

ABSTRACT
Autophagic flux is a critical cellular process that is vastly under-appreciated in terms of its importance to human health. Preclinical studies have demonstrated that reductions in autophagic flux cause cancer and exacerbate chronic diseases, including heart disease and the pathological hallmarks of dementia. Autophagic flux can be increased by targeting nutrition-related biochemical signaling. To date, translation of this knowledge has been hampered because there has been no way to directly measure autophagic flux in humans. In this study we detail a method whereby human macroautophagic/autophagic flux can be directly measured from human blood samples. We show that whole blood samples can be treated with the lysosomal inhibitor chloroquine, and peripheral blood mononuclear cells isolated from these samples could be used to measure autophagic machinery protein LC3B-II. Blocking of autophagic flux in cells while still in whole blood represents an important advance because it preserves genetic, nutritional, and signaling parameters inherent to the individual. We show this method was reproducible and defined LC3B-II as the best protein to measure autophagic flux in these cells. Finally, we show that this method is relevant to assess intra-individual variation induced by an intervention by manipulating nutrition signaling with an ex vivo treatment of whole blood that comprised leucine and insulin. Significantly, this method will enable the identification of factors that alter autophagic flux in humans, and better aid their translation in the clinic. With further research, it could also be used as a novel biomarker for risk of age-related chronic disease.
Abbreviations: AMPK: AMP-activated protein kinase; ACTB: actin beta; ATG5: autophagy related 5; BAF: bafilomycin A1; CQ: chloroquine; DMSO: dimethyl sulfoxide; DPBS: Dulbecco’s phosphate-buffered saline; EDTA: ethylenediaminetetraacetic acid; KO: knockout; MAP1LC3A/LC3A: microtubule associated protein 1 light chain 3 alpha; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MAP1LC3C/LC3C: microtubule associated protein 1 light chain 3 gamma; MTOR: mechanistic target of rapamycin kinase; NBR1: NBR1 autophagy cargo receptor; PBMCs: peripheral blood mononuclear cells; PMNs: polymorphonuclear cells; RPMI: Roswell Park Memorial Institute; SQSTM1: sequestosome 1; TBST: Tris-buffered saline containing 0.1% (v:v) Tween 20; TEM: transmission electron microscopy.
KEYWORDS: Autophagy, biomarker, blood, chloroquine, human, lc3b, lysosome, pbmc
Introduction
Autophagic flux is the acquisition, transport, and degradation of unwanted or damaged material in the lysosomal system. The efficient execution of this entire series of events is important for two aspects of physiology: the first is cellular quality control, which is critical for healthy tissue function; the second is nutrient recycling, which allows an organism to adapt to starvation [1]. Accumulating human and preclinical research also shows that inefficient autophagic flux plays a major and direct role in prevalent diseases such as dementia [2,3], cancer [4,5], and heart disease [6]. Further, lysosomal system function supports both healthy proteostasis and mitochondrial function, the dysfunction of which are hallmarks of aging [7]. Consistent with this, mice with higher levels of autophagic activity live longer [8].
As such, lysosomal system function (the degradative capacity of the lysosome coupled with efficient cargo trafficking from supply pathways including autophagy, endocytosis, and phagocytosis) is important to human health, and interventions that modify it (including nutrition, exercise, or pharmacological agents) should be translated into clinical practice. It is well known that caloric restriction increases autophagy [9], and more translatable nutritional interventions such as reduction of protein consumption may also promote this process [10]. Aerobic exercise [11,12], inhibitors of MTOR (mechanistic target of rapamycin kinase) such as rapamycin, or compounds that augment nicotinamide adenine dinucleotide-dependent protein deacetylase sirtuin-1 activity such as resveratrol, also augment autophagy [13]. In addition, metformin – a widely-prescribed diabetes medication – enhances autophagy through AMP-activated protein kinase (AMPK) activation and galectin-mediated responses to mild endomembrane damage [14]. Pharmacological inhibitors of lysosomal function or autophagic initiation are also important for human health as they are being trialed to inhibit autophagy in cancers, thereby removing important nutritional support and damage repair mechanisms from the cancer [15].
Many studies have measured lysosomal system proteins or morphological features in human samples as a proxy for autophagic flux. This does not measure the autophagic flux, which is defined as the rate of protein degradation through the entire autophagy pathway [16,17], and is an unreliable measure of lysosomal system activity [18]. The gold standard test for autophagic flux is western blot for MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) without and with inhibition of lysosomal proteolysis and analysis of its lipidated form, LC3B-II [16]. This technique is commonly applied to cells in culture.
Unfortunately, there is no method that directly measures autophagic flux in humans, and this is recognized as a barrier to translating interventions that target autophagy [19–21]. Although important in their own right, the methods that have been developed for analysis of human autophagic flux so far have only assessed autophagic flux in primary cells cultured in nutrient-rich media [22,23]. The impact of nutrient-signaling on autophagy represents an important and obvious bias in the interpretation of the results from those studies. Commonly used synthetic culture media such as Dulbecco’s modified Eagle’s medium (DMEM) or Roswell Park Memorial Institute (RPMI) 1640 are very different to physiological conditions (e.g. human plasma, human cerebrospinal fluid) that typically see lower concentrations of amino acids and glucose [24]. Measurement of autophagic flux has not yet been successfully adapted to organotypic human samples that reflect both the nutritional and endocrine status of an individual; both factors directly impact MTOR signaling and thus lysosomal system function.
Because of a lack of physiologically relevant measures of autophagic flux, we do not know what risk factors impact autophagic flux in a human population, or what important co-variates are. In the absence of such a measure, disease-specific endpoints would need to be used, and the impact of treatments that aim to increase or decrease autophagic flux will remain unclear [19]. This gap in knowledge represents an urgent unmet need that is currently hampering translation of a wealth of available scientific data on the lysosomal system.
Here, for the first time, we present quantitative measurement of autophagic flux in an organotypic human sample. We use whole blood which is cultured and treated with pharmacological lysosomal blockade for a short period of time and measure autophagic proteins in isolated peripheral blood mononuclear cells (PBMCs). This study tests different clinically relevant factors that could cause measurement variation. We describe a protocol that is easy to perform and well within the capability of most biochemical research laboratories. This test will be important for measuring the effect of lifestyle or pharmacological interventions on autophagic flux. Further, this test could provide an important biomarker for diseases of aging.
Results
Measurement of autophagic flux in primary cultures of human leukocytes
First, as proof of concept, we assessed autophagic flux in different white blood cell populations in culture. Blood from three individuals was subjected to two types of cell isolation. Whole blood was fractionated using lymphoprep to isolate either PBMCs or polymorphonuclear cells (PMNs) (Figure 1A). After isolation, each cell type sample was split into two and each cultured in media without or with 150 µM chloroquine (CQ) for one h for analysis of autophagic flux (Figure 1B). These populations were verified by flow cytometry (Figure 1C). Autophagic flux was assessed by western blotting for LC3B and SQSTM1 (sequestosome 1) (Figure 1D). As expected, LC3B-II degradation was inhibited by CQ treatment, as observed by the increase in the lipidated isoform of LC3B, LC3B-II. Autophagic flux could therefore be investigated in these two cell populations. SQSTM1 did not change after CQ treatment in PBMCs cultured in vitro, and was not detected in PMNs, making this autophagy receptor unsuitable for further investigation in these cell types (Figure 1D). Given that PBMC isolation is much faster compared to PMN isolation, we pursued our experiments in PBMCs only. Faster isolation of PBMCs compared with PMNs is important because more samples can be handled at once, and artifacts due to processing are less likely to occur.
Figure 1.

Autophagic flux can be measured in primary culture of human leukocytes in vitro. (A) Human blood was fractionated using lymphoprep, and (B) PBMCs and PMNs were cultured without and with CQ in RPMI 1640 (C). Cell populations derived from lymphoprep fractionation were determined using flow cytometry. (D) Western blots for lysosomal system proteins (LC3B and SQSTM1) across PBMCs and PMNs are shown (N = 3, 2 subjects shown in figure). Mr: molecular weight marker
LC3B-II and SQSTM1 increase in PBMCs with increasing concentration of CQ added to whole blood
Having observed that measurement of autophagic flux was possible in primary cultures of PBMCs, we next determined whether CQ could be added to whole blood before PBMC isolation. The purpose of this experiment was to determine if flux could be blocked by CQ in PBMCs while they were still in whole blood, and then measured. This is important as autophagy in these cells would be influenced by the physiological status (e.g. nutrient content, insulin level) of the blood itself. Thus, CQ was added directly to fresh whole blood across a concentration range from 0 µM through to 200 µM. This titration was performed on blood from four subjects. CQ-treated blood samples were incubated at 37°C for one h while being rotated. PBMCs were then isolated from the blood using lymphoprep (Figure 2A, B). PBMCs were processed for western blotting and samples were probed for the autophagic proteins MAP1LC3A/LC3A (microtubule associated protein 1 light chain 3 alpha), LC3B, MAP1LC3C/LC3C (microtubule associated protein 1 light chain 3 gamma), SQSTM1, and the loading control protein ACTB (actin beta) (Figure 2C, Fig. S1A-C).
Figure 2.
