

ABSTRACT
Lipid droplets (LDs), which are neutral lipid storage organelles, are important for lipid metabolism and energy homeostasis. LD lipolysis and interactions with mitochondria are tightly coupled to cellular metabolism and may be potential targets to buffer the effects of excessive toxic lipid species levels. Acetylcholine (ACh), the major neurotransmitter of the vagus nerve, exhibits cardioprotective effects. However, limited research has focused on its effects on LD lipolysis and the LD-mitochondria association in fatty acid (FA) overload models. Here, we reveal that palmitate (PA) induces an increase in expression of the FA transport protein cluster of differentiation 36 (CD36) and LD formation; remarkably reduces the expression of lipases involved in triacylglycerol (TAG) lipolysis, such as adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL) and monoacylglycerol lipase (MGL); impairs LD-mitochondria interaction; and decreases perilipin 5 (PLIN5) expression, resulting in LD accumulation and mitochondrial dysfunction, which ultimately lead to cardiomyocyte apoptosis. ACh significantly upregulates PLIN5 expression and improved LD lipolysis and the LD-mitochondria association. Moreover, ACh reduces CD36 expression, LD deposition and mitochondrial dysfunction, ultimately suppressing apoptosis in PA-treated neonatal rat ventricular cardiomyocytes (NRVCs). Knockdown of PLIN5, which plays a role in LD-mitochondria contact site formation, abolishes the protective effects of ACh in PA-treated NRVCs. Thus, ACh protects cardiomyocytes from PA-induced apoptosis, at least partly, by promoting LD lipolysis and activating LD-mitochondria interactions via PLIN5. These findings may aid in developing novel therapeutic approaches that target LD lipolysis and PLIN5-mediated LD-mitochondria interactions to prevent or alleviate lipotoxic cardiomyopathy.
KEYWORDS: palmitate, acetylcholine, lipid droplet lipolysis, lipid droplet-mitochondria interaction, perilipin 5, cardiomyocyte apoptosis
1. Introduction
Recently, high levels of saturated free fatty acids (FFAs), such as palmitate (PA), in the heart have been identified as critical mediators of obesity-induced cardiac remodeling and heart failure [1]. Increased myocardial lipid deposition occurs when cardiomyocyte fatty acid (FA) uptake outpaces FA oxidation. Increased FA levels may provoke accumulation of lipid intermediates, such as diacylglycerol (DAG), or enhance FA condensation with serine to generate ceramides, which interfere with cellular signaling and eventually lead to cellular dysfunction. This phenomenon is designated lipotoxicity and is thought to contribute to cardiomyopathies associated with the FA overload often present in diabetic and obese patients [2]. Lipid droplets (LDs) are intracellular organelles specialized for energy storage in the form of neutral lipids, such as triacylglycerols (TAGs) and sterol esters, and are bounded by a protein-decorated phospholipid monolayer. LDs are found in virtually all cell types and play a pivotal role in lipid homeostasis [3]. Additionally, excessive LD accumulation in cardiomyocytes is a hallmark of lipotoxic cardiomyopathies in metabolic syndrome and heart failure [4]. However, the mechanisms underlying abnormal LD deposition in cardiomyocyte apoptosis are still unclear, but the balance between lipid uptake and utilization appears to be crucial. Elucidation of the mechanisms of LD metabolism would enhance our understanding of the pathogenesis of PA-related cardiovascular diseases.
LDs are highly dynamic. TAG in LDs can be subsequently hydrolyzed by a subset of lipases, namely, adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL) and monoglyceride lipase (MGL). FAs released during lipolysis can further be used for mitochondrial β-oxidation in mitochondria to provide energy or substrate for cellular lipid synthesis [5]. Recently, several studies have documented tight anchoring between LDs and mitochondria in tissues with FA storage and oxidation capacities, including heart tissues, brown adipose tissue (BAT) and other tissues [6–8]. Such close connections between the two organelles ensure efficient transfer of FAs for β-oxidation without requiring them to move long distances through the cytosol, which may be an important way to prevent FA toxicity caused by the overabundance of FFAs in the cytoplasm or in mitochondria. LD-mitochondria associations have been shown to be highly regulated by perilipin 5 (PLIN5), an LD-associated protein that is highly expressed on the surface of LDs and in mitochondria in fat-oxidizing tissues and involved with LD metabolism and FA oxidation by mitochondria [9,10,11]. Nevertheless, the roles of LD lipolysis and PLIN5-mediated LD-mitochondria associations in different pathologies, specifically in metabolic disorder-induced cardiovascular diseases such as PA-induced cardiomyocyte apoptosis, remain poorly defined.
Accumulating evidence has indicated that the presence of an autonomic imbalance is characterized by sympathetic nerve overactivity and diminished vagus nerve activity in obesity and metabolic syndrome-induced cardiovascular diseases [12–14]. Our previous data revealed that pyridostigmine (PYR) increases serum acetylcholine (ACh) levels and improves cardiac function in obese rats [15]. In addition, ACh improves mitochondrial cristae remodeling to defend against PA-induced cardiomyocyte hypertrophy, presumably by increasing mitofilin expression and activating AMP-activated protein kinase to form the mitochondrial contact site and cristae organizing system complex through the muscarinic ACh receptor [16]. However, the effects of ACh on LD lipolysis and the LD-mitochondria interaction in metabolic disorder models, such as those induced by PA, have not been illuminated. In this study, we explored whether ACh protects against PA-induced cardiomyocyte apoptosis by enhancing LD lipolysis and PLIN5-mediated LD-mitochondria interactions. The findings provide mechanistic insight into therapeutic strategies for cardiomyopathy in metabolic disorder models, such as those induced by PA.
2. Materials and Methods
2.1. Chemicals and reagents
ACh (#A2661), sodium PA (#P9767) and FA-free bovine serum albumin (BSA, #A7030) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sodium PA was dissolved in phosphate-buffered saline (PBS) at 70°C and then complexed with FA-free BSA in a 6:1 molar ratio. Drug concentrations are expressed as their final molar concentrations in the culture dishes.
2.2. Neonatal rat ventricular cardiomyocyte (NRVC) isolation and culture
All experiments performed in this study were in accordance with the Guidelines on the Care and Use of Laboratory Animals and were approved by the Ethics Committee of Xi’an Jiaotong University. NRVCs were obtained from 1- to 3-day-old Sprague-Dawley rats by collagenase (0.1%) digestion according to the methodology described in previous studies [16]. The culture medium consisted of low-glucose Dulbecco’s modified Eagle’s medium (DMEM), 10% (v/v) fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. All cells were maintained in a humidified 5% CO2/95% air incubator at 37°C. After being cultured for 72 h, NRVCs were stained with α-actinin antibody to evaluate cell purity and then used for experiments.
2.3. Treatment protocol
The NRVCs were serum-starved in FBS-free DMEM and randomly divided into four groups: (1) the control (Con) group, in which cells were treated with FA-free BSA alone for 24 h; (2) the ACh group, in which cells were treated with 10−6 M ACh for 2 h and then co-incubated with FA-free BSA for 24 h; (3) the PA group, in which cells were treated with PA-BSA complex (FA-free BSA with 800 μM PA) for 24 h; and (4) the ACh-treated PA (PA+ACh) group, in which cells were treated with 10−6 M ACh for 2 h and then co-incubated with PA-BSA complex (FA-free BSA with 800 μM PA) for 24 h.
2.4. Western blot
Western blot of protein lysates from whole-cell homogenates and mitochondrial fractions was performed. Immunoblot analysis was performed as previously described [17]. The primary antibodies and their dilutions were as follows: B-cell lymphoma-2 (Bcl-2, 1:500 dilution; Abcam, UK), Bcl-2-like protein 4 (Bax, 1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA), cleaved caspase-3 (1:500 dilution; Cell Signaling Technology), cluster of differentiation 36 (CD36, 1:1000 dilution; Invitrogen, Carlsbad, CA, USA), ATGL (1:1000 dilution; Invitrogen), HSL (1:2000 dilution; Invitrogen), MGL (1:2500 dilution; Invitrogen), PLIN5 (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), perilipin 2 (PLIN2, 1:1000 dilution; Absci), voltage dependent anion channel (VDAC, 1:1000 dilution; Cell Signaling Technology) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:5000 dilution; CMCTAG, Milwaukee, WI, USA). The relative intensity of the bands was measured with ImageJ.
