

ABSTRACT
Oropharyngeal squamous cell carcinoma (OPSCC) is an increasing world health problem with a more favorable prognosis for patients with human papillomavirus (HPV)-positive tumors compared to those with HPV-negative OPSCC. How HPV confers a less aggressive phenotype, however, remains undefined. We demonstrated that HPV-positive OPSCC cells display reduced macroautophagy/autophagy activity, mediated by the ability of HPV-E7 to interact with AMBRA1, to compete with its binding to BECN1 and to trigger its calpain-dependent degradation. Moreover, we have shown that AMBRA1 downregulation and pharmacological inhibition of autophagy sensitized HPV-negative OPSCC cells to the cytotoxic effects of cisplatin. Importantly, semi-quantitative immunohistochemical analysis in primary OPSCCs confirmed that AMBRA1 expression is reduced in HPV-positive compared to HPV-negative tumors. Collectively, these data identify AMBRA1 as a key target of HPV to impair autophagy and propose the targeting of autophagy as a viable therapeutic strategy to improve treatment response of HPV-negative OPSCC.
Abbreviations: AMBRA1: autophagy and beclin 1 regulator 1; CDDP: cisplatin (CDDP); FFPE: formalin-fixed paraffin-embedded (FFPE); HNC: head and neck cancers (HNC); HPV: human papillomavirus (HPV); hrHPV: high risk human papillomavirus (hrHPV); OCSCC: oral cavity squamous carcinomas (OCSSC); OPSCC: oropharyngeal squamous cell carcinoma (OPSCC); OS: overall survival (OS); qPCR: quantitative polymerase chain reaction; RB1: RB transcriptional corepressor 1; ROC: receiver operating characteristic curve (ROC).
KEYWORDS: AMBRA1, autophagy, calpains, hpv-E7, oropharyngeal squamous cell carcinoma
Introduction
OPSCC represents one quarter of total head and neck cancers (HNC) causing around 97,000 deaths/year worldwide [1]. The high risk human papillomavirus (hrHPV) represents the etiological agent of a subset of OPSCC [2]. In particular, hrHPV proteins E6 and E7 play a major role in tumorigenesis through their ability to modulate stability or activity of cellular proteins for viral benefit [3]. E6 promotes the degradation of both tumor suppressors and proto-oncogenes (TP53/p53 and MYC/c-Myc) through E3-ligase UBE3A/E6-AP-mediated ubiquitination thus ensuring proliferation and apoptosis inhibition required for viral replication [4]. HPV protein E7 promotes the degradation of the tumor suppressor RB1/Rb (RB transcriptional corepressor 1) through both calpain-dependent cleavage and proteasomal degradation triggered by CUL2 (cullin 2)-mediated ubiquitination [5]. In addition, HPV E7 stimulates growth factor signaling pathways by binding and inhibiting PPP2/PP2A, a negative regulator of AKT/PKB [6].
Although HPV promotes tumorigenesis, patients with HPV-positive OPSCC have a more favorable prognosis and treatment response compared to their HPV-negative counterparts [7,8], stimulating recent debate over treatment de-escalation for HPV-positive OPSCC.
Randomized controlled trials of chemo-radiation (standard of care: cisplatin and radiotherapy) versus cetuximab and radiotherapy for patients with HPV-related OPSCC were conceived on the premise that biologic therapy would be less toxic than cisplatin, while maintaining survivorship. However, these studies recently reported no reduced toxicity and inferior survival [7,8], likely due to the heterogeneity within this molecularly defined group of tumors, and emphasizing the unmet need for credible biomarkers to identify genuinely low risk HPV-positive tumor subsets to support de-escalated treatment regimens.
Autophagy is the principle catabolic mechanism for the lysosomal-mediated degradation and recycling of intracellular components to sustain cellular energy and homeostasis. Activated by nutrient or oxidative stress, autophagy impairment or blockade drives tumorigenesis, whereas it plays a pro-tumoral role in advanced solid tumors by guaranteeing their survival 9. In this context, a key role for autophagy in distinguishing between HPV-positive and -negative OPSCCs has been recently proposed, as highlighted in a bioinformatics study identifying autophagy as a principle pathway altered in HPV positive OPSCC [9]. Moreover, increased expression of the autophagy markers SQSTM1/p62 and LC3-II are associated with reduced overall and disease specific survival in oral cavity squamous carcinomas (OCSSC), a HNC with distinct anatomic localization [10]. The mechanisms by which HPV oncoviral proteins mediate autophagy down regulation and the impact on tumor survival or chemo sensitivity, however, remain undefined. Studies of autophagy modulation in vitro have also shown that the concomitant treatment of chemo-resistant OCSSC cell lines with cisplatin and chloroquine results in increased apoptosis, suggesting that autophagy inhibition may be a useful adjuvant therapy [10]. Nevertheless, the potential for autophagy biomarkers to distinguish between high and low risk HPV-positive OPSCC tumor subsets or how the modulation of autophagy may be harnessed for the therapeutic benefit of OPSCC remains unexplored.
AMBRA1 is a key regulator of both autophagy and cell proliferation, positively regulating autophagy through interaction and activation of ULK1 and BECN1-PIK3C3/VPS34 complexes via non-degradative ubiquitination [11]. Tightly regulated at the post-translational level, AMBRA1 has been reported to be inhibited by i) MTOR-mediated phosphorylation, ii) interaction with the dynein motor complex, iii) degradative ubiquitination by the E3-ubiquitin ligase CUL4 (cullin 4) and RNF2 or iv) calpain mediated cleavage upon apoptosis induction [12]. In addition, AMBRA1 acts as a negative regulator of cell proliferation as well as a tumor suppressor, a feature reported to depend on its ability to bind and inhibit MYC/c-Myc through the recruitment of its negative regulator PPP2/PP2A [13]. Accordingly, ambra1 KO mice die during embryonic development through uncontrolled proliferation of neural progenitors [14] whereas Ambra1 heterozygous mice are susceptible to tumorigenesis, primarily affecting liver and lungs [13]. Studies in a cutaneous environment and more specifically in the epidermis overlying primary melanomas show loss of AMBRA1 expression is associated with loss of epidermal differentiation/integrity, increased keratinocyte proliferation and tumor ulceration [15]; an additional effect of AMBRA1 independent of its role in autophagy. Conversely the retention and even increased expression of AMBRA1 in advanced prostate cancer reflect pro-survival autophagy [16], highlighting a more complex relationship between AMBRA1 expression levels between differing cancer types.
The aim of the present study was to define the role of AMBRA1 in OPSCC, how HPV influences its expression and function and to explore the potential for autophagy modulation as a therapeutic strategy for OPSCC.
