

ABSTRACT
Macroautophagy/autophagy is a cellular process to recycle damaged cellular components, and its modulation can be exploited for disease treatments. A key autophagy player is the ubiquitin-like protein MAP1LC3B/LC3B. Mutations and changes in MAP1LC3B expression occur in cancer samples. However, the investigation of the effects of these mutations on MAP1LC3B protein structure is still missing. Despite many LC3B structures that have been solved, a comprehensive study, including dynamics, has not yet been undertaken. To address this knowledge gap, we assessed nine physical models for biomolecular simulations for their capabilities to describe the structural ensemble of MAP1LC3B. With the resulting MAP1LC3B structural ensembles, we characterized the impact of 26 missense mutations from pan-cancer studies with different approaches, and we experimentally validated our prediction for six variants using cellular assays. Our findings shed light on damaging or neutral mutations in MAP1LC3B, providing an atlas of its modifications in cancer. In particular, P32Q mutation was found detrimental for protein stability with a propensity to aggregation. In a broader context, our framework can be applied to assess the pathogenicity of protein mutations or to prioritize variants for experimental studies, allowing to comprehensively account for different aspects that mutational events alter in terms of protein structure and function.
Abbreviations: ATG: autophagy-related; Cα: alpha carbon; CG: coarse-grained; CHARMM: Chemistry at Harvard macromolecular mechanics; CONAN: contact analysis; FUNDC1: FUN14 domain containing 1; FYCO1: FYVE and coiled-coil domain containing 1; GABARAP: GABA type A receptor-associated protein; GROMACS: Groningen machine for chemical simulations; HP: hydrophobic pocket; LIR: LC3 interacting region; MAP1LC3B/LC3B microtubule associated protein 1 light chain 3 B; MD: molecular dynamics; OPTN: optineurin; OSF: open software foundation; PE: phosphatidylethanolamine, PLEKHM1: pleckstrin homology domain-containing family M 1; PSN: protein structure network; PTM: post-translational modification; SA: structural alphabet; SLiM: short linear motif; SQSTM1/p62: sequestosome 1; WT: wild-type.
KEYWORDS: Autophagy, cancer mutations, MAP1LC3B, molecular dynamics, protein structure network, structural alphabets
Introduction
Autophagy is a highly conserved pathway in eukaryotes that allows the recycling of multiple cellular components. Autophagy is active at a basal level and can be further activated in response to different types of stimuli, such as starvation [1,2]. Autophagy mediates the sequestration of proteins and organelles within double-membrane vesicles, known as autophagosomes, which then mature and fuse to lysosomes, leading to the degradation of their cargo [3]. Selective autophagy occurs in diverse forms depending on the target organelles or components [1,4]. A common trait of selective autophagy is that a receptor protein binds the cargo and links it to the LC3/GABARAP family of Atg8 homologs [5] through the interaction with scaffold proteins [1].
One of the most studied proteins of the LC3/GABARAP family is MAP1LC3B/LC3B, which is frequently used as a marker for the assessment of autophagy activity in cellular assays [6,7]. The first study on LC3B in autophagy was published in 2000 [8] and has been cited more than 5000 times ever since. The structure of LC3B includes two α-helices at the N-terminal of the protein and a ubiquitin-like core [9]. LC3B is a versatile protein that serves as a platform for protein-protein interactions [9,10]. LC3/GABARAP proteins, more broadly, recruit the autophagy receptors through the binding of a specific short linear motif (SLiM) known as the LC3-interacting region (LIR) [9,11–13]. The first LIR was discovered in the mammalian SQSTM1/p62 autophagy receptor in 2007 [14]. The consensus sequence of the core LIR motif includes an aromatic residue (W/F/Y) and a hydrophobic one (L/I/V), separated by two other residues [9,12,13,15]. Flanking regions, which are located N- or C-terminal to the core LIR motif, influence the specificity and binding affinity to LC3/GABARAP proteins [9,16–18].
Selective autophagy pathways affect a broad range of diseases including cancer [19–22]. Autophagy can exert protective functions against cancer development, but it can also contribute to cancer progression and resistance to treatments [23–27]. The same patterns are observed for LC3B proteins, which has been mostly linked to tumor progression and adverse outcomes [28–32]. Modulation of autophagy could provide means for new disease treatments and the identification of prognostic factors or disease-related markers [33]. Apart from evidence regarding changes in the expression level of MAP1LC3B in different cancer types, genomic alterations of LC3/GABARAP family members and their relation to cancer have not been thoroughly explored [34].
Several X-ray or NMR three-dimensional (3D) structures of LC3B are available in the free (unbound) state or in complex with different biological partners [9]. This provides a valuable source of information for studies with structure-based approaches, which also account for protein dynamics. The studies published so far focused on specific aspects of LC3B dynamics using all-atom molecular dynamics (MD) simulations with a single physical model (i.e., force field) or coarse-grained approaches, as we recently summarized in a review article [9].
Here we applied methods that combine structural ensembles derived by all-atom and coarse-grained MD simulations, free energy calculations, and graph theory [35], to study the LC3B structure-dynamics-function relationship and the effect of missense mutations. As a first step, we selected the best physical description for LC3B by assessing nine different state-of-the-art MD force fields. Indeed, the choice of the force field affects the quality of the simulated ensemble of conformations and the results are highly protein-dependent. Similar assessments have been extensively carried out for ubiquitin [36–46] but not for LC3/GABARAP proteins.
With meaningful structural ensembles in hand for LC3B, we turned our attention to the study of the impact of mutations identified in genomic cancer studies. In our study, we accounted for the many layers that a mutation could alter: i) protein stability, ii) interaction with biological partners, iii) long-range communication between sites distant from the functional ones (which is often at the base of allostery), and iv) the interplay with post-translational modifications or functional motifs. We also experimentally validated our findings on a selection of six variants, using co-immunoprecipitation, measurement of cellular protein levels, and tendency to form aggregates.
Results and discussion
State-of-the-art force fields for all-atom molecular dynamics consistently describe microsecond dynamics of LC3B
To compare the quality and sampling of the MD simulations of LC3B carried out with the nine state-of-the-art force fields selected for this study, we integrated different and complementary metrics. An extended and more technical discussion of these results is in the Text S1.
In brief, we estimated the atomic resolution (R) for each of the MD ensembles as a measurement of structural quality [47]. The resulting median R values for the different MD ensembles of LC3B7-116 are in reasonable agreement with each other, and R values are mostly below the threshold for good structures, which is 1.5 Å (Figure 1A, Table S1). Conformations collected with RSFF1 feature the highest median value and more outliers.
Figure 1.
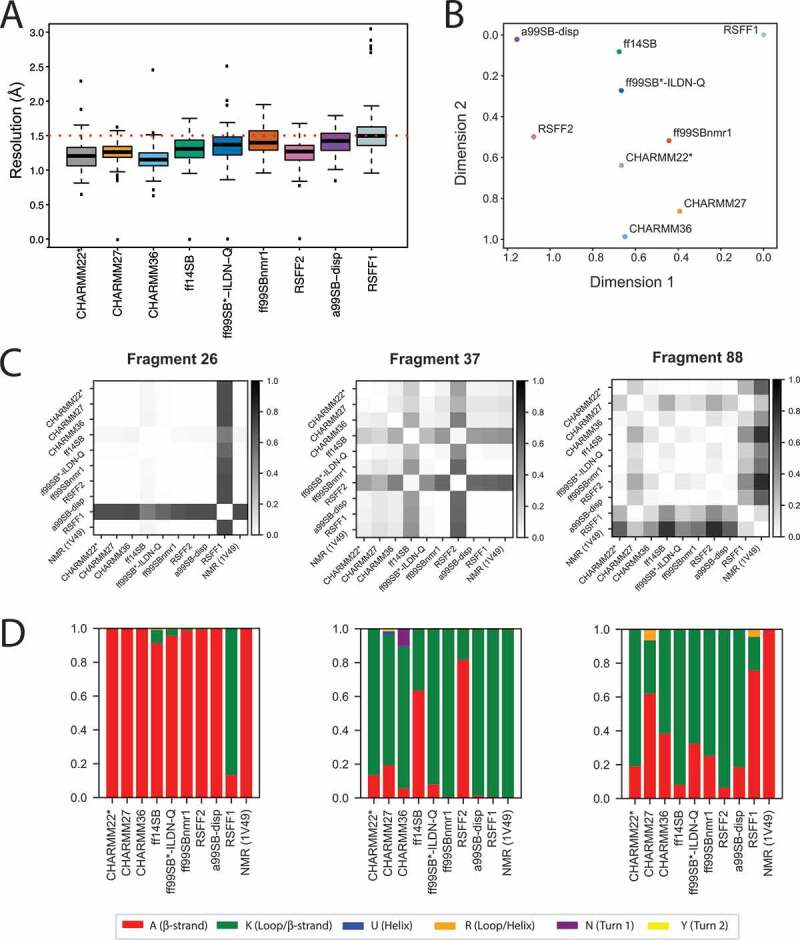
Comparison of MD ensembles of LC3B. (A) Prediction of structural resolution values for different MD ensembles of LC3B generated using different force fields. All the MD force fields generally refine the structure to a resolution close to an X-ray structure deposited in the entry 3VTU and are in reasonable agreement with each other according to this parameter. The only (not pronounced) differences are observed for RSFF1, ff99SBnmr1, and a99SB-disp, according to the statistical test reported in Table S1. (B) Comparison of MD ensembles from different force fields using the clustering-based ensemble similarity (CES) method implemented in ENCORE. The two-dimensional plot for the clustering of the of the ten force field ensembles is based on the pairwise similarities calculated by CES using the tree preserving embedding method. The higher the distance between two force-fields is in the plot, the more different the sampling was between the respective simulations. The corresponding heatmap is provided in Figure S2. The analysis shows that RSFF1, RSFF2 and a99SB-disp were the ensembles more distant from the others. (C and D) These figures showcase the results obtained with the reduced structural alphabet (SA) method for three different SA fragments as representative example (i.e., fragments 26,37 and 88). The full datasets are available in the GitHub repository. Here, independently for each fragment, we have calculated the frequency of each letter in each force-field simulation, as shown in panel D. The frequencies of the samples have been used as an estimate of the underlying probability distributions and compared using the Jensen-Shannon divergence (dJS) between each pair of simulations, as shown in panel C. Higher values in this plot mean a more different sampling of SA letters between a given pair of force fields for a specific fragment
LC3B has been studied by NMR spectroscopy, a technique which provides data on protein dynamics in solution, such as backbone chemical shifts [48]. Chemical shifts entail information about motions on different time scales and can be calculated from an ensemble of MD structures [49]. We thus calculated the backbone chemical shifts from each MD ensemble and compared them to the available experimental values. The calculated chemical shifts from our simulations were in fair agreement with the experimental data (Table S2) [49]. The only relevant exceptions are RSFF1 and RSFF2 in the case of Cα atoms.
As a complementary approach, the structural ensembles can be compared in terms of the overlap of the conformational space sampled by the different simulations. The conformational space described by all-atom simulations of a protein is rather complex and entail a multi-dimensional landscape. Therefore, we applied a variety of methods for dimensionality reduction [40,50] to compare the simulations (S1, S2, and Figures 1B). Using a method based on structural clustering [50], we observed that the MD ensembles collected with the RSFF1, RSFF2, and a99SB-disp sample different LC3B structures compared to the other simulations (Figure 1B, S2 and GitHub repository).
We then wondered if these differences could be due to local conformational variabilities in the structures sampled by each simulation. To this aim, we exploited the structural alphabet paradigm [51] (Figure 1C -D and GitHub). In detail, we estimated the probability distribution of the states for each protein fragment in the different MD ensembles and compared them (Figure 1D and GitHub). The structural alphabet analysis confirmed the diversity of the structures explored by RSFF1 in different areas of the protein, including the LIR-binding interface. We also observed local differences for CHARMM36, CHARMM27, ff14SB and RSFF2 when compared to other force fields (Figure 1D and GitHub).
In summary, all the force fields provided a structural ensemble of reasonable quality for LC3B. However, using methods for comparison of ensembles of structures and their local states, we have been able to identify local differences. Overall, CHARMM22* seemed to be among the most robust force fields in the description of LC3B according to the properties here analyzed. We selected CHARMM22* for the following analyses for the investigation of mutation sites of LC3B from genomic cancer studies.
An atlas of LC3B missense mutations in cancer and their interplay with post-translational modifications and functional motifs
We aim to investigate the structural mechanisms related to mutations of LC3B in cancer. Thus, we retrieved 28 missense mutations identified in LC3B from an aggregation of cancer studies (Figure 2A -B). At first, we verified if the dataset contained possible natural polymorphisms in healthy individuals. To this end, we used the ExAC database [52] to verify if any of the mutations are found at high frequency in the healthy population. We did not find high frequency mutations and only three variants from our dataset (R37Q, K65E, and R70H) are reported in ExAC, but with low frequency (< 1/10000). Thus, we retained all the 28 mutations in 23 residue sites and found in 13 different cancer types (Table S3).
Figure 2.
