

Abstract
Chloride channels play an important role in regulating smooth muscle contraction and proliferation, and contribute to the enhanced constriction of pulmonary arteries (PAs) in pulmonary hypertension (PH). The intracellular Cl− concentration ([Cl−]i), tightly regulated by various Cl− transporters, determines the driving force for Cl− conductance, thereby the functional outcome of Cl− channel activation. This study characterizes for the first time the expression profile of Cl− transporters/exchangers in PA smooth muscle and provides the first evidence that the intracellular Cl− homeostasis is altered in PA smooth muscle cells (PASMCs) associated with chronic hypoxic PH (CHPH). Quantitative RT-PCR revealed that the endothelium-denuded intralobar PA of rats expressed Slc12a gene family-encoded Na-K-2Cl cotransporter 1 (NKCC1), K-Cl cotransporters (KCC) 1, 3, and 4, and Slc4a gene family-encoded Na+-independent and Na+-dependent Cl−/HCO3− exchangers. Exposure of rats to chronic hypoxia (10% O2, 3 wk) caused CHPH and selectively increased the expression of Cl−-accumulating NKCC1 and reduced the Cl−-extruding KCC4. The intracellular Cl− concentration ([Cl−]i) averaged at 45 mM and 47 mM in normoxic PASMCs as determined by fluorescent indicator MEQ and by gramicidin-perforated patch-clamp technique, respectively. The ([Cl−]i was increased by ∼10 mM in PASMCs of rats with CHPH. Future studies are warranted to further establish the hypothesis that the altered intracellular Cl− homeostasis contributes to the pathogenesis of CHPH.
Keywords: Cl− homeostasis, Cl− transporters, intracellular Cl− concentration, pulmonary hypertension
INTRODUCTION
Chloride is the most abundant anion in the biological system. It plays a crucial role in the regulation of many fundamental physiological processes, such as maintaining cell volume, pH, and ionic homeostasis (1, 2). Unlike Na+, K+, or Ca2+, the intracellular Cl− concentration ([Cl−]i) differs in different cell types and is the main determinant of the Cl− equilibrium potential (ECl) given that the plasma Cl− concentration remains relatively constant. In the majority of mammalian cells, the Cl− is not passively distributed across the plasma membrane but actively regulated such that the [Cl−]i is maintained above (i.e., ECl being more positive than the membrane potential Em: ECl > EM) or below the equilibrium (ECl < EM), which in turn determines the functional outcomes of the transmembrane Cl− movements via Cl− transporters and channels (3). In the vascular smooth muscle cells (VSMCs) that accumulate Cl− intracellularly (i.e., ECl > EM), activation of Cl− channels will lead to Cl− efflux and membrane depolarization followed by an increased intracellular Ca2+ and contraction (4).
The electroneutral cation-chloride cotransporters (CCCs) are the major regulators of the intracellular Cl− concentrations in most mammalian cells. CCCs are encoded by members of SLC12A gene family (SLC12A1-7) and divided into two subfamilies: the Na+-coupled Na+-K+-Cl− cotransporters (NKCC1, NKCC2) and Na+-Cl− cotransporter (NCC); and K+ gradient-driven K+-Cl− cotransporters (KCC1-4). NCC and NKCC2 are mainly expressed in kidneys whereas NKCC1 is ubiquitously expressed (5, 6). Although both NKCC1 and KCCs are bidirectional, NKCC1 utilizes the inward Na+ and Cl− chemical gradients to move 1Na+, 1K+, and 2Cl− into the cell whereas KCCs facilitate the efflux of 1K+ and 1Cl− using outward K+ gradient under resting, physiological conditions. Because Na+ and K+ are quickly returned to the extracellular or intracellular space, respectively, by the powerful Na+-K+-ATPase, the results of these cotransport activities are changes in the [Cl−]i (7). In addition to CCCs, the Cl−/HCO3− exchangers (also referred to as anion exchangers, AEs) encoded by members from SLC4A and SLC26A gene families may also mediate the Cl− transport across the plasma membrane although their major function is to regulate the intracellular and plasma pH (8–11).
It is well known that Cl− currents play a crucial role in the agonist-induced constriction of pulmonary arteries (PAs) (12, 13). Moreover, we and others have previously demonstrated that the TMEM16A-mediated Ca2+-activated Cl− channel in pulmonary arterial smooth muscle cells (PASMCs) is upregulated in animal models of pulmonary hypertension (PH) and contributes to the enhanced pulmonary vasoconstriction associated with PH (14, 15). Despite the physiological and pathophysiological importance of Cl− channels in the pulmonary vasculature, little is known about the plasma membrane Cl− transporters and intracellular Cl− homeostasis in PASMCs that determine the functional outcome of Cl− channel activations. No information is available on whether the [Cl−]i in PASMCs is altered in disease states such as chronic hypoxic pulmonary hypertension (CHPH), which occurs in many chronic lung diseases (16). Indeed, our current knowledge about transmembrane Cl− transports and the [Cl−]i in VSMCs has been acquired from the extensive studies carried out in the smooth muscles of various systemic arteries (4, 17–19). In this study, we performed a comprehensive survey on the mRNA expression of Cl− transporters/exchangers in endothelium-denuded intralobar PA and determined the [Cl−]i in PASMCs derived from control and chronic hypoxia-induced pulmonary hypertensive rats. Our results show that the [Cl−]i is increased in chronic hypoxic PASMCs accompanied by an increase in NKCC1 expression and a reduced KCC4 mRNA levels, suggesting that the altered Cl− homeostasis in PASMCs likely plays a role in the pathogenesis of PH.
METHODS
Ethical Approval
All animal procedures conform to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health and were approved by the Johns Hopkins University Animal Care and Use Committee.
Animal Model
The chronic hypoxic pulmonary hypertension (CHPH) was generated by placing male Wistar rats (150–200 g) in a hypoxic chamber and exposed to normobaric hypoxia (10 ± 0.5% O2) for 3–4 wk, as previously described and characterized in our laboratory (20). The O2 level inside the chamber was servo-controlled and continuously monitored using PRO:OX 110 gas analyzers (Reming Bioinstruments Co., Redfield, NY). Rat were exposed to room air for 10 min twice a week to clean the cages, and to replenish food and water supplies. Age-matched animals housed in room air were used as normoxic controls.
Cell Isolation
Rats were anesthetized with sodium pentobarbital (130 mg/kg ip). Lungs were removed quickly and immersed in ice-cold HEPES-buffered salt solution (HBSS) containing (in mM) 130 NaCl, 5 KCl, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, and 10 glucose (pH 7.2 with NaOH). The third generation of intralobar PAs (o.d.: 300–800 µm) were dissected out and cleaned of connective tissue. After removal of endothelium by rubbing the luminal surface with a cotton swab, the arteries were digested at 37°C for 20 min in 20 µmol-Ca2+ HBSS containing type I collagenase (1,750 U/mL), papain (9.6 U/mL), bovine serum albumin (2 mg/mL), and DTT (1 mmol/L). After washing with nominal Ca2+-free HBSS, the PASMCs were mechanically dispersed and kept in Ham’s F-12 medium (with 0.5% of fetal calf serum) overnight before use under 4% O2/5% CO2 and 21% O2-5% CO2 (37°C) for cells isolated from hypoxic and normoxic rats, respectively.