2.5. Terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNEL) assay
To assess NRVC apoptosis, cells were labeled with the DeadEnd Fluorometric TUNEL system (Promega, Madison, WI, USA) according to the manufacturer’s standards and protocols. Briefly, cells were washed with PBS, fixed in 4% paraformaldehyde for 30 min at room temperature, rinsed with PBS, permeabilized using 0.2% Triton X-100 for 20 min, labeled with TUNEL reaction reagents, and stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000 dilution in PBS; Beyotime, Shanghai, China) for 15 min to visualize the nuclei. Fluorescence images were taken under a confocal microscope (Nikon, Tokyo, Japan) and analyzed with ImageJ. The number of TUNEL-positive cells is presented as the percentage of positive nuclei relative to total nuclei.
2.6. LD and mitochondrial staining
For LD analysis, cells were stained with Nile red (Invitrogen) and observed by confocal microscopy. Briefly, NRVCs were first washed three times with PBS and then fixed in 4% paraformaldehyde in PBS for 30 min. After three washes, the fixed cells were stained with Nile red solution (1 μg/ml) for 15 min in the dark at room temperature and then washed three times with PBS. The cells were then incubated with 5 μg/ml DAPI (Beyotime) for 15 min at 37°C in the dark to label the nuclei and washed again with PBS. Fluorescent images were acquired using a confocal microscope (Nikon) with a 200 objective lens and analyzed using ImageJ software.
To stain mitochondria in NRVCs, cells were washed twice with PBS and incubated and loaded with 200 nM MitoTracker Green (Beyotime) in PBS at 37°C in the dark after LD staining. Fluorescence images were taken under a confocal microscope (Nikon) with a 63× oil immersion objective lens. Mitochondria were stained green, LDs appeared red, and nuclei were stained blue.
2.7. Assessment of TAG content
Intracellular TAG content was determined using a TAG assay kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China) according to the manufacturer’s instructions. The optical densities (ODs) of the samples were determined by measuring absorbance at 510 nm using a Stat Fax 2100 spectrophotometer (Awareness Technology, Palm City, FL, USA).
2.8. Analysis of mitochondrial morphology
After treatment, NRVCs were washed three times with PBS and then fixed in 4% paraformaldehyde in PBS for 30 min. cells were washed twice with PBS and incubated and loaded with 200 nM MitoTracker Green (Beyotime) in PBS at 37°C in the dark. Mitochondrial morphology was observed via fluorescence microscopy (Nikon, Japan). Cells that displayed a network of filamentous mitochondria were classified as normal, whereas cells with fragmented or partially truncated mitochondria were classified as fragmented.
2.9. Measurement of the mitochondrial membrane potential
The mitochondrial membrane potential was determined using a JC-1 assay kit (Beyotime). Cells were stained with JC-1 staining solution for 30 min at 37°C in the dark and rinsed twice with the buffer provided in the kit. Fluorescence images were acquired using a confocal microscope (Nikon). JC-1 exists either as a green fluorescent monomer at depolarized membrane potentials or as a red fluorescent J-aggregate at hyperpolarized membrane potentials. The ratio of red and green fluorescence intensities was measured to evaluate changes in the mitochondrial membrane potential.
2.10. Measurement of mitochondrial reactive oxygen species (ROS)
Mitochondrial ROS levels were detected using MitoSOX Red (Invitrogen) according to the manufacturer’s instructions. Briefly, NRVCs were loaded with 5 µM MitoSOX Red for 30 min at 37°C in the dark and washed twice with PBS. Images were acquired using a confocal microscope (Nikon) and analyzed with ImageJ.
2.11. Intracellular adenosine triphosphate (ATP) determination
ATP content in NRVCs was assessed using an enhanced ATP assay kit (Beyotime) according to the manufacturer’s instructions. The ATP content was detected using a multimode microplate reader with a luminometer (FLUOstar Omega, Germany).
2.12. Transmission electron microscopy (TEM)
NRVCs were harvested, fixed with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2–7.4) for 2 h at 4°C and then post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer for 2 h. Following fixation, the cells were dehydrated in a graded series of ethanol, infiltrated with propylene oxide embedding media, and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate. Images of the cardiomyocyte ultrastructure were captured by TEM (Hitachi, Tokyo, Japan).
2.13. Mitochondrial isolation
Mitochondrial fractions were isolated from NRVCs using a mitochondria isolation kit for cultured cells (Beyotime) according to the manufacturer’s instructions. Briefly, cells were harvested, mixed with a mitochondria extraction reagent and homogenized. The homogenates were centrifuged at 600 × g at 4°C for 10 min. Then, the supernatants were gently collected and centrifuged at 11,000 × g at 4°C for 10 min. The resultant supernatants were designated cytosolic fractions, and the pellets were considered the mitochondrial fractions.
2.14. Small interfering RNA (siRNA)
SiRNA oligonucleotides specific for the PLIN5 protein and negative control (NC) siRNA oligonucleotides were provided by GenePharma (Shanghai, China). NRVCs were transfected with siRNAs using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s instructions. The efficiency of PLIN5 protein knockdown was evaluated by western blot.
2.15. Cell viability assay
Cell viability was detected with the Cell Counting Kit-8 (CCK-8; Beyotime) according to the manufacturer’s instructions. Briefly, NRVCs were cultured in a 96-well plate with or without treatment. Next, the cells were incubated with 10 μl of CCK-8 solution for 1 h at 37°C. Then, the OD value of each well was read at a wavelength of 450 nm on a Stat Fax 2100 spectrophotometer (Awareness Technology). The cells in the control group were considered 100% viable.
2.16. Statistical analysis
The data are presented as the mean ± standard error of the mean (SEM). For normally distributed datasets, statistical significance was determined by Student’s t-test (two-tailed) or one-way or two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison post hoc test using GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant.
3. Results
3.1. ACh decreased PA-induced cardiomyocyte apoptosis and LD accumulation
Obesity and increased FFA levels are tightly linked and lead to the development of cardiovascular disorders. Furthermore, cell apoptosis is one of the characteristics of obesity-induced cardiac remodeling [18]. Therefore, our current study evaluated the mechanisms mediating cardiomyocyte apoptosis in obesity or under hyperlipidemia conditions using saturated FFA (PA)-stimulated NRVCs in vitro (Figure 1(a)). We first determined the dose-dependent effect of PA on cell death. Different concentrations of PA were added to NRVCs for 24 h to assess the expression of apoptosis-related markers. As shown in Figure 1(b), NRVCs cultured with PA showed lower Bcl-2 levels and higher cleaved caspase-3 levels than control NRVCs. At a concentration of 800 μM, PA induced maximal apoptosis in NRVCs. However, the expression of Bax was not changed after PA treatment. Based on these results and previously published studies, we chose 800 μM palmitate as the lipotoxic concentration used in all further experiments [19–22].
Figure 1.