Results
Lower autophagy levels in HPV-positive compared to HPV-negative OPSCC cell lines
We analyzed basal and nutrient starvation-induced autophagy in two human HPV-positive (SCC90 and SCC154) and two HPV-negative OPSCC cell lines (SCC89 and SCC25) by measuring lipidated LC3 (LC3-II) and SQSTM1/p62 expression by western blotting (Figure 1A and S1A). HPV-positive cells displayed lower LC3-II and higher SQSTM1/p62 levels compared to HPV-negative cells, suggesting that autophagy is inhibited. LC3 and SQSTM1/p62 mRNA expression was measured by real-time PCR to assess transcription does not account for different protein expression levels (Fig. S1B). To evaluate if these differences corresponded to alterations in autophagy activity/flux, LC3-II and SQSTM1/p62 levels were compared following treatment with bafilomycin A1 to inhibit lysosomal activity. As shown in Figure 1B and S1C, HPV-negative cells have increased autophagy induction compared to HPV-positive OPSCC cells. To confirm that autophagy is differentially regulated in HPV-negative and positive cells, the amount of autolysosomes was evaluated by analyzing the co-localization between LC3 and the lysosomal marker LAMP1 by confocal microscopy. As shown in Figure 1C and D, HPV-negative SCC89 cells triplicate the number of autolysosomes following starvation, with the addition of the lysosomal inhibitor bafilomycin A1 further increasing LC3-LAMP1 colocalization. In contrast, HPV-positive SCC90 cells show a very limited accumulation of autolysosome following starvation, confirming the impairment of autophagy.
Figure 1.

Autophagy is impaired in HPV-positive OPSCC. (A) LC3 and SQSTM1/p62 western blotting analysis of HPV-negative (SCC89 and SCC25) and HPV-positive (SCC90 and SCC154) starved cells. (B) Analysis of the autophagic flux (LC3 and SQSTM1/p62 blot) of HPV-negative and -positive cells deprived of nutrients without bafilomycin A1 for 1 h at 5 nM (n = 4, mean ± SEM. ANOVA 2-way test, *P < 0.05, **P < 0.005, ****P < 0.0005). (C) Analysis of LC3 and LAMP1 colocalization expressed as the number of dots per cell. HPV-negative SCC89 and HPV-positive SCC90 cells were subjected to nutrient deprivation for 2 h (starvation) and treated or not with the lysosomal inhibitor bafilomycin A1 (bafA1). Numbers under the x-axis represent the fold of change compared to control of each cell line (n = 30 cells, mean ± SEM. ANOVA 2-way test, *P < 0.05, **P < 0.005). Statistical analysis are limited to untreated/bafA1-treated paired samples as an indication of autophagy flux modulation
HPV-positive primary OPSCCs have lower levels of AMBRA1 compared to HPV-negative OPSCCs
In order to investigate the molecular players responsible for the differential autophagy activity between these cells, we monitored the expression level of a panel of autophagy proteins. Interestingly, the pro-autophagic protein AMBRA1 was the only autophagic protein tested to be expressed at lower levels in HPV16-positive OPSCC cells (Figure 2A and S2A), paralleling studies reporting the reduced expression of RB1/Rb, a protein known to be targeted by HPV16 E7 for proteasomal and calpain-dependent degradation [5]. Our initial observations of reduced AMBRA1 expression in HPV-positive OPSCC cells prompted us to analyze AMBRA1 expression in a cohort of 83 primary OPSCC cases derived from a sub cohort of the UK HPV prevalence study [17]. Consistent with current literature, a correlation of HPV status with overall survival (OS) revealed a significant reduction in OS for patients with HPV-negative OPSCC to just 32% compared to 63% for patients with HPV-positive OPSCC (Kaplan Meier, Log rank test, P = 0.0012 HR 2.87 (95% CI 1.54–5.35, Figure 2B). Using a clinically validated recombinant antibody to AMBRA1, we performed semi quantitative automated immunohistochemical expression analysis in triplicate 1 mm tissue microarrays (Figure 2C and D). Results revealed significantly reduced expression of AMBRA1 in HPV-positive OPSCC compared to HPV-negative cases. An inverse correlation between AMBRA1 levels and HPV positivity was also observed in tissue sections from a cohort of 40 OPSCC patients (Regina Elena National Cancer Institute, Rome, Italy) analyzed by immunohistochemistry using a different commercial AMBRA1 antibody (Fig. S2B, Table 1 and S1). Collectively these findings confirmed the association of reduced AMBRA1 in HPV-positive OPSCC.
Figure 2.

Relationship between HPV status and AMBRA1 expression in OPSCC. (A) Western blotting analysis of several autophagic proteins comparing HPV-negative SCC89 and HPV-positive SCC90 starved cells. Lower graph: densitometric analysis of AMBRA1 protein levels (n = 3, mean ± SEM. ANOVA 2-way test, ****P < 0.00005). (B) Overall survival rates of Newcastle OPSCC cohort (83) determined by Kaplan-Meier analysis and compared by Log-Rank test in HPV positive versus HPV negative OPSCC samples (OS = 63% versus 32.7%; P = 0.0012 HR 2.87 (95% CI 1.54–5.35). (C) Representative photomicrographs of AMBRA1 expression in a cohort of 83 Newcastle OPSCC TMA i. AMBRA1 H-Score = 300 ii. H-score = 180 iii. H-Score = 120 iv. H-score = 20. Scale bars = 300 µm. (D) Median tumoral expression of AMBRA1 in the Newcastle cohort of 45 HPV negative or 38 HPV positive OPSCCs revealing significantly greater levels of expression (h score) in HPV negative OPSCC (H-score = 150 (IQR 100)) compared to HPV positive OPSCC (H-score = 100 (IQR 117.75). Mann-Whitney P = 0.049)
Table 1.
Expression of AMBRA1 in 40 OPSCCs tumors from the Italian cohort
| Total n (%) |
HPV/p16 negative n (%) | HPV/p16 positive n (%) | p | |
|---|---|---|---|---|
| AMBRA1 | ||||
| Negative | 22 (55.0) | 10 (41.7) | 12 (75.0) | p = 0.04 |
| Positive | 18 (45.0) | 14 (58.3) | 4 (25.0) | |
HPV16 E7 promotes AMBRA1 protein degradation through calpains
To understand how AMBRA1 protein levels are decreased in HPV-positive OPSCC cells, we first analyzed the expression levels of AMBRA1 mRNA by qPCR (Figure 3A), and observed that changes in transcript levels do not parallel those observed at the protein level. We then investigated whether AMBRA1 protein stability was compromised in HPV-positive cells. To this aim, we treated HPV-negative and positive OPSCC cells with the proteasomal inhibitor MG132, the CAPN1 (calpain 1)-CAPN2 (calpain 2) inhibitor ALLN or the lysosomal inhibitor bafilomycin A1. Interestingly, we found a significant increase in AMBRA1 protein levels in HPV-positive OPSCC cells when calpain activity was inhibited (Figure 3B and FigureS1C). The known ability of HPV16 E7 to trigger calpain-mediated degradation of RB1/Rb [5], prompted us to further test whether HPV16 E7 also affects AMBRA1 stability. To this aim, we expressed increasing amounts of HPV16 E7 in 293 T cells and observed an HPV16 E7 dose dependent-downregulation of AMBRA1 protein levels (Figure 3C). Remarkably, the reduction of AMBRA1 induced by HPV16 E7 expression was reverted by the addition of the calpain inhibitor ALLN (Figure 3D), indicating that HPV16 E7 mediates the degradation of AMBRA1 in a calpain-dependent manner.
Figure 3.