Conventional RT-PCR
Tissues (brain, kidney, and de-endothelialized intralobar PAs) were harvested, snap frozen in liquid nitrogen, and homogenized using mortar and pestle. Total RNA was extracted using RNeasy Mini Kit from Qiagen including DNase I treatment and quantified with Nanodrop. The quality of RNA samples was checked by using 260/280 and 260/230 ratios. cDNA was synthesized with SuperScript III First-Strand Synthesis from Invitrogen. Primers specific to the Cl− transporters were used (Table 1). Hot-start Taq DNA polymerase from Denville Scientific was used for PCR. The reaction was run at 94°C for 2 min, then for 35 cycles at 94°C for 30 s, 60°C for 45 s, and 72°C for 1 min, followed by the final extension at 72°C for 10 min. PCR products were analyzed by electrophoresis with 1.8% agarose gel and visualized by ethidium bromide staining using a Kodak Gel logic 200 imaging system. Brain and kidney samples were used as positive controls.
Table 1.
RT-PCR primer sequences
| Gene | Accession No. | Primers | Sequence (5′–3′) | Predicted Size, bp |
|---|---|---|---|---|
| Slc12a1 | NM_001270617.1 | Forward | AGGCACGATCGATGTTTGGT | 457 |
| Reverse | CGCCACTGGAAGGCTCAATA | |||
| Slc12a2 | NM_031798.1 | Forward | CTGGTATTCTAGCTGGGGCG | 781 |
| Reverse | CCCCAGTTCACATCTGGCTT | |||
| Slc12a3 | NM_019345.3 | Forward | CAGCACCTTGTACCTACGCA | 496 |
| Reverse | AGGTCCCAGACTCCGAGAAA | |||
| Slc12a4 | NM_019229.2 | Forward | AAGACTCGGATGGACAGGGT | 486 |
| Reverse | ATTCTGGTCCCAAAGAGCGG | |||
| Slc12a5 | NM_134363.1 | Forward | GGACCCCCGCATACAAAGAA | 570 |
| Reverse | CCTCCAGACCTTGTGGTGAC | |||
| Slc12a6 | NM_001109630.1 | Forward | TCCAGAAACCACCACCAAGAT | 689 |
| Reverse | ACATTGTACAGCAGCAGCAGAT | |||
| Slc12a7 | NM_001013144.2 | Forward | AGCCATGCCCACGAACTTTA | 509 |
| Reverse | TGGCTGTCAGCATTGTACAGG | |||
| Slc4a1 | NM_012651.2 | Forward | GGCCCCTTCTGAACCTCTTC | 339 |
| Reverse | ACGCGGAACACTCTTTCTGT | |||
| Slc4a2 | NM_017048.2 | Forward | CCCAGGGTCCCTACTGATGA | 700 |
| Reverse | GCTCACGCTCTCTATCCACC | |||
| Slc4a3 | NM_017049.1 | Forward | AGAAGATCCCTGAGGACGCT | 679 |
| Reverse | TTCTCTCCTAGCAGACCCCC | |||
| Slc4a8 | NM_199497.2 | Forward | GGGACCAGCACGATTCTCAA | 323 |
| Reverse | TTCCATTCGGCTTCTTCCCC |
Real-Time Quantitative PCR
Primer pairs were designed preferably to span exon-exon junction where possible with the product size between 100 and 150 bp (Table 2). Primers were then validated with quantitative RT-PCR, using either rat brain or kidney as the positive control. The PCR protocol consisted of initial enzyme activation at 94°C for 2 min, followed by 40 cycles at 94°C for 30 s, 60°C for 45 s, and 72°C for 1 min. Using the same protocol, we generated standard curves from serial dilutions of purified PCR products with known copy numbers determined by Nanodrop with absorbance at 260 nm. The absolute copy number of mRNA of interest was calculated by interpolation of the standard curve with the threshold cycle value of each sample. To confirm the specificity of PCR products, we obtained a melting curve at the end of each run. Standard gel electrophoresis also was performed to ensure the end product generated a single band with the predicted size (100–150 bases). Cyclophilin A was used as the housekeeping gene to normalize the data. SYBR Green detection was used to perform real-time quantitative PCR with BioRad iQCycler.
Table 2.
Real-time PCR primer sequences
| Gene | Accession No. | Primers | Sequence (5′–3′) | Predicted Size, bp |
|---|---|---|---|---|
| Slc12a1 | NM_001270617.1 | Forward | AATGCAGTGGCTGTCGCTAT | 131 |
| Reverse | ATCACCACCGTGATGGAACC | |||
| Slc12a2 | NM_031798.1 | Forward | GGAGCGATTCTCTGCTGCAT | 122 |
| Reverse | CCCCAGTTCACATCTGGCTT | |||
| Slc12a3 | NM_019345.3 | Forward | CAGCACCTTGTACCTACGCA | 138 |
| Reverse | TTGAGGAACGAGTGCAGGTC | |||
| Slc12a4 | NM_019229.2 | Forward | TCGGATGGACAGGGTAACCA | 150 |
| Reverse | AAGCTTGCCCAGGAGAGATG | |||
| Slc12a5 | NM_134363.1 | Forward | AAGGACCCCCGCATACAAAG | 146 |
| Reverse | GACAGAGCCCACAATGGTCA | |||
| Slc12a6 | NM_001109630.1 | Forward | TGCCAGAAACAAGCCGTAGT | 134 |
| Reverse | GTTCTGATTCGGGTCCTCGG | |||
| Slc12a7 | NM_001013144.2 | Forward | ATGCCCACGAACTTTACGGT | 135 |
| Reverse | GTTTCCATCTCCCGGCGTAG | |||
| Slc4a1 | NM_012651.2 | Forward | GCGCCGGTATCCTTACTACC | 130 |
| Reverse | CTTTTCTCCCAGGAGACCGC | |||
| Slc4a2 | NM_017048.2 | Forward | AAACACAGCCACCCAAGTGA | 137 |
| Reverse | CCGATGAGAGGCTCAGTGAC | |||
| Slc4a3 | NM_017049.1 | Forward | TTGGGGAATCACCACCCAAC | 122 |
| Reverse | AGGGGCTTCTCCTTTGCATC | |||
| Slc4a8 | NM_199497.2 | Forward | AAGCCAACGAGGGATGATCG | 148 |
| Reverse | TCCGGTTAATGATGACGGCG |
Gramicidin-Perforated Patch Recording and Calculation of [Cl−]i
Gramicidin-perforated patch clamp technique was used to record the caffeine-induced Ca2+-activated Cl− current (ICl.Ca) with undisturbed [Cl−]i in PASMCs. Caffeine-induced Ca2+ release was simultaneously monitored using fluo-3 loaded into the cells in the form of acetoxymethyl ester. Membrane currents elicited by four consecutive voltage ramps (800 ms each with 1-s interval) from −45 to +45 mV during Ca2+ release were sampled at 10 kHz and filtered at 5 kHz using an Axopatch 200B amplifier driven by pClamp 10 software (Molecular Devices Co., Sunnyvale, CA). Contamination by other currents was minimized by replacing K+ ions with Cs+ in both bath and pipette solutions, and by adding tetraethylammonium (TEA) chloride and nifedipine (2 µM) in bath solutions. Bath solution contained (in mM): NaCl 135, CsCl 5.4, TEA-Cl 5, MgCl2 1, CaCl2 1, NaHPO4 0.33, HEPES 10, and glucose 10 (pH 7.35 adjusted with NaOH). Pipette solution contained (in mM): Cs-methane-sulfonate 125, CsCl 20, and HEPES 20 (pH 7.2 adjusted with CsOH). Freshly prepared gramicidin was added to the pipette solution (100 µg/mL) before experiments. Pipette resistance ranged from 2 to 3 MΩ when filled with the pipette solution with gramicidin. After seal formation, the access resistance was continuously monitored, and all recordings were performed when it was between 20 and 40 MΩ. All experiments were performed at room temperature.
