Leukocytes and Plasma From Diabetic Mice Compared to Non-Diabetic Mice
We used EnzChek Elastase Assay kit to quantify NE activity in white blood cells (100,000 cells) and plasma (10 µL) isolated from non-diabetic and diabetic mice and found that in both types of samples protease activity was higher in diabetic animals when compared to non-diabetic animals (Fig. 1A). We also used an AAT ELISA kit to quantify the amount of the endogenous protease inhibitor AAT in the plasma from the same animals, and found that there was no significant difference between non-diabetic and diabetic groups (Fig. 1B). To determine if the increased protease activity observed in white blood cells (WBCs) from diabetic animals was mainly driven by NE, we used the same NE activity kit in the presence of sivelestat, an NE inhibitor, and found that independent of assay incubation time, the protease activity in WBCs from diabetic animals was totally inhibited (Fig. 1C), indicating that the increased protease activity measured in leukocyte samples from diabetic mice probably is due to the increased expression and/or release of NE, and not due to reduced levels of AAT in the plasma.
Figure 1.

Protease activity in diabetes. Protease activity was increased in (A) leukocytes and plasma of mice diabetic for 2 months. The increase (B) could not be attributed to a change in plasma hAAT level, but (C) could be inhibited in vitro by sivelestat (100 µM). The protease activity in WBCs and plasma was determined using the EnzCheck Elastase Assay Kit, and the amount of AAT in plasma was determined by ELISA. N, non-diabetic; D, diabetic. Data are expressed as mean ± SD. Graphs represent the combined results of 2 to 3 experiments; n = 4 to 6 per group; P < 0.05 is significant.
Endothelial Cytotoxicity Caused by Leukocytes and Neutrophils From Diabetic Mice is Inhibited by Selective Deletion or Pharmacological Inhibition of NE, but not by Overexpression of hAAT
Previous studies from our group have shown that co-culture of leukocytes from diabetic animals or patients with retinal endothelial cells leads to death of the endothelial cells.28,29 We hypothesized that NE on the surface, or released from, neutrophils while in close contact with the retinal endothelium might have a cytotoxic effect on the endothelial cells. To test our hypothesis, we evaluated if acute exposure to NE blockers inhibited diabetes-induced leukocyte-mediated endothelial cytotoxicity. In all studies, we added leukocytes from diabetic animals (WT, Elane−/− or hAAT expressing mice) to endothelial cultures for 6 hours, and in some experiments a general protease inhibitor or sivelestat was added to the medium for the incubation duration, we then assessed endothelial cell death by flow cytometry (Fig. 2). We found that cytotoxicity to endothelial cells caused by leukocytes isolated from animals that were diabetic for 2 months was inhibited as a result of the addition of the general protease inhibitor or specific NE inhibitor (sivelestat) in the culture medium (see Fig. 2A). Leukocytes or neutrophils from diabetic Elane−/− mice showed a significant inhibition of the leukocyte- or neutrophil-mediated cytotoxicity to endothelial cells when compared to leukocytes or neutrophils from WT diabetic mice (see Figs. 2B, 2C). Leukocytes from diabetic hAAT+ mice did not inhibit the diabetes-induced leukocyte-mediated cytotoxicity to endothelial cells (see Fig. 2B). Inhibition of neutrophil-mediated cytotoxicity to endothelial cells by genetic deletion of NE was also observed in long-term (8 months) diabetes (see Fig. 2D), indicating that NE derived from neutrophils of diabetic animals contributed to the cytotoxicity of endothelial cells.
Figure 2.
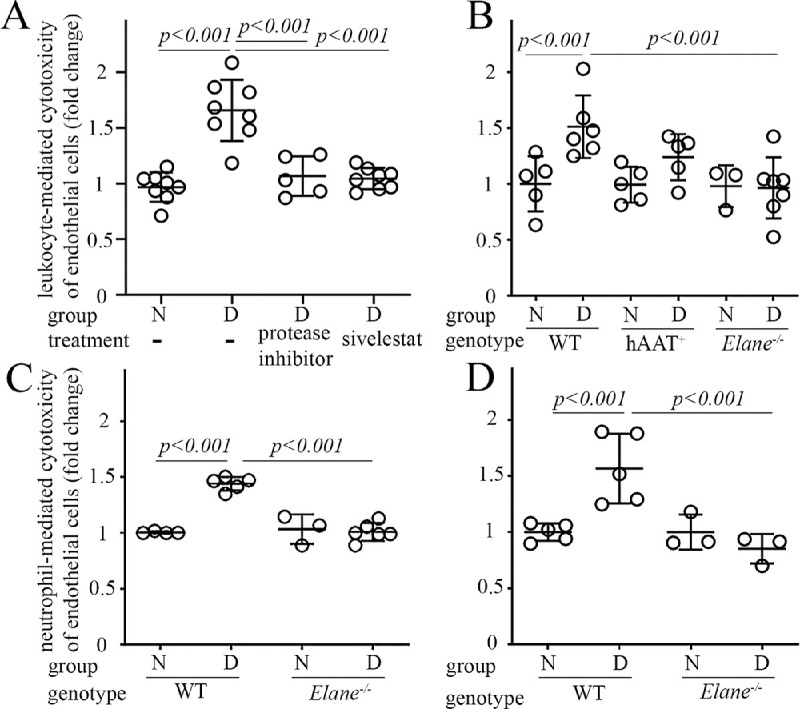
The diabetes-induced increase in cytotoxicity of leukocytes (A, B) or neutrophils (C, D) against retinal endothelial cells is mitigated by inhibition of proteases or NE in vitro for 6 hours or deletion of NE (Elane−/−). Damage to retinal endothelial cells was determined by flow cytometry. The duration of diabetes was 2 months in panels A to C or 8 months in panel D. The Y-axis legend in A and C applies for B and D graphs. N, nondiabetic; D, diabetic. Data are normalized to N and expressed as mean ± SD. Graphs represent the combined results of 2 to 3 experiments; n = 6–12 per group; P < 0.05 is significant.