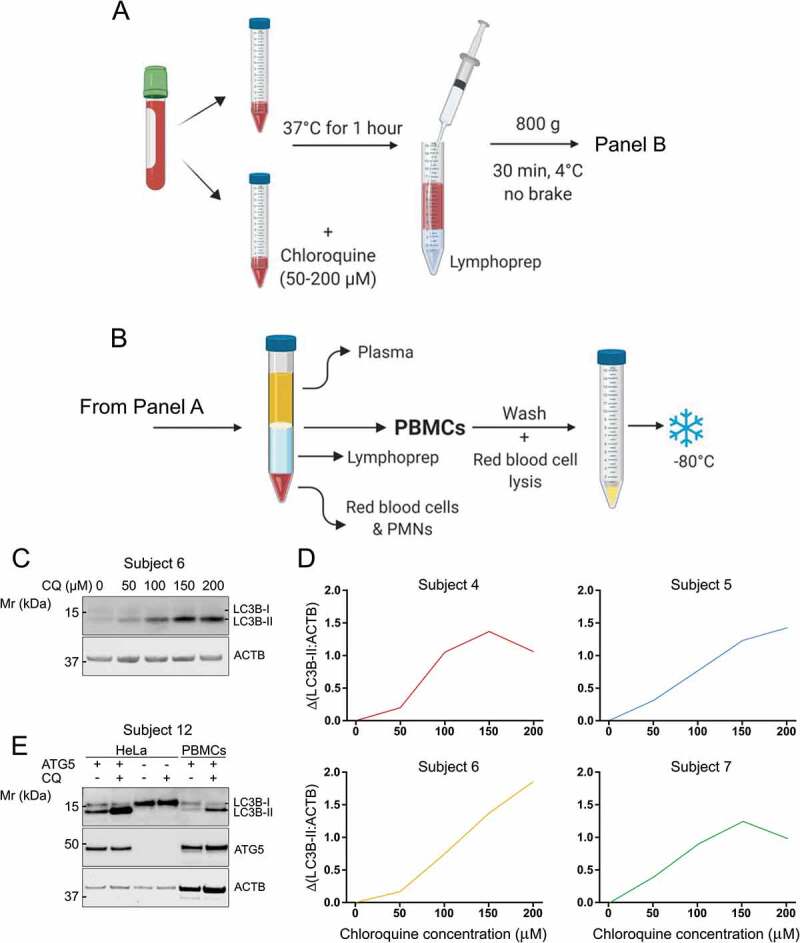
Autophagic flux in PBMCs can be measured by lysosomal inhibition in whole human blood. (A) CQ was titrated into whole blood samples at final concentrations ranging from 0–200 µM and incubated at 37°C for 1 h before (B) PBMCs were extracted for analysis of LC3B-II by western blot. (C) Western blot for LC3B from PBMCs that were exposed to CQ while in whole blood. (D) Blood from four subjects was processed according to the diagram in panels A and B and is displayed as ΔLC3B-II normalized to ACTB. (E) ATG5 KO HeLa cells that cannot lipidate LC3B-I to form LC3B-II were used to determine that the LC3B antibody used in this study was specific. Mr: molecular weight marker
Of the proteins analyzed, only LC3B-II was useful as a marker of autophagic flux in PBMCs that were exposed to CQ in whole blood (Figure 2D). Indeed, while CQ clearly blocked autophagic flux in PBMCs when added to whole blood, not all markers correlated in a linear relationship with CQ concentration. LC3B-II displayed a robust, directly proportional increase in response to increasing CQ concentration in whole blood up to 150 µM, after which a ceiling effect was observed (Figure 2D). Autophagic flux in Figure 2D is quantified as change in LC3B-II, displayed as:
Δ(LC3B-II:ACTB) = (LC3B-II:ACTB with CQ) – (LC3B-II:ACTB without CQ)
The correlation between concentration of CQ added to whole blood and Δ(LC3B-II:ACTB) was statistically significant (p = 0.0062; R2 = 0.9411; N = 4). LC3A did not clearly respond to CQ treatment and was unreliable between different subjects; LC3A-II was barely detectable in some samples (Fig. S1A); LC3C was almost undetectable by western blot in PBMCs (Fig. S1B). Interestingly, SQSTM1 did not respond to CQ as well as LC3B, although increased protein abundance with CQ treatment was observed in three out of four subjects (Fig. S1B, C). As SQSTM1 abundance can adapt to change through changes in gene expression [25], we used analysis by quantitative polymerase chain reaction to determine whether this was the case. The abundance of SQSTM1 transcript did not change during the 1 h of CQ treatment (Fig. S1D), showing that compensatory changes in expression were not likely to influence the abundance of SQSTM1. Samples were also probed for NBR1 (NBR1 autophagy cargo receptor) after being treated for 1 h with 150 µM CQ. NBR1 protein was barely detected, although a slight increase after CQ treatment may be distinguishable (Fig. S1E). Thus, we chose to treat whole blood with a final concentration of 150 µM CQ and analyze LC3B-II for the rest of the study.
In order to verify the specificity of the LC3B antibody that was used for this study, ATG5 (autophagy-related 5) knockout (KO) cells were generated by Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 genome editing. Whereas LC3B-II was clearly visible in wild-type HeLa cells, it was missing in ATG5 KO HeLa cells and, in line with a lipidation defect in the ATG5 KO cells, LC3B-I was more abundant. These bands were identical to bands detected in PBMCs confirming the specificity of this LC3B antibody for its use in human PBMCs (Figure 2E). LC3B-II and ACTB were also shown to be in the linear range of detection for western blotting with the amounts of protein and detection systems used in this study (Fig. S2).
LC3B-II increases in a linear fashion with time after incubation of CQ in whole blood
Although the method presented here assesses the number of autophagosomes only indirectly based on the abundance of LC3B-II in the PBMC lysate, it offers the possibility of measuring autophagic flux as a rate. Indeed, measuring accumulation of LC3B-II over multiple time-points after lysosomal inhibition has been described as a better measure of flux [26], and has been previously demonstrated in animal systems [27]. Therefore, individual blood samples from given subjects were split into seven tubes and incubated without or with CQ for different durations (Figure 3A) before LC3B-II was measured by western blot. Due to the nature of the method, it was unpractical to treat all the samples with CQ at the same time and to collect the samples at the different timepoints. Hence, we incubated the samples for a period of 2 h and treated them at different timepoints. One sample was processed immediately without incubation nor treatment with CQ in order to assess the effect of the two-h incubation and evaluate if the system was at steady state. The 2 h incubation did not affect LC3B-II readout when compared to the sample incubated for 2 h without CQ treatment (p = 0.3231; paired t-test; N = 4; data not shown; representative western blot displayed in Figure 3B). We observed an increase in the amount of LC3B-II after treatment of blood with CQ for every subject when plotted over time (Figure 3B–D). The slope of increase appeared different between the different subjects (Figure 3C) and may provide a more sensitive measure of autophagic activity than a single timepoint, although a 1 h timepoint still allows for determination of inter-individual variation.
Figure 3.
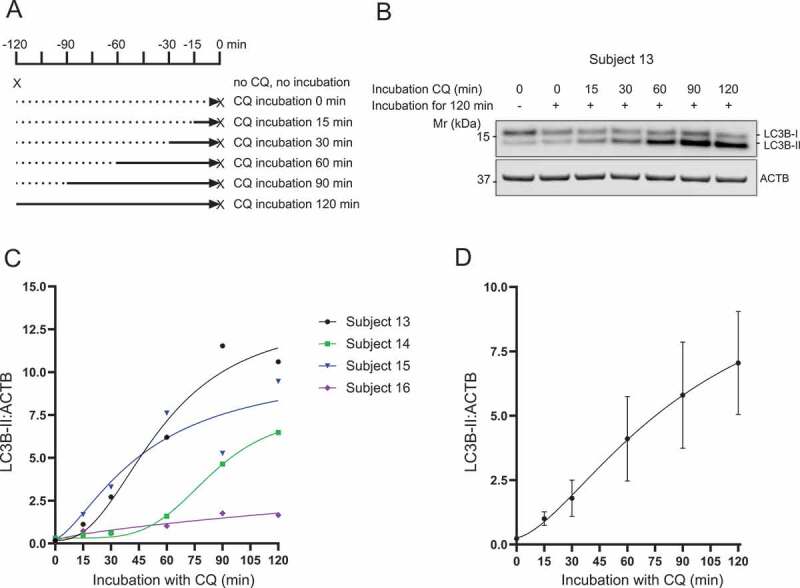
Accumulation of LC3B-II in PBMCs over time with incubation of CQ in whole blood. (A) Human blood was incubated for up to 120 min with addition of CQ (150 µM) after 0, 30, 60, 90, 105 min leading to a total incubation time with CQ of 120, 90, 60, 30 and 15 min respectively. One sample was not treated with CQ and incubated for 120 min. One sample was processed immediately to assess the effect of the 2 h incubation. The X symbol represents the time of samples processing. (B) Western blot for LC3B from PBMCs exposed to CQ for different durations while in whole blood. (C) LC3B-II normalized to ACTB is displayed for four different subjects with the best-fit non-linear sigmoidal regression. (D) LC3B-II normalized to ACTB averaged for four subjects showing the accumulation of LC3B-II with increasing incubation time with CQ. Data represent mean ± SEM (N = 4). Mr: molecular weight marker
LC3B immuno-positive area increases in lymphocytes that are isolated from whole blood treated with CQ
Fluorescent microscopy has been described as a suitable method to quantify autophagic vesicles at a single-cell level [28]. The nature of whole blood, and our intention to use it as a physiologically intact sample, does not allow the use of fluorescent probes or living-cell imaging. Therefore, we decided to assess the effect of CQ treatment on LC3B puncta in PBMCs by immunofluorescence. As PBMCs can easily be visually differentiated by microscopy, we did not proceed with the PBMC isolation step and we extracted all leukocytes. Blood samples were treated for one h without or with CQ before leukocytes were isolated (red blood cell lysis), fixed and smeared on slides for immunofluorescent staining for LC3B (Figure 4A–D). Interestingly, this method of cell isolation allows the use of a very small amount of blood (only 200 µL of blood required per condition for leukocyte isolation). Lymphocytes were analyzed for LC3B-positive staining by an experimenter blinded to the treatment groups and subjects (Figure 4E,F). Although this method did not allow for quantification of the number of LC3B puncta (due to puncta overlapping – mainly observed in CQ-treated samples as shown in Figure 4C,D), we quantitively measured LC3B-positive area per cell. As expected, we observed a statistically significant increase in LC3B-positive area after CQ treatment for the four subjects assessed (Figure 4E,F).
Figure 4.