The reversal potential of caffeine-induced ICl.Ca (ECl) was measured as the voltage at which the current equals to zero, and corrected offline for the liquid junction potential (10.8 mV). The liquid junction potential was estimated using Clampex’s Junction Potential Calculator and expressed in the direction of the bath with respect to the pipette. The corrected ECl was then used to calculate the [Cl−]i according to Nernst equation: [Cl−]i = [Cl−]o × eEClF/RT, where [Cl−]o was the Cl− concentration in bath solution, and F, R, and T had their conventional meanings.
MEQ Epifluorescence Measurement
MEQ (Invitrogen) was reduced to a membrane-permeable form, 6-methoxy-N-ethyl-1,2-dihydroquinoline (diH-MEQ), using 12% sodium borohydride under constant N2 flow according to manufacturer’s instruction (21). The diH-MEQ was prepared freshly on each experiment day and loaded into cells by incubating the cells with 100 µM dye dissolved in Ca2+-containing HBSS. After 30 min’ incubation at room temperature, the extracellular dye was washed out and the cells were perfused with Tyrode solution for 30–45 min before experiments to allow for the conversion of intracellular diH-MEQ into membrane-impermeable MEQ by cytoplasmic oxidases. The Tyrode contained (mM) NaCl 135, KCl 5.4, CaCl2 1, MgCl2 1, HNaPO4 0.33, HEPES 10, and glucose 10 (pH 7.35 adjusted with NaOH). Intracellular MEQ was excited at 360 nm using UV light emitted from a xenon short arc lamp, directed through a series of neutral density filters (OD: 5) and reflected into a ×40 long-working distance objective. The emitted fluorescence filtered by an interference filter centered at 455 (±50) nm was detected by a photomultiplier (Photon Technology International) and digitized by the Digidata 1440A driven by pClamp 10 software (Molecular Devices).
Intracellular Calibration of MEQ
The fluorescence of all quinolinium-derived Cl− indicators including MEQ is quenched collisionally by Cl−, which can be described by the Stern–Volmer equation: F0/F = 1 + Kq[Cl−], where F0 is the total quenchable fluorescence, F the intensity of signal at a given Cl− concentration ([Cl−]) and Kq the Stern–Volmer constant (in M−1) (22). To quantify the [Cl−]i in PASMCs, the MEQ signal was calibrated against the intracellular Cl− set equal to the external Cl− concentrations by using a high K+ bath solution containing the K/H exchange ionophore nigericin (10 µM) and the Cl/OH exchange ionophore tributyltin (12.5–25 µM) as described by Chao et al. (23). The high K+ buffer contained (mM) KNO3 or KCl 130, NaNO3 10, MgSO4 1, Ca-acetate 0.1, HEPES 10, and glucose 10 (pH adjusted to 7.2 with KOH). The final Cl− concentration in the buffer was set to 0, 20, 40, and 80 mM by appropriately mixing the KCl- and KNO3-containing high K+ solutions. Due to the narrow time windows between a reliable equilibrium of intra-extracellular Cl− and loss of cell membrane integrity reflected by MEQ leakage induced by ionophores in PASMCs, the MEQ signal was only measured at 0 mM Cl− that gave the maximal fluorescent intensity (Fmax), and at an additional Cl− concentration in each cell during calibration. The minimal fluorescence signal (Fmin) was determined at the end of each experiment by switching to a solution containing 150 mM of KSCN and 25 µM of valinomycin to completely quench the intracellular MEQ. The difference between Fmax and Fmin is defined as F0.The ratio of F0 over the fluorescence measured at other [Cl−]i (FCl) was then plotted against the Cl− concentrations and fitted to the Stern–Volmer equation to determine the intracellular Kq.
Data Analyses and Statistics
Data were analyzed with Clampfit 10 and Sigmaplot softwares. Group data were expressed as means ± SD. The differences between two groups were assessed by unpaired Student’s t test. The differences between multiple groups were assessed with one-way ANOVA followed by Holm–Sidak test for post hoc pairwise comparisons.
RESULTS
Expression Profile of CCC-Encoding Slc12a Genes in PA Smooth Muscles of Normoxic and Hypoxic Rats
Slc12a gene family has nine members and seven of which encode for cation-chloride cotransporters: Slc12a1 encoding for NKCC2, Slc12a2 for NKCC1, Slc12a3 for NCC, Slc12a4–7 for KCC1-4 (6). Using the RT-PCR technique and cDNA synthesized from brain and kidney tissues as the positive controls, we noted a strong expression of Slc12a2, 4, 6, and 7; very low level of expression of Slc12a5 and no expression of Slc12a1 and Slc12a3 in the endothelium-denuded intralobal PAs of normoxic rats. Slc12a5 was most abundantly expressed in the brain, and Slc12a1 and Slc12a3 were mainly expressed in the kidneys (Fig. 1A), consistent with previous reports (5). To examine the relative expression levels of CCC transcripts in PA smooth muscle and to determine whether they are altered in rats exposed to chronic hypoxia, quantitative real-time PCR assay was employed. As shown in Fig. 1, B and C, intralobal PAs expressed a substantial amount of Slc12a2/NKCC1, Slc12a4/KCC1, Slc12a6/KCC3, and Slc12a7/KCC4. The mRNA levels of Slc12a1/NKCC2, Scl12a3/NCC, and Slc12a5/KCC2 were extremely low. Among the three main KCC isoforms expressed in PA smooth muscle, the KCC4 transcript was lower than that of KCC1 and KCC3. Chronic hypoxia doubled the Slc12a2/NKCC1 mRNA level and significantly reduced the Slc12a7/KCC4 expression by ∼60% in PA smooth muscle.
Figure 1.

