

ABSTRACT
The innate immune restriction factor SAMHD1 can inhibit diverse viruses in myeloid cells. Mechanistically, SAMHD1 inhibits lentiviral replication including HIV-1 by depleting the nucleotide pool to interfere with their reverse transcription. Equine infectious anemia virus (EIAV) is an ancient lentivirus that preferentially attacks macrophages. However, the mechanism by which EIAV successfully establishes infection in macrophages with functional SAMHD1 remains unclear. Here, we demonstrate that while equine SAMDH1 can limit EIAV replication in equine macrophages at the reverse transcription stage, the antiviral effect is counteracted by the well-known transcriptional regulator Rev, which downregulates equine SAMHD1 through the lysosomal pathway. Remarkably, Rev hijacks BECN1 (beclin 1) and PIK3C3 to mediate SAMHD1 degradation in a canonical macroautophagy/autophagy-independent pathway. Our study illustrates that equine lentiviral Rev possesses important functions in evading cellular innate immunity in addition to its RNA regulatory function, and may provide new insights into the co-evolutionary arms race between SAMHD1 and lentiviruses.
Abbreviations:3-MA: 3-methyladenine; AA: amino acid; ACTB: actin beta; AD: activation domain; ATG: autophagy related; Baf A1: bafilomycin A1; BD: binding domain; BECN1: beclin 1; BH3: BCL2-homology-3 domain; BiFC: bimolecular fluorescence complementation; CCD: coiled-coil domain; class III PtdIns3K: class III phosphatidylinositol 3-kinase; CQ: chloroquine; Co-IP: co-immunoprecipitation; dNTPase: dGTP-stimulated deoxynucleoside triphosphate triphosphohydrolase; ECD: evolutionarily conserved domain; EIAV: equine infectious anemia virus; eMDMs: equine monocyte-derived macrophages; GFP: green fluorescent protein; HD: histidine-aspartic; HIV-1: human immunodeficiency virus-1; hpi: hours post infection; hpt: hours post transfection; KO: knockout; LAMP2: lysosomal associated membrane protein 2; LMB: leptomycin B; PMA: phorbol 12-myristate 13-acetate; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; ND: unknown non-essential domain; NES: nuclear export signal; NLS: localization signal; NS: statistically non-significant; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; RBD: RNA binding domain; RT: reverse transcriptase; siRNAs: small interfering RNAs; SAMHD1: SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1; SIV: simian immunodeficiency virus; VN: C-terminal residues of Venus 174 to 238; VC: N-terminal residues 2 to 173 of Venus
KEYWORDS: BECN1, EIAV, HIV-1, innate immunity, lysosome, non-canonical autophagy, PIK3C3, retrovirus, Rev, SAMHD1
Introduction
SAMHD1 (SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1) is a host restriction factor against viral infection in myeloid cells [1]. This protein was initially identified as a dGTP-stimulated deoxynucleoside triphosphate triphosphohydrolase (dNTPase) to maintain low levels of intracellular dNTP, which limits human immunodeficiency virus-1 (HIV-1) replication in non-dividing myeloid cells [2–5]. SAMHD1 is composed of a sterile alpha domain and a histidine-aspartic (HD) domain. The HD domain has dNTPase activity that converts dNTPs to deoxynucleoside and inorganic triphosphate [4,6,7]. The dNTPase activity of SAMHD1 protein was demonstrated to be crucial for its antiviral function, and is regulated by post-translational modifications such as phosphorylation and acetylation [5,8–13].
With its enzymatic activity, SAMHD1 is able to restrict a diverse range of retroviruses and several DNA viruses including human cytomegalovirus, murine cytomegalovirus, and herpes simplex virus 1 [12,14–18]. On the other hand, to achieve successful replication, viruses have evolved various strategies to evade the antiviral activity of SAMHD1. HIV-2 and multiple simian immunodeficiency virus (SIV) strains are able to antagonize SAMHD1 antiviral activity through Vpx/Vpr-mediated proteasomal degradation. Mechanistically, both Vpx and Vpr directly load human SAMHD1 (HsSAMHD1) onto the cullin ring-finger ubiquitin ligase 4 E3 ubiquitin ligase complex, where it is polyubiquitinated and subsequently degraded [3,19–21]. Moreover, herpes viruses are able to overcome SAMHD1 inhibition by modulating SAMHD1 phosphorylation [15,16,18].
Of the lentiviruses in the Retroviridae family, non-primate lentiviruses such as equine infectious anemia virus (EIAV) and feline immunodeficiency virus share a similar genome structure and myeloid cell tropism to the primate lentiviruses [22–24]. All lentiviruses are considered to originate from one non-primate lentivirus, based on the presence or absence of a dUTPase gene and its sequence diversity [25]. EIAV possesses the smallest and genetically simplest genome [26]. In addition to three major structural genes gag, pol and env, the EIAV genome contains three open reading frames that encode three accessory proteins (Tat, Rev, and S2). Notably, EIAV is the sole member of the lentivirus family that primarily infects equine macrophages in vivo [27,28]. Since SAMHD1 depletes dNTPs in non-dividing cells, the process by which the seemingly simplistic EIAV is able to successfully replicate deserves further investigation.
In this study, we found that equine SAMHD1 (EfSAMHD1) limits EIAV replication in equine macrophages and its dNTPase activity is required for its antiviral effect. EIAV infection in equine macrophages leads to reduction of EfSAMHD1 expression. Interestingly, EIAV Rev, a well-documented viral accessory protein that mediates viral RNA transportation, physically interacts with EfSAMHD1, leading to the translocation of EfSAMHD1 from the nucleus to the cytoplasm for degradation in a lysosomal-dependent manner. We also identified that Rev commandeers BECN1 (beclin 1), a key member of class III phosphatidylinositol 3-kinase (class III PtdIns3K) complex, to facilitate the degradation of SAMHD1 without inducing macroautophagy/autophagy. Knockout of either PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) or BECN1 completely rescued the expression of EfSAMHD1 in the presence of EIAV Rev. Taken together, we characterize here a novel role for Rev in the EIAV life cycle, and present a unique strategy employed by EIAV to mitigate inhibition by EfSAMHD1 in its primary target cells.
Results
Equine SAMHD1 restricts EIAV infection
To verify the antiviral effect of EfSAMHD1 on EIAV replication in equine monocyte-derived macrophages (eMDMs), small interfering RNA (siRNA) targeting EfSAMHD1 were employed to knockdown EfSAMHD1 expression in eMDMs. Two days following siRNA knockdown, eMDMs transfected with EfSAMHD1-specific siRNA showed reduced level of the target protein (Figure 1A). To investigate the effect of SAMHD1 depletion on EIAV infection, a low dose (10 ng reverse transcriptase [RT]) of EIAV luciferase reporter virus was used to infect eMDM cells. The luciferase reporter assay showed that viral infectivity was up to 7.1-fold higher in eMDMs depleted of EfSAMHD1, compared to those in the cells transfected with scrambled siRNA, or in untreated eMDMs (Figure 1B). To explore the mechanism by which EfSAMHD1 inhibits EIAV infection in eMDMs, the products of early and late reverse transcription of EIAV over the course of infection from 6 h to 24 h were quantified in siRNA-transfected eMDMs following EIAV infection. We found that knockdown of EfSAMHD1 in eMDMs slightly increased the number of early minus strand reverse transcription products at the indicated time points (Figure 1C), while EIAV late reverse transcription products significantly increased in eMDMs depleted of EfSAMHD1 (Figure 1D). Taken together, these results suggested that EfSAMHD1 restricts EIAV replication in eMDMs by limiting EIAV reverse transcription.
Figure 1.
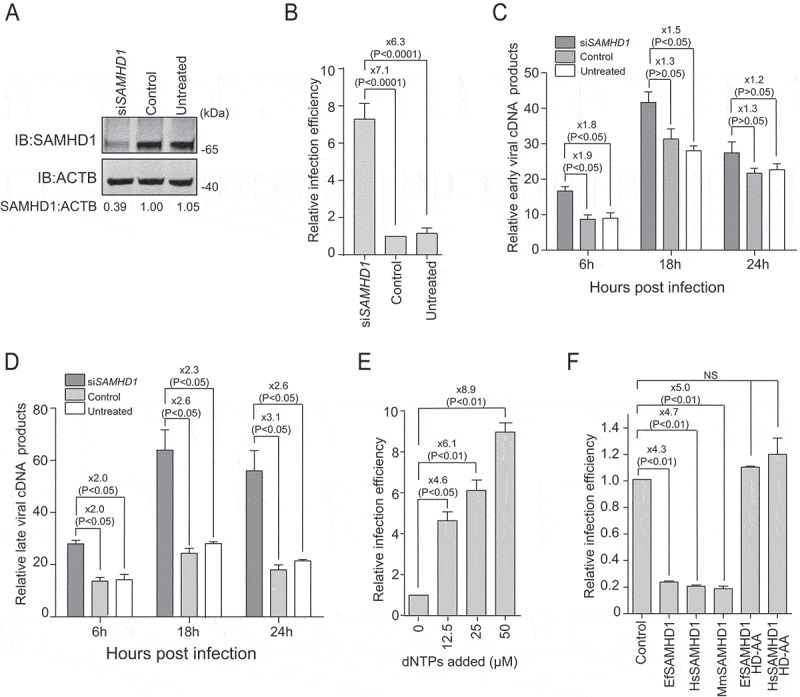
Equine SAMHD1 restricts EIAV infection. (A) Knockdown of the protein expression levels of EfSAMHD1. eMDMs were transfected with EfSAMHD1-specific siRNA or scrambled siRNA control. At 48 hpt, EfSAMHD1 protein levels were quantified by western blotting using ACTB as an internal control. The densities of EfSAMHD1 bands were analyzed with the Odyssey CLx Image Studio to calculate the values relative to that of ACTB. Results were normalized to control cells, which were set as 1. (B) Knockdown of EfSAMHD1 increases EIAV replication in equine macrophages. eMDMs transfected with the indicated siRNA were infected with EIAV luciferase reporter virus (RT = 10 ng). Cells were lysed, and luciferase activity in the cell lysates was measured at 48 hpi. Results were normalized to control cells. P > 0.05 was considered NS, P < 0.05 was considered statistically significant. (C and D) Knockdown of EfSAMHD1 increases viral reverse transcription products in eMDMs. EIAV luciferase reporter viruses were first treated with DNase and then used to infect eMDMs. Total DNA was collected, and the viral early (C) and late reverse transcripts (D) were quantified by real-time PCR using specific primers at 6, 12 and 18 hpi. ACTB was measured as an endogenous control. Data represent means and SD of three independent experiments. P > 0.05 was considered NS, P < 0.05 was considered statistically significant. (E) Adding dNTPs into eMDMs enhances the infection of EIAV in a dose-dependent manner. eMDMs were exposed to deoxynucleosides at different concentration, and then infected with EIAV luciferase reporter virus. Cells were lysed, and luciferase activity in the cell lysates was measured at 48 hpi. Results were normalized to control cells, which were set as 1. Data represent means and SD of three independent experiments. P > 0.05 was considered NS, P < 0.05 was considered statistically significant. (F) HD domain of EfSAMHD1 is essential for its antiviral activity. Stable U937 cell lines expressing wild-type SAMHD1 or SAMHD1 mutants were inoculated with EIAV luciferase reporter viruses. Cells were lysed, and luciferase activity in the cell lysates was measured at 48 hpi. Results were normalized to control cells, which were set as 1. Data represent means and SD of three independent experiments. P > 0.05 was considered NS, P < 0.05 was considered statistically significant
Since the antiviral effect of HsSAMHD1 mainly relies on its dNTPase activity, we assessed the dNTPase activity of EfSAMHD1. The intracellular dNTP levels in eMDMs were quantified using a previously developed fluorescence-based primer extension assay [29]. As shown in Figure S1A, the intracellular concentrations of the four dNTPs were less than 100 fmol per 106 eMDM cells. Following depletion of EfSAMHD1 expression, the dNTPs level dramatically increased up to 4-fold, suggesting that EfSAMHD1 has functional dNTPase activity to maintain relatively low dNTP concentrations in eMDMs. To verify that the cellular dNTP pool is a critically limiting factor for EIAV replication, eMDMs cells were cultured in presence of deoxynucleosides at different concentrations, and then infected with single-cycle EIAV. Viral infection was measured by assessing luciferase activity. Exogenous addition of deoxynucleosides resulted in a dose-dependent enhancement of EIAV infection in eMDMs (Figure 1E). The enhancement of viral infection mediated by addition of deoxynucleosides was compromised in cells depleted of EfSAMHD1, compared to those in the cells transfected with scrambled siRNA, or in untreated eMDMs (Figure S1B). Altogether, these results further confirmed that EfSAMHD1 restricts EIAV infection in eMDMs by lowering cellular dNTP levels.