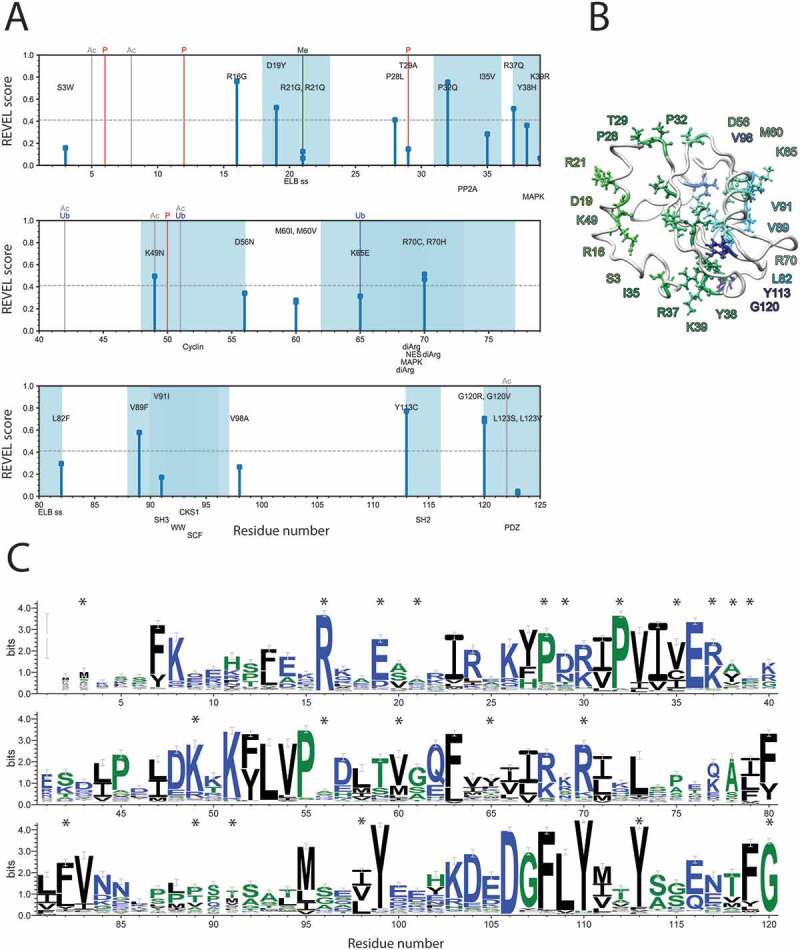
Sequence-based assessment of the LC3B mutations sites found in cancer genomic studies. (A) Schematic representation of the identified cancer-related mutations and the analysis of: i) REVEL score, ii) overlap with PTMs and, iii) overlap with short linear motifs (SLiMs). In this plot, the amino acid sequence of LC3B, according to the main Uniprot isoform of the protein, runs on the X-axis. The missense mutations are shown as sticks, whose height is proportional to the associated REVEL pathogenicity score. PTMs from literature annotations are shown as vertical lines spanning the height of the plot (P for phosphorylation, Ubq for ubiquitination, Me for methylation, Ac for acetylation). SLiMs predicted for the protein sequence are shown below the plot, and the associated residue intervals colored in blue shades. Only the SLiMs overlapping with mutation sites are shown. The full set of data and annotation is provided as Table S3. We observed a substantial overlap with PTMs and functional motifs for the LC3B mutations under investigation and 11 mutations with a high pathogenicity score. (B) LC3B is a ubiquitin-like protein, characterized by two α-helices at the N-terminal followed by a ubiquitin (Ub)-like core. Localization in the structure of LC3B of the 28 residues identified as targets of missense mutations from cancer genomic studies. The structure of LC3B (PDB entry 1V49) is shown as white cartoon while the target residues are indicated as colored stick, using a color gradient from the N-terminal (green) to the C-terminal (dark blue). (C) The logo plot obtained by the multiple sequence alignment calculated by Gremlin is shown to evaluate the conservation and tolerated substitutions for the mutation sites of LC3B, which are marked by a *
We analyzed the mutation sites in the context of different aspects, which could compromise MAP1LC3B function, described step by step below. We verified that the mutation was the only one targeting the MAP1LC3B gene in that specific sample (for samples with available information).
We carried out a first annotation of the potential pathogenic impact for each mutation using a pathogenicity score based on sequence analysis REVEL [53] (Figure 2A). We identified 11 predicted pathogenic variants according to this sequence-based prediction: R16G, D19Y, P28L, P32Q, R37Q, K49N, R70C/H, V89F, Y113 C, and G120 R/V.
In the second annotation step, we evaluated if the mutations could have a functional impact in abolishing functional SLiMsor PTMs, along with the likelihood that the mutant variants could harbor new PTM sites (Figure 2A). The mutation T29A is, for example, expected to abolish a phosphorylation site for the protein kinase C [54]. The analysis of the multiple sequence alignment and the associated scores showed that at this site prefers negatively charged residues or other phosphorylatable residues (i.e., serine), emphasizing the functional importance of a negative charge at this position (Figure 2C). S3W, P32Q, and K49N are in the flanking region of T29 or the other two phosphorylation sites (T6 [54] and T50 [55,56]) and could impair the binding of the kinases/phosphatases. The R21G/Q, K49N, and K65E mutations might abolish methylation, acetylation [57] and ubiquitination sites [58], respectively (Figure 2A). In particular, acetylation of K49 on the cytoplasmic form of LC3B is pivotal for nuclear transport and the maintenance of the LC3B reservoir, deacetylation is, on the contrary, necessary for the translocation to the cytoplasm where the protein interacts with the autophagy machinery [57,59]. Moreover, LC3B acetylation abrogates the LC3B binding with SQSTM1/p62 and prevent proteasomal degradation of LC3B [60]. Deacetylation/acetylation cycles maintain the proper pools of LC3B in the cell and a mutation impairing this modification, such as K49N, could increase the cytoplasmic pool of LC3B and its interactions with SQSTM1/p62, resulting in uncontrolled autophagy.
D19Y, on the contrary, is predicted to introduce a phosphorylatable residue for the TK and EGFR families of kinases by predictors of phosphosites [61,62]. The residue is in a solvent exposed region on the protein surface and its substitution to tyrosine could make it available for post-translational modification, introducing a new level of regulation absent in the wild-type variant. D19 is also tightly coupled to R16 according to the coevolution analysis (GitHub) and the only conserved substitutions are, to glutamate or to lysine, respectively (Figure 2B), suggesting a key contribution of charged residues at these positions, which might be compromised by the mutations of arginine to glycine and of aspartate to tyrosine.
The mutations D19Y and R21Q/G are also likely to impair the signal motif (i.e., Endosome-Lysosome-Basolateral sorting signals, ELB) to direct the protein to the endosome and lysosome compartments with a possible impact on the autophagy pathway (Figure 2A). Several LC3B mutations could affect docking or recognition motifs for phosphatases or kinases, thus impacting on the regulation of LC3B activity and stability by its upstream regulators (Table S3). Mutations at the residue G120 could abolish the ATG4B cleavage site, impairing the activity of LC3B, as confirmed by the experiments on the G120A variants [48,63]. Additional information on sequence comparisons are in Text S1 and Figure S3.
Assessment of the impact on protein stability upon LC3B missense mutations
Structural methods can help to achieve a more profound understanding of the impact of missense mutations on a protein [64–66]. A deleterious effect of a mutation could, for example, be related to changes in protein structural stability, causing local misfolding and a higher propensity of the protein to be degraded in the cell. Thus, we estimated the changes of folding free energy for each of the LC3B mutations as a measure of structural stability [67]. We implemented this procedure in a high-throughput manner [68] so that all the possible single mutations at each LC3B site can be analyzed. Our high-throughput approach evaluates the impact of mutations in a protein without being limited to the mutations currently available from cancer studies. Indeed, we investigated, more broadly, if the cancer mutation sites are sensitive hotspots to substitutions. Moreover, we could predict if other sites of the protein when mutated could impact on LC3B stability, providing groundwork for the prediction of future LC3B mutations, which might arise from other cancer studies or the profiling of new cancer samples (Figure 3A and GitHub). In addition, we verified the predictions with another method for calculations of free energy changes based on Rosetta energy functions that have the advantage of modeling flexibility of the protein backbone (Figure S4).
Figure 3.
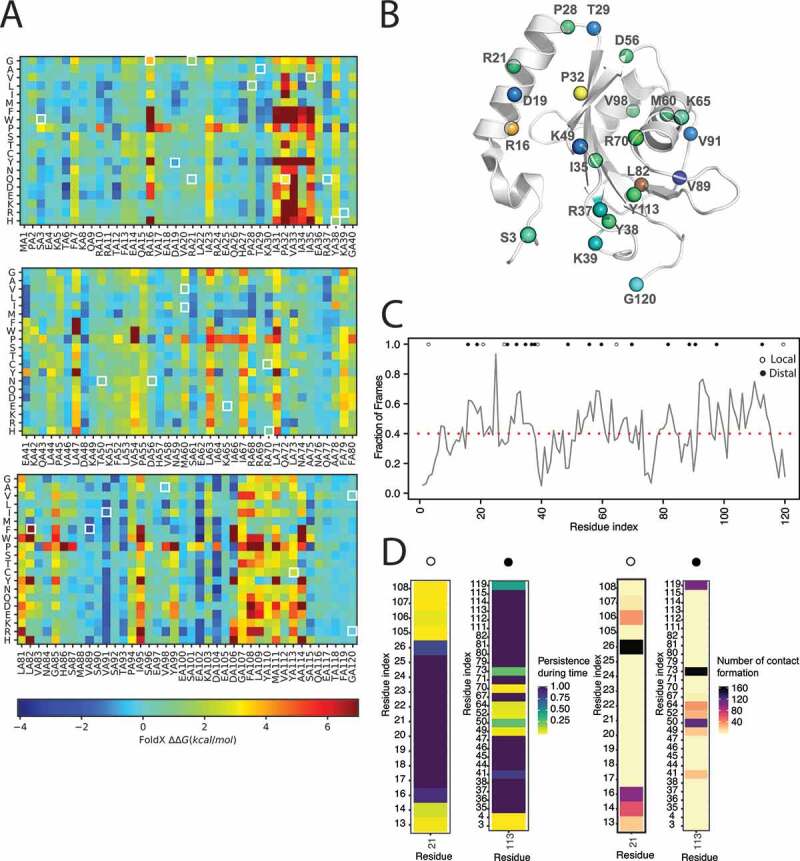
Assessment of the impact on stability of missense mutations of LC3B found in cancer genomic studies. (A) The figure illustrated the heatmap of the saturation mutational scan performed using the 1V49 PDB entry of LC3B. Values of ΔΔG higher than 0 indicates destabilizing mutations. (B) Mapping on the 3D structure of LC3B (PDB entry 1V49) of the ΔΔG associated to protein stability of the 26 missense mutations of LC3B under investigation. In case of different mutations at the same site the highest ΔΔG is shown. The mutations were color-coded according to the corresponding heatmap color for sake of clarity. (C and D) The contact-based analyses on the LC3B MD-derived ensemble with the CHARMM22* force field is illustrated in panels C and D and File S13. The analyses were carried out with CONAN. (C) The average local interaction time (avLIT) profile of the LC3B points out several mutation sites (R16, D19, P32, L82, V98 and Y113), with avLIT values higher than the mean of the distribution (0.4 fraction of the simulation frames), as possible sensitive hotspots for protein stability. (D) Persistence and number of encounters in the CHARMM22* MD ensemble for each interaction of the mutation site R21 and Y113, as examples. The persistence of each contact is normalized on the total time length of the simulation and represented with a color gradient, from low (yellow) to high (blue) persistence. The number of formation events for each contact is represented with a color gradient, from low (light brown) to high (black). Analogous plots for each residue in the LC3B are reported in File S13. We identified two classes of mutation sites: local and distal, indicated with white and black dots, respectively. The “local” class accounts for mutation sites (e.g. R21) forming strong atomic contacts with only residues contiguous in the sequence and they are mostly predicted to be neutral for stability. The “distal” class accounts for mutation sites (e.g. Y113) forming strong atomic contacts with also residues distant in the sequence and they are mostly predicted to be relevant for stability
The mutational scans showed that R16, P32, I35, and Y113 are sensitive hotspots for protein stability, in general, and they cannot tolerate most of the amino-acid substitutions. Regarding the atlas of somatic mutations found for LC3B, five of them are altering the structural stability of the protein according to both the predictors (R16G, R21G, P28L, P32Q, Y38H, and Y113C). Other mutations have mild or neutral effects except for V89F with a stabilizing effect (Figure 3B and GitHub). Nevertheless, if we used an X-ray structure of LC3B (PDB entry 3VTU) as initial structure, the effect of this mutation is less pronounced, likely due to a better residue packing originally present in the X-ray structure at this site with respect to the NMR structure (GitHub). This result points out the importance of verifying predictions using different structures from different experimental sources to fully appreciate conformation-dependent changes in the surrounding of the mutation site.
To account for dynamics, we also employed a method to estimate atomic contacts [69] and their lifetime in the MD ensembles. In particular, we calculated the average local interaction time (avLIT) for each residue of the protein during the simulation (Figure 3C). High values of avLIT indicated residues of LC3B forming highly persistent local contacts during the simulations, possibly suggesting a role for the maintenance of the protein architecture. The mean of the distribution of avLIT values was of 0.4 (fraction of frames), and the mutations sites with avLIT values higher than this threshold were R16, D19, P32, L82, V98 and, Y113, reflecting the results of the mutational scans. We estimated the strength and location of the interactions for each mutation site over time, along with the associated number of encounters (Figure 3D and GitHub). We identified two macro-groups: i) mutation sites with only contacts with residues contiguous in the sequence, and ii) mutation sites also involved in contacts with distal residues in the sequence space. In the first class, we mostly found the mutation sites predicted neutral for stability, whereas the second group accounts for residues such as D19, P32, L82, and Y113.
Local effects of the mutations on binding of LIR motifs and other interactors
The LC3B interactome is large [10], and the primary function of LC3B is to recruit many different proteins to the phagophore, the precursor to the autophagosome. Thus, one should also consider in the same samples the alterations of the LC3B interactors. To this goal, we retrieved LC3B interactors mining the IID protein-protein interactions database [70] and integrated them with interactors reported in a recent publication [10]. We identified overall 95 LC3B partners, and for each of them, we verified if a LIR motif was reported in the literature as experimentally validated. For cases where no information was available on the mode of interaction, we predicted LIR motifs with iLIR [71] (Table S4).
We identified 70 interactors as either experimentally validated LIR-containing proteins or having a predicted LIR motif with a significant score by the predictor. We then retrieved the mutational status of the LIR-containing interactors in the same samples where the LC3B missense mutations were identified to explore the possibility of co-occurrence of mutations (Table S4). We evaluated if the mutation was in the proximity of the experimentally validated or predicted LIRs (Figure 4A -B). We found 39 mutations in 27 LIR-containing interactors occurring in samples where LC3B was mutated (Table S4, highlighted in red in Figure 4A). In particular, 17 mutations are truncations that abolish all or most of the LIR motifs in the interactors (Figure 4B). The remaining mutations are located in the core motif or its proximity, along with in the N- and C-terminal regions (Figure 4B). We noticed that the mutations that are in the proximity of the LIR region or in the core motifs affected mostly charged residues, which are likely to stabilize the binding of the wild-type complexes through changes in electrostatic interactions.
Figure 4.