The NE-Mediated Cytotoxicity of Retinal Endothelial Cells Involves Apoptosis
Apoptosis is a predominant form of endothelial cell death in diabetic retinas,30 then we investigated if the cytotoxicity driven by NE on retinal endothelial cells triggers this type of regulated cell death. Using mouse retinal endothelial cell cultures, human NE was added to the media with or without the irreversible NE inhibitor GW311616A and incubated for 1, 6, and 12 hours. We found that human NE significantly increased endothelial cell apoptosis in vitro, and administration of GW311616A was able to completely abolish the effect at 6 and 12 hours (Fig. 3). Although we also found a small percentage of unregulated cell death (necrosis), this was not modified by the addition of the NE inhibitor at any time point, suggesting that this type of cell death was secondary to other factors besides NE.
Figure 3.

Effect of human NE on mRECs apoptosis. (A) Plots along with gating strategies to determine early and late apoptosis in endothelial cells are depicted. Top panels – tunicamycin (10 µg/mL) was used as positive control to define the quadrants. FSC-H versus SSC-H dot plot (Main) are gated to eliminate debris and then singlet cell (Singles) was selected on FSC-A versus FSC-H. Endothelial cells were gated on CD31 to confirm cell population. Annexin V was used to determine apoptosis and 7-ADD was used to determine cell viability. Bottom panels – Representative flow dot plots from non-treated, hNE treated cells (50 nmol/L) and hNE plus GW311616A inhibitor (GW 150 µMol/L) with quadrants representing endothelial cells in various stages. (B) Early apoptosis is summarized in the graph. All conditions were performed in triplicates. Results are the combination of two experiments. Data are normalized to non-treated cells and expressed as mean ± SD, n = 6 per condition; *P < 0.05, **P < 0.01.
The Diabetes-Induced Increase in Retinal Superoxide, Expression of Inflammatory Proteins, Leukostasis, and Capillary Degeneration are Inhibited in Diabetic Elane−/− Mice
Retinal production of superoxide and induction of inflammatory proteins have been implicated in the pathogenesis of DR,31,32 and thus we determined whether or not genetic deletion of NE would inhibit the molecular defects associated to the development of DR. To determine whether proteases (in general) or a specific protease (NE) plays an important role in diabetes-induced retinal oxidative stress and inflammation, Elane−/− mice and hAAT+ mice were used. Compared to diabetic WT mice, the diabetes-induced increase in retinal superoxide was significantly inhibited in Elane−/− mice. Overexpression of hAAT had the opposite effect, increasing the diabetes-induced superoxide generation (Fig. 4A). Both iNOS and ICAM-1 expression in the retina were significantly inhibited in Elane−/− diabetic mice compared to WT control diabetic mice, whereas levels of iNOS, but not ICAM-1, were significantly inhibited in diabetic hAAT+ mice (see Figs. 4B, 4C). The diabetes-induced increase in leukostasis within the retinal vasculature was inhibited in Elane−/− diabetic mice (Fig. 4D and image in right panel). These data indicate that the diabetes-induced increase of NE activity, but apparently not all proteases, plays a significant role in the development of molecular and physiological changes observed in the retina in diabetes.
Figure 4.

Effects of Elane deletion, and overexpression of hAAT on diabetes-induced increases in superoxide and inflammatory markers in the retina, leukostasis within the retinal vasculature, and degeneration of retinal capillaries. Retinal superoxide (A) was detected by lucigenin luminescence using freshly isolated retinas, and expression of iNOS and ICAM-1 (B, C) were evaluated by Western blots. X-axis legend in panel C applies also to panels A and B. Leukostasis in retinal microvessels (D) was assessed by injection of fluorescein-coupled concanavalin A lectin, followed by perfusion to remove unbound blood cells, and then quantitation of adherent cells via fluorescence microscopy. A representative image of leukostasis from diabetic animals is shown as an inset panel in D (white arrows). (E, F) Show the effects of diabetes and Elane−/– on retinal vascular pathology, with representative micrographs of retinal vasculature for each group. Black arrows indicate typical degenerated capillaries. (F) summarizes the numbers of degenerated (acellular) capillaries in the various groups at 8 months of diabetes (10 months of age). Duration of diabetes was 2 months for panels A to D, and 8 months for panels E and F. N, nondiabetic; D, diabetic. Data are expressed as mean ± SD. Graphs represent the combined results of 2 to 3 experiments; n = 4 to 7 per group; P < 0.05 is significant.
Having demonstrated that retinal oxidative stress and inflammation were attenuated by selective deletion of NE, we next sought to determine whether diabetes-induced degeneration of retinal capillaries was inhibited in Elane−/− mice. Diabetes of 32 weeks of duration significantly increased the number of degenerated capillaries in retinas of WT mice, as expected, and genetic deletion of Elane significantly inhibited capillary degeneration compared to that in WT diabetic mice (Figs. 4E, 4F). The number of degenerated retinal capillaries in diabetic Elane−/− mice was not significantly different than that in non-diabetic WT animals, indicating that capillary degeneration is driven substantially by NE. There were no significant differences in retinal capillary loss between non-diabetic WT and Elane−/− mice.