The HD domain within SAMHD1 is highly conserved across different species. Importantly, it has been reported that the catalytic and phosphohydrolase activities of the protein are conferred by the HD domain of HsSAMHD1 [30,31]. To further investigate the antiviral function of the HD domain, we generated five U937 cell lines, each stably expressing one of the following SAMHD1 constructs: EfSAMHD1, HsSAMHD1, macaque SAMHD1 (MmSAMHD1), EfSAMHD1 catalytically inactive mutant EfSAMHD1 HD-AA, and HsSAMHD1 catalytically inactive mutant HsSAMHD1 HD-AA. SAMHD1 expression levels were confirmed by western blotting analysis (Figure S1C). These cell lines were further differentiated into macrophages with phorbol 12-myristate 13-acetate (PMA) treatment and then infected with EIAV reporter virus. Consistent with the results obtained from eMDMs, ectopic expression of equine, human and macaque SAMHD1 in U937 cells resulted in reduced EIAV infectivity by 3 to 5-fold in each case, while the mutants with a defective HD domain completely lost their ability to restrict EIAV infection, compared to the wild type (Figure 1F).
We next quantified the levels of intracellular dNTPs in differentiated U937 cells expressing the different SAMHD1 constructs. Cells expressing wild-type SAMHD1 had 2 to 3-fold lower levels of dNTPs compared to the parental cells, whereas SAMHD1 mutants with a defective HD domain were unable to hydrolyze the cellular dNTPs (Figure S1D). Together, these data suggested that the conserved HD domain of EfSMAHD1 is crucial for its antiviral activity.
EIAV Rev specifically downregulates expression of equine SAMHD1
Vpx or Vpr encoded by HIV-2 and certain SIV lineages can abolish the antiviral function of SAMHD1 in myeloid cells by loading SAMHD1 for proteasome-dependent degradation. Since macrophages with active SAMHD1 are the primary target cells of EIAV in vivo, we speculated that EIAV has evolved strategies to counteract the antiviral activity of EfSAMHD1 in these cells. To this end, eMDMs were infected with high dose (RT = 200 ng) of EIAVDLV36 (a replication-competent EIAV strain) [32] to achieve a saturated infection, and cells were harvested to detect SAMHD1 expression at 12 and 24 h post infection (hpi). Intriguingly, the expression level of EfSAMHD1 protein, but not of mRNA, gradually decreased with time following infection with EIAV (Figure 2A,B). Additionally, EIAV infection diminished SAMHD1 levels in a dose-dependent manner (Figure 2C), and knockdown of EfSAMHD1 in eMDMs further exaggerated infection-induced downregulation of SAMHD1 (Figure S2A).
Figure 2.
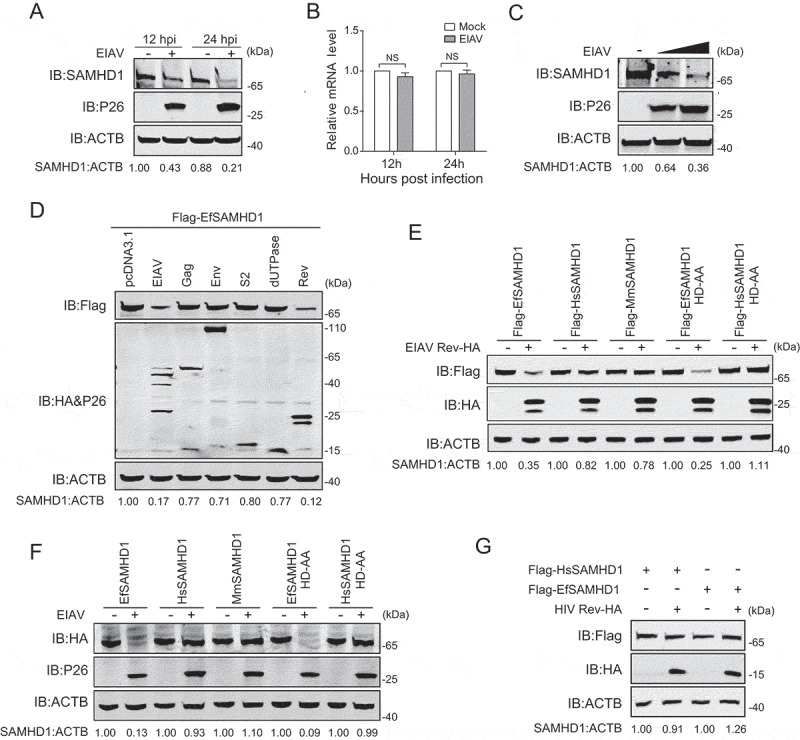
EIAV Rev decreases equine SAMHD1 expression. (A) EIAV infection decreases levels of EfSAMHD1 expression at the protein level. eMDM cells were infected with EIAVDLV36 (RT = 200 ng). EfSAMHD1 protein levels were monitored at 12 and 24 hpi. Western blotting was performed with indicated antibodies. The densities of EfSAMHD1 bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to uninfected cells as control group. (B) EIAV infection does not decrease levels of EfSAMHD1 mRNA. eMDM cells were infected with EIAVDLV36 (RT = 200 ng). EfSAMHD1 mRNA levels were monitored at 12 and 24 hpi. mRNA levels were analyzed using real-time PCR. Data represent means and SD of three independent experiments. P > 0.05 was considered NS, P < 0.05 was considered statistically significant. (C) EIAV infection decreases levels of EfSAMHD1 protein in a dose-dependent manner. eMDMs were mock-infected or infected with EIAVDLV36 for 24 h (RT = 200 ng; RT = 400 ng). (D) EIAV Rev decreases protein levels of EfSAMHD1. HEK293T cells were transfected with the indicated plasmids. Protein expression was analyzed using western blotting with indicated antibodies at 48 hpt. The densities of EfSAMHD1 bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to control cells. (E) Immunoblot analysis of SAMHD1 expression from the EIAV Rev and SAMHD1mutant co-expression experiment. HEK293T cells were transfected with indicated plasmids. Protein expression was analyzed using western blotting with indicated antibodies at 48 hpt. The densities of SAMHD1 mutant bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to respective control cells. (F) The effect of EIAV infection on different SAMHD1 mutants expression. EIAV with VSV-G (RT = 400 ng) was used to infect stable U937 cell lines expressing different SAMHD1 mutants. Protein expression was analyzed using western blotting with indicated antibodies at 48 hpi. The densities of SAMHD1 mutant bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to respective control cells. (G) HIV-1 Rev does not decrease expression levels of EfSAMHD1 or huSAMHD1 proteins
To identify the viral proteins required for SAMHD1 downregulation, HEK293T cells were co-transfected with expression vectors of EIAV proviral DNA, gag, env, dUTPase, S2 or rev gene, along with EfSAMHD1 expression vectors. The protein expression was determined using western blotting with indicated antibodies at 48 h post transfection (hpt). Interestingly, we found that transfection of either EIAV proviral DNA or the rev gene was capable of decreasing levels of EfSAMHD1 (Figure 2D). Rev is a conserved regulatory protein involved in transportation of incompletely spliced structural protein-encoding viral mRNAs [33,34]. To confirm this specific downregulation, we evaluated the expression of SAMHD1 from human, macaque and horse in the context of EIAV Rev expression. We found that EIAV Rev, specifically mediates the downregulation of EfSAMHD1 and its catalytically inactive mutant, rather than HsSAMHD1, HsSAMHD1 HD-AA or MmSAMHD1 (Figure 2E). To further corroborate these results, we also examined the expression of these SAMHD1 variants upon infection with high dose of VSV-G pseudotyped EIAV (RT = 400 ng) which achieved high infection efficiency in U937 cells. As expected, the results showed that EIAV infection led to reduction of EfSAMHD1 and its HD mutant, but not the HsSAMHD1, HsSAMHD1 HD mutant, or MmSAMHD1 (Figure 2F). Moreover, HIV-1 Rev failed to downregulate EfSAMHD1 or HsSAMHD1 expression (Figure 2G), and HIV-2 Vpx was unable to reduce EfSAMHD1 expression (Figure S2B). Collectively, these results suggested that EIAV overcomes SAMHD1-mediated restriction through its encoded Rev protein in a species-specific manner.
EIAV Rev interacts with equine SAMHD1
To further elucidate the mechanism by which EIAV Rev reduces EfSAMHD1 expression, we assessed the physical interaction of EfSAMHD1 with Rev by GST affinity isolation and co-immunoprecipitation (Co-IP). Rev-GST is able to pull down EfSAMHD1 tagged with Flag in an RNA-independent manner (Figure 3A,B), whereas the interaction between Rev-GST with HsSAMHD1, HsSAMHD1 HD mutant, or MmSAMHD1 was barely detected (Figure S3A). These data further indicate that the binding between EfSAMHD1 and Rev is necessary for Rev-mediated degradation of EfSAMHD1. We next performed confocal imaging to analyze the subcellular localization of EIAV Rev and EfSAMHD1. Both EfSAMHD1 and EIAV Rev were predominately localized at the nucleus of cells when expressed alone, however, both of them translocated to the cytosol, resulting in the formation of puncta in the cells co-expressing EfSAMHD1 and EIAV Rev (Figure 3C). To further confirm the confocal results, a bimolecular fluorescence complementation (BiFC) assay that could directly visualize protein-protein interactions in living cells [35] was utilized to analyze the specific interaction of EfSAMHD1 and EIAV Rev. In brief, two constructs were generated: Rev-VC that EIAV Rev-HA was fused with C-terminal residues of Venus 174 to 238 (VC), and VN-EfSAMHD1 that Flag-EfSAMHD1 was fused with N-terminal residues 2 to 173 of Venus (VN) (Figure 3D). We first confirmed that the Rev-VC retained the ability to downregulate VN-EfSAMHD1 expression (Figure S3B). After co-transfection of Rev-VC and VN-EfSAMHD1 into HeLa cells, a specific BiFC fluorescent signal was readily detected in the cytoplasmic compartment as observed by immunostaining (Figure 3E), whereas, no specific signal was detected in the cells co-transfected with HIV-1 Rev-VC and VN-EfSAMHD1 (Figure 3F), suggesting that EfSMAHD1 specifically interacts with EIAV Rev. Therefore, our data clearly demonstrated that EIAV Rev specifically interacts with EfSAMHD1 to drive the redistribution of EfSAMHD1 from the nucleus to the cytosol.
Figure 3.