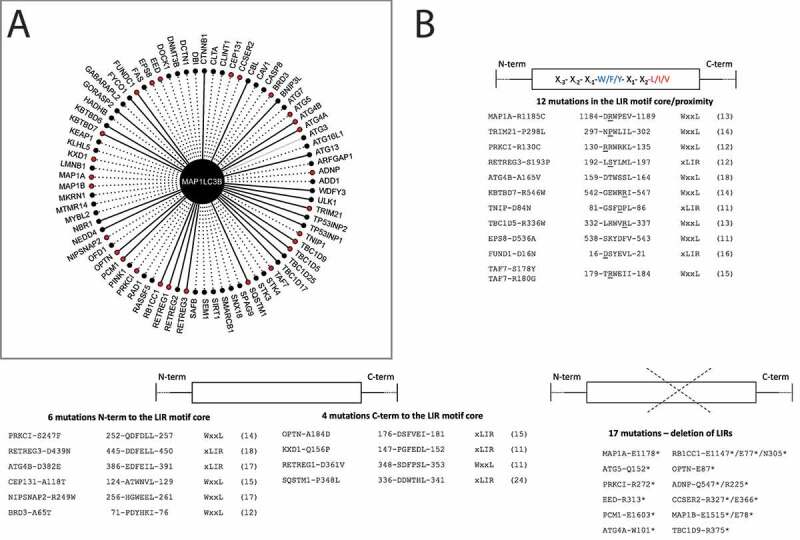
Co-occurrence of mutations in LC3B and LIR-containing interactors and local effects of mutations on intermolecular interactions. (A) Network of the 70 LIR-containing proteins that interact with LC3B. The plot shows all the LC3B interactors which harbor an experimentally validated (solid line) or predicted LIR with PSSM score > 11 (dashed line). The 39 mutations found in 27 LIR-containing proteins in the proximity or within the LIR motif and co-occurring with LC3B mutations are highlighted in red. Only in one case, we identified an experimentally validated interactions for which no LIR motif could be predicted within the significance threshold used for the prediction, i.e., ATG3, which is showed as a gray edge. (B) Among the 39 co-occurring mutations, 12 were located in the LIR core motif or its proximity (top figure), 6 and 4 were N- and C-terminal to the LIR motif core (middle figure), and 17 resulted in truncations abolishing all or most of the LIR motifs of the interactors (bottom figure)
Next, we aim to provide an assessment of the effects of the mutations on the protein-protein interactions mediated by LC3B. To this end, we used the known 3D structures of the complexes between LC3B and three LIR-containing proteins, along with a complex between LC3B and ATG4B. We then applied the in silico approaches for deep mutational scans described above to estimate, in this case, the changes in binding free energy associated with the mutations.
At first, we focused on LIR mutations in co-occurrence with LC3B mutations (Figure 5A): A184D of OPTN and R37Q of LC3B, along with D16N of FUNDC1 and R70C of LC3B. For OPTN, we considered the phosphorylated, phosphomimetic, and wild-type structure of the complex with LC3B (GitHub). A184D-R37Q do not affect, individually or in combination, the binding to any of the OPTN-LC3B complexes. The combination of D16N (FUNDC1) and R70C (LC3B) causes a destabilization of the protein fold (average ΔΔG = 1.07 kcal/mol), which is due to R70C with no additional effect from D16N (Figure 5A).
Figure 5.
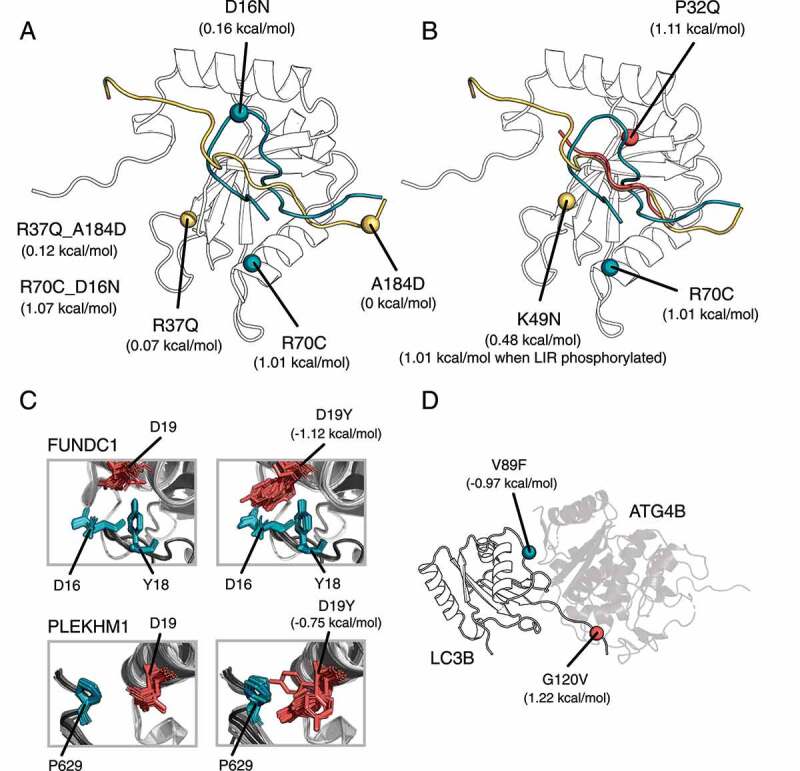
Changes in ΔΔG of binding for LC3B-LIR and LC3B-ATG4B complexes (Rosetta, talaris2014 energy function). (A) We illustrate the location and effect of the co-occurring mutations in the complex LC3B-OPTN (mutations and LIR in yellow), and in the complex LC3B-FUNDC1 (mutations and LIR in blue). Mutations of the first complex are neutral, whereas the mutations in LC3B-FUNDC1 complex destabilize the binding. (B) Mutations of LC3B can have a LIR-specific effect, as illustrated for P32Q in the complex LC3B-SQSTM1/p62 (mutation and LIR in red), K49N in the complex LC3B-OPTN (mutation and LIR in yellow) and for R70C in the complex LC3B-FUNDC1 (mutation and LIR in blue). (C) Mutations of LC3B could change specificity. We illustrate the effect of D19Y which may increase the binding affinity of PLEKHM1 and FUNDC1 to LC3B (D) V89F and G120V stabilizes and destabilizes the interaction between ATG4B and LC3B, respectively
Moreover, we further estimated local effects induced by the LC3B mutations on the binding to their partners of interaction, calculating the changes in binding free energies for the complexes of LC3B with SQSTM1/p62, FUNDC1, and OPTN, as prototypes of different binding modes of LIR-containing proteins (Figure 4B, GitHub). Most of the mutations have neutral effects on the local binding. We observed LIR-specific effects for the remaining mutations: i.e., P32Q as destabilizing mutations for the SQSTM1/p62-LC3B complex and the phosphorylated variant of OPTN. Moreover, K49N has also an effect only with the phosphorylated variant of OPTN. K49 is especially important since it is a gatekeeper that regulates the binding of the LIR to the LC3/GABARAP pocket and undergoes conformational changes upon binding [72]. R70C, on the contrary, seems to target the interaction with FUNDC1 (Figure 5B).
Another mechanism that can be induced by LC3B mutations is to change the specificity toward different members of the LC3/GABARAP family. Indeed, swapping mutations in which residues of LC3B are replaced by residues of GABARAP or vice versa can tune the preferences of LIR motifs for one of the two LC3/GABARAP subfamilies [9,17]. To test this hypothesis, we estimated the changes in binding free energy induced by the missense mutations of LC3B on the binding of the PLEKHM1 LIR, which is more specific toward GABARAP [73]. We observed a conformation-dependent effect by D19Y which seems to improve the binding of PLEKHM1 toward LC3B (Figure 5C). A mutation at D19 (D19N) was recently characterized in the context of the binding of FUNDC1 with LC3B [74]. We observed a mild stabilizing effect of the LC3B-FUNDC1 interaction upon D19Y mutation (Figure 5C). D19 is located in proximity of the inhibitory LIR phospho-site Y18 of FUNDC1 LIR and is responsible for the stabilization of the phosphorylated state of FUNDC1 [74]. Our results could suggest that D19Y might increase the binding affinity and thus partially overcome the inhibitory effect of Y18 phosphorylation on FUNDC1 activity.
LC3B does not interact with LIR motifs exclusively, but also with other proteins of the core autophagy machinery, such as ATG4B [63]. We estimated the changes in binding free energy upon mutations also for the LC3B-ATG4B complex (Figure 5D). Only two mutations (i.e., V89F and G120V) specifically altered the interaction with ATG4B in our calculations with opposing effects (i.e., stabilizing and destabilizing the binding, respectively). Y113C was also showed experimentally to compromise the interaction with ATG4B [34], but was not identified as destabilizing for the interaction by our local mutation scan, a point that might require future investigations.
LC3B ensemble under the lens of protein structure network: hubs for stability and long-range induced effects
The mutational scan described in the previous section only captures local effects for mutations in residues in the proximity of the interface. We used the Protein Structure Network (PSN) framework combined to MD simulations [35] to assess more distal or complex effects.
In detail, a PSN employs the graph formalism to identify a network of interacting residues in a given protein from the number of non-covalent contacts in the protein or other intramolecular interactions. Two main properties of a PSN are the hub residues, i.e., residues that are highly connected within the network and the connected components which are clusters of inter-connected residues that do not interact with residues in other clusters. Hubs in a PSN could have both the role of shortening the communication between distal residues or they can have a structural role thanks to their contribution to the robustness of the network. Indeed, substitutions occurring on the nodes with small degree are likely not to have a large effect on the network (and thus the structure) integrity. On the contrary, if hubs are altered, the network integrity can be compromised. We calculated three PSNs, based on sidechain contacts [75,76], hydrogen bonds [75] and salt-bridges [75,77], respectively.
We calculated the hubs and connected components from the contact-based and hydrogen-bond based PSNs from the MD ensemble of LC3B. Among the LC3B cancer mutation sites, I35, Y38, and V89 have a hub behavior in the contact-based PSN (Figure 6A). These residues also belong to the second connected component of the contact-based network together with other hydrophobic residues, highlighting their importance for protein stability (Figure 6B). Moreover, many mutation sites are located at hub positions in the hydrogen bond network (such as R16, D19, R21, Y38, K65, R70, and Y113) (Figure 6C -D). Overall, due to their pivotal role to mediate different classes of intramolecular interactions, and the introduction of substitutions which would not conserve these interactions, the mutations R16G, D19Y, R21G, P32Q, R70C, Y113C might impact on the structural stability of LC3B.
Figure 6.
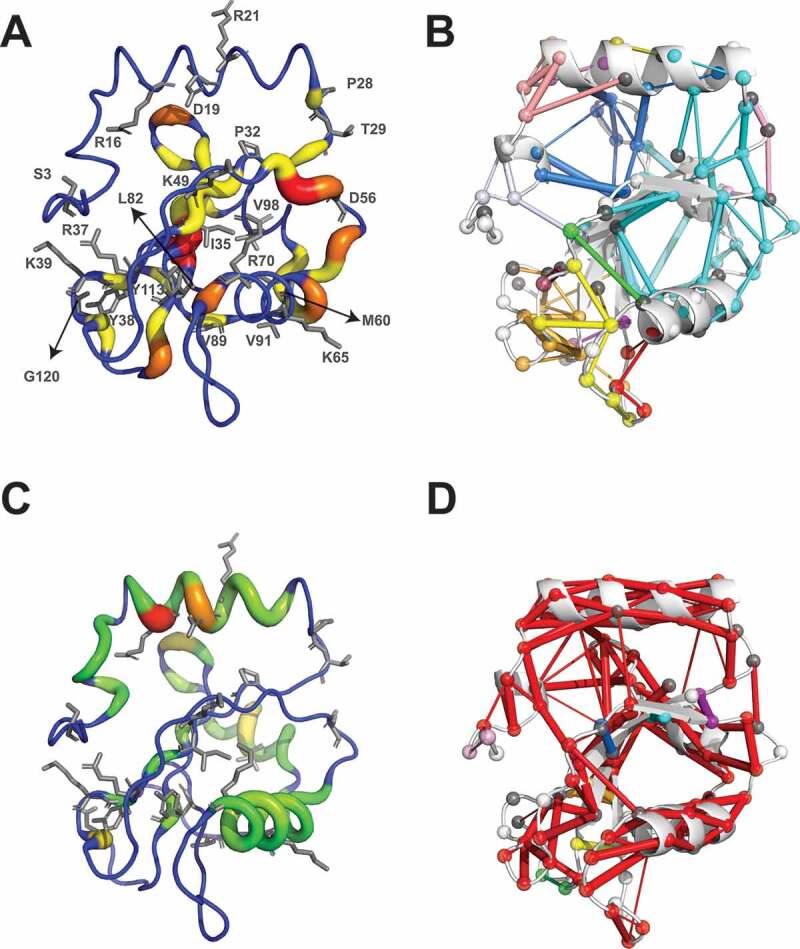
Assessment of long-range effects induced by LC3B missense mutations found in cancer genomic studies. Schematic representation of hubs and connected components for the protein structure network (PSN) based on side-chain contacts (A and B) and hydrogen bonds (C and D), calculated from the CHARMM22* MD ensemble of LC3B. (A and C) Hubs are residues that are highly connected within a PSN. The hubs showed in the figure are color-coded with green (node degree = 3), yellow (4), orange (5), dark orange (6) and red (7), the ribbon thickness indicates the node degree of each hub. (B and D) Connected components are clusters of linked nodes with no edges in common with the nodes belonging to other clusters of the PSN. The results showed in panels A and B refer the contact-based PSN calculations based on a 5.125 Å cutoff
Because 11 mutations occur in charged residues of LC3B, we also calculated the network of electrostatic interactions between positively (arginine and lysine) and negatively charged (aspartate and glutamate) residues in the MD ensemble (Figure S5). D19 is central to a small network of salt bridges with K51 and R11 (which are important for the LIR binding), R16 is on one side of the four-residue network with D106, K8 and D104, which constraint a loop of the protein. K65 and R21 are only involved in local intra-helical salt bridges, whereas R70 shows a persistent interaction with D48. The other charged mutation sites have either low persistent electrostatic interactions or they are not involved in salt bridges and in solvent exposed positions.
To predict effects promoted from distal sites to the LIR binding region, we also calculated the shortest paths of communication from the contact-based PSN between each of the mutation sites and the LIR binding interface, i.e., R10, R11 [78,79], K49, K51, L53, H57 and R70 [78–80], which could be disrupted or weakened by the mutations (Table 1). We identified long-range communication between the LC3B mutation sites only for the interface residues K49, L53, and H57. We observed that I35 is crucial for the communication from the core of the protein to the LIR binding interface at several different sites, spanning different areas of the LIR binding groove. I35 is often an intermediate residue in other paths mediated by different mutation sites, although its substitution to valine is likely not to have a marked effect on these properties (Table 1). K49, apart from playing an important local role in mediating the LIR binding, can also communicate long range with H57 on the LIR binding groove important for the binding of the C-terminal part of the LIR. A similar behavior is observed for M60 and K65 which are pivotal for long range communication to all the three LIR binding sites (K49, L53 and H57, Table 1). In addition, K65 and I35 are part of the same long-range communication spine from the surface of the protein to the LIR binding interface, suggesting that two site communication can occur between the ATG4B binding site to which K65 belongs and the LIR binding site. Long-range effects can also be exerted by P32 to H57 (passing through L53), along with the three valine residues at positions 89, 91 and 98, which seems to act more as intermediate nodes of more complex paths (Table 1).