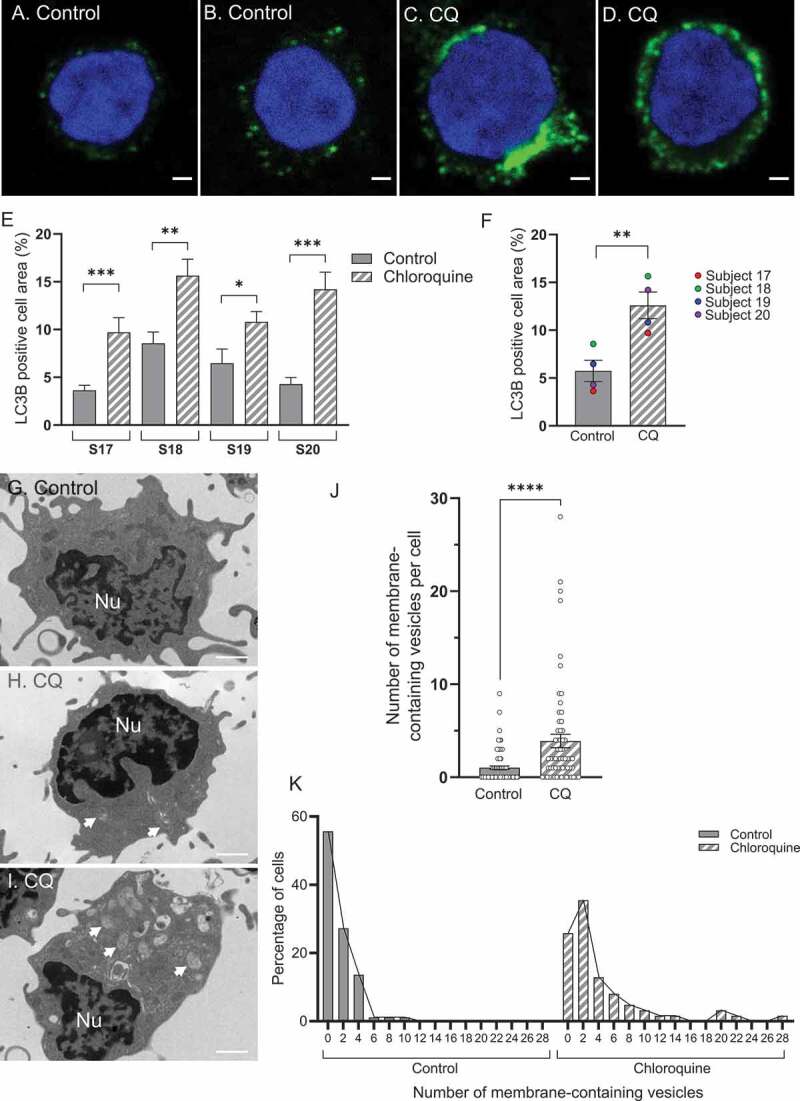
Image-based analysis shows an increase in autophagic vesicles after addition of CQ to whole blood. (A-B) Representative immunofluorescent staining for LC3B in lymphocytes from whole blood samples untreated or (C-D) treated with 150 µM CQ for one h. (E) Immunofluorescent staining quantification for LC3B is displayed for four different subjects and showed an increase in LC3B-positive cell area in CQ treated samples (p = 0.0010; Mann-Whitney; subject 17; p = 0.0072, 0.0334 and 0.0001 for subjects 18, 19 and 20 respectively; Unpaired t-test; N = 6–10 cells per condition). (F) LC3B-positive cell area increases in lymphocytes treated with CQ versus untreated controls (p = 0.0099; paired t-test; N = 4 subjects). (G) Transmission electron microscopy images of PBMCs for untreated control blood showing no vesicles (median control group = 0 vesicles), and for (H-I) CQ-treated blood samples (H: median CQ group = 2 vesicles; I: cell with high number of vesicles). Arrows pointing at membrane-containing vesicles. (J) The number of membrane-containing vesicles, quantified as autophagosomes, increased in PBMCs from CQ-treated blood samples compared to untreated samples (p < 0.0001; Mann-Whitney test; n = 88 control cells, n = 62 cells from whole blood treated with CQ pooled from 3 subjects). (K) Distribution of the number of vesicles per cell as percentage of cells for the same data presented in J. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Error bars are SEM. Scale bars = 1 µm. Nu: nucleus. S: subject
Transmission electron microscopy shows an increase in autophagosomes after addition of CQ to whole blood
In order to further validate that CQ added to whole blood was indeed leading to the accumulation of autophagic material in PBMCs, we quantitatively assessed the number of autophagosomes in PBMCs by transmission electron microscopy (TEM) (Figure 4G–I). Images of PBMCs were assessed blinded to whether or not they came from control or chloroquine treated samples. Vesicles that contained membranous material were scored as autophagosomes and were counted per cell. Vesicles that contained no material (likely pinosomes) were not counted. Cells from CQ-treated blood samples (n = 62 cells) displayed significantly more autophagosomes than cells from untreated blood samples (n = 88 cells) (p < 0.0001; Mann-Whitney test; cells analyzed from 3 subjects) (Figure 4J). The median number of autophagosomes from control cells was 0 per cell (Figure 4G), whereas the median number from CQ-treated cells was 2 per cell (Figure 4H). CQ-treated cells displayed significantly more cells with more than 5 vesicles compared to control cells (p = 0.0043; Fisher’s exact test for frequency of cells with more than 5 vesicles; n = 62 total cells for CQ including 16 cells with more than 5 vesicles, n = 88 cells for control including 3 cells with more than 5 vesicles; N = 3 subjects) (Figure 4J,K). Further, whereas CQ-treated cells sometimes displayed very high numbers of autophagosomes (above 10 per cell) (Figure 4I), control cells did not. (Figure 4J,K).
Bafilomycin A1 added to whole blood causes an increase in LC3B-II abundance in PBMCs
While we chose to use the lysosomotropic drug CQ to inhibit lysosomal degradation in our method to measure autophagic flux in whole blood, other pharmacological agents such as the v-type ATPase inhibitor bafilomycin A1 (BAF), are also commonly used. CQ and BAF are often used interchangeably although their mechanism of action differs [29]. It is worth noting, however, that autophagy inhibitors can have pleiotropic effects [29,30]. Thus, we decided to test BAF as another pharmacological inhibitor of lysosomal function. BAF titration in PBMCs in culture indicated a dose of 200 nM was sufficient to induce LC3B-II accumulation (data not shown). For addition of BAF to whole blood, ethanol was used as a vehicle as dimethyl sulfoxide (DMSO) caused significant hemolysis when added to whole blood (Fig. S3A) and impacted western blot quantitation (Fig. S3B). Addition of BAF to whole blood led to LC3B-II accumulation in PBMCs after one h of treatment, similar to the effect of CQ in whole blood (Fig. S4A, B). Further, the effect of CQ and BAF was not additive, indicating that CQ does not induce autophagy in this system. The effect of CQ on accumulation of LC3B-II appeared lower compared to BAF, although this difference was not statistically significant (p = 0.1918; repeated measure one-way ANOVA; N = 4) (Fig. S4A). It is important to note here that this experiment was conducted using ethanol as a vehicle, which may affect autophagic flux or cell integrity.
Determining the reproducibility of autophagic flux measurements in blood
As autophagic flux is a dynamic process that responds to different kinds of cell stress, we investigated the reproducibility of this autophagic flux measurement in human blood. We therefore designed three experiments to test three different sources of variation – variation from the measurement system, variation from the biological system, and variation caused by the experimenter conducting the measurement.
To test variation from the measurement system, we collected two vials of blood each from four subjects on the same occasion (Figure 5A). The paired blood samples were processed (incubation for one h with 150 µM of CQ followed by PBMC isolation; Figure 2A,B) in parallel and autophagic flux was measured. Paired samples from the same subject were then compared to determine the repeatability of the flux measurement (Figure 5B,C). Paired samples from the same subject processed in parallel on the same day gave similar autophagic flux values for both samples (replicate effect p = 0.2847; paired t-test between replicates; N = 4).
Figure 5.
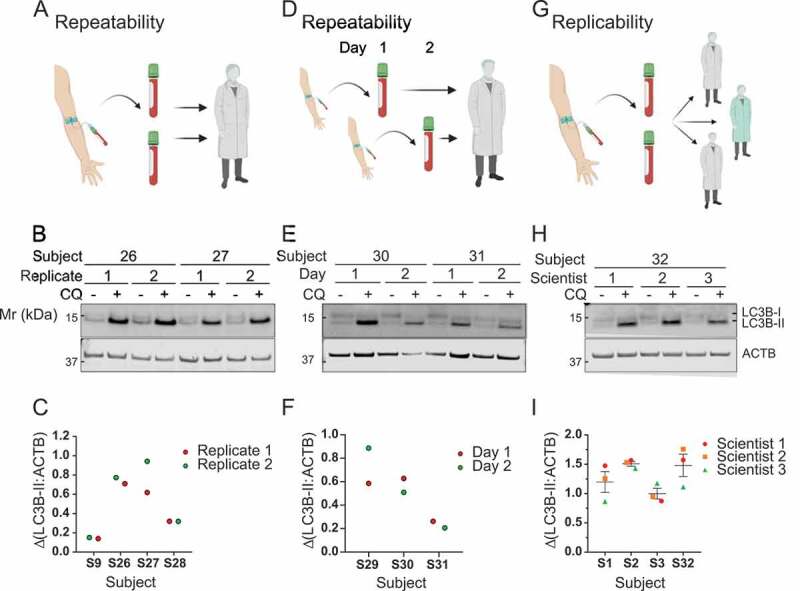
Measurement of autophagic flux in whole blood is repeatable and replicable. (A) To determine if measurement of autophagic flux in PBMCs were repeatable when processed in parallel, two vials of blood from the same subject were given to one scientist to process. (B) Western blot analysis of LC3B (2 replicates ± CQ for 2 subjects displayed). (C) ΔLC3B-II normalized to ACTB for the two replicates from four subjects is shown. (D) To determine stability of autophagic flux measurement blood was collected from the same fasted subject on two consecutive days. (E) Samples were analyzed for LC3B by western blot (2 samples collected on consecutive days ± CQ for 2 subjects displayed). (F) ΔLC3B-II normalized to ACTB is shown for two consecutive days for three subjects. (G) To determine replicability of autophagic flux measurement, two vials of whole blood were taken from subjects, pooled, and split between three different scientists to independently process. (H) LC3B was analyzed by western blot (triplicates ± CQ performed by 3 different scientists for one subject displayed). (I) ΔLC3B-II normalized to ACTB is shown, as determined by three different scientists working in parallel on blood from four subjects. Error bars are SEM. S: subject. Mr: molecular weight marker
Variation from the biological system was measured by taking blood from the same subject (N = 3) on two consecutive days under the same conditions (Figure 5D). Blood samples were processed under the same conditions each day and autophagic flux was measured. Paired samples taken from the same subject on two consecutive days were compared (Figure 5E,F). We observed good repeatability for autophagic flux measurements (day effect p = 0.7786; paired t-test between days; N = 3).
