

ABSTRACT
Although molecular targeted therapies have recently displayed therapeutic effects in acute myeloid leukemia (AML), limited response and acquired resistance remain common problems. Numerous studies have associated autophagy, an essential degradation process involved in the cellular response to stress, with the development and therapeutic response of cancers including AML. Thus, we review studies on the role of autophagy in AML development and summarize the linkage between autophagy and several recurrent genetic abnormalities in AML, highlighting the potential of capitalizing on autophagy modulation in targeted therapy for AML.
Abbreviations: AML: acute myeloid leukemia; AMPK: AMP-activated protein kinase; APL: acute promyelocytic leukemia; ATG: autophagy related; ATM: ATM serine/threonine kinase; ATO: arsenic trioxide; ATRA: all trans retinoic acid; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; BET proteins, bromodomain and extra-terminal domain family; CMA: chaperone-mediated autophagy; CQ: chloroquine; DNMT, DNA methyltransferase; DOT1L: DOT1 like histone lysine methyltransferase; FLT3: fms related receptor tyrosine kinase 3; FIS1: fission, mitochondrial 1; HCQ: hydroxychloroquine; HSC: hematopoietic stem cell; IDH: isocitrate dehydrogenase; ITD: internal tandem duplication; KMT2A/MLL: lysine methyltransferase 2A; LSC: leukemia stem cell; MDS: myelodysplastic syndromes; MTORC1: mechanistic target of rapamycin kinase complex 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; NPM1: nucleophosmin 1; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PML: PML nuclear body scaffold; ROS: reactive oxygen species; RB1CC1/FIP200: RB1 inducible coiled-coil 1; SAHA: vorinostat; SQSTM1: sequestosome 1; TET2: tet methylcytosine dioxygenase 2; TKD: tyrosine kinase domain; TKI: tyrosine kinase inhibitor; TP53/p53: tumor protein p53; ULK1: unc-51 like autophagy activating kinase 1; VPA: valproic acid; WDFY3/ALFY: WD repeat and FYVE domain containing 3.
KEYWORDS: Acute myeloid leukemia, autophagy, autophagy modulator, genetic abnormalities, molecular targeted therapy
Introduction
As a common type of acute leukemia with poor survival and prognosis, acute myeloid leukemia (AML) originates from aberrant alterations of hematopoietic cells, which result in the blockage of myeloid differentiation and the suppression of hematopoietic functions [1]. The poor prognosis and clinical response of patients with AML are closely associated with the molecular genetic characteristics of this disease, which are illustrated by chromosomal translocations and recurrent mutations in the genes related to hematopoietic functions [2]; this correlation has provoked research interest in molecular targeted therapy for patients suffering from AML. Recently, targeted therapies have gradually enriched the current pattern of clinical treatment for AML. Small molecule agents targeting altered proteins or signal pathways such as FLT3 (fms related receptor tyrosine kinase 3), IDH (isocitrate dehydrogenase) and BCL2 (BCL2 apoptosis regulator) have shown benefits [3–5]. However, treatment failures caused by limited clinical response and acquired resistance have restricted the development and clinical applications of molecular targeted agents. Recently, emerging evidence has revealed that autophagy has a critical role in AML development and the response to targeted therapies, suggesting that autophagy modulation holds promise for enhancing the therapeutic benefit of AML treatment.
Extensive evidence has shown that disordered autophagy regulation is necessarily associated with cancer and other diseases. Autophagy has been acknowledged as a metabolic process to digest intracellular contents, and is involved in important cellular responses to external or internal stimuli arising from hypoxia, genomic instability, metabolic stress, energy demand and chemotherapy in cancer [6,7]. Autophagy may support cell survival and assist cancer cells in resisting against metabolic and therapeutic stress [8]. Moreover, the effects of the contents degraded by the autophagy-lysosome pathway on cancer development need to be taken into consideration. It is universally acknowledged that autophagy exerts complicated affects on the generation and progression of cancers including AML. Thus, this review discusses the role of autophagy in AML development and explains how autophagy may be manipulated to strengthen therapeutic benefits of targeted therapy for AML.
The role of autophagy in AML development
Macroautophagy is acknowledged as the major autophagic process; other common forms of autophagy include microautophagy and chaperone-mediated autophagy (CMA). Hence, we will discuss how the distinct types of autophagy participate in the initiation and progression of AML, which will shed some light on the exploitation of targeted strategies for AML (summarized in Table 1).
Table 1.
The role of different forms of autophagy in AML development
| Autophagy modulation | autophagy-modulating method | leukemic model | key finding | role of autophagy in AML | Refs |
|---|---|---|---|---|---|
| autophagy suppression | shRNA against ATG7 | mice with OCI-AML3 cells | Autophagy suppression prolongs survival after chemotherapy | promoting leukemia | [9] |
| autophagy suppression | Spautin-1 | mice with OCI-AML3 cells | Autophagy suppression prolongs survival after chemotherapy | promoting leukemia | [10] |
| autophagy suppression | atg5 or atg7-floxed | mice with BM transplantation of BM cells transduced with MLL-ENL | Autophagy suppression delays AML progression and decreases frequencies of leukemia initiating cells | promoting leukemia | [15] |
| autophagy suppression | shRNA against ATG5 or ATG7 | umbilical cord blood CD34+ cells | Autophagy suppression reduces the frequencies of hematopoietic stem cells and progenitor cells | suppressing leukemogenesis | [131] |
| mitophagy suppression | sqstm1−/− | mice with BM transplantation of ldMBM cells transduced with MN1 | Loss of SQSTM1 restored leukemia development | promoting leukemia | [19] |
| mitophagy suppression | shRNA against FIS1 | MOLM-13 and primary AML cells | Absence of FIS1 attenuates self-renewal capacity of leukemia stem cells and induce myeloid differentiation | promoting leukemia | [14] |
| mitophagy suppression | Chloroquine, Lys05, and bafilomycin A1 | MOLM-13 cells | Targeting mitophagy contributes to enhanced anti-leukemic effects of autophagy inhibitors to AML cells under hypoxia | promoting leukemia | [20] |
| CMA improvement | LAMP2 expression plasmid | OCI-AML2 cells | Overexpression of LAMP2 restores sensitivity to chemotherapy and increase cell death | suppressing leukemia | [28] |
| CMA improvement | HSP90 inhibitor 17-AAG | NB4 cells | CMA degrades mutant TP53 under metabolic stress | suppressing leukemia | [30] |
Table 2b.
(continued)
| AML subtype | Autophagy Regulation | Autophagy Modulator | Molecular Targeted Therapy Combined with | leukemic model |
Benefits from Autophagy Regulation | Refs | ||
|---|---|---|---|---|---|---|---|---|
| cell line in vitro | cell lines/xenografted animals | |||||||
| TP53 | TP53-WT | autophagy suppression | shRNA against ATG5 or ATG7 | – | AML cell lines (HL60, MOLM13, OCIM3, NB4) | patient AML CD34+ cells | acts as a potential strategy for TP53-WT AML therapy | [55,56] |
| autophagy suppression | lysosomal autophagy inhibitor HCQ | – | AML cell lines (HL60, MOLM13, OCIM3, NB4), patient AML CD34+ cells | – | acts as a potential strategy for TP53-WT AML therapy | [55,56] | ||
| TP53-mutated | autophagy improvement | HSP90 inhibitor 17-AAG | – | NB4 with TP53-R248Q | – | serves as a potential means for mutant-p53 elimination in AML therapy | [30] | |
| Epigenetic dysregulated | autophagy suppression | lysosomal autophagy inhibitor CQ | HDAC inhibitor valproic acid (VPA) | t(8;21) positive AML cell lines (Kasumi-1, SKNO-1), primary t(8;21) AML cells | – | serves as a combination therapy for t(8;21) AML | [95] | |
| autophagy suppression | AMPK inhibitor compound C | BET inhibitor JQ1 | KG-1, KG-1a | – | overcomes resistance to BET inhibitors in AML | [36] | ||
| balanced rearrangements | PML-RARA | autophagy improvement | ATRA, ATO | – | NB4 | – | contributes to effective PML/RARα eradication | [100] |
| MLL fusion | autophagy improvement | shRNA against LAMP5 and DOT1L inhibitor EPZ5676 | – | – | MV4-11 | serves as a potential strategy for MLL leukemia treatment | [108] | |
Macroautophagy
In the process of macroautophagy (autophagy), a double-membrane structure called the phagophore engulfs intracellular components including proteins and organelles. This transient compartment matures into a completed autophagosome. Subsequently, the fusion of autophagosomes and lysosomes produces autolysosomes, leading to the lysosomal digestion of vesicle contents for recycling. Various investigations have linked macroautophagy with AML development. The high expression of key genes involved in autophagic processes, such as ATG7 (autophagy related 7), SIRT1 (sirtuin 1), STK11/LKB1 (serine/threonine kinase 11) and BECN1 (beclin 1), are correlated with poor clinical outcome and short remission duration in AML patients [9,10]. Besides, multiple proteins deregulated in AML, such as TRPM2 (transient receptor potential cation channel subfamily M member 2) [11], VMP1 (vacuole membrane protein 1) [12] and CXCR4 (C-X-C motif chemokine receptor 4) [10], elevate basal autophagy levels in leukemia cells, and thus facilitate cell survival and leukemia progression. These investigations indicated that heightened autophagy activity is required for malignant progression in AML. Notably, accumulating evidence has shown that intrinsic autophagy activity supports the maintenance, pluripotency and self-renewal capacity of cancer stem cells [13], leading to malignant progression in various cancer types including AML. AML LSCs (leukemia stem cells) intrinsically retain high mitophagy activity through AMP-activated protein kinase (AMPK) activation to sustain the reactive oxygen species (ROS)-low physiological state, which is critically required for the maintenance of their self-renewal potential [14]. Furthermore, the genetic inhibition of these essential autophagy-associated genes including Atg5 and Atg7 can prolong the survival of murine leukemia models and eliminate leukemia-initiating cells [15]. These findings have highlighted the significance of autophagy activity in AML development.