Table 1.
Shortest paths of communication between the mutation sites and the residues at the interaction interface between the LIR and LC3B in the MD ensembles derived by the CHARMM22* force field
| MUTATION SITE | END NODE: K49 | END NODE: L53 | END NODE: H57 |
|---|---|---|---|
| P32 | L53-I23-P32 (av.weight = 41.2) | H57-P55-K30-L53-I23-P32 (av.weight = 43.8) | |
| I35 | K49-F52-I35 (av.weight = 64.7) | L53-K30-P55-V54-V33-M111-I35 (av.weight = 44) |
H57-V58-V54-V33-M111-I35 (av.weight = 41.1) |
| K49* | H57-V58-V54-V33-M111-I35-F52-K49 (av.weight = 47.8) | ||
| M60 | K49-F52-I35-M111-V83-V89-M60 (av.weight = 53.1) | L53-K30-P55-V54-V33-M111-V83-V89-M60 (av.weight = 51.6) | H57-V58-V54-V33-M111-V83-V89-M60 (av.weight = 50.7) |
| K65 | K49-F52-I35-M111-V33-V54-V58-E62-K65 (av. weight = 45.9) | L53-K30-P55-V58-E62-K65 (av. weight = 45.5) | H57-V58-E62-K65 (av.weight = 38.7) |
| V89 | K49-F52-I35-M111-V83-V89 (av.weight = 56.3) | L53-K30-P55-V54-V33-M111-V83-V89 (av.weight = 53.7) | H57-V58-V54-V33-M111-V83-V89 (av.weight = 52.9) |
| V91 | K49-F52-I35-M111-V33-V54-V58-E62-S61-V91 (av.weight = 48.3) | L53-K30-P55-V58-E62-S61-V91 (av.weight = 49.3) | H57-V58-E62-S61-V91 (av.weight = 46.0) |
| V98 | K49-F52-I35-M111-V83-V89-V98 (av.weight = 60.6) | L53-K30-P55-V54-V33-M111-V83-V89-V98 (av.weight = 57.2) | H57-V58-V54-V33-M111-V83-V89-V98 (av.weight = 57.8) |
Only communication to K49, L53 and H57 was identified for the mutation sites: P32, I35, K49, M60, K65, V89, V91 and V98. * This position is both a LIR-interacting site and a mutation site.
For all these residues that we found critical in distal communication, we speculate that their mutations could alter the native structural communication of LC3B protein from distal sites to the binding sites for different interactors.
Assessment of the mutations studying the structure of LC3B in its membrane-bound state
The active form of LC3B is conjugated to phosphatidylethanolamine (PE) at its conserved C-terminal G120 (i.e., II-form or LC3B–PE). The PE lipid inserts into the autophagic membranes. Therefore, we also investigated the structural properties of the LC3B–PE form when associated with biological membranes and compared them to the data collected on the non-lipidated LC3B membrane-unbound variants described above. At first, we explored the possibility of spontaneous insertion of LC3B–PE inside a lipid bilayer with four MD simulations using a coarse-grained force field, which allow to sample long timescales (Figure 7A). The number of lipid atoms in the proximity of the PE-conjugated G120 was recorded during the simulation time (GitHub). In all the simulations, we observed the spontaneous insertion of LC3B–PE into the membrane, through the PE lipid group within 4 μs of simulation time (Figure 7A,GitHub). After LC3B–PE forms contact with the membrane surface, the PE lipid inserts into the hydrophobic bilayer membrane until its acyl chains are fully buried in the membrane, forming stable interactions with the acyl chains of POPC lipids. This is in agreement with the timescale and the interaction mechanisms observed in a previous work with LC3B from Rattus norvegicus [81].
Figure 7.
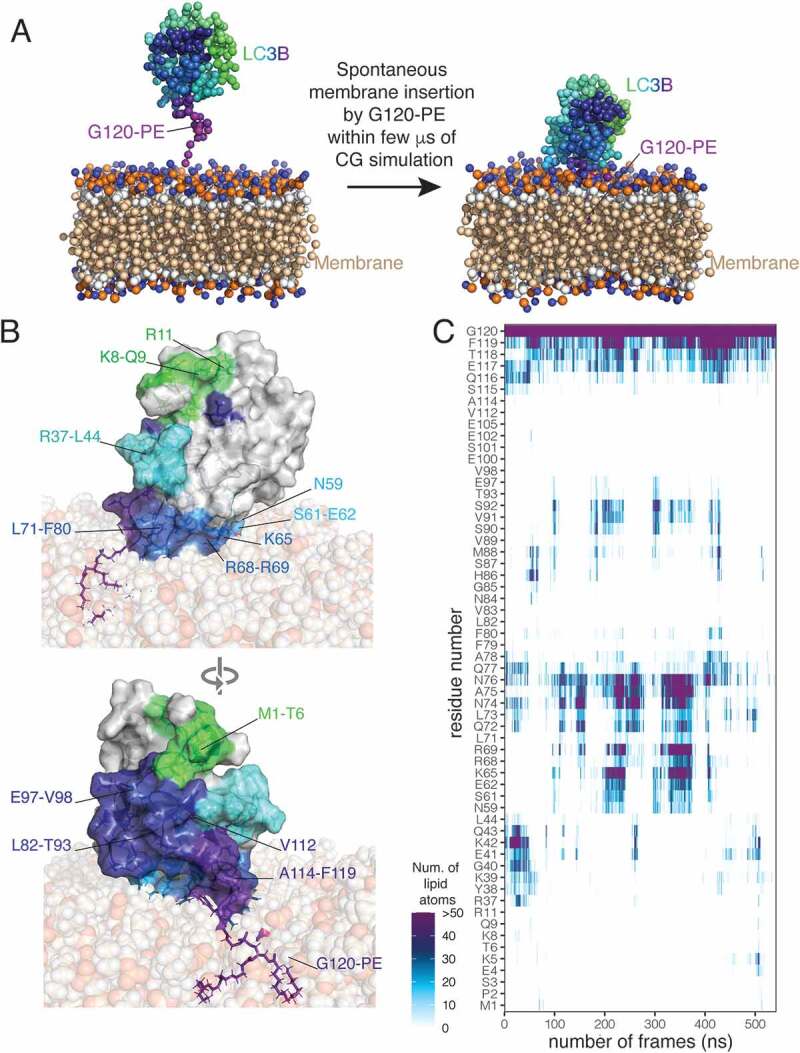
PE-conjugated LC3B spontaneously inserts into bilayer membranes and associates with the membrane lipids through different interaction interfaces. (A) The sphere and stick representation shows the LC3B–PE with a gradient of colors from N- (green) to C-terminal G120-PE (purple) and the bilayer membrane composed by 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). The left panel shows the starting system, after the preparation steps, for the coarse-grained (CG) simulations (the replicate 2) with LC3B–PE in the solvent not making extensive preformed contacts with the bilayer membrane. The right panel shows the system after a few μs of CG simulations in which LC3B–PE spontaneously inserts inside the membrane, by anchoring with the PE lipid. (B) The surface and cartoon representation shows a conformation of LC3B–PE anchored to the bilayer membrane from the full-atom simulation (replicate 2) while the sphere representation shows the membrane. The upper and lower panels show the two sides of LC3B–PE. The gradient of colors from N- (green) to C-terminal G120–PE (purple) on the surface representation indicates the residues identified to form contacts with the membrane for at least 20% of the simulation time of at least one full-atom replicate (C) The plot shows the number of protein-lipid contacts during the simulation time of the full-atom replicate 2, calculated as the number of lipid atoms inside a spherical surround with 6 Å of radius around each residue of LC3B–PE. For clarity sake, we show only residues forming contacts for at least 20% of the simulation time of at least one full-atom replicate
We then used the structural ensemble of LC3B in its membrane-bound form to verify the results that we collected so far with PSN for the protein in its membrane-free state. Hence, we selected three frames from the coarse-grained simulations in which LC3B–PE is inside the membrane and forms contacts with the membrane by a patch of basic residues experimentally validated for the LC3B-membrane recognition [81] and performed all-atom simulations. During the all-atom simulations, LC3B–PE stably interacts with the membrane, remaining anchored by the PE lipid insertion. We performed a scan of the protein-lipid contacts for each protein residue (Figure 7B -C). The interaction interfaces often include positively charged residues that might play a role in the recognition between the protein and the membrane (Figure 7B -C). Possible contributions are provided by residue on helix α1 (residues M1-T6, K8, R11), β1-β2 loop (residues K38 and K42), and helix α3 (i.e., N59, S61-E62, K65, and R68-R69). The helix α1 has been suggested to interact with membranes and promote tethering and fusion during autophagy [82]. K65, R68, and R69 in helix α3 can cause a reduction of autophagosome formation if mutated [81]. We also observed regions of LC3B–PE that are often in the proximity of the membrane during the simulations but do not present patches of positively charged residues. The loops α3-β3 and β3-α4 and the strand β3 (especially L71-F80 and L82-T93) constitute a mostly hydrophobic area close to the C-terminal and the PE-conjugated G120. In summary, we identified multiple possible hotspots to mediate the interactions between LC3B–PE and the membrane, including patches of positively charged or hydrophobic residues.
We then analyzed the solvent accessibility of the mutation sites in the all-atom MD of its free and membrane-bound to evaluate if any of the mutation sites will be masked by the membrane. We found that only R70 can be affected in terms of accessibility by the presence of the membrane, in agreement with its location in proximity of one of the charged hotspots for membrane recognition (Figure 7). R70C was not interested by marked roles in long range communication according to the PSN analysis so this behavior is not affecting the results of the PSN predictions.
We used the structural ensemble of the LC3B–PE form in the membrane-bound state to carry out the same PSN presented above. The hub behavior of the contact-based PSN changed depending on the orientation in the membrane, with T29, I35, M60, L82, V89, P32 as potential hubs (GitHub). Y38, which we first identified in the simulations of the free state is no longer relevant in the network analyses of the membrane-bound states of the protein. The properties of the hydrogen-bond and salt-bridge networks are more conserved than the contact based PSN. In terms of allosteric communication, most of the observation above hold but I35 is not predicted as an important residue for allosteric effect. Moreover, K49 seems to have a marginal effect as end point of communication.
Classification and impact of LC3B missense mutations
We integrated all the data collected in this study to map the different effects that the cancer-related mutations of LC3B could exert, providing a comprehensive view of the many aspects that they can alter and that are ultimately linked to protein function at the cellular level, i.e., protein stability, regulation, abolishment/formation of PTMs or functional motifs for protein-protein interactions, local and distal effects influencing the binding to the partner of interactions (Figure 8A). We then ranked the mutations according to the properties that they alter to help in the classification of potential damaging and neutral ones (Figure 8B). The ranking allowed the selection of mutations to prioritize for validation of their neutral or pathogenic potential, along with planning the proper experimental readout for the validation. For example, if a mutation is predicted damaging in relation to stability, experiments tailored to estimate the cellular protein level and half-life could be used as we recently did for other disease-related proteins [83,84]. On the other side, if the impact is more related to the protein activity or introduction/abolishment of functional motifs, binding assays for example based on peptide arrays, isothermal titration calorimetry, NMR spectroscopy, co-immunoprecipitation can be used together with assays of the related biological readouts in cellular models as for example we recently combined to the study of a LIR-containing scaffolding protein [18]. Assays with upstream modifiers, such as ubiquitination, or phosphorylation assays can instead be used to validate the interplay with PTMs, both in the direction of introduction of new layers of regulation upon mutation or their abolishment.
Figure 8.
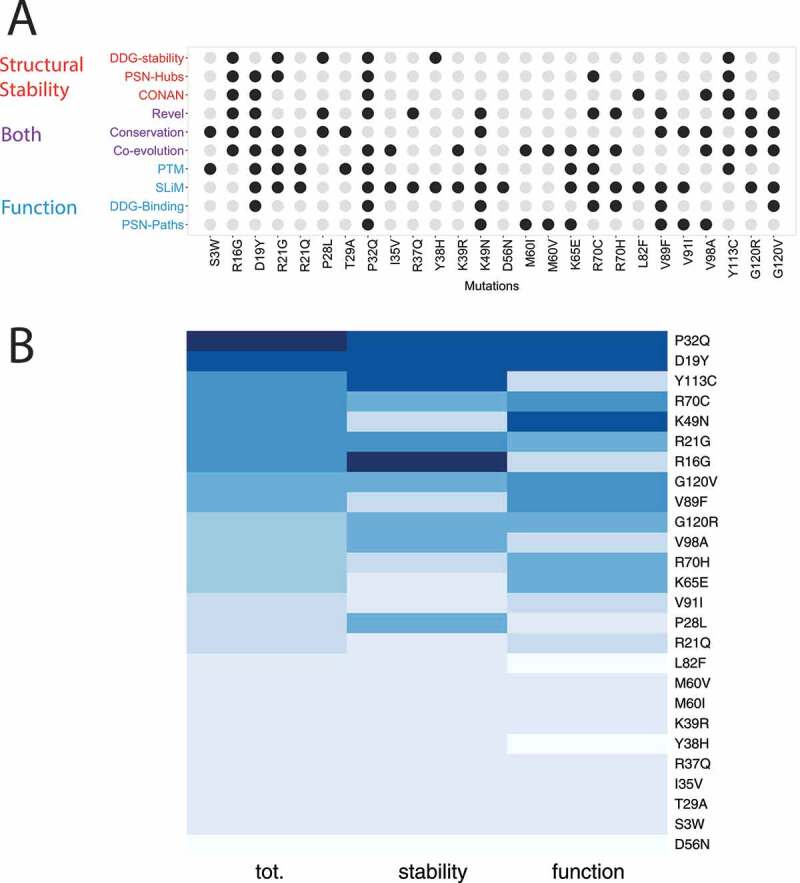
Classification of LC3B mutations found in cancer genomic studies. (A) The different analyses carried out in our study have been aggregated to associate the potential of damaging (driver) or neutral (passenger) effect of each mutation. We used descriptors that account for protein stability (red), function (blue) or implicitly for both (purple). Mutations altering one of these properties are highlighted as black dots. The diagram in panel A allow to link each mutation to a specific effect which could guide the selection of the best set up for experimental validation and further studies. (B) The heatmap is the results of a ranking on a collective score of damaging potential for the mutation (first column). The results for the ranking only according stability and function is showed as a reference in the second and third column of the heatmap. Darker the color more damaging the mutation is
In our case study, we identified three potential classes of detrimental mutations: i) mutations that alter both stability and activity (D19Y, P32Q, and Y113C); ii) detrimental mutations for protein stability only (R16G); iii) mutations neutral for the stability but altering the protein function (mostly K49N and partially G120V, V89F, and R70C).