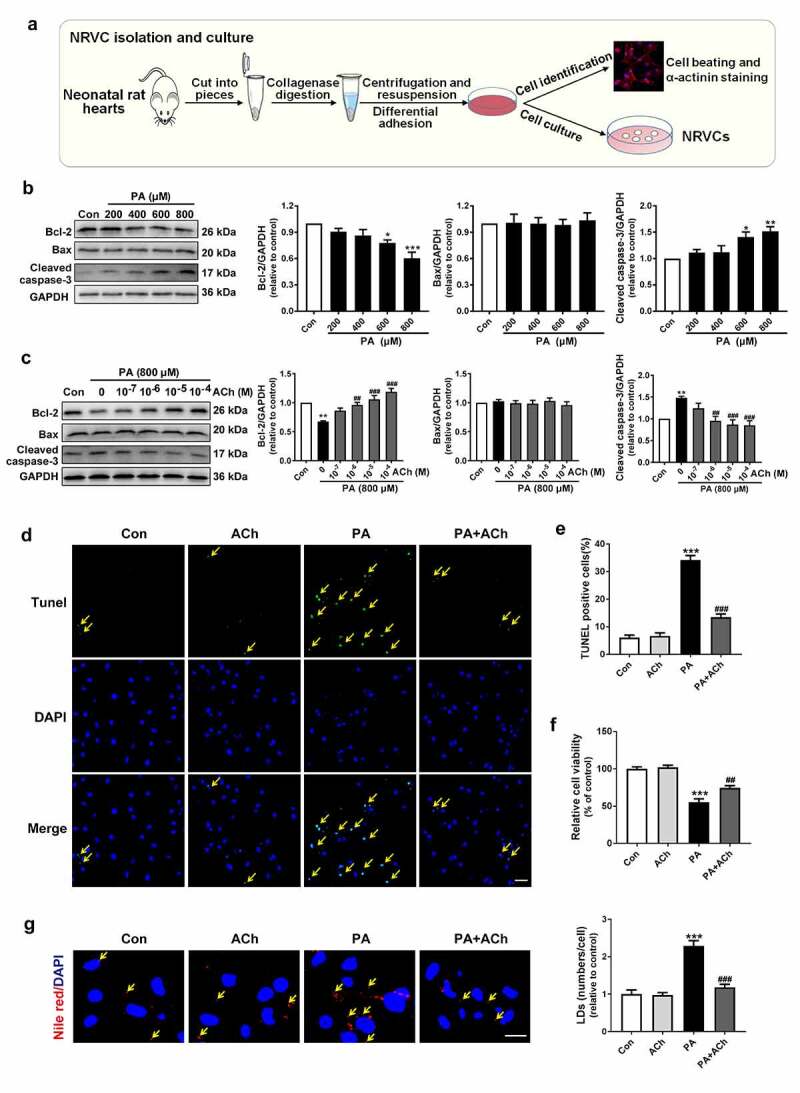
ACh reduced PA-induced apoptosis and LD accumulation in NRVCs. (a) Schematic diagram of NRVC isolation and culture. (b) PA decreased Bcl-2 expression and increased cleaved caspase-3 expression in a dose-dependent manner. n = 6 independent experiments. (c) ACh treatment attenuated the changes in Bcl-2 expression and cleaved caspase-3 expression in PA-treated NRVCs in a concentration-dependent manner. n = 6 independent experiments. (d-e) TUNEL assay was used to measure cell death in the different groups. Yellow arrows indicate TUNEL-positive nuclei. Scale bar, 50 μm. The experiments were repeated three times with cells obtained from three independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (f) ACh administration attenuated the PA-induced decrease in cell viability; n = 6 independent experiments. (g) ACh inhibited PA-induced LD accumulation. NRVCs were stained with Nile red (LDs, red) and DAPI (nuclei, blue). Yellow arrows indicate LDs. Scale bar, 20 μm. The experiments were repeated three times with cells obtained from three independent cultures, and static fluorescence images of four visual fields were analyzed by counting red lipid bodies in the merged images using ImageJ software. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Con; ##P < 0.01, ###P < 0.001 vs. PA
To examine whether ACh can decrease PA-induced cell apoptosis, NRVCs were treated with ACh at concentrations of 10−7 to 10−4 M. Pre-treatment with ACh attenuated PA-induced cell apoptosis in a concentration-dependent manner, evidenced by the elevation of Bcl-2 expression, as well as a reduction in cleaved caspase-3 expression. Interestingly, the alteration of Bax expression was also not obvious after ACh treatment. (Figure 1(c)). According to the results, 10−6 M ACh was chosen for the subsequent experiments.
Moreover, cell apoptosis was measured using the TUNEL assay. As shown in Figure 1(d, e), the number of TUNEL-positive cells was significantly increased in the PA group compared with the control group, whereas ACh treatment significantly attenuated the number of PA-induced TUNEL-positive cells. Furthermore, the viability of NRVCs was decreased in response to PA administration, while this change was inhibited by pretreatment with ACh. (Figure 1(f)). In addition, PA treatment also induced LD accumulation in NRVCs compared to control cardiomyocytes, while this change was inhibited by pretreatment with ACh (Figure 1(g)). These data suggest that ACh decreased PA-induced cardiomyocyte apoptosis and LD accumulation.
3.2. ACh decreased LD formation and reversed LD lipolysis in PA-treated cardiomyocytes
To further characterize the intimate correlation between PA-induced cell death and LD accumulation, we evaluated LD production in NRVCs after treatment with 800 μM PA for 0 h, 2 h, 4 h, 8 h, 12 h, 24 h and 36 h by Nile red staining. Our results showed that the LD content of cardiomyocytes increased in a time-dependent manner in response to PA. We found that treating NRVCs with 800 μM PA for 24 h caused a marked increase in LD number compared with that in the 0 h group. In addition, PA induced the maximal increase in LD number in NRVCs at 36 h. However, treatment with ACh decreased LD formation and LD number in PA-treated cardiomyocytes (Figure 2(a, b)). Furthermore, we found that the concentration of TAGs in the PA group was significantly higher than that in the control group, and this effect was partially corrected by ACh administration (Figure 2(c)). Next, to evaluate the involvement of FA uptake in LD accumulation and TAG levels, as well as the effect of ACh, we detected the expression of CD36, an FA transporter, in NRVCs. Compared to control treatment, PA treatment led to upregulation of CD36 expression, whereas ACh reversed the PA-induced increase in CD36 expression (Figure 2(d, e)), suggesting that ACh inhibited PA-induced cardiac lipid uptake and deposition. We also examined the effect of ACh on LD lipolysis in NRVCs via western blot. Because lipolysis of TAG in LDs occurs through the actions of a series of lipases (such as ATGL, HSL and MGL), we determined the effects of ACh on the expression of lipases involved in TAG hydrolysis in PA-treated NRVCs. As shown in Figure 2(f-h), PA supplementation reduced the expression of ATGL, HSL and MGL, and these effects were diminished by ACh. In general, these data suggest that ACh suppressed PA-induced LD formation and promoted TAG hydrolysis (Figure 2(i)).
Figure 2.
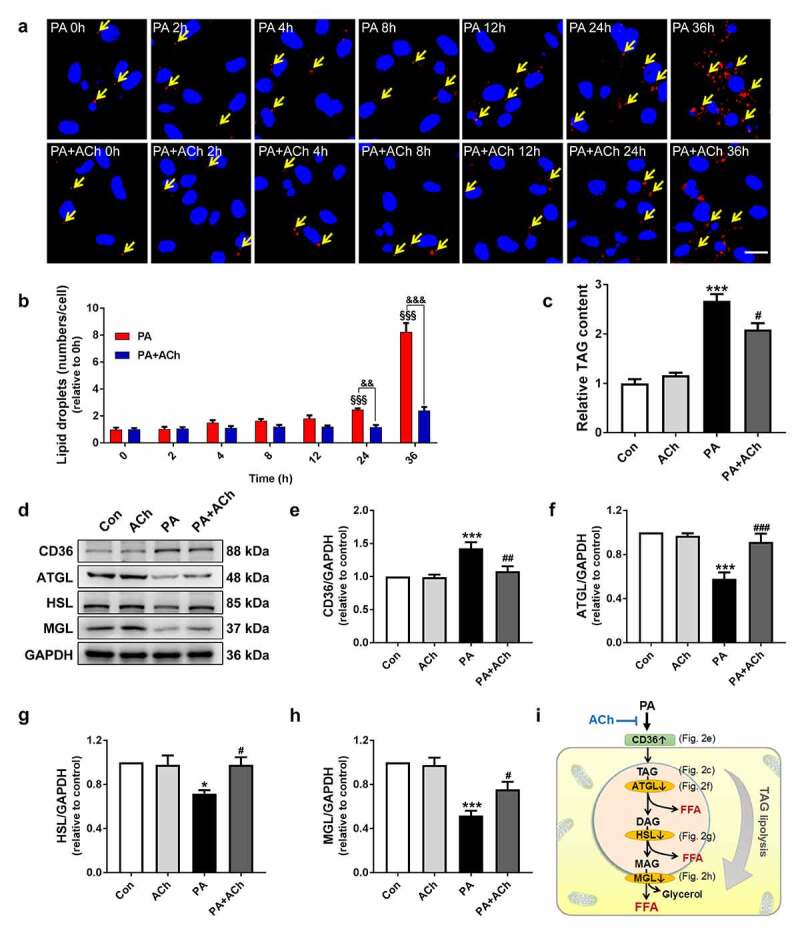
ACh inhibited LD formation and ameliorated LD lipolysis in PA-treated NRVCs. (a-b) ACh inhibited PA-induced LD formation. NRVCs were stained with Nile red (LDs, red) and DAPI (nuclei, blue). Yellow arrows indicate LDs. Scale bar, 20 μm. The experiments were repeated three times with cells obtained from three independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. LD quantification was performed by counting red lipid bodies in the merged images using ImageJ software. (c) Intracellular TAG content; n = 4 independent experiments. (d-h) The effect of ACh on CD36, ATGL, HSL and MGL expression in PA-treated NRVCs; n = 6 independent experiments. (i) ACh suppressed PA-induced LD deposition and promoted TAG lipolysis. DAG, diacylglycerol; MAG, monoacylglycerol. §§§P < 0.001 vs. 0 h; &&P < 0.01, &&&P < 0.001 vs. PA-treated NRVCs treated without ACh; *P < 0.05, ***P < 0.001 vs. Con; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. PA
3.3. ACh ameliorated the PA-induced mitochondrial dysfunction
Mitochondria adapt their morphology to support their cellular functions [23]. To determine the morphological changes in the mitochondrial network during NRVC apoptosis, cultured NRVCs were treated and then stained with MitoTracker Green. As shown in Figure 3(a), PA-treated cardiomyocytes exhibited a less connected mitochondrial network than control cardiomyocytes, which was inhibited by pretreatment with ACh. These mitochondrial morphological changes were further confirmed by the percentage of NRVCs displaying a fragmented mitochondrial morphology. PA significantly increased the percentage of NRVCs with fragmented mitochondria; however, ACh treatment abolished this phenomenon (Figure 3(b)). Moreover, to elucidate whether mitochondrial dysfunction is involved in LD accumulation after PA treatment and determine the effects of ACh, we used JC-1 staining to detect changes in mitochondrial membrane potential. A collapse of the mitochondrial membrane potential was indicated by a reduction in the ratio of red fluorescence: green fluorescence. As shown in Figure 3(c), ACh relieved the changes in the mitochondrial membrane potential of NRVCs exposed to PA, as indicated by a significant increase in the red/green fluorescence ratio in the PA + ACh group compared to the PA group. To evaluate the mitochondrial ROS levels, we used MitoSOX Red staining. As shown in Figure 3(d), a robust increase in mitochondrial ROS content was observed in the PA group compared to the control group. However, ACh reversed the effect of PA on mitochondrial ROS production. Next, we examined the expression of carnitine palmitoyl transferase 1B (CPT1B), the main regulatory enzyme of mitochondrial FA oxidation. Western blot showed that CPT1B expression in the PA-treated group was decreased compared with that in the control group. ACh treatment increased CPT1B expression in PA-treated NRVCs (Figure 3(e)). Furthermore, pretreatment with ACh prevented the decrease in ATP content induced by PA treatment (Figure 3(f)). Taken together, these results suggest that ACh protected against mitochondrial dysfunction in PA-treated cardiomyocytes.