Chimeric Mice Lacking NE Only From Myeloid-Derived Cells Were Protected From the Diabetes-induced Increase in Retinal Superoxide and Leukocyte-mediated Cytotoxicity to Endothelial Cells
To investigate if the observed contribution of NE to the diabetes-induced oxidative stress in the retina and leukocyte-mediated killing of endothelial cells was indeed due to leukocytes (as opposed to the potential induction of NE-like activity in non-myeloid cells in diabetes), NE-deficient chimeric mice were generated (see Fig. 5). In some diabetic mice (D), NE was deleted only from bone marrow-derived cells whereas the rest of the animal cells retained the NE activity (Elane−/− D → WT D), and, in other cases, NE was present only in the myeloid cells whereas the rest of the animal cells were NE-deficient (WT D → Elane−/− D). As expected, the expression of NE in myeloid cells isolated from Elane−/− D → WT D mice was significantly less than that in WT D → Elane−/− D mice and WT D → WT D mice at 2 months of diabetes (see Fig. 5A, Supplementary Fig. S2A). Consistent with earlier data demonstrating that diabetes increased leukocyte-mediated killing of retinal endothelial cells and retinal superoxide production in unirradiated WT mice, we found that diabetes (2 months duration) in WT D → WT D controls likewise significantly increased leukocyte-mediated cytotoxicity of endothelial cells ex vivo (see Fig. 5B) and retinal superoxide production in vivo (see Fig. 5C). Incubation of freshly isolated leukocytes from Elane−/− D → WT D mice with retinal endothelial cells resulted in a significant inhibition of the cytotoxicity ex vivo and decreased retinal superoxide production in vivo, whereas these defects were not inhibited in chimeric diabetic mice in which only marrow-derived cells contained NE. Thus, NE in myeloid cells is responsible for the adverse effects of diabetes on these molecular defects that contribute to the development of DR.
Figure 5.
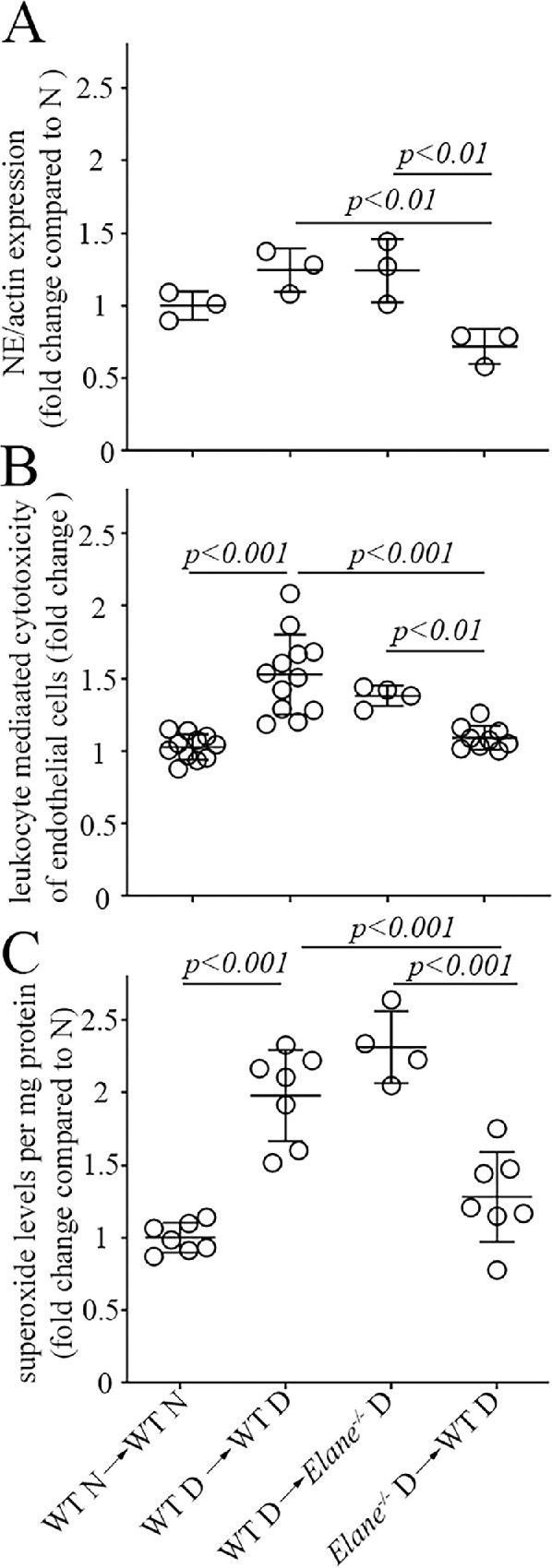
Chimeric animals constructed to lack Elane only in bone marrow-derived cells successfully reduced expression of NE in leukocytes (A) and inhibited the diabetes-induced increase in leukocyte-mediated endothelial cell cytotoxicity (B) and in retinal superoxide (C). None of these changes were detected when only marrow-derived cells still expressed NE. The expression of NE was determined by Western blotting, leukocyte-mediated cytotoxicity toward endothelial cells was quantitated by flow cytometry, and retinal superoxide was determined by lucigenin luminescence. WT N → WT N indicates nondiabetic WT controls in which WT marrow is injected into irradiated WT N animals. WT D → WT D indicates diabetic WT controls in which WT marrow is injected into irradiated WT D animals. WT D → Elane−/− D indicates mice in which marrow from WT diabetic mice is injected into irradiated diabetic Elane−/− mice. Elane−/− D → WT D indicates mice in which marrow from diabetic Elane−/− mice is injected into irradiated WT diabetic mice. The legend for the X axis in C applies also to A and B. Duration of diabetes was 3 months (duration after irradiation 2–3 months). Data are normalized to WT N → WT N and expressed as mean ± SD; n = 4 to 12 per group; P < 0.05 is significant.