HPV16 E7 promotes a calpain-mediated degradation of AMBRA1. (A) Quantitative PCR of AMBRA1 mRNA comparing HPV-negative SCC89 and HPV-positive SCC90 cells. (B) Western blot analysis of AMBRA1 protein levels in HPV-negative SCC89 and HPV-positive SCC90 cells, where proteasomal, calpains or lysosomal activities were blocked using MG132 at 5 mM, ALLN at 10 mM and bafilomycin A1 at 5 nM for 5 h, respectively. Lower panel shows the densitometric analysis of 3 independent experiment reported as the fold of changes compared the each untreated cell line (mean ± SEM, ANOVA 2-way test, *P < 0.05, **P < 0.005, ***P < 0.005). (C) AMBRA1 protein levels following the expression of HA/FLAG-tagged HPV16 E7 in 293 T cells (n = 3), (D) in presence or absence of ALLN at 10 mM for 5 h (n = 3)
To confirm that HPV16 E7 acts directly on AMBRA1, we tested whether the two proteins interact with each other. As reported in Figure 4A, HPV16 E7 binds AMBRA1, while no interaction was observed with BECN1, a component of the pro-autophagic PIK3C3/VPS34 complex that interacts with AMBRA1 [14]. The interaction of HPV16 E7 with RB1/Rb was also included as a positive control. Immunoprecipitation of endogenous HPV16 E7 was also performed in HPV-positive OPSCC cells, showing that E7 binds AMBRA1 in a physiological HPV system (Figure 4B). To map the protein domains of AMBRA1 that mediate the interaction with HPV16 E7, we expressed different MYC-tagged AMBRA1 deletion mutants and HPV16 E7 in an OPSCC-unrelated cell system. The immunoprecipitation of AMBRA1 fragments showed that the central part of the protein is essential for the interaction with E7 (Figure 4C and D). Considering that the central region of AMBRA1 is involved in the binding to BECN1 [14], we tested whether HPV16 E7 also binds to BECN1 and whether HPV16 E7 interferes with the BECN1-AMBRA1 interaction. The immunoprecipitation of BECN1 revealed that, despite the association with high levels of AMBRA1, no binding with HPV16 E7 was observed; suggesting the pool of AMBRA1 binding BECN1 differs from the one associating with HPV16 E7 (Figure 4E). We next compared the levels of BECN1 bound to AMBRA1 in the presence or absence of HPV16 E7. As shown in Figure 4F, HPV16 E7 overexpression reduced the interaction of AMBRA1 with BECN1, suggesting HPV16 E7 and BECN1 compete for the same binding region on the AMBRA1 protein.
Figure 4.

HPV16 E7 interacts with AMBRA1 to compete with BECN1 interaction and to promote CAPN2 association. (A) Immunoprecipitation of expressed HA/FLAG-tagged HPV16 E7 in 293 T cells revealed for AMBRA1, Rb and BECN1 interaction by western blot. (B) Immunoprecipitation of endogenous HPV16 E7 from HPV-positive OPSCC protein extracts probed with anti-AMBRA1 antibody. (C) Schematic representation of different AMBRA1 deletion mutants. (D) Immunoprecipitation of different MYC-tagged AMBRA1 deletion mutants in presence of expressing HA/FLAG-tagged HPV16 E7 in 293 T cells. (E) Immunoprecipitation of BECN1 using a specific antibody in 293 T cells expressing both Myc-tagged AMBRA1 and HA/FLAG-tagged E7. (F) Analysis of AMBRA1-BECN1 interaction in presence or not of HPV16 E7 performed by immunoprecipitation of endogenous AMBRA1 revealed for BECN1 by western blotting, the lower panel represents the amount of BECN1 co-eluted and normalized on immunoprecipitated AMBRA1 (n = 3, mean ± SEM. Two-tailed Student’s t-test, ****P < 0.00005). (G) Immunoprecipitation of FLAG-tagged AMBRA1 expressed in 293 T cells revealed with anti-CAPN2 antibody. (H) Immunoprecipitation FLAG-tagged AMBRA1 expressing 293 T in presence or not of HA-tagged HPV16 E7, CAPN2 interaction was detected by western blot. The right panel represents the amount of CAPN2 co-eluted and normalized on immunoprecipitated AMBRA1 (n = 4, mean ± SEM. Two-tailed Student’s t-test, *P < 0.05)
We then attempted to identify which calpain is responsible for AMBRA1 degradation. To this aim, we took advantage of data obtained in our previous study where, combining tandem affinity purification and LC-MS using SILAC approach [11,18], we found CAPN2 as an AMBRA1 binding partner (data not shown). Interestingly, CAPN2 was previously shown to negatively impact on autophagy by degrading two essential proteins, ATG3 and ATG7 [12]. We validated the interaction of AMBRA1 with CAPN2 (Figure 4G), and then test whether HPV16 E7 modulates the interaction between AMBRA1 and CAPN2 (Figure 4H). Western blot analysis of the AMBRA1 co-immunoprecipitated proteins confirmed CAPN2 as an AMBRA1 interactor. Moreover, HPV16 E7 expression increased AMBRA1-CAPN2 interaction, suggesting that AMBRA1 degradation is triggered through CAPN2.
Autophagy confers resistance of HPV-negative OPSCC cells to cytotoxic cell death
Since AMBRA1 expression was retained and basal autophagy was increased in HPV-negative OPSCC cells, we hypothesized, as in cutaneous squamous cell carcinoma [19,20] that autophagy contributes to the chemo-resistance of HPV-negative OPSCC. To question this possibility both HPV-negative and positive OPSCC cells were exposed to increasing concentrations of cisplatin (CDDP), within a clinically achievable dose range for 48 h. Apoptosis was analyzed by western blotting for cleaved PARP1 and flow cytometry for ANXA5/annexin V staining. Results revealed significant dose dependent CDDP-induced PARP1 cleavage in HPV-positive cells (by up to a 17 fold increase) compared to that observed in HPV-negative OPSCC cells (Figure 5A and B). We then verified whether the increase in cleaved PARP1 observed in HPV-positive OPSCC cells was prevented by reducing E7 triggered AMBRA1 degradation using the calpain inhibitor ALLN. As shown in Figure 5C, CDDP-induced PARP1 cleavage was decreased by ALLN in HPV-positive cells to a similar extent observed in HPV-negative cells, which correlated with increased AMBRA1 and LC3-II levels, while no major changes were detected in other upstream autophagy proteins, such as BECN1.
Figure 5.