Figure 3.
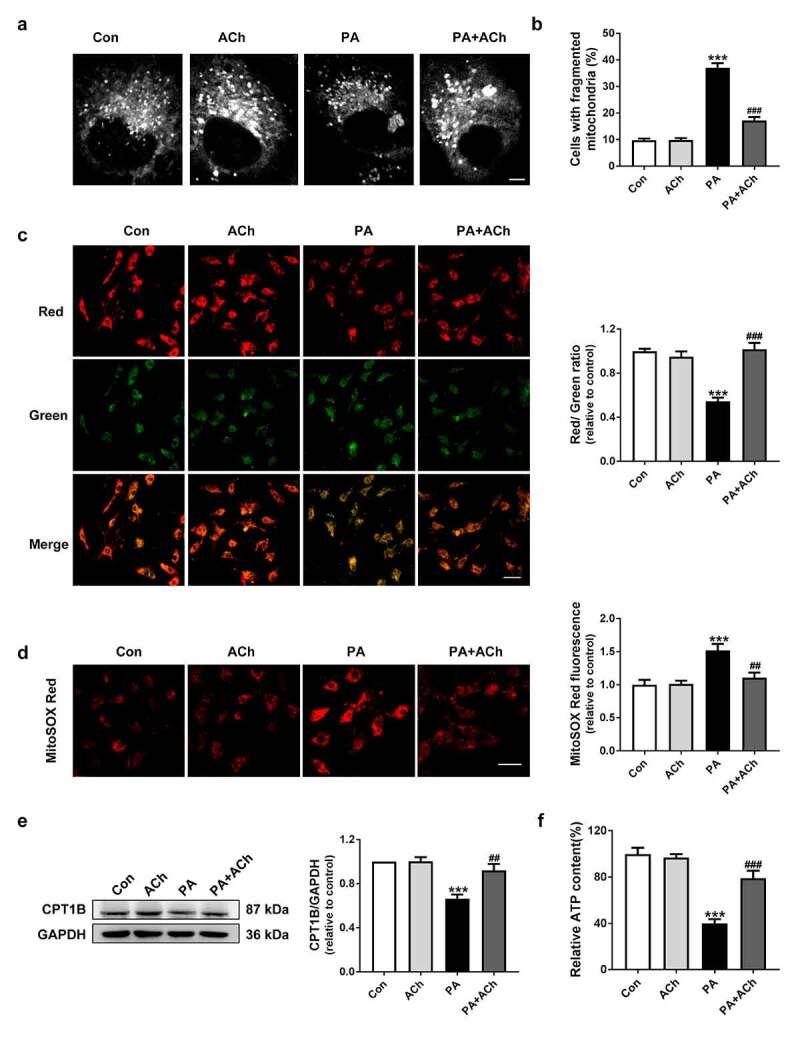
ACh protected mitochondrial function in PA-treated NRVCs. (a) Representative confocal images showing mitochondrial morphology in NRVCs. (b) The percentage of cells displaying fragmented mitochondria in each group, quantified in four randomly chosen fields of view (more than 200 cells) per experiment; n = 3 independent experiments. Scale bar, 10 μm. (c) JC-1 staining. Changes in the red/green fluorescence ratio were quantified. Scale bar, 50 μm. The experiments were repeated three times with cells obtained from three independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (d) ACh administration attenuated the PA-induced increase in mitochondrial ROS levels. Scale bar, 50 μm. The experiments were repeated four times with cells obtained from four independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (e) Expression of CPT1B in cardiomyocytes; n = 6 independent experiments. (f) Intracellular ATP content; n = 6 independent experiments. ***P < 0.001 vs. Con; ##P < 0.01, ###P < 0.001 vs. PA
3.4. ACh protected contacts between LDs and mitochondria in PA-treated cardiomyocytes
LDs are dynamic cytoplasmic organelles capable of forming stable interactions with other cell organelles, including mitochondria. Recent studies have proposed that the connections between LDs and mitochondria facilitate FA trafficking directly from LDs to mitochondria during lipolysis, which might be an efficient method for preventing high concentrations of cytotoxic FAs in the cell [9]. Therefore, we first detected the LD-mitochondrial interaction via TEM. As shown in Figure 4(a-c), LDs predominantly positioned themselves in contact with mitochondria, with approximately 80% of LDs being in immediate contact with mitochondrial membranes in the control group, but direct contacts between mitochondria and LDs were less frequent in the PA group. In addition, the ratio of mitochondria that directly contacted LDs to the total number of mitochondria was decreased in PA-treated cardiomyocytes. Pre-treatment of PA-treated NRVCs with ACh significantly increased LD-mitochondria apposition. The association of mitochondria with LDs was further confirmed by LD and mitochondrial staining (Figure 4(d,e)). In line with the TEM and confocal microscopy analyses, the LD-mitochondria interaction was further corroborated using mitochondrial isolation and western blot. As shown in Figure 4(f), the LD structural/resident protein PLIN2, was observed in mitochondrial fractions of NRVCs. The presence of PLIN2 in mitochondrial extracts was significantly reduced in the PA group compared with the control group, further suggesting that the LD-mitochondria interaction was impaired; however, ACh treatment significantly ameliorated this impairment. In summary, these results indicate that ACh can upregulate the LD-mitochondria interaction to protect against cell apoptosis in PA-induced cardiomyocytes.
Figure 4.
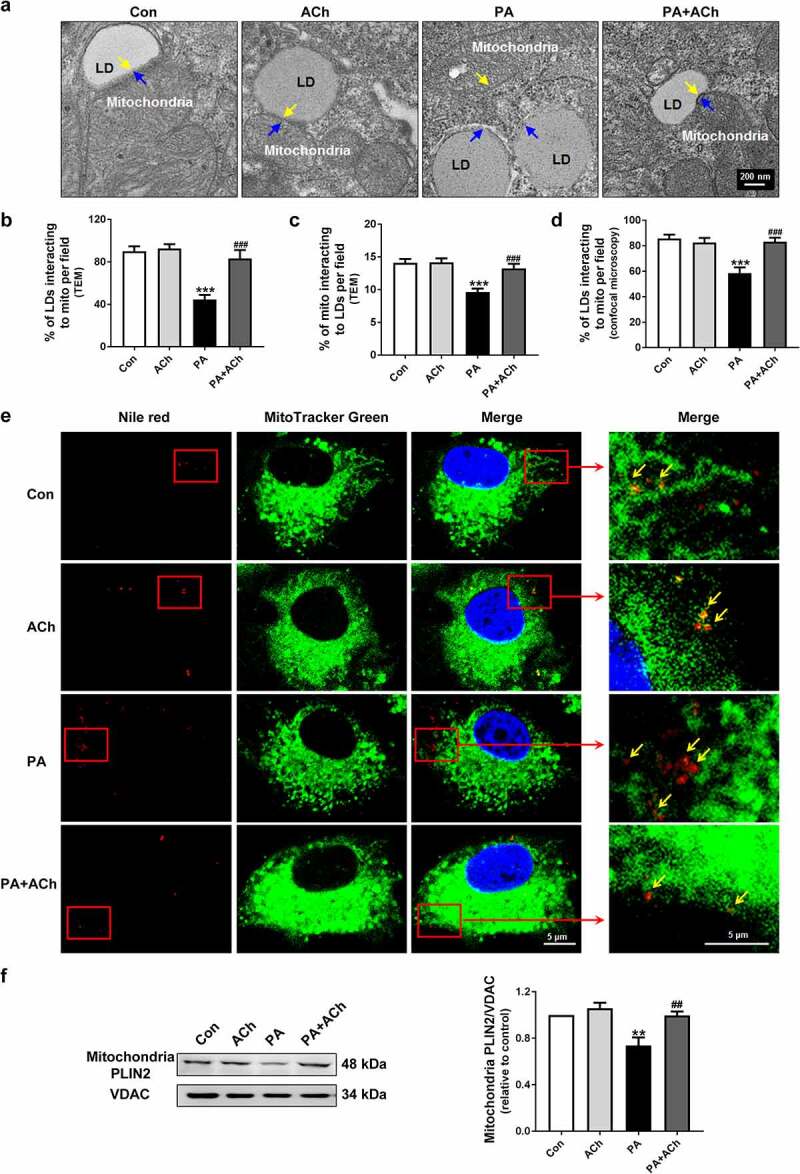
ACh reversed the PA-induced LD-mitochondria interaction. (a) Representative TEM images of NRVCs. Yellow arrows indicate individual mitochondria, and blue arrows indicate LDs. Scale bar, 200 nm. LD-mitochondria interactions in TEM images were evaluated by measuring (b) the ratio of LDs that directly contacted mitochondria to the total number of LDs, and (c) the ratio of mitochondria that directly contacted LDs to the total number of mitochondria. The experiments were repeated four times with cells obtained from four independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (d) LD-mitochondria interactions in confocal images (e) were evaluated by measuring the ratio of LDs that directly contacted mitochondria to the total number of LDs. The experiments were repeated four times with cells obtained from four independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (e) Confocal imaging of NRVCs. Scale bar, 5 μm. Cardiomyocytes were stained with Nile red (LDs, red), MitoTracker Green (mitochondria, green), and DAPI (nuclei, blue). Yellow arrows indicate LDs. (f) The effect of ACh on PLIN2 expression in mitochondrial fractions of PA-treated NRVCs; n = 6 independent experiments. **P < 0.01, ***P < 0.001 vs. Con; ##P < 0.01, ###P < 0.001 vs. PA. Mito, mitochondria
3.5. PLIN5 played an important role in LD formation
PLIN5 is important for maintaining intracellular lipid homeostasis and plays a critical role in regulation of LD-mitochondria interactions (Figure 5(a)). In our experiments, PLIN5 expression was markedly decreased during PA-induced cell apoptosis, and this decrease was reversed by ACh treatment (Figure 5(b)). To determine the role of PLIN5 in LD formation, PLIN5 expression in NRVCs was inhibited by transfection with siRNA (Figure 5(c)). The toxicity of transfection was detected by cell viability assay. Neither NC siRNA nor PLIN5 siRNA had significant effects on cell viability (Figure 5(d)). To provide direct evidence of the involvement of PLIN5 in the formation of LDs in the anti-apoptotic effect of ACh, we evaluated LD production by Nile red staining. As shown in Figure 5(e), in NRVCs transfected with NC siRNA, ACh reduced the increase in LD content induced by PA. However, under PLIN5 knockdown, ACh failed to regulate PA-induced LD production.
Figure 5.
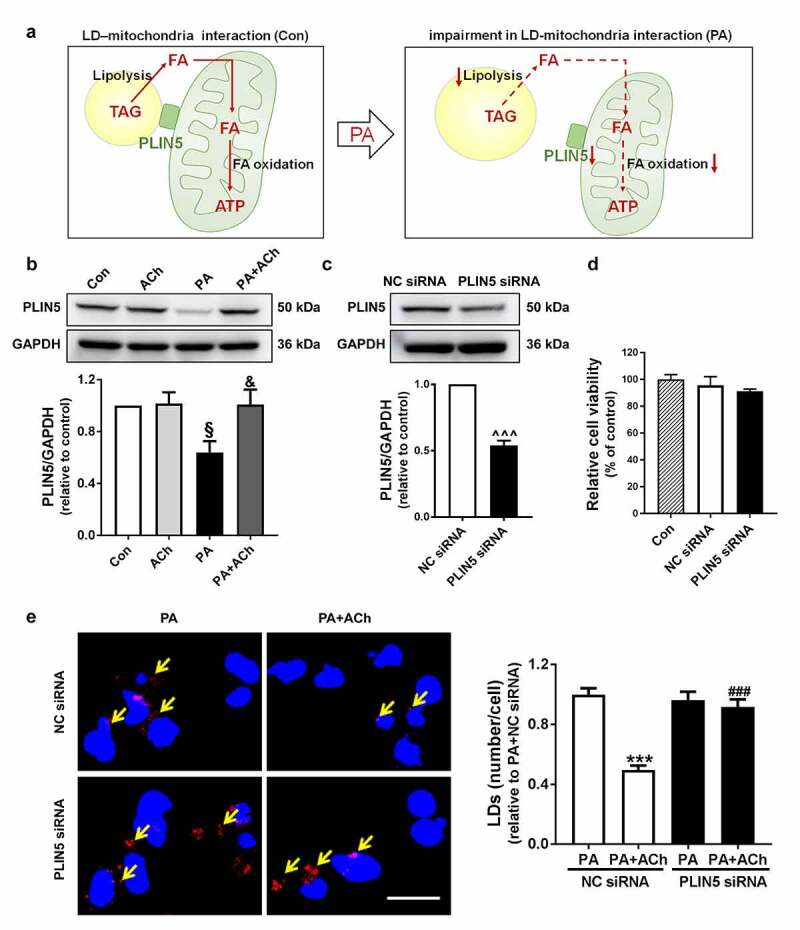
PLIN5-regulated LD formation. (a) Schematic diagram of the role of PLIN5 in LD-mitochondria interactions in PA-treated NRVCs. (b) Expression of PLIN5 in cardiomyocytes; n = 6 independent experiments. (c) Silencing efficiency of PLIN5, as determined by western blot; n = 6 independent experiments. (d) Cell viability was measured with a CCK-8 assay; n = 6 independent experiments. (e) PLIN5 knockdown in NRVCs inhibited the effects of ACh on PA-induced LD production. NRVCs were stained with Nile red (LDs, red) and DAPI (nuclei, blue). Yellow arrows indicate LDs. Scale bar, 20 μm. LD quantification was performed by counting red lipid bodies in the merged images using ImageJ software. The experiments were repeated four times with cells obtained from four independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. §P < 0.05 vs. Con; &P < 0.05 vs. PA; ^^^P < 0.001 vs. NC siRNA; ***P < 0.001 vs. PA+NC siRNA; ###P < 0.001 vs. PA+ACh+NC siRNA
3.6. Knockdown of PLIN5 abolished the protective effects of ACh against mitochondrial dysfunction and cell apoptosis in PA-treated NRVCs
We further transfected PA-treated NRVCs with PLIN5 siRNA to examine the effect of ACh on mitochondrial dysfunction and apoptosis in these cells. Our results showed that compared with control treatment, treatment with ACh decreased mitochondrial ROS production and increased ATP production. Transfection with PLIN5 siRNA inhibited the protective effects of ACh (Figure 6(a-c)). In addition, cell apoptosis induced by PA was also reversed by treatment with ACh, the effects of which were lost in NRVCs following PLIN5 knockdown (Figure 6(d-e)). Our results also showed that in NRVCs transfected with NC siRNA, ACh ameliorated the decrease in viability induced by PA. However, transfection with PLIN5 siRNA inhibited the protective effects of ACh (Figure 6 (f)). Taken together, these results confirmed that PLIN5 is activated by ACh in PA-induced NRVCs.
Figure 6.