We then searched in literature for experimental data that could validate our predictions, and we found results in agreement with the functional impact for mutations at R70, D19, G120, and K49, supporting our results. Mutations at R70 showed no accumulation of the pro-forms for LC3B [85], slower kinetics for ATG4B-mediated cleavage [86] and reduced binding for more that 20 interactors [80,87,88]. G120 is fundamental for a proper C-terminal cleavage, which is impaired when this glycine is mutated to alanine [89] and also G120 substitution with alanine has been shown to impair the binding of LMNB1 (lamin B1) with LC3B [90]. Y113C was recently shown to inhibit the enzymatic activity of ATG7 (E1-like enzyme) but not the E2-like activity [34]. Mutations at K49 alter the binding to the phosphorylated variants of the LIR-containing LC3B interactor, FUNDC1 [91], whereas if this residue is mutated to alanine, it can increase the binding of another LIR-containing protein, i.e., Nix [92]. D19N altered the selectivity for phosphorylation and unphosphorylated variants of FUNDC1 [74].
Biochemical validation of LC3B mutations effect on protein stability and SQSTM1/p62 interaction
To experimentally validate our predictions, we also generated six GFP-LC3B mutants: P32Q, I35V, K49N, M60V, K65E and Y113C. Next, we performed transient transfections on HEK 293 cells and we checked the wild-type and mutant variant expression levels (Figure 9). Strikingly, the mutant P32Q showed a strong expression decrease in respect to the wild-type form, validating our predictions on the effect of P32Q mutation on LC3B stability and the neutral effects of I35V, M60V, K65E and K49N. Our results on the variants Y113C and K49N were also in line with recent papers, indicating that these mutations do not have a strong effect on protein stability [34,60,93]. Our scans predict Y113C as destabilizing the protein architecture, thus we wonder if this discrepancy could be related to the fact that the high-throughput mutational scans do not provide enough conformational sampling to model this mutation. Therefore, we used a MD simulation of the Y113C variant to verify if this variant could provide compensatory intermolecular interactions due to local rearrangements induced by the substitution of tyrosine with cysteine. Indeed, we identified new hydrogen bonds and contacts which are likely to compensate for the loss of some of the original native contacts (Table S5).
Figure 9.
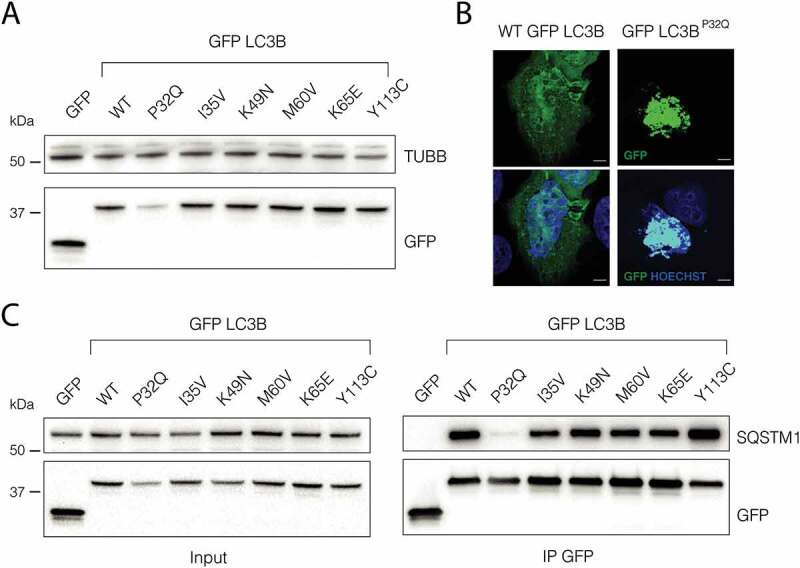
P32Q mutation affects LC3B stability and its ability to interact with SQSTM1/p62. (A) Immunoblot of HEK293 cells transfected with GFP, GFP-LC3B WT or GFP-LC3B mutants n = 3 (B) Representative confocal image of U2OS cells transfected with GFP-LC3B and GFP-LC3BP32Q plasmids. n = 3 Scale bar: 5 µm (C) Immunoblot of GFP and SQSTM1/p62 from GFP immunoprecipitation in HEK293 cells transfected with GFP or GFP-LC3B WT or mutants. n = 3
The other mutants did not show clear differences of expression levels respect to the wild-type form (Figure 9). However, we cannot completely rule out that these mutations could have an effect in a more physiological context since strong transient overexpression could not allow detecting milder effects.
To further investigate the effect of P32Q mutation, we transfected U2OS cells and we checked the protein localization by fluorescence. Strikingly the mutant P32Q showed large aggregates inside the cell, a common feature of protein misfolding (Figure 9B).
To evaluate if the selected mutations could impact on LC3B-LIR interaction, we performed GFP-LC3 co-immunoprecipitations and checked for SQSTM1/p62. Interestingly, the P32Q mutation causes a clear decrease of SQSTM1/p62, supporting the validity of our models (Figure 9B -C). Also, in this case, the other mutants did not show significant differences between the mutants in line with what observed in the in silico mutational scans. The mutant Y113C has been previously shown to impair the binding with ATG7 [34] and to affect LC3B lipidation. Indeed, we confirmed an alteration in LC3BY113C localization following starvation (Figure S6).
Overall our analyses confirmed that P32Q mutation has a profound impact on LC3B protein stability and on its ability to interact with SQSTM1/p62, highlighting the relevance of this mutation in autophagy and cancer.
Conclusions
We unveiled the effects exerted by missense mutations found in cancer genomic studies for the key autophagy ubiquitin-like protein, LC3B, providing a solid computational framework that allows to assess in parallel the impact on the most important properties that define its function and stability (Figure 10). We classified as damaging for function, four mutation sites (R70, D19, G120, and K49) that were experimentally proved to alter the protein activity, supporting our approach. Moreover, we validated experimentally six variants which highlighted the marked effect of the P32Q mutation on both protein stability and interaction with LIR-containing proteins. Moreover, our study, thanks to the collection of MD simulations with nine different force fields, can also guide the selection of physical models for MD simulations of LC3/GABARAP family, here illustrated on LC3B as a prototype of the family.
Figure 10.
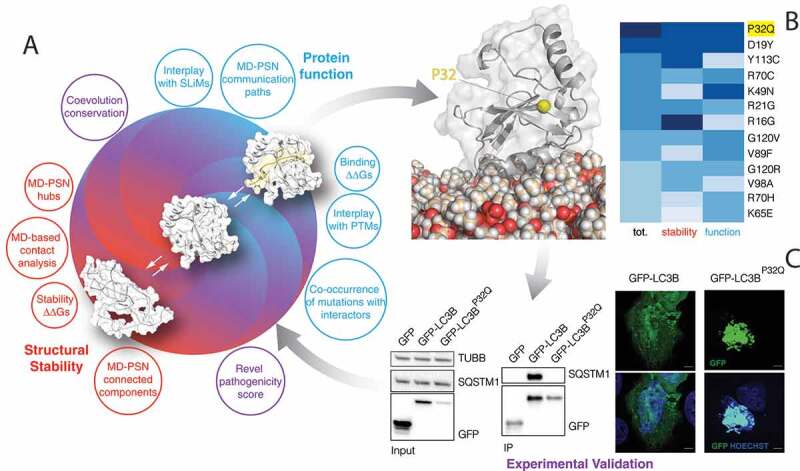
The framework used to obtain a comprehensive classification and analysis of the missense mutations in the coding region of LC3B. (A) We used a collection of different computational approaches to analyze the simulation ensembles and evaluate the impacts that a missense mutation could have on the protein structural stability (i.e., contact analysis, folding ΔΔGs, shown in red), protein function (i.e., binding ΔΔGs, interplay with SLiMs and PTMs, co-occurrence of mutations with interactors, shown in blue) or both of them (e.g. Revel pathogenicity score, coevolution conservation, shown in purple). (B) We used these approaches to classify the missense mutations of LC3B found in cancer genomic studies. The heatmap shows the mutations predicted to be the most damaging (dark blue), ranked on the basis of a collective score (first column) or only according to the predicted impact on stability (second column) or function (third column). The white cartoon shows LC3B anchored to the membrane while the yellow sphere indicates the position of P32, whose mutation into glutamine is predicted to be highly damaging. (C) We experimentally validated the impact of mutations by overexpressing LC3B mutants and checking their expression, interaction, and cellular localization
Our framework provides the groundwork to better understand the impact of mutations found in high-throughput cancer genomics data on a group of proteins that are key players of the autophagy pathway. More in general, it can be applied to the study of cancer proteins to prioritize variants for experimental validation of their damaging or neutral effects (Figure 10). In such cases, our approach can suggest the most convenient experimental methodologies for the validation, depending if the impact of the mutations is likely to be on the protein structural stability or its activity or even more specific aspects such as changes in post-translational regulation or allosteric mechanisms.
Materials and methods
Data availability
All the raw data, inputs, outputs and scripts associated with this publication are available in two repositories: GitHub https://github.com/ELELAB/lc3b_cancer_paper.git and OSF https://osf.io/4zxym/.
Molecular Dynamics (MD) simulations
One-μs MD simulations in explicit solvent of LC3B monomer were collected starting from the free state NMR structure with PDB entry 1V49 [48] using GROMACS [94,95]. We employed nine force fields: ff14SB [96], ff99SBnmr1 [97], ff99SB*-ILDN-Q [98–100], a99SB-disp [101], RSFF2 [102,103], CHARMM22* [104], CHARMM27 [105], CHARMM36 [39] and RSFF1 [106]. An additional 500-ns simulation for the Y113C LC3B variant was collected with CHARMM22*.
We used, as solvent models, TIP3P adjusted for CHARMM force fields [107], TIP3P [108] for AMBER force fields and TIP4P-Ew [109] water model for the RSFF1 force field. We used a dodecahedral box with a minimum distance between protein and box edges of 12 Å applying periodic boundary conditions and a concentration of NaCl of 150 mM, neutralizing the charges of the system. In the simulations, we used the Nε2-H tautomer for all the histidine residues. The system was equilibrated in multiple steps. We carried out productive MD simulations in the canonical ensemble at 300 K and a time-step of 2 fs. We calculated long-range electrostatic interactions using the particle-mesh Ewald summation scheme, whereas we truncated Van der Waals and Coulomb interactions at 10 Å. Other details are provided in the input files in the OSF and GitHub repositories.
Structural assessment of the MD ensembles
We selected a subset of structures from the MD ensembles, taking 100 frames (equally spaced in time) from each of the different simulations for the prediction of chemical shifts and atomic resolution.
In particular, we calculated the backbone and proton side-chain chemical shift values for each MD ensemble with PPM_One [49]. We estimated the root mean square error (RMSE) between the predicted chemical shift values (from the simulations) and the experimentally measured chemical shifts from Biological Magnetic Resonance Bank (BMRB entry 5958) [48] to assess the ability of the force fields in describing a conformational ensemble close to the experimental one. We used for the comparison 119 Cα, 116 C, 119 Hα, 113 H and 114 N chemical shifts.
We predicted the atomic resolutions with ResProx [47] to assess the structural quality of the MD conformational ensembles representing each trajectory. We also verified that the ResProx results and their distribution were not affected by the approach used to select the subset of 100 MD frames. To this aim, we carried out the ResProx calculations on a set of equally-spaced 1000 frames selected from the one-μs CHARMM36 trajectory. Moreover, we carried out structural clustering on the 1000-frame ensemble of CHARMM36 trajectory using the GROMOS algorithm for clustering [110]. In the clustering, we used a mainchain root mean square deviation (RMSD) cutoff of 2.4 Å. We retained only the most populated cluster (which accounts for 888 structures) and estimated the atomic resolution of each structure of the cluster and the corresponding distribution. The two approaches gave results similar to calculations performed on the 100-frame ensembles, featuring similar distributions and median values. The statistical tests that are performed to assess the differences of the atomic resolution data for each MD ensemble for the different force field simulations are detailed in the Statistical Analysis section.
Principal component analysis of the MD trajectories
Principal component analysis (PCA) can be used to extract the essential motions relevant to the function of the protein through the eigenvectors (principal components, PCs) of the covariance matrix of the positional fluctuations observed in MD trajectories, leaving out the irrelevant physically constrained local fluctuations of the protein [111]. We performed all-atom PCA on a concatenated trajectory (including all the trajectories for the nine force fields) superposing the protein using the Cɑ coordinates to compare them in the same subspace. Prior to the calculation, we discarded the six N-terminal and four C-terminal residues from our analyses, to prevent their motion to mask the important motion in the remainder of the protein.
MD ensemble comparison with ENCORE
We have used ENCORE [50] as implemented in the MDAnalysis package [112] to calculate ensemble similarity scores between each pair of ensembles. ENCORE estimates the probability distributions of the conformations that underlie each ensemble and calculates a probability similarity measures between each pair of them. To compare the LC3B simulations, we used the clustering ensemble similarity (CES) approach. The method calculates the ensemble similarity as the Jensen-Shannon divergence between the estimated probability densities. CES partitions the whole conformational space in clusters and uses the relative populations of different ensembles in the clusters as an estimate of probability density. The CES values range between 0 and ln(2), where 0 indicates completely superimposable ensembles and ln(2) means non-overlapping ensembles. The clustering process is carried out using the affinity propagation method [113] The calculation of the similarity score was carried out using 1000 frames for each simulation, on Cα only and excluding the flexible N- and C-terminal tails, as done in the PCA. The pairwise divergence values were visualized in heat maps and visualized as scatter plots using the tree preserving embedding method [114] on the similarity matrices.
Structural alphabets
We compared differences in the sampling of local conformations by analyzing the trajectories using the M32K25 structural alphabet (SA) [115]. This particular alphabet describes the local conformation of the protein by means of unique fragments made of Cα atoms of four consecutive residues, which were originally described by means of three angles. The 25 conformations or letters of the SA represent a set of canonical states describing the most probable local conformations (i.e., conformational attractors) in a set of experimentally derived protein structures. For every simulation, we have used GSATools [116] to encode the conformation of each frame into a SA string, composed of 117 letters for our 120-residue protein. For all the encodings, we transformed the SA representation to that of a reduced structural alphabet (rSA), according to the mapping defined between these two alphabets [117]. The rSA is a reduced representation of the original alphabet in which each letter corresponds to a macro-region of the conformational space. As we will be comparing distributions derived from the structural alphabet (see below), this ensures that the observed differences depend on significant differences between different states rather than the minor differences existing between letters of the original SA.