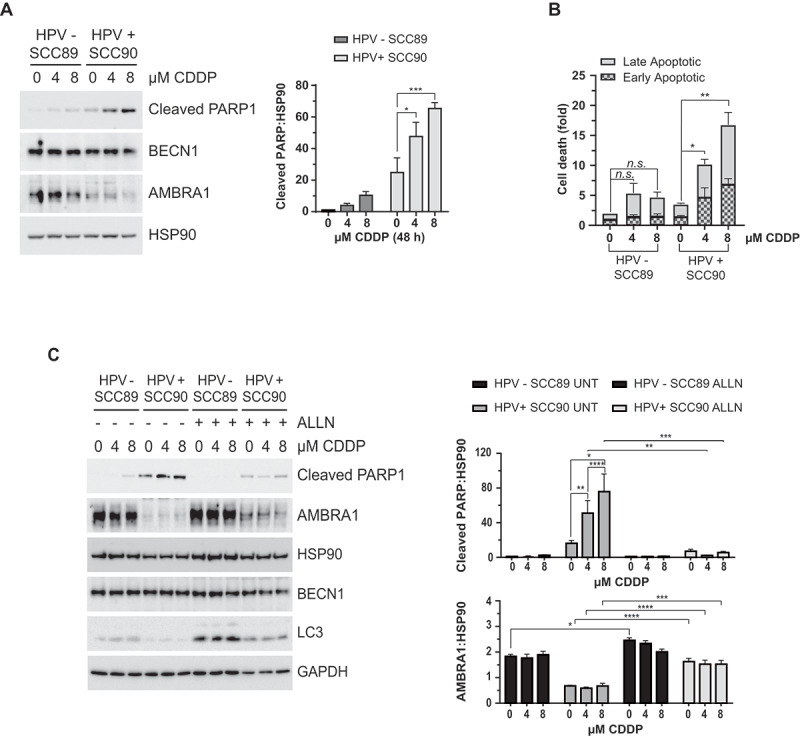
Calpains are responsible for the increased susceptibility of HPV-positive OPSCC cells to Cisplatin-induced cell death. (A) Analysis of cell death comparing HPV-negative and positive OPSCC cells in response Cisplatin (CDDP) treatment for 4 or 8 M (48 h) analyzed by western blotting of cleaved PARP1, (B) and evaluating PI/Annexin V incorporation by FACS (n = 3, mean ± SEM. ANOVA 2-way test, *P < 0.05 **P < 0.005). (C) Cleaved PARP1 western blotting of HPV-negative and positive OPSCC cells treated with increasing amount of Cisplatin in presence or absence of ALLN and relative densitometric analysis; several autophagic proteins were evaluated as control (n = 4, mean ± SEM. ANOVA 2-way test, P** P < 0.005 *** P < 0.0005)
Prompted by these results, we next tested whether AMBRA1-dependent autophagy contributes to the chemo-resistance of HPV-negative OPSCC cells. To this aim, AMBRA1 expression was downregulated using both shRNA and CRISPR-Cas9 approaches prior to assessing the effect on CCDP-induced PARP1 cleavage or apoptosis (as assessed by PI ANXA5 staining). Remarkably, AMBRA1 downregulation (both by shRNA, Figure 6A and C, and using CRISPR-Cas9, Figure 6B) resulted in the significantly enhanced sensitivity of HPV-negative OPSCC cells to CDDP-induced apoptosis. Similarly, CCDP-induced PARP1 cleavage was also increased in HPV-negative OPSCC cells following shRNA-mediated downregulation of BECN1 (a main component of the PIK3C3/VPS34 complex to which AMBRA1 associates to promote autophagy, Figure 6D). In addition, we evaluated the effect of pharmacological inhibition of autophagy in HPV-negative OPSCC cells using 3-methyladenine (3-MA), a pan inhibitor of phosphoinositide 3-kinases (PI3Ks), and PIK-III, a specific inhibitor for PIK3C3/VPS34 [21]. To note, these treatments result in a block of autophagy flux rather than a reduction autophagy initiation in OPSCC cells (Fig. S2D). Results clearly showed that both 3-MA (Figure 6E) and PIK-III (Figure 6F and G) significantly enhanced CDDP-induced PARP1 cleavage, confirming that targeting the autophagy initiation complex leads to enhanced apoptotic response of HPV-negative OPSCC cells to CDDP.
Figure 6.

Autophagy is responsible for cisplatin-resistance of HPV-negative OPSCC cells. (A) Lentiviral infected HPV-negative OPSCC cells for shRNA of AMBRA1 using two different vectors and (B) stable AMBRA1 KO HPV-negative OPSCC cells, tested for Cisplatin-induced apoptosis by western blotting, and (C) by FACS. (D) HPV-negative OPSCC cells infected with lentivirus for BECN1 shRNA, tested for PARP1 cleavage by western blotting. (E-G) HPV-negative OPSCC cells where autophagy was inhibited or not using (E) 3-MA at 2.5 mM or, (F) and (G) PIK-III at 2.5 M, and cell death stimulated by cisplatin treatment for 48 h. n = 3, mean ± SEM. ANOVA 2-way test, * P < 0.05, ** P < 0.005, **** P < 0.00005
Discussion
OPSCCs are divided in two major sub-types with distinct clinical features based on HPV status, with HPV-positive tumors displaying a better prognosis and treatment response compared to patients with HPV negative OPSCC [22]. Understanding how HPV influences the response of OPSCC to chemotherapy can provide crucial information to establish more efficient precision based treatment strategies.
Here, we show that autophagy activity and expression of the pro-autophagy protein AMBRA1 are low in two independent HPV-positive OPSCC cell lines. Although we cannot rule out that the observed autophagy differences are due to the different genetic background of OPSCC cell lines, this finding was mirrored by expression analysis of AMBRA1 in primary HPV-positive OPSCC. Characterization of the molecular mechanism responsible for AMBRA1 downregulation in HPV-positive OPSCC cells revealed that AMBRA1 protein is degraded by calpains. We have previously reported the regulation of AMBRA1 protein stability by either the ubiquitin/proteasome system through the CUL4-DDB1 E3-ligase upon nutrient starvation [23] or by both caspases and calpains following apoptotic induction [24]. Since HPV16 E7 is known to promote the degradation of cellular RB1/Rb by calpains [5]; we questioned this possibility in HPV-positive OPSCC cells, demonstrating the interaction of HPV16 E7 with AMBRA1 and the competition with BECLIN1 for the same binding region. In addition, we demonstrated that HPV16 E7 favors the association of AMBRA1 with CAPN2 and is sufficient to promote AMBRA1 degradation also when expressed alone. Proteasome and caspase-mediated degradation of AMBRA1 does not appear to be further stimulated by HPV in OPSCC cells (Figure 3B and data not shown). However, the observation that the proteasome inhibitor has also a reduced but significant effect on AMBRA1 levels suggests the presence of a residual pool of AMBRA1 protein that escapes from E7-mediated calpain degradation, possibly preserving specific AMBRA1 activities in HPV positive OPSCC cells. Importantly, the in vivo relevance of these findings was confirmed by analyzing AMBRA1 expression in two independent tissue cohorts of OPSCC, revealing an association of low AMBRA1 levels in HPV-positive cases.