To test variation caused by the experimenter (replicability), blood was collected from four subjects on the same day and provided to three scientists for independent processing and measurement of autophagic flux (Figure 5G). The results were similar between experimenters for each subject (scientist effect p = 0.6528; Friedman test between scientists; N = 4) (Figure 5H,I).
As expected, these three tests of repeatability and replicability showed intra-individual variability (between replicates) was lower than inter-individual variability (between subjects), allowing further experiments to evaluate factors that impact individual autophagic activity.
Autophagic flux is affected by short-term storage of blood in the laboratory
To determine the impact of short-term processing delays on autophagic flux measurement, we took two vials of whole blood each from three subjects. One vial was processed immediately after collection (delay < 30 min) and the other vial was kept at room temperature for four h. This experiment was also conducted on blood collected from three additional subjects to investigate the impact of four h of storage of whole blood on ice. Blood was processed as described above: 1 h incubation at 37°C with or without CQ (150 µM) followed by PBMC isolation. LC3B was analyzed by western blot.
We observed different responses to storage of whole blood samples prior to CQ treatment for different individuals. In blood that had been stored at room temperature (Figure 6A,B) and on ice for four h (Figure 6C,D), divergence from paired samples was larger than variation observed between replicates in Figure 5.
Figure 6.

Measurement of autophagic flux in different storage and collection conditions. (A) Whole blood was taken from subjects and stored at room temperature for four h and then processed for measurement of autophagic flux by western blot for LC3B (2 subjects displayed). (B) ΔLC3B-II normalized to ACTB was determined for blood taken from three subjects and processed for autophagic flux after storage at room temperature (RT) for 0 h or 4 h. (C) Whole blood was taken from subjects and stored on ice for four h and then processed for measurement of autophagic flux by western blot for LC3B (2 subjects displayed). (D) ΔLC3B-II normalized to ACTB was determined for blood taken from three subjects and processed for autophagic flux after storage on ice for 0 h or 4 h. (E) Whole blood was collected from subjects in different tubes – lithium heparin- (Li-Hep) or EDTA-containing tubes were tested. Blood was then processed for analysis of autophagic flux by western blot for LC3B (2 subjects displayed). (F). ΔLC3B-II normalized to ACTB was determined for blood taken from three subjects using lithium heparin- (Li-Hep) or EDTA-containing tubes. S: subject. Mr: molecular weight marker
This experiment showed it is better to process blood as soon as possible after collection as autophagic activity does change over a period of hours, and it does so somewhat unpredictably. Maintaining blood on ice does not reliably preserve the flux. In any case, it is critical to process blood consistently and within the shortest amount of time as possible.
Autophagic flux measurement is differentially affected by blood collection tubes containing lithium heparin or EDTA
As blood can be collected in tubes containing different anti-coagulation factors, it was important to determine whether they impacted autophagic flux measurement. To test this, we collected blood from three subjects in tubes either containing EDTA (ethylenediaminetetraacetic acid) or lithium heparin as anti-coagulation reagents. Whole blood was treated with or without 150 µM CQ and incubated for one h at 37°C before isolation of the PBMCs. LC3B-II was analyzed by western blot. We found autophagic flux ∆(LC3B-II:ACTB) was greater in samples collected in tubes with lithium heparin compared with EDTA. LC3B-II signal was globally lower and ACTB signal was higher in EDTA tubes, suggesting that EDTA may either reduce autophagy or LC3B-II readout, or increase ACTB readout (Figure 6E,F). Conversely, lithium heparin may also increase autophagic flux or LC3B-II readout, or reduce ACTB readout. Interestingly, although displaying large differences between tube types, the flux appeared to follow a similar profile between the three subjects (Figure 6F). Based on these data, lithium heparin tubes were used to complete experiments because autophagic flux was more reliably detected in blood taken using these tubes. However, this result showed EDTA tubes can also be used to measure autophagic flux. Although both types of collection tubes allow measurement of autophagic flux, it is critical to process blood consistently and to only compare results obtained from the same type of blood collection tube.
Autophagic flux measurement in blood allows detection of changes induced by nutrient signaling
After determining that autophagic flux in PBMCs can be reliably measured by lysosomal inhibition in whole blood, we determined whether we could measure a predicted outcome with an autophagy-relevant intervention. Autophagy is highly responsive to nutrition and subject to regulation via the MTOR pathway. In the presence of amino acids, particularly leucine, and insulin, MTORC1 (mechanistic target of rapamycin kinase complex 1) is activated, which in turn inhibits autophagy [31,32]. Thus, we pre-treated blood ex vivo with L-leucine and insulin.
Blood was collected from 10 subjects and cultured with or without a combination of L-leucine (200 µM) and insulin (400 nM) for three h at 37°C before addition of CQ for one h. PBMCs were then isolated as described above and LC3B-II was analyzed by western blot (Figure 7). As expected, we observed a significant decrease in LC3B-II accumulation with CQ after the samples had been treated with leucine and insulin (p = 0.0455 vehicle + CQ- treated samples vs [leucine + insulin] + CQ-treated samples; repeated measure two-way ANOVA followed by Fisher’s LSD test; N = 10) (Figure 7C,D). ∆LC3B-II between non-treated (vehicle) and [leucine + insulin]-treated samples was significantly decreased (p = 0.0280, paired t-test; N = 10) indicating a decrease in autophagic flux (Figure 7E). However, two blood samples out of the 10 did not respond to this treatment, with flux slightly increasing. Importantly, leucine and insulin did not change LC3B-II expression level in the control (no CQ) condition (Figure 7C) emphasizing the importance of measuring the autophagic flux. The result confirms this method of autophagic flux measurement can detect differences or changes in autophagy in human blood.
Figure 7.

Addition of leucine and insulin to whole blood reduces autophagic flux. (A-B) Whole blood was taken and incubated without or with a mixture of leucine and insulin for three h. Whole blood was then processed for measurement of autophagic flux. (C) LC3B-II normalized to ACTB was determined for blood taken from N = 10 subjects. CQ increased LC3B-II in both vehicle and [leucine + insulin] treatments (p < 0.0001 control – vehicle vs CQ – vehicle, p < 0001 control – [leucine + insulin] vs CQ – [leucine + insulin]; p = 0.0455 CQ-vehicle vs CQ – [leucine + insulin]; repeated measures two-way ANOVA followed by Fisher’s LSD test; N = 10). Error bars are SEM. (D) Western blots showing analysis of LC3B in blood cultured without or with leucine and insulin and then treated without or with CQ (2 subjects displayed). (E) ΔLC3B-II = (LC3B-II:ACTB with CQ) – (LC3B-II:ACTB without CQ) was determined for blood taken from N = 10 subjects. The effect of leucine and insulin on the autophagic flux was statistically significant (p = 0.0280; paired t-test; N = 10). * p < 0.05, **** p < 0.0001. Mr: molecular weight marker
Discussion
Here we present an optimized protocol to measure autophagic flux in organotypic human blood samples. Importantly, this method reports autophagic flux measurement in a system with endocrine signaling, nutritional status, and genetic make-up inherent to the individual. Using human blood, we demonstrated that the amount of LC3B-II in PBMCs increases in a manner that is directly proportional to the amount of CQ added to whole blood up to 150 µM. At a concentration of 150 µM, CQ also causes a linear increase in LC3B-II in PBMCs over time up to 120 min. We further confirmed that CQ added to whole blood blocks lysosomal function using immunocytochemical staining for LC3B, which accumulated upon treatment. TEM analysis of autophagic material in PBMCs was previously used to determine the biological activity of hydroxychloroquine on the lysosomal system in humans (but not to determine autophagic flux) [33]. We used this technique to provide additional confirmation that CQ inhibits lysosomal activity when incubated in whole blood samples. We further showed that the method displays low variation and responds in a predictable way to an ex vivo treatment (leucine + insulin). Thus, the method is suitable for measurement of autophagic flux in humans in response to experimental interventions, or for determining the impact of important parameters such as age, body composition, or disease status on autophagic flux.
Several other studies have attempted to create a similar measure, but the technique presented here differs in critical ways. Firstly, whereas others have added lysosomal inhibitors to PBMCs isolated from human blood and grown in artificial media that is nutrient-rich (primary cultures) [22,34], we expose PBMCs to a lysosomal inhibitor (CQ) while still in whole blood. This is important as an individual’s physiological characteristics that impact autophagy (such as blood glucose, circulating amino acids, and insulin) remain intact, important for faithful measurement of individual autophagic flux. Furthermore, one study attempted to inhibit autophagic flux in primary culture of PBMCs isolated from human blood using leupeptin as a lysosomal inhibitor in vitro but failed to demonstrate autophagic flux measurement by western blot [34]. Whereas CQ can diffuse across lipid membranes, leupeptin cannot [35]. It instead depends on fluid phase uptake for delivery to the lysosome, significantly increasing the time needed for this compound to inhibit lysosomal proteolysis.