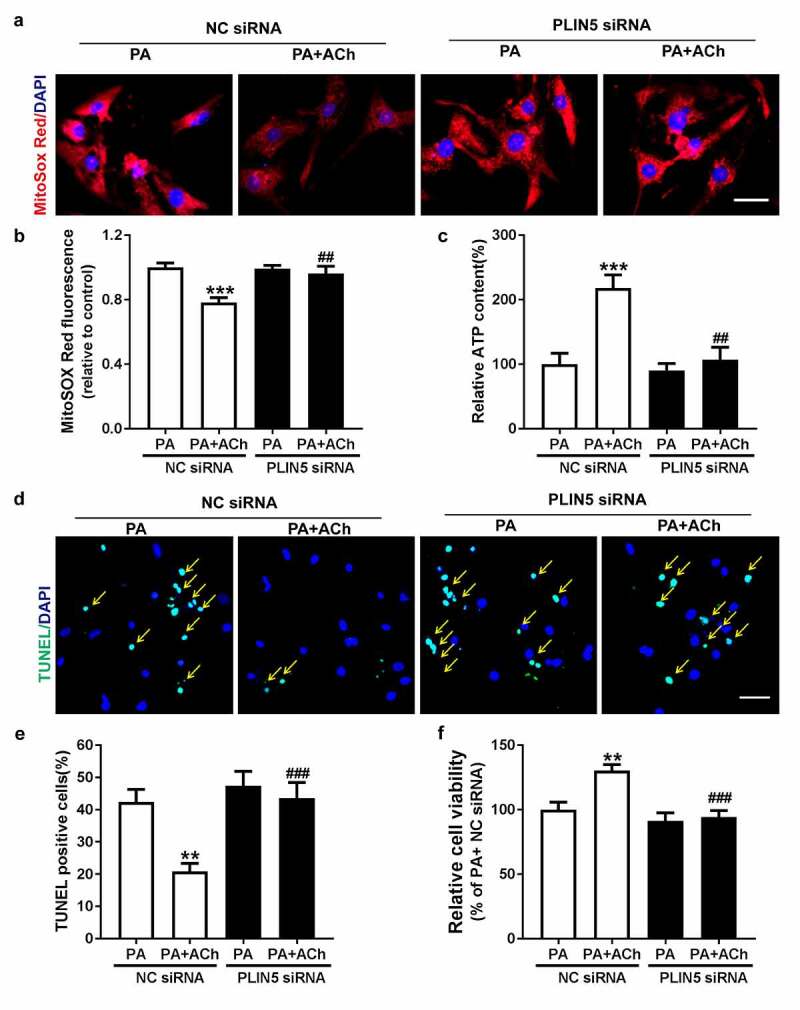
PLIN5 activation is responsible for the protective effect of ACh against mitochondrial dysfunction and cell apoptosis in PA-treated NRVCs. (a-b) The effect of PLIN5/NC siRNA transfection on mitochondrial ROS production in PA-treated NRVCs treated with or without ACh. Scale bar, 50 μm. The experiments were repeated three times with cells obtained from three independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (c) Intracellular ATP content in PA-treated NRVCs treated with or without ACh after transfection with PLIN5/NC siRNA; n = 6 independent experiments. (d-e) Changes in apoptosis of in PA-treated NRVCs treated with or without ACh after transfection with PLIN5/NC siRNA. Yellow arrows indicate TUNEL-positive nuclei. Scale bar, 50 μm. The experiments were repeated four times with cells obtained from four independent cultures, and static fluorescence images of four visual fields were analyzed for each sample. (f) Viability of PA-treated NRVCs treated with or without ACh after transfection with PLIN5/NC siRNA; n = 6 independent experiments. **P < 0.01, ***P < 0.001 vs. PA+NC siRNA; ##P < 0.01, ###P < 0.001 vs. PA+ACh+NC siRNA
4. Discussion
Elevated FFA levels resulting from obesity are accompanied by cardiac lipid metabolism dysfunction, which leads to accumulation of LDs and potentially toxic lipid species and thus an increased risk of cardiovascular disease. LD lipolysis and interactions with mitochondria might be potential effective targets to protect cells from lipotoxicity caused by an overabundance of FFAs [24]. Here, the role of LD lipolysis and LD-mitochondria interactions in PA-induced NRVCs apoptosis was investigated, and the beneficial effects of ACh were explored. The main findings of this study were as follows: PA administration led to an increase in lipid uptake, LD formation and a remarkable reduction in lipolysis, accompanied by impairment of LD-mitochondria interactions and decreased PLIN5 expression, resulting in LD accumulation and mitochondrial dysfunction, which eventually induced cardiomyocyte apoptosis. ACh, the principal neurotransmitter of the vagus nerve, significantly normalized changes in lipid uptake, LD production and LD lipolysis and improved LD-mitochondria associations and PLIN5 expression, thereby reducing the excess deposition of LDs and mitochondrial dysfunction and ultimately suppressing apoptosis of PA-treated NRVCs. In addition, PLIN5 siRNA abolished the ACh-induced attenuation of LD accumulation, mitochondrial damage and cardiomyocyte apoptosis. Taken together, these findings show that ACh ameliorates PA-induced cardiomyocyte apoptosis, at least in part, by promoting LD lipolysis and activating LD-mitochondria interactions via PLIN5 (Figure 7). Thus, improving vagal activity could be a new therapeutic strategy to prevent or alleviate cardiac dysfunction related to obesity, possibly through protection of LD lipolysis and LD-mitochondria interactions.
Figure 7.
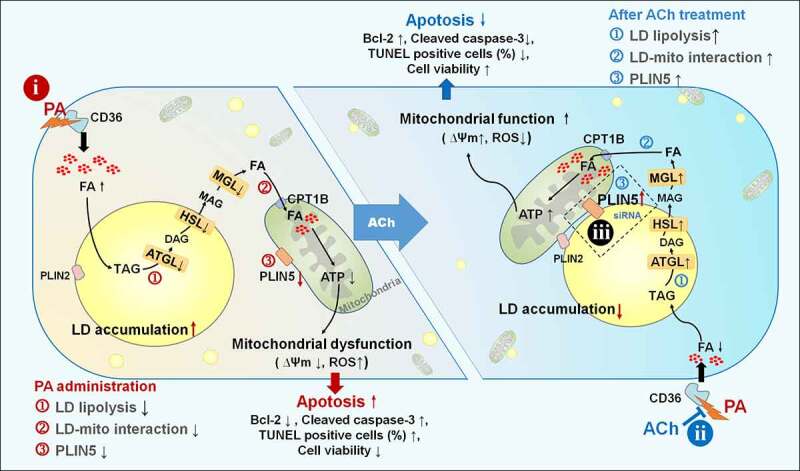
ACh improved LD lipolysis and PLIN5-mediated LD-mitochondria interaction in PA-induced cardiomyocyte apoptosis. In cultured NRVCs, (i) PA induces an increase in lipid uptake, LD formation and a remarkable reduction in lipolysis, accompanied by impairments in LD-mitochondria interactions and decreased PLIN5 expression, resulting in LD accumulation and mitochondrial dysfunction, which eventually induce cardiomyocyte apoptosis. (ii) ACh normalizes changes in lipid uptake, LD production and LD lipolysis and improves LD-mitochondria associations and PLIN5 expression, thereby reducing the excess deposition of LDs and mitochondrial dysfunction and ultimately suppressing apoptosis of PA-treated NRVCs. (iii) PLIN5 siRNA abolishes the ACh-induced attenuation of LD accumulation, mitochondrial damage, and cardiomyocyte apoptosis. ACh, acetylcholine; ATGL, adipose triglyceride lipase; ATP, adenosine triphosphate; CD36, cluster of differentiation 36; CPT1B, carnitine palmitoyl transferase 1B; DAG, diacylglycerol; FA, fatty acid; HSL, hormone-sensitive lipase; LD, lipid droplet; MAG, monoacylglycerol; MGL, monoacylglycerol lipase; PA, palmitate; PLIN, perilipin; ROS, reactive oxygen species; TAG, triacylglycerol; Ψm, mitochondrial membrane potential
An excessive accumulation of cellular FFAs may cause lipotoxicity in non-adipose tissues, and such a phenomenon is considered a hallmark of metabolic disease [2,25]. In particular, the accumulation of saturated FAs in the heart has been proposed to play a role in the development of heart failure and cardiomyopathy [26]. In our study, we utilized a specific saturated FFA, PA, to reduce complexity and investigate specific signaling pathways. We chose this FFA because it is the most common saturated FFA in circulation (approximately 28% of the total FFAs [27], with a plasma concentration range of 300–4100 μM in humans [19]). Thus, our studies at 200–800 μM fall within the pathologic range of FFA concentrations. We first test the effect of 200, 400, 600 and 800 μM PA on cardiomyocyte death. In this study, our results indicated that PA induced lower Bcl-2 levels and higher cleaved caspase-3 levels in a dose-dependent manner, and PA at 800 μM induced maximal apoptosis in NRVCs. PA supplementation significantly also increased the number of TUNEL-positive cells. However, ACh treatment significantly attenuated apoptosis in PA-treated NRVCs. However, compared with that in the control group, the expression of Bax was not changed after PA or ACh treatment. Similarly, previous studies have demonstrated that treatment with PA (100, 250 and 500 μM) for 48 and 72 h resulted in a significant decrease in Bcl-2 expression, but Bax expression was not affected by PA treatment in human osteoblasts [28]. In studies on HepG2 cells, the cells were treated with 50 μM dihydromyricetin for 3, 6, 12, 24, and 48 h, and the expression of Bcl-2 was reduced significantly in a time-dependent manner, but the alteration of Bax expression was not obvious [29]. These phenomena may be due to the fact that apoptotic pathways are not universal and may differ depending on the death stimulus and the cell type. In some models of apoptosis, Bax expression is induced [30]; whereas in others, only a significant increase in Bax activation is observed, and total Bax is not changed [31]; in some cases, only a change in Bax localization is observed [32]. Overall, our results have shown that ACh ameliorated the apoptosis of PA-treated NRVCs possibly by regulating the expression of Bcl-2 and cleaved caspase-3 but not the expression of Bax.