Autophagy can trigger the selective elimination of impaired or extra organelles, protein aggregates and other contents [8]. For example, mitophagy is the selective autophagic elimination of mitochondria [16]. Mitochondrial translation, mitochondrial DNA copy number and other characteristics of mitochondria are regulated differently in AML cells and normal hematopoietic cells [14]. Additionally, several drugs targeting mitochondria are under research for the treatment of AML [17,18]. The loss of SQSTM1 (sequestosome 1), a selective autophagy receptor that binds to mitochondria and mediates mitophagy, induces the accumulation of injured mitochondria and mitochondrial superoxide, thus impairing leukemia cell survival [19]. The overexpression of the mitophagy regulator FIS1 (fission, mitochondrial 1), is observed in AML cells, and FIS1 depletion impairs mitophagy, weakening the self-renewal capacity of leukemia stem cells and resulting in myeloid differentiation induction through GSK3 (glycogen synthase kinase 3) inactivation [14]. These findings implicate mitophagy as a regulatory mechanism of AML progression [19] and provide the rationale for mitophagy-targeting strategies in AML treatment. Treatment with several classical macroautophagy inhibitors targeting lysosomes such as chloroquine (CQ), Lys05 and bafilomycin A1, are thought to attenuate mitophagy in AML cells and enhance anti-leukemic effects, specifically when mitophagy activity increases under hypoxia stress [20]. This result broadens the further clinical development of autophagy inhibitors for AML therapy.
Moreover, aggrephagy is also involved in autophagic degradation of oncoprotein aggregates in AML. Aggrephagy is an autophagic pathway specialized in the selective degradation of protein aggregates, which tend to accumulate aberrantly and perturb normal cellular functions. For instance, the PML (PML nuclear body scaffold)-RARA/RARα (retinoic acid receptor alpha) fusion protein, inducing acute promyelocytic leukemia (APL), can be degraded through aggrephagy mediated by SQSTM1 [21] and WDFY3/ALFY (WD repeat and FYVE domain containing 3) [22], which sequentially modulates granulocytic differentiation.
Pexophagy is another selective autophagic pathway that mediates the elimination of excessive peroxisomes. Specificity for the selective autophagic degradation of peroxisomes requires the involvement of ATM (ATM serine/threonine kinase) [23,24]. ROS activates cytoplasmic ATM kinase, and activated ATM kinase phosphorylates PEX5 (peroxisomal biogenesis factor 5) and subsequently leads to the monoubiquitination of PEX5, which is recognized by autophagy receptor protein SQSTM1, thus targeting peroxisomes to selective lysosomal degradation [23]. Notably, several widely used DNA-damaging agents in AML clinical treatment, such as doxorubicin, mitoxantrone and etoposide, induce DNA damage response and ATM activation. And some reports have shown that activated ATM resulting from DNA damage can be exported from the nucleus to the cytoplasm [25], which indicates that the treatment of these DNA-damaging agents may trigger pexophagy. However, whether pexophagy affects malignant progression and therapeutic response of AML needs to be further studied.
Other types of autophagy
Emerging evidence shows that other types of autophagy participate in the regulation of AML development. Chaperone-mediated autophagy allows selective degradation of proteins recognized by chaperone proteins. These proteins can be directly transported to LAMP2A (lysosomal associated membrane protein 2A), and ultimately degraded by lysosomes [26]. Although the upregulation of CMA has been found in the majority of cancers, CMA deficiency has recently been reported in hematological malignancies [27]. A significant defect in CMA caused by the lack of LAMP2 expression is correlated with resistance to azacytidine and poor survival of patients with myelodysplastic syndromes (MDS)-AML, and AML cells with LAMP2 deficiency display sensitivity to lysosomal autophagy inhibitors such as CQ [28]. The CMA pathway mediates the degradation of MLLT11/AF1Q (MLLT11 transcription factor 7 cofactor), which is closely linked to poor prognosis in patients with pediatric AML [29]. In addition, CMA is also partially responsible for the elimination of mutant TP53/p53 (tumor protein p53) [30]. The molecular mechanism of oncoprotein degradation via CMA and the crosstalk between CMA and macroautophagy should be studied further.
The association between autophagy and therapies for different molecular types of AML
Currently, conventional chemotherapy constitutes the mainstay of clinical treatment for AML, and it has been well documented that several chemotherapeutic agents widely used in AML treatment such as cytarabine and daunorubicin are able to induce autophagy as a survival mechanism to resist cytotoxic stress and counteract the therapeutic effects of these drugs [31,32]. In addition, pharmacological inhibition of autophagy synergized with traditional cytotoxic agents would help to overcome drug resistance, improve clinical outcomes and alleviate drug toxicity for AML therapy [31,33,34]. Moreover, autophagy activity also serves as a cytoprotective adaptive mechanism against cellular stress, such as chemotherapy, in leukemia stem cells. Autophagy activation by antileukemic agents is regarded as a prosurvival response contributing to the drug resistance of AML LSCs, including deoxycytidine analogs [15,35], BET inhibitors [36], dual MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1)-MTORC2 inhibitors [37], histone methyltransferase inhibitors [38] and BCL2 inhibitors [12]. In summary, autophagy has a critical role in AML therapy, and targeting autophagy may represent a feasible strategy to fight against AML.
In recent years, the development of an accurate classification of AML into specific molecular subtypes according to the genome landscape of AML has promoted a deeper understanding of the associations between autophagy and AML therapy from a subtype-specific perspective. Next, we propose distinct associations between autophagy and certain genetic alterations and specific applications of autophagy modulation to molecular targeted therapies for different subtypes of AML, including AML with mutated FLT3, mutated NPM1, wild-type/mutated TP53, epigenetic dysregulation and balanced rearrangements. Preclinical studies that have validated the therapeutic efficacy of autophagy regulation are summarized in Table 2.
Table 2.
The preclinical application of autophagy modulation in targeted therapies for different subtypes of AML
| AML subtype | Autophagy Regulation | Autophagy Modulator | Molecular Targeted Therapy Combined with | leukemic model |
Benefits from Autophagy Regulation | Refs | |
|---|---|---|---|---|---|---|---|
| cell line in vitro | cell lines/ xenografted animals |
||||||
| FLT3-mutated | autophagy suppression | PIK3C3/VPS34 inhibitor SAR405 | – | MOLM-14 with FLT3-ITD and FLT3-D835Y | – | overcomes acquired resistance to FLT3 inhibitors | [40] |
| autophagy suppression | shRNA against ATG12 | – | – | MOLM-14 with FLT3-D835Y | overcomes acquired resistance to FLT3 inhibitors | [40] | |
| autophagy suppression | lysosomal autophagy inhibitor Lys05 | FLT3 TKI inhibitor Quizartinib | MV4-11, MOLM-13 with FLT3-ITD | – | enhances sensitivity of FLT3-ITD+ AML cells to TKI treatment | [41] | |
| autophagy improvement | Proteasome inhibitor bortezomib | FLT3 TKI inhibitor Quizartinib | MOLM-14 with FLT3-ITD and FLT3-D835Y | MOLM-14 with FLT3-ITD and FLT3-D835Y | overcomes acquired resistance to FLT3 inhibitors | [45] | |
| autophagy improvement | RET inhibitor vandetanib | FLT3 TKI inhibitor crenolanib | MV4-11, MOLM-13 with FLT3-ITD; MONO-MAC-6 with FLT3-V592A | – | enhances sensitivity of FLT3-ITD+ AML cells to TKI treatment | [46] | |
| NPM1-mutated | autophagy suppression | shRNA against PML | – | OCI-AML3 with NPM1 mutation type A (NPM1-mA) |
– | serves as a potential strategy for NPM1-mutated AML therapy | [51] |
FLT3-mutated AML
As a receptor tyrosine kinase mainly expressed by hematopoietic progenitor cells, FLT3 plays a critical role in the normal development of the hematopoiesis system [3,39]. Mutations in FLT3 are commonly found in AML, and there are two main types of FLT3 mutations: FLT3-ITD (internal tandem duplications) and FLT3-TKD (point mutations generally involving the tyrosine kinase domain). Both types of FLT3 mutations overactivate FLT3 kinase activity and downstream pathways, resulting in high hematopoietic malignancy burdens and poor clinical outcomes in AML patients [3].
The associations of FLT3 with autophagy in AML have been gradually revealed. FLT3-ITD mutations are found to increase autophagic flux in AML cells [40,41]. The identical phenomenon was also observed in sorafenib-resistant AML cell lines bearing FLT3-TKD mutations [42]. Consequently, enhanced autophagy activity, required for leukemic cell survival and proliferation, participates in AML initiation and progression. In addition, increased autophagy levels are also related to FLT3 inhibitor resistance. However, the molecular mechanisms by which FLT3 mutations enhance autophagic flux have not been demonstrated in detail. It is highly improbable that FLT3 mutations increase autophagy in a kinase-independent manner [41]. It was reported that the transcription factor ATF4 (activating transcription factor 4) is a crucial mediator of autophagy activity stimulated by FLT3-ITD [40]. Consequently, targeting autophagy and potential regulators of the autophagic response induced by FLT3 mutants will likely be combined with FLT3 inhibitors to enhance the effects of FLT3 inhibitors and overcome resistance, because poor treatment outcomes and drug resistance have hindered the development of effective FLT3 inhibitors. For example, combinatorial treatment with quizartinib, an FLT3 inhibitor with more specific and potent inhibitory activity, and the novel autophagy inhibitor Lys05, of MV4-11 and MOLM13 cells, achieved markedly improved efficacy of proliferation inhibition and apoptosis induction in comparison with quizartinib alone [41]. In addition, in solid tumors, quizartinib combined with autophagy inhibitors such as spautins or TAK-165 (an ERBB2/HER-2 antagonist inhibiting autophagy) synergistically exert antitumor effects in various types of cancer cells [43,44]. The growth of MOLM-14 cells with the FLT3D835Y mutation, which confers resistance to quizartinib, is significantly inhibited by autophagy suppression through treatment with the PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3) inhibitor SAR405 [40]. These studies suggest that targeting autophagy is a promising approach to enhance sensitivity to tyrosine kinase inhibitor (TKI) treatment and overcome acquired resistance to FLT3 inhibitors in FLT3-mutated AML (Figure 1).