Expression profile of Slc12a genes that encode for cation-chloride cotransporters (CCCs). A: gel image showing the RT-PCR results for Slc12a1-7 obtained in a normoxic pulmonary artery (PA) (Pa). PCR amplicons obtained from brain (B) and/or kidney (K) cDNA are given as positive controls. B and C: relative expression levels of Cl−-accumulating cotransporter genes and Cl−-extruding cotransporter genes, respectively, in normoxic (Nx) and chronic hypoxic (CH) PAs determined by quantitative real-time PCR assay (n = 6 rats per group). The error bars show the values of SD. *P < 0.01 vs. normoxic PAs (unpaired t test). †P < 0.05 vs. normoxic Slc12a4/KCC1 and Slc12a6/KCC3 (one-way ANOVA followed by Holm–Sidak test for pairwise comparisons).
Expression Profile of Cl−/HCO3− Exchanger-Encoding Slc4a Genes in PA Smooth Muscle of Normoxic and Chronic Hypoxic Rats
Previous studies have documented the functional activity of Cl−/HCO3− exchangers and the expression of AE2 in the smooth muscle of both systemic (8, 24) and pulmonary arteries (25, 26). Here, we quantified the mRNA expression of Slc4a genes that encode for the Na+-independent AE1-3 (Slc4a1-3) and Na+-dependent Cl−/HCO3− exchanger, NDCBE (Slc4a8), in the endothelium-denuded PAs and evaluated whether their expression levels were altered in rats with CHPH. We found that the intralobal PAs expressed most abundantly Slc4a2/AE2 transcript, followed by Slc4a3/AE3, then Slc4a8/NDCBE mRNA. The expression level of Slc4a1/AE1 was very low. Exposure of rats to chronic hypoxia did not alter the expression of these Slc4a genes (Fig. 2).
Figure 2.

Expression profile of Slc4a genes that encode for Cl−/HCO3− exchangers. A: examples of gel images showing the RT-PCR results for Slc4a1-3 and Slc4a8 detected in endothelium-denuded pulmonary arteries (Pas) (Pa). Results obtained from brain (B) and kidney (K) cDNA were used as positive controls. B: relative expression levels of Slc4a1, 2, 3, and 8 in PAs derived from normoxic (Nx) and chronic hypoxic (CH) rats determined by quantitative PCR assay (n = 6 rats per group). The error bars give the values of SD.
Measurement of [Cl−]i in Normoxic and Chronic Hypoxic PASMCs by Fluorescent Cl− Indicator
To evaluate the functional impact of altered NKCC1 and KCC4 expression on the intracellular Cl− homeostasis of PA smooth muscles derived from rats with CHPH, we measured the [Cl−]i in PASMCs isolated from normoxic and chronic hypoxic rats using the epifluorescent method with MEQ as the Cl− indicator. Since the quenching of MEQ fluorescence by Cl− does not cause any spectral shift, which renders a ratiometric measurement impossible, the amplitude of total quenchable fluorescence F0 measured from one wavelength was determined in every experiment. An example is shown in Fig. 3A. After a 3-min recording in a cell loaded with MEQ and perfused with Tyrode, the bath solution was switched to a Cl−-free high K+ buffer containing nigericin and tribultyltin. The fluorescence signal raised as the intracellular Cl− was gradually depleted. When the signal reached the plateau, KSCN and valinomycin were applied to quench completely the MEQ fluorescence. The F0 was then determined as the difference between the maximal and minimal MEQ fluorescence. In this study, only the relaxing cells showing adequate MEQ loading and having stable signal for at least 5–10 min were used for the measurement. To analyze quantitatively the MEQ signal measured from single PASMCs, the intracellular calibration of MEQ fluorescence was performed as depicted in methods. The Stern–Volmer plot of the calibration data obtained in five cells for each [Cl−]i (Fig. 3B) yielded a Kq of 27 M−1, corresponding to a 50% MEQ quenching by 37 mM of intracellular Cl− in the PASMCs. Using this Kq value and the values of F0 and F measured from the same cell, the [Cl−]i was calculated. The mean [Cl−]i was found to be 45.0 ± 14.4 mM in normoxic PASMCs (n = 30). A significant increase in the [Cl−]i was detected in PASMCs of chronic hypoxic rats (56.1 ± 16.5 mM, n = 26, Fig. 3C).
Figure 3.

MEQ epifluorescent measurement of [Cl−]i in normoxic and hypoxic pulmonary arterial smooth muscle cells (PASMCs). A: representative recording of MEQ fluorescence signal obtained from a normoxic PASMC initially perfused with Tyrode solution. The bath solution was then switched to a Cl−-free high K+ buffer containing nigericin (10 µM) and tributyltin (25 µM) followed by a KSCN solution containing 25 µM valinomycin to quench completely the intracellular MEQ signal. B: Plot of F0/FCl against the [Cl−]i set equal to the external Cl− concentrations. FCl was measured at 20, 40, and 80 mM of [Cl−]i (n = 5). The solid line represents a fit of data points to the Stern–Volmer equation (R = 0.99). C: box-and-whisker plot for the values of [Cl−]i obtained in normoxic (Nx, n = 30 cells) and chronic hypoxic (CH) PASMCs (n = 26 cells). Horizontal lines of boxes represent 25th percentile, median, and 75th percentile. Whiskers represent 10th/90th percentile. The gray lines inside the box represent the mean values. *P < 0.01 by unpaired t test.
[Cl−]i in Normoxic and Chronic Hypoxic PASMCs Determined by Gramicidin-Perforated Patch Recording
To confirm the findings obtained with the fluorescent Cl indicator MEQ, the [Cl−]i in PASMCs was measured using a different technique, the gramicidin-perforated patch-clamp technique. Gramicidin is an uncharged polypeptide antibiotic that incorporates in cell membrane to form pores permeable only to monovalent cations (27). Thus, the whole cell patch clamp recording can be performed without altering the [Cl−]i by the pipette solution. Here, we made use of this technique to measure the ECl of caffeine-induced Ca2+-activated Cl− current (ICl.Ca) in PASMCs, which can then be used to calculate the [Cl−]i with Nernst equation. Figure 4A shows the typical recordings and data analyses obtained from a normoxic PASMC. The cell was held at −45 mV. The voltage clamp protocol consisting of four identical voltage ramps from −45 to +45 mV elicited small outward currents in the absence of increase in intracellular Ca2+ (Fig. 4Aa). Rapid application of 10 mM caffeine evoked a large Ca2+ transient accompanied by an inward current. The voltage ramps applied during caffeine-induced Ca2+ release elicited significant outward currents at depolarizing voltages. Both inward and outward currents diminished as the Ca2+ signal declined (Fig. 4Ab). The subsequent application of caffeine in the presence of Cl− channel blocker niflumic acid (100 µM) induced a Ca2+ release of similar amplitude while the current was significantly suppressed (Fig. 4Ac). Digital subtraction of membrane current recorded before caffeine from that obtained during caffeine application gave the caffeine-induced current (ICaff); and the niflumic acid-sensitive ICaff (ICaff-NFA, Fig. 4Ba, lower trace), which has been shown to be ICl.Ca (14), was obtained by subtracting the caffeine-induced current recorded in the presence of niflumic acid from that recorded in the absence of the blocker. A plot of the I-V relationship of ICaff and ICaff.NFA elicited by the first voltage ramp from the normoxic PASMC shown in Fig. 4A is presented in Fig. 4B, top. The ICaff and ICaff.NFA reverse at nearly the same voltage, suggesting that the caffeine-induced currents recorded under our experimental conditions are predominantly ICl.Ca. The same experiments were performed in PASMCs of chronically hypoxic rats. The I-V relationship of ICaff and ICaff.NFA elicited by the first voltage ramp from a typical chronic hypoxic PASMC is plotted in Fig. 4B, bottom. The reversal potentials of ICaff and ICaff.NFA are also very close in the hypoxic cell although they are more positive compared with those obtained in the normoxic cell. In both normoxic and hypoxic PASMCs, however, the ICl.Ca reverses at voltages very different from the predicted ECl (−51 mV) based on the bath and pipette solutions used in this study, strongly indicating that the native [Cl−]i was not altered or clamped in these experiments.