The duration of incubation of whole blood with a lysosomal inhibitor is also a factor that we considered when designing this method. Whereas other research that used primary culture to assess flux chose long incubation times (such as 24 h with CQ [22]), we chose only a 1 h incubation time. This is because lysosomal inhibitor drugs can inhibit MTOR activity and cause nuclear localization of the transcription factor TFEB (transcription factor EB) [36,37], which is discussed in more detail below. However, an incubation period of just one h with CQ is too short for nuclear localization of TFEB and should minimize unwanted changes in autophagy through altered gene expression [37].
Other research groups have developed fluorescent reporters of autophagic flux that do not depend on the use of lysosomal inhibitors such as CQ or leupeptin. The use of fluorescent probes relies on differential sensitivity to pH between EGFP, which is quenched by the low pH in the autolysosome lumen, and a red fluorescent protein, which are typically pH insensitive at a similar pH range [16]. This approach is advantageous as lysosomal inhibitors can change lysosomal system physiology as discussed above. Fluorescent probes for the measurement of flux have seen continual improvement since the introduction of EGFP-LC3B, that could be used to image autophagosomes. Ratiometric probes, such as RFP-EGFP-LC3B [38] have made it possible to estimate flux in cells [39] and even in tissues [40,41]. Subsequent improvements to ratiometric fluorescent probes have further improved on this concept [42]. However, their use relies on transgenesis that would negate the use of whole human blood, which was the aim of this study. To this end, measurement of flux in PBMCs that experience the unique nutritional and signaling environment of an individual’s blood is only possible using pharmacological inhibition of the lysosome with compounds such as chloroquine. In saying this, use of fluorescent probes in cells such as PBMCs taken from an individual and maintained in vitro could yield insights on the impact of a person’s age or genetic background on autophagy.
Our assay assesses the number of autophagosomes indirectly based on the presence of LC3B-II in the lysate. We showed that this approach can also be used to capture the kinetic dynamics of the autophagic process over time. Although measuring the rate of LC3B-II accumulation over time after CQ treatment offers a better understanding of the system, it requires more resources (blood volume, incubation time, processing time) and is therefore less adapted to large-scale clinical studies. Measurement of the rate of autophagic flux has been previously demonstrated at a single-cell level by fluorescence live-cell imaging using EGFP-LC3 and lysotracker dye (which reveals the acidic compartment). This approach was developed to assess autophagosome and autolysosome pool size, autophagosome flux, and the transition time required to turn over the intracellular autophagosome pool [26,28]. Although this is a very precise technique for measuring autophagic flux, single-cell imaging of PBMCs in blood would not be possible without cell isolation and extensive manipulation, which would remove the cell from its physiological context and alter its autophagy. In our study, we assessed changes in LC3B-positive vesicles by immunofluorescence and demonstrated a significant increase after CQ treatment, similar to the accumulation of autophagosomes observed when du Toit and colleagues inhibited lysosomal function with BAF [28].
Our investigation of variability in autophagic flux in human blood showed that intra-individual variation remained low whereas inter-individual or ex vivo treatment-induced variations were detectable. This means that adding CQ to whole blood can be used to measure inter-individual or inter-treatment differences. In vivo, we expect variation between individuals to be caused by both genetic and environmental factors. This could explain why two subjects out of 10 did not respond to the ex vivo treatment with leucine and insulin. Those physiological inter-individual differences in autophagy activity are also reflected in Figure 3C where we assessed LC3B-II accumulation over time after CQ-treatment. Participants in this study were fasted before blood was taken, which will produce a measurement with less day-to-day variation (as measured in Figure 5D–F). Fasting likely removed nutritional status as a factor that influenced this inter-individual variation. The wide range of physiological autophagic flux observed in Figure 3C will require further research and will be important to future study design. This future research will need to determine major influencing genetic and environmental factors for autophagic flux.
Human variation in autophagy genes has been investigated. Overall lysosomal system performance, measured by autophagic flux, is the product of hundreds of different genes [43]. These genes display significant heterogeneity [43], which also shows association with Alzheimer and Parkinson diseases [3]. Environmental factors will also alter flux. Factors such as age, adiposity, exercise, treatment with drugs such as metformin, and circulating hormones (such as insulin and sex hormones) are believed to impact autophagy and should also induce variation in human autophagy [11–13,44-47]. This was directly demonstrated by adding leucine and insulin to whole blood, and the observation that this decreased autophagic flux (Figure 7). This occurs because amino acids, particularly leucine, work with insulin to activate MTORC1, and this will inhibit autophagy [31,32]. Conversely, inhibition of MTOR in whole blood may reveal an individual’s autophagic responsivity. Studies that reveal which factors influence human autophagy will provide exciting new insight into the development of age-related disease.
The most widely studied autophagy-regulating system centers around MTOR in the context of MTOR complex 1 (MTORC1). Nutrients (through the RAGULATOR complex) and insulin and growth factors (through AKT1 [AKT serine/threonine kinase 1]-TSC1 [TSC complex subunit 1]; TSC2 [TSC complex subunit 2]-RHEB [Ras homolog, mTORC1 binding] signaling) activate MTOR [48] to suppress autophagy through two known mechanisms. MTOR phosphorylates and suppresses ULK1 (unc-51 Like autophagy activating kinase 1), which inhibits AMPK-mediated activation of ULK1 and the initiation of autophagy [32]. MTOR also phosphorylates and inhibits MITF (microphthalmia-associated transcription factor)-family transcription factors such as TFEB, which promote transcription of lysosomal system genes through binding to the Coordinated Lysosomal Expression and Regulation (CLEAR) element in promoters of genes such as LAMP1 (lysosomal associated membrane protein 1) [49]. In this way, nutrition, through MTOR, controls autophagy. This chain of events likely accounts for the reduction in autophagy we observed when adding insulin and leucine to whole blood (Figure 7). MTOR independent mechanisms of autophagy regulation also exist. For example, the autophagic system is regulated by acetylation of multiple proteins, such as ATG5, ATG7 (autophagy related 7), and ATG12 (autophagy related 12) [50]. This mode of control is effected by acetyltransferases and acetylases that respond to various pharmacological compounds such as spermidine and resveratrol.
It is not just nutritional status that could alter the autophagic flux that is measured with this assay. Autophagy plays many different roles in the cell, including in cell death. Autophagic machinery is required for autophagic cell death, which plays important roles in development [51] and pathology [52], and also in a distinct form of cell death called autosis which is stimulated by autophagy inducing peptides, nutrient restriction, and hypoxia [53]. We also expect the presence of neoplasms to dramatically impact autophagy measurement [54]. Numerous examples show cancerous cells rely upon, or increase lysosomal system function to increase access to nutrients [55,56]. Relevant to the test outlined in this study, leukemias have a complex relationship with autophagy, where autophagy appears to confer resistance to cell death induced by therapeutic agents [57,58]. It is highly likely that the presence of a blood cancer would alter the measurement of flux performed using this assay. However, the assay presented here could also be adapted to faithfully measure flux in neoplastic blood cells.
The method presented here has some limitations. Although it allows measurement of autophagic flux in organotypic human samples, it must be performed immediately on fresh blood, as storage at room temperature or on ice for four h led to increased variation in the abundance of LC3B-II in both basal and CQ-treated samples (Figure 6). Consistent with this observation, blood cultures are often used for detection and identification of microorganisms circulating in the bloodstream of patients. It is recommended that these specimens are not refrigerated or frozen, and that they should be held at room temperature for no more than a few hours [59]. The nutritional status of the subjects is a very important factor to consider, given the responsiveness of PBMCs to their nutritional environment, highlighted by the change in autophagy activity after treatment of blood with leucine and insulin (Figure 7). We would recommend using blood samples from fasted subjects as this will reduce variation associated with the subject’s diet. PMBCs have cell-type specific limitations as well. Viral infection [60] and inflammatory cues [61] in PMBCs (or derivative cell types) are known to alter autophagy, suggesting that at least a recent history of infection may be important data for any study assessing human autophagy using PMBCs. These critical technical aspects (time to process the samples, storage, nutritional and perhaps infection status of the subjects) should be considered carefully when implementing this approach at higher throughput scale.
Although we demonstrated autophagy can be suppressed using leucine and insulin, we were not able to test a more conventional autophagy inducer or MTOR inhibitor such as rapamycin. The reason for this is that those drugs require DMSO as a vehicle, which induces hemolysis, platelet aggregation, and reduces PBMC viability [62,63]. Furthermore, DMSO has previously been shown to activate autophagy [64]. We tested the addition of DMSO in whole blood samples and followed the PBMC isolation protocol described in this paper and observed significant dose-dependent hemolysis (Fig. S3A), and a detrimental impact on quantitation by western blot (Fig. S3B). Ethanol is another vehicle used as a solvent for drugs. However, it has been shown that cell exposure to a very low level of ethanol (0.4%) increases autophagy marker expression and autophagic flux [65]. This is why we decided to use water-soluble compounds (chloroquine, leucine and insulin) in our study and, although we showed that BAF could technically be used, why chloroquine represents a better choice as an autophagy inhibitor.
Anti-coagulant agents used in blood collection also influence autophagy. That is true for both lithium [66] (although millimolar concentrations are required for this effect) and EDTA [67,68], both of which were tested in this study. This means that the type of blood collection tube used for a study must be consistently used across groups so as not to introduce experimental artifacts. Furthermore, analysis by western blot is semi-quantitative and, in general, is both too variable and low-throughput to scale up to very large cohorts. Adapting this method for a quantitative analysis such as using ELISA to detect LC3B-II will be important for large studies. Finally, whether autophagic flux in PBMCs correlates well with flux in other tissues also requires investigation under a variety of conditions, therefore caution must be used when generalizing autophagic flux measurements from human blood cells to other human tissues. However, the method presented in this article is important because PBMCs can be sampled easily from living humans, and flux can be measured using a lysosomal inhibitor in their organotypic state, unlike any other tissue.
In conclusion, we show that adding CQ to whole blood permits measurement of autophagic flux in PBMCs while still experiencing physiologically relevant cues that are faithful to the environmental factors experienced by that individual. This test will be useful to measure the effect of important factors that are likely to impact autophagic flux such as age, obesity, and diseases such as diabetes and Alzheimer disease. Autophagic flux measurement in human blood could be used as a new biomarker to help to detect the presence and monitor progression of autophagy-related disease, develop and monitor interventions that have the potential to alter the course of a disease, or identify individuals that are at risk of disease. This is the first demonstration of autophagic flux measurement in a human biological sample where the cells remain in their physiological environment, and development of this method will permit use of autophagic flux as an endpoint in clinical trials in and of itself.
Materials and methods
Human ethics approval
The use of human samples in this study was approved by the Royal Adelaide Hospital Human Ethics Committee: HREC/18/CALHN/171; CALHN Ref. number: R20180319.
Blood collection
Blood was collected in lithium heparin (Vacuette tube 9 mL lithium heparin; Greiner Bio-One, 455,084), or EDTA (Vacuette tube 9 mL K3EDTA; Greiner Bio-One, 455,036) blood collection tubes from adult subjects who were fasted for a minimum of 12 h. Lithium heparin tubes were used for all experiments and EDTA tubes were used for comparison between blood collection tubes experiment (Figure 6E,F). A total of 45 subjects provided blood in this study up to three times from May 2019 to August 2020. Samples were de-identified and referenced as ‘subjects 1–45ʹ in this manuscript.
PBMC and PMN isolation for primary culture
After blood collection, PBMCs and PMNs were isolated following standard procedures. To each blood sample, 8 mL of Dulbecco’s phosphate buffered saline (DPBS, pH 7.0–7.3; GIBCO, Thermo Fisher Scientific, 14,190,136) was added to 8 mL of blood (1:1) in a 50 mL conical centrifuge tube and mixed by gently inverting 3–4 times. Twelve mL of lymphoprep (Stemcell Technologies, 07811) was carefully underlaid beneath the blood-DPBS mixture using a 20 mL syringe and a sterile canula and centrifuged for 30 min at 800 x g at room temperature, with brake off.
PBMCs (white layer at the interface of plasma [upper phase] and lymphoprep [translucent phase]) were carefully aspirated and dispensed in a 50 mL conical centrifuge tube (approximatively 4 mL). PMNs (red bottom phase containing PMNs and erythrocytes) were collected and dispensed in a 50 mL conical centrifuge tube (4 mL). PBMCs and PMNs were then diluted with DPBS to a final volume of 20 mL and mixed gently by inverting 4–5 times, then pelleted by centrifugation at 600 x g for 10 min (with brake). The supernatant was discarded.
PBMCs were resuspended with 2 mL of red blood cell lysis buffer (1X, BD Biosciences; 555,899) and mixed by gentle pipetting and incubated for 2 min, after which they were pelleted by centrifugation at 600 x g for 5 min.
The phase containing the PMNs required three red blood cell lysis steps to eliminate all erythrocyte contamination. PMNs were resuspended with 10 mL of red blood cell lysis buffer and mixed by gentle pipetting, incubated for 15 min, after which they were pelleted by centrifugation at 600 x g for 5 min at 4°C. The supernatant was discarded and PMNs were washed by resuspension in 20 mL of DPBS. PMNs were pelleted again by centrifugation at 600 x g for 5 min. The supernatant was discarded. PMNs were then washed twice in 20 mL DPBS and resuspended in 2 mL of red blood cell lysis buffer, then mixed by gentle pipetting and incubated for 2 min, after which they were pelleted by centrifugation at 600 x g for 5 min. This last red blood cell lysis step was repeated a second time.
After red blood cell lysis, PBMCs and PMNs were washed twice in 20 mL DPBS and resuspended in 2.5 mL RPMI 1640 medium (Life Technologies, R8758) containing 10% fetal bovine serum (Life Technologies, 10,099–141) and 1% L-glutamine (Sigma Aldrich, 59,202 c); 0.5 mL of cell suspension was retained for cytometry analysis. Each sample was then split into two wells (1 mL per well in a six-well plate) and each well was topped up with RPMI 1640 to a final volume of 3 mL. After one h of incubation in a humidified incubator at 37°C, 5% CO2 for each cell population, one well was treated with 150 µM CQ (chloroquine diphosphate; Sigma Aldrich, C6628) (3 µL per mL of media of 50 mM CQ solution diluted in sterile water) and the other with sterile water (3 µL per mL of media). Plates were incubated for one h in a humidified incubator at 37°C, 5% CO2. After incubation, cells were harvested and centrifuged at 600 x g for 5 min at 4°C. The pellets were washed by resuspension in 5 mL of cold DPBS. Cells were pelleted again by centrifugation at 600 x g for 5 min at 4°C and resuspended in 1 mL of cold DPBS and transferred to a 1.5 mL microcentrifuge tube. This tube was centrifuged at 2,000 x g for 10 min at 4°C. The supernatant was discarded and the pellet containing PBMCs or PMNs was snap frozen on dry ice and stored at −80°C for biochemical analysis.
Flow cytometry
Whole leukocytes, PBMCs and PMNs (N = 3 of each) were analyzed by flow cytometry on the BD LSR Fortessa X20 Analyzer (BD Bioscience, USA) to confirm purity. Single cells were gated on FSC-H v FSC-A. Lymphocyte, monocyte and granulocyte populations were plotted based upon their size and granularity (FSC-A and SSC-A) using BD FACSDiva software version 8.0 (BD Bioscience, USA).
Whole-blood incubation with chloroquine
Autophagic flux measurement at 1 h
After collection, blood samples were split into two (3 mL each) and pipetted into 10 mL conical centrifuge tubes. One tube was treated with 150 µM final concentration of CQ (3 µL per mL of 50 mM CQ solution diluted in sterile water). Blood was mixed gently by inverting both tubes 3–4 times. Both tubes (±CQ) were incubated at 37°C for one h with rotation (10 revolutions per min; Thermo Fisher Scientific Tube Revolver, 88,881,002).
Chloroquine titration
After collection, blood samples were split into five (3 mL each) and pipetted into 10 mL conical centrifuge tubes. Samples were treated as described above with CQ at concentrations ranging from 0 to 200 µM.
Assessment of LC3B-II accumulation with CQ in whole blood over time
After collection, blood samples were split into seven (3 mL each) and pipetted into 10 mL conical centrifuge tubes. One sample was immediately processed for PBMC isolation without incubation. Other samples were incubated at 37°C for two h with rotation and treated with 150 µM final concentration of CQ after 0, 30, 60, 90, 105 min leading to total incubation time with CQ of 120, 90, 60, 30, 15 min respectively (Figure 3A). One sample was incubated for 2 h at 37°C but not treated with CQ (referred to as 0 min with CQ).
Treatment with chloroquine and bafilomycin A1
After collection, blood samples were split into four (3 mL each) and pipetted into 10 mL conical centrifuge tubes. One tube was treated with ethanol (2 µL per mL of blood + 3 µL per mL of sterile water), one tube with ethanol (2 µL per mL of blood) and 150 µM final concentration of CQ (3 µL per mL of 50 mM CQ solution diluted in sterile water), one tube with BAF 200 nM (bafilomycin A1 from Streptomyces griseus; Sigma, B1793-10UG; 2 µL per mL of 100 µM BAF solution diluted in ethanol + 3 µL per mL of sterile water), and one tube treated with a combination of CQ 150 µM and BAF 200 nM. The four tubes (± BAF, ± CQ) were incubated at 37°C for one h with rotation.
Ex vivo nutritional intervention
After collection, blood samples were split into four wells (4 x 3.5 mL) in a six-well culture plate. Two wells were treated with 200 µM final concentration L-leucine (Sigma-Aldrich, L8912) (2 µL per mL of blood of 100 mM leucine) and 400 nM of insulin (Sigma-Aldrich, I6634-50 MG) (4 µL per mL of blood of 100 µM insulin solution). The two other wells were treated with sterile water (6 µL per mL of blood). Plates were incubated for three h in a humidified incubator at 37°C, 5% CO2. After incubation, [leucine + insulin]-treated samples (2 x 3.5 mL) were pipetted into one 10 mL conical centrifuge tube and vehicle-treated samples (2 x 3.5 mL) into another 10 mL conical centrifuge tube. Samples were then split into two (2 x 3 mL) for each treatment (vehicle or leucine + insulin). One sample for each treatment was treated with CQ and the other was not, as described above, followed by incubation for one h at 37°C with rotation.
PBMC isolation from treated blood samples
After incubation with the different treatments described above, blood samples were maintained cold on ice or at 4°C during centrifugation, and cold buffers were used to stop or prevent vesicle trafficking during further processing.
PBMCs were isolated after the 1 h incubation using the following procedure. To each sample tube containing blood incubated without or with CQ, 3 mL of cold DPBS was added to 3 mL of blood (1:1) and samples were mixed by gently inverting tubes 3–4 times. Four mL of lymphoprep was carefully underlaid beneath the blood-DPBS mixture using a 10 mL syringe and a canula (sterile), and tubes were centrifuged for 30 min at 800 x g, with brake off, at 4°C.
PBMCs (white layer) were carefully aspirated and dispensed in a 10 mL conical centrifuge tube (2 mL). PBMCs were then diluted with cold DPBS to a final volume of 5 mL. PBMCs and DPBS were mixed gently by inverting tubes 3–4 times and pelleted by centrifugation at 600 x g for 10 min at 4°C (with brake). The supernatant was discarded.
PBMCs were resuspended with 1 mL of cold red blood cell lysis buffer and mixed by gentle pipetting, incubated on ice for 2 min, then pelleted by centrifugation at 600 x g for 5 min at 4°C. PBMC pellets were washed by resuspension in 5 mL of cold DPBS and pelleted again by centrifugation. The pelleted cells were resuspended in 1 mL of cold DPBS and transferred to a 1.5 mL microcentrifuge tube. This tube was centrifuged at 2,000 x g for 10 min at 4°C. The supernatant was discarded, and the pellet containing PBMCs was snap frozen on dry ice and stored at −80°C until analysis.