Figure 1.
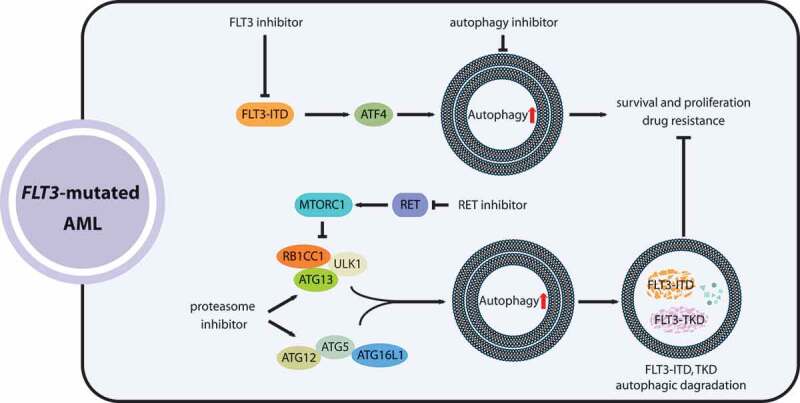
The roles of autophagy in the development and targeted therapy response of AML with FLT3 mutations. FLT3-ITD mutations promote autophagy in AML cells via ATF4, which benefits leukemia cell survival and acquired resistance to FLT3 inhibitors. Coupling FLT3-inhibiting agents with autophagy inhibitors enhances the therapeutic effectiveness for FLT3-mutated AML. Proteasome inhibitors and RET suppression (RET inhibits autophagy by activating MTORC1) can stimulate mutated-FLT3 degradation by enhancing autophagy activity, holding promise as a combinatorial treatment for FLT3-mutated AML
Moreover, the participation of autophagy in the posttranslational degradation of FLT3-ITD was revealed. For example, proteasome inhibitors such as bortezomib can activate autophagy and consequently induce the degradation of FLT3 proteins with both ITD and TKD D835Y mutations in AML cells [45]. In addition, a study also found that receptor tyrosine kinase RET (ret proto-oncogene), highly activated in AML with pro-survival functions, can drive MTORC1 activation then inhibit autophagic degradation of FLT3 proteins [46]. Thus, RET suppression by small-molecule inhibitors, such as vandetanib or danusertib, combined with crenolanib, a selective FLT3 inhibitor, synergistically attenuates the viability and proliferation of FLT3-mutated AML cells [46]. Additionally, ATO (arsenic trioxide) can also induce autophagic degradation of the FLT3-ITD proteins [47]. Accordingly, the induction of autophagy by proteasome inhibitors, RET inhibitors or ATO combined with FLT3 TKI inhibitors provides a therapeutic opportunity and prevents drug resistance acquired after TKI treatments in AML patients with FLT3 mutations (Figure 1).
Similarly, an activating mutation in another receptor tyrosine kinase, KIT (KITD816V), which is associated with AML, was reported to increase basal autophagy levels in a STAT3-dependent manner, contributing to cell survival in AML. Furthermore, autophagy suppression through ATG12 knockdown inhibits KITD816V-AML burden in vivo [48].
NPM1-mutated AML
NPM1 (nucleophosmin 1) acts as a chaperone protein that shuttles between the nucleus and the cytoplasm. The shuttling capability of NPM1 and its interaction with other proteins are involved in several cellular processes, including centrosome duplication, ribosome biogenesis, ribosomal protein transport and the regulation of tumor suppressors such as TP53. NPM1 mutations positioned in the NPM1 nuclear localization domain, frequently detected in AML patients, disturb the subcellular localization and functions of NPM1 protein, thus promoting hematopoietic malignant transformationb [49].
Several findings have revealed that autophagy contributes to the survival and growth of leukemia cells with NPM1 mutations. According to an analysis of cancer-related alterations in the autophagy pathway in multiple cancer types harboring recurrent mutations, elevated mRNA levels of autophagy-associated genes were discovered in NPM1-mutated AML [50]. Mutated NPM1 enhances autophagic activity, which confers a survival benefit onto leukemia cells [51]. Mutant NPM1 binds to PML and results in the abnormal cytoplasmic localization and accumulation of PML protein, which promotes autophagy activation and cell survival via AKT signaling. Treatment with 3-methyladenine (3-MA), an autophagy inhibitor, counteracts the cell survival promoted by mutant-NPM1-mediated autophagy induction [51]. In addition, PKM/PKM2 (pyruvate kinase M1/2) was also reported to phosphorylate BECN1 and activate autophagy in NPM1-mutated AML [52]. In light of the significant effects of heightened autophagy activity on NPM1-mutated AML, pharmacological inhibitors of autophagy and/or crucial mediators, including PML, may supply potential opportunities for the development of therapies for NPM1-mutated AML (Figure 2).
Figure 2.
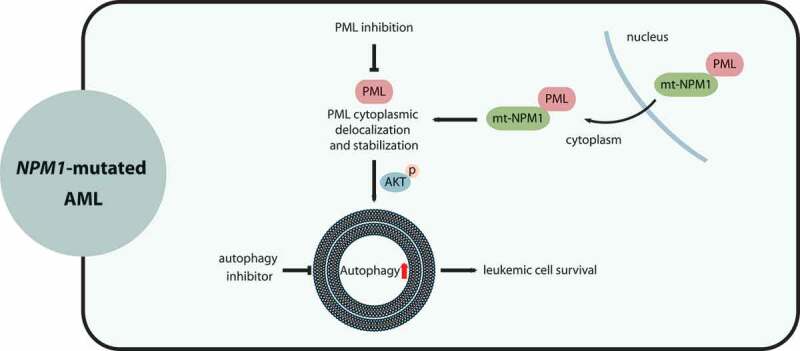
The interactions of autophagy with NPM1-mutated (mt-NPM1) AML. NPM1 interacts with PML in the nucleus. Mutated NPM1 abnormally localizes at the cytoplasm, leading to PML cytoplasmic delocalization and stabilization. Aberrantly-accumulated PML enhances autophagy levels via AKT and promotes leukemic cell survival and the progression of AML with NPM1 mutations. Pharmacological repression of autophagy and/or PML may be a promising approach for treating NPM1-mutated AML patients
TP53-WT and -mutated AML
Acting as a tumor suppressor, TP53 has a critical role in genome integrity preservation and oncogenesis suppression. Mutations in the TP53 gene are commonly identified in therapy-related AML [53,54]. It has been proposed that the role of autophagy in the development of AML may be determined by TP53 status.
For AML with wild-type TP53, research showed that pharmacological blockage of autophagy achieves therapeutic benefit, whereas AMLs harboring TP53 mutations fail to respond to autophagy inhibition by hydroxychloroquine (HCQ) [55,56]. Consistent with these observations, the deletion of key genes related to the autophagy pathway in pancreatic cancer blocks the tumor progression to high-grade carcinoma when TP53 functions are intact [57]. In hereditary breast cancer, autophagy impairment through the ablation of BECN1 restrains tumorigenesis in wild-type TP53 but does not affect tumor development with TP53 deletion [58]. Another study on lymphoma revealed that CQ induces lysosomal stress and subsequently promotes lymphoma cell death in a TP53-mediated manner [59]. These findings can be partially explained by TP53 induction caused by autophagy suppression. atg7Δ/Δ trp53Δ/Δ mice have significantly prolonged survival periods compared with atg7Δ/Δ mice, and atg7Δ/Δ trp53Δ/Δ mice also exhibit reduced apoptosis and DNA damage in liver and brain tissues, suggesting that TP53/TRP53 mediates death induction by autophagy impairment [60].
In a model of tumor-bearing mice, genetic deletion of Atg7 promotes atypical accumulation of impaired mitochondria, which results in TRP53 induction and proliferation inhibition, leading to relieved tumor burden. Importantly, the antitumor effects of Atg7 ablation are partially reversed by the loss of Trp53 [61,62]. In light of the aforementioned evidence, it can be reasonably deduced that intact functions of TP53 are required for the tumor-suppressing effects of autophagy inhibition. Similarly, in AML cells with wild-type TP53, blocking autophagy by silencing ATG5 or ATG7 or by pharmacological inhibition, such as applying HCQ treatment, stimulates the apoptotic response, which is accompanied by the enhanced activity of TP53 and the downstream genes BAX (BCL2 associated X, apoptosis regulator) and BBC3/PUMA (BCL2 binding component 3) with proapoptotic functions [55,56]. These findings indicate that pharmacological inhibition of autophagy holds potential to be a therapeutic strategy, particularly for wild-type TP53 AML (Figure 3).
Figure 3.