Figure 4.
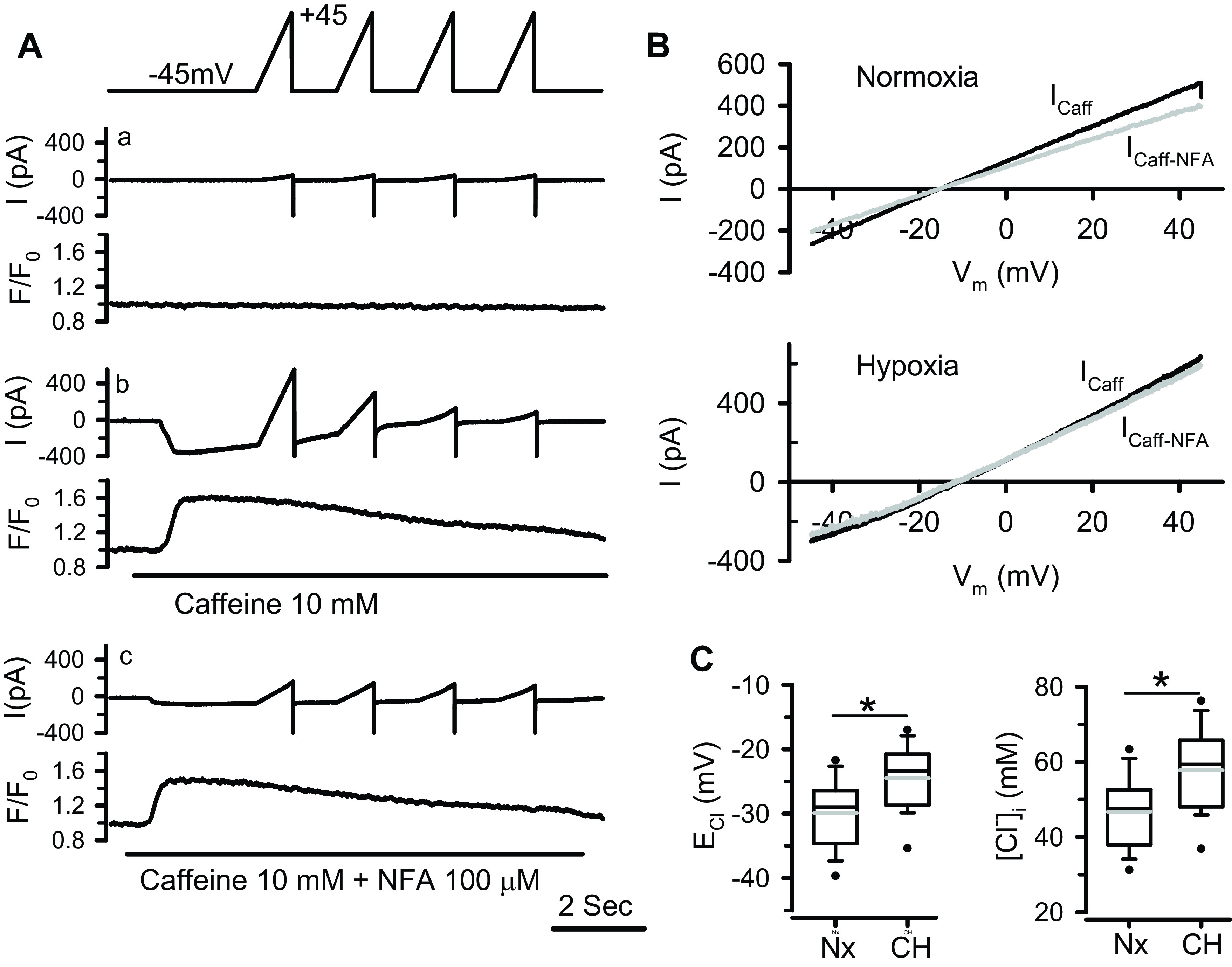
Determination of [Cl−]i by measuring the ICl.Ca reversal potential (ECl) in normoxic and chronic hypoxic (CH) pulmonary arterial smooth muscle cells (PASMCs) with gramicidin-perforated patch clamp technique. A: simultaneously recorded membrane currents and Ca2+ signals from a normoxic cell under control conditions (without drug application, a), during rapid application of 10 mM caffeine (b), or combined 10 mM caffeine and 100 µM niflumic acid (NFA, c). Membrane currents were elicited by four consecutive voltage ramps from −45 to +45 mV shown on the top. B (top): shows the I-V relationship of caffeine-induced current (Icaff) obtained by subtracting the background current (Aa) from the current recorded during caffeine-induced Ca2+ transient (Ab), and NFA-sensitive caffeine-induced current (Icaff-NFA) obtained by subtracting the current recorded during combined caffeine and NFA application (Ac) from that recorded during caffeine application (Ab). The Icaff and Icaff-NFA elicited by the first voltage ramp were plotted. B (bottom): shows the I-V relationship of Icaff and Icaff-NFA elicited by the first voltage ramp from a chronic hypoxic PASMC. C, left: box-and-whisker plot for the values of ECl (the reversal potential of Icaff-NFA) measured from normoxic (Nx, n = 33 cells) and hypoxic (CH, n = 36 cells) PASMCs. The values were corrected for the junction potential (10.8 mV). C, right: box-and-whisker plot for the values of [Cl−]i derived from the measured ECl for normoxic and hypoxic groups. Horizontal lines of boxes represent 25th percentile, median, and 75th percentile. Whiskers represent 10th/90th percentile. The gray lines inside the box represent the mean values. *P < 0.001 (unpaired t test).
The gramicidin-perforated patch recording on 33 normoxic and 36 hypoxic PASMCs revealed that the caffeine-induced ICl.Ca reversed at significantly more positive voltages in hypoxic cells compared with normoxic group (−24.5 ± 5.0 vs. −29.9 ± 5.3 mV after correction for the junction potential, P < 0.001, Fig. 4C, left). Given the same Cl− concentration in the bath solution used for these experiments, the more positive ECl is translated to a significant increase in the [Cl−]i in hypoxic PASMCs (57.8 ± 10.9 vs. 46.7 ± 9.4 mM, P < 0.001, Fig. 4C, right). These values are closely similar to those obtained with fluorescent Cl− indicator MEQ shown in Fig. 3. Moreover, we found that the [Cl−]i exhibited a normal distribution in both normoxic and hypoxic PASMCs although it varied considerably among individual PASMCs (Fig. 5).