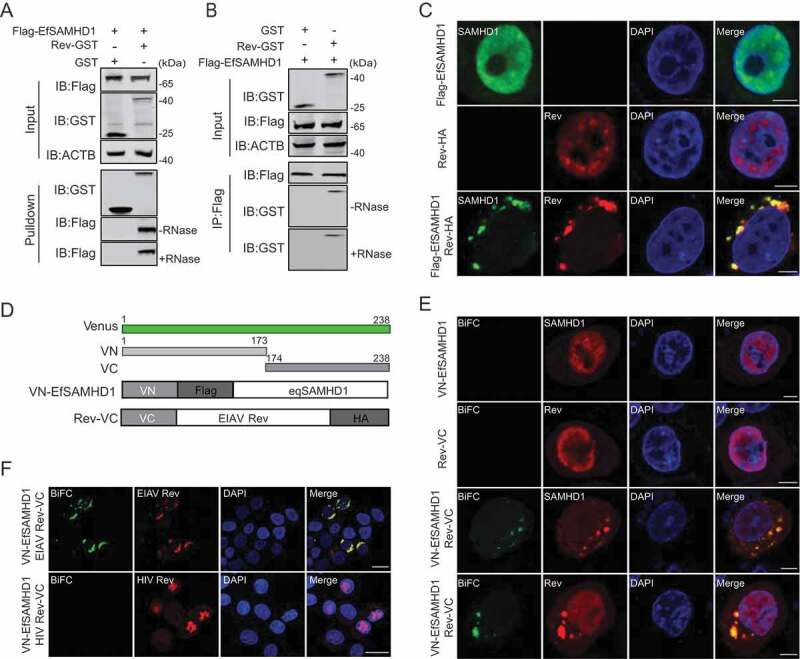
EIAV Rev interacts with equine SAMHD1. (A) GST affinity-isolation analysis of EfSAMHD1 and Rev. Flag-SAMHD1 was co-transfected with VR-GST or Rev-GST. Whole-cells lysates were prepared, and GST affinity isolations were performed with glutathione magbeads and analyzed using western blotting. (B) Co-IP analysis of EfSAMHD1 and Rev. HEK293T cells were transfected with indicated plasmids. Whole-cell lysates were prepared at 48 hpt, and immunoprecipitations were performed with anti-Flag beads and analyzed using western blotting. (C) EIAV Rev relocalizes EfSAMHD1 from the nucleus to cytoplasmic compartments. Flag-EfSAMHD1 and Rev-HA were expressed individually or together in HeLa cells. Cells were fixed, permeabilized and incubated with primary and secondary antibodies at 36 hpt, EfSAMHD1 and Rev expression were observed using confocal microscopy. Scale bar: 5 μm. (D) Schematic of the BiFC fusion proteins. (E) Detection of EfSAMHD1 and Rev interaction using BiFC assay. VN-Flag-EfSAMHD1 and Rev-VC were expressed individually or together in HeLa cells and EfSAMHD1 and Rev fusion proteins were stained with rabbit anti-Flag or anti-HA polyclonal antibodies followed by Alexa Fluor 647-conjuated goat anti-rabbit antibodies. BiFC green fluorescent signals together with the expression of EfSAMHD1 or Rev expression were visualized using confocal microscopy. Scale bar: 5 μm. (F) HIV-1 Rev does not produce BiFC green fluorescent signals with EfSAMHD1. RevHIV-VC-HA was used as a negative control. Scale bar: 20 μm
EIAV Rev promotes the lysosomal degradation of equine SAMHD1
The ubiquitin-proteasome pathway and the lysosome pathway are the two major protein degradation pathways in eukaryotic cells [36]. To illustrate the pathways involved in EfSAMHD1 downregulation mediated by EIAV Rev, HEK293T cells co-transfected with the expression vectors of both Flag-EfSAMHD1 and Rev-HA were treated with various inhibitors that block protein degradation via the two different pathways. Intriguingly, the degradation of Flag-EfSAMHD1 mediated by Rev-HA was blocked both by lysosome inhibitors, including chloroquine (CQ) and bafilomycin A1 (Baf A1), and by class III PtdIns3K inhibitors, including 3-methyladenine (3-MA) and wortmannin. However, the proteasome inhibitor MG132 treatment was unable to rescue EfSAMHD1 expression in the presence of EIAV Rev expression (Figure 4A). These results suggested that Rev may promote EfSAMHD1 degradation via the lysosomal pathway. To further explore the function of lysosomal pathway in EIAV Rev-mediated degradation of EfSAMHD1, HeLa cells expressing the VN-EfSAMHD1 and Rev-VC BiFC pair were transfected with either LAMP2 (lysosomal associated membrane protein 2)-mCherry or mCherry, and their subcellular colocalization was detected by confocal microscopy. As shown in Figure 4B, mCherry was diffused in the cytoplasm, whereas LAMP2, a lysosome marker, almost completely colocalized with SAMHD1-Rev BiFC complex. Furthermore, we found that treatment with Baf A1 or CQ, which disrupts intra-lysosomal degradation by inhibiting acidification, caused the formation of larger SAMHD1-Rev puncta. Conversely, 3-MA or wortmannin, which inhibits class III PtdIns3k and thereby prevents initial recruitment of effector proteins governing protein degradation [37–39], displayed the opposite effect (Figure 4C,D). Taken together, these results suggested that EIAV Rev mediated the degradation of EfSAMHD1 via class III PtdIns3K-dependent lysosomal pathway.
Figure 4.
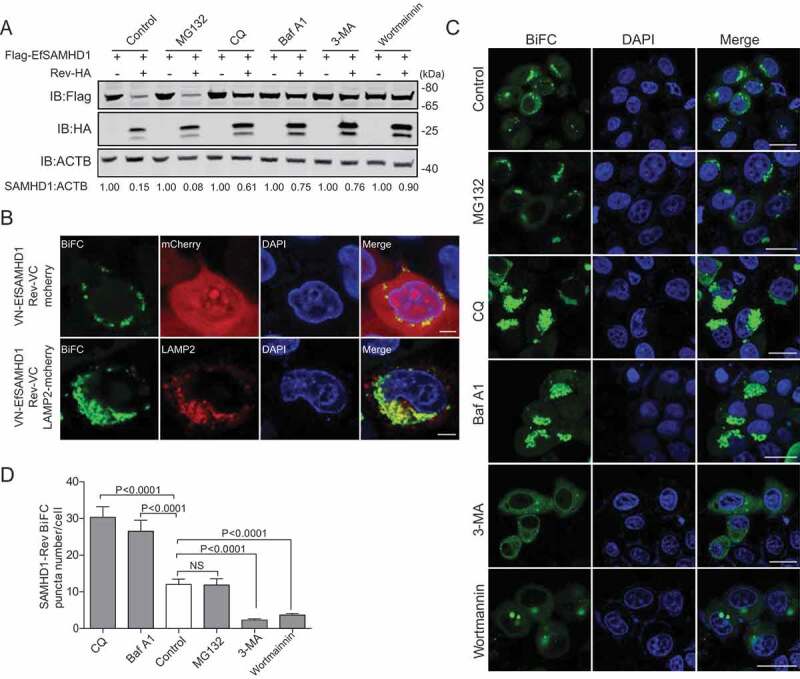
EIAV Rev promotes the lysosomal degradation of equine SAMHD1. (A) Lysosome pathway inhibitors block Rev-induced EfSAMHD1 degradation. HEK293T cells were transfected with indicated plasmids. At 20 hpt, cells were treated with MG132 (10 μM), 3-MA (5 mM), CQ (150 μM), Baf A1 (200 nM), or wortmannin (200 nM). After incubation for 16 h, the cells were analyzed using western blotting with indicated antibodies. The densities of SAMHD1 bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to respective control cells. (B) EfSAMHD1 is targeted to lysosomes for degradation. VR1012-mcherry or VR1012-LAMP2-mcherry was expressed in HeLa cells with the VN-Flag-EfSAMHD1 and Rev-VC pair. Colocalization of these fluorescent signals was visualized by confocal microscopy. Scale bar: 5 μm. (C and D) Analysis of the effect of protein degradation inhibitors on the SAMHD1-Rev BiFC punctate number. VN-Flag-EfSAMHD1 and Rev-VC were co-expressed in HeLa cells. At 20 hpt, cells were further treated with indicated inhibitors. After Incubation for 16 h, the cells were observed using confocal microscopy (C). Scale bar: 20 μm. The number of SAMHD1-Rev BiFC puncta was statistically analyzed (D)
BECN1 is involved in Rev-mediated degradation of equine SAMHD1
To further elucidate the mechanism underlying the degradation of EfSAMHD1 mediated by EIAV Rev, potential cellular partners of EIAV Rev were then identified using a yeast two hybrid screening. A yeast two hybrid cDNA library of eMDMs was generated using SMART technology for identification of the partners of EIAV Rev. Surprisingly, equine BECN1 (EfBECN1), a core component of class III PtdIns3K complex [40], was identified to specifically interact with Rev (Figure 5A). To further confirm these results, we performed GST affinity-isolation and Co-IP assays, and found that Flag-BECN1 coimmunoprecipitated with Rev-GST in an RNA-independent manner (Figure 5B,C). BECN1 is composed of three motifs, including a BCL2-homology-3 domain (BH3), a coiled-coil domain (CCD), and an evolutionarily conserved domain (ECD) [41]. To determine the region of EfBECN1 essential for its interaction with EIAV Rev, we constructed a series of Flag-BECN1 truncated mutants and checked the interactions between these variants and EIAV Rev using GST affinity isolation. Deletion of CCD domain of EfBECN1 completely abolished the interaction between BECN1 and EIAV Rev, while the CCD domain itself was able to bind with EIAV Rev (Figure S4A, S4B). These results suggested that EIAV Rev specifically interacts with EfBECN1 through binding with the CCD domain of BECN1.
Figure 5.