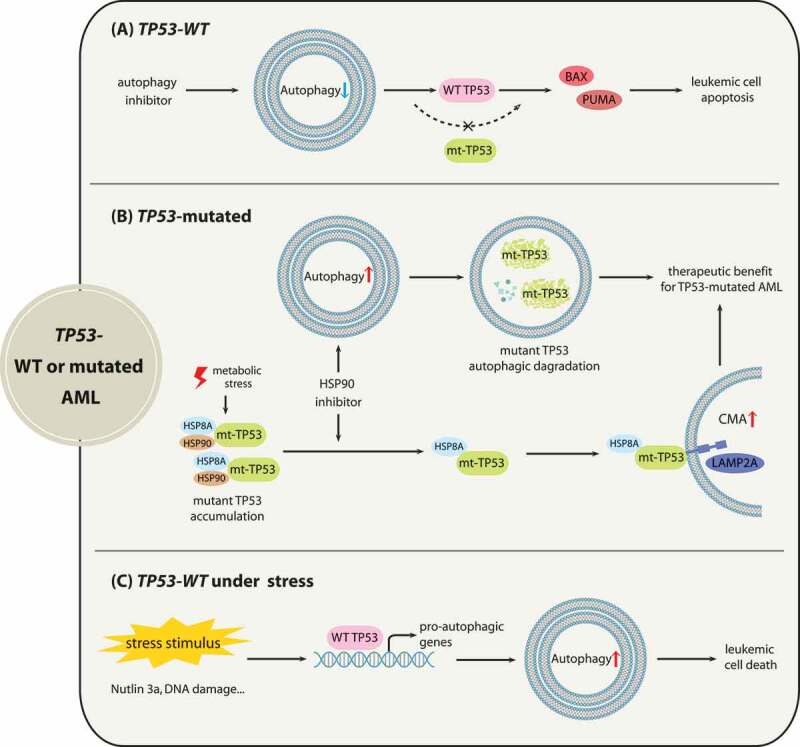
The associations between autophagy with AML depending on TP53 status. (A) for AML with wild-type TP53, autophagy suppression activates TP53 to increase the efficacy of promoting apoptosis. (B) for AML with TP53 mutations (mt-TP53), HSP90 inhibitor 17-AAG induces macroautophagy to promote the autophagic degradation of TP53R248Q. When metabolic stress suppresses macroautophagy, 17-AAG can mediate the CMA-dependent degradation of TP53R248Q in AML cells. (C) for AML with wild-type TP53 under cellular stresses, activated TP53 by cellular stress promotes autophagy induction to induce cell death
For AML with TP53 mutations, because mounting evidence has shown that TP53 gain-of-function mutants contribute to malignancy progression [63], eliminating mutant TP53 through autophagy pathways may offer therapeutic opportunities. Research has shown that macroautophagy stimulation by the HSP90 inhibitor 17-AAG mediates the degradation of TP53R248Q in AML cells, and 17-AAG may trigger autophagic flux by enhancing the transcription of autophagy-associated genes [30]. Moreover, when metabolic stress arises and results in macroautophagy repression followed by TP53R248Q accumulation, 17-AAG also promotes the elimination of the TP53R248Q protein via the CMA pathway [30]. As for the mechanism of HSP90 inhibitor-induced TP53R248Q degradation, mutant TP53 may be associated with chaperone proteins including HSP90 and HSPA8/HSC70 in cancer cells, which prevents TP53 mutants from undergoing degradation [64,65]. Treatment with17-AAG disrupts the interaction of HSP90 with TP53R248Q but does not affect the binding of HSPA8 [30], thus leading to TP53R248Q degradation through CMA (Figure 3).
In addition, accumulating evidence indicates that activated TP53 by a variety of cellular stresses can trigger autophagy through transactivating pro-autophagic genes, such as DRAM1 (DNA damage regulated autophagy modulator 1), SESN1 (sestrin 1) and SESN2 (sestrin 2) [66–69]. Autophagy induction by activated TP53 under cellular stress may result in cell death. For instance, TP53 activation by DNA damage was reported to upregulate ULK1 (unc-51 like autophagy activating kinase 1) and promote sustained autophagy activation, which is critical for the cell death induced by genotoxic stress [70]. Consistently, TP53 inactivation is frequently present in AML due to the overexpression of its E3 ubiquitin ligase MDM2 [71], and it has been reported that MDM2 inhibitors can restore TP53 activity to enhance autophagy by the transcriptional activation of AMPK, contributing to the cytotoxic effect of MDM2 antagonist Nutlin 3a in AML cells with wild-type TP53 [72,73].
Epigenetic dysregulated AML
The major forms of epigenetic modifications include DNA methylation, histone posttranslational modifications and chromatin remodeling [74]. Numerous studies have linked epigenetic alterations to leukemogenesis and disease development in AML. Epigenetic dysregulation has been regarded as a feasible target for AML treatment because these changes are pharmacologically reversible and do not involve DNA sequence alterations [74].
IDH proteins encoded by the IDH1 and IDH2 genes are related to diverse processes of epigenetic regulation, including DNA and histone demethylation. IDH mutations are present in approximately 20% of adult patients with AML [75]. IDH mutants obtain a neomorphic function to catalyze the conversion of alpha-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG), which impairs the activities of TET2 (tet methylcytosine dioxygenase 2) and histone demethylases, promoting the hypermethylation of DNA and histones. This hypermethylation phenotype causes gene expression alterations and blocks hematopoietic progenitor cell differentiation [75,76]. Drugs directly or indirectly targeting mutant IDH are currently under clinical investigation. Interestingly, recent studies have suggested the potential associations between IDH alterations and autophagy. IDH1 mutants or 2-HG product can induce autophagy, as confirmed by increased autophagosome formation in glioma cells [77]. Moreover, the autophagy inhibitor CQ suppresses GLUD (glutamate dehydrogenase), an enzyme that catalyzes the conversion of glutamate to α-KG, disrupting the mutant IDH metabolic pathway, because cells with IDH1/2 mutations require α-KG for the production of 2-HG [78]. These results suggest that inhibiting autophagy might benefit mutated-IDH targeting therapy for AML treatment. And the association of autophagy and IDH alterations need to be further studied.
TET functions in DNA demethylation by catalyzing the conversion of 5-methylcytosine/5mC to 5-hydroxymethylcytosine/5hmC, 5-formylcytosine/5fC and 5-carboxylcytosine/5caC [79]. TET2 mutations in AML, which cause hypermethylation profiles and inactivation of protein functions, have been identified to alter hematopoietic stem cell functions and development [80]. Several studies found evidence that TET2 may participate in cellular autophagy regulation. The downregulation of TET2 during the development of atherosclerosis induces the methylation of the BECN1 promoter, which results in impaired autophagic flux in endothelial cells [81]. Similarly, impaired TET2 expression decreases the expression levels of autophagy-associated genes BECN1 and MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) to downregulate endothelial cell autophagy during the atherogenic process [82]. In addition, TET2 activity can be recovered under treatment with vitamin C. Vitamin C is able to enhance the generation of 5-hydroxymethylcytosine, which results in DNA hypomethylation, thus blocking leukemia progression [83]. In addition, vitamin C combined with the hypomethylating agent decitabine holds therapeutic promise for patients with MDS or AML [84]. Interestingly, vitamin C has been demonstrated to trigger autophagy in pancreatic cancer [85]. These investigations suggest that autophagy induction of vitamin C partially results from the restoration of TET2 functions. Thus, it seems that TET2 alterations affect autophagy by regulating the transcription of autophagy-associated genes.
There are also several studies indicating that DNMTs (DNA methyltransferases) are involved in the regulation of autophagic flux. DNMT3A mutations are frequently found in myeloid malignancies with negative effects on clinical outcome [86]. The upregulation of DNMT3A is involved in rapamycin-induced autophagic responses through a decrease of Mir200b (microRNA 200b) in cardiac fibroblasts [87]. In contrast, some reports demonstrated that treatment with DNMT-inhibiting agents may induce autophagy activity. A DNMT-inhibiting phthalimido-alkanamide derivative, MA17, enhances autophagic flux in glioblastoma cells [88]. Similarly, treatment with the DNMT2 inhibitor EGCG or siRNA targeting Dnmt2 upregulates the expression of Atg5 and Lc3 in macrophages derived from aged mice [89]. In summary, the interactions between DNA methylation dysregulation and autophagy in AML development need to be further studied.
Numerous reports have shown that a number of HDAC (histone deacetylase) inhibitors (HDACis) such as VPA (valproic acid), SAHA (vorinostat), TSA (trichostatin A), panobinostat and givinostat can enhance autophagy levels in a variety of cancer types [90–94]. In AML1-ETO-positive AML cells, autophagy is also stimulated by SAHA and VPA, which facilitates cell survival and weakens the pro-apoptotic effects exerted by HDAC inhibitors. The synergistic combination of VPA with autophagy inhibitors promises to provide a therapeutic opportunity to patients with AML1-ETO-positive leukemia (Figure 4A) [95]. However, several recent publications demonstrated that HDAC inhibition represses autophagy in acute megakaryoblastic leukemia (AKML), which is a peculiar type of pediatric AML [96,97]. Treatment of Down syndrome-associated (DS-) AMKL cells with TSA, SAHA or VPA leads to autophagy suppression, because DS-AMKL cells display low basal autophagy levels owing to MTOR activation (Figure 4A). Autophagy repression results in mitochondrial mass accumulation along with ROS production, and contributes to the apoptotic effects of HDAC inhibitors [97]. These findings suggested that a low degree of autophagic flux might reflect a susceptibility to HDAC inhibitors, whereas heightened autophagy activity contributes to therapy resistance, which was confirmed by the correlation analysis of autophagy levels and treatment responses to HDACis in multiple AML cell lines and pediatric AML patient specimens [96]. In addition, reducing basal autophagy levels can reverse resistance to HDACi-induced apoptosis [95,96].
Figure 4.