We devised a per-fragment protocol to estimate the difference in sampling between different simulations. The following procedure was carried out independently for each fragment. For each simulation, we calculated the frequency of each letter over the frames and used this as an estimate of the discrete probability distribution of the letters for that fragment and simulation. We then pairwise compared these distributions using the Jensen-Shannon (JSd) divergence. In this way we obtained a JSd value for every pair of simulations, which accounts for the difference in sampling of different letters. We also included in the comparison the experimental structure (PDB entry 1V49). It should be noted however that since only one frame is available per structure in this case, this does not represent a real probability distribution, just one available conformation. As this conformation has been obtained by modeling tools with constraints from NMR data, we have to consider it as a model itself and not as a reference of the expected final results of SA.
Network analyses of the MD Ensembles
We applied PSN analysis to the MD ensemble as implemented in PyInteraph [75]. We defined as hubs those residues of the network with at least three edges, as commonly done for networks of protein structures [35]. We used the node inter-connectivity to calculate the connected components, which are clusters of connected residues in the graph. For the contact-based PSN, we tested four different distance cutoffs to define the existence of a link between the nodes (5, 5.125, 5.25 and 5.5 Å). Then we selected the cutoff of 5.125 Å as the best compromise between an entirely connected and a sparse network, according to our recent work on PSN cutoffs [76]. The distance was estimated between the center of mass of the residues side chains (except glycines). Since MD force fields are known to have different mass definitions, we thus used the PyInteraph mass databases for each of the MD ensembles.
We also calculated other two PSNs to reflect other classes of intramolecular interactions, i.e., hydrogen bonds and salt bridges. For salt bridges, all of the distances between atom pairs belonging to charged moieties of two oppositely charged residues were calculated, and the charged moieties were considered as interacting if at least one pair of atoms was found at a distance shorter than 4.5 Å. In the case of aspartate and glutamate residues, the atoms forming the carboxylic group were considered. The NH3- and the guanidinium groups were employed for lysine and arginine, respectively. A hydrogen bond was identified when the distance between the acceptor atom and the hydrogen atom was lower than 3.5 Å and the donor-hydrogen-acceptor atom angle was greater than 120°.
To obtain contact, salt bridges or hydrogen bond-based PSNs for each MD ensemble, we retained only those edges which were present in at least 20% of the simulation frames (pcrit = 20%), as previously applied to other proteins [75,118]. We applied a variant of the depth-first search algorithm to identify the shortest path of communication. We defined the shortest path as the path in which the two residues were non-covalently connected by the smallest number of intermediate nodes. All the PSN calculations have been carried out using the PyInteraph suite of tools [75], whereas we used the xPyder plugin for PyMOL [119] the mapping of the connected components on the 3D structure.
Contact analysis with CONAN
We performed the analysis of intramolecular contacts using the software CONtact ANalysis (CONAN) [69]. CONAN allows statistical analyses of contacts in proteins and to study their time evolution in MD trajectories. Inter-residue contacts in CONAN are computed using different cutoff of distances, that can be defined by the user. We used as cutoffs rcut value of 10 Å, and rinter and rhigh-inter values to 5 Å, as previously employed for simulations of ubiquitin [69] and a timestep of one ns for the analyses. The output data from CONAN were further analyzed: i) to estimate the fraction of each contact formation in the simulation time, calculated as the frame in which the contact is identified divided by the total number of frames in the trajectory, and to ii) estimate the number of encounters, as the number that a contact is formed and broken during the trajectory.
Identification of cancer missense mutations and their annotation
We collected and aggregated a subset of cancer-related missense somatic mutations found in LC3B from cBioPortal [120] and COSMIC version 86 [121], considering all cancer types and excluding those mutations classified as natural polymorphisms. Moreover, we collected annotations on PTMs at the mutation sites using a local version of the PhosphoSitePlus database [122], downloaded on 04/05/2018. Additional PTMs have been manually annotated through a survey of the literature on LC3B. We collected short linear motifs (SLiMs) located in proximity of the identified mutations using predictions from the Eukaryotic Linear Motif (ELM) server [123]. Those SLiMs for which an interaction is not compatible with their localization on the LC3B structure have been discarded by further analyses, such as a PP2A docking site. Moreover, for each cancer samples where the mutation was identified we verified (when the information was available): i) the expression level of the LC3B gene; ii) if other mutations were occurring in the LC3B gene; and iii) if any of the interactors (see below) was mutated in the same sample. We also verified that any of the mutations was reported with high frequency in the healthy population, by searching in the ExAC database [52]. We predicted if each of the mutant variant could harbor new SLiMs querying ELM with the sequence of each mutant variant.
We annotated each variant with the REVEL score [53], as available on the MyVariant.info web resource [124]. REVEL is an ensemble method for predicting the pathogenicity of missense variants from the scores generated by other individual prediction tools, which was found to be among the top performing pathogenicity predictors in a recent benchmarking study [125]. The REVEL score can range from 0 to 1, with higher values indicating a stronger indication of pathogenicity. As done in the benchmarking study, we classified as pathogenic those variants having a score ≥ 0.4, which represents the best trade-off between sensitivity and specificity. All the analyses have been done in October 2018.
Coevolution analysis
We used two different parameters estimated by Gremlin [126] to analyze the mutation sites. In particular, we employed: i) the conservation score estimated by the coupling matrix for the wild-type and the mutated residue at a certain position; ii) the residues that are coupled to the wild-type residue with a scaled score higher than one. We also derived a logo plot from the Gremlin sequence alignment with WebLogo [127].
LC3B interaction network and identification of LIR-containing candidates
We retrieved the known LC3B interactors through the Integrated Interaction Database (IID) version 2018–05 [70]. We retained only those interactions identified by at least two of the studies annotated in the database. We then predicted LIR motifs for each of the interactors with iLIR [71] and retained only those with a score higher than 11. This threshold was selected as it allows for a higher sensitivity (92%) at price of slightly lower specificity [71]. We also verified through literature search if any of the interactors include one or more already experimentally verified LIR motifs. For each interactor with at least one LIR motif, we annotated the occurrence of cancer mutations in the same samples where a mutation of LC3B was found. We then retained only the interactors for which this mutation was abolishing a LIR motif or has mutations in its proximity. The resulting LC3B interactors were displayed as a network plot, using the igraph R package and in-house developed code.
Model of the interaction between LC3B and PLEKHM1 including LIR flanking regions
We used Modeller version 9.15 [128] to generate models of LC3B1-120-PLEKHM1 627–643 complexes, using the crystallographic structure with PDB entry 3X0W [129] as a starting structure. Only the flanking regions to the LIR core motif for which the coordinates were not available in the PDB file have been modeled. We generated 100 models. We then calculated the solvent-accessible surface area (SASA) of the side-chains of the HP1 and HP2 residues using GROMACS tools, discarding models having a SASA larger than 7.5 Å 2 for the HP1 or larger than 5 Å 2 for the HP2 residues. These cutoff values are based on SASA values calculated on a set of reference experimental structures of LC3/GABARAP-LIR complexes. Finally, the models were ranked on the radius of gyration of the LIR peptide and the model with the highest radius for each complex (i.e., a more extended structure) was selected as the final models.
Structure-based prediction of impact on protein stability and binding free energies
We employed the FoldX energy function [67] to perform in silico saturation mutagenesis using a Python wrapper that we recently developed. We used the same protocol that we recently applied to another protein [64]. Calculations with the wrapper resulted in an average ΔΔG (differences in ΔG between mutant and wild-type variant) for each mutation over five independent runs performed using the NMR structure of LC3B (PDB entry 1V49 [48]) and the X-ray crystallographic structure 3VTU [130]. We used the same pipeline to estimate the effect of mutations on the interaction free energy with the LIR domains using the structure of LC3B in complex with SQSTM1/p62, FUNDC1, FYCO1, OPTN, PLEKHM1 and ATG4B. This was performed by using the AnalyseComplex FoldX command on the mutant variant and the corresponding wild-type conformation, and calculating the difference between their interaction energies.
Moreover, we also accounted for a correction to the FoldX energy values related to protein stability, as defined by Tawfik’s group [131] to make the ΔΔG FoldX values more comparable to the expected experimental values, as previously described [64]. The experimental value of folding ΔG for LC3B is unknown to the best of our knowledge. Nevertheless, values in the range of −5/–15 kcal/mol are generally expected for the net free energy of folding of proteins [132,133]. Since LC3B has a ubiquitin-like fold, we used, as a reference, the free energy of folding measured for ubiquitin [134] which is −7.2 kcal/mol.
Moreover, we predicted the ΔΔG of stability upon mutation also using a protocol based on the Rosetta relax and cartesian_ddg applications using version 3.11 [135]. The protocol uses a single initial structure which is relaxed in the Cartesian space and further optimizes the structure with sampling methods in the Cartesian space. We ran the protocol with the same parameters used in the original publication. The only exception is the relaxation script used by cartesian_ddg, which is hard-coded and changed since the protocol was developed. We calculated the ΔΔG values by averaging over the values predicted for the mutant and wild-type structures produced by three iterations of cartesian_ddg.
We also collected predictions of the ΔΔGs of binding upon mutation using the Rosetta-based Flex ddG protocol [136] on the complexes between LC3B and LIRs or ATG4B mentioned above. This protocol couples standard side-chains repacking and minimization with a backrub approach to produce an ensemble of structures sampling backbone degrees of freedom. Flex ddG returns ΔΔG scores in Rosetta Energy Units (REUs). We ran the protocol for each point mutation setting 35000 backrub trials, 5000 maximum iterations per minimization and an absolute score threshold for minimization convergence of 1.0 REUs. We generated ensembles of 35 different structures for each mutant and calculated the average ΔΔGs. For both protocols, we performed the scans using the ref2015 and talaris2014 energy functions. Talaris2014 has been shown to perform slightly better in the benchmarking of Flex ddG [136]. Both protocols return ΔΔG values in REUs, which we converted into kcal/mol using the conversion factors provided for each energy function by Park et al [135].
Molecular dynamics simulations with membranes
We performed coarse-grained (CG) MD simulations to spontaneously observe the insertion inside the membrane of LC3B–PE. We carried out CG MD simulations of LC3B–PE in explicit solvent with GROMACS version 5.1.2 using the MARTINI force field version 2.2 together with an Elastic Network in Dynamic approach [137,138,139]. The elastic network had a force constant of 500 kJ/mol and allowed to maintain the tertiary structure of LC3B–PE. We derived the parameters of the C-terminal PE-conjugated G120 for the MARTINI force field from the already existing parameters of the glycine and 1-palmitoyl-2-oleoly-sn-phosphoethanolamine (POPE) lipid. We designed four different CG systems (i.e., replicate1-4) by using CHARMM-GUI, each including a single lipidated LC3B–PE and a bilayer membrane composed of 238 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid molecules. In each CG system, we localized LC3B–PE in the aqueous solvent, at a different distance from the center of the bilayer membrane. We used the NMR structure of LC3B (PDB entry 1V49) as starting structure and modeled the PE lipid covalently bound to the C-terminal G120 with PyMOL. To evaluate the effect of the starting orientation of the PE lipid and the C-terminal tail of LC3B on the insertion inside the bilayer membrane, we reoriented the PE lipid toward the bilayer membrane in the starting structures. We employed periodic boundary conditions, setting the distance between periodic images of the membrane to be at least ~110 Å in height to allow the diffusion of LC3B–PE in the solvent. We gradually equilibrated the CG systems through a series of energy minimization, solvent and membrane equilibration, thermalization, and pressurization. We performed four CG productive simulations at 300 K and an integration time step of 20 fs (from 5 μs to 14.5 μs). Other parameters can be found in the input files in the OSF repository.
We then extracted three frames from the CG replicate 2–4 in which LC3B–PE is associated with the membrane. We mapped the three CG frames (each composed by LC3B–PE, bilayer membrane, solvent, and ions) to a full-atom description using the CHARMM36 force field and a modified version of the initram-V5 and backmapping scripts obtained from http://cgmartini.nl/. We derived the parameters of the C-terminal PE-conjugated G120 for the CHARMM36 force field from the already existing parameters of the glycine and POPE lipid. We used these three systems as starting structure for full-atom MD simulations, employing the CHARMM36 force field and the TIP3P solvent model. We gradually equilibrated the systems through a series of energy minimization and equilibrations. For more details the parameter files are available in the OSF repository. We carried out productive simulations with a time-step of 2 fs in periodic boundary conditions. We collected three full-atom simulations (named replicate 1–3) with different time length (from 500–600 ns).
We calculated possible contacts between LC3B–PE and the lipids by calculating during the simulation time the number of lipid atoms inside a spherical surround with 6 Å of radius centered in the center of mass of each residue. We filtered only the residues that are involved in forming contact with the lipids for at least 20% of the frames of at least one full-atom simulation. We also calculated the solvent accessibility of the mutation sites in the all-atom MD simulations of the free and membrane-bound state of LC3B to evaluate possible masking effects by the interaction with the membrane.
Cell lines and culture
Cell lines were grown at 37°C in a humidified incubator containing 5% CO2. U2OS and HEK293 cells were purchased from the American Type Culture Collection, ATCC (CRL-11268) and cultured in Dulbecco’s modified eagle’s medium (DMEM) GlutaMAX (Gibco, 31966–021) supplemented with 10% fetal bovine serum (FBS; Gibco, 10091148) and antibiotics.
Transfections and treatments
Transient transfections were performed using polyethylenimine linear MW 25000 (PEI; Polysciences, 23966–2) for HEK293 cells and GeneJuice transfection reagent (Merck-Millipore, 70967) for U2OS cells following to the producers’ protocols. Earle’s Balanced Salt Solution (EBSS; Sigma-Aldrich, E2888) was used for starvation treatments (2 h).
DNA constructs and primers
EGFP-LC3 plasmid was purchased by Addgene (11546; deposited by Karla Kirkegaard [139]). mCherry-GFP and EGFP plasmids were kindly provided by Francesco Cecconi’s laboratory.