The Circulating Neutrophil Population was Decreased in Non-Diabetic Elane−/− Mice, but not in Diabetic Elane−/− Mice
It has been reported that Elane mutations cause neutropenia in patients,33,34 so flow cytometry was used to quantify the effect of Elane−/− on circulating neutrophils in mice. Flow cytometry plots along with gating strategies for neutrophils in the blood of different mouse models are provided in Figure 6. Consistent with prior reports, diabetes of 8 months duration in both control and Elane−/− animals caused a significant increase in the fraction of leukocytes represented by neutrophils, and non-diabetic Elane−/− mice showed a reduction in the fraction of circulating neutrophils (see Fig. 6 bottom panels and graph). Surprisingly, however, the neutrophil fraction was not subnormal in diabetic Elane−/− mice. Neutrophil population is reported as percentage of total leukocytes in Figure 6, but even if neutrophils are reported in absolute numbers, there was not a significant difference between the WT nondiabetic and the diabetic Elane−/− group (2.8 ± 1.0, 4.3 ± 0.8, 2.2 ± 1.5, and 2.8 ± 1.7 neutrophils × 105/mL for nondiabetic and diabetic WT, and nondiabetic and diabetic Elane−/- mice, respectively; n = 6 per group).
Figure 6.

Peripheral blood phenotyping by flow cytometry. (A) Plots along with gating strategies for neutrophils in the blood of WT and Elane−/− mice are depicted. Top panels – FSC-A versus SSC-A dot plot (P1) are gated to eliminate debris and then singlet cell (P2) was selected on FSC-A versus FSC-H. Non-lymphocyte population was gated on CD11b+ (P3 and P4) to eliminate T cells (CD4+ and CD8+), B cells (CD45R+), and NK cells (NK1.1+), then Ly6G and Ly6C antibodies were used to define the neutrophil (Ly6G+Ly6Cinterm) where classical monocytes (Ly6G−Ly6Chigh) and nonclassical monocytes (Ly6G−Ly6Clow) were eliminated. Bottom panels – Flow dot plots represent neutrophils in various groups. (B) Graph summarizes the neutrophil population per genotype. Mice are 8 months of diabetes; n = 6 per group.
The Diabetes-Induced Increase of Retinal Superoxide Production, Inflammatory Proteins and Leukocyte-Mediated Cytotoxicity to Endothelial Cells are Inhibited by Daily Administration of NE Inhibitors
The genetic deletion of NE is not a suitable therapeutic option to treat DR in patients, so we further investigated if the molecular defects studied above could be inhibited using pharmacologic agents. Daily administration of three different NE inhibitors – sivelestat, GW311616A, and alvelestat – for 2 months to diabetic animals each inhibited the diabetes-induced increase in retinal superoxide, the expression of inflammatory proteins ICAM-1 and iNOS, the altered expression of the ratio of phospho-IkB to total IkB, and the leukocyte-mediated retinal endothelial cell cytotoxicity (Figs. 7A–I, Supplementary Figs. S2B–D).
Figure 7.

Pharmacologic inhibition of NE mitigates diabetes-induced abnormalities in retinal superoxide (A) and expression of pro-inflammatory proteins (B–D), as well as leukocyte-mediated cytotoxicity to retinal endothelial cells (E). Sivelestat was administered IP daily at a dosage of 0.3 or 2.0 mg/kg/day. Similar results were shown in diabetic mice treated with other two NE inhibitors – GW311616A (GW) at a dosage of 4 mg/kg/injection twice a week and alvelestat (Alv) at a dosage of 2 mg/kg/day (F–I). Total duration of diabetes was 10 weeks, drug treatment was 8 weeks. Data are expressed as mean ± SD. Graphs represent the combined results of 2 to 3 experiments; n = 4 to 8 per group; P < 0.05 is significant.
Because of potentially undesirable effects, there could be reluctance to administer protease inhibitors systemically as a long-term therapy.35–39 In our studies, other than the neutropenia noted above in nondiabetic NE-deficient animals, however, we did not detect changes in general appearance, activity, nest building, interaction with cage mates, or breeding pattern secondary to inhibiting or deleting NE. Nevertheless, we conducted initial studies to determine if local delivery of an NE inhibitor to the eyes might be an alternative approach to inhibiting NE and DR. Administration of sivelestat (3%) by eye drops multiple times per day for 2 months likewise inhibited the diabetes-induced increase in retinal superoxide (Fig. 8).
Figure 8.