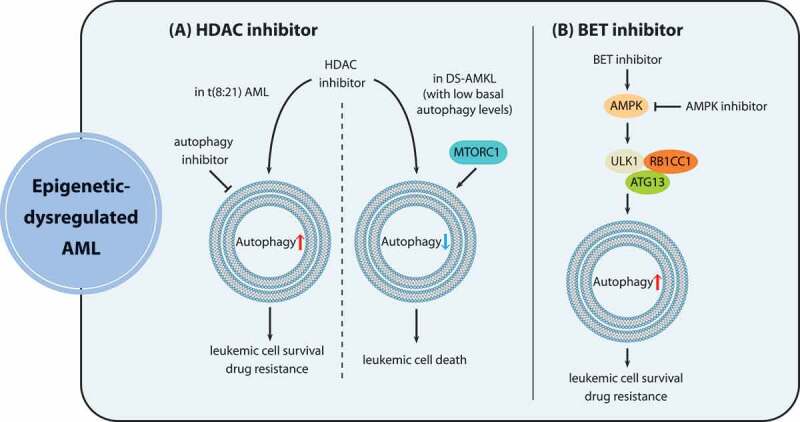
The role of autophagy in epigenetic dysregulation in AML. (A) HDAC inhibitors repress autophagic flux in DS-AMKL cells exhibiting low basal autophagy levels because of MTORC1 activation, contributing to apoptotic effects of HDAC inhibition. In contrast, t(8:21) AML cells acquire resistance against HDAC inhibitors due to autophagy induction, and the combination of HDAC inhibitors with pharmacological autophagy suppression represents a promising approach to overcoming resistance of t(8:21) AML. (B) BET inhibitors enhance autophagy through the activation of the AMPK-ULK1 pathway, thus conferring drug resistance to leukemia stem cells, which can be overcome by synergistic treatment with AMPK-inhibiting agents
BET protein (bromodomain and extra-terminal domain family) inhibitors, targeting bromodomain proteins that bind acetylated chromatin marks, show therapeutic potential especially in AML treatment [98]. However, resistance against BET protein inhibitors in leukemia stem cells is viewed as the major cause of treatment failure. BET protein inhibitor JQ1 enhances autophagy by activating the AMPK-ULK1 pathway, thus conferring the ability to antagonize the apoptotic effects of JQ1 to LSCs [36]. Therefore, autophagy inhibition holds promise as an effective means of eliminating resistance against BET protein inhibitors in AML (Figure 4B).
AML with balanced rearrangements
APL is classified as AML-M3 and presents the chromosome rearrangement t(15;17), which generates the aberrant fusion protein PML-RARA. The PML-RARA oncoprotein causes transcriptional dysregulation and differentiation disruption, leading to malignant transformation [99]. Early studies suggested that affecting the stability of PML-RARA is an important approach for APL treatment, and that the ubiquitin-proteasome pathway is mainly responsible for PML-RARA degradation induced by medical treatment. However, mounting recent evidence has shown that autophagy participates in the degradation of PML-RARA. In detail, ATRA (all trans retinoic acid) and ATO, two classical differentiation inducers that achieve ideal therapeutic effects, can stimulate autophagy through the MTOR pathway in APL cells, which contributes to PML-RARA degradation [100] (Figure 5A). Silencing of genes associated with the autophagy pathway including ATG1, ATG5 or those encoding components of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, and pharmacological inhibition by 3-MA can block PML-RARA degradation and impede the process of myeloid differentiation. Conversely, the MTOR kinase inhibitor rapamycin promotes PML-RARA elimination and subsequent myeloid differentiation by enhancing autophagy levels [21].
Figure 5.
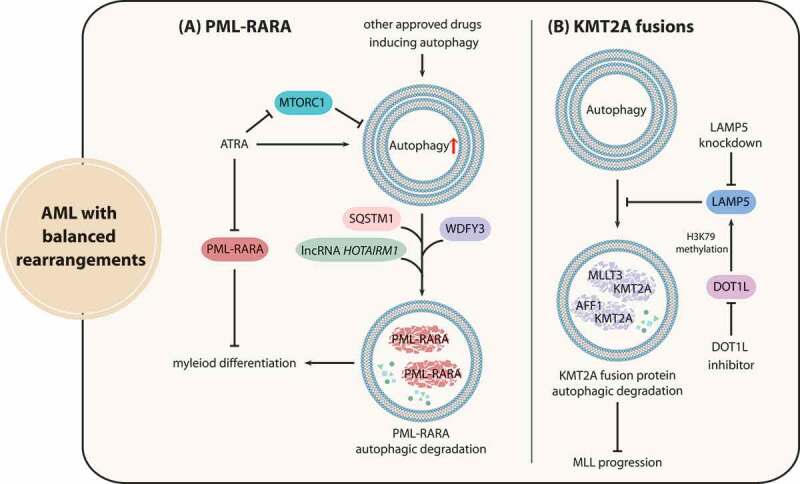
Interactions between autophagy and fusion oncoproteins caused by chromosome rearrangements in AML. (A) The differentiation-inducing agent ATRA can enhance autophagy through MTORC1 repression, and stimulated autophagy activity promotes PML-RARA autophagic degradation via a variety of mechanisms. (B) The stability of KMT2A-MLLT3 and KMT2A-AFF1 fusion proteins is maintained by LAMP5 through the suppression of selective autophagic degradation, and DOT1L mediates the activation of LAMP5. LAMP5 knockdown can be applied to synergize with DOT1L inhibitors to promote KMT2A fusion eradication for KMT2A treatment
Researchers have delineated that autophagic degradation of PML-RARA is induced through multiple mechanisms (Figure 5A). SQSTM1, an autophagy receptor protein, can bind to PML-RARA and trigger PML-RARA degradation, thus contributing to differentiation induction [21]. Additionally, PML-RARA can also interact with WDFY3/ALFY, which facilitates SQSTM1 function in promoting autophagy-dependent PML-RARA degradation [22]. Furthermore, the long noncoding RNA (lncRNA) HOTAIRM1 (HOXA transcript antisense RNA, myeloid-specific 1) also participates in PML-RARA degradation and myeloid differentiation by activating the autophagy pathway [101]. Therefore, enhancing autophagy activity in combination with classical differentiation-inducing agents constitutes an attractive strategy for APL differentiation therapy and holds potential for enhancing the sensitivity of other AML subtypes to ATRA and ATO. Promising improvement for autophagy induction can be realized by several available drugs approved by the FDA, including rapamycin analogs (sirolimus, temsirolimus, and everolimus), calcium channel blockers (verapamil, loperamide, and pimozide), lithium and dasatinib [102–104].
KMT2A/MLL (lysine methyltransferase 2A) gene translocation leads to the fusion of KMT2A and multiple partner genes, which drives gene transcription dysregulation implicated in poor prognosis of AML patients [105]. Atg5-mediated autophagy activity plays a critical role in the leukemogenesis of Kmt2a-Mllt3/Af9-driven murine AML [106]. Homozygous Atg5 ablation significantly delays Kmt2a-Mllt3-induced AML initiation and progression in vivo [107]. These results revealed the possibility that autophagy modulators may be effectively applied to the treatment of AML with KMT2A rearrangements. However, the ablation of Atg5 in AML cells during secondary transplantation had no impact on the chemotherapeutic sensitivity of mice with leukemic burdens, which indicated that Atg5-dependent autophagy may not influence chemotherapy outcome of KMT2A-rearranged AML [106].
Similar to PML-RARA, targeting the autophagic degradation process of KMT2A fusion proteins serves as a potential therapy for AML with KMT2A rearrangements, because the ubiquitin-proteasome pathway does not seem to be responsible for the degradation of KMT2A fusion proteins due to their domain defects and/or the degradation resistance of the fusion partners. As an autophagic repressor, LAMP5 sustains the stability of KMT2A-MLLT3 and KMT2A-AFF1/AF4 fusion proteins by suppressing selective autophagic degradation. It was further demonstrated that H3K79 histone methyltransferase DOT1L (DOT1 like histone lysine methyltransferase) mediates the activation of LAMP5, and the therapeutic effectiveness of DOT1L inhibitors coupled with LAMP5 knockdown was confirmed in vivo, underscoring the potential of promoting KMT2A fusion degradation via the autophagic pathway for KMT2A leukemia treatment (Figure 5B) [108].
In addition, the SQSTM1-NUP214 (nucleoporin 214) fusion protein has been discovered in AML and reported to promote leukemia development in mice [109]. Also, the fusion of SQSTM1 with NUP214 may lead to impaired autophagy activity [110]. However, whether this defect in autophagy is involved in leukemogenesis is still lacking evidence.
The application of autophagy modulators to targeted therapy for AML
The involvement of autophagy modulation in targeted therapy for AML has achieved remarkable progress in preclinical studies. Multiple early-phase clinical trials have been conducted to combine conventional autophagy inhibitors with cytotoxic anticancer agents or molecular targeted drugs to improve clinical outcomes. These combination strategies were tested in patients with other cancer types including glioblastoma, non-small cell lung cancer, myeloma, melanoma and other solid tumors, where HCQ or CQ was combined with vorinostat, bortezomib, erlotinib or other oncological therapeutic agents. These trials provided important lessons showing that the validation of predictive biomarkers would facilitate the identification of AML patient subpopulations that are likely to respond to autophagy modulation treatment. Several clinical trials have already started to validate or utilize some biomarkers of estimating the dependency on autophagy in cancers [111]. A clinical trial in glioblastoma is assessing EGFRvIII as a marker to recognize patients that may benefit from the treatment of CQ coupled with chemotherapy and radiation. Thus, research on the molecular biology mechanisms to elucidate how autophagy associates with distinct genetic alterations is of great importance to select molecular subgroups of AML highly dependent on autophagy.
Bringing the concept of genetic diversity into autophagy modulation therapy
It is widely acknowledged that gene mutations and chromosomal rearrangements provide cell growth advantages and/or disrupt hematopoietic differentiation, thus leading to AML initiation and progression. The distinctive pathogenesis, prognosis and clinical outcomes of AML greatly depend on different gene alterations and/or chromosomal abnormalities. Many studies suggest that changes in autophagy levels derived from genetic defects vary based on the diverse molecular subgroups. Furthermore, responses to pharmacological regulation of autophagy also differ in various AML subtypes. Thus, different types of autophagy modulators, including autophagy inhibitors and autophagy inducers, should be applied to different molecular subgroups of AML under diverse therapeutic conditions.