Figure 5.

Normal distribution of [Cl−]i in normoxic and hypoxic pulmonary arterial smooth muscle cells (PASMCs) measured with gramicidin-perforated patch clamp technique. The solid curves represent the fit of data to a Gaussian function. The curves peak at 45.3 and 58.5 mM for normoxic (n = 33 cells) and hypoxic (n = 36 cells) groups, respectively.
DISCUSSION
To the best of our knowledge, this is the first report on the gene expression profile of plasma membrane Cl− transporters in the pulmonary arterial smooth muscle and on the assessment of [Cl−]i in single living PASMCs. This study also provides the first evidence that the intracellular Cl− homeostasis in PASMCs is altered in chronic hypoxic pulmonary hypertension.
Our results showed that the rat intralobar PA smooth muscle mainly expresses the Cl−-accumulating NKCC1/Slc12a2 (but not NKCC2/Slc12a1 or NCC/Slc12a3) and Cl−-extruding KCC1/Slc12a4, KCC3/Slc12a6, and KCC4/Slc12a7. The expression profile of KCC isoforms we observed in PA smooth muscles is very similar to that reported in rat aortic smooth muscles, i.e., the mRNA level of KCC4 is ∼50% of that for KCC1 or KCC3, and a very low level of KCC2 transcript can be detected (19, 28). Our study further showed that chronic exposure of rats to hypoxia caused an increase in the NKCC1 and a decrease in the KCC4 mRNA levels in the PA smooth muscle associated with pulmonary hypertension. Increased NKCC1 expression and/or activities have also been observed in the aorta and resistance arteries of spontaneously hypertensive rats (29, 30) and deoxycorticosterone acetate/salt-(31) or angiotensin II-induced hypertension (32). Whether the systemic hypertension altered the KCC expression had not been examined in these studies. The upregulation of NKCC1 in the aortas of spontaneous hypertensive and angiotensin II-induced hypertensive animals has been shown to result from the hypomethylation of Slc12a2 promoter during the development of hypertension (30, 32–34). Whether the same epigenetic modification underlies the increased NKCC1 expression in PA smooth muscle of CHPH rats remain to be elucidated. The upregulation of vascular smooth muscle NKCC1 expression in various hypertensive models raises the question of whether the epigenetically upregulated NKCC1 expression is the cause or result of hypertension. Jiang et al. addressed this issue by using a rat model of descending aortic coarctation that produced both hypertensive and hypotensive segments of the aorta (proximal and distal to the coarctation, respectively) in the same animal. They reported that the NKCC1 activity of the hypertensive segment was increased along with increased NKCC1 mRNA levels compared with hypotensive segments in the same animals and corresponding aortic segments of sham-operated rats (35). This study clearly demonstrated that the NKCC1 expression in vascular smooth muscle is regulated by blood pressure. Thus, the increased [Cl−]i and altered NKCC1 and KCC4 expressions found in PA smooth muscle associated with CHPH in this study may reflect the PH-induced remodeling of intracellular Cl− regulation in PASMCs. In addition, it has been shown that hypoxia enhanced the NKCC1 cotransport activity in cerebral microvascular endothelial cells (36, 37) and increased NKCC1 mRNA expression in brain tissue (38). The hypoxia-inducible factor-1 (HIF-1), that is expressed under hypoxic condition, upregulates the transcription of Slc12a2/NKCC1 in hippocampal neurons (39), but decreases NKCC1 expression in intestinal epithelial cells (40). There is currently no report about the influence of hypoxia or HIF-1 on the expression and function of NKCC1 in VSMCs. It remains, however, possible that hypoxia contributes to the altered Cl− homeostasis and transporter expression in PASMCs observed in this study. Future investigations using a different PH model, such as monocrotaline-induced PH model, will help to clarify this important issue.
The Cl−/HCO3− anion exchangers are one class of bicarbonate transporters encoded by members of two distinct gene families: Slc4a and Slc26a (10). Here, we focused on the Slc4a gene family members since the Slc26a genes that encode for the Cl−/anions exchangers are mainly expressed in the epithelial cells of kidneys, digestive tracts and airways, and in the heart whereas AE2 and AE3 have been found in the smooth muscles (10, 41, 42). Four of ten members of the Slc4a gene family encode for Cl−/HCO3− exchangers (42). A previous study has reported the expression of AE2 in the rat PA smooth muscle (26). Here, our quantitative PCR results revealed that the rat PA smooth muscle also expresses Slc4a3/AE3 and Slc4a8/NDCBE in addition to the most abundant Slc4a2/AE2. These findings are consistent with previous functional studies showing the presence of both Na+-independent and Na+-dependent Cl−/HCO3− exchange activities in PASMCs (11, 25, 43). Under physiological ionic conditions, AE2 and AE3 transport one Cl− into the cells in exchange for one HCO3− whereas NDCBE moves Cl− out from the cytosol and brings in 1 Na+ and 2 HCO3− electroneutrally (9, 10). It has been shown that the mRNA level of Cl−-accumulating AE2 is increased in the small cerebral arteries of stroke-prone spontaneously hypertensive rats (44). We did not detect any changes in the expression of Slc4a genes-encoded Cl−/HCO3− exchangers in the PA smooth muscle of rats with chronic hypoxic PH in this study. A previous study has also shown that the AE2 mRNA levels were unaltered in the PAs of rats exposed to hypoxia for upto 7 days (26).
Previous measurements of [Cl−]i in vascular SMCs have been almost exclusively performed in systemic arterial smooth muscles, except for one early study that has estimated the [Cl−]i in PA smooth muscle to be 51 mM by measuring the total amount of 36Cl loaded into the rabbit main PA tissue at steady state (45). Here, we provide the first assessment of [Cl−]i in single living PASMCs. Currently available approaches that provide relatively direct measurements of [Cl−]i in single living cells are double-barreled Cl−- selective microelectrodes, gramicidin-perforated patch clamp recording, and Cl−-sensitive fluorescent indicators. The technically demanding double-barreled microelectrode technique allows simultaneous monitoring of intracellular Cl− activities and Em. Due to the imperfect selectivity of Cl−-sensing resin, however, the accuracy of [Cl−]i measurements is compromised by the intracellular interference from other ions that needs to be estimated and corrected for (3). The gramicidin-perforated patch clamp technique has been successfully used to determine the [Cl−]i in neurons and cardiac myocytes by measuring the ECl of a specific Cl− conductance (27). The accuracy of this method relies on the ability to isolate a specific Cl− conductance and may be limited by the need to estimate the liquid junction potential under a particular experimental condition. To the best of our knowledge, the present study is the first application of this technique to SMCs, making use of a well-isolated caffeine-induced ICl.Ca. Fluorescent Cl− indicators provide a noninvasive approach to monitor the changes in the [Cl−]i. Available fluorescent Cl− indicators are a class of N-substituted quinoline compounds, the fluorescence of which can be quenched by halide ions via a diffusion-limited collisional quenching mechanism (22). Due to the lack of spectral shift upon quenching and the presence of intracellular non-Cl− quenchers, these indicators need to be calibrated intracellularly. The intracellular calibration of MEQ in rat PASMCs in this study has yielded a KSV of 27 M−1, which is between the 19 M−1 found in fibroblasts (21) and the 32 M−1 (46) and 52 M−1 (47) reported in the brain slice. Because of the intrinsic limitations with each available technique, we used two different approaches to measure the [Cl−]i in rat intralobal PASMCs and obtained closely similar readings, i.e., around 45 and 47 mM in normoxic PASMCs by fluorescent Cl− indicator MEQ and gramicidin-perforated patch clamp technique, respectively. These values appear to be slightly greater than the [Cl−]i reported for aortic (24–38 mM) (48, 49), femoral arterial (31–44 mM) (50, 51), human umbilical arterial (34 mM), and placental arterial SMCs (35 mM) (52). This difference could be due to intrinsic differences in Cl− regulation in pulmonary and systemic arterial smooth muscles. It could also be due to the different methods used and/or different experimental conditions. For instance, most of the aforementioned [Cl−]i measurements in various systemic VSMCs have been obtained using Cl−-selective microelectrodes.