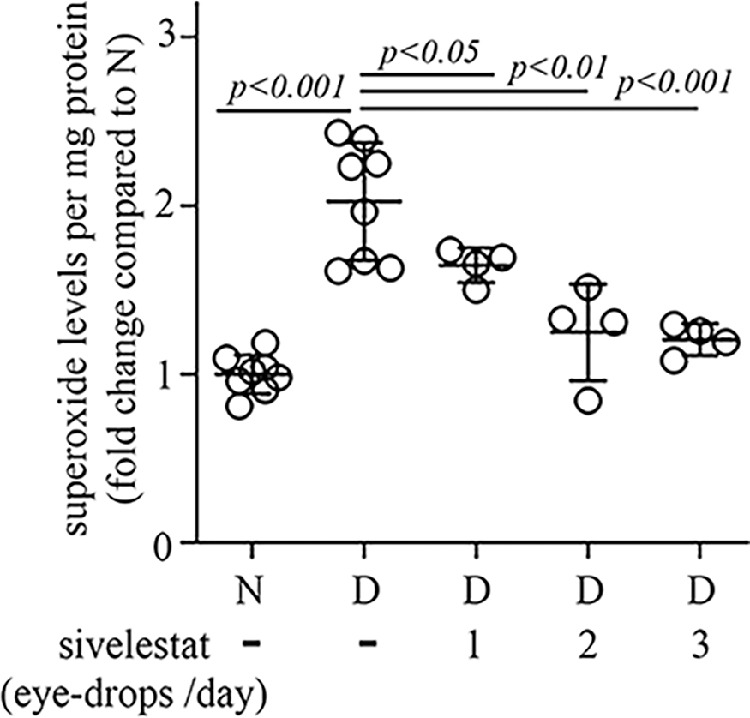
Retinal superoxide was inhibited in diabetic mice receiving daily treatment with 3% sivelestat eye drops (once, twice, or 3 times per day) compared to nontreated diabetic mice. Total duration of diabetes was 10 weeks, drug treatment was 8 weeks. Data are expressed as mean ± SD; n = 4 to 8 per group; P < 0.05 is significant.
Discussion
Neutrophils and other granulocytes have previously been implicated in the pathogenesis of DR,20,40,41 and their ability to kill microvascular endothelial cells in vitro likely contribute to the retinal capillary degeneration observed in diabetes.20,42–45 In an effort to identify the mechanism by which circulating neutrophils exert this action, we focused on NE, a serine protease released by neutrophils. In the current study, we found that the enzymatic activity of NE was greater than normal in circulating leukocytes and plasma from diabetic animals. This increase in activity was not due to a reduction in the amount of circulating AAT, although remains possible that the functional activity of AAT in diabetes is impaired.46
We found that the diabetes-induced increase in retinal capillary degeneration was significantly (although partially) inhibited in Elane−/− mice, suggesting that proteases released from circulating cells play a previously unrecognized role in the development of DR, and that NE is an important contributor to the degeneration of retinal capillaries. Such contribution was confirmed in experiments incubating leukocytes or neutrophils from experimental animals with mRECs. We observed that the cytotoxic effect of the leukocytes from diabetic mice was significantly inhibited by acutely adding a selective NE inhibitor into the media. Inhibition of the leukocyte-mediated cytotoxicity to mRECs was demonstrated also in diabetic Elane−/− mice, but not in hAAT+ mice, indicating that the leukocyte-mediated cytotoxicity in diabetes depends on NE, but not on proteases in general.
Previously, it has been reported that serine proteases cause oxidative stress in cells, and that AAT inhibits the oxidative stress in pre-eclampsia.47 Thus, the increase in oxidative stress in diabetic animals expressing hAAT+ was unexpected. Perhaps, the protease inhibitory activity of AAT was decreased due to nonenzymatic glycation in diabetes.48 Furthermore, it has been reported that cell surface-bound NE remains catalytically active and remarkably resistant to inhibition by naturally occurring proteinase inhibitors, especially that fraction in the cleft between the neutrophils adhering to endothelial cells.49 Thus, NE on the surface of neutrophils might interact with (and damage) the surface of endothelial cells in diabetes without being inhibited by circulating protease inhibitors.50–53 The NE that does interact with the endothelial cell surface can proteolytically damage the endothelial cell surface,54,55 and can even enter endothelial cells,56 where its proteolytic actions (that can trigger apoptosis) likely would be unrestrained. This NE-based mechanism for capillary damage in diabetes is consistent with prior evidence that the retinal capillary degeneration is inhibited in diabetic animals in which leukocytes cannot bind to adhesion molecules on endothelial cells due to the presence of neutrophil inhibitory factor or to a deficiency of ICAM-1 or CD-18.3,44
In the current study, we also found that leukostasis was significantly increased in WT diabetic mice, but not in Elane−/− diabetic mice, suggesting that NE can influence leukocyte adherence to the retinal endothelium in diabetes. ICAM-1 is the adhesion molecule that plays a major role in leukocyte adhesion to endothelial cells, and our data shows that inhibiting or eliminating NE mitigates the diabetes-mediated increase of ICAM-1 expression in the retina. Significant variability of ICAM-1 expression was observed between the animals of different study groups, and although we do not know why this occurs, it is possible that the nature of the in vivo model leads to hyperglycemia fluctuations during the day between and in the diabetic animal itself and such difference might impact the expression of ICAM-1 directly.57 In addition, the circadian pattern in the expression of ICAM-1 might contribute to the observed variability.58 In any case, this is an intriguing observation that requires additional investigation.
Multiple studies have reported that NE is generated by neutrophils, but mRNA for the elastase also has been detected in monocytes and mast cells.59,60 It was not clear if diabetes might induce other cells in the body to express NE, so we generated chimeric mice to evaluate the possible contribution of nonmyeloid cells in the body to the NE-mediated alteration in diabetes. Our studies demonstrated that diabetes increased retinal superoxide and leukocyte-mediated killing of endothelial cells when NE was present in myeloid-derived cells but not in the rest of the body, whereas diabetes did not cause these abnormalities if myeloid cells lacked NE. These data suggest that only myeloid-derived cells are causing the NE-mediated abnormalities detected in retinas of diabetic mice. How leukocytes regulate retinal oxidative stress in diabetes is not known.
Studies have reported that mutations of the Elane gene cause congenital neutropenia and cyclic neutropenia in patients.33,34 Because we have shown that neutrophils contribute to the development of DR,41,44 it seemed possible that beneficial reduction in capillary degeneration in elastase-deficient mice might have been due merely to fewer neutrophils. To examine this possibility, we measured the neutrophil population (CD11b+Ly6G+Ly6Cinterm) in the circulating blood by flow cytometry. Compared to appropriate nondiabetic control, diabetes increased the fraction of leukocytes that expressed markers for neutrophils in both WT and Elane−/− mice, and we saw a reduction of neutrophils in the non-diabetic Elane−/- mice. Interestingly, however, this neutropenia did not develop in diabetic Elane−/− mice. The mechanism by which NE influences the number of neutrophils in the circulation remains under investigation but might involve a role of NE in mobilization of the cells from the bone marrow.61 Nevertheless, the beneficial effect of NE deletion on the inhibition of retinal capillary degeneration and other molecular changes in the retina induced by diabetes in our experiments apparently was not due to a reduction in the number of cytotoxic neutrophils in the circulation.
Genetic deletion of Elane is not likely to be adopted as a therapeutic option to inhibit DR, so we investigated whether we could reproduce the beneficial effects of NE deletion by pharmacological inhibition of NE. In 2-month studies of diabetic mice, administration of sivelestat (either systemically or locally via eye drops) or other NE inhibitors (GW311616A and alvelestat) significantly inhibited the diabetes-induced increase in retinal superoxide. In addition, the increased expression of inflammatory proteins in the retina, and leukocyte-mediated cytotoxicity to endothelial cells were also inhibited in diabetic mice treated with sivelestat, GW311616A, and alvelestat. Thus, pharmacologic inhibition of NE is a meaningful therapeutic target for future studies related to DR.
In summary, NE plays an important role in capillary degeneration in the early stage of DR, as well as in the diabetes-induced retinal oxidative stress and inflammation that contribute to the development of the retinopathy. These findings have important implications for inhibition of NE as a potential therapy to inhibit DR and possibly other degenerative processes. It seems that targeting NE in diabetes may achieve good therapeutic effects on the retina via multiple mechanisms.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank Eli Lewis from the Ben Gurion University of the Negev in Israel who provided the hAAT+ mice, Dr. Nader Sheibani from the University of Wisconsin for the mREC cells, and Chieh Allen Lee, Katie Franke, and Heather Butler for mouse breeding and maintenance. Dawn Smith (Case Western Reserve University, Cleveland, OH) and Huajun Yan (University of California Irvine, Irvine, CA) maintained the mouse retinal endothelial cells.
Supported by National Institutes of Health (NIH) Grants EY022938 and R24 EY024864 to T.S.K. and EY022938-S1 minority supplement to E.M.L. and Department of Veterans Affairs Grants BX002754 and BX003604 to T.S.K., and a core grant P30 EY011373 to Case Western Reserve University. The authors acknowledge an unrestricted grant from Research to Prevent Blindness to the Gavin Herbert Eye Institute at the University of California – Irvine.
Author contributions: E.M.L., H.L., A.S., Y.D., J.T., and J.K. performed molecular analyses and analyzed data. J.T. evaluated histopathology. E.M.L. and H.L. wrote and edited the manuscript. T.S.K. reviewed and edited the manuscript. T.S.K. is the guarantor of this work and, as such, had full access to all the data in the study.
Data and Resource Availability: The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request. Antibodies: A rabbit polyclonal antibody against iNOS (Transduction Laboratories Cat # 610328, RRID:AB_2314673), a rabbit polyclonal antibody against ICAM1 (Proteintech Group Cat # 16174-1-AP, RRID: AB_2248702) were used for Western blots.
Disclosure: E.M. Lessieur, None; H. Liu, None; A. Saadane, None; Y. Du, None; J. Tang, None; J. Kiser, None; T.S. Kern, None
References
- 1. Ogurtsova K, da Rocha Fernandes JD, Huang Y, et al.. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017; 128: 40–50. [DOI] [PubMed] [Google Scholar]
- 2. Lee R, Wong TY, Sabanayagam C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis (Lond). 2015; 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joussen AM, Poulaki V, Le ML, et al.. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004; 18: 1450–1452. [DOI] [PubMed] [Google Scholar]
- 4. Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007; 2007: 95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pavlovic D, Leteux C, Ovchinnikova T, Tsvetkov Y, Nifant'ev N, Feizi T. Chemically synthesized solid phase oligosaccharide probes for carbohydrate-binding receptors. Interactions of the E-, L- and P-selectins with sialyl-Le(x) and O-sulphated forms linked to biotin or to polyacrylamide. J Immunologic Methods. 2002; 264: 53–58. [DOI] [PubMed] [Google Scholar]
- 6. Tennenberg SD, Weller JJ. Endotoxin-induced, neutrophil-mediated endothelial cytotoxicity is enhanced by T-lymphocytes. J Surg Res. 1997; 69: 11–13. [DOI] [PubMed] [Google Scholar]
- 7. Varani J, Ginsburg I, Schuger L, et al.. Endothelial cell killing by neutrophils. Synergistic interaction of oxygen products and proteases. Am J Pathol. 1989; 135: 435–438. [PMC free article] [PubMed] [Google Scholar]
- 8. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006; 6: 541–550. [DOI] [PubMed] [Google Scholar]
- 9. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacologic Rev. 2010; 62: 726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ge Y, Weygant N, Qu D, et al.. Alternative splice variants of DCLK1 mark cancer stem cells, promote self-renewal and drug-resistance, and can be targeted to inhibit tumorigenesis in kidney cancer. Int J Cancer. 2018; 143: 1162–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang C, Shu W, Zhou G, et al.. Anti-TNF-alpha Therapy Suppresses Proinflammatory Activities of Mucosal Neutrophils in Inflammatory Bowel Disease. Mediators Inflamm. 2018; 2018: 3021863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respiratory Crit Care Med. 2001; 164: 896–904. [DOI] [PubMed] [Google Scholar]
- 13. Moraes TJ, Chow CW, Downey GP. Proteases and lung injury. Crit Care Med. 2003; 31: S189–S194. [DOI] [PubMed] [Google Scholar]
- 14. Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chronic Obstructive Pulmon Dis. 2008; 3: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shapiro SD. Proteinases in chronic obstructive pulmonary disease. Biochem Soc Trans. 2002; 30: 98–102. [DOI] [PubMed] [Google Scholar]
- 16. Devaney JM, Greene CM, Taggart CC, Carroll TP, O'Neill SJ, McElvaney NG. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Letters. 2003; 544: 129–132. [DOI] [PubMed] [Google Scholar]
- 17. Sun R, Iribarren P, Zhang N, et al.. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J Immunol. 2004; 173: 428–436. [DOI] [PubMed] [Google Scholar]
- 18. Walsh DE, Greene CM, Carroll TP, et al.. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. J Biologic Chem. 2001; 276: 35494–35499. [DOI] [PubMed] [Google Scholar]
- 19. Liu H, Lessieur EM, Saadane A, Lindstrom SI, Taylor PR, Kern TS. Neutrophil elastase contributes to the pathological vascular permeability characteristic of diabetic retinopathy. Diabetologia. 2019; 62: 2365–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li G, Veenstra AA, Talahalli RR, et al.. Marrow-derived cells regulate the development of early diabetic retinopathy and tactile allodynia in mice. Diabetes. 2012; 61: 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Veenstra A, Liu H, Lee CA, Du Y, Tang J, Kern TS. Diabetic Retinopathy: Retina-Specific Methods for Maintenance of Diabetic Rodents and Evaluation of Vascular Histopathology and Molecular Abnormalities. Curr Protoc Mouse Biol. 2015; 5: 247–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu H, Tang J, Du Y, et al.. Retinylamine Benefits Early Diabetic Retinopathy in Mice. J Biol Chem. 2015; 290: 21568–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Su X, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003; 9: 171–178. [PubMed] [Google Scholar]
- 24. Cummings BS, Schnellmann RG. Measurement of Cell Death in Mammalian Cells. Curr Protoc. 2021; 1: e210. [DOI] [PubMed] [Google Scholar]
- 25. Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007; 48: 361–367. [DOI] [PubMed] [Google Scholar]
- 26. Veenstra A, Liu H, Lee CA, Du Y, Tang J, Kern TS. Diabetic Retinopathy: Retina-Specific Methods for Maintenance of Diabetic Rodents and Evaluation of Vascular Histopathology and Molecular Abnormalities. Curr Protoc Mouse Biol. 2015; 5: 247–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mattapallil MJ, Wawrousek EF, Chan CC, et al.. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012; 53: 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tian P, Ge HY, Liu HT, et al.. Leukocytes from diabetic patients kill retinal endothelial cells: Effects of berberine. Molec Vis. 2013; 19: 2092–2105. [PMC free article] [PubMed] [Google Scholar]
- 29. Saliba A, Du YP, Liu HT, et al.. Photobiomodulation Mitigates Diabetes-Induced Retinopathy by Direct and Indirect Mechanisms: Evidence from Intervention Studies in Pigmented Mice. PLoS One. 2015; 10: e0139003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feenstra DJ, Yego EC, Mohr S. Modes of Retinal Cell Death in Diabetic Retinopathy. J Clin Exp Ophthalmol. 2013; 4: 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007; 2007: 43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011; 30: 343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dale DC, Makaryan V. ELANE-Related Neutropenia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, Eds. GeneReviews((R)), Seattle, WA: GeneReviews; 1993. [PubMed] [Google Scholar]