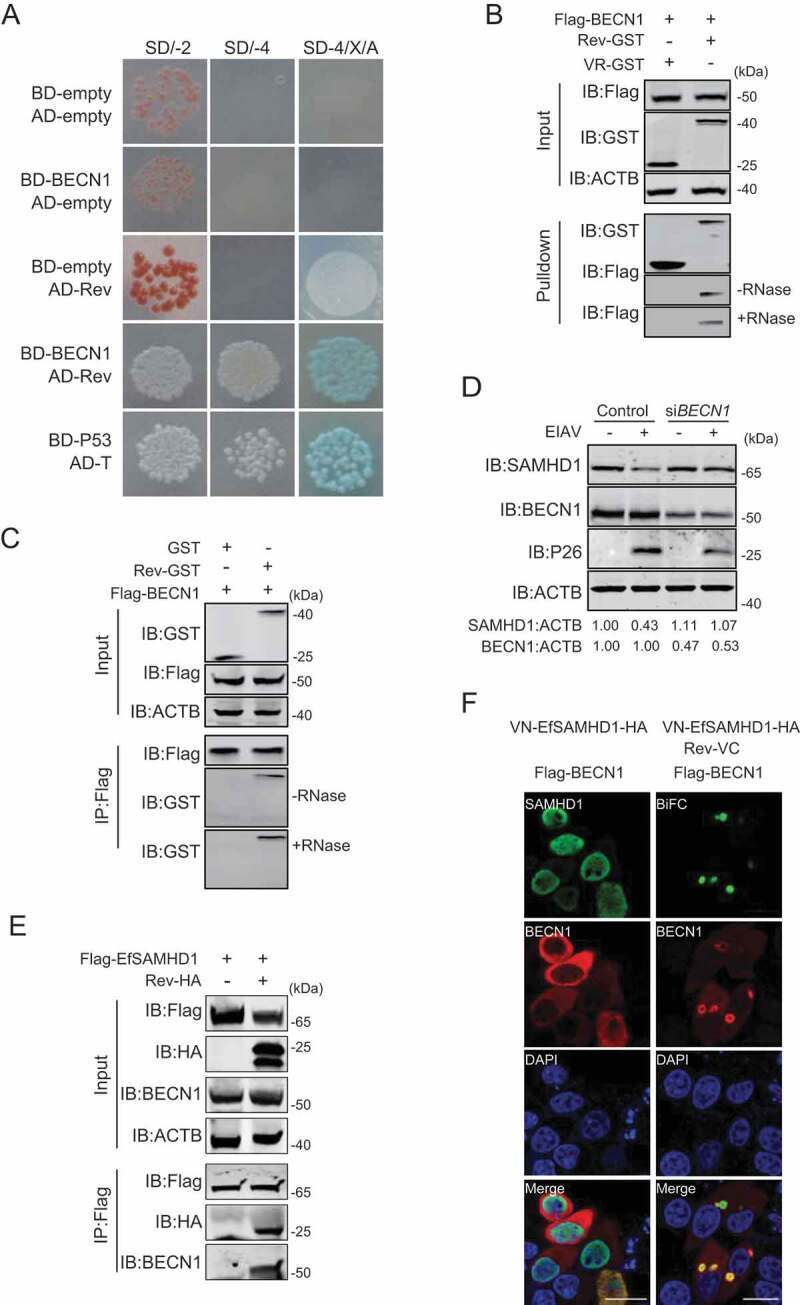
BECN1 is involved in Rev-mediated equine SAMHD1 degradation. (A) Yeast two-hybrid analysis of EfBECN1 and Rev. The indicated plasmids were co-transformed into the Y2HGold yeast strain. (B) GST affinity-isolation analysis of EfBECN1 and Rev. Flag-BECN1 was co-transfected with VR-GST or Rev-GST. Whole-cells lysates were prepared, and GST affinity isolations were performed with glutathione magbeads and analyzed using western blotting. (C) Co-IP analysis of EfSAMHD1 and Rev. HEK293T cells were transfected with indicated plasmids. Whole-cell lysates were prepared at 48 hpt, and immunoprecipitations were performed with anti-Flag beads and analyzed using western blotting. (D) Knockdown of EfBECN1 blocks EIAV-induced EfSAMHD1 degradation. eMDMs were transfected with BECN1-specific siRNA or scrambled siRNA control. At 48 hpt, eMDMs were infected with EIAVDLV36. BECN1 and EfSAMHD1 protein levels were visualized using western blotting after additional 24 h. The densities of EfSAMHD1 and BECN1 bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to uninfected cells of control group. (E and F) BECN1 is recruited to the SAMHD1-Rev complex. (E) Flag-EfSAMHD1 was transiently expressed with or without Rev-HA in HEK293T cells. At 48 hpt, anti-Flag immune complexes were immunoblotted against endogenous BECN1 and ectopically expressed SAMHD1 and Rev. (F) Flag-BECN1 was co-transfected in HeLa cells with the VN-EfSAMHD1-HA and Rev-VC pair or with VN-EfSAMHD1-HA alone. Ectopic BECN1 was stained with rabbit anti-Flag antibodies, followed by Alexa Fluor 647-conjugated secondary antibodies. Colocalization of these fluorescent signals was visualized by confocal microscopy. Scale bar: 20 μm
BECN1 is a multifunctional protein and plays a key role in the class III PtdIns3K activity [38,42,43], we reasoned that EIAV Rev likely recruit BECN1 to mediate EfSAMHD1 degradation in the lysosomes. To this end, eMDMs transfected with siRNA targeting EfBECN1 were infected with EIAVDLV36, and the EfSAMHD1 expression was analyzed using western blotting at 24 hpi. As shown in Figure 5D, depletion of EfBECN1 blocked EIAV infection-induced EfSAMHD1 degradation. Interestingly, EfSAMHD1 only formed a complex with BECN1 in the presence of EIAV Rev, whereas EfSAMHD1 alone did not bind to endogenous BECN1 (Figure 5E). Furthermore, we found that EfBECN1 was strongly recruited to SAMHD1-Rev BiFC complexes to form a ring-like or omegasome-like structure, further suggesting that EfBECN1 is recruited to the EfSAMHD1-Rev complex to mediate the degradation of EfSAMHD1 (Figure 5F). Collectively, all of these results indicated that EIAV Rev may act as a scaffold to bridge SAMHD1 and BECN1 for lysosomal degradation.
BECN1 and PIK3C3, but not all components of the autophagy pathway, are required for Rev-mediated degradation of equine SAMHD1
BECN1 and PIK3C3 are recognized key regulators of autophagy, and also participate in vacuolar protein sorting, endocytic trafficking, and autophagy independent protein degradation [37–39]. It is unclear whether Rev promotes SAMHD1 degradation directly, or through induction of the autophagy pathway (Figure 6A). Thus, we assessed the autophagic flux in EIAV infected eMDMs by monitoring the turnover of endogenous MAP1LC3/LC3-II (microtubule associated protein 1 light chain 3)-II in the presence or absence of Baf A1, an accurate indicator to determine autophagy activation [44,45]. As shown in Figure 6B, LC3-II levels were comparable in infected cells and mock-infected cells. Treatment with Baf A1, an inhibitor of lysosomal acidification that blocks degradation of autophagosomes by the lysosome led to the accumulation of LC3-II in both infected and mock-infected cells; however, there were no observed differences in relative LC3-II levels between infected cells and mock-infected cells, indicating that there was no cumulative increase in autophagosome formation as infection progressed. To corroborate this result, we transfected either EIAV proviral DNA or EIAV Rev expressing vector into HEK293T cells, and then treated these cells with or without Baf A1. We found that neither expression of EIAV proviral DNA or EIAV Rev has an effect on turnover of LC3 II, compared with the control (Figure 6C). To further confirm this result, we utilized green fluorescent protein (GFP)-LC3 in conjunction with Baf A1 to detect autophagic flux by fluorescence microscopy. GFP-LC3 was recruited to autophagosomes forming punctate structures upon starvation as typically observed [44,45]. As expected, expression of EIAV proviral DNA or EIAV Rev has no effect on puncta formation of GFP LC3-labeled vacuoles, compared to the control (Figure S5A, S5B). Collectively, these results suggest that EIAV facilitates the degradation of SAMHD1 by Rev through the lysosome pathway without inducing autophagosome formation.
Figure 6.
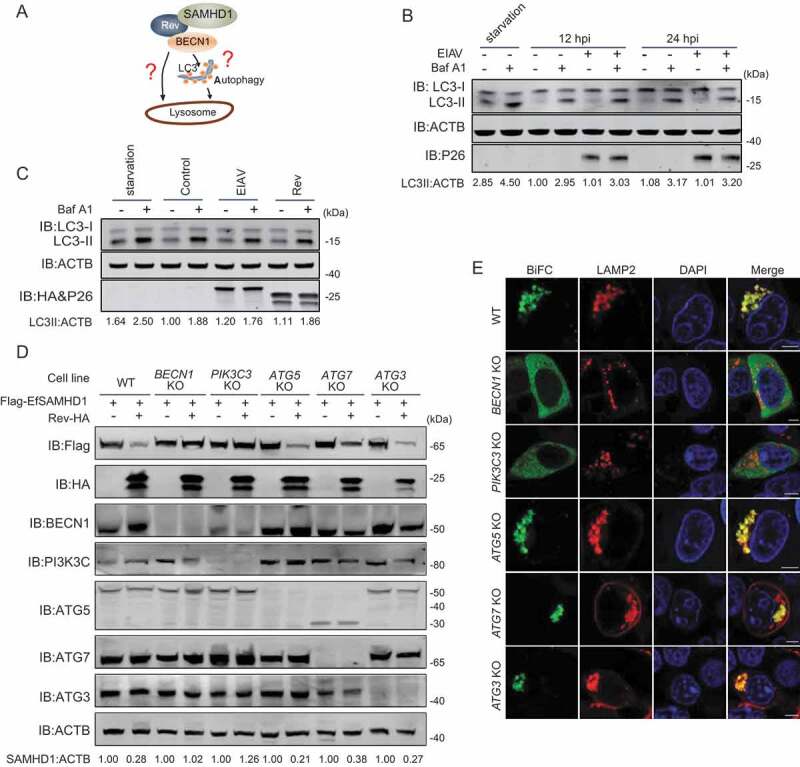
BECN1 and PIK3C3, but not all components of the autophagy pathway, are required for Rev-mediated equine SAMHD1 degradation. (A) Schematic overview of the lysosomal degradation related to BECN1. (B) EIAV infection does not trigger autophagy in eMDMs. eMDMs were mock-infected or infected with EIAVDLV36 for 12 h and 24 h (RT = 200 ng). Cells were washed three times with PBS and then cultured in EBSS medium for 2 h for inducing starvation as a positive control. eMDMs were treated with Baf A1 (50 nM) or DMSO for 6 h. The cell lysates were analyzed using western blotting. The densities of LC3 II bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to the control cells with DMSO treatment at 12 hpi. (C) Overexpression of EIAV proviral DNA or Rev does not trigger autophagy. HEK293T cells were co-transfected with plasmids encoding EIAV proviral DNA and Rev for 36 h, and HEK293T cells starved in EBSS medium for 2 h were used as a positive control. HEK293T cells were treated with Baf A1 (200 nM) or DMSO for 6 h. The cell lysates were analyzed using western blotting. The densities of LC3 II bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to the control cells with DMSO treatment. (D) Knockout of BECN1 or PIK3C3 genes block Rev-induced EfSAMHD1 degradation. WT and ATG KO HEK293T cells were transfected with indicated plasmids, the lysates were analyzed using western blotting. The densities of SAMHD1 bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to respective control cells. (E) Detection of BiFC signals from the SAMHD1 and Rev co-expression in WT and ATG KO HEK293T cells. LAMP2-mCherry was expressed in WT and ATG KO HEK293T cells with the VN-EfSAMHD1-HA and Rev-VC pair. Colocalization of these fluorescent signals was visualized using confocal microscopy at 36 hpt. Scale bar: 5 μm
To further corroborate the involvement of autophagy in EfSAMHD1 degradation mediated by EIAV Rev, a series of autophagy-related (ATG) gene knockout (KO) HEK293T cell lines, ATG3 KO, ATG5 KO, ATG7 KO, BECN1 KO, and PIK3C3 KO, were generated using CRISPR/Cas9 technology. The knockout efficiency and EfSAMHD1 degradation were assessed by western blotting. We found that the degradation of EfSAMHD1 by EIAV Rev was abolished in BECN1 KO and PIK3C3 KO cells; however, EIAV Rev was still able to mediate EfSAMHD1 degradation in ATG3 KO, ATG5 KO and ATG7 KO cells (Figure 6D). Agreeingly, SAMHD1-Rev BiFC complexes were diffused in the cytoplasm of BECN1 KO and PIK3C3 KO cells, but exhibited a puncta localization pattern and strong colocalization with LAMP2 in ATG3 KO, ATG5 KO and ATG7 KO cells (Figure 6E). Knockout of BECN1 was unable to impair the interaction between EfSAMHD1 and Rev (Figure S5C). In addition, knockdown of BECN1 in eMDMs slightly decreased EIAV replication (Figure S5D, S5E). Collectively, these results demonstrate that EIAV Rev induced degradation of SAMHD1 by hijacking BECN1, and this process is independent of the canonical autophagy pathway. BECN1 and PIK3C3, but not all components of the autophagy pathway, are required for EfSAMHD1 degradation mediated by EIAV Rev.
Identification of Rev domains crucial for the degradation of equine SAMHD1
The widely accepted function of lentiviral Rev is to mediate transportation of unspliced or partially spliced pre-mRNA from the nucleus to the cytoplasm [33,34]. Thus, the role of EIAV Rev in counteracting the restriction of SAMHD1 is novel and unconventional. EIAV Rev contains four domains: a nuclear localization signal (NLS), an RNA binding domain (RBD), a nuclear export signal (NES), and a functionally unknown non-essential domain (ND). The conserved hydrophobic residues (L36, W41, L45, L49) are required for its nuclear export function. Two short, non-contiguous arginine-rich motifs including a central RRDRW motif (76–80 amino acids [AA]) and a C-terminal KRRRK motif (159–163 AA) are necessary for RNA binding. The KRRRK motif is also essential for the nuclear localization. A Leu residue at position 95 plays a role in dimerization and is required for RNA-binding activity [33,46]. To map the key domain of Rev responsible for EfSAMHD1 degradation, we constructed five EIAV Rev mutants, including a Rev NES mutant (Rev-ΔNES), a RBD mutant (Rev-AADAA), a dimerization domain mutant (Rev-L95D), a NLS/RBD mutant (Rev-KAAAK) and a non-essential domain mutant (Rev-ΔND) for the analysis (Figure 7A). Upon co-expression of SAMHD1 with each individual Rev mutant, reduction of SAMHD1 expression was observed in Rev-L95D and Rev-KAAAK, and comparable to Rev-WT. Conversely, the SAMHD1 degradation activities of Rev-ΔNES, Rev-AADAA, and Rev-ΔND mutants were impaired (Figure 7B). BiFC assay results also displayed measurable variation in SAMHD1-Rev distribution among the Rev mutants (Figure 7C). Rev-L95D and Rev-KAAAK signals were found in the same perinuclear compartments as Rev-WT, whereas Rev-ΔNES and Rev-AADAA signals were found in the nucleus. Furthermore, Rev-ΔND signal was diffused in the cytoplasm and poorly colocalized with LAMP2. GST affinity-isolation results showed that all the Rev mutants pulled down EfSAMHD1 protein, while all but Rev-ΔND also pulled down with BECN1 (Figure 7D,E). These data demonstrated that the ND domain (122–148 AA) of EIAV Rev that is required for binding to BECN1 as well as the NES and RBD domains that control the shuttle between nucleus and cytoplasm, are important for Rev-mediated EfSAMHD1 degradation. To further confirm the role of nucleocytoplasmic transport of EIAV Rev in EfSAMHD1 degradation, we used Leptomycin B (LMB) to block nuclear export factor exportin 1-mediated export from the nucleus. We found that Rev-mediated EfSAMHD1 degradation was significantly inhibited following treatment with LMB, and SAMHD1-Rev BiFC signals were primarily located in the nucleus (Figure 7F).
Figure 7.
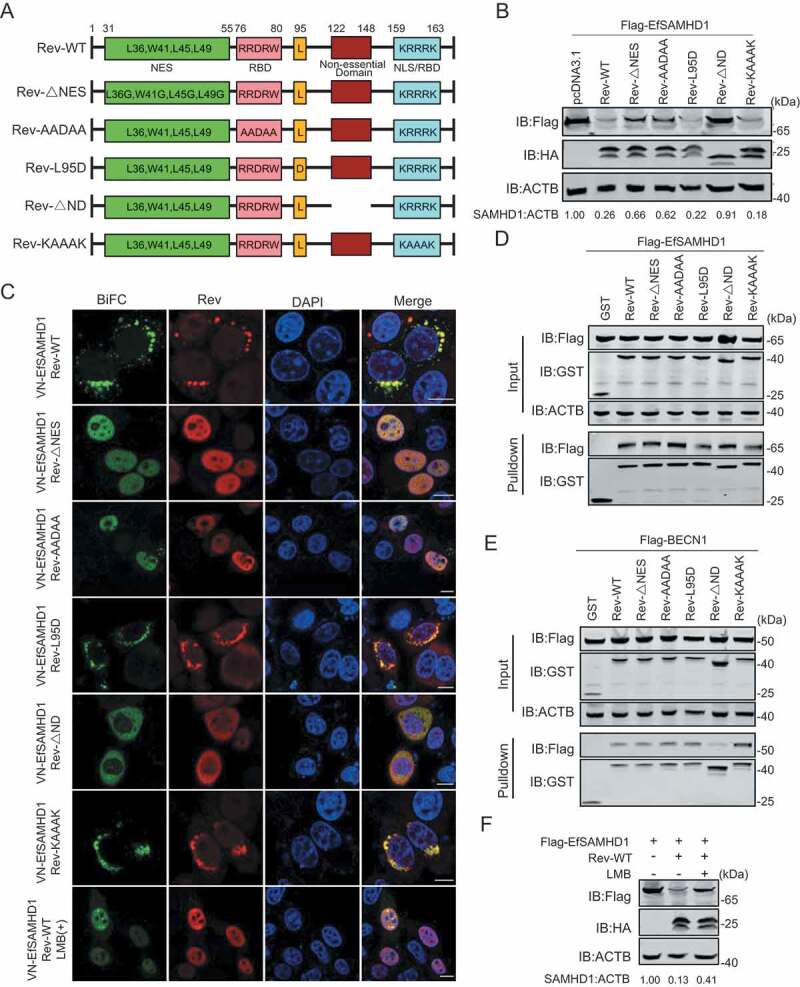
Identification of Rev domains crucial for equine SAMHD1 degradation. (A) Schematic diagram of wild-type EIAV Rev and structures of respective mutants. (B) Immunoblot analysis of EfSAMHD1 expression from the EfSAMHD1 and EIAV Rev mutant co-expression experiment. (C) Detection of BiFC green fluorescent signals from the EfSAMHD1 and EIAV Rev mutants co-expression experiment. HeLa cells were transfected with indicated plasmids. At 20 hpt, cells were either treated with LMB (5 nM) or not. After Incubation for 16 h, ectopic Rev proteins were stained with rabbit anti-HA antibodies, followed by Alexa Fluor 647-conjugated secondary antibodies. Colocalization of these fluorescent signals was visualized by confocal microscopy. Scale bar: 10 μm. (D) GST affinity-isolation analysis of EfSAMHD1 and EIAV Rev mutants. (E) GST affinity-isolation analysis of EfBECN1 and EIAV Rev mutants. (F) LMB blocks Rev-induced EfSAMHD1 degradation. HEK293T cells were transfected with indicated plasmid. At 20 hpt, cells were treated with LMB (5 nM) or not. After Incubation for 16 h, the cell lysates were analyzed by western blotting. The densities of SAMHD1 bands were analyzed to calculate the values relative to that of ACTB. Results were normalized to control cells
To determine the specific amino acids in Rev responsible for BECN1 binding and subsequent EfSAMHD1 degradation, we generated a series of mutants with internal 9-AA deletions (Rev-Δ122-130, Rev-Δ131-139 and Rev-Δ140-148). Upon co-transfection of HEK293T cells with plasmids encoding Flag-EfSAMHD1 and each individual Rev mutant, we found that loss of any 9 AA within the 122–148 AA range abolished its ability to activate SAMHD1 degradation (Figure S6A,S6B). Consistent with these results, the interactions between BECN1 and these Rev mutants were also impaired (Figure S6C). When the 9-AA regions flanking Rev 122–148 were deleted (Rev-Δ113-121 and Rev-Δ149-158), interaction between Rev and BECN1 was unaffected (Figure S6D). These results indicated that deletions within amino acids 122–148, but not deletions of similar length in adjacent regions, abolished the interaction between Rev and BECN1.
Discussion
SAMHD1 limits lentiviral infections in non-dividing myeloid cells by maintaining low levels of dNTPs required for reverse-transcription [2,4]. Unlike other lentiviruses that primarily replicate in CD4 + T cells, the equine lentivirus EIAV exclusively infects macrophages harboring functional SAMHD1 [27,28]. In this study, we provide evidence that EfSAMHD1 inhibits EIAV infection in macrophages as does HsSAMHD1 in non-dividing cells. However, upon infection, EIAV was able to circumvent the host antiviral defense. Remarkably, we found EIAV Rev is the key player promoting lysosomal degradation of SAMHD1 in a BECN1-dependent manner, the function of which was previously unknown.
Lentiviruses utilize cellular dNTPs for reverse transcription in infected host cells. The low dNTPs concentrations in macrophages result in the limited replication kinetics of HIV-1 [47,48]. Our data demonstrate that the concentration of dNTPs in eMDMs is extremely low (Figure S1A). Depletion of endogenous SAMHD1 protein in eMDMs was able to increase intracellular dNTP concentrations, and thereby enhance EIAV replication in eMDMs (Figure 1A-D). In contrast, overexpression of equine SAMHD1 in PMA-treated U937 cells efficiently inhibited EIAV infection (Figure 1F), indicating that the dNTPase activity of SAMHD1 is highly conserved across different species. Thus, the dNTPase activity of SAMHD1 is highly correlated with its antiviral function in macrophages.
SAMHD1 has been characterized as a critical restriction factor that blocks a broad range of retroviruses and DNA viruses as well as enterovirus 71 [49]. These viruses may have evolved weaponry to counteract the restriction of SAMHD1. Herpes virus, for example, may use viral kinase to inactivate SAMHD1 by modifying SAMHD1 phosphorylation. Enterovirus 71 is proposed to indirectly use TRIM21 as a modulator to degrade SAMHD1. Only certain primate lentiviruses such as HIV-2 and several SIV strains are able to productively infect macrophages, due to evolutionarily acquired countermeasures (Vpx or Vpr) that enable them to degrade HsSAMHD1 [3,19]. Other non-primate lentiviruses that display macrophage tropism overcome SAMHD1 restriction by mechanisms that have yet to be determined. EIAV exclusively and successfully infect macrophages which contain extremely low dNTP concentrations, yet previous literature reported that EIAV and other non-primate lentiviruses failed to proteasomally degrade SAMHD1 proteins [50]. Thus, in the present study, it was surprising to find EIAV was able to induce EfSAMHD1 lysosomal degradation though the virally encoded accessory protein Rev. EIAV Rev is a well-studied essential regulatory protein that is highly involved in the mediation of nucleocytoplasmic transportation of incompletely spliced viral mRNA, but its functions beyond this role are poorly understood. Here, we present a novel function of EIAV Rev in degradation and antagonization of SAMHD1 in a species-specific manner. EIAV Rev was unable to degrade HsSAMHD1 and MmSAMHD1, while HIV-1 Rev and HIV-2 Vpx failed to degrade EfSAMHD1. These species-specific limitations suggest evolutionary selection and adaptation in lentiviruses to their hosts.
We investigated the mechanism by which EIAV Rev mediates EfSAMHD1 degradation, and found that EIAV Rev actually recruited SAMHD1 into the lysosome to undergo degradation in a BECN1-dependent manner. BECN1 is a multifunctional protein involved in many biological processes, including autophagy, endocytic trafficking, adaptation to stress, cytokinesis control and more [38,51,52]. BECN1 is also a component of the class III PtdIns3K complex that can play a central role in autophagy. We found that Rev-mediated SAMHD1 degradation was only abolished in PIK3C3 and BECN1 KO cells, but not ATG3, ATG5 or ATG7 KO cells, indicating that EIAV infection or Rev expression does not induce autophagy. Recent literature supports the role of BECN1 in the autophagy-independent maintenance of cellular homeostasis. The BECN1-PIK3C3 complex can recruit retromers to phagosomal membranes, facilitating recycling of the phagocytic receptor CD36 molecule in microglia [53]. In addition, the BECN1-PIK3C3 complex regulates neuronal transforming growth factor beta signaling by mediating recycling of transforming growth factor beta receptor1, but ATG7 and ATG14L have no role in this process [54]. BECN1 and PIK3C3 are also involved in the internalization and degradation of the epidermal growth factor receptor in HeLa cells, whereas Atg14L knockdown does not affect the downregulation of epidermal growth factor receptor [42]. Consistently, our data demonstrated that BECN1, but not the factors involved in the elongation of the autophagosomal membrane, is required for EIAV Rev-mediated EfSAMHD1 degradation. Given that ATG3, ATG5 and ATG7 are not essential for EfSAMHD1 degradation, we cannot exclude that a BECN1-dependent non-canonical autophagy pathway is hijacked by EIAV Rev to degrade SAMHD1. These non-canonical autophagy pathways bypass several canonical steps such as elongation and closure (ATG7, ATG5 and LC3), initiation (unc-51 like autophagy activating kinase 1), and nucleation (PIK3C3-BECN1) [55–57]. Indeed, our data demonstrate that EIAV infection does not induce canonical autophagy. However, the yet unknown host factors and mechanism by which SAMHD1 is recruited to the complex to mediate fusion with the lysosome for degradation remains to be investigated.
SAMHD1 is mainly localized in the nucleus, but EIAV Rev is able to relocalize EfSAMHD1 to the cytoplasm, and target EfSAMHD1 for lysosomal destruction. Our data showed that the ability of EIAV Rev to shuttle between the nucleus and cytoplasm is essential for Rev-mediated EfSAMHD1 degradation as evidenced by loss of SAMHD1 degradation mediated by Rev-ΔNES and Rev-AADAA that exhibit low nuclear export activity [58]. Previously, it has been shown that Arg to Gly mutations at positions 161 and 162 in the KRRRK motif abolished nuclear localization of Rev-GFP fusion proteins [46]. In our study, we observed that Rev-KAAAK, the NLS mutant, was still located in nucleus and yet could efficiently mediate EfSAMHD1 degradation. Therefore, the KRRRK motif of Rev may not be necessary for targeting Rev to the nucleus.
Since Rev is an essential viral protein for EIAV replication, we were unable to generate an infectious clone lacking the entire Rev gene to assess the role of Rev in SAMHD1 degradation in the context of viral infection. However, we have mapped the region within Rev that is responsible for SAMHD1 degradation. The BECN1-binding-deficient Rev mutant bearing 122–148 AA deletion in the non-essential domain of Rev failed to target SAMHD1 for lysosomal degradation, and loss of any 9 AA in the non-essential domain (region 122–148 AA) of Rev abolished its ability to interact with BECN1. Dimerization and the ability for RNA binding of Rev are not likely to be important for Rev-mediated EfSAMHD1 degradation. Our ongoing studies are to map the key residues that are essential for SAMHD1 degradation only, but have no effect on its ability to promote nuclear export of the Rev-mRNA complex.
In conclusion, our study revealed that equine SAMHD1 limits EIAV replication by its dNTPase activity, and that EIAV Rev degrades equine SAMHD1 through a BECN1-dependent lysosome pathway to overcome equine SAMHD1 restriction (Figure 8). Despite numerous studies regarding the role of BECN1 and canonical autophagy in viral infection, usurpation of BECN1 by a viral protein to counteract a restriction factor through a canonical autophagy-independent pathway has never been reported. Therefore, this study highlights a novel mechanism in which non-primate lentivirus overcomes an intrinsic viral defense process. These findings may expand our understanding of the evolutionary conflicts between lentiviruses and restriction factors.
Figure 8.
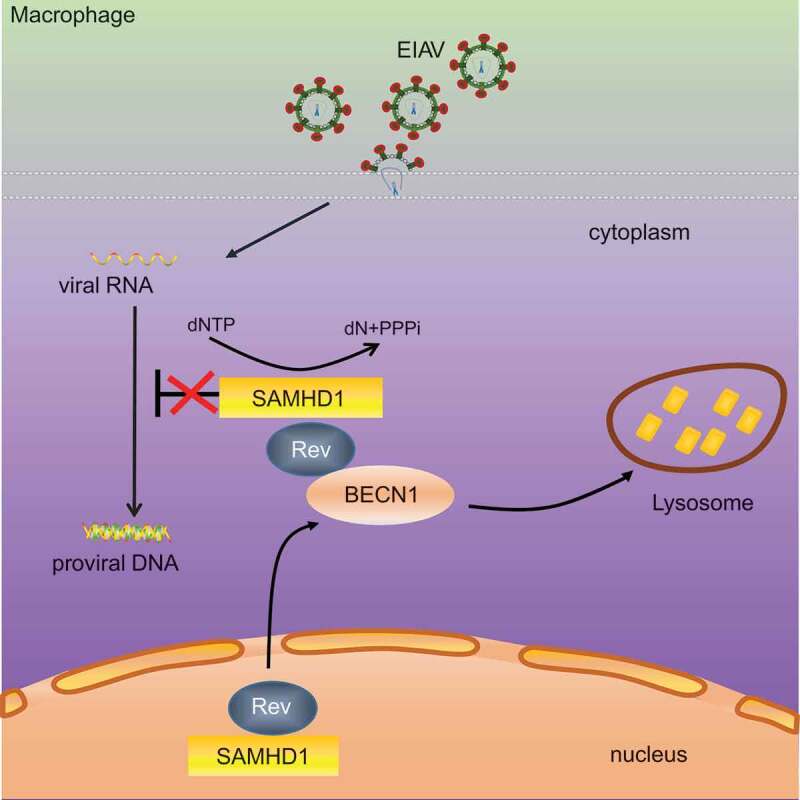
Schematic representation shows the model of Rev-mediated equine SAMHD1 degradation. Equine SAMHD1 limits EIAV replication by its dNTPase activity. However, EIAV Rev degrades equine SAMHD1 through a BECN1-dependent lysosome pathway to overcome equine SAMHD1 restriction
Materials and methods
Cell lines and culture conditions
eMDMs were prepared from equine peripheral blood mononuclear cells as described previously [59], and maintained in RPMI 1640 (Sigma-Aldrich, R8758) supplemented with 30% horse serum (HyClone, SH30074.03) and 30% newborn bovine serum (Ausbian, VS500N). The human cell lines HEK293T and Hela were maintained in Dulbecco’s high-glucose modified Eagle’s medium (Sigma-Aldrich, D6429) supplemented with 10% fetal bovine serum (Sigma-Aldrich, F2442) and 1% penicillin-streptomycin (Gibco, 15140122). U937 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum. All primary cells and cultured cell lines were maintained at 37°C in a humidified incubator containing 5% carbon dioxide.
Plasmids
SAMHD1 genes derived from horse, human, and rhesus macaque were amplified from cDNA using RT-PCR, and cloned into the pcDNA3.1 expression vector (Invitrogen, V79020) with either a 3× Flag at the N terminus or a 2× HA at the C terminus. Equine BECN1 was amplified from cDNA of eMDMs using RT-PCR and inserted into the pCMV-3× Flag expression vector (Sigma-Aldrich, E7658). The human LAMP2 gene was amplified from cDNA and fused to the VR1012 expression vector (a gift kindly provided by Dr. Xiaofang Yu at First Hospital of Jilin University) with mCherry tag. pEGFP-LC3 was constructed by inserting human LC3 encoding fragment into the pEGFP-N1 (Clontech, HG-VYC0086). The pCMV 3–8 vector (maintained in our laboratory) expressed full-length EIAV proviral genome of the EIAVFDDV13 strain [60]. Genes encoding viral structural (dUTPase and Env) and non-structural proteins (Tat and S2) were amplified from pCMV 3–8 and cloned into the pcDNA3.1–2× HA. Gag was amplified from pCMV 3–8 and cloned into the VR1012. The VR-Rev-GST was constructed by cloning the Rev sequence from pcDNA3.1-RevEIAV-HA and fusing it to the VR1012 vector with GST at the C terminus. The plasmids pcDNA3.1-VN-Flag-EfSAMHD1, pcDNA3.1-VN-EfSAMHD1-HA, and pcDNA3.1-RevEIAV-VC-HA, were constructed by fusing the N-terminal residues 2–173 of Venus and C-terminal residues 174–239 to the N-termini of either SAMHD1 or EIAV Rev, respectively. pcDNA3.1-RevHIV-VC, pcDNA3.1-RevHIV-HA were reported previously [61]. A series of Flag-tagged truncated EfBECN1 constructs were generated using site-directed mutagenesis. HIV-2 Vpx was codon-optimized for expression in human cells using services provided by Life Technologies Corporation (Rockville, USA). The codon optimized cDNAs were cloned into pcDNA3.1–2× HA. All constructed plasmids were analyzed and verified by DNA sequencing.
Chemical reagents
Puromycin (P9620), PMA (P1585), MG132 (C2211), chloroquine (PHR1258), bafilomycin A1 (B1793), wortmannin (12–338), and 3-MA (M9281) were purchased from Sigma-Aldrich. Leptomycin B (S1726) and DAPI (C1002) were purchased from Beyotime.
Transfection and western blotting
HEK293T cells were cultured in 6-well plates and transiently transfected with the indicated plasmids using a standard calcium phosphate method or with PolyJet DNA reagent (SignaGen Laboratories, SL100688), following the manufacturer’s instructions. At 48hpt, the cells were harvested, lysed in lysis buffer (50 mM Tris-HCl [Biosharp, 0234], pH 7.5, 50 mM NaCl [Sigma-Aldrich, S7653], 5 mM EDTA [Sigma-Aldrich, E6758], 1% Triton X-100 [Sigma-Aldrich, T7878]), and then centrifuged at 10,000 × g for 5 min to remove the cell nuclei. The proteins in the cell lysates were separated using SDS-PAGE and then transferred onto nitrocellulose membranes (Millipore, HATF00010). Membranes were blocked with 5% fat-free dry milk (BD, 232100) in Tris-buffered saline (20 mM Tris-HCl, 150 mM NaCl) for 2 h at room temperature, and then incubated with the indicated primary and secondary antibodies. The mouse anti-Flag (F1804), mouse anti-HA (H9658), mouse anti-actin beta (ACTB; A1978) monoclonal antibodies, rabbit anti-SAMHD1 (SAB2102077), rabbit anti-Flag (F7425), and rabbit anti-HA (H6908) were purchased from Sigma-Aldrich; the rabbit anti-BECN1 (ab207612), rabbit anti-ATG3 (ab108282), rabbit anti-ATG5 (ab108327), and rabbit anti-ATG7 (ab52472) antibodies were purchased from Abcam; the rabbit anti-class III PtdIns3K (4263) and rabbit anti-LC3B (2775) antibodies were purchased from Cell Signaling Technology; and the rabbit anti-GST (10000-0-AP) antibodies were purchased from Proteintech. Monoclonal antibodies against P26, which is the capsid protein of EIAV, were prepared in our laboratory [62]. DyLight™ 800-labeled goat anti-mouse (5230–0415) and DyLight™ 680-labeled goat anti-rabbit (5230–0403) secondary antibodies were purchased from KPL. Bands were analyzed by scanning blots using the Odyssey Imaging System (Li-Cor, Lincoln, NE, USA).
Generation of stable expression cell lines
Stable cell lines expressing wild-type SAMHD1 or SAMHD1 mutants were generated through retroviral transduction. The cDNAs of SAMHD1 derived from horse, human and rhesus macaque were cloned separately into pLPCX retroviral vectors (Clontech, 3558616) with 3× HA tags at the C terminus. pLPCX-EfSAMHD1 HD-AA and pLPCX-HsSAMHD1 HD-AA were generated using site-directed mutagenesis and confirmed using DNA sequencing. The resultant plasmids were co-transfected with the MLV Gag-Pol (Addgene, 35614; deposited by Patrick Salmon at University of Geneva) and VSV-G helper plasmids (Addgene, 8454; deposited by Bob Weinberg at Massachusetts Institute of Technology) respectively, to generate pseudotyped viruses in HEK293T cells using calcium phosphate. Transduced U937 cells that stably expressed SAMHD1 were infected with the lentiviral expression vectors and were selected in 0.4 μg/mL puromycin. Clonal U937 cell lines were generated by limiting dilution and finally identified using western blotting. U937 were differentiated for 24 h with PMA.
Establishment of knockout cell lines
HEK293T cells were used to generate ATG gene knockout cells using CRISPR-Cas9 technology. The gRNAs were designed using the Broad Institute Zhang Lab Guide Design Resources. The sequences targeting each gene were as follows: BECN1, ATTTATTGAAACTCCTCGCC; ATG5, AACTTGTTTCACGCTATA; ATG3, GTGAAGGCATACCTACCAAC; ATG7, GAAGCTGAACGAGTATCGGC; PIK3C3, CTACATCTATAGTTGTGACC. DNA fragments that contained the gRNAs specific for each host factor, a guide RNA scaffold, a U6 promoter and a U6 termination signal sequence were synthesized and subcloned into the pMD18-T backbone vector (Clontech, 6011). The Cas9-eGFP expression plasmid (pMJ920) was kindly provided by Dr. Jennifer Doudna at University of California, Berkeley. Briefly, HEK293T cells in 6-well plates were transfected with 1.0 μg of gRNA expression plasmids and 1.0 μg of pMJ920 plasmids with PolyJet DNA reagent (Signagen Laboratories, SL100688). GFP-positive cells were sorted by fluorescence-activated cell sorting at 36 hpt. The positive clones were validated by DNA sequencing and western blotting.
RNA interference
Equine SAMHD1 siRNA and control scrambled siRNA, were designed and synthesized by Sigma-Aldrich. The sequences targeting each gene were as follows: SAMHD1, GAAUCAUUGACACACCUCA;BECN1, GACAAUUUGGCACAAUCAA. 2.0 × 105 eMDMs were seeded into 6-well plates and cultivated for 48 h, and then were transfected with SAMHD1 or scrambled siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778100). Briefly, 100 pmol siRNA in 100 μL serum-free Opti-MEM medium (Gibco, 31985070) and 6 μL Lipofectamine RNAiMAX in 100 μL of Opti-MEM were mixed and incubated for 5 min at room temperature. The mixtures were then added dropwise to each well. The knockdown efficiency was determined at 48 hpt by western blotting analysis.
Quantification of cellular dNTPs pools
The protocol for cellular dNTP extraction from cells was similar to that previously described [28]. Briefly, macrophages and PMA-treated U937 cells (2.0 × 107) were resuspended in 200 μL ice-cold 60% methanol. Samples were then heated at 95°C for 3 min, prior to centrifugation at 16,000 × g for 5 min. The supernatants were harvested and evaporated under centrifugal vacuum. The resultant pellet was then resuspended in 20 μL nuclease-free water and ready to assay. Quantification of dNTPs was carried out using a fluorescence-based primer extension assay as previously described [63]. Reaction mixtures contained 2.5 μL cell extract or dNTP standard, 1 μL dNTP mix (2.5 mmol/L) exlcuding the dNTP to be assayed, 1 μL primer NDP1 (10 μmol/L), 1 μL probe (10 μmol/L), 1 μL template (10 μmol/L), 2 μL MgCl2 (25 mmol/L), 2.5 μL 10× PCR Buffer II and 0.8175 μL AmpliTaq Gold DNA polymerase (ABI, N8080241), with a final volume of 25 μL. Thermal profiling and fluorescence detection were carried out using an isothermal program on Applied Biosystems 7500 Real-Time PCR system (Life Technologies, Grand Island, NY, USA). Data were collected at 30 s intervals for 15 min. The concentration of each dNTP was determined by comparison to the standard curves analyzed by linear regression and corrected to give fmol quantities for 106 cells.
Co-IP assay
HEK293T cells were transfected with the indicated plasmids as described above. At 48 hpt, the cellular extracts were incubated with 30 μL anti-Flag beads (Sigma-Aldrich, M8823) at 4°C for 8 h. The beads were then washed six times with phosphate-buffered saline (PBS; Thermo Fisher Scientific, 10010023), boiled in sample buffer (Beyotime, P0015L), and subjected to SDS-PAGE followed by immunoblot analysis with indicated antibodies.
GST affinity isolation
Whole-cell extracts were prepared after transfection, followed by incubation with 100 μL glutathione magbeads (GenScript, L00327) at 4°C for 8 h. The beads were then washed six times with PBS, boiled in sample buffer, and subjected to SDS-PAGE followed by immunoblot analysis with indicated antibodies.
Confocal microscopy
HEK293T and HeLa cells were transfected with indicated vectors using PolyJet DNA reagent. At 36hpt, the cells were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 for 15 min at room temperature, and blocked in 5% fat-free milk in PBS for 1 h. For the immunofluorescence assay, cells were incubated with primary antibodies against HA or Flag tags at a 1:500 dilution in blocking solution for 2 h, and then stained using Alexa Fluor 488-conjugated goat anti-mouse antibody (Invitrogen, A32723), Alexa Fluor 647-conjugated goat anti-mouse antibody (Invitrogen, A32738), or Alexa Fluor 647-conjugated goat anti-rabbit antibody (Invitrogen, A32733) at a 1:500 dilution for 1 h. Finally, the nuclei were stained with DAPI for 5 min, and cells were observed and imaged on a confocal microscope (ZEISS LSM880, Thornwood, NY, USA).
Virus production and infection
EIAV luciferase reporter virus was produced through co-transfection of pVSV-G, pUKgp (maintained in our laboratory) [64] and pONY8.1-Luc (a gift kindly provided by Dr. Carsten Münk), at a ratio of 3:3:1 using standard calcium phosphate transfection. The virus was titrated by detecting the virion-associated viral reverse transcriptase using a Reverse Transcriptase Assay Colorimetric Kit (Roche, 11468120910) according to the manufacturer’s instructions. EIAV reporter viruses were then used to infect indicted cells and the viral infectivity was measured by detecting the luciferase activity in cytoplasmic lysates using a luciferase assay according to the manufacturer’s instructions (Promega, E1500) at 48 hpi. EIAVDLV36, a replication-competent EIAV strain, was sourced from this laboratory. To achieve high infection efficiency in U937 cells, EIAV with VSV-G was used to infect U937 cells. Psuedotyped EIAV with VSV-G was produced through co-transfection of pVSV-G and pCMV 3–8 at a ratio of 10:1 using PolyJet DNA reagent.
Quantitative real-time PCR
Total RNA was extracted from eMDMs using an RNeasy mini kit (Qiagen, 74106) according to the manufacturer’s instructions, and then subjected to reverse transcription using Prime-Script RT reagent kit with a gDNA Eraser (Takara, RR047B). The expression levels of equine SAMHD1 mRNA were quantified using SYBR-Green (Takara, RR430A)-based real-time quantitative PCR analysis on Agilent Mx3000P, according to the manufacturer’s protocols. Real-time RT-PCR was performed using the SAMHD1 primers: SAMHD1-forward (5ʹCCACTTGCTCGCCCAGAC3ʹ) and SAMHD1-reverse (5ʹAGAATTAACCAGGTGCTCAAACATC3ʹ). ACTB was used as a housekeeping control to normalize the number of living cells. The ACTB primers were ACTB-forward (5ʹ ACGGCATCGTCACCAACTG3ʹ) and ACTB-reverse (5ʹCAAACATGATCTGGGTCATCTTCTC3ʹ).
To measure cDNA levels of early and late EIAV reverse transcription products in infected cells, virus stocks from transfected HEK293T cells were first treated with 50 units/mL RQ1-DNase (Promega, M6101) for 1 h to remove any plasmid DNA. eMDMs were then challenged with EIAV luciferase reporter virus and collected at the indicated time points. The total cellular DNA was extracted using DNeasy kits (Qiagen, 51306), and was further treated with DpnI (NEB, R0176V) at 37°C for 2 h to remove any plasmid DNA contamination. Equal amounts of cellular DNAs were then measured for early and late viral reverse transcripts using SYBR-Green-based real-time quantitative PCR. The primer pair used to detect early reverse transcription products of EIAV luciferase reporter virus were Early-EIAV-forward (5ʹGATTCTGCGGTCTGAGTC3ʹ) and Early-EIAV-reverse (5ʹTAGGATCTCGAACAGACAAAC3ʹ), and the primer pair used to detect EIAV late reverse transcription products of EIAV luciferase reporter virus were Late-EIAV-forward (5ʹAGGTGACGGTACAAGGGTCTC3ʹ) and 3ʹ-Late-EIAV-reverse (5ʹATGGAATGACATCCCTCAGC3ʹ). ACTB was used as a housekeeping control to normalize the number of living cells.
Yeast two-hybrid screening
Plasmids encoding a DNA binding domain (BD) fused with EIAV Rev were transformed into Saccharomyces cerevisiae AH109 and used as a bait protein to screen an eMDMs cDNA library cloned into a pGADT7-Rec vector, according to the manufacturer’s protocol. The specificity of the interaction was confirmed by retransforming activation domain (AD)-BECN1 into Y187 yeast cells and remating with BD-Rev expressed in AH109 yeast cells. The mating cultures were coated onto SD-minus Trp/Leu plate for diploid cell growth and onto SD-minus Trp/Leu/Ade/His plate containing X-α-Gal and aureobasdin A for detecting blue colony growth to select protein interaction.
Autophagic flux measurements
Turnover of LC3-II in the presence or absence of lysosome inhibitor Baf A1 was analyzed by western blot as an indicator of the autophagic flux [44,45]. To analyze autophagic flux in EIAV infected eMDMs, cells were infected or mock-infected with EIAV (RT = 200 ng) for 12 h and 24 h, and then treated with Baf A1 (50 nM) or dimethyl sulfoxide (DMSO; Sigma-Aldrich, D4540) for 6 h. To analyze the effect of overexpression of EIAV proviral DNA or Rev on induction of autophagic flux, HEK293T cells were transfected with expression vectors of EIAV proviral DNA or Rev gene for 30 h, and then treated with Baf A1 (200 nM) or DMSO for 6 h. For the starvation assay, cells were washed three times with PBS and then cultured in Earle’s balanced salt solution (EBSS; Sigma-Aldrich, E2888) medium for 2 h. Cells were harvested and probed with anti-LC3B, anti-P26, anti-HA and anti-ACTB antibodies. Microscopy-based GFP-LC3 puncta formation assays were performed as described previously [44,45]. Briefly, HEK293T cells were co-transfected with expression vectors for GFP-LC3 and EIAV proviral DNA or Rev for 30 h, and then treated with Baf A1 (200 nM) or DMSO for 6 h. HEK293T cells starved in EBSS medium for 2 h were used as a positive control. Cells were fixed, permeabilized, incubated with primary antibodies against HA or P26, and then stained using Alexa Fluor-647-conjuated goat anti-mouse antibodies.
Animals
The horses used in this study for macrophage preparation were 2–8 years old and EIAV-free or without relevant pathogens. Peripheral blood was obtained from these horses using a protocol approved by the Ethics Committees of Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences.