Another important finding in this study is that the [Cl−]i was increased in the PASMCs of chronically hypoxic rats, suggesting a dynamic regulation of [Cl−]i in pulmonary arterial myocytes which may be altered under pathological conditions. Three Cl−-accumulating mechanisms, the Na+-K+-Cl− cotransporter, the Cl−/HCO3− exchanger, and pump III (3), responsible for maintaining an [Cl−]i above equilibrium in smooth muscle have been documented. Pump III has been described in the systemic resistance arteries by Harper’s group as a small acetazolamide-sensitive inward Cl− transport (52, 53). Its molecular basis and transport mechanism remain, however, undefined. This study therefore focused on Cl− cotransporters and exchangers, and showed that chronic hypoxia selectively increased the NKCC1 and decreased the KCC4 transcript levels in the PA smooth muscle, both of which favor the net influx of Cl− and increase in the [Cl−]i. The role of NKCC1 in regulating the [Cl−]i of various cell types including vascular smooth muscle cells is well recognized (3, 7). It has been shown that inhibition of NKCC1 by loop diuretics bumetanide reduced [Cl−]i in the smooth muscle of rat femoral arteries (50, 51, 54) and aorta (55, 56). Moreover, noradrenaline that stimulates NKCC1 increased the [Cl−]i and this increase was largely suppressed by bumetanide (51). With respect to KCCs, the KCC2 isoform is a well-established key player in maintaining a low [Cl−]i required for synaptic inhibitions in mature neurons, whereas the other three KCC isoforms that coexpress in many cell types including PASMCs shown in this study are mainly known for their role in the cell volume regulation (57). Due to the lack of isoform-specific inhibitors, the function of individual KCC isoforms in the VSMCs remains ill-defined. KCC3 knockout led to a slight but significant increase in the [Cl−]i in saphenous arterial smooth muscles (58). The effect of inactivating KCC1 or KCC4 on intracellular Cl− homeostasis has not been reported in smooth muscle.
It has been shown that the NKCC1 activity in cultured VSMCs and intact arterial smooth muscle is enhanced by vasoconstrictors such as angiotensin II, phenylephrine, and endothelin-1, by hypertonic shrinkage and by raising intracellular Ca2+ with calcium ionophores (59–61). Inhibition of NKCC1 by bumetanide suppresses the myogenic tone of mesenteric and renal afferent arteries (62, 63), and attenuates the contractile response of aorta and various resistance arteries to an array of vasoconstrictors (e.g., phenylephrine, endothelin-1, serotonin, angiotensin II, and UTP) (34, 63–65). These effects of bumetanide are absent in NKCC1-KO mice (63), indicating that the NKCC1 contributes to the regulation of vascular tone. It is generally believed that the cotransporter functions to maintain a high [Cl−]i, thereby an appropriate driving force for Cl− efflux via agonist-activated Cl− channels to depolarize the VSMCs, which in turn activates the voltage-gated Ca2+ channels and contraction (3, 63). Pulmonary vascular tone and PASMCs are constantly under the influence of various circulating and locally released vasoconstrictors and vasodilators. Increased plasma levels and pulmonary local release of endothelin-1, angiotensin II, and serotonin, as well as overexpression of their respective receptors in the lung have been reported in patients with PH and in animal models of CHPH (66–70). Moreover, the [Ca2+]i is elevated in PASMCs associated with CHPH (20). These changes due to pulmonary hypertension are expected to increase the NKCC1 activity in PASMCs derived from rats with CHPH, enhancing intracellular accumulation of Cl−. The increased expression of NKCC1 observed in this study may further amplify these effects, leading to an increased [Cl−]i in PASMCs. In support of this view, greater bumetanide-sensitive contractile responses of mesenteric arteries to phenylephrine, accompanied by increased NKCC1 mRNA levels, have been observed in rats with spontaneous hypertension or angiotensin II-induced hypertension compared with their normotensive controls (30, 32, 34). Future studies making use of NKCC1-KO and KCC4-KO mice may help to identify whether increased NKCC1 expression and/or reduced KCC4 play a role in the alteration of intracellular Cl− homeostasis in PASMCs associated with pulmonary hypertension.
Our results show that the smooth muscle cells of resistive PA have a fairly high [Cl−]i (averaged 45 and 47 mM determined by two different methods) which places the ECl at −28 to −30 mV, nearly 30 mV more depolarized than the resting potential of 57 mV in PASMCs (45). This provides a strong driving force for Cl− efflux via activated Cl− channels, leading to membrane depolarization, increase in intracellular Ca2+, and contraction of PASMCs. Increased [Cl−]i by chronic hypoxia pushes the ECl to an even more depolarized value which can potentiate the membrane depolarization of PASMCs following Cl− channel activation and contribute to the enhanced agonist-induced PA constriction observed in CHPH (14). Future studies are warranted to further examine this hypothesis and elucidate the role of altered PASMC Cl− homeostasis in the pathogenesis of chronic hypoxic pulmonary hypertension associated with many chronic lung diseases.
GRANTS
This work is supported by American Heart Association Grant 11SDG7440069 (to H. Sun) and National Institutes of Health Grant R01 HL-075134 (to J. S. K. Sham).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.S. and J.S.K.S. conceived and designed research; H.S. and O.P. performed experiments; H.S. and O.P. analyzed data; H.S. and J.S.K.S. interpreted results of experiments; H.S. prepared figures; H.S. drafted manuscript; H.S. and J.S.K.S. edited and revised manuscript; H.S., O.P., and J.S.K.S. approved final version of manuscript.