Generation of ATG5 KO cells
HeLa cells were co-transfected with plasmids that expressed sgATG5-Cas9 (LentiCRISPRv2-ATG5; Addgene, 99,573 [69]; deposited by Edward Campbell) and GFP (pUltrahot-GFP, modified from pUltrahot; Addgene, 24,130; deposited by Malcolm Moore) using Lipofectamine-2000 (Thermo Fisher Scientific, 11,668,027). After two days, a single GFP-positive cell was plated per well in a 96-well plate using florescence-activated cell sorting (FACS). Monoclonal lines were amplified and screened for ATG5 KO using rabbit anti-ATG5 (1:1,000, D5F5U; Cell Signaling Technologies, 12,994), and functional inhibition of autophagy by western blot using rabbit anti-MAP1LC3B (1:1,000; Novus Biologicals, NB100-2220).
Western blotting
PBMC, PMN, HeLa and ATG5 KO HeLa cell pellets were resuspended in a lysis buffer containing protease and phosphatase inhibitors (10 mM Tris, pH 7.0, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100 [Sigma Aldrich, X100], 0.1% sodium deoxycholate [BDH, 430353P], 0.1% SDS [Sigma Aldrich, L3771], 140 mM NaCl, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM β-glycerophosphate, EDTA-free Protease Inhibitor Cocktail [Sigma-Aldrich, 4,693,132,001]). Cell suspensions were then sonicated (twice for PBMC and PMN pellets and once for HeLa and ATG5 KO HeLa cell pellets), on ice for 20 sec. Cell lysates were centrifuged at 16,000 x g for 10 min at 4°C and the supernatant collected. Total protein was measured using a micro BCA protein assay kit (Thermo Fisher Scientific, 23,235).
Western blot analysis was performed using 10 μg of total protein of each of the homogenates that were electrophoresed at 130 V for 70 min through 4–12% Bis-Tris Plus SDS-PAGE gels (pH 6.4), 10-, 12- or 15-well (Bolt™; Invitrogen, Thermo Fisher Scientific, NW04120BOX, NW04122BOX and NW04125BOX). The gels were transferred to a methanol-activated PVDF membrane (Perkin Elmer, NEF1002001PK) at 35 V for 70 min. The membranes were then incubated in blocking solution (Tris-buffered saline [TBS; 20 mM Tris [Merck Millipore, 648,310], 250 mM NaCl [Chem-Supply, SA046], pH 7.0, containing 0.1% [v:v] Tween 20 [Merck Millipore, 8,221,840,500; TBST] and either 2% [w:v] bovine serum albumin [BSA; Sigma Aldrich, A9647] or 5% [w:v] skim powder milk) for one h at room temperature, with rocking.
The membranes were cut at different sizes to allow for analyze of different proteins on the same membrane and incubated overnight at 4°C with primary antibodies diluted in blocking solution on a tube roller. The following primary antibodies were used: rabbit anti-MAP1LC3A (1:1,000; Abcam, ab62720), rabbit anti-MAP1LC3B (1:1,000; Novus Biologicals, NB100-2220), rabbit anti-MAP1LC3C (1:1,000, clone D1R8V; Cell Signaling Technology, 14,723), mouse anti-SQSTM1 (1:1,000, clone 2 C11; Abnova, H00008878-M01), rabbit anti-ATG5 (1:1,000, D5F5U; Cell Signaling Technology, 12,994), rabbit anti-NBR1 (1:1000; Abcam, ab126175).
The following day the membranes were washed three times for 5 min each in TBST and then incubated for one h at room temperature, with rocking, with HRP-conjugated goat anti-rabbit (Merck Millipore, AP307P) or sheep anti-mouse (Merck Millipore, AC111P) diluted 1:10,000 in blocking solution. The membranes were washed three times for 5 min each in TBST and then developed using the WestFemto ECL blotting system (Thermo Fisher Scientific, 34,095) and detected using the LAS4000 Luminescent Image Analyzer (Fujifilm Life Science). Mouse anti-ACTB (1:5,000; Cell Signaling Technology, 3700S) diluted in blocking solution was assessed as loading control. For ACTB, membranes were either used on the same day or left to dry out then reactivated in methanol before being processed as described hereafter. The membranes were incubated in blocking solution (TBST and 5% [w:v] milk powder) for one h at room temperature, with rocking. The membranes were incubated overnight at 4°C with ACTB primary antibody diluted in blocking solution on a tube roller. The following day the membranes were washed three times for 5 min each in TBST and then incubated for one h at room temperature, with rocking, in the dark, with a goat anti-Mouse IgG (H and L) secondary antibody, DyLight 800 4X PEG (1:20,000, Thermo Fisher Scientific, SA5-35,521). The membranes were washed twice for 5 min in TBST and once in TBS before being developed using the Li-Cor Odyssey CLx Imaging system (Li-Cor).
Quantification of western blots
Western blot images were analyzed using ImageJ software.
To determine that western blot detection was occurring within the linear range, 0; 1.5; 2; 3; 5; 7.5; 10; 12.5; 15; and 20 µg of protein from PBMC lysate were loaded for detection of ACTB, and 0; 1.5; 3; 5; 7.5; 10; 12.5; 15; and 20 µg of protein from PBMC lysate were loaded for detection of LC3B. Western blots were performed as described above. Bands corresponding to LC3B protein were detected at a range of exposure times (10 sec to 3 min) on the LAS4000 Luminescent Image Analyzer and quantified using ImageJ software and assessed for linearity. Bands corresponding to ACTB protein were detected on the Li-Cor Odyssey CLx Imaging system and quantified using ImageJ software and assessed for linearity. Using these data, we found LC3B-II and ACTB were within the linear range for quantification at 10 µg of protein that was required to robustly image LC3B-II. ACTB was therefore chosen as the loading control for quantification of LC3B. We confirmed that bands for LC3B-II were also in the linear range of detection for CQ-treated samples (150 µM CQ for one h, loaded protein concentrations 0–20 µg) (Fig. S2). ACTB was shown to not respond to CQ treatment.
Quantitative real-time PCR
Gene expression of SQSTM1 was measured by quantitative real-time PCR. RNA was extracted from PBMC pellets using the PureLink RNA Mini kit (Life Technologies, 1218301A) following manufacturer instructions. cDNA was synthesized from 0.3 µg of RNA using SuperScript III First Strand Synthesis System (Invitrogen/Life technologies, 18,080–051) following manufacturer instructions. Quantitative real-time PCR was performed using PowerUp SYBR Green Master Mix (Life Technologies, A25742) and with the following PCR conditions: 95°C for 10 min followed by 40 cycles of 95°C for 15 sec, 60°C for 1 min on the StepOnePlus Real-Time PCR System (v2.3, Applied Bioscience). B2M (β-2-microglobulin) was used as the reference gene. The forward- and reverse-primer sequences for SQSTM1 and B2M were the following: human SQSTM1 Forward: 5ʹ-ATCGGAGGATCCGAGTGTG-3ʹ, Reverse: 5ʹ-GCTCTTCTCCTCTGTGCTG-3ʹ; human B2M Forward: 5ʹ-GAGTATGCCTGCCGTGTGAAC-3ʹ, Reverse: 5ʹ-CCAATCCAAATGCGGCATCTTC-3ʹ.
Immunofluorescence staining for LC3B, confocal microscopy and quantification
After incubation of blood (3 mL) with or without CQ for 1 h, 200 µL of blood was pipetted into 1.5 mL Eppendorf tubes and maintained cold on ice or at 4°C during centrifugation, and cold buffers were used to stop or prevent vesicle trafficking during further processing. Blood was mixed with 1 mL of red blood cell lysis buffer and incubated on ice for 10 min and vortexed at 0, 5 and 10 min of incubation. The samples were then pelleted by centrifugation at 600 x g for 5 min at 4°C. The supernatant was discarded, and this red blood cell lysis was repeated a second time to eliminate remaining erythrocyte contamination. The supernatant was discarded and the white blood cells pellet was washed twice by resuspension in 1 mL of DPBS and pelleted by centrifugation at 600 x g for 5 min. The final pellet containing white blood cells was resuspended in 20 µL of 5% neutral buffered formalin solution (LabServ, BSPFS426.2.5) and mixed by pipetting. Five µL of the cell solution was smeared (1.5 x 1.5 cm circle) on a microscope slide (Superfrost Plus; Thermo Fisher Scientific, J1800AMNT) and air dried at room temperature before immunofluorescence staining.
Fixed cells were permeabilized with DPBS containing 0.1% saponin for 15 min and blocked with DPBS containing 3% BSA and 0.01% saponin for 1 h. Cells were immunostained with rabbit anti-MAP1LC3B (1:1,000, Novus Biologicals, NB100-2220) primary antibody diluted in blocking solution overnight and then washed four-times with DPBS. Fluorophore-conjugated secondary antibody AlexaFluor 488 donkey anti-rabbit IgG H + L, (LifeTechnologies, A21206) was diluted 1:500 in blocking solution and used to immunostain cells for 1 h in the dark prior to washing four times with DPBS. Coverslips were mounted onto glass slides with mounting medium containing DAPI (VectaShield HardSet + DAPI, Abacus DX, H-1500) and sealed with nail polish. Slides were kept in the dark at 4°C until confocal microscopy analysis. Images were captured by a blinded experimenter using a TCS SP8X multi-photon confocal microscope with LASX software (Leica, Germany) using identical acquisition conditions between samples. Images were processed and quantified by a blinded experimenter using FIJI ImageJ. LC3B puncta area was quantified using “analyze particle” of thresholded images. Only lymphocytes were analyzed. Lymphocytes were determined based on their morphology (nucleus size and shape, cell size and shape).