Autophagy is induced as a primary antiviral response by infected cells, while many viruses attempt to block autophagy to avoid degradation or exploit it for viral replication [25]. It is therefore conceivable that HPV has evolved multiple strategies to alter the autophagy process. For example, it has been observed that in keratinocytes the functional interaction of HPV16 pseudovirions with EGFR (epidermal growth factor receptor) during viral entry leads to a rapid activation of PIK3CA/PI3K-AKT-MTOR axis and consequent autophagy inhibition, which favors HPV infection [26]. On the other hand, ectopic expression of HPV11 E6 in keratinocyte cell lines has been reported to induce autophagy by inhibiting AKT-MTOR and MAPK/ERK-MTOR pathways [27]. In apparent contrast to our results, HPV16 E7 overexpression has been shown to increase the amount of autophagosomes in normal keratinocytes, which is, however, associated to enhanced cell death susceptibility upon serum deprivation [28]. A proautophagic role of HPV16 E6 and E7 has been also proposed in cervical cancer cells, where E7 was found to associate with ATG9B and to promote expression of ATG9B and LAMP1 [29]. Further studies are required to elucidate whether and how E7 have pro- and anti-autophagic roles depending on the type of epithelial cells infected by HPV. Interestingly, among several proteins analyzed AMBRA1 appears to be the only autophagy factor targeted for degradation in HPV positive OPSCC tumors. Previous reports indicate AMBRA1 not only positively regulates autophagy, but also acts as anti-oncogene affecting cell proliferation [13,14]. AMBRA1 degradation may therefore confers a double benefit for HPV by blocking autophagy-mediated viral degradation and promoting cell proliferation to favor viral replication, both events contributing to tumor development. Evidence that autophagy is altered in HPV-positive tumors comes from studies of uterine cervical carcinoma, where BECN1 levels inversely correlated to the tumor differentiation state [30]. The precise contribution of autophagy to HNC development and progression however, remains largely undefined, although recent studies in oral and OPSCC report a correlation between elevated LC3B levels and poor prognosis in HPV-negative patients [31], supporting the known association of increased autophagy activity with chemo and radiotherapy resistance. Alternatively, the observed reduced autophagy in HPV-positive OPSCC cells may also partially explain their increased chemo-sensitivity.
Further investigating the role of AMBRA1 in the response to chemotherapy, we found both genetic downregulation of AMBRA1 and pharmacological inhibition of the BECN1-PIK3C3/VPS34 complex sensitized HPV-negative OPSCC cells to the cytotoxic effects of cisplatin as well as the pharmacological inhibition of the BECN1/VPS34 complex. In this regard, it should be noted that the two PI3K inhibitors we have tested result in an accumulation of LC3-II rather than a decrease (Fig. S2D), which is not further increased upon bafilomycin A1 treatment, suggesting that, in these cell lines, they mainly affect PIK3C3/VPS34 complex II, resulting in a block of autophagosome maturation. An effect of these inhibitors at the late step of the autophagy process has been also reported in other cell lines [21,32]. Collectively these data suggest autophagy modulation as viable therapeutic strategy to improve the clinical outcome of HPV-negative OPSCC.
The de-escalation of treatment for HPV-positive OPSCC is hot topic in cancer medicine with efforts focussed on strategies to maintain therapeutic efficacy, while reducing toxicity and improving the quality of life of these patients. Nevertheless, as a heterogeneous group of tumors, treatment de-escalation of HPV positive OPSCC is a contentious issue in the absence of biomarkers able to identify genuinely high-risk subsets. Our observation that AMBRA1 levels are directly regulated by HPV in OPSCCs paves the way to test AMBRA1 as a prognostic biomarker enabling the identification of high risk tumor-subsets as well as appropriately stratifying patients for treatment de-escalation.
Materials and Methods
Cell culture
UPCI-SCC-089 (cell line derived from HPV-negative AJCC stage IVa OPSCC), UPCI-SCC-090 (cell line derived from HPV16-positive AJCC stage II OPSCC) [33] and 293 T cells (ATCC, CRL-3216) were cultured in DMEM (Sigma-Aldrich, D6546) supplemented with 10% or 20% (UPCI-SCCs) FBS (Gibco, 10,270), 2 mM L-glutamine (Sigma-Aldrich, G7513), and 1% penicillin-streptomycin solution (Sigma-Aldrich, P0781) at 37°C under 5% CO2. HPV-negative SCC25 cells [34] (ATCC, CRL1628) were cultured using DMEM-F12 (Sigma-Aldrich, D8437) supplemented with 10% FBS; HPV-positive UPCI:SCC154 [33] (ATCC, CRL3241) were cultured using EMEM (Sigma-Aldrich, M2279) supplemented with heat-inactivated FBS to a final concentration of 10%. Reached 80% confluence, cells were washed twice with PBS (Sigma-Aldrich, D8537) and split using trypsin (Sigma-Aldrich, T3924). Periodically, cells were screened to exclude mycoplasma contamination (ABMGood, G238).
Cell Treatments
To evaluate autophagy, UPCI-SCCs cells were deprived of nutrients in EBSS medium (Sigma-Aldrich, E2888) and treated with 5 nM bafilomycin A1 (Sigma-Aldrich, B1793), for the indicated time. ALLN (Santa Cruz Biotechnology, sc-29,119) was added at final concentration of 10M for 5 h and proteasome activity was inhibited by MG132 (Calbiochem, 474,790) at the concentration of 5M for 5 h. To inhibit autophagy 3-methyladenine (3-MA; Sigma-Aldrich, M9281) was used at 2.5 mM and PIK-III at 2.5 M (APExBio, B6160) for 24 h, where indicated. Cell death was induced by cisplatin (Santa Cruz Biotechnology, sc-200896A) at 4 or 8 M for 48 h with an equal volume of DMSO (Sigma-Aldrich, D2650) or PBS used as vehicle negative controls. The calcium phosphate method was used to transfect 293 T cells with expression vectors. Lentiviral production was performed using 293 T as previously described [35], and UPCI-SCC-089 cells infected using 20 L of viral suspension in a medium supplemented with 4 μg/ml polybrene (Sigma-Aldrich, TR-1003) for 8 h.
Plasmids
CMV Flag/HA 16 E7 plasmid was purchased from Addgene (13,734; deposited by Karl Munger); retroviral pLPCX vector (Clontech, 63,151) was modified to encode FLAG-, MYC-AMBRA1 full-length and MYC-AMBRA1 deletion mutants (aa 1–532, 533–751, 761–1269, 533–617,610-751) and BECN1 were previously described [14,35]. CRISPR-Cas9 All-in-one lentiviral vector specific for human AMBRA1 (HsAMBRA1 sgRNA, K0079606, K0079608) and control sgRNA (Scramble sgRNA, K010) were purchased from ABMGood.
Antibodies
The primary antibodies for western blotting experiments were: rabbit anti-LC3 (Cell Signaling Technology, 27,755), rabbit anti-SQSTM1/p62 (MBL, P045), mouse anti-SQSTM1/p62 (Santa Cruz Biotechnology, sc-28,359), rabbit anti-AMBRA1 (Millipore, ABC131), mouse anti-RB1/Rb (Santa Cruz Biotechnology, sc-74,562), rabbit anti-BECN1 H-300 (Santa Cruz Biotechnology, sc-11,427), mouse anti-BECN1 E-8 (Santa Cruz Biotechnology, sc-48,341), rabbit anti-ATG4B (Sigma-Aldrich, A2981), rabbit anti-ATG5 (Santa Cruz Biotechnology, sc-33,210), goat anti-ATG7 (Santa Cruz Biotechnology, sc-8668), mouse anti-HPV16 E7 NM2 (Santa Cruz Biotechnology, sc-65,711), mouse anti-CAPN2 (Santa Cruz Biotechnology, sc-373,967), rabbit anti-PARP1 (Cell Signaling Technology, #9542), rabbit anti-PARP1 cleaved Asp214 (Cell Signaling Technology, #9541), mouse anti-MYC 9E10 (Santa Cruz Biotechnology, sc-40), rat anti-FLAG (Novus Biologicals, NBP1-06712), mouse anti-HSP90A-HSP90B/Hsp90 03B1α/β Santa Cruz Biotechnology, sc-13,119), mouse anti-TUBA/alpha tubulin (Santa Cruz Biotechnology, sc-32,293). HRP – conjugated secondary antibodies used were purchased from Jackson immune research: anti-rabbit (JI 711–036-152), anti-mouse (JI 715–036-150), anti-goat (JI 705–036-147), anti-rat (JI 712–036-150). Antibodies for Immunofluorescence: Cy3-conjugated anti-mouse secondary antibodies (Jackson ImmunoResearch, 715–166-150), anti-rabbit Alexa Fluor 488-conjugated (Thermo Fischer, A21206), rabbit anti-LC3B (Sigma-Aldrich, L7543), mouse anti-LAMP1 (Abcam, 25,630).