- 34. Makaryan V, Zeidler C, Bolyard AA, et al.. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol. 2015; 22: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watanabe F, Sato M, Kato A, Murakami T, Higashi Y, Yata N. First-pass metabolism of ONO-5046 (N-[2-[4-(2,2-dimethylpropionyloxy) phenylsulfonylamino]benzoyl]aminoacetic acid), a novel elastase inhibitor, in rats. Biolog Pharmaceut Bull. 1997; 20: 392–396. [DOI] [PubMed] [Google Scholar]
- 36. Tamakuma S, Ogawa M, Aikawa N, et al.. Relationship between neutrophil elastase and acute lung injury in humans. Pulmon Pharmacol Therap. 2004; 17: 271–279. [DOI] [PubMed] [Google Scholar]
- 37. Zeiher BG, Matsuoka S, Kawabata K, Repine JE. Neutrophil elastase and acute lung injury: prospects for sivelestat and other neutrophil elastase inhibitors as therapeutics. Critic Care Med. 2002; 30: S281–287. [DOI] [PubMed] [Google Scholar]
- 38. Zeiher BG, Artigas A, Vincent JL, et al.. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit Care Med. 2004; 32: 1695–1702. [DOI] [PubMed] [Google Scholar]
- 39. Miyoshi S, Hamada H, Ito R, et al.. Usefulness of a selective neutrophil elastase inhibitor, sivelestat, in acute lung injury patients with sepsis. Drug Design, Develop Ther. 2013; 7: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Woo SJ, Ahn SJ, Ahn J, Park KH, Lee K. Elevated systemic neutrophil count in diabetic retinopathy and diabetes: a hospital-based cross-sectional study of 30,793 Korean subjects. Invest Ophthalmol Vis Sci. 2011; 52: 7697–7703. [DOI] [PubMed] [Google Scholar]
- 41. Kim SY, Johnson MA, McLeod DS, Alexander T, Hansen BC, Lutty GA. Neutrophils are associated with capillary closure in spontaneously diabetic monkey retinas. Diabetes. 2005; 54: 1534–1542. [DOI] [PubMed] [Google Scholar]
- 42. Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001; 158: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smedly LA, Tonnesen MG, Sandhaus RA, et al.. Neutrophil-mediated injury to endothelial cells. Enhancement by endotoxin and essential role of neutrophil elastase. J Clin Invest. 1986; 77: 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Veenstra AA, Tang J, Kern TS. Antagonism of CD11b with neutrophil inhibitory factor (NIF) inhibits vascular lesions in diabetic retinopathy. PLoS One. 2013; 8: e78405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goldstein IM, Ostwald P, Roth S. Nitric oxide: a review of its role in retinal function and disease. Vis Res. 1996; 36: 2979–2994. [DOI] [PubMed] [Google Scholar]
- 46. Lisowska-Myjak B, Pachecka J, Kaczynska B, Miszkurka G, Kadziela K. Serum protease inhibitor concentrations and total antitrypsin activity in diabetic and non-diabetic children during adolescence. Acta Diabetologica. 2006; 43: 88–92. [DOI] [PubMed] [Google Scholar]
- 47. Feng YL, Yin YX, Ding J, et al.. Alpha-1-antitrypsin suppresses oxidative stress in preeclampsia by inhibiting the p38MAPK signaling pathway: An in vivo and in vitro study. PLoS One. 2017; 12: e0173711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hashemi M, Naderi M, Rashidi H, Ghavami S. Impaired activity of serum alpha-1-antitrypsin in diabetes mellitus. Diabetes Res Clin Pract. 2007; 75: 246–248. [DOI] [PubMed] [Google Scholar]
- 49. Yu X, Akbarzadeh R, Pieper M, et al.. Neutrophil Adhesion Is a Prerequisite for Antibody-Mediated Proteolytic Tissue Damage in Experimental Models of Epidermolysis Bullosa Acquisita. J Invest Dermatol. 2018; 138: 1990–1998. [DOI] [PubMed] [Google Scholar]
- 50. Preston GA, Zarella CS, Pendergraft WF 3rd, et al.. Novel effects of neutrophil-derived proteinase 3 and elastase on the vascular endothelium involve in vivo cleavage of NF-kappaB and proapoptotic changes in JNK, ERK, and p38 MAPK signaling pathways. J Am Soc Nephrol: JASN. 2002; 13: 2840–2849. [DOI] [PubMed] [Google Scholar]
- 51. Oltmanns U, Sukkar MB, Xie S, John M, Chung KF. Induction of human airway smooth muscle apoptosis by neutrophils and neutrophil elastase. Am J Respir Cell Molec Biol. 2005; 32: 334–341. [DOI] [PubMed] [Google Scholar]
- 52. Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol. 1995; 131: 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukocyte Biol. 1999; 65: 137–150. [DOI] [PubMed] [Google Scholar]
- 54. Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiol. 1998; 275: H385–H392. [DOI] [PubMed] [Google Scholar]
- 55. Jerke U, Hernandez DP, Beaudette P, Korkmaz B, Dittmar G, Kettritz R. Neutrophil serine proteases exert proteolytic activity on endothelial cells. Kidney Int. 2015; 88: 764–775. [DOI] [PubMed] [Google Scholar]
- 56. Grechowa I, Horke S, Wallrath A, Vahl CF, Dorweiler B. Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J: Official Publication of the Federation of American Societies for Experimental Biology. 2017; 31: 3868–3881. [DOI] [PubMed] [Google Scholar]
- 57. Takami S, Yamashita S, Kihara S, Kameda-Takemura K, Matsuzawa Y. High concentration of glucose induces the expression of intercellular adhesion molecule-1 in human umbilical vein endothelial cells. Atherosclerosis. 1998; 138: 35–41. [DOI] [PubMed] [Google Scholar]
- 58. Gao Y, Meng D, Sun N, et al.. Clock upregulates intercellular adhesion molecule-1 expression and promotes mononuclear cells adhesion to endothelial cells. Biochem Biophys Res Commun. 2014; 443: 586–591. [DOI] [PubMed] [Google Scholar]
- 59. Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infection. 2003; 5: 1317–1327. [DOI] [PubMed] [Google Scholar]
- 60. Shapiro SD, Campbell EJ, Senior RM, Welgus HG. Proteinases secreted by human mononuclear phagocytes. J Rheumatol Supp. 1991; 27: 95–98. [PubMed] [Google Scholar]
- 61. Pruijt JF, Verzaal P, van Os R, et al.. Neutrophils are indispensable for hematopoietic stem cell mobilization induced by interleukin-8 in mice. Proc Natl Acad Sci USA. 2002; 99: 6228–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.