REFERENCES
- 1.Berend K, van Hulsteijn LH, Gans RO. Chloride: the queen of electrolytes? Eur J Intern Med 23: 203–211, 2012. doi: 10.1016/j.ejim.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch TJ, Pusch M. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev 98: 1493–1590, 2018. doi: 10.1152/physrev.00047.2017. [DOI] [PubMed] [Google Scholar]
- 3.Chipperfield AR, Harper AA. Chloride in smooth muscle. Prog Biophys Mol Biol 74: 175–221, 2000. doi: 10.1016/s0079-6107(00)00024-9. [DOI] [PubMed] [Google Scholar]
- 4.Hubner CA, Schroeder BC, Ehmke H. Regulation of vascular tone and arterial blood pressure: role of chloride transport in vascular smooth muscle. Pflugers Arch 467: 605–614, 2015. doi: 10.1007/s00424-014-1684-y. [DOI] [PubMed] [Google Scholar]
- 5.Hebert SC, Mount DB, Gamba G. Molecular physiology of cation-coupled Cl- cotransport: the SLC12 family. Pflugers Arch 447: 580–593, 2004. doi: 10.1007/s00424-003-1066-3. [DOI] [PubMed] [Google Scholar]
- 6.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85: 423–493, 2005. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 7.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev 80: 211–276, 2000. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 8.Kahn AM, Cragoe EJ Jr, Allen JC, Seidel CL, Shelat H. Effects of pHi on Na(+)-H+, Na(+)-dependent, and Na(+)-independent C1(−)-HCO3-exchangers in vascular smooth muscle. Am J Physiol Cell Physiol 261: C837–C844, 1991. doi: 10.1152/ajpcell.1991.261.5.C837. [DOI] [PubMed] [Google Scholar]
- 9.Choi I. SLC4A transporters. Curr Top Membr 70: 77–103, 2012. doi: 10.1016/B978-0-12-394316-3.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonar PT, Casey JR. Plasma membrane Cl−/HCO3− exchangers: structure, mechanism and physiology. Channels 2: 337–345, 2008. doi: 10.4161/chan.2.5.6899. [DOI] [PubMed] [Google Scholar]
- 11.Madden JA, Keller PA, Kleinman JG. Changes in smooth muscle cell pH during hypoxic pulmonary vasoconstriction: a possible role for ion transporters. Physiol Res 49: 561–566, 2000. [PubMed] [Google Scholar]
- 12.Yuan XJ. Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol Lung Cell Mol Physiol 272: L959–L968, 1997. doi: 10.1152/ajplung.1997.272.5.L959. [DOI] [PubMed] [Google Scholar]
- 13.Leblanc N, Forrest AS, Ayon RJ, Wiwchar M, Angermann JE, Pritchard HA, Singer CA, Valencik ML, Britton F, Greenwood IA. Molecular and functional significance of Ca(2+)-activated Cl(−) channels in pulmonary arterial smooth muscle. Pulm Circ 5: 244–268, 2015. doi: 10.1086/680189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun H, Xia Y, Paudel O, Yang XR, Sham JS. Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol 590: 3507–3521, 2012. doi: 10.1113/jphysiol.2012.232520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, Singer CA, Valencik ML, Greenwood IA, Leblanc N. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 303: C1229–C1243, 2012. doi: 10.1152/ajpcell.00044.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: D34–D41, 2013. [Erratum in J Am Coll Cardiol 63: 746, 2014]. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 17.Orlov SN, Koltsova SV, Tremblay J, Baskakov MB, Hamet P. NKCC1 and hypertension: role in the regulation of vascular smooth muscle contractions and myogenic tone. Ann Med 44: S111–S118, 2012. doi: 10.3109/07853890.2011.653395. [DOI] [PubMed] [Google Scholar]
- 18.Garneau AP, Marcoux AA, Slimani S, Tremblay LE, Frenette-Cotton R, Mac-Way F, Isenring P. Physiological roles and molecular mechanisms of K+ -Cl− cotransport in the mammalian kidney and cardiovascular system: where are we? J Physiol 597: 1451–1465, 2019. doi: 10.1113/JP276807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adragna NC, Ferrell CM, Zhang J, Di Fulvio M, Temprana CF, Sharma A, Fyffe RE, Cool DR, Lauf PK. Signal transduction mechanisms of K+-Cl− cotransport regulation and relationship to disease. Acta Physiol 187: 125–139, 2006. doi: 10.1111/j.1748-1716.2006.01560.x. [DOI] [PubMed] [Google Scholar]
- 20.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 21.Biwersi J, Verkman AS. Cell-permeable fluorescent indicator for cytosolic chloride. Biochemistry 30: 7879–7883, 1991. doi: 10.1021/bi00246a001. [DOI] [PubMed] [Google Scholar]
- 22.Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol Cell Physiol 259: C375–C388, 1990. doi: 10.1152/ajpcell.1990.259.3.C375. [DOI] [PubMed] [Google Scholar]
- 23.Chao AC, Dix JA, Sellers MC, Verkman AS. Fluorescence measurement of chloride transport in monolayer cultured cells. Mechanisms of chloride transport in fibroblasts. Biophys J 56: 1071–1081, 1989. doi: 10.1016/S0006-3495(89)82755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brosius FC, Pisoni RL, Cao X, Deshmukh G, Yannoukakos D, Stuart-Tilley AK, Haller C, Alper SL. AE anion exchanger mRNA and protein expression in vascular smooth muscle cells, aorta, and renal microvessels. Am J Physiol Renal Physiol 273: F1039–F1047, 1997. doi: 10.1152/ajprenal.1997.273.6.F1039. [DOI] [PubMed] [Google Scholar]
- 25.Madden JA, Ray DE, Keller PA, Kleinman JG. Ion exchange activity in pulmonary artery smooth muscle cells: the response to hypoxia. Am J Physiol Lung Cell Mol Physiol 280: L264–L271, 2001. doi: 10.1152/ajplung.2001.280.2.L264. [DOI] [PubMed] [Google Scholar]
- 26.Nozik-Grayck E, Huang YC, Carraway MS, Piantadosi CA. Bicarbonate-dependent superoxide release and pulmonary artery tone. Am J Physiol Heart Circ Physiol 285: H2327–H2335, 2003. doi: 10.1152/ajpheart.00507.2003. [DOI] [PubMed] [Google Scholar]
- 27.Akaike N. Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Prog Biophys Mol Biol 65: 251–264, 1996. doi: 10.1016/s0079-6107(96)00013-2. [DOI] [PubMed] [Google Scholar]
- 28.Di Fulvio M, Lincoln TM, Lauf PK, Adragna NC. Protein kinase G regulates potassium chloride cotransporter-4 [corrected] expression in primary cultures of rat vascular smooth muscle cells. J Biol Chem 276: 21046–21052, 2001. [Erratum in J Biol Chem 281: 21575, 2006]. doi: 10.1074/jbc.M100901200. [DOI] [PubMed] [Google Scholar]
- 29.Orlov SN, Resink TJ, Bernhardt J, Buhler FR. Na(+)-K+ pump and Na(+)-K+ co-transport in cultured vascular smooth muscle cells from spontaneously hypertensive and normotensive rats: baseline activity and regulation. J Hypertens 10: 733–740, 1992. [PubMed] [Google Scholar]
- 30.Cho HM, Lee HA, Kim HY, Han HS, Kim IK. Expