

Abstract
Although multiple components of the cell membrane modulate the stability and activation of G protein coupled receptors (GPCRs), the insights on the dynamics of GPCR structures comes from biophysical studies conducted in detergents. This is because of the challenges to study activation in multi-component lipid bilayer. To understand the role of cellular membrane lipids and cations on GPCR activation, we performed multi-scale molecular dynamics simulations(56μs) on three different conformational states of adenosine receptor A2AR, in both cell membrane like lipid bilayer and in detergent micelles. MD simulations show that the Phosphatidylinositol bisphosphate (PIP2) interacts with the basic residues in the intracellular regions of the A2AR thereby reducing the flexibility of the receptor in the inactive state and limiting the transition to the active-intermediate state. In the G protein coupled fully active state, PIP2 stabilizes the GPCR:G protein complex. Such stiffening effects are absent in non-ionic detergent micelles and therefore more transitions have been observed in detergents. The inter-residue distances that show significant changes upon GPCR activation are known as activation microswitches. The activation microswitches show different level of activation in cell membrane, pure POPC bilayer and in detergents. Thus, the temporal heat map of different activation microswitches calculated from the MD simulations suggests a rheostat model of GPCR activation microswitches rather than the binary switch model. These simulation results connect the chemistry of cell membrane lipids to receptor activity useful to design detergents mimicking cell membrane.
Keywords: GPCR activation, cell membrane, PIP2, GM3, microswitches
Graphical Abstract

Introduction
G protein coupled receptors (GPCRs) are seven helical transmembrane (TM) proteins that transduce a variety of cell signaling pathways. Agonist binding leads to coupling of the receptor to trimeric G proteins which initiates a cascade of signaling events resulting in intracellular biological response. Pioneering structural studies on agonists and G proteins bound GPCRs using crystallography, electron microscopy, NMR and DEER techniques have shown that GPCRs are inherently dynamic proteins adopting multiple conformations, such as inactive, active intermediate and G protein coupled fully active conformational states 1–8. These studies have highlighted the structural changes in GPCRs associated with the different conformational states leading to the activation of GPCRs. The dominant structural changes involve movement of transmembrane helices 5, 6 and 7 (TM5, TM6 and TM7) as well as changes in the intracellular loops. Comparison of three-dimensional structures of active state to that of inactive state of several class A GPCRs have shed light on the inter-residue distances that show distinct changes upon activation. These inter-residue distances are called “activation microswitches”9–11. The activation microswitches are thought of as binary “on or off “ switches, a concept that stems from analysis of static structures. All these structural studies were conducted in detergent micellar solution or in nanodiscs containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids 12–15. On the other hand, multiple studies have shown that different lipids in the cell membrane play an active role in modulating the activity of GPCRs15–22. It is also known that cations such as Ca2+, and Mg2+ play an important role as allosteric modulators of GPCR activity23–26. NMR studies of β2-adrenergic receptor in detergent micelles compared to reconstituted high density lipoprotein environment showed that the basal activity and exchange rates between inactive and active conformations is higher in detergents and in phospholipid bilayer containing HDL particles compared to cell membranes15,27,28.
Although we know that the cellular environment has a definitive effect on GPCR stability and activity, and even the selective coupling of G proteins by GPCRs 19, there is a serious gap in our understanding, at an atomic level, of the effect of the chemical nature of different lipids and cations on the GPCR conformation dynamics. More importantly, the spatiotemporal component (the persistence time) of lipid-receptor contacts that play an important role in activation mechanism has not been considered while analyzing static structural contacts with lipids. In summary, the similarities and differences of the GPCR activation mechanism in cell membranes compared to detergents is unknown. The effect of cell membrane components on the activation microswitches is also poorly understood. An understanding of these fundamental phenomenon would (i) lead to insights into GPCR dynamics and activation in physiological cell membranes that is critical to cell signaling and (ii) allow us to design detergents that mimic cell membrane behavior more closely, or add lipids that modulate GPCR activity in cells, to biophysical studies conducted in detergents or nanodiscs. Studying GPCR dynamics in cellular environment is challenging experimentally. However, multi-scale Molecular Dynamics (MD) simulations offer a suitable tool to map the conformation ensemble of different conformational states of GPCRs and to probe the role of multiple lipid and cation components of cell membrane on GPCR dynamics.
In this study we have used a combination of coarse grain MD (30 μs) followed by 26 μs of all-atom MD simulations in a hierarchical workflow, to uncover the mechanisms of how different lipids in the cell membrane affect the dynamics and conformation ensemble of GPCRs in different conformational states. For comparisons, we have also performed MD simulations in detergent micelles that is often used to study GPCR activation mechanism and pure POPC bilayer that is often used in MD simulation studies of GPCRs. The MD simulations were performed on human adenosine A2AR receptor in its agonist bound inactive (R), active intermediate (R’) and miniG protein bound fully active (R*.G) states in cell membrane mimicking bilayer. We chose A2AR for this work due to availability of crystal structures in all the three above mentioned conformational states. Our results highlight the fact that not only the nature of lipid interaction with the receptor, but also the temporal persistence of these interactions during the dynamics in cell membrane conditions, play a critical role in modulating the receptor flexibility and activity. MD simulation data show that the detergent environment increases the flexibility of the TM helices and therefore promote transitions between the inactive and active-intermediate state when bound to an agonist. This is in stark contrast to the cell membrane environment especially Phosphatidylinositol bisphosphate (PIP2) and calcium ions that together maintain a balance between the receptor flexibility and stability and therefore limit the transitions between inactive state and active intermediate states.
Analysis of static structures of the inactive and active state of several GPCRs showed that the activation microswitches show a binary on and off switch like behavior9–11. Our dynamics study that takes the temporal persistence of activation microswitches into account, suggesting that the activation microswitches behave like rheostats in cell membrane like conditions, detergent and in pure POPC bilayer (showing different extent of activation) rather than binary on and off switches. Our data shows that the combination of microswitches activated in cell membrane like conditions are different than those activated in detergents or pure POPC bilayer during the early events of activation. Additionally, the extent to which the microswitches are activated are different in cell membrane and detergents. Taken together, our work provides mechanistic insights into the role of the chemistry of lipids in modulating the GPCR dynamics that can be used for designing detergents that better mimic the cell membrane.