Autophagy inhibitors
Some common genetic defects in AML, such as FLT3-ITD and NPM1 mutations were found to promote autophagy and thus facilitate the survival and proliferation of leukemia cells, suggesting that autophagy inhibition is a promising approach to promote the therapeutic effectiveness of targeted therapies. For instance, FLT3-ITD mutants can enhance autophagic flux to support cell survival in AML, and, correspondingly, autophagy inhibitors combined with FLT3 inhibitors demonstrate significant synergistic efficacy. Additionally, several studies have established that TKIs such as sorafenib and imatinib induce autophagy as a protective cellular response in various cancers, including leukemia [112–114]. Autophagy stimulation has been reported to contribute to resistance against imatinib treatment in chronic myeloid leukemia (CML) [115,116]. These results indicated that autophagy inhibitors can also overcome autophagy-related resistance for therapeutic advantage and antagonize autophagy induction by genetic abnormalities.
Moreover, because the response to autophagy inhibitors is affected by TP53 status, autophagy inhibitors such as HCQ holds potential to be a therapeutic strategy for wild-type TP53 AML rather than AML with TP53 alterations. These phenomena indicate that the treatment with autophagy inhibitors may not be applicable to certain subtypes of AML, further underscoring the necessity of identifying molecular biomarkers that can predict the therapeutic outcome of autophagy inhibitors.
Autophagy inducers
Eliminating oncoproteins through the autophagic pathway represents a promising approach for AML therapy. Autophagy induction by available drugs to promote autophagic degradation of oncogenic proteins including FLT3-ITD, mutant TP53, PML-RARA and KMT2A fusion proteins in AML, has displayed remarkable anti-leukemia effects in multiple preclinical studies for the development of novel therapies. These findings are apparently contradictory to the fact that autophagy activity enhanced by FLT3-ITD facilitates cell survival and promotes AML development. These conflicting observations may be attributed to the differences in the extent of autophagy manipulation and/or the stage of disease progression.
In conclusion, the roles of autophagy in the process of disease development and targeted therapy show differences in various AML subtypes. These findings further emphasize the necessity of performing specific and detailed molecular analysis on the associations between autophagy and each AML subtype with certain genetic alterations. The results of these studies are required for providing accurate guidance on whether and how autophagy manipulation can be applied to targeted therapy for individual AML patients.
Development of autophagy modulators
Currently, preclinical findings and clinical studies applying pharmacological modulation of autophagy to cancer therapies have exhibited encouraging results, which has instigated a demand for novel autophagy modulators with higher efficacy and safety. The further exploitation of autophagy modulators is expected to facilitate the practicality of using these therapeutic approaches and offer more opportunities to AML patients.
Autophagy inhibitors
Currently, CQ and HCQ are the only autophagy inhibitors approved for clinical application [117]. These agents block the autophagic progress by deacidifying the lysosome and impairing its fusion with autophagosomes. A crucial limit to the clinical applications of CQ and HCQ is the high concentrations required for effective autophagy inhibition in vitro, which are difficult to be achieved in patients [118]. The lack of selectivity and the existence of side effects also impede the development and clinical usage of CQ and HCQ. Apart from the canonical function of lysosome inhibition through raising the lysosomal pH, CQ was also reported to disrupt the endocytosis processes requiring low pH, as well as the exiting process out of the Golgi [119]. In addition, CQ can also diminish the transcription of inflammatory cytokines such as TNF/TNF-α by a nonlysosmotropic mechanism [120]. In addition, several studies have found that CQ can facilitate the normalization of tumor vasculature through enhanced NOTCH1 signaling [121]. Moreover, several studies have reported that CQ inhibits survival and proliferation of cancer cells, and this effect cannot be imitated by the knockdown of autophagy-related genes, which indicates that the antitumor effects of CQ may not entirely result from lysosomal inhibition [122,123]. Based on these findings, the utilization of CQ and its analogs as tool compounds in cancer research should be treated cautiously. The limitation of CQ prompts a demand for novel autophagy inhibitors with higher efficacy and specificity. A novel lysosomal autophagy inhibitor, Lys05, with relatively high safety, can target lysosomes potently [124], and it holds greater promise for utilization in medical applications in cancer therapy [125,126]. Lys05 has already been used in AML preclinical research and achieved good effects. Another novel lysosomal inhibitor ROC-325 displays significantly higher potency than HCQ as well as heightened therapeutic efficacy in combination with azacitidine in AML [127]. Furthermore, multiple steps in the autophagy pathway can be targeted to provide novel approaches for inhibiting autophagy in the clinic. Compounds targeting autophagy modulators such as ULK1 [128], the BECN1-PIK3C3/VPS34 complex [116], and ATG4 [129] have been reported in early preclinical anticancer research. The development of novel autophagy inhibitors provides strong support for the application of autophagy modulation in AML therapy.
Autophagy inducers
To date, autophagy inducers applied to AML therapeutics have mostly been used to induce autophagic degradation of proteins promoting AML development. At present, the majority of autophagy inducers with the potential to be used in combination with AML therapies are currently approved drugs, including proteasome inhibitors (bortezomib), rapamycin and its analogs, kinase inhibitors (vandetanib, danusertib, dasatinib) and so on. The investigation of autophagy inducers among approved drugs would contribute to studies of their therapeutic potential for medical application in vivo. Further exploration of the specific molecular mechanisms regulating autophagic degradation of oncoproteins is still needed to develop novel therapies for AML patients.
Concluding remarks and perspective
Mounting evidence has expanded the scope of the significant role of autophagy in the development of AML. Associations between autophagy and recurrent genetic alterations in AML revealed the potential of applying autophagy modulation to therapeutic treatment for different AML subtypes, to enhance therapeutic efficacy and overcome drug resistance. Researchers have a long road to the discovery of the specific molecular mechanisms involved and to combine autophagy manipulation accurately with specific classifications of AML molecular subgroups, to further render autophagy modulation as an effective strategy in molecular targeted therapy for AML.
Notably, while autophagy may be a therapeutic target in established AML, autophagy also plays significant roles in the maintenance and functioning of normal HSCs (hematopoietic stem cells). Several investigations have found that the ablation of autophagy-related genes such as RB1CC1/FIP200 (RB1 inducible coiled-coil 1), ATG5 or ATG7 reduces HSC frequencies and impairs the reconstituting function of normal HSCs [130,131]. Moreover, ATG7 or RB1CC1 deficiency in the hematopoietic system results in aberrant myeloid expansion, coinciding with ROS accumulation and genomic instability, which is responsible for the development of aggressive phenotypes [130,132]. And it has been shown that mice harboring the deletion of Atg7 or Rb1cc1 in HSC display symptoms similar to MDS-AML such as anemia, lymphopenia and splenomegaly [130,132,133]. Similarly, mutated U2AF1 (U2 small nuclear RNA auxiliary factor 1), which associates with MDS, promotes malignant transformation through autophagy inhibition [134]; the defect in autophagy may be attributed to diminished ATG7 levels due to the abnormally altered 3ʹ UTR of ATG7. These findings suggested that autophagy impairment may facilitate malignant transformation during the initiation period of AML, whereas enhanced autophagy activity may contribute to leukemia progression and poor therapeutic outcomes in advanced stages. Thus, these findings deserve attention as they show that the roles of autophagy in AML may vary according to the stage of disease development.