Single site mutagenesis was performed by using the following primers:
P32fw ccaaccaaaatccAggtgataatagaacgatacaag
P32rv caccTggattttggttggatgctgctc
I35V fw ccggtgataGtagaacgatacaagggtgagaag
I35V rv tcgttctaCtatcaccgggattttggttggatg
K49N fw ctggataaCacaaagttccttgtacctgaccatg
K49N rv aggaactttgtGttatccagaacaggaagctg
M60V fw ccatgtcaacGtgagtgagctcatcaagataattag
M60V rv ctcactcaCgttgacatggtcaggtacaagg
K65E fw ctcatcGagataattagaaggcgcttacagctc
K65E rv cgccttctaattatctCgatgagctcactcatgttgac
Y113C fw tggtctGtgcctcccaggagacg
Y113C rv tgggaggcaCagaccatgtacaggaatcc
Antibodies
GFP (Santa Cruz Biotechnology, sc8334; WB 1:1000), TUBB/beta tubulin (Cell Signaling Technology, T4026; WB 1:1000), SQSTM1/p62 (MBL International, PM045; WB 1:1000), LAMP2 (Abcam, H4B4; IF 1:400).
Co-immunoprecipitation
Cells were collected 24 h after the transfection, washed in PBS (Gibco, 14040–091) and lysed in lysis buffer containing 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.5% Triton X-100 (Sigma-Aldrich, T8787) with protease and phosphatase inhibitors (PhosSTOP, Sigma-Aldrich, 4906837001, cOmplete™, EDTA-free Protease Inhibitor Cocktail, Sigma-Aldrich, 4693132001). Lysates (300 µg) were incubated with GFP-trap MA beads (Chromotek, gtma-20) for 1 h at 4°C. The beads were washed 5 times in IP lysis buffer with protease and phosphatase inhibitors and the precipitates were eluted in 4× Laemmli sample buffer for 10 min at 95°C. The eluates were than resolved by SDS-PAGE and western blotting.
Immunoblotting
Cell were incubated for 30 min on ice with lysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.5% Triton X-100) with protease and phosphatase inhibitors. Supernatants were collected after 15 min centrifugation at 16000 g and protein extracts were quantified using the DC protein assay (Bio-Rad, 5000112), and denatured in NuPAGE® LDS Sample Buffer (ThermoFisher, NP0007). Proteins were separated on acrylamide gradient gels (Bio-Rad, 4561083) and blotted onto Nitrocellulose membranes (Bio-Rad, 1704158) using the Trans-Blot turbo system (Bio-Rad). Blocking was performed in 5% nonfat dry milk in PBS plus 0.1% Tween-20 (Fisher Bioreagents, BP337). Membranes were incubated in primary antibodies in 5% nonfat dry milk in PBS plus 0.1% Tween-20 at 4°C overnight followed by incubation in secondary horseradish-peroxidase (HRP)-conjugated antibodies (1:10000; Bio-Rad, 1706515, 1706516) for 1 h at room temperature. Image acquisition was performed with ChemiDoc Imaging Systems (Bio-Rad).
Immunofluorescence
Cells were grown on plastic coverslip and fixed with 4% formaldehyde for 10 min. For LAMP2 immunofluorescence cells were permeabilized in ice-cold MeOH for 3 min at −20°C. Blocking was performed with Buffer 1 (PBS, 1% BSA [VWR Chemicals, 0332] and 0.3% Triton X-100] + 5% goat serum [EMD Millipore, S26] for 60 min). The slides were than incubated for 1 h with the primary antibody in Buffer 1. Following 3 washes in PBS, slides were incubated for 1 h with secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, A-11001) diluted in Buffer 2 (PBS containing 0.25% BSA and 0.1% Triton X-100). DNA was stained with Hoechst 3342 (ThermoFisher Scientific, H3570; 1:1000 in PBS) and slides were mounted on coverslips with fluorescence mounting medium (Dako, S3023). Image acquisition was performed with laser scanning confocal microscopes (LSM700 Carl Zeiss A/S). 4 to 6 consecutive Z-stacks (distance between planes = 0.29 μm) have been acquired and projected using the Max. Intensity projection.
Statistical analysis
All the wet lab experiments were repeated three times.
To assess whether the ResProx data for the different force field simulations were significantly different, we first considered the distribution of the atomic resolution data for each MD ensemble and performed the Shapiro-Wilk test to evaluate if our samples come from a normal distribution. According to this test, most of the ResProx data distributions do not come from a normally-distributed population. In fact, the distribution of the sampled R values is either left- or right-skewed, due to outlier structures. The only exceptions are the data of the MD ensembles obtained with ff99SBnmr1 and a99SB-disp. We performed a pairwise Wilcoxon (Mann-Whitney U) rank sum test with continuity correction adjusting the p-value for multiple testing with the Holm-Bonferroni method on the R sets obtained from each force field pair to test whether the samples were selected from populations having the same distributions. The samples of R values from different force field simulation pairs that have p-values higher than 0.05 are not significantly different from each other under the Holm-Bonferroni correction (Table S1). R functions shapiro.test and pairwise.wilcox.test implemented in the stats R package version 3.6.1 were used for the calculations.
Supplementary Material
Acknowledgments
The authors would like to thank Giuseppe Filomeni and Lipi Thukral for fruitful comments and discussion.
Funding Statement
This project was supported by LEO fondet grants (LF17006, LF17024), Carlsberg fondet Distinguished Fellowship (CF18-0314), Danmarks Frie Forskningsfond, Natural Science, Project 1 (102517), Danmarks Grundforskningsfond (DNRF125). Moreover, the project has been supported by a KBVU pre-graduate fellowship to MM and a Netaji Subhash ICAR international fellowship (Govt. of India) to MK to work in EP group. BAF is partially supported by COST-STSM-BM1405-34558. The calculations described in this paper were performed using the DeiC National Life Science Supercomputer Computerome at DTU (DK), a DeiC Pilot grant OULC3P62 on Abacus (DK) DECI-PRACE 14th and 15th HPC Grants for calculations on Archer (UK), and ISCRA-CINECA HP10C0T58M and HP10C0T58.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Gatica D, Lahiri V, Klionsky DJ.. Cargo recognition and degradation by selective autophagy. Nat Cell Biol [Internet]. 2018;20:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. Embo J [Internet]. 2017;36:1811–1836. Available from: http://emboj.embopress.org/lookup/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2013;20:460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol [Internet]. 2016;428:1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schaaf MBE, Keulers TG, Vooijs MA, et al. LC3/GABARAP family proteins: autophagy-(un)related functions. Faseb J. 2016;30:3961–3978. [DOI] [PubMed] [Google Scholar]
- [6].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell [Internet]. 2010;140:313–326. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20144757%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2852113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J [Internet]. 2000;19:5720–5728. Available from: http://emboj.embopress.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sora V, Kumar M, Maiani E, et al. Structure and dynamics in the ATG8 family from experimental to computational techniques.Front. Cell Dev. Biol. 2020;8:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci. 2014;127:3–9. [DOI] [PubMed] [Google Scholar]
- [11].Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol [Internet]. 2014;21:336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alemu EA, Lamark T, Torgersen KM, et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem. 2012;287:39275–39290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Noda NN, Kumeta H, Nakatogawa H, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–1218. [DOI] [PubMed] [Google Scholar]
- [14].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy*[S]. J Biol Chem. 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- [15].Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci [Internet]. 2013;126:3552–3562. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3744024&tool=pmcentrez&rendertype=abstract [DOI] [PubMed] [Google Scholar]
- [16].Wirth M, Zhang W, Razi M, et al. Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins. Nat Commun [Internet]. 2019;10:2055. Available from: http://www.nature.com/articles/s41467-019-10059-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Holdgaard SG, Cianfanelli V, Pupo E, et al. Selective autophagy maintains centrosome integrity and accurate mitosis by turnover of centriolar satellites. Nat Commun. 2019;10:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Di Rita A, Peschiaroli A, D′Acunzo P, et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun [Internet]. 2018;9:3755. [cited 2019 Feb 28]. Available from: http://www.nature.com/articles/s41467-018-05722-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol [Internet]. 2018;20:521–527. [DOI] [PubMed] [Google Scholar]
- [20].Mancias JD, Kimmelman AC. Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol [Internet]. 2016;428:1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rybstein MD, Bravo-San Pedro JM, Kroemer G, et al. The autophagic network and cancer. Nat Cell Biol [Internet]. 2018;20:243–251. [DOI] [PubMed] [Google Scholar]
- [22].Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb Perspect Biol [Internet]. 2012;4:a008821. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16874083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thorburn A. Autophagy and its effects: making sense of double-edged swords. PLoS Biol. 2014;12:10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer [Internet]. 2012. DOI: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Singh SS, Vats S, Chia AYQ, et al. Dual role of autophagy in hallmarks of cancer. Oncogene [Internet]. 2018;37:1142–1158. [DOI] [PubMed] [Google Scholar]
- [28].Lazova R, Camp RL, Klump V, et al. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].He Y, Zhao X, Subahan NR, et al. The prognostic value of autophagy-related markers beclin-1 and microtubule-associated protein light chain 3B in cancers: a systematic review and meta-analysis. Tumour Biol [Internet]. 2014;35:7317–7326. [cited 2019 May 9]. Available from: http://link.springer.com/10.1007/s13277-014-2060-4 [DOI] [PubMed] [Google Scholar]
- [30].Tang JY, Hsi E, Huang YC, et al. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum Pathol [Internet]. 2013;44:2558–2562. [DOI] [PubMed] [Google Scholar]
- [31].Zhao H, Yang M, Zhao J, et al. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. 2013;30: 475. [DOI] [PubMed] [Google Scholar]
- [32].Wu DH, Jia CC, Chen J, et al. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumor Biol. 2014;35:12225–12233. [DOI] [PubMed] [Google Scholar]
- [33].Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine [Internet]. 2016;14:15–23. Available from: http://linkinghub.elsevier.com/retrieve/pii/S2352396416304959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nuta GC, Gilad Y, Gershoni M, et al. A cancer associated somatic mutation in LC3B attenuates its binding to E1-like ATG7 protein and subsequent lipidation. Autophagy [Internet]. 2018;15548627.2018.1525476. Available from: https://www.tandfonline.com/doi/full/10.1080/15548627.2018.1525476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Papaleo E. Integrating atomistic molecular dynamics simulations, experiments, and network analysis to study protein dynamics: strength in unity. Front Mol Biosci [Internet]. 2015;2:1–6. [cited 2015 Oct 8]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4445042&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lindorff-Larsen K, Maragakis P, Piana S, et al. Systematic validation of protein force fields against experimental data. PLoS One [Internet]. 2012;7:e32131. [cited 2015 Aug 27]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3285199&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lange OF, van der Spoel D, de Groot BL. Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data. Biophys J [Internet]. 2010;99:647–655. [cited 2015 Jul 8]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2905107&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bowman GR. Accurately modeling nanosecond protein dynamics requires at least microseconds of simulation. J Comput Chem. 2016;37:558–566. [DOI] [PubMed] [Google Scholar]
- [39].Huang J, Rauscher S, Nawrocki G, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods [Internet]. 2017;14:71–73. [cited 2018 Apr 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27819658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Martín-García F, Papaleo E, Gomez-Puertas P, et al. Comparing molecular dynamics force fields in the essential subspace. PLoS One [Internet]. 2015;10:e0121114. [cited 2015 Oct 17]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4374674&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maragakis P, Lindorff-Larsen K, Eastwood MP, et al. Microsecond molecular dynamics simulation shows effect of slow loop dynamics on backbone amide order parameters of proteins. J Phys Chem B [Internet]. 2008;112:6155–6158. [cited 2015 Oct 17]. Available from: https://pubs.acs.org/doi/10.1021/jp077018h [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sultan MM, Wayment-Steele HK, Pande VS. Transferable Neural Networks for Enhanced Sampling of Protein Dynamics. J. Chem. Theory Comput. 2018; 14(4):1887–1894 [DOI] [PubMed] [Google Scholar]
- [43].Showalter SA, Brüschweiler R. Validation of molecular dynamics simulations of biomolecules using NMR spin relaxation as benchmarks: application to the AMBER99SB force field. J Chem Theory Comput [Internet]. 2007;3:961–975. [cited 2019 May 10]. Available from: https://pubs.acs.org/doi/10.1021/ct7000045 [DOI] [PubMed] [Google Scholar]
- [44].Long D, Li DW, Walter KFA, et al. Toward a predictive understanding of slow methyl group dynamics in proteins. Biophys J. 2011;101:910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lindorff-Larsen K, Maragakis P, Piana S, et al. Picosecond to millisecond structural dynamics in human ubiquitin. J Phys Chem B [Internet]. 2016;120:8313–8320. Available from: http://pubs.acs.org/doi/abs/10.1021/acs.jpcb.6b02024 [DOI] [PubMed] [Google Scholar]
- [46].Li DW, Brüschweiler R. Certification of molecular dynamics trajectories with NMR chemical shifts. J Phys Chem Lett. 2010;1:246–248. [Google Scholar]
- [47].Berjanskii M, Zhou J, Liang Y, et al. Resolution-by-proxy: a simple measure for assessing and comparing the overall quality of NMR protein structures. J Biomol NMR [Internet]. 2012;53:167–180. [cited 2016 Jul 13]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22678091 [DOI] [PubMed] [Google Scholar]
- [48].Kouno T, Mizuguchi M, Tanidal I, et al. Solution structure of microtubule-associated protein light chain 3 and identification of its functional subdomains. J Biol Chem. 2005;280:24610–24617. [DOI] [PubMed] [Google Scholar]
- [49].Li D, Brüschweiler R. PPM-One: A static protein structure based chemical shift predictor. J Biomol NMR [Internet]. 2015;62:403–409. [cited 2016 Jul 13]. Available from: http://link.springer.com/10.1007/s10858-015-9958-z [DOI] [PubMed] [Google Scholar]
- [50].Tiberti M, Papaleo E, Bengtsen T, et al. ENCORE: software for quantitative ensemble comparison. de Groot BL, editor. PLoS Comput Biol [Internet]. 2015;11:e1004415. [cited 2019 May 9]. Available from: http://dx.plos.org/10.1371/journal.pcbi.1004415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Craveur P, Joseph AP, Esque J, et al. Protein flexibility in the light of structural alphabets. Front Mol Biosci. 2015;2:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kobayashi Y, Yang S, Nykamp K, et al. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet [Internet]. 2016;99:877–885. [cited 2019 Mar 6]. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002929716303706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jiang H, Cheng D, Liu W, et al. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem Biophys Res Commun [Internet]. 2010;395:471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wilkinson DS, Hansen M. LC3 is a novel substrate for the mammalian Hippo kinases, STK3/STK4. Autophagy. 2015;11:856–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wilkinson DS, Jariwala JS, Anderson E, et al. Phosphorylation of LC3 by the hippo kinases STK3/STK4 is essential for autophagy. Mol Cell [Internet]. 2015;57:55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Huang R, Xu Y, Lippincott-schwartz J, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell [Internet]. 2015;57:456–466. [DOI] [PubMed] [Google Scholar]