Methods
Coarse Grain (CG) simulation followed by All atom MD simulations:
The hierarchical workflow combining coarse grain MD with all-atom MD simulations described in this section is shown in Fig. S1. To study the effect of multiple lipids on the GPCR conformation ensemble, we used mixed lipid bilayer to mimic the cell membrane. The outer leaflet of the membrane bilayer consists of POPC, DOPC, POPE, DOPE, sphingomyelin(Sph), ganglioside(GM3) and cholesterol in the ratio of 20:20:5:5:15:10:25, while the inner leaflet contains POPC, DOPC, POPE, DOPE, POPS, DOPS, phosphatidylinositol 4,5-bisphosphate(PIP2), and cholesterol in the ratio of 5:5:20:20:8:7:10:25 and all simulations were neutralized by adding 0.15M of NaCl or CaCl2 (see Fig. 1A and Table S1). The mix-bilayer membrane of a given composition was generated using CHARMM-GUI 29,30, coarse grain (CG) membrane builder program 31,32. The 50 nm × 50 nm simulation box generated for coarse grain MD simulations, contains nine GPCR units that cover three units of each agonist bound inactive R state, active-intermediate (R’) state and G-protein bound fully active R*.G state. The nine units were placed 10~12 nm apart in the simulation box. The lipid bilayer was built three times in the same composition given above, to get statistically significant random distribution of lipids that can be used as starting structures for coarse grain simulations. After equilibration of each of the 3 simulation boxes, we performed 3*10μs of coarse grain MD (CGMD) simulations with Martini2.2 forcefield 33. We extracted 5 different cell membrane bilayer configurations from the coarse grain simulations for each conformational state of the receptor (R, R’ and R*.G). The lipid environment surrounding the GPCR units were cut out into a 12 nm × 12 nm box. We chose those lipid configurations where the receptor remained a monomer (without dimerization) during the coarse grain MD simulations (see box in the middle in Fig. S1). The CG models were converted to all-atom models using the script “backward.py” from the Martini website.34 Thus we extracted 5 different cell membrane lipid configurations for each of R, R’ and R*.G states as starting structures for all-atom MD simulations. We discarded the receptor structure from coarse grain MD simulations and retained just the cell membrane lipid configuration. We then placed the crystal structures (pdb code of 4EIY for R, 2YDV for R’ and 5G53 for R*.G) in this equilibrated cell membrane lipid bilayer from coarse grain MD simulations, and solvated with water and neutralized with 0.15M of NaCl or 0.15M of CaCl2. The thermostabilizing mutations in the crystal structure were mutated back to the wild type residues using Maestro9.2. Residues within 5Å of the sites of mutation were minimized using MacroModel with position restraints on all backbone atoms and all residues further than 5Å from the site of mutation. The disulfide bonds were built according to the disulfide bonds listed in the pdb structure file. The minimization-heating-equilibration-production was carried out using the protocol used in the previous work35–38. Each of the 5 extracted all-atom simulation boxes for each conformational state, was minimized and equilibrated using 50 ns long NPT equilibration simulation protocol. Equilibration was performed starting with position restraints placed on receptor and NECA heavy atoms and on lipid heavy atoms in the head group of the lipid. The force constant on the position restraints were gradually reduced from 5 to 0 kcal/mol by 1 kcal/mol interval per 10 ns simulation window. The last 10ns of equilibration simulations were performed with no restraints. Starting from the 5 different equilibrated all-atom MD simulation starting structures we performed all-atom MD simulation runs each 400ns long with NPT ensemble at 310 K with 2 fs time step using GROMACS with CHARMM36FF39. Thus, we generated 5*400ns = 2 μs of all-atom simulation trajectories for each R, R’ and R*.G state. For each of the three conformational states we performed all-atom simulations in two environments namely: cell membrane, cell membrane with calcium. A total of 2*6= 12μs of all-atom MD simulations in cell membrane with and without calcium were done. We repeated this procedure in cell membrane without calcium for the antagonist ZMA241687 bound inactive state R (2 μs). A total of 14 μs of all-atom MD simulations (see Table S2) were performed in cell membrane environment. We stored the MD snapshots every 20ps. For the all atom simulations the non-bond interactions were calculated with cutoff of 12 Å, particle mesh Ewald method was applied for van der Waals interactions calculation. The temperature was maintained at 310K using Nose-Hoover thermostat40 and pressure at 1 atmosphere using Parrinello-Rahman method41. We used the last 200ns of each simulation run aggregated to 1μs over 5 runs, for all the analysis of properties from MD simulation trajectories.
Figure 1 A.
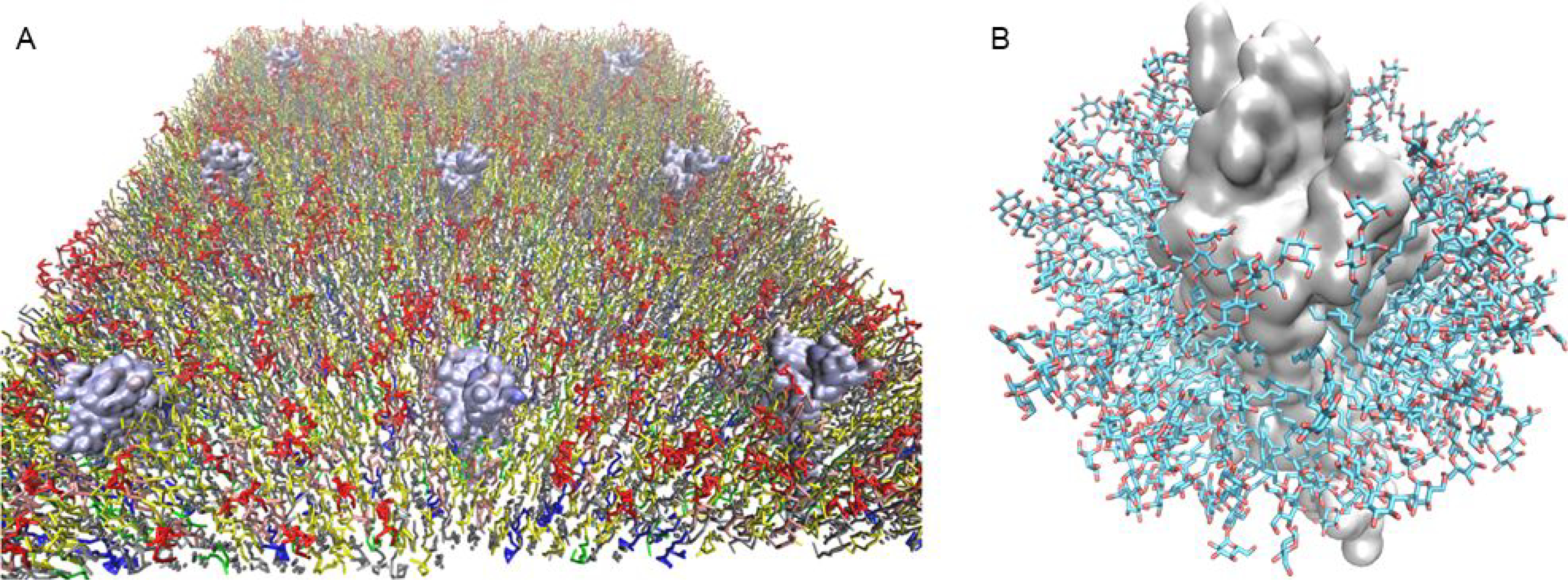
Starting model of the cell membrane consisting of multiple lipids used in the coarse grain simulations and all-atom molecular dynamics simulations. The colors denote different types of lipids. The surface rendering are of the A2AR crystal structures in the agonist NECA bound inactive state R, NECA bound active intermediate state R’ and mini Gs protein and NECA bound fully active R*.G conformation state. B. Starting model of the DDM detergent micelle with A2AR used for MD simulations.
CG simulation validation:
Following the procedure outlined in reference42, we validated the cell membrane coarse grain MD simulations using metrics such as: convergence of radial distribution function of different lipids, surface area per lipid in the top and bottom leaflet of the lipid bilayer to be within 1% (Fig. S2), also, the number of lipid molecules within 10 Å (first shell) of GPCR remains stable (Fig. S3). The radial distribution function (RDF) was used a metric to identify the lipids that accumulate close to the GPCR during the coarse grain simulations. RDF was calculated using VMD 43, as a function of the distance between mass centers of GPCR TM region and heavy atoms of each lipid. The counting of the number of lipids counting was done for every type of lipid within 10 Å of the mass center of GPCR TM region. Area per lipid is calculated using membrane plugin, a plugin in VMD by choosing the head group beads from CG model44. The coarse grain MD simulations showed clustering of cholesterol, PIP2 and GM3 close to the receptor in all the three conformation states (Fig. S4A), as evident from the radial distribution function and the number of each type of lipid calculated around the receptor (see Fig. S4B and S4C of the Supporting Information).
All atom MD simulations in detergent DDM environment:
We have performed all-atom MD simulations on the wild type A2AR bound to the agonist NECA in the inactive state (R), active-intermediate (R’), and fully active state of A2AR in DDM detergent micelles. The simulations were performed using GROMACS MD simulation package45 with CHARMM36FF39. The initial coordinates of A2AR in the R, R’ and R*.G were taken from the PDB code of 4EIY, 2YDV and 5G53, respectively. All thermostable mutants were mutated back to the wild type sequence using Maestro9.2. Residues within 5Å of the sites of mutation were minimized using MacroModel (Schrödinger Release 2020–3: Maestro, Schrödinger, LLC, New York, NY, 2020.) with position restraints on all backbone atoms and all residues further than 5Å from the site of mutation. The disulfide bonds were built according to the disulfide bonds listed in the pdb structure file. We used 192 DDM molecules to build each receptor-micelle complex using CHARMM-GUI/Micelle Builder module. We used the TIP3 force field for describing the waters. The receptor-micelle complexes were equilibrated using the same MD simulations as described in above mixed-bilayer membrane systems. We performed 5 production runs with different starting velocities each 400ns long. The trajectories of the all-atom MD simulations in the LMNG series of detergents were taken from our previous work46 and used here for comparison. We used the TIP3 force field for describing the waters. For all the analysis described below, we took the second half of each trajectory for each state (R, R’, and R*.G) and each environment (cell membrane, cell membrane with calcium, detergent and POPC) from 200ns to 400ns and concatenated them for 5 runs (totaling to 1 μs) to perform analyses.
All atom MD simulations in pure POPC bilayer:
We performed all-atom MD simulations in POPC bilayer for comparison to cell membrane simulations since most of the MD simulations for GPCRs are done in POPC bilayer. The simulation box containing POPC bilayer was built using CHARMM-GUI. We then placed the A2AR structures prepared above in the R, R’ and R*.G in the POPC bilayer, and solvated the system with water and neutralized with 0.15M of NaCl. This solvated system energy was minimized, and the system was equilibrated using 50 ns long NPT equilibration simulation protocol. Equilibration was performed starting with position restraints placed on receptor and NECA heavy atoms and on the P atom in the head group of POPC. The force constant on the position restraints were gradually reduced from 5 to 0 kcal/mol by 1 kcal/mol interval per 10 ns simulation window. The last 10ns of equilibration simulations were performed with no restraints. Starting from the 5 different equilibrated all-atom MD simulation starting structures we performed all-atom MD simulation runs each 400ns long with NPT ensemble at 310 K with 2 fs time step using GROMACS with CHARMM36FF39. Thus, we generated 5*400ns = 2 μs of all-atom simulation trajectories for each R, R’ and R*.G state. We stored the MD snapshots every 20ps. For the all atom simulations the non-bond interactions were calculated with cutoff of 12 Å, particle mesh Ewald method47 was applied for van der Waals interactions calculation. The temperature was maintained at 310K using Nose-Hoover thermostat40 and pressure at 1 atmosphere using Parrinello-Rahman method41. We used the last 200ns of each simulation run aggregated to 1μs over 5 runs, for all the analysis of properties from MD simulation trajectories.
Inter-residue distance analysis:
The distances between Cα atoms of residues at positions 3.50 and 6.34, 3.50 and 7.53 were calculated combining using the aggregated trajectories for each system. The distances are plotted as 2D heatmap using 80 bins after normalizing the population in each bin.
Extracellular loop interactions with GM3 or DDM:
We calculated the heat map for the persistence of interactions of GM3 with the residues in the extracellular loops. To this end, we first calculated the distance between all heavy atoms from the residues located in the extracellular (ECL)loops and the heavy atoms of GM3, in the cell membrane and DDM environment. The ECL residues in ECL are: ECL1 residue number 68 to 73, ECL2 (residues 142 to 172) and ECL3 (residues 258 to 264). The interaction is considered made, if any of the distance between a residue and any heavy atom of GM3 is smaller than 4.5 Å. The persistence frequency was then calculated as the percentage of MD snapshots that showed a prescribed GM3-residue contact.
Ligand binding pocket volume analysis:
The trajectories for the NECA bound inactive R state of A2AR in both DDM and cell membrane were clustered by NECA RMSD after aligning the MD snapshots by the residues in the TM region. The representative structure from the most populated conformational cluster in both environments (cell membrane and DDM) was extracted and used for binding pocket volume analysis. The software “trj_cavity” was used to calculate binding pocket volume, with a solvent probe radius of 1.7 Å 48. To calculate the fluctuation in the NECA binding site volume, we averaged the NECA binding site volume calculated for 10 frames extracted from the aggregated trajectory every 100 ns. In this analysis the average and standard deviation of ligand binding pocket volume in cell membrane is 237.2 ±36.4 Å3 and 383.6 ± 150.7 Å3 in DDM.
Ligand binding pose and pocket residues flexibility analysis:
The flexibility of the ligand was assessed by calculating the number of distinct conformation clusters sampled during the MD simulations of NECA bound A2AR in the inactive R state, in DDM and cell membrane environment. We clustered the NECA conformations by first aligning the MD snapshots by the coordinates of the receptor to the reference structure and then clustered based on the RMSD in coordinates of the heavy atoms in NECA. DBscan method in MDanalysis49 was used to cluster the ligand poses based on RMSD matrix with a 1.5 Å as cutoff used for clustering. To assess the flexibility of the residues in the receptor, we used the RMSF (root mean square fluctuation in coordinates) calculations of the backbone atoms of the receptor. RMSF for every residue was calculated using gmx rmsf module in GROMACS.
Ligand contact frequency analysis:
The ligand-residues contact frequency was calculated for residues whose heavy atoms are initially within 4.5 Å cutoff of NECA heavy atoms. NECA was partitioned into two organic groups as shown in Figure S2A. The ligand-residues contact frequency was calculated as the percentage of frames over total frames in which one residue is within 4.5 Å cutoff of the corresponding organic group of NECA from crystal binding conformation.
Spatio-temporal Heatmap of Activation Micro-switches:
We used all the activation microswitches characterized by Zhou et al by comparing several inactive state crystal structures of GPCRs to active state crystal or EM structures 10. The distance between centers of mass of each residue pair was calculated. The cutoff distance for defining if an activation microswitch is on or off has to be established. To this end we used an ensemble of conformations populated by the antagonist bound inactive state R. We performed MD simulations on the antagonist bound inactive state R of A2AR. We calculated the distance distribution of all the pairs of activation microswitches from these trajectories. We fitted a Gaussian function to each of the activation microswitch distance distribution and extracted the mean distance from the distribution and the standard deviation. For a given activation microswitch, we used the mean value of the distance distribution minus one standard deviation for the activation microswitch pairs that are known to shrink in distance upon activation. For those activation microswitch pairs that are known to expand upon activation, the mean plus one standard deviation was used as cutoff distance to define the active state. This cutoff distance was used to determine if an activation microswitch is in an “on” or “off” state in each MD snapshot. The same cutoff distance was applied to simulations in cell membrane, detergent micelle and POPC bilayer bound with NECA, for R, R’ and R*.G states. Once the cutoff distance was determined, the distance histograms for all microswitch pairs from all the simulations were calculated. The population of conformations below the cutoff distance for the shrinking activation microswitches (above the cutoff for the expanding pairs) were counted and the percentages over whole population were calculated to arrive at the percentage of activation for each microswitch.
Interaction energy of PIP2 with the receptor:
The interaction energy between PIP2 and the receptor was calculated as average of the sum of electrostatic energy and van der Waal interaction energy between all PIP2 molecules and all the residues in A2AR, averaged over the concatenated trajectory for each system.
Cholesterol persistence frequency:
We identified the cholesterol molecules within 4.5 Å of every A2AR residue and calculated the persistence frequency of these contacts as the percentage of MD snapshots that showed such contacts. The contact frequencies were set as B-factor for each residue and colored as heatmap on GPCR structure. This was also plotted as a bar graph for each of R/R’/R*.G states.
Free energy perturbation method for calculating the Ligand binding free energy:
The binding free energy (ΔG) of NECA in three conformational states (R, R’, and R*.G) and two environments (DDM micelle and cell membrane) was calculated using the BAR (Bennett acceptance ratio) algorithm available in the GROMACS package 50. The MD simulations were conducted at different values of λ (coupling parameter) in the BAR method. We performed MD simulations from ligand-bound state where λ=0 to ligand–free state where λ=1, using equidistance increment of λ values of 0.05. We performed 10ns of all-atom MD simulations for each λ value. The total simulation time for NECA in each of the three conformational states, R, R’ and R*.G in each environment is 200ns.
Results
Multi-scale MD simulations in mixed lipid bilayer
Previous coarse grain MD simulation studies on A2AR receptor in various conformational states showed accumulation of GM3, PIP2 and cholesterol close to the receptor depending on the conformational state of the receptor20. However, there is very little knowledge on the atomic level mechanism of these interactions and the effect of the chemistry of the lipids and cations, on GPCR conformational dynamics in cell membrane compared to detergents. To study the effect of multiple lipids on the GPCR conformation ensemble, we used multi-component lipid bilayer to mimic the cell membrane as described in the Methods section. Since this mixed lipid bilayer mimics the cell membrane, we refer to this as “cell membrane” hereafter in the paper. We started the coarse grain MD simulation from the crystal structures of the agonist 5’-N ethylcarboxamidoadenosine (NECA) bound human adenosine receptor A2AR in the inactive state R, the active-intermediate state R’ and the mini-G-protein bound fully active state of the receptor R*.G in the cell membrane model 3,9,51 (see Methods for more details). We performed 30μs of coarse-grained MD simulations to obtain an equilibrated multi lipid cell membrane mimic system. We extracted the receptor conformations in all the three states from coarse grain simulations and converted them to all-atom models (see Methods) and performed all-atom MD simulations. The list of systems simulated in this study, the notations used to represent different conformational states, and other details of these systems are given in Table S2.
MD simulations in pure POPC bilayer and Detergents
To compare the effect of the detergent environment on receptor dynamics, to that of cell membrane we also performed all-atom MD simulations on NECA bound R, R’ and R*.G states of A2AR in DDM detergent micelle (Fig. 1B) using procedures described in the Methods section. For comparisons, we have also used analysis of our previous MD simulations using branched detergent Lauryl Maltose Neopentylglycol, LMNG46. Since most of the MD simulation studies on GPCRs are performed in pure POPC bilayer, for comparison we also performed all-atom MD simulations using a pure POPC lipid bilayer.
Agonist binds with higher affinity in cells compared to detergent micelles
Binding affinity of the agonist NECA is an order of magnitude stronger (Kd = 2.4 ± 0.1 nM)52 in cell based measurements compared to that in DDM detergent solution (23.4 ± 0.11 nM) 53. This translates to a difference of 1.4 kcals/mol in free energy at 310K. Using free energy perturbation alchemy methods (Bennett Acceptance Ratio method – see Methods section) we computed the binding free energy of NECA in R, R’ and R*.G states both in cell membrane and in detergent DDM micelles. The calculated binding free energy of NECA is 2.1 kcal/mol and 0.8kcal/mol better in the cell membrane compared to DDM detergent micelle in the inactive R and active-intermediate R’ states respectively with no difference in the R*.G state (Fig. 2A). We analyzed the structural basis for this enhanced agonist binding affinity for the R state in the cell membrane compared to detergents. We identified the receptor ligand interactions that cause this difference in binding free energies. More importantly, even for the receptor ligand interactions that are common to both detergent and cell membrane environments, the difference in their “persistence frequencies” contribute to the difference in binding energies. The persistence frequency of a residue-residue contact or a receptor-ligand contact is defined as the percentage of MD snapshots that contain that contact. The list of receptor-NECA contacts and their persistence frequencies in the NECA bound R state in cell membrane and in DDM detergent are shown in Fig. 2B (see also Fig. S5A). The persistence frequencies of the NECA-receptor contacts are higher in the cell membrane environment compared to the detergent environment for the R state. This shows that the cell membrane components tighten the binding site residue-NECA contacts even in the inactive R state conformation similar to that of the binding site of NECA in the R’ state. This finding demonstrates the importance of dynamics simulations in cell membrane conditions to recapitulate the observed differences between cell membrane and detergents. In the persistence frequency plot shown in Fig. S5B, the adenine ring moves between making contact with Asn253 but more persistently with Phe168 for over 80% of the MD simulation snapshots in cell membrane conditions. Similarly, in the POPC bilayer the adenine ring makes more persistent contact with Phe168 and to a lesser extent with Asn253. However, in the detergent environment, the adenine ring stays close to both Asn253 and Phe168. Mutation experiments have shown that Phe168 to Alanine abolishes NECA binding in cells 54. The agonist NECA shows higher flexibility in the agonist binding site in the R state in detergent environment compared to cell membrane environment as shown by the 3 distinct conformations in detergent shown in Fig. 2C. The receptor also shows higher flexibility in the R state around the agonist binding site, in detergent environment compared to cell membrane environment as seen in the heat map of flexibility shown in Fig. 2C. This shows that the lipids in the cell membrane have a pronounced effect on agonist binding even in the inactive state compared to detergents. Next we identified the lipid components of the cell membrane that bring about this favorable effect on agonist binding affinity in the extracellular ligand binding region in the inactive state.
Figure 2.

Structural basis for the higher binding affinity of NECA in cell membrane compared to detergent DDM. A. Calculated binding free energy of NECA in R, R’ and R*.G conformational states of A2AR shown with the respective standard deviations. The free energies were computed using free energy perturbation - BAR method (see Methods section). B. Persistence frequency of the NECA-residue contacts in the inactive R state, in cell membrane (grey) and in DDM detergent (blue). The persistence frequency is the percentage of MD snapshots that contain the NECA-residue contacts. C. The conformational flexibility of the agonist NECA in the R state in cell membrane and in detergent. Top 3 representative structures from clustering analysis of NECA are shown. The representative structures of NECA from cell membrane simulations are shown in purple (51%), blue(22%) and green (7%). The respective population of these agonist conformation clusters are given within parenthesis. The representative structures of NECA and the population of the conformation clusters from the detergent DDM simulations are shown in purple (43%), blue (15%), green (11%). The population of the top three occupied clusters of NECA are shown in parenthesis. The RMSF values that measure the flexibility of the Cα atoms of the residues in the NECA binding site, are shown in a heat map overlaid on the A2AR inactive R state structure. Blue to red color indicates increasing flexibility of the receptor.
GM3 tightens the ligand binding site in the extracellular leaflet of the cell membrane
GM3 is present in the outer leaflet of the cell membrane. Its polar head group consists of sugars and has a long hydrophobic tail. During the simulations of all the three conformational states of A2AR in cell membrane, we observed that the hydrophobic tail of GM3 binds in exterior crevices between the extracellular regions of TM6 and TM7. The polar head groups interact with the residues G152, K153 and S156 in the extracellular loop2 (ECL2). Additionally, residues in both ECL1 and ECL2 show persistent hydrogen bond interaction with the head group of GM3 as shown in Fig. 3A (and Fig. S6A). Gangliosides have been shown to regulate GPCR function and coarse grain MD simulations have shown that they interact with extracellular loops55,56. As a result of these persistent interactions of GM3 with ECL2 residues, we observed a large movement in ECL2 (4 to 8Å) that covers the NECA binding site as shown by the grey arrow in Fig. 3B (and Fig. S6B). Such interactions of DDM with ECL2 residues were not observed in the detergent micelles (Fig. 3A) and the ECL2 actually shifted 3Å away from ligand binding pocket (Fig. 3B blue arrow). The residues G152, K153 and S156 in ECL2 showed stable interactions with GM3 head group in R state as shown in Fig. S6B. Calculation of persistence frequency (percentage of MD snapshots that show contact between GM3 and residues in ECL2) of GM3 with these residues show that the GM3 contacts with these residues is most persistent in the R state compared to R’ and R*.G state as shown in Fig. S6C. The closing of ECL2 over the NECA binding site leads to contraction of the volume of the NECA binding site in cell membrane simulations as shown in Fig. 3C. The average volume of ligand binding pocket calculated using the MD snapshots from the most populated conformation cluster decrease from 333.4 Å3 in DDM to 279 Å3 in cell membrane. The cell membrane components such as GM3 constrain the structural elements around the NECA binding site thus leading to improved interactions with the receptor even in the inactive state. Song et al20 reported the effect of GM3 on gating the on and off rate of agonist using the persistence of contact of GM3 with the receptor calculated from coarse grain MD simulation study. Here, from the atomistic level simulations we calculated the binding free energy of NECA in cell membrane and in detergents. We also show how GM3 enhances the receptor-agonist interactions in the binding site by tightening these interactions.
Figure 3:

GM3 pulls the ECL2 to cover and shrink the agonist binding site. A. The persistence frequency of NECA-residue contacts in the extracellular loops ECL1, ECL2, ECL3 in the R state with GM3 is shown as a heat map on the structure of R state of A2AR in cell membrane and in DDM detergent. B. Overlay of representative structures extracted from the top two populated conformation clusters extracted from the MD simulations of the inactive R state in cell membrane and in DDM detergent. The starting structure of the simulations is shown in white in these two figures, and the representative structure from the most populated conformation cluster are shown in grey (cell membrane) and cyan (detergent) color. The structure shown in green is the representative structure from the second most occupied cluster in cell membrane. Both these conformations show a closing of ECL2 over the NECA binding site which is not seen in detergent. C. The NECA binding pocket shrinks in volume, caused by GM3 closing the ECL2 over the ligand binding site and enhancing the NECA-residue contact frequency for the R state. The ligand binding site volume is larger in the DDM environment.