LDs are intracellular compartments that serve as fat reservoirs. Upon exposure to excessive FA amounts, cells can esterify FAs into TAG-rich LDs to prevent the buildup of toxic lipid intermediates and/or oxidized lipids [33]. Transient formation of LDs may protect mitochondria from a lipotoxic flux of FAs released as a result of autophagic degradation during prolonged periods of nutrient deprivation [34]. However, in the presence of chronically excessive FA levels, as observed in metabolic syndrome, LDs not only cease to be protective but may also be a characteristic signature of lipotoxic cardiomyopathy [35]. Accompanied by abnormally excessive LD deposition, a number of various lipid intermediates (diacylglycerol (DAG), ceramides, long-chain acyl CoA, and acylcarnitine) begin to accumulate in cardiomyocytes [1]. A further consequence is activation of downstream signaling cascades, which may lead to cell death, resulting in cardiomyopathy [2]. In our study, we showed that a high level of PA upregulated CD36 expression. PA also led to an increase in LD and TAG content, whereas expression of lipases involved in TAG lipolysis was decreased in response to PA, thereby inducing LD accumulation and cardiomyocyte apoptosis. Treatment with ACh not only suppressed the PA-induced increase in FA uptake and LD formation but also promoted the process of TAG hydrolysis that was disturbed by PA, which ultimately attenuated LD deposition and cell apoptosis.
Myocellular LDs provide energy through hydrolysis of TAG by the canonical lipases ATGL, HSL and MGL. Subsequently, FFAs are metabolized by mitochondrial β-oxidation to yield large amounts of ATP [36]. Dysregulation of TAG utilization can result in inadequate energy supplies and accumulation of TAG and/or esterified species, and these changes may be responsible for significant cardiac dysfunction in a variety of disease states [37]. The importance of efficient cardiac TAG turnover for cardiac health is particularly evident in patients carrying mutated alleles of the gene encoding ATGL, the principal enzyme in TAG lipolysis, who show severe cardiac steatosis and heart dysfunction [38]. In addition, lower intramyocellular TAG synthesis and oxidation rates have been reported in obese and prediabetic males than in healthy males, which suggests that the intramyocellular TAG pool loses its dynamic character in patients with obesity and type 2 diabetes [39]. However, studies in diabetic mouse models with cardiac-specific overexpression of HSL or ATGL have shown that both transgenic mouse model types are protected from diabetes-induced structural and metabolic cardiac remodeling events, such as the buildup of cardiac TAG, palmitoyl CoA and ceramide accumulation and chronically increased oxidative stress [40,41]. Consistently, our results suggest that PA supplementation reduced the expression of the TAG lipolysis-related lipases ATGL, HSL and MGL and triggered a decrease in intracellular ATP production and mitochondrial dysfunction, which in turn led to TAG accumulation, LD deposition, mitochondrial dysfunction and cell death in NRVCs. Similarly, in our study, treatment with ACh improved TAG lipolysis, increased intracellular ATP production and mitochondrial function, and ultimately reduced PA-induced LD deposition and cell death. Taken together, these studies support the conclusion that accelerated cardiac TAG turnover can inhibit lipid accumulation and ameliorate the metabolic aberrations and functional decline in the myocardium in metabolic disease.
Close LD and mitochondria localization have been proposed to facilitate the exchange of FAs and to couple lipolytic and FA oxidation processes [42]. Recent studies have provided intriguing insight into the physiological signals that regulate LD-mitochondria associations. Transient (kiss-and-run) and stable LD-mitochondria contacts are increased during starvation and depend on microtubule-mediated dispersion of these organelles to the cell periphery, which is required for LD consumption and FA β-oxidation [43]. Additionally, data reported by Yu et al. have provided new clues to the molecular mechanisms that underpin BAT-dependent heat production under cold stress; LDs adapt their protein complements to increase TAG hydrolysis and maintain a tight association with activated mitochondria [44]. However, LD-mitochondria interactions in lipotoxic cardiomyopathy have been poorly explored. Our studies have shown that the presence of excessive FFAs (PA) leads to impairments in mitochondria-LD associations, evidenced by less frequent direct contacts between mitochondria and LDs, as well as a reduction in PLIN2 expression in mitochondrial extracts. In addition, the expression of PLIN5 (the most well-studied effector of the LD-mitochondria interaction) was found to be decreased. These changes further triggered mitochondrial dysfunction (e.g. decreased mitochondrial membrane potential, CPT1B expression and ATP production and increased mitochondrial ROS production) and LD accumulation, which eventually induced NRVC apoptosis. However, ACh treatment significantly upregulated PLIN5 expression and LD-mitochondria interactions and reduced mitochondrial dysfunction, LD accumulation and cell death in PA-treated NRVCs. Taken together, these results suggest that targeting LD-mitochondria interactions to promote lipid oxidation may be an effective way to relieve cell toxicity caused by lipid overload. Understanding the mechanism underlying the interactions between mitochondria and LDs is essential for the development of therapies to treat metabolic diseases.
PLIN5 is considered to be important for coordinating LDs with FA oxidation in mitochondria and maintaining intracellular lipid homeostasis. In mitochondria and at LD-mitochondria interaction sites, PLIN5 may serve to facilitate direct delivery of FAs produced by lipolysis in LDs to mitochondria for oxidation [10]. A recent study showed that PLIN5 protects against cellular oxidative stress by enhancing mitochondrial function in HepG2 cells. Overexpression of PLIN5 promotes LD-mitochondria contacts, reduces cellular ROS levels, upregulates mitochondrial function-related genes and reduces the apoptosis rate of HepG2 cells, whereas knockdown of PLIN5 induces the opposite phenotypes [8]. Additionally, PLIN5 reduces PA-induced lipotoxicity in normal human liver cells by regulating lipid metabolism-related factors [45]. PLIN5 deficiency reduces mitochondrial function and membrane depolarization in the mouse heart [46]. Our results showed that PA treatment resulted in obviously reduced PLIN5 protein levels and decreased LD lipolysis and LD-mitochondria interactions, which ultimately induced LD deposition, mitochondrial dysfunction and apoptosis in cardiomyocytes. However, ACh intervention markedly inhibited these changes induced by PA, and PLIN5 knockdown significantly attenuated the protective effects of ACh. In summary, these findings indicate that the PLIN5-mediated LD contact with mitochondria could be an important protection mechanism for avoiding lipid overload-incuced cell injury and the protective effects of ACh on PA-treated NRVCs are mediated in part through PLIN5 activation.
In conclusion, our study shows that PA decreased LD-mitochondria interactions and LD lipolysis, as well as increased lipid uptake, resulting in LD accumulation and mitochondrial dysfunction, which eventually induced cardiomyocyte apoptosis. ACh reduces lipid uptake and the deposition of LDs and improves mitochondrial function in PA-treated cardiomyocytes, which eventually inhibits cardiomyocyte apoptosis. These protective effects of ACh are related to enhance LD lipolysis and upregulated LD-mitochondria associations via activation of PLIN5 expression. Our findings provide novel insight into the molecular mechanisms underlying the cardioprotective effects of ACh that target LD lipolysis and PLIN5-mediated LD-mitochondria interactions and may inform the development of future therapeutic strategies for treatment of lipotoxic cardiomyopathy.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (No. 81970221, No. 81770293).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Zlobine I, Gopal K, Ussher JR.. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim Biophys Acta. 2016;1861:1555–1568. [DOI] [PubMed] [Google Scholar]
- [2].D’Souza K, Nzirorera C, Kienesberger PC.. Lipid metabolism and signaling in cardiac lipotoxicity. Biochimica Et Biophysica Acta (BBA) - Mol Cell Biol Lipids. 2016;1861:1513–1524. [DOI] [PubMed] [Google Scholar]
- [3].Welte MA, Gould AP. Lipid droplet functions beyond energy storage. Biochimica Et Biophysica Acta (BBA) - Mol Cell Biol Lipids. 2017;1862:1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Krahmer N, Farese RV, Walther TC. Balancing the fat: lipid droplets and human disease. Embo Mol Med. 2013;5:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu S, Zhang X, Liu P. Lipid droplet proteins and metabolic diseases. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2018;1864(5 Pt B): 1968-1983. [DOI] [PubMed] [Google Scholar]
- [6].Drevinge C, Dalen KT, Mannila MN, et al. Perilipin 5 is protective in the ischemic heart. Int J Cardiol. 2016;219:446–454. [DOI] [PubMed] [Google Scholar]
- [7].Benador IY, Veliova M, Mahdaviani K, et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 2018;27:869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tan Y, Jin Y, Wang Q, et al. Perilipin 5 protects against cellular oxidative stress by enhancing mitochondrial function in HepG2 cells. Cells-Basel. 2019;8:1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schuldiner M, Bohnert M. A different kind of love - lipid droplet contact sites. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:1188–1196. [DOI] [PubMed] [Google Scholar]
- [10].Gemmink A, Daemen S, Kuijpers HJH, et al. Super-resolution microscopy localizes perilipin 5 at lipid droplet-mitochondria interaction sites and at lipid droplets juxtaposing to perilipin 2. Biochimica Et Biophysica Acta (BBA) - Mol Cell Biol Lipids. 2018;1863:1423–1432. [DOI] [PubMed] [Google Scholar]
- [11].Bosma M, Minnaard R, Sparks LM, et al. The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem Cell Biol. 2012;137:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: from pathophysiology to prevention and management. Pharmacol Therapeut. 2016;161:52–66. [DOI] [PubMed] [Google Scholar]
- [13].Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res. 2015;(2015):341583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xue R, Yu X, Zhao M, et al. Pyridostigmine alleviates cardiac dysfunction via improving mitochondrial cristae shape in a mouse model of metabolic syndrome. Free Radical Bio Med. 2019;134:119–132. [DOI] [PubMed] [Google Scholar]
- [15].Lu Y, Wu Q, Liu L, et al. Pyridostigmine protects against cardiomyopathy associated with adipose tissue browning and improvement of vagal activity in high-fat diet rats. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2018;1864:1037–1050. [DOI] [PubMed] [Google Scholar]
- [16].Xue R, Zhao M, Wu Q, et al. Regulation of mitochondrial cristae remodelling by acetylcholine alleviates palmitate-induced cardiomyocyte hypertrophy. Free Radical Bio Med. 2019;145:103–117. [DOI] [PubMed] [Google Scholar]
- [17].He X, Bi XY, Lu XZ, et al. Reduction of mitochondria-endoplasmic reticulum interactions by acetylcholine protects human umbilical vein endothelial cells from hypoxia/reoxygenation injury. Arterioscler Thromb Vasc Biol. 2015;35(7):1623-1634. [DOI] [PubMed] [Google Scholar]
- [18].Zhong P, Quan D, Peng J, et al. Role of CaMKII in free fatty acid/hyperlipidemia-induced cardiac remodeling both in vitro and in vivo. J Mol Cell Cardiol. 2017;109:1–16. [DOI] [PubMed] [Google Scholar]
- [19].Abdelmagid SA, Clarke SE, Nielsen DE, et al. Comprehensive profiling of plasma fatty acid concentrations in young healthy canadian adults. Plos One. 2015;10:e116195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fang CX, Dong F, Thomas DP, et al. Hypertrophic cardiomyopathy in high-fat diet-induced obesity: role of suppression of forkhead transcription factor and atrophy gene transcription. Am J Physiol Heart C. 2008;295:H1206–H1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Natarajan SK, Ingham SA, Mohr AM, et al. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology. 2014;60(6):1942-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Natarajan SK, Stringham BA, Mohr AM, et al. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J Lipid Res. 2017;58:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Eisner V, Picard M, Hajnóczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barbosa AD, Siniossoglou S. Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim Biophys Acta, Mol Cell Res. 2017;1864:1459–1468. [DOI] [PubMed] [Google Scholar]
- [25].Mancini A, Imperlini E, Nigro E, et al. Biological and nutritional properties of palm oil and palmitic acid: effects on health. Molecules. 2015;20:17339–17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haffar T, Bérubé-Simard F, Bousette N. Impaired fatty acid oxidation as a cause for lipotoxicity in cardiomyocytes. Biochem Biophys Res Commun. 2015;468:73–78. [DOI] [PubMed] [Google Scholar]
- [27].Fraser DA, Thoen J, Rustan AC, et al. Changes in plasma free fatty acid concentrations in rheumatoid arthritis patients during fasting and their effects upon T-lymphocyte proliferation. Rheumatology (Oxford). 1999;38:948–952. [DOI] [PubMed] [Google Scholar]
- [28].Gunaratnam K, Vidal C, Boadle R, et al. Mechanisms of palmitate-induced cell death in human osteoblasts. Biol Open. 2013;2:1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu S, Liu B, Zhang Q, et al. Dihydromyricetin reduced Bcl-2 expression via p53 in human hepatoma HepG2 cells. Plos One. 2013;8:e76886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen X, Li H, Wang K, et al. Aerobic exercise ameliorates myocardial inflammation, fibrosis and apoptosis in high-fat-diet rats by inhibiting P2X7 purinergic receptors. Front Physiol. 2019;10:1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang F, Wang Y, Zhang D, et al. Fatty acid-induced nuclear translocation of heparanase uncouples glucose metabolism in endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:406–414. [DOI] [PubMed] [Google Scholar]
- [32].Han CW, Kang ES, Ham SA, et al. Antioxidative effects of Alisma orientale extract in palmitate-induced cellular injury. Pharm Biol. 2012;50:1281–1288. [DOI] [PubMed] [Google Scholar]
- [33].Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nguyen TB, Louie SM, Daniele JR, et al. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev Cell. 2017;42:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goldberg IJ, Reue K, Abumrad NA, et al. Deciphering the role of lipid droplets in cardiovascular disease: a report from the 2017 national heart, lung, and blood institute workshop. Circulation. 2018;138:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Heier C, Haemmerle G. Fat in the heart: the enzymatic machinery regulating cardiac triacylglycerol metabolism. Biochimica Et Biophysica Acta (BBA) - Mol Cell Biol Lipids. 2016;1861:1500–1512. [DOI] [PubMed] [Google Scholar]
- [37].Evans RD, Hauton D. The role of triacylglycerol in cardiac energy provision. Biochimica Et Biophysica Acta (BBA) - Mol Cell Biol Lipids. 2016;1861:1481–1491. [DOI] [PubMed] [Google Scholar]
- [38].Guenter Haemmerle ALRZ. Defective Lipolysis and Altered Energy Metabolism in Mice Lacking Adipose Triglyceride Lipase. Science. 2006;312(5774):734-737. [DOI] [PubMed] [Google Scholar]
- [39].Perreault L, Bergman BC, Hunerdosse DM, et al. Altered intramuscular lipid metabolism relates to diminished insulin action in men, but not women, in progression to diabetes. Obesity. 2010;18:2093–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pulinilkunnil T, Kienesberger PC, Nagendran J, et al. Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes. 2013;62:1464–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ueno M, Suzuki J, Zenimaru Y, et al. Cardiac overexpression of hormone-sensitive lipase inhibits myocardial steatosis and fibrosis in streptozotocin diabetic mice. Am J Physiol Endocrinol Metab. 2008;294:1109–1118. [DOI] [PubMed] [Google Scholar]
- [42].Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Herms A, Bosch M, Reddy BJN, et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun. 2015;6. DOI: 10.1038/ncomms8176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yu J, Zhang S, Cui L, et al. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim Biophys Acta, Mol Cell Res. 2015;1853:918–928. [DOI] [PubMed] [Google Scholar]
- [45].Ma X, Cheng F, Yuan K, et al. Lipid storage droplet protein 5 reduces sodium palmitate‑induced lipotoxicity in human normal liver cells by regulating lipid metabolism‑related factors. Mol Med Rep. 2019;20:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Andersson L, Drevinge C, Mardani I, et al. Deficiency in perilipin 5 reduces mitochondrial function and membrane depolarization in mouse hearts. Int J Biochem Cell Biol. 2017;91:9–13. [DOI] [PubMed] [Google Scholar]