Immunofluorescence analysis
HPV-negative SCC89 or HPV-positive SCC90 cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS for 15 min at room temperature and then permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, T9284) in PBS for 10 min at room temperature. After washings, cells were incubated with anti-LC3B (Sigma-Aldrich, L7543) and with anti-LAMP1 (Abcam, 25,630) primary antibodies for 1 h, washed and, then incubated with anti-rabbit Alexa Fluor 488-conjugated (Thermo Fischer, A21206) and anti-mouse Cy3 (Jackson ImmunoResearch, 715–166-150) for an additional 45 min at 37°C. Immunofluorescences were examined with a LSM 900, Airyscan SR Zeiss confocal microscopy. The number of LC3 and LAMP1 colocalizing dots was measured using ZEN 3.0 Blue edition software. A minimum of 30 cells/sample was analyzed and the statistical analysis was performed using ANOVA 2-way test for repeated samples by using Graphpad Prism, p values of less than 0.05 were considered significant.
Immunoprecipitation assays
Coimmunoprecipitation was performed by lysing cells in Tris buffer (10 mM Tris Base pH 8.0 [Merk Millipore, 648,611], 150 mM NaCl [Sigma-Aldrich, S7653], 10% glycerol [Sigma-Aldrich, 67,757], 0.5% NP-40/IGEPAL [Sigma-Aldrich, 56,741-F]) complemented with protease and phosphatase inhibitors (Protease inhibitor cocktail plus [Sigma-Aldrich, P8340], 5 mM sodium fluoride [Sigma-Aldrich, S-7920], 0.5 mM sodium orthovanadate [Sigma-Aldrich, S6508], 1 mM sodium molibdate [Sigma-Aldrich, S-6646], 50 mM 2-chloroacetamide [Sigma-Aldrich, C0267], 2 mM 1,10-phenanthroline monohydrate [Sigma-Aldrich, 320,056], 0.5 mM PMSF [Sigma-Aldrich, P7626]). For endogenous immunoprecipitation, 2 mg protein extracts were incubated overnight with 2 g of antibody and immunocomplexes recovered using 25 l protein G or recombinant protein A Sepharose (GE Healthcare, 17–0618-01 and 17–1279-01, respectively). For immunoprecipitation of expressed protein, 1 mg of protein extracts were incubated with 25 l anti-FLAG or anti-MYC antibodies conjugated to agarose beads (Sigma-Aldrich, A2220 and E6654, respectively) for 2 h. Immunocomplexes were solved on NuPAGE Bis-Tris gels (Life Technologies, 3 to 8%, Tris-Acetate [EA03785BOX], 4 to 12% Bis-Tris [NW04120BOX]) and blotted on nitrocellulose (Whatman Amersham, 10,600,041) or PVDF (Millipore, IPVH20200) membranes. HRP-secondary antibodies (Jackson ImmunoResearch Laboratories: anti-rabbit [JI 711–036-152], anti-mouse [JI 715–036-150], anti-goat [JI 705–036-147], anti-rat [JI 712–036-150]) and ECL (Millipore, WBLUC0500 or WBLUR0500) were used to detect the conjugation with the primary antibody using the ChemiDoc Imaging Touch System (Bio-Rad).
Real Time PCR
Trizol (Invitrogen, 15,596–018) was used to extract RNA and cDNA synthesis was obtained using a RT-kit from Promega, as recommended. qPCR were performed with the Rotor-Gene 6000 (Corbett Research Ltd) thermocycler, as previously described [36]. Primers: human LC3B forward (5ʹ-AAAGACCCTGGAGAAAGAGTGGCA-3ʹ) and reverse (5ʹ-ACTGGTACACTGCTGC TTTCCGTA-3ʹ); human SQSTM1 forward (5ʹ-ACAGATGCCAGAATCCGAAG-3ʹ) and reverse (5ʹ-TGGGAGAGGGACTCAATCA-3ʹ), human AMBRA1 forward (5ʹ- AACCCTCCACTGCGAGTTGA-3ʹ) and reverse (5ʹ- TCTACCTGTTCCGTGGTTCTCC-3ʹ) and hL34 forward (5ʹ- GTCCCGAACCCCTGGTAATAGA-3ʹ) and reverse (5ʹ- GGCCCTGCTGACATGTTTCTT-3ʹ), RPL34 mRNA level was used as an internal control.
Flow cytometry
ANXA5/annexin V-FITC and PE-A propidium iodide staining were performed using Abcam apoptosis detection kit (ab14085). Stained UPCI-SCC-089 or 090 cells were analyzed using BD Canto I cytometer, and the occurrence of cell death was recorded as the percentage of both early and late apoptotic cells on the entire population; graphs report fold of increase respect to the control (NS), defined as 1.00.
OPSCC patient cohorts and immunohistochemical analysis of AMBRA1
UK COHORT: Retrospective OPSCC patient samples were identified between August 2002 and December 2008 as part of the UK HPV Prevalence Study [37]. 170 cases of oro-pharyngeal squamous cell carcinomas were identified at Newcastle Hospital NHS Foundation Trust and tissue microarrays (TMA) were constructed from formalin-fixed paraffin embedded tissue blocks. Three 1 mm cores per case were transferred to the TMA recipient block. The HPV status of the samples was determined by p16 immunohistochemistry, high-risk HPV DNA in situ hybridization and HPV PCR as previously described [37]. Overall survival data (time from diagnosis to last follow up appointment or death) were available for 91 patients and 84 samples provided sufficient tissue for AMBRA1 immunohistochemistry. AMBRA1 was stained on a Benchmark XT autostainer (Ventana Medical Systems Ltd) using a clinically validated primary recombinant peptide antibody to human AMBRA1 (AMLo Biosciences Ltd), and images scanned for analysis using an Aperio AT2 slide scanner and e slide manager software (Leica Biosystems Ltd). Staining was scored by a pathologist (MR) and independent investigator (RE) blinded to HPV status using an H score [38].