Quantification and statistical analysis
All experiments were repeated independently at least three times. Data were presented as means ± SD. GraphPad Prism (Graph Pad Software Inc., San Diego, CA, USA) was used for statistical tests. An unpaired two-tailed Student’s t test was used to evaluate the significance of differences between samples. P > 0.05 was considered statistically non-significant (NS), P < 0.05 was considered statistically significant. Western blotting and confocal microscopy assays were performed independently at least three times, with representative experiments being shown. Quantification of the bands in the immunoblot was performed using Odyssey CLx Image Studio.
Supplementary Material
Acknowledgments
We thank Dr. Carsten Münk at Heinrich Heine University for providing the pONY8.1-luc plasmid, Dr. Jennifer Doudna for the Cas9-eGFP expression plasmid (pMJ920), and Dr. Xiaofang Yu for providing the VR1012 vector. We thank Wdee Thienphrapa and Yuan Pu (Sanford Burnham Prebys Medical Discovery Institute, USA) for suggestions and critical reading of the manuscript. We thank the Core Facility of the Harbin Veterinary Research Institute, the Chinese Academy of Agricultural Sciences for providing the technical support.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81561128010]; National Natural Science Foundation of China [31672578]; National Natural Science Foundation of China [31072133]; National Natural Science Foundation of China [31222054]; Natural Science Foundation of Heilongjiang Province [C2017076]; Natural Science Foundation of Heilongjiang Province [C2016064]; Natural Science Foundation of Heilongjiang Province [JC2018010].
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ballana E, Este JA.. SAMHD1: at the crossroads of cell proliferation, immune responses, and virus restriction. Trends Microbiol. 2015;23(11):680–692. [DOI] [PubMed] [Google Scholar]
- [2].Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474(7353):654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hrecka K, Hao CL, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474(7353):658–U137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Goldstone DC, Ennis-Adeniran V, Hedden JJ, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480(7377):379–U134. [DOI] [PubMed] [Google Scholar]
- [5].Lahouassa H, Daddacha W, Hofmann H, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012;13(3):223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ayinde D, Casartelli N, Schwartz O. Restricting HIV the SAMHD1 way: through nucleotide starvation. Nat Rev Microbiol. 2012;10(10):675–680. [DOI] [PubMed] [Google Scholar]
- [7].Ji XY, Wu Y, Yan JP, et al. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat Struct Mol Biol. 2013;20(11):1304–U232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Coiras M, Bermejo M, Descours B, et al. IL-7 induces SAMHD1 phosphorylation in CD4+T lymphocytes, improving early steps of HIV-1 life cycle. Cell Rep. 2016;14(9):2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kyei GB, Cheng XG, Ramani R, et al. Cyclin L2 is a critical HIV dependency factor in macrophages that Controls SAMHD1 abundance. Cell Host Microbe. 2015;17(1):98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim B, Nguyen LA, Daddacha W, et al. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem. 2012;287(26):21570–21574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee EJ, Seo JH, Park JH, et al. SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget. 2017;8(40):68517–68529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gramberg T, Kahle T, Bloch N, et al. Restriction of diverse retroviruses by SAMHD1. Retrovirology. 2013;10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].White TE, Brandariz-Nunez A, Valle-Casuso JC, et al. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe. 2013;13(4):441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim ET, White TE, Brandariz-Nunez A, et al. SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J Virol. 2013;87(23):12949–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Businger R, Deutschmann J, Gruska I, et al. Human cytomegalovirus overcomes SAMHD1 restriction in macrophages via pUL97. Nat Microbiol. 2019;4(12):2260–2272. [DOI] [PubMed] [Google Scholar]
- [16].Deutschmann J, Schneider A, Gruska I, et al. A viral kinase counteracts in vivo restriction of murine cytomegalovirus by SAMHD1. Nat Microbiol. 2019;4(12):2273–2284. [DOI] [PubMed] [Google Scholar]
- [17].Hollenbaugh JA, Gee P, Baker J, et al. Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. Plos Pathog. 2013;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang K, Lv DW, Li RF. Conserved herpesvirus protein kinases target SAMHD1 to facilitate virus replication. Cell Rep. 2019;28(2):449-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ahn J, Hao CL, Yan JP, et al. HIV/Simian Immunodeficiency Virus (SIV) Accessory Virulence Factor Vpx Loads the Host Cell Restriction Factor SAMHD1 onto the E3 Ubiquitin Ligase Complex CRL4(DCAF1). J Biol Chem. 2012;287(15):12550–12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schwefel D, Boucherit VC, Christodoulou E, et al. Molecular determinants for recognition of divergent SAMHD1 proteins by the lentiviral accessory protein Vpx. Cell Host Microbe. 2015;17(4):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hofmann H, Logue EC, Bloch N, et al. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J Virol. 2012;86(23):12552–12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brunner D, Pedersen NC. Infection of peritoneal macrophages in vitro and in vivo with feline immunodeficiency virus. J Virol. 1989;63(12):5483–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].de Pablo-maiso L, Domenech A, Echeverria I, et al. Prospects in innate immune responses as potential control strategies against non-primate lentiviruses. Viruses. 2018;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Du C, Duan Y, Wang XF, et al. Attenuation of equine lentivirus alters mitochondrial protein expression profile from inflammation to apoptosis. J Virol. 2019;93:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Foley B. An overview of the molecular phylogeny of lentiviruses. Los Alamos National Laboratory. Los Alamos, NM.: Theoretical Biology and Biophysics Group; 2000. (Kuiken C MF, Foley B, JW Mellors, Hahn B, Mullins J, Marx P, Wolinsky S., editor. HIV Sequence Compendium 2000). [Google Scholar]
- [26].Leroux C, Cadore JL, Montelaro RC. Equine Infectious Anemia Virus (EIAV): what has HIV’s country cousin got to tell us? Vet Res. 2004;35(4):485–512. [DOI] [PubMed] [Google Scholar]
- [27].Oaks JL, McGuire TC, Ulibarri C, et al. Equine infectious anemia virus is found in tissue macrophages during subclinical infection. J Virol. 1998;72(9):7263–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sellon DC, Perry ST, Coggins L, et al. Wild-type equine infectious anemia virus replicates in vivo predominantly in tissue macrophages, not in peripheral blood monocytes. J Virol. 1992;66(10):5906–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Diamond TL, Roshal M, Jamburuthugoda VK, et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279(49):51545–51553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Powell RD, Holland PJ, Hollis T, et al. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286(51):43596–43600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zimmerman MD, Proudfoot M, Yakunin A, et al. Structural insight into the mechanism of substrate specificity and catalytic activity of an HD-domain phosphohydrolase: the 5ʹ-deoxyribonucleotidase YfbR from Escherichia coli. J Mol Biol. 2008;378(1):215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lin Y, Wang XF, Wang Y, et al. Env diversity-dependent protection of the attenuated equine infectious anaemia virus vaccine. Emerg Microbes Infect. 2020;9(1):1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Carpenter S, Dobbs D. Molecular and biological characterization of equine infectious anemia virus Rev. Curr HIV Res. 2010;8(1):87–93. [DOI] [PubMed] [Google Scholar]
- [34].Ma J, Zhang Z, Yao Q, et al. Regulation of Rev expression by the equine infectious anaemia virus tat-rev mRNA Kozak sequence and its potential influence on viral replication. J Gen Virol. 2016;97(9):2421–2426. [DOI] [PubMed] [Google Scholar]
- [35].Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37:465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224. [DOI] [PubMed] [Google Scholar]
- [37].Funderburk SF, Wang QJ, Yue ZY. The Beclin 1-VPS34 complex - at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wirawan E, Lippens S, Vanden Berghe T, et al. Beclin 1 A role in membrane dynamics and beyond. Autophagy. 2012;8(1):6–17. [DOI] [PubMed] [Google Scholar]
- [39].Mei Y, Glover K, Su MF, et al. Conformational flexibility of BECN1: essential to its key role in autophagy and beyond. Protein Sci. 2016;25(10):1767–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saglimbene VM, Wong G, Craig JC, et al. The association of mediterranean and DASH diets with mortality in adults on hemodialysis: the DIET-HD multinational cohort study. J Am Soc Nephrol. 2018;29(6):1741–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thoresen SB, Pedersen NM, Liestol K, et al. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res. 2010;316(20):3368–3378. [DOI] [PubMed] [Google Scholar]
- [43].McKnight NC, Zhong Y, Wold MS, et al. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014;10(10):e1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee JH, Murphy SC, Belshan M, et al. Characterization of functional domains of equine infectious anemia virus Rev suggests a bipartite RNA-binding domain. J Virol. 2006;80(8):3844–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].O’Brien WA. HIV-1 entry and reverse transcription in macrophages. J Leukoc Biol. 1994;56(3):273–277. [DOI] [PubMed] [Google Scholar]
- [48].O’Brien WA, Namazi A, Kalhor H, et al. Kinetics of human immunodeficiency virus type 1 reverse transcription in blood mononuclear phagocytes are slowed by limitations of nucleotide precursors. J Virol. 1994;68(2):1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li ZL, Huan C, Wang H, et al. TRIM21-mediated proteasomal degradation of SAMHD1 regulates its antiviral activity. EMBO Rep. 2020;21(1):e47528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mereby SA, Maehigashi T, Holler JM, et al. Interplay of ancestral non-primate lentiviruses with the virus-restricting SAMHD1 proteins of their hosts. J Biol Chem. 2018;293(42):16402–16412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Seaman MN, Marcusson EG, Cereghino JL, et al. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J Cell Biol. 1997;137(1):79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. [DOI] [PubMed] [Google Scholar]
- [53].Lucin KM, O’Brien CE, Bieri G, et al. Microglial Beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79(5):873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].O’Brien CE, Bonanno L, Zhang H, et al. Beclin 1 regulates neuronal transforming growth factor-beta signaling by mediating recycling of the type I receptor ALK5 [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S.]. Mol Neurodegener. 2015;10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13(1):7–12. [DOI] [PubMed] [Google Scholar]
- [56].Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy (vol 461, pg 654, 2009). Nature. 2016;533:7601. [DOI] [PubMed] [Google Scholar]
- [57].Kim JY, Zhao H, Martinez J, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154(2):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Harris ME, Gontarek RR, Derse D, et al. Differential requirements for alternative splicing and nuclear export functions of equine infectious anemia virus rev protein. Mol Cell Biol. 1998;18(7):3889–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lin YZ, Cao XZ, Li L, et al. The pathogenic and vaccine strains of equine infectious anemia virus differentially induce cytokine and chemokine expression and apoptosis in macrophages. Virus Res. 2011;160(1–2):274–282. [DOI] [PubMed] [Google Scholar]
- [60].Wang XF, Bai B, Lin Y, et al. High-efficiency rescue of equine infectious anemia virus from a CMV-driven infectious clone. Virol Sin. 2019;34(6):725–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang Y, Zhang H, Na L, et al. ANP32A and ANP32B are key factors in the Rev-dependent CRM1 pathway for nuclear export of HIV-1 unspliced mRNA. J Biol Chem. 2019;294(42):15346–15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hu Z, Chang H, Chu X, et al. Identification and characterization of a common B-cell epitope on EIAV capsid proteins. Appl Microbiol Biotechnol. 2016;100(24):10531–10542. [DOI] [PubMed] [Google Scholar]
- [63].Wilson PM, Labonte MJ, Russell J, et al. A novel fluorescence-based assay for the rapid detection and quantification of cellular deoxyribonucleoside triphosphates. Nucleic Acids Res. 2011;39(17):e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yin X, Hu Z, Gu QY, et al. Equine tetherin blocks retrovirus release and its activity is antagonized by equine infectious anemia virus envelope protein. J Virol. 2014;88(2):1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.