- [58].Wagner SA, Beli P, Weinert BT, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics [Internet]. 2011;10:M111.013284. Available from: http://www.mcponline.org/lookup/doi/10.1074/mcp.M111.013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Huang R, Liu W. Identifying an essential role of nuclear LC3 for autophagy. Autophagy [Internet]. 2015;11:852–853. [cited 2019 May 10]. Available from: http://www.tandfonline.com/doi/full/10.1080/15548627.2015.1038016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Song T, Su H, Yin W, et al. Acetylation modulates LC3 stability and cargo recognition. FEBS Lett. 2019;593:414–422. [DOI] [PubMed] [Google Scholar]
- [61].Xue Y, Ren J, Gao X, et al. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics [Internet]. 2008;7:1598–1608. [cited 2019 May 10]. Available from: http://www.mcponline.org/lookup/doi/10.1074/mcp.M700574-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Blom N, Sicheritz-Pontén T, Gupta R, et al. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. [DOI] [PubMed] [Google Scholar]
- [63].Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J [Internet]. 2009;28:1341–1350. [cited 2019 May 10]. Available from: http://emboj.embopress.org/cgi/doi/10.1038/emboj.2009.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nygaard M, Terkelsen T, Olsen AV, et al. The mutational landscape of the oncogenic MZF1 SCAN domain in cancer. Front Mol Biosci. 2016;3:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Riera C, Lois S, de la Cruz X. Prediction of pathological mutations in proteins: the challenge of integrating sequence conservation and structure stability principles. Wiley Interdiscip Rev Comput Mol Sci [Internet]. 2014;4:249–268. [Google Scholar]
- [66].Stein A, Fowler DM, Hartmann-Petersen R, et al. Biophysical and mechanistic models for disease-causing protein variants. Trends Biochem Sci [Internet]. 2019;1–14. DOI: 10.1016/j.tibs.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. [DOI] [PubMed] [Google Scholar]
- [68].Tiberti M, Terkelsen T, Canter Cremers T, et al. MutateX: an automated pipeline for in-silico saturation mutagenesis of protein structures and structural ensembles. bioRxiv 2019. 10.1101/824938 [DOI] [PubMed] [Google Scholar]
- [69].Mercadante D, Gräter F, Daday C. CONAN: a tool to decode dynamical information from molecular interaction maps. Biophys J. 2018;114:1267–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kotlyar M, Pastrello C, Malik Z, et al. IID 2018 update: context-specific physical protein-protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2019;47:D581–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jacomin AC, Samavedam S, Promponas V, et al. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy [Internet]. 2016;12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Suzuki H, Tabata K, Morita E, et al. Structural basis of the autophagy-related LC3/Atg13 LIR complex: recognition and interaction mechanism. Structure [Internet]. 2014;22:47–58. [DOI] [PubMed] [Google Scholar]
- [73]. Rogov VV, Stolz A, Ravichandran AC, et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep [Internet]. 2017;18:1382–1396. [cited 2018 Dec 20]. Available from: http://embor.embopress.org/lookup/doi/10.15252/embr.201643587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kuang Y, Ma K, Zhou C, et al. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy [Internet]. 2016;12:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tiberti M, Invernizzi G, Lambrughi M, et al. PyInteraph : a framework for the analysis of interaction networks in structural ensembles of proteins. J Chem Inf Model. 2014;54:1537–1551. [DOI] [PubMed] [Google Scholar]
- [76].Viloria JS, Allega MF, Lambrughi M, et al. An optimal distance cutoff for contact-based protein structure networks using side-chain centers of mass. Sci Rep. 2017;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jónsdóttir LB, Ellertsson B, Invernizzi G, et al. The role of salt bridges on the temperature adaptation of aqualysin I, a thermostable subtilisin-like proteinase. Biochim Biophys Acta - Proteins Proteomics [Internet]. 2014;1844:2174–2181. [DOI] [PubMed] [Google Scholar]
- [78].Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. [DOI] [PubMed] [Google Scholar]
- [79].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. [DOI] [PubMed] [Google Scholar]
- [80].Olsvik HL, Lamark T, Takagi K, et al. FYCO1 contains a C-terminally extended, LC3A/B-preferring LC3-interacting region (LIR) motif required for efficient maturation of autophagosomes during basal autophagy. J Biol Chem. 2015;290:29361–29374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Thukral L, Sengupta D, Ramkumar A, et al. The molecular mechanism underlying recruitment and insertion of lipid-anchored LC3 protein into membranes. Biophys J [Internet]. 2015;109:2067–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Weidberg H, Shpilka T, Shvets E, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell [Internet]. 2011;20:444–454. [cited 2020 Jan 9]. Available from: https://linkinghub.elsevier.com/retrieve/pii/S153458071100075X [DOI] [PubMed] [Google Scholar]
- [83].Nielsen SV, Stein A, Dinitzen AB, et al. Predicting the impact of Lynch syndrome-causing missense mutations from structural calculations. Nichols KE, editor. PLOS Genet [Internet]. 2017;13:e1006739. [cited 2018 Apr 8]. Available from: http://dx.plos.org/10.1371/journal.pgen.1006739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Scheller R, Stein A, Nielsen SV, et al. Toward mechanistic models for genotype-phenotype correlations in phenylketonuria using protein stability calculations. Hum Mutat [Internet]. 2019;40(4):444–457. [cited 2019 Mar 6]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30648773 [DOI] [PubMed] [Google Scholar]
- [85].Liu C, Ma H, Wu J, et al. Arginine68 is an essential residue for the C-terminal cleavage of human Atg8 family proteins. BMC Cell Biol [Internet]. 2013;14:27. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3686597&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Costa JR, Prak K, Aldous S, et al. Autophagy gene expression profiling identifies a defective microtubule-associated protein light chain 3A mutant in cancer. Oncotarget. 2016;7:41203–41216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature [Internet]. 2010;466:68–76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20562859%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2901998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kraft LJ, Nguyen TA, Vogel SS, et al. Size, stoichiometry, and organization of soluble LC3-associated complexes. Autophagy. 2014;10:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B Is cleaved at its carboxyl-terminal Met 121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279:47704–47710. [DOI] [PubMed] [Google Scholar]
- [90].Dou Z, Xu C, Donahue G, et al. Autophagy mediates degradation of nuclear lamina. Nature [Internet]. 2015;527:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lv M, Wang C, Li F, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein & Cell [Internet]. 2017 8:25–38 Available from: http://link.springer.com/10.1007/s13238-016-0328-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rogov VV, Suzuki H, Marinković M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jia R, Bonifacino JS. Negative regulation of autophagy by uba6-birc6–mediated ubiquitination of lc3. Elife. 2019;8:1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hess B, Kutzner C, van der Spoel D, et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. [DOI] [PubMed] [Google Scholar]
- [95].Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX [Internet]. 2015;2:19–25. [cited 2019 Mar 6]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S2352711015000059 [Google Scholar]
- [96].Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Li DW, Brüschweiler R. NMR-based protein potentials. Angew Chem Int Educ. 2010;49:6778–6780. [DOI] [PubMed] [Google Scholar]
- [98].Lindorff-Larsen K, Piana S, Palmo K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins [Internet]. 2010;78:1950–1958. [cited 2016 Jul 12]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20408171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Best RB, De Sancho D, Mittal J. Residue-specific α-helix propensities from molecular simulation. Biophys J [Internet]. 2012;102:1462–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B [Internet]. 2009;113:9004–9015. [cited 2015 Aug 9]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3115786&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Robustelli P, Piana S, Shaw DE. Developing a molecular dynamics force field for both folded and disordered protein states. Proc Natl Acad Sci. 2018;115:E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhou CY, Jiang F, Wu YD. Residue-specific force field based on protein coil library. RSFF2: modification of AMBER ff99SB. J Phys Chem B. 2015;119:1035–1047. [DOI] [PubMed] [Google Scholar]
- [103].Li S, Elcock AH. Residue-Specific Force Field (RSFF2) improves the modeling of conformational behavior of peptides and proteins. J Phys Chem Lett. 2015;6:2127–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Piana S, Lindorff-Larsen K, Shaw DE. How robust are protein folding simulations with respect to force field parameterization? Biophys J. 2011;100:L47–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Bjelkmar P, Larsson P, Cuendet MA, et al. Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction Maps, virtual interaction sites, and water models. J Chem Theory Comput [Internet]. 2010;6:459–466. [cited 2016 Jul 12]. Available from: https://pubs.acs.org/doi/10.1021/ct900549r [DOI] [PubMed] [Google Scholar]
- [106].Jiang F, Zhou C-Y, Wu Y-D. Residue-specific force field based on the protein coil library. RSFF1: modification of OPLS-AA/L. J Phys Chem B [Internet]. 2014;118:6983–6998. [cited 2016 Jul 12]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24815738 [DOI] [PubMed] [Google Scholar]
- [107].MacKerell AD, Bashford D, Dunbrack RL, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. [DOI] [PubMed] [Google Scholar]
- [108].Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- [109].Horn HW, Swope WC, Pitera JW, et al. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J Chem Phys [Internet]. 2004;120:9665–9678. [cited 2016 Jul 12]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15267980 [DOI] [PubMed] [Google Scholar]
- [110].Daura X, Gademann K, Jaun B, et al. Peptide folding: when simulation meets experiment. Angew Chem Int Educ. 1999;38:236–240. [Google Scholar]
- [111].Amadei A, Linssen AB, Berendsen HJ. Essential dynamics of proteins. Proteins. 1993;17:412–425. [DOI] [PubMed] [Google Scholar]
- [112].Michaud-Agrawal N, Denning EJ, Woolf TB, et al. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011;32:2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Frey BJ, Dueck D. Clustering by passing messages between data points. Science. 2007; 315:972–976. [DOI] [PubMed] [Google Scholar]
- [114].Shieh AD, Hashimoto TB, Airoldi EM. Tree preserving embedding. Proc Natl Acad Sci [Internet]. 2011;108:16916–16921. [cited 2019 May 10]. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1018393108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Pandini A, Fornili A, Kleinjung J, et al. Structural alphabets derived from attractors in conformational space. BMC Bioinformatics [Internet]. 2010;11:97. Available from: http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Pandini A, Fornili A, Fraternali F, et al. GSATools: analysis of allosteric communication and functional local motions using a structural alphabet. Bioinformatics. 2013;29:2053–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Pandini A, Fornili A. Using local states to drive the sampling of global conformations in proteins. J Chem Theory Comput. 2016;12:1368–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Papaleo E, Renzetti G, Tiberti M. Mechanisms of intramolecular communication in a hyperthermophilic acylaminoacyl peptidase: a molecular dynamics investigation. Fraternali F, editor. PLoS One [Internet]. 2012;7:e35686. [cited 2016 Jul 13]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22558199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Pasi M, Tiberti M, Arrigoni A, et al. xPyder: a PyMOL plugin to analyze coupled residues and their networks in protein structures. J Chem Inf Model. 2012;279:1–6. [DOI] [PubMed] [Google Scholar]
- [120].Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hornbeck PV, Zhang B, Murray B, et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res [Internet]. 2015;43:D512–D520. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Van Roey K, Dinkel H, Weatheritt RJ, et al. The switches.ELM resource: a compendium of conditional regulatory interaction interfaces. Sci Signal [Internet]. 2013;6:rs7. [cited 2016 Aug 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23550212 [DOI] [PubMed] [Google Scholar]
- [124].Xin J, Mark A, Afrasiabi C, et al. High-performance web services for querying gene and variant annotation. Genome Biol [Internet]. 2016;17:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Li J, Zhao T, Zhang Y, et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018;46:7793–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. Elife [Internet]. 2014;3:1–21. Available from: http://elifesciences.org/lookup/doi/10.7554/eLife.02030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Crooks G, Hon G, Chandonia J, et al. WebLogo: a sequence logo generator. Genome Res [Internet]. 2004;14:1188–1190. Available from: ftp://ftp.ncbi.nih.gov/genomes/Bacteria [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2:Unit2.9. [DOI] [PubMed] [Google Scholar]
- [129].McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell [Internet]. 2015;57:39–54. [DOI] [PubMed] [Google Scholar]
- [130].Rogov VV, Suzuki H, Fiskin E, et al. Structural basis for phosphorylation-triggered autophagic clearance of Salmonella. Biochem J [Internet]. 2013;454:459–466. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23805866 [DOI] [PubMed] [Google Scholar]
- [131].Tokuriki N, Stricher F, Schymkowitz J, et al. The stability effects of protein mutations appear to be universally distributed. J Mol Biol. 2007;369:1318–1332. [DOI] [PubMed] [Google Scholar]
- [132].Privalov PL. Stability of proteins small globular proteins. Adv Protein Chem. 1979;33:167–241. [DOI] [PubMed] [Google Scholar]
- [133].Fersht AR, Serrano L. Principles of protein stability derived from protein engineering experiments. Curr Opin Struct Biol. 1993;3:75–83. [Google Scholar]
- [134].Khorasanizadeh S, Peters ID, Roder H. Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nat Struct Biol [Internet]. 1996;3:193–205. [cited 2019 May 10]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8564547 [DOI] [PubMed] [Google Scholar]
- [135].Park H, Bradley P, Greisen PJ, et al. Simultaneous optimization of biomolecular energy function on features from small molecules and macromolecules Hahnbeom. J Chem Theory Comput. 2016;12:6201–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Barlow KA, Conchúir Ó, Thompson S, et al. Flex ddG: Rosetta ensemble-based estimation of changes in protein-protein binding affinity upon mutation. J Phys Chem B. 2018;122:5389–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Marrink SJ, Risselada HJ, Yefimov S, et al. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B [Internet]. 2007;111:7812–7824. [cited 2018 May 30]. Available from: http://pubs.acs.org/doi/abs/10.1021/jp071097f [DOI] [PubMed] [Google Scholar]
- [138].Monticelli L, Kandasamy SK, Periole X, et al. The MARTINI coarse-grained force field: extension to proteins. J Chem Theory Comput [Internet]. 2008;4:819–834. [cited 2020 Jan 13]. Available from: https://pubs.acs.org/doi/10.1021/ct700324x [DOI] [PubMed] [Google Scholar]
- [139].Jackson WT, Giddings TH, Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:0861–0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw data, inputs, outputs and scripts associated with this publication are available in two repositories: GitHub https://github.com/ELELAB/lc3b_cancer_paper.git and OSF https://osf.io/4zxym/.