Cell membrane environment constrains the conformational flexibility of GPCRs compared to detergents
The movement of transmembrane helix TM6 away from the core of the receptor and movement of TM7 towards TM3 and TM5 has been shown to be major conformational changes in the active state of the receptor compared to the inactive state 57. To assess the extent of conformational sampling and flexibility of the receptor in the intracellular regions, we calculated the Cα-Cα distances between residues R1023.50-E2286.34 on TM3 and TM6 and between R1023.50-Y2887.53 on TM3 and TM7 for every snapshot in the MD simulation trajectories. We projected the snapshots from MD simulations in these two distance space, for the R, R’ and R*.G states in the cell membrane (Fig. 4A), in cell membrane with Ca2+ (Fig. 4B), in DDM detergent (Fig. 4C) and in pure POPC bilayer (Fig. 4D). In the NECA bound inactive R state, the receptor conformation ensemble shifts away from the starting crystal structure and the number of transitions between R and R’ states are more populated in the DDM environment (Fig. 4C) than in cell membrane environment (Figs. 4A and B). On the other hand, there are no transitions between the R’ and R*.G states observed within the time scale of the all-atom MD simulations in both cell membrane and detergent environment. In the pure POPC bilayer (Fig. 4D), the number of transitions between R and R’ state are fewer than in detergent, but more than in cell membrane. The spread of the receptor conformation ensemble in cell membrane is more constrained in each R, R’ and R*.G states (Fig. 4A) compared to the detergent DDM environment (Fig. 4C). As detailed in the next section, this is due to the charged lipid PIP2 present in the inner leaflet of the cell membrane accumulating close to the basic residues (Arg/Lys) in the intracellular regions of the receptor in all the three conformational states.
Figure 4:

PIP2 and intracellular calcium modulate receptor flexibility and activity. A-D. The extent of receptor conformation sampling of NECA bound A2AR in R, R’ and R*.G states in four different environments, namely, the cell membrane(A), cell membrane with calcium ions (B) and DDM detergent (C) and pure POPC bilayer (D). The MD snapshots are projected on inter-residue distance measures. The distances are measured between the Cα-Cα atoms of the residue pairs R1023.50-E2286.34 on TM3 and TM6 and between R1023.50-Y2887.53 on TM3 and TM7. These two distances in the starting crystal structures of A2AR in R, R’ and R*.G states are shown as red (R), yellow (R’) and green (R*.G) dots respectively. E-P. The rows in this set of figures show the representative structures in R, R’ and R*.G states. The four columns are the four environments as indicated in the heading of each column. In figures E and I, PIP2 forms bifurcated salt bridge interactions with the Lys/Arg in the intracellular region of A2AR, bridging the TM helices, in cell membrane conditions, in the inactive R state (E) and in active intermediate R’ state (I). In figure M, PIP2 forms salt bridge interactions with basic residues both in the fully active state of A2AR and in mini-G protein thus stapling the receptor with mini-G protein. PIP2 is shown in grey and orange sticks representation. Figures F, J and N show Calcium ions (shown in black spheres) interactions with PIP2 and yanks the PIP2 away from the Lys/Arg residues in the receptor R state (F), R’ state (J) and R*.G state (N). Calcium ions coordinate with PIP2 freeing up the TM helices thus promoting transitions between R and R’ state seen in figure B. In figures G, K and O, DDM forms weak hydrogen bonds with Arg/Lys in the intracellular region in the R state (G), R’ state (K) and R*.G state (O). This results in transitions between R and R’ states in DDM as shown in figure C. In figures H, L and P, the POPC lipid forms salt bridge interactions with the Arg/Lys in the intracellular region in the R state (H), R’ state (L) and in R*.G state (P). However, POPC does not form stapling interactions between A2AR and mini-G protein. All the structures shown in this figure are representative structures from the most occupied conformation cluster. The conformation clustering was done using RMSD of the receptor backbone with a cutoff of 1.5Å.
PIP2 reduces movement of TM helices in the intracellular regions
PIP2 accumulates close to the receptor in all the three R, R’ and R*.G conformational states (Fig S4B). In the R and R’ states, as shown in Figs. 4E and 4I, PIP2 forms salt bridge interactions to the Arg/Lys residues in the intracellular region of TM5, TM6 and TM7. These bifurcated salt bridges of PIP2 with the basic residues, bridges the TM helices thereby restricting the movement of the receptor in the intracellular regions. However, in the DDM environment, in the R and R’ states, although DDM head group forms hydrogen bonds with basic residues in the intracellular regions (Figs. 4G and 4K), they are not as strong as the salt bridges formed by PIP2, and do not bridge the helices and hence do not restrict the flexibility of the TM helices (Fig. 4C). The residues K7.56, K6.35, R5.66, R222 on ICL3 and R293 in the C-terminus of the receptor are involved in forming salt bridges with PIP2. This increased PIP2 interaction with the intracellular regions of the receptor is also borne out by the calculated non-bonded interaction energy between PIP2 and receptor in R and R’ states shown in Fig. S7. In the MD simulations of A2AR in the R and R’ states in pure POPC bilayer, the negative charged phosphate group in POPC forms salt bridge interactions with Arg/Lys in the intracellular region (Figs. 4H and 4L) but not as many as the more negatively charged PIP2.
PIP2 staples the GPCR:G protein complex:
PIP2 in the R*.G state acts as a “stapler” between the active state A2AR and the mini-Gs protein in the R*.G complex. The hydrophobic tail of the PIP2 interacts with the hydrophobic regions of the TM helices in R*.G state, and the charged head group of PIP2 makes salt bridge contacts with Arg/Lys residues in the miniGs protein (Fig. 4M). As shown in Fig. S7, this leads to a much stronger interaction energy of PIP2 with the R*.G complex compared to its interaction energies with R and R’ states of the receptor. Thus, PIP2 increases the stability of the R*.G complex by interacting with both these moieties. Such stapling interactions that strengthens the A2AR-miniGs complex were not present in either of the simulations in DDM or POPC bilayer (Fig. 4O and 4P respectively) environment.
Intracellular calcium ions ease the receptor stiffening by PIP2 to modulate the basal activity of GPCRs
Cations such as Ca2+ are known to play an important role as allosteric modulators of GPCR activity in cells 23–26. In our MD simulations in cell membrane with calcium, the Ca2+ ions make strong salt bridges with the phosphate groups of PIP2 resulting in breaking the PIP2 salt bridges with the intracellular Lys and Arg residues in the R and R’ states (Figs. 4F and 4J). Ca2+ coordinating with PIP2 thus frees up the Arg/Lys residues in the receptor. Consequently, the interaction energy between A2AR and PIP2 is weakened by Ca2+ ions and this trend is true for R, R’ and R*.G states (Fig. S7). This results in an increase in the flexibility of the TM helices which facilitates a few transitions between R and R’ states that was not seen in the cell membrane simulations without Ca2+ (Fig. 4B). Based on this data we speculate that the localized concentration of PIP2, and Ca2+ in lipid rafts together modulate the basal activity of the GPCRs. Calcium ions also weaken the interaction energies of PIP2 with the A2AR-miniGs complex (Fig. 4N). In an NMR study of the basal activity of β2-adrenergic receptor Staus et al speculated that the detergent as well as lipid nanodisc environments promote more transitions among the R, R’ and R*.G states compared to cell membrane15. Thus, our findings suggest a mechanism by which cell membranes could lead to restraining receptor conformations and thus modulate their basal activity. Calcium weakens the interaction energy of PIP2 with the receptor in all the three conformation states including the R*.G state (Fig. S7).
Branched detergents mimic the behavior of PIP2 compared to unbranched detergents
In a previous study we examined the differences in the dynamic behavior of GPCRs in branched detergent micelles from the maltose-neopentylglycol (MNG) series compared to their unbranched counterparts such as DDM46. We observed that LMNG tucks its branched hydrophobic tails between the TM helices in the intracellular region of the GPCR, while its two branched polar head groups make hydrogen bonds with the polar and basic residues in the intracellular region (Fig. S8A). The two head groups in LMNG being covalently linked through a central carbon atom aids the formation of the bifurcated hydrogen bonds and reduce the flexibility of the receptor in the intracellular region. This behavior of LMNG detergent is a mimic of the role of PIP2 in the cell membrane although the bifurcated hydrogen bonds are not as strong as the salt bridges made by PIP2 with the Arg/Lys in the intracellular part of the TM helices (Fig. S8B).