ITALY COHORT: A retrospective cohort of 40 formalin-fixed paraffin-embedded (FFPE) primary OPSCCs was derived from the archives of the Pathology Department of the Regina Elena National Cancer Institute of Rome, Italy. All FFPE samples had been routinely characterized for HPV DNA presence by using INNO-LiPA HPV Genotyping Extra II kit (Fujirebio) and p16 expression by using CINtec® Histology Kit (Roche Diagnostics) as previously described [39,40]. Inclusion criteria were: 1. concordant results for HPV DNA and p16 immunostaining (either both positive or negative); 2. for the HPV DNA positive cases presence of HPV16 genotype; 3. sufficient tissue material to perform immunohistochemical analysis. The study was approved by the Ethics Committee of the Regina Elena National Cancer Institute (N. 1118/18). Sections of 2–3 μm were processed for automated immunohistochemical evaluation of AMBRA1 expression and a third section stained with hematoxylin-eosin for confirmation of the histological diagnosis and presence of sufficient tumor cells by a certified pathologist. Antigen retrieval was carried out using a citrate buffer (Bond™ Epitope Retrieval Solution 1, Leica Biosystems). Automated immunohistochemical analysis for AMBRA1 was performed on an automated autostainer (BOND-III; Leica, Milan, Italy) using a primary rabbit polyclonal anti-AMBRA1 (Novus Biologicals, NBP1-07124), and The Bond™ Polymer Refine Detection (Leica Biosystems) was used to reveal primary antibody binding. Staining was reported by visual analysis of the proportion of positive tumor cells and staining intensity (scored as none, weak, moderate and strong), with the assessment of biomarkers blinded to HPV status. AMBRA1 expression was considered positive when immunostaining was apparent in ≥ 50% of the cancer cells with moderate or strong staining intensity.
Statistical analysis
Statistical analysis of western blotting, PCR and FACs data were performed using unpaired, two-tailed Student’s t-test. Values are shown as mean ± SEM of at least 3 independent experiments. P-values (P) <0.05 were marked by *, <0.005 by **, <0.0005 by ***, <0.00005 by ****). Densitometric analysis of western blotting was performed using the Image Lab 5.2.1 from Biorad. The control ratio was arbitrarily defined as 1.00. All statistical analysis were undertaken using GraphPad Prism 8 software. For western blotting analysis, ANOVA 2-way test for repeated samples was used with multiple comparisons within the same group. For immunohistochemical expression analysis of AMBRA1 in primary OPSCC samples derived from the UK COHORT, a receiver operating characteristic curve (ROC) was built using the continuous classification of AMBRA1 H-score as the discrimination variable for overall survival [38]. The optimum cut-point was identified at an H-score of 140 with a sensitivity of 55% (95% CI 39.83% – 69.29%), specificity of 62.79% (95% CI 47.86% – 75.62%) and a likelihood ratio of 1.48. Binary scores of 0 for low-risk (H-score <140) and 1 for high-risk (H-score > or = to 140) were applied for further Kaplan-Meier survival curve analysis15 Other variables assessed included age, sex, HPV status and AMBRA1 tumor positivity.
For the analysis of samples from the ITALY COHORT, Chi-square test was used to evaluate the statistical significance of differences in positivity rate of biomarkers between HPV related and unrelated OPSCC. Differences were considered significant when p < 0.05. The MedCalc Statistical Software (version 18.9; MedCalc Software bvba, Ostend, Belgium; http://www.medcalc.org; 2018) was used.
Supplementary Material
Acknowledgments
This work was supported by Fondazione Umberto Veronesi trough individual-fellowship (2017) to MA; FRIAS COFUND Fellowship Programme (University of Freiburg, Germany) and People Programme (Marie Curie Actions) of the European Union’s 7th Framework Programme (FP/2007-2013) under REA grant agreement n [609305] to MA; Transautophagy COST action CA15138 to MA, PL and GMF; the Italian Ministry of Health to GMF, MP (Ricerca Corrente) and TV (Ricerca Finalizzata, GR-2013-02359524); Fondazione Fibrosi Cistica (Progetto FFC#8/2018 to MP), Regione Lazio (progetto “Gruppi di Ricerca” 2018 to MP), AIRC (IG2015 n. 17404 to GMF, and IG2018 no. 21880 to MP), the Russian Government Program for the Recruitment of the leading scientists into the Russian Institutions of Higher Education 14. W03.31.0029 to M.P.; the Italian Ministry of University and Research (PRIN 2015 20152CB22L). Work in the UK was supported by a clinical bursary from Cancer Research UK to BC and PL, an AMLo Biosciences project grant to PL, GMF, RE and MR and the Medical Research Council (MRC)/Engineering and Physical Sciences Research Council (EPSRC) Molecular pathology Node (PL & MR).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported by the Associazione Italiana per la Ricerca sul Cancro [17404]; Associazione Italiana per la Ricerca sul Cancro [21880]; Cancer Research UK [clinical bursary]; Cancer Research UK [clinical bursary]; European Cooperation in Science and Technology [CA15138]; FP7 People: Marie-Curie Actions [Fellowship]; Fondazione Umberto Veronesi [Fellowship]; Fondazione per la Ricerca sulla Fibrosi Cistica [FFC#8/2018]; Medical Research Council [Molecular pathology Node]; Medical Research Council [Molecular pathology Node]; Ministero della Salute [GR-2013-02359524]; Ministero dell’Istruzione, dell’Università e della Ricerca [20152CB22L]; Regione Lazio [“Gruppi di Ricerca” 2018].
Disclosure statement
Dr Rob Ellis, Dr Marie Labus and Prof. Penny Lovat are directors of AMLo Biosciences Ltd. The other authors declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Amin MB, Greene FL, Edge SB, et al. The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin [Internet] 2017. [cited 2019 Apr 4]; 67(2):93–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28094848 [DOI] [PubMed] [Google Scholar]
- [2].IARC working group on the evaluation of carcinogenic risks to humans. Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr Eval Carcinog Risks Hum [Internet] 2012. [cited 2019 Aug 5]; 100:1–441. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23189750 [PMC free article] [PubMed] [Google Scholar]
- [3].Vescovo T, Pagni B, Piacentini M, et al. Regulation of autophagy in cells infected with oncogenic human viruses and its impact on cancer development. Front Cell Dev Biol. 2020;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schiffman M, Doorbar J, Wentzensen N, et al. Carcinogenic human papillomavirus infection. Nat Rev Dis Prim. [Internet] 2016. [cited 2019 Jul 23]; 2:16086. Available from: http://www.nature.com/articles/nrdp201686 [DOI] [PubMed] [Google Scholar]
- [5].Darnell GA, Schroder WA, Antalis TM, et al. Human Papillomavirus E7 requires the protease calpain to degrade the retinoblastoma protein. J Biol Chem [Internet] 2007. [cited 2019 Apr 4]; 282(52):37492–37500. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17977825 [DOI] [PubMed] [Google Scholar]
- [6].Pim D, Massimi P, Dilworth SM, et al. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene [Internet] 2005. [cited 2019 Apr 4]; 24(53):7830–7838. Available from: http://www.nature.com/articles/1208935 [DOI] [PubMed] [Google Scholar]
- [7].Gillison ML, Trotti AM, Harris J, et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet. Internet] 2019. [cited 2019 Jul 23]; 393(10166):40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mehanna H, Robinson M, Hartley A, et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): an open-label randomised controlled phase 3 trial. Lancet. Internet 2019. [cited 2019 Jul 23]; 393(10166):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rybstein MD, Bravo-San Pedro JM, Kroemer G, et al. The autophagic network and cancer. Nat Cell Biol Internet] 2018. [cited 2019 Jul 23]; 20(3):243–251. Available from: http://www.nature.com/articles/s41556-018-0042–2 [DOI] [PubMed] [Google Scholar]
- [10].Cosway B, Lovat P.. The role of autophagy in squamous cell carcinoma of the head and neck. Oral Oncol. Internet] 2016. [cited 2019 Apr 4]; 54:1–6. [DOI] [PubMed] [Google Scholar]
- [11].Antonioli M, Di Rienzo M, Piacentini M, et al. Emerging mechanisms in initiating and terminating autophagy [Internet]. Trends Biochem Sci. cited 2020 Apr 24 2017;42(1):28–41. [DOI] [PubMed] [Google Scholar]
- [12].Fimia GM, Corazzari M, Antonioli M, et al. 1 at the crossroad between autophagy and cell death. Oncogene Internet] 2013. [cited 2019 Jul 25]; 32(28):3311–3318. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23069654 [DOI] [PubMed] [Google Scholar]
- [13].Cianfanelli V, Fuoco C, Lorente M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015. cited 2014 Dec 2;17:(1):20–30. . Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. Internet] 2007. [cited 2019 Apr 4]; 447:1121–1125. Available from: http://www.nature.com/doifinder/10.1038/nature05925 [DOI] [PubMed] [Google Scholar]
- [15].Ellis R, Tang D, Nasr B, et al. Epidermal autophagy and beclin 1 regulator 1 and loricrin: a paradigm shift in the prognostication and stratification of the American Joint Committee on Cancer stage I melanomas. Br J Dermatol. Internet 2020. [cited 2020 Apr 24]; 182:156–165. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31056744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Falasca L, Torino F, Marconi M, et al. AMBRA1 and SQSTM1 expression pattern in prostate cancer. Apoptosis. Internet 2015. [cited 2019 Apr 4]; 20(12):1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].New J, Arnold L, Ananth M, et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. Internet 2017. [cited 2019 Apr 15]; 77(23):6679–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Antonioli M, Ciccosanti F, Dengjel J, et al. Methods to study the BECN1 interactome in the course of autophagic responses. Methods Enzymol. Internet] 2017. [cited 2019 Apr 4]; 587:429–445. Available from: https://www.sciencedirect.com/science/article/pii/S0076687916303421 [DOI] [PubMed] [Google Scholar]
- [19].Wright TJ, McKee C, Birch-Machin MA, et al. Increasing the therapeutic efficacy of docetaxel for cutaneous squamous cell carcinoma through the combined inhibition of phosphatidylinositol 3-kinase/AKT signalling and autophagy. Clin Exp Dermatol Internet] 2013. [cited 2019 Jul 23]; 38(4):421–423. Available from: http://doi.wiley.com/10.1111/ced.12138 [DOI] [PubMed] [Google Scholar]
- [20].Claerhout S, Verschooten L, Van Kelst S, et al. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer Internet 2010. [cited 2019 Jul 23]; 127(12):2790–2803. Available from: http://doi.wiley.com/10.1002/ijc.25300 [DOI] [PubMed] [Google Scholar]
- [21].Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. Internet 2014. [cited 2019 Apr 4]; 16(11):1069–1079. [DOI] [PubMed] [Google Scholar]
- [22].Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. JNCI J Natl Cancer Inst Internet] 2008. [cited 2019 Apr 4]; 100(4):261–269. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18270337 [DOI] [PubMed] [Google Scholar]
- [23].Antonioli M, Albiero F, Nazio F, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell. Internet] 2014. [cited 2017 Aug 18]; 31(6):734–746. [DOI] [PubMed] [Google Scholar]
- [24].Pagliarini V, Wirawan E, Romagnoli A, et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. Internet] 2012. [cited 2019 Apr 16]; 19:1495–1504. Available from: http://www.nature.com/articles/cdd201227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol Internet 2018. [cited 2019 Apr 15]; 16(6):341–354. Available from: http://www.nature.com/articles/s41579-018-0003–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Surviladze Z, Sterk RT, DeHaro SA, et al. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-Kinase/Akt/mTOR Pathway and inhibition of autophagy. J Virol Internet 2013. [cited 2019 Apr 15]; 87(5):2508–2517. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23255786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang B, Song Y, Sun S, et al. 11 early protein E6 activates autophagy by repressing AKT/mTOR and Erk/mTOR. J Virol Internet] 2019. [cited 2020 Aug 24]; 93(12). doi: 10.1128/JVI.00172-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou X, Münger K. Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology Internet 2009. [cited 2015 Mar 2]; 385(1):192–197. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2673705&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tingting C, Shizhou Y, Songfa Z, et al. Human papillomavirus 16E6/E7 activates autophagy via Atg9B and LAMP1 in cervical cancer cells. Cancer Med Internet 2019. [cited 2020 Aug 24]; 8(9):4404–4416. Available from: /pmc/articles/PMC6675746/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cheng HY, Zhang YN, Wu QL, et al. Expression of beclin 1, an autophagy-related protein, in human cervical carcinoma and its clinical significance. Eur J Gynaecol Oncol. Internet 2012. [cited 2019 Apr 15]; 33:15–20. Available from:http://www.ncbi.nlm.nih.gov/pubmed/22439399 [PubMed] [Google Scholar]
- [31].Lai K, Matthews S, Wilmott JS, et al. Differences in LC3B expression and prognostic implications in oropharyngeal and oral cavity squamous cell carcinoma patients. BMC Cancer. Internet 2018. [cited 2019 Apr 15]; 18(1):624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu Y, Wang X, Guo H, et al. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy Internet] 2013. [cited 2020 Oct 7]; 9(4):595–603. Available from: /pmc/articles/PMC3627673/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].White JS, Weissfeld JL, Ragin CCR, et al. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. Internet] 2007. [cited 2019 Jul 23]; 43(7):701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981;41(5):1657-63. [PubMed] [Google Scholar]
- [35].Di Rienzo M, Antonioli M, Fusco C, et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci Adv. Internet 2019. [cited 2019 May 23]; 5(5):eaau8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Refolo G, Ciccosanti F, Di Rienzo M, et al. Negative regulation of mitochondrial antiviral signaling protein-mediated antiviral signaling by the mitochondrial protein LRPPRC during hepatitis C virus infection. Hepatology Internet] 2019. [cited 2019 Apr 4]; 69(1):34–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30070380 [DOI] [PubMed] [Google Scholar]
- [37].Schache AG, Powell NG, Cuschieri KS, et al. HPV-related oropharynx cancer in the United Kingdom: an evolution in the understanding of disease etiology. Cancer Res. Internet] 2016. [cited 2019 Jul 23]; 76(22):6598–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Robinson M, James J, Thomas G, et al. Quality assurance guidance for scoring and reporting for pathologists and laboratories undertaking clinical trial work. J Pathol Clin Res. Internet 2019. [cited 2019 Jul 23]; 5(2):91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Donà MG, Rollo F, Pichi B, et al. Evaluation of the Xpert® HPV assay in the detection of Human Papillomavirus in formalin-fixed paraffin-embedded oropharyngeal carcinomas. Oral Oncol. Internet 2017. [cited 2019 Jul 23]; 72:117–122. [DOI] [PubMed] [Google Scholar]
- [40].Donà MG, Spriano G, Pichi B, et al. Human papillomavirus infection and p16 overexpression in oropharyngeal squamous cell carcinoma: a case series from 2010 to 2014. Future Microbiol Internet 2015. [cited 2019 Jul 23]; 10(8):1283–1291. http://www.ncbi.nlm.nih.gov/pubmed/26228531 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.