Taken together, the potential toxicity induced in HSCs even during development of hematological malignant phenotypes should be considered before introducing autophagy inhibitors to AML treatment. To overcome this potential toxicity, further studies should focus on determining the therapeutic index of autophagy inhibitors. Additionally, novel drug delivery system specifically targeting leukemia cells (including LSCs) is also a possible route to achieve successful clinical application of autophagy inhibitors.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81973354]; National Natural Science Foundation of China [81830107].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Short NJ, Konopleva M, Kadia TM, et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 2020. Apr;10(4):506–525. [DOI] [PubMed] [Google Scholar]
- [2].Benard B, Gentles AJ, Kohnke T, et al. Data mining for mutation-specific targets in acute myeloid leukemia. Leukemia. 2019. Apr;33(4):826–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Antar AI, Otrock ZK, Jabbour E, et al. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia. 2020. Jan 9;34(3):682–696. [DOI] [PubMed] [Google Scholar]
- [4].Liu X, Gong Y.. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark Res. 2019;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Konopleva M, Letai A. BCL-2 inhibition in AML: an unexpected bonus? Blood. 2018. Sep 6;132(10):1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Long M, McWilliams TG. Monitoring autophagy in cancer: from bench to bedside. Semin Cancer Biol. 2019. Jul 15. DOI: 10.1016/j.semcancer.2019.05.016 [DOI] [PubMed] [Google Scholar]
- [7].Wen X, Klionsky DJ. At a glance: A history of autophagy and cancer. Semin Cancer Biol. 2019. Nov 7. DOI: 10.1016/j.semcancer.2019.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019. Sep;9(9):1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Piya S, Kornblau SM, Ruvolo VR, et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood. 2016. Sep 1;128(9):1260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu X, Mei S, Meng W, et al. CXCR4-mediated signaling regulates autophagy and influences acute myeloid leukemia cell survival and drug resistance. Cancer Lett. 2018. Jul 1;425:1–12. [DOI] [PubMed] [Google Scholar]
- [11].Chen SJ, Bao L, Keefer K, et al. Transient receptor potential ion channel TRPM2 promotes AML proliferation and survival through modulation of mitochondrial function, ROS, and autophagy. Cell Death Dis. 2020. Apr 20;11(4):247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Folkerts H, Wierenga AT, van den Heuvel FA, et al. Elevated VMP1 expression in acute myeloid leukemia amplifies autophagy and is protective against venetoclax-induced apoptosis. Cell Death Dis. 2019. May 29;10. DOI: 10.1038/s41419-019-1648-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nazio F, Bordi M, Cianfanelli V, et al. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019. Apr;26(4):690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pei S, Minhajuddin M, Adane B, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018. Jul 5;23(1):86–100 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sumitomo Y, Koya J, Nakazaki K, et al. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood. 2016. Sep 22;128(12):1614–1624. [DOI] [PubMed] [Google Scholar]
- [16].Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018. Sep;20(9):1013–1022. [DOI] [PubMed] [Google Scholar]
- [17].Lee EA, Angka L, Rota SG, et al. Targeting mitochondria with avocatin B induces selective leukemia cell death. Cancer Res. 2015. Jun 15;75(12):2478–2488. [DOI] [PubMed] [Google Scholar]
- [18].Basak NP, Banerjee S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion. 2015. Mar;21:41–48. [DOI] [PubMed] [Google Scholar]
- [19].Nguyen TD, Shaid S, Vakhrusheva O, et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood. 2019. Jan 10;133(2):168–179. [DOI] [PubMed] [Google Scholar]
- [20].Fay H, Dykstra K, Johnson M, et al. Mitophagy plays a key role in the anti-leukemic activity of autophagy inhibitors under hypoxia in acute myeloid leukemia. Blood. 2019. 11/13;134:1278-1278. DOI: 10.1182/blood-2019-127024 [DOI] [Google Scholar]
- [21].Wang Z, Cao LZ, Kang R, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RAR alpha oncoprotein. Autophagy. 2011. Apr;7(4):401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schlafli AM, Isakson P, Garattini E, et al. The autophagy scaffold protein ALFY is critical for the granulocytic differentiation of AML cells. Sci Rep-Uk. 2017. Oct 11;7:12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang JW, Tripathi DN, Jing J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015. Oct;17(10):1259-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tripathi DN, Zhang JW, Jing J, et al. A new role for ATM in selective autophagy of peroxisomes (pexophagy). Autophagy. 2016;12(4):711–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hinz M, Stilmann M, Arslan SC, et al. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappa B activation. Mol Cell. 2010. Oct 8;40(1):63–74. [DOI] [PubMed] [Google Scholar]
- [26].Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Bio. 2018. Jun;19(6):365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Arias E, Cuervo AM. Pros and cons of chaperone-mediated autophagy in cancer biology. Trends Endocrinol Metab. 2020. Jan;31(1):53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dubois A, Furstoss N, Calleja A, et al. LAMP2 expression dictates azacytidine response and prognosis in MDS/AML. Leukemia. 2019. Jun;33(6):1501–1513. [DOI] [PubMed] [Google Scholar]
- [29].Li P, Ji M, Lu F, et al. Degradation of AF1Q by chaperone-mediated autophagy. Exp Cell Res. 2014. Sep 10;327(1):48–56. [DOI] [PubMed] [Google Scholar]
- [30].Allende-Vega N, Villalba M. Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci Rep. 2019. Apr 4;9(1):5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Auberger P, Puissant A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 2017. Feb 2;129(5):547–552. [DOI] [PubMed] [Google Scholar]
- [32].Qiu L, Zhou G, Cao S. Targeted inhibition of ULK1 enhances daunorubicin sensitivity in acute myeloid leukemia. Life Sci. 2020. Feb 15;243:117234. [DOI] [PubMed] [Google Scholar]
- [33].Piya S, Andreeff M, Borthakur G. Targeting autophagy to overcome chemoresistance in acute myleogenous leukemia. Autophagy. 2017. Jan 2;13(1):214–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Putyrski M, Vakhrusheva O, Bonn F, et al. Disrupting the LC3 interaction region (LIR) binding of selective autophagy receptors sensitizes AML Cell lines to cytarabine. Front Cell Dev Biol. 2020. Mar 31;8. DOI: 10.3389/fcell.2020.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dykstra KM, Hanekamp DW, Johnson M, et al. Inhibition of the late stages of autophagy overcomes hypoxia-induced chemoresistance and targets leukemic stem cells in acute myeloid leukemia. Cancer Res. 2018. Jul;78(13):2864. [Google Scholar]
- [36].Jang JE, Eom JI, Jeung HK, et al. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy. 2017. Apr 3;13(4):761–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Altman JK, Szilard A, Goussetis DJ, et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014. May 1;20(9):2400–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jang JE, Eom JI, Jeung HK, et al. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J Exp Clin Canc Res. 2020. Apr 15;39(1). DOI: 10.1186/s13046-020-01565-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Daver N, Schlenk RF, Russell NH, et al. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019. Feb;33(2):299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Heydt Q, Larrue C, Saland E, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. 2018. Feb 8;37(6):787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Qiu SW, Paterson AJ, Abraham A, et al. Role of enhanced autophagy in resistance of FLT3-ITD AML stem cells to FLT3 TKI treatment. Blood. 2018. Nov 29;132(Supplement 1):1358-1358. [Google Scholar]
- [42].Zhang WG, Yu GP, Zhang HY, et al. Concomitant targeting of FLT3 and BTK with CG’806 overcomes FLT3-inhibitor resistance through inhibition of autophagy. Blood. 2018. Nov 29;132:2635. [Google Scholar]
- [43].Ouchida AT, Li YB, Geng JF, et al. Synergistic effect of a novel autophagy inhibitor and Quizartinib enhances cancer cell death. Cell Death Dis. 2018. Jan 26;9. DOI: 10.1038/s41419-017-0170-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xia HG, Najafov A, Geng JF, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. 2015. Aug 31;210(5):705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Larrue C, Saland E, Boutzen H, et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016. Feb 18;127(7):882–892. [DOI] [PubMed] [Google Scholar]
- [46].Rudat S, Pfaus A, Cheng YY, et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia. 2018. Oct;32(10):2189–2202. [DOI] [PubMed] [Google Scholar]
- [47].Liu XJ, Wang LN, Zhang ZH, et al. Arsenic trioxide induces autophagic degradation of the FLT3-ITD mutated protein in FLT3-ITD acute myeloid leukemia cells. J Cancer. 2020;11(12):3476–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Larrue C, Heydt Q, Saland E, et al. Oncogenic KIT mutations induce STAT3-dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis. 2019. Jul 16;8(8):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kunchala P, Kuravi S, Jensen R, et al. When the good go bad: mutant NPM1 in acute myeloid leukemia. Blood Rev. 2018. May;32(3):167–183. [DOI] [PubMed] [Google Scholar]
- [50].Lebovitz CB, Robertson AG, Goya R, et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015. Sep;11(9):1668–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zou Q, Tan S, Yang Z, et al. NPM1 mutant mediated PML delocalization and stabilization enhances autophagy and cell survival in leukemic cells. Theranostics. 2017;7(8):2289–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang L, Yang LY, Yang ZL, et al. Glycolytic enzyme PKM2 mediates autophagic activation to promote cell survival in NPM1-mutated leukemia. Int J Biol Sci. 2019;15(4):882–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015. May 8;8. DOI: 10.1186/s13045-015-0139-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood. 2017. Aug 10;130(6):699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Folkerts H, Hilgendorf S, Wierenga AT, et al. Blocking the autophagy pathway as potential target for the treatment of wild type P53 AMLs. Blood. 2016. Dec 2;128(22):770-770. [Google Scholar]
- [56].Folkerts H, Hilgendorf S, Wierenga ATJ, et al. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017. Jul;8(7):e2927–e2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013. Dec 12;504(7479):296-+. [DOI] [PubMed] [Google Scholar]
- [58].Huo YY, Cai H, Teplova I, et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013. Aug;3(8):894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Maclean KH, Dorsey FC, Cleveland JL, et al. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008. Jan;118(1):79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yang Y, Karsli-Uzunbas G, Poillet-Perez L, et al. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Gene Dev. 2020. May 1;34(9–10):688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Guo JY, Karsli-Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Gene Dev. 2013. Jul 1;27(13):1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Strohecker AM, Guo JY, Karsli-Uzunbas G, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013. Nov;3(11):1272–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014. Mar 17;25(3):304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005. Oct;5(10):761–772. [DOI] [PubMed] [Google Scholar]
- [65].Vakifahmetoglu-Norberg H, Kim M, Xia HG, et al. Chaperone-mediated autophagy degrades mutant p53. Gene Dev. 2013. Aug 1;27(15):1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Maiuri MC, Galluzzi L, Morselli E, et al. Autophagy regulation by p53. Curr Opin Cell Biol. 2010. Apr;22(2):181–185. [DOI] [PubMed] [Google Scholar]
- [67].Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006. Jul 14;126(1):121–134. [DOI] [PubMed] [Google Scholar]
- [68].Feng Z, Zhang H, Levine AJ, et al. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005. Jun 7;102(23):8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Budanov AV, Karin M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling (vol 134, pg 451, 2008). Cell. 2009. Jan 23;136(2):378-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gao W, Shen Z, Shang L, et al. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011. Oct;18(10):1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005. Nov 1;106(9):3150–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Borthakur G, Duvvuri S, Ruvolo V, et al. MDM2 inhibitor, nutlin 3a, induces p53 dependent autophagy in acute leukemia by AMP kinase activation. Plos One. 2016. Oct 6;128(22):e0139254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Duvvuri S, Ruvolo V, Mak DH, et al. MDM2 inhibitor nutlin-3A triggers autophagic cell death in addition to apoptosis in leukemia cell lines with wild-type p53. Blood. 2010. Nov 19;116(21):1352-1352. [Google Scholar]
- [74].Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019. Mar;25(3):403–418. [DOI] [PubMed] [Google Scholar]
- [75].Medeiros BC, Fathi AT, DiNardo CD, et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017. Feb;31(2):272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Amaya ML, Pollyea DA. Targeting the IDH2 pathway in acute myeloid leukemia. Clin Cancer Res. 2018. Oct 15;24(20):4931–4936. [DOI] [PubMed] [Google Scholar]
- [77].Gilbert MR, Liu YX, Neltner J, et al. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014. Feb;127(2):221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Peterse EFP, Niessen B, Addie RD, et al. Targeting glutaminolysis in chondrosarcoma in context of the IDH1/2 mutation. Br J Cancer. 2018. Apr;118(8):1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lio CWJ, Yuita H, Rao A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood. 2019. Oct 31;134(18):1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yamashita M, Dellorusso PV, Olson OC, et al. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat Rev Cancer. 2020. Jul;20(7):365–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Peng J, Yang Q, Li AF, et al. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE(-/-) mice. Oncotarget. 2016. Nov 22;7(47):76423–76436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yang Q, Li XH, Li RQ, et al. Low shear stress inhibited endothelial cell autophagy through TET2 downregulation. Ann Biomed Eng. 2016. Jul;44(7):2218–2227. [DOI] [PubMed] [Google Scholar]
- [83].Cimmino L, Dolgalev I, Wang YB, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017. Sep 7;170(6):1079–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Welch JS, Klco JM, Gao F, et al. Combination decitabine, arsenic trioxide, and ascorbic acid for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a phase I study. Am J Hematol. 2011. Sep;86(9):796–800. [DOI] [PubMed] [Google Scholar]
- [85].Cullen JJ. Ascorbate induces autophagy in pancreatic cancer. Autophagy. 2010. Apr 1;6(3):421–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Prada-Arismendy J, Arroyave JC, Rothlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2017. Jan;31(1):63–76. [DOI] [PubMed] [Google Scholar]
- [87].Zhao XD, Qin RH, Yang JJ, et al. DNMT3A controls miR-200b in cardiac fibroblast autophagy and cardiac fibrosis. Inflamm Res. 2018. Aug;67(8):681–690. [DOI] [PubMed] [Google Scholar]
- [88].Wee CW, Kim JH, Kim HJ, et al. Radiosensitization of glioblastoma cells by a novel DNA methyltransferase-inhibiting phthalimido-alkanamide derivative. Anticancer Res. 2019. Feb;39(2):759–769. [DOI] [PubMed] [Google Scholar]
- [89].Khalil H, Tazi M, Caution K, et al. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics-Us. 2016;11(5):381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Francisco R, Perez-Perarnau A, Cortes C, et al. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012. May 1;318(1):42–52. [DOI] [PubMed] [Google Scholar]
- [91].Chiao MT, Cheng WY, Yang YC, et al. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy. 2013. Oct 1;9(10):1509–1526. [DOI] [PubMed] [Google Scholar]
- [92].Gandesiri M, Chakilam S, Ivanovska J, et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis. 2012. Dec;17(12):1300–1315. [DOI] [PubMed] [Google Scholar]
- [93].Angeletti F, Fossati G, Pattarozzi A, et al. Inhibition of the autophagy pathway synergistically potentiates the cytotoxic activity of givinostat (ITF2357) on human glioblastoma cancer stem cells. Front Mol Neurosci. 2016. Oct 27;9. DOI: 10.3389/fnmol.2016.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Xia QH, Zheng Y, Jiang W, et al. Valproic acid induces autophagy by suppressing the Akt/mTOR pathway in human prostate cancer cells. Oncol Lett. 2016. Sep;12(3):1826–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Torgersen ML, Engedal N, Boe SO, et al. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood. 2013. Oct 3;122(14):2467–2476. [DOI] [PubMed] [Google Scholar]
- [96].Stankov MV, El Khatib M, Kumar Thakur B, et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia. 2014. Mar;28(3):577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Stankov MV, Heitmann K, Li Z, et al. mTOR pathway links suppressed autophagy to HDAC inhibitor-induced apoptosis in myeloid leukemia. Blood. 2011. Nov 18;118(21):1544-1544. [Google Scholar]
- [98].Fong CY, Gilan O, Lam EYN, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015. Sep 24;525(7570):538-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].de The H, Pandolfi PP, Chen Z. Acute promyelocytic leukemia: a paradigm for oncoprotein-targeted cure. Cancer Cell. 2017. Nov 13;32(5):552–560. [DOI] [PubMed] [Google Scholar]
- [100].Isakson P, Bjoras M, Boe SO, et al. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010. Sep 30;116(13):2324–2331. [DOI] [PubMed] [Google Scholar]
- [101].Chen ZH, Wang WT, Huang W, et al. The lncRNA HOTAIRM1 regulates the degradation of PML-RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ. 2017. Feb;24(2):212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jin J, Britschgi A, Schlafli AM, et al. Low autophagy (ATG) gene expression is associated with an immature AML blast cell phenotype and can be restored during AML differentiation therapy. Oxid Med Cell Longev. 2018;2018:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Orfali NM, O’Donovan T, Nyhan M, et al. Autophagy as a target for differentiation therapy in acute myeloid Leukemia. Blood. 2012. Nov 16;120(21):2464-2464. [Google Scholar]
- [104].Xie N, Zhong LK, Liu L, et al. Autophagy contributes to dasatinib-induced myeloid differentiation of human acute myeloid leukemia cells. Biochem Pharmacol. 2014. May 1;89(1):74–85. [DOI] [PubMed] [Google Scholar]
- [105].Kotani S, Yoda A, Kon A, et al. Molecular pathogenesis of disease progression in MLL-rearranged AML. Leukemia. 2019. Mar;33(3):612–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Liu Q, Chen LG, Atkinson JM, et al. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016. Sep;7(9):e2361–e2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Liu Q, Chen LG, Wang HG. Autophagy facilitates leukemogenesis in a murine model of MLL-AF9-driven AML. Cancer Res. 2016. Jul;76(14):3531. [Google Scholar]
- [108].Wang WT, Han C, Sun YM, et al. Activation of the lysosome-associated membrane protein LAMP5 by DOT1L serves as a bodyguard for MLL fusion oncoproteins to evade degradation in leukemia. Clin Cancer Res. 2019. May 1;25(9):2795–2808. [DOI] [PubMed] [Google Scholar]
- [109].Lavau CP, Aumann WK, Sze SGK, et al. The SQSTM1-NUP214 fusion protein interacts with Crm1, activates Hoxa and Meis1 genes, and drives leukemogenesis in mice. Plos One. 2020. Apr 28;15(4):e0232036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Mendes A, Fahrenkrog B. NUP214 in Leukemia: it’s More than Transport. Cells. 2019. Jan;8(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017. Sep;17(9):528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Shi YH, Ding ZB, Zhou J, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011. Oct;7(10):1159–1172. [DOI] [PubMed] [Google Scholar]
- [113].Zheng B, Zhu H, Gu DH, et al. MiRNA-30a-mediated autophagy inhibition sensitizes renal cell carcinoma cells to sorafenib. Biochem Biophys Res Commun. 2015. Apr 3;459(2):234–239. [DOI] [PubMed] [Google Scholar]
- [114].Kamitsuji Y, Kuroda J, Kimura S, et al. The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and different modes of cell death execution in Bcr-Abl-positive leukemias. Cell Death Differ. 2008. Nov;15(11):1712–1722. [DOI] [PubMed] [Google Scholar]
- [115].Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009. May;119(5):1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Shao S, Li S, Qin YW, et al. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int J Oncol. 2014. May;44(5):1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zheng K, He ZD, Kitazato K, et al. Selective autophagy regulates cell cycle in cancer therapy. Theranostics. 2019;9(1):104–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Yang YP, Hu LF, Zheng HF, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013. May;34(5):625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kliosnky D. Guidelines for the Use and interpretation of assays for monitoring autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy. 2016;12(2):443-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Weber SM, Levitz SM. Chloroquine interferes with lipopolysaccharide-induced TNF-alpha gene expression by a nonlysosomotropic mechanism. J Immunol. 2000. Aug 1;165(3):1534–1540. [DOI] [PubMed] [Google Scholar]
- [121].Maes H, Kuchnio A, Peric A, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014. Aug 11;26(2):190–206. [DOI] [PubMed] [Google Scholar]
- [122].Maycotte P, Aryal S, Cummings CT, et al. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012. Feb;8(2):200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Eng CH, Wang ZC, Tkach D, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016. Jan 5;113(1):182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Amaravadi RK, Winkler JD. Lys05 A new lysosomal autophagy inhibitor. Autophagy. 2012. Sep;8(9):1383–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011. Feb 15;17(4):654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012. May 22;109(21):8253–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Nawrocki ST, Han YC, Visconte V, et al. The novel autophagy inhibitor ROC-325 augments the antileukemic activity of azacitidine. Leukemia. 2019. Dec;33(12):2971–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Egan DF, Chun MGH, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015. Jul 16;59(2):285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Akin D, Wang SK, Habibzadegah-Tari P, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014. Nov;10(11):2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Liu F, Lee JY, Wei HJ, et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010. Dec 2;116(23):4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gomez-Puerto MC, Folkerts H, Wierenga AT, et al. Autophagy proteins ATG5 and ATG7 are essential for the maintenance of human CD34(+) hematopoietic stem-progenitor cells. Stem Cells. 2016. Jun;34(6):1651–1663. [DOI] [PubMed] [Google Scholar]
- [132].Mortensen M, Soilleux EJ, Djordjevic G, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011. Mar 14;208(3):455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Ali AM, Raza A. Two different “tales” of ATG7: clinical relevance to myelodysplastic syndromes. Mol Cell Oncol. 2016;3(5):e1212686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Park SM, Ou JH, Chamberlain L, et al. U2AF35(S34F) promotes transformation by directing aberrant ATG7 Pre-mRNA 3 ‘ end formation. Mol Cell. 2016. May 19;62(4):479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]