Spatiotemporal heat map of GPCR activation microswitches reveal rheostat behavior rather than binary switches in cell membrane
Conserved residue pairs that show significant change in the inter-residue distance upon activation were defined as activation microswitches 10,53,57–59. However, more recently a comprehensive analysis of the change in the inter-residue distances upon activation was performed by Zhou et al by comparing several inactive state and active state static structures of multiple class A GPCRs10. This study elucidated the inter-residue distances (Figs. 5A and 5B) that show a distinct change going from inactive to active states and are known as activation “microswitches”. Comparison of static structures reveal a binary view (“on” or “off” states) of the activation microswitches. To perform a comprehensive analysis, we took the list of activation microswitches from this study (see Table S3) and analyzed the dynamic behavior of each of these activation microswitches in cell membrane, POPC bilayer and detergent environments. However, in the cell membrane environment the receptor exists in an ensemble of conformational states and the activation microswitches show differences in persistence frequency of each of these microswitches, that calls for a continuum view of activation microswitches as in a rheostat. In order to examine this concept, we calculated the spatio-temporal heat map of the persistence frequency (percentage of MD snapshots showing these contacts made or broken within a cutoff distance – see Methods) of the activation microswitches made or broken from the MD simulation trajectories in cell membrane and in DDM detergent (shown in Fig. S9). As seen in Fig. 5B, there are four layers across the GPCR structure in which the activation microswitches are distributed. Layer 1 which is below the agonist binding site, involves the PIF motif, CWxP motif and the sodium binding site. The major conformational changes upon activation in this layer are the collapse of the sodium binding site (D2.50, S3.39 and N7.45) and repacking of P5.50 and F6.44 in the PIF motif (P5.50, F6.44 and I3.40). Layer 2, which is one step further down towards the intracellular region involves the opening of hydrophobic lock of L3.43, V6.40 and V6.41. Layer 3 involves the rewiring and compression of residues R3.50 and I3.46 and also expanding L6.37 and R3.50. Another important conformational change is the collapse of Y7.53 of the NPxxY motif, to its nearby residues L3.43, I3.46 and R3.50. Layer 4 which is close to the G-protein binding site undergoes releasing of R3.50 from D3.49 in the DRY motif and A3.53 motif upon activation. All the microswitches are in their active state in R*.G state in DDM detergent, in cell membrane with or without calcium and in POPC bilayer (Fig. 5E). However, it should be noted that the percentage of activation is not the same for all the microswitches in all the four environments. The interesting differences are in the heat map for NECA bound inactive state R and active-intermediate state R’. In the inactive R state (Fig. 5C), more activation microswitches in layer 1 close to the NECA binding site, and also microswitches centered at Y7.53 in layers 3 and 4 are active in detergent environment. Comparison between cell membrane with and without calcium, shows that the presence of calcium improves activation around Y7.53 and around F6.44, similar to detergent micelle but to a lesser degree. In the cell membrane with and without calcium, the activation microswitches are active only in layer 1 close to NECA and not in layers 3 and 4 as in the detergent. The activation of microswitches centered at F6.44 in layer 1 and centered at L3.43 in layer 2 are active in the R state in detergent. All the four environments show similar activation profile in the R’ state (Fig. 5D), with exception of PIF motif that shrinks more and I6.40 moves away from N7.49 in detergent micelle compared to cell membrane. Cell membrane with and without calcium show similar activation profile in R’ state. The shrinking of the sodium binding site typified by the distance D2.50 - S3.39, is not complete in cell membrane without calcium, while calcium enhances the extent of this shrinking and detergent micelle in the R state. The POPC bilayer shows the highest level of inactivation in the NECA bound R state. Taken together, these results show that activation microswitches indeed function as rheostats. Cell membrane conditions limit the flexibility of the activation microswitches thus reducing the basal activity of the receptor. On the other hand, the detergent environment increases the flexibility of the receptor. While adding Ca2+ to cell membrane environment increase the receptor flexibility thus tuning some of the microswitches earlier than others, but to a less degree than detergent.
Figure 5:

GPCR dynamic ensemble shows that activation microswitches behave like rheostats in cell membrane, cell membrane with Ca2+, POPC bilayer, and DDM detergent environments. A. Positions of residues labeled as activation microswitches by analysis of active and inactive state crystal structures. The agonist NECA is shown in grey, red and blue stick representation for reference. The microswitch residues are colored differently based on their positions relative to ligand binding site. Residues in layer 1 are colored in dark green (close to the agonist binding site), layer 2 in light green, layer 3 in yellow, layer 4 in red. B. A schematic showing the location of the inter-residue distances that are used as activation microswitches, and their respective TM helices shown as grey bars. The residue positions are shown by their Ballesteros-Weinstein numbering scheme commonly used for class A GPCR residue numbering. The solid line indicates that this inter-residue distance decreases upon activation, and the dashed line indicates the expansion of the inter-residue distance upon activation. C. The spatio-temporal heat map of the activation microswitches in the inactive R state in cell membrane, cell membrane with calcium, DDM detergent and pure POPC bilayer. The heat scale from red to green shows the percentage of MD snapshots where each of the inter-residue distance is in the active state (see Methods for more details). D-E. The spatio-temporal heat map of the microswitches in the four environments for the R’ state (D) and R*.G state (E).
Cholesterol binding sites
The persistence frequency of contacts between cholesterol and A2AR residues within a cutoff distance of 4.5Å were calculated from MD trajectories in cell membrane for all of R/R’/R*.G states. The persistence frequency for each residue is shown colored in a gray scale heat map on A2AR structure in Fig. S10. We identified three cholesterol binding sites: the first site is located in the extracellular region between TM1, TM2, the second site is located in the intracellular region around TM2, TM3 and TM4, and the third site is located in the extracellular region around TM6 and TM7. The first and third binding sites were also observed in the A2AR crystal structure 4EIY, where the binding site was lined with residues L2.56, F2.60, G3.24, C3.25, F3.27, I3.28, F6.57, I6.53, and L6.49 9. The cholesterol binding site in the crystal structure was also observed in our simulations with >50% persistence contacts with all the residues listed above except G3.24, C3.25, F3.27 and I3.28. The second binding site located in TM2 and TM4 intracellular region was similar to the binding site identified in β2-adrenergic receptor crystal structure (pdb id:3D4S) 60. The residues lining this binding site are I1.47, L1.48, I2.51, W4.50, I4.48, I4.45 and V4.51. We observed cholesterol in this binding site with persistence frequency greater than 70% for the key residue contacts. The cholesterol binding sites are relatively conserved in all of R/R’/R*.G states, but they show differences in the persistent frequency of the contacts. The R’ state has highest persistence frequency with cholesterol in the TM1, TM2 site as well as the TM6, TM7 site. In the R*.G state, cholesterol binding site in the TM4, TM5 region is more persistent than the other two sites. In all the three states, TM3 has lowest persistence frequency with cholesterol, especially for R*.G state.
Discussion
Although it is known that lipids and cations in the cell membrane play a functional role in modulating GPCR activity 16–18, the mechanism by which lipids and cations together modulate the receptor activity in comparison to detergents is unknown. We have used multi-scale MD simulations to decipher the mechanistic insights into the role of lipids and cations present in cell membrane on the flexibility and activity of agonist NECA bound A2AR in three different conformation states, R, R’ and R*.G. We have compared the dynamics of A2AR in these three states in cell membrane to that in DDM and LMNG detergent micelle environment that are commonly used for biophysical studies on GPCRs.