Transmission electron microscopy
Following a 1 h incubation with or without CQ, PBMCs were isolated from blood samples following the protocol described above. Before the last wash with DPBS, 3 samples from the same subject were pooled together by resuspension in 1 mL of cold DPBS, and centrifuged at 2,000 x g for 10 min at 4°C. The supernatant was discarded, and the pellet containing PBMCs was resuspended in fixative solution (PBS [4.9 mM Na2HPO4, 1.2 mM KH2PO4, 145.5 mM NaCl, pH 7.2] containing 1.25% glutaraldehyde [ProSciTech, EMS16210], 4% paraformaldehyde [ProSciTech, C007]), and kept at 4°C. Cells were washed once with PBS containing 4% sucrose (Chem-Supply Pty Ltd, SA046) and stained with 2% osmium tetroxide (ProSciTech, EMS19102) for 1 h with rotation. Cells were re-washed, then dehydrated with increasing concentrations of ethanol (70%, 90%, 95% and 100% for ~30 min per concentration). Cells were treated with propylene oxide (ProSciTech, 20,412) for 30 min, before incubation in a 1:1 solution of 100% propylene oxide:100% resin from the Procure 812 embedding kit (ProSciTech, C038) for 1 h and infiltration for two days, before embedding in fresh resin and polymerization at 70°C for 24 h. Sections of 80 nm thickness were mounted on mesh copper grids. Images were captured using a Tecnai G2 Spirit TWIN Transmission Electron Microscope (FEI, USA). PBMCs were imaged such that one cell filled the field of view (approximately 2,900 x magnification) and images were then randomized and de-identified. De-identified images were assessed and autophagosomes were then scored per cell. If a vesicle contained membranous material, it was scored as an autophagosome. If the vesicle was empty, it was not counted. Scores of the number of autophagosomes per cell were then identified using a file generated by the person who blinded the images and scores were assigned to their original experimental groups for further statistical assessment.
Statistical analysis
Graphs and statistical analyzes were generated using GRAPHPAD PRISM, version 8.2.0, for Windows (GraphPad Software, La Jolla, CA, USA). Data normality was assessed using the Shapiro-Wilk test. In case the normality was not reached non-parametric tests were used. Statistical tests are described in the results section and in the figure legends. Data in the figures are expressed as mean values with their standard errors (SEM). Results were considered significantly different when p < 0.05.
Illustration
Diagrams were created with BioRender (BioRender.com) and figures were compiled using INKSCAPE 0.92 (www.inkscape.org).
Supplementary Material
Acknowledgments
Authors acknowledge Malcolm Moore as provider of pUltrahot; and Edward Campbell as provider of LentiCRISPRv2-ATG5 via Addgene. Authors acknowledge Bo Liu and Julia Liu for their assistance with sample collection. Authors acknowledge Prof. John Hopwood for helpful discussions, and Prof. Christopher Proud for provision of insulin. We thank Ruth Williams at Adelaide Microscopy, University of Adelaide, for electron microscopy.
Disclosure statement
Authors JB and TJS hold a patent: “Methods and products for assessing lysosomal system flux”; International Patent Application PCT/AU2020/050908.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Karsli-Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gao S, Casey AE, Sargeant TJ, et al. Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain. 2018;141(9):2711–2720. [DOI] [PubMed] [Google Scholar]
- [4].Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. [DOI] [PubMed] [Google Scholar]
- [6].Castaneda D, Gabani M, Choi SK, et al. Targeting autophagy in obesity-associated heart disease. Obesity (Silver Spring). 2019;27(7):1050–1058. [DOI] [PubMed] [Google Scholar]
- [7].Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fernandez AF, Sebti S, Wei Y, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558(7708):136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Morselli E, Maiuri MC, Markaki M, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grumati P, Coletto L, Sabatelli P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16(11):1313–1320. [DOI] [PubMed] [Google Scholar]
- [11].He C, Sumpter Jr. R, Levine B.. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. 2012;8(10):1548–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Boland B, Yu WH, Corti O, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17(9):660–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jia J, Bissa B, Brecht L, et al. AMPK, a Regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020;77(5):951–969. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mulcahy Levy JM, Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020;27(3):843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Loos B, Klionsky DJ, Du Toit A, et al. On the relevance of precision autophagy flux control in vivo – points of departure for clinical translation. Autophagy. 2020;16(4):750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91. [DOI] [PubMed] [Google Scholar]
- [19].Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018;14(6):356–376. [DOI] [PubMed] [Google Scholar]
- [22].Alonzi T, Petruccioli E, Vanini V, et al. Optimization of the autophagy measurement in a human cell line and primary cells by flow cytometry. Eur J Histochem. 2019;63(2). DOI: 10.4081/ejh.2019.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Papagiannakis N, Xilouri M, Koros C, et al. Autophagy dysfunction in peripheral blood mononuclear cells of Parkinson’s disease patients. Neurosci Lett. 2019;704:112–115. [DOI] [PubMed] [Google Scholar]
- [24].McKee TJ, Komarova SV. Is it time to reinvent basic cell culture medium? Am J Physiol Cell Physiol. 2017;312(5):C624–C626. [DOI] [PubMed] [Google Scholar]
- [25].Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10(3):431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Loos B, Du Toit A, Hofmeyr JH. Defining and measuring autophagosome flux-concept and reality. Autophagy. 2014;10(11):2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Haspel J, Shaik RS, Ifedigbo E, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. 2011;7(6):629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Du Toit A, Hofmeyr JS, Gniadek TJ, et al. Measuring autophagosome flux. Autophagy. 2018;14(6):1060–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Klionsky DJ, Elazar Z, Seglen PO, et al. Does bafilomycin A 1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4(7):849–850. [DOI] [PubMed] [Google Scholar]
- [31].Wang L, Wang X, Proud CG. Activation of mRNA translation in rat cardiac myocytes by insulin involves multiple rapamycin-sensitive steps. Am J Physiol Heart Circ Physiol. 2000;278(4):H1056–68. [DOI] [PubMed] [Google Scholar]
- [32].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pietrocola F, Demont Y, Castoldi F, et al. Metabolic effects of fasting on human and mouse blood in vivo. Autophagy. 2017;13(3):567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mehdi S. Cell-penetrating inhibitors of calpain. Trends Biochem Sci. 1991;16(4):150–153. [DOI] [PubMed] [Google Scholar]
- [36].Fedele AO, Proud CG. Chloroquine and bafilomycin A mimic lysosomal storage disorders and impair mTORC1 signalling. Biosci Rep. 2020;40(4). DOI: 10.1042/BSR20200905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhitomirsky B, Yunaev A, Kreiserman R, et al. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 2018;9(12):1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. [DOI] [PubMed] [Google Scholar]
- [39].Hein LK, Apaja PM, Hattersley K, et al. A novel fluorescent probe reveals starvation controls the commitment of amyloid precursor protein to the lysosome. Biochim Biophys Acta Mol Cell Res. 2017;1864(10):1554–1565. [DOI] [PubMed] [Google Scholar]
- [40].Li L, Wang ZV, Hill JA, et al. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2014;25(2):305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee JH, Rao MV, Yang D-S, et al. Transgenic expression of a ratiometric autophagy probe specifically in neurons enables the interrogation of brain autophagy in vivo. Autophagy. 2019;15(3):543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. [DOI] [PubMed] [Google Scholar]
- [43].Di Fruscio G, Schulz A, De Cegli R, et al. Lysoplex: an efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy. 2015;11(6):928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Barbosa MC, Grosso RA, Fader CM. Hallmarks of aging: an autophagic perspective. Front Endocrinol (Lausanne). 2018;9:790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Congdon EE. Sex differences in autophagy contribute to female vulnerability in Alzheimer’s disease. Front Neurosci. 2018;12:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21(4):183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bánréti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy. 2013;9(6):819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Denton D, Shravage B, Simin R, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19(20):1741–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Koike M, Shibata M, Tadakoshi M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172(2):454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu Y, Shoji-Kawata S, Sumpter RM, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Perera RM, Stoykova S, Nicolay BN, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524(7565):361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22(2):165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Auberger P, Puissant A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 2017;129(5):547–552. [DOI] [PubMed] [Google Scholar]
- [58].Torgersen ML, Engedal N, Bøe S-O, et al. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood. 2013;122(14):2467–2476. [DOI] [PubMed] [Google Scholar]
- [59].Kirn TJ, Weinstein MP. Update on blood cultures: how to obtain, process, report, and interpret. Clin Microbiol Infect. 2013;19(6):513–520. [DOI] [PubMed] [Google Scholar]
- [60].Nardacci R, Amendola A, Ciccosanti F, et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy. 2014;10(7):1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yi X, Liu M, Luo Q, et al. Toxic effects of dimethyl sulfoxide on red blood cells, platelets, and vascular endothelial cells in vitro. FEBS Open Bio. 2017;7(4):485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kloverpris H, Fomsgaard A, Handley A, et al. Dimethyl sulfoxide (DMSO) exposure to human peripheral blood mononuclear cells (PBMCs) abolish T cell responses only in high concentrations and following coincubation for more than two hours. J Immunol Methods. 2010;356(1–2):70–78. [DOI] [PubMed] [Google Scholar]
- [64].Song YM, Song S-O, Jung Y-K, et al. Dimethyl sulfoxide reduces hepatocellular lipid accumulation through autophagy induction. Autophagy. 2012;8(7):1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chen G, Ke Z, Xu M, et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8(11):1577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sarkar S, Floto RA, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170(7):1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Richie DL, Fuller KK, Fortwendel J, et al. Unexpected link between metal ion deficiency and autophagy in Aspergillus fumigatus. Eukaryot Cell. 2007;6(12):2437–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Richie DL, Askew DS. Autophagy: a role in metal ion homeostasis? Autophagy. 2008;4(1):115–117. [DOI] [PubMed] [Google Scholar]
- [69].Imam S, Talley S, Nelson RS, et al. TRIM5alpha degradation via autophagy is not required for retroviral restriction. J Virol. 2016;90(7):3400–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.