In this study, we observed that PIP2 accumulates close to the receptor in all the three R, R’ and R*.G states of A2AR but, play a different role in how they modulate the receptor activity and stability in each conformational state. PIP2 forms bifurcated salt bridges with Arg/Lys in the intracellular regions of the receptor in the R and R’ states. These bifurcated salt bridges act as interhelical clamps holding the TM helices in place thus making them less flexible and more stable in membrane (left hand side figure in Fig. 6). This reduces the number of transitions between inactive state and active-intermediate state in cell membrane. Presence of intracellular Ca2+ ions in the cell membrane breaks the salt bridges of PIP2 with the Arg/Lys in the TM helices and in the intracellular loops. This loosens the interhelical rigidity and promotes transitions between R and R’ states. These observations are in line with the enhanced exchange rate between active and inactive conformations observed in NMR experiments15,22 in HDL nanodiscs and detergents. These authors further speculated that the lower level of basal activity in cells could come from cellular mechanisms that control basal activity. Here we provide insights into how the cell membrane components such as PIP2 and calcium maintain a balance between flexibility and stability in membranes. Such bifurcated interactions that reduce the interhelical flexibility were also observed in branched detergents such as LMNG and not in unbranched detergent DDM. However, the stiffness provided by these bifurcated hydrogen bonds in LMNG are not as strong as the bifurcated salt bridges with PIP2. MD simulations in pure POPC bilayer also shows salt bridge interactions of POPC with basic residues in the intracellular region of the receptor in the R and R’ states. However, the interactions with POPC are weaker than with PIP2. In the G protein bound fully active state of A2AR, PIP2 acts as a stapler between the A2AR and the miniGs protein thus enhancing the stability of the R*.G complex in its fully active state as shown in the righthand side figure in Fig. 6.
Figure 6:

A model summarizing the findings from this study. On the left is the inactive state of A2AR showing the interactions of PIP2 with Arg/Lys in the intracellular region. The interaction of GM3 (blue and red stick representation) with ECL2 residues shrinks the receptor-NECA binding site and enhances the binding affinity of agonist in the inactive R state shown on the left. PIP2 (orange stick representation) reduces the receptor flexibility in cell membranes by interacting with Arg/Lys in the intracellular region of A2AR in both inactive and active states. Shown on the right is the agonist NECA and mini-Gs protein bound fully active state of A2AR. PIP2 interacts with basic residues in A2AR and in the mini-G protein. Intracellular calcium ions (shown as spheres) interact with the PIP2. The activation microswitches located at different layers in the receptor structure show different levels of activation as shown by the heat scale indicating the percentage of the snapshots in the conformation ensemble that is activated in cell membrane. In the inactive state, the activation microswitch closest to the agonist NECA is activated while those farther away from the agonist is less activated as shown by their colors. The activation microswitches (green spheres in the figure on the right) in the fully active state of A2AR when bound to G protein are all activated.
Although experiments have shown that the binding affinity of the agonist NECA is an order of magnitude better in cells compared to detergents, there was no structural basis for this observation. Our ligand free energy calculations recapitulated this difference in binding affinity and showed that the enhanced binding affinity of NECA in cell membrane is due to the effect of GM3 enhancing the agonist receptor interactions and shrinking the NECA binding site, that does not happen in the detergent DDM environment. We also observed that GM3 pulls the extracellular loops down closer to the ligand binding site. Such strong modulatory interactions are absent in the detergent micelle. These differences between detergent and cell membrane environments on GPCR activity could yield different structural and biophysical properties when studied in detergents.
It is known that GPCRs exist in conformation ensembles whether it be an inactive or active state. We show that these ensembles contain a mixed population of “on or off” states for each activation microswitch which we call as rheostat like behavior. The results of our study suggest that activation microswitches involved in GPCR activation function as rheostats rather than binary on and off switches whether it be in cell membrane or detergent environment. The spatio-temporal heat map of persistence frequency of the activation microswitches show that the cell membrane environment stabilizes a different combination of the activation microswitches compared to the detergent environment. The propagation of the activation microswitches in different layers of the GPCR structure is different in cell membrane compared to detergents (Fig. 6). MD simulations play a unique and critical role in delineating such mechanistic insights in modulating the differential effects of these activation microswitches compared to detergents.
In summary, our work exemplifies the role of GPCR structural dynamics and the effect of persistence frequency of lipids, cations or detergent interactions with the receptor in modulating the activity of GPCRs. The direct comparison between the cell membrane and detergent conditions allows us to draw insights into how cell membrane modulates the receptor activity in some cases similar to the detergent and many other cases differently from the detergent environment. These insights directly provide clues to design detergents or identify additives to detergent solutions that could better mimic the cell membrane properties. The observation that the components of the cell membrane constrain the receptor in various conformational states shows the synergy between sequence evolution and the endogenous environment that together provide the balance between the stability and the activity for GPCRs.
Supplementary Material
Acknowledgements:
We thank Dr. Supriyo Bhattacharya for the insightful discussions. Funding for this work was provided by NIH R01 grants GM097261 and GM117923 to N.V.
Footnotes
Supporting Information
1) Figures S1 to S10 that supports the data and conclusions from the figures shown in the main manuscript
References
- (1).Weis WI, and Kobilka BK (2018) The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Vaidehi N, and Bhattacharya S (2011) Chapter 7 - Multiscale computational methods for mapping conformational ensembles of G-protein-coupled receptors, in Computational chemistry methods in structural biology (Christov CBT-A in P. C. and S. B., Ed.), pp 253–280. Academic Press. [DOI] [PubMed] [Google Scholar]
- (3).Carpenter B, Nehmé R, Warne T, Leslie AGWW, and Tate CG (2016) Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Audet M, and Bouvier M (2012) Restructuring G-Protein- Coupled Receptor Activation. Cell 151, 14–23. [DOI] [PubMed] [Google Scholar]
- (5).Carpenter B, and Tate CG (2017) Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr. Opin. Struct. Biol. 45, 124–132. [DOI] [PubMed] [Google Scholar]
- (6).Schafer CT, and Farrens DL (2015) Conformational Selection and Equilibrium Governs the Ability of Retinals to Bind Opsin. J. Biol. Chem. 290, 4304–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, Javitch JA, Weinstein H, Kobilka BK, and Blanchard SC (2017) Single-molecule analysis of ligand efficacy in β2AR–G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kim TH, Chung KY, Manglik A, Hansen AL, Dror RO, Mildorf TJ, Shaw DE, Kobilka BK, and Prosser RS (2013) The Role of Ligands on the Equilibria Between Functional States of a G Protein-Coupled Receptor. J. Am. Chem. Soc. 135, 9465–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, Cherezov V, and Stevens RC (2012) Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science (80-. ). 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhou Q, Yang D, Wu M, Guo Y, Guo W, Zhong L, Cai X, Dai A, Jang W, Shakhnovich EI, Liu Z-JJ, Stevens RC, Lambert NA, Babu MM, Wang M-WW, and Zhao S (2019) Common activation mechanism of class A GPCRs. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi H-J, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, and Kobilka BK (2011) Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 469, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chung KY, Kim TH, Manglik A, Alvares R, Kobilka BK, and Prosser RS (2012) Role of Detergents in Conformational Exchange of a G Protein-coupled Receptor. J. Biol. Chem. 287, 36305–36311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gao Y, Erickson JW, Cerione RA, and Ramachandran S (2019) Reconstitution of the Rhodopsin–Transducin Complex into Lipid Nanodiscs BT, in Protein Lipidation: Methods and Protocols (Linder ME, Ed.), pp 317–324. Springer New York, New York, NY. [DOI] [PubMed] [Google Scholar]
- (14).Mitra N, Liu Y, Liu J, Serebryany E, Mooney V, DeVree BT, Sunahara RK, and Yan ECY (2013) Calcium-Dependent Ligand Binding and G-protein Signaling of Family B GPCR Parathyroid Hormone 1 Receptor Purified in Nanodiscs. ACS Chem. Biol. 8, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Staus DP, Wingler LM, Pichugin D, Prosser RS, and Lefkowitz RJ (2019) Detergent- and phospholipid-based reconstitution systems have differential effects on constitutive activity of G-protein–coupled receptors. J. Biol. Chem. 294, 13218–13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dijkman PM, and Watts A (2015) Lipid modulation of early G protein-coupled receptor signalling events. Biochim. Biophys. Acta - Biomembr. 1848, 2889–2897. [DOI] [PubMed] [Google Scholar]
- (17).Dawaliby R, Trubbia C, Delporte C, Masureel M, Van Antwerpen P, Kobilka BK, and Govaerts C (2016) Allosteric regulation of G protein–coupled receptor activity by phospholipids. Nat. Chem. Biol. 12, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sengupta D, Prasanna X, Mohole M, and Chattopadhyay A (2018) Exploring GPCR–Lipid Interactions by Molecular Dynamics Simulations: Excitements, Challenges, and the Way Forward. J. Phys. Chem. B 122, 5727–5737. [DOI] [PubMed] [Google Scholar]
- (19).Yen H-Y, Hoi KK, Liko I, Hedger G, Horrell MR, Song W, Wu D, Heine P, Warne T, Lee Y, Carpenter B, Plückthun A, Tate CG, Sansom MSP, and Robinson CV (2018) PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Song W, Yen H-YY, Robinson CV, and Sansom MSP (2019) State-dependent Lipid Interactions with the A2a Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 27, 392–403.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sejdiu BI, and Tieleman DP (2020) Lipid-Protein Interactions Are a Unique Property and Defining Feature of G Protein-Coupled Receptors. Biophys. J. 118, 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ueda T, Kofuku Y, Okude J, Imai S, Shiraishi Y, and Shimada I (2019) Function-related conformational dynamics of G protein–coupled receptors revealed by NMR. Biophys. Rev. 11, 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).van der Westhuizen ET, Valant C, Sexton PM, and Christopoulos A (2015) Endogenous Allosteric Modulators of G Protein–Coupled Receptors. J. Pharmacol. Exp. Ther. 353, 246–260. [DOI] [PubMed] [Google Scholar]
- (24).Birnbaumer L, and Zurita AR (2010) On the roles of Mg in the activation of G proteins. J. Recept. Signal Transduct. 30, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rodriguez FD, Bardaji E, and Traynor JR (1992) Differential Effects of Mg 2+ and Other Divalent Cations on the Binding of Tritiated Opioid Ligands. J. Neurochem. 59, 467–472. [DOI] [PubMed] [Google Scholar]
- (26).Williams LT, Mullikin D, and Lefkowitz RJ (1978) Magnesium dependence of agonist binding to adenylate cyclase-coupled hormone receptors. J. Biol. Chem. 253, 2984–2989. [PubMed] [Google Scholar]
- (27).Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Mizumura T, Suzuki S, and Shimada I (2014) Functional Dynamics of Deuterated β 2 -Adrenergic Receptor in Lipid Bilayers Revealed by NMR Spectroscopy. Angew. Chemie Int. Ed. 53, 13376–13379. [DOI] [PubMed] [Google Scholar]
- (28).Tsukamoto H, and Farrens DL (2013) A Constitutively Activating Mutation Alters the Dynamics and Energetics of a Key Conformational Change in a Ligand-free G Protein-coupled Receptor. J. Biol. Chem. 288, 28207–28216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jo S, Kim T, Iyer VG, and Im W (2008) CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. [DOI] [PubMed] [Google Scholar]
- (30).Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, and Im W (2016) CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Qi Y, Ingólfsson HI, Cheng X, Lee J, Marrink SJ, and Im W (2015) CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. [DOI] [PubMed] [Google Scholar]
- (32).Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, and Im W (2014) CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).de Jong DH, Singh G, Bennett WFD, Arnarez C, Wassenaar TA, Schäfer LV, Periole X, Tieleman DP, and Marrink SJ (2013) Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 9, 687–697. [DOI] [PubMed] [Google Scholar]
- (34).Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, and De Vries AH (2007) The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- (35).Lee S, Bhattacharya S, Tate CG, Grisshammer R, and Vaidehi N (2015) Structural dynamics and thermostabilization of neurotensin receptor 1. J. Phys. Chem. B 119, 4917–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lee S, Devamani T, Song HD, Sandhu M, Larsen A, Sommese R, Jain A, Vaidehi N, and Sivaramakrishnan S (2017) Distinct structural mechanisms determine substrate affinity and kinase activity of protein kinase Cα. J. Biol. Chem. 292, 16300–16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lee S, Nivedha AK, Tate CG, and Vaidehi N (2019) Dynamic Role of the G Protein in Stabilizing the Active State of the Adenosine A 2A Receptor. Structure 27, 703–712.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ma N, Lippert LG, Devamani T, Levy B, Lee S, Sandhu M, Vaidehi N, and Sivaramakrishnan S (2018) Bitopic Inhibition of ATP and Substrate Binding in Ser/Thr Kinases through a Conserved Allosteric Mechanism. Biochemistry 57, 6387–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huang J, and Mackerell AD (2013) CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Martyna GJ, Klein ML, and Tuckerman M (1992) Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643. [Google Scholar]
- (41).Parrinello M, and Rahman A (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190. [Google Scholar]
- (42).Koldsø H, Shorthouse D, Hélie J, and Sansom MSP (2014) Lipid Clustering Correlates with Membrane Curvature as Revealed by Molecular Simulations of Complex Lipid Bilayers. PLoS Comput. Biol. (Fradin C, Ed.) 10, e1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Humphrey W, Dalke A, and Schulten K (1996) VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (44).Guixà-González R, Rodriguez-Espigares I, Ramírez-Anguita JM, Carrió-Gaspar P, Martinez-Seara H, Giorgino T, and Selent J (2014) MEMBPLUGIN: studying membrane complexity in VMD. Bioinformatics 30, 1478–1480. [DOI] [PubMed] [Google Scholar]
- (45).Berendsen HJC, van der Spoel D, and van Drunen R (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56. [Google Scholar]
- (46).Lee S, Ghosh S, Jana S, Robertson N, Tate CG, and Vaidehi N (2020) How Do Branched Detergents Stabilize GPCRs in Micelles? Biochemistry 59, 2125–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Darden T, York D, and Pedersen L (1993) Particle mesh Ewald: An N ·log( N ) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- (48).Paramo T, East A, Garzón D, Ulmschneider MB, and Bond PJ (2014) Efficient Characterization of Protein Cavities within Molecular Simulation Trajectories: trj_cavity. J. Chem. Theory Comput. 10, 2151–2164. [DOI] [PubMed] [Google Scholar]
- (49).Michaud-Agrawal N, Denning EJ, Woolf TB, and Beckstein O (2011) MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shirts MR, and Pande VS (2005) Comparison of efficiency and bias of free energies computed by exponential averaging, the Bennett acceptance ratio, and thermodynamic integration. J. Chem. Phys. 122, 144107. [DOI] [PubMed] [Google Scholar]
- (51).Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGWW, and Tate CG (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Borea PA, Dalpiaz A, Varani K, Guerra L, and Gilli G (1995) Binding thermodynamics of adenosine A2a receptor ligands. Biochem. Pharmacol. 49, 461–469. [DOI] [PubMed] [Google Scholar]
- (53).White KL, Eddy MT, Gao Z-GG, Han GW, Lian T, Deary A, Patel N, Jacobson KA, Katritch V, and Stevens RC (2018) Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 26, 259–269.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Jaakola V-P, Lane JR, Lin JY, Katritch V, IJzerman AP, and Stevens RC (2010) Ligand Binding and Subtype Selectivity of the Human A 2A Adenosine Receptor. J. Biol. Chem. 285, 13032–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jacques F, and Yahi N (2015) Brain Lipids in Synaptic Function and Neurological Disease 1 st, pp 163–181. Elsevier. [Google Scholar]
- (56).Prasanna X, Jafurulla M, Sengupta D, and Chattopadhyay A (2016) The ganglioside GM1 interacts with the serotonin 1A receptor via the sphingolipid binding domain. Biochim. Biophys. Acta - Biomembr. 1858, 2818–2826. [DOI] [PubMed] [Google Scholar]
- (57).Tehan BG, Bortolato A, Blaney FE, Weir MP, and Mason JS (2014) Unifying Family A GPCR Theories of Activation. Pharmacol. Ther. 143, 51–60. [DOI] [PubMed] [Google Scholar]
- (58).Yao X, Parnot C, Deupi X, Ratnala VRP, Swaminath G, Farrens D, and Kobilka B (2006) Coupling ligand structure to specific conformational switches in the β2-adrenoceptor. Nat. Chem. Biol. 2, 417–422. [DOI] [PubMed] [Google Scholar]
- (59).Trzaskowski B, Latek D, Yuan S, Ghoshdastider U, Debinski A, and Filipek S (2012) Action of Molecular Switches in GPCRs - Theoretical and Experimental Studies. Curr. Med. Chem. 19, 1090–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EYT, Velasquez J, Kuhn P, and Stevens RC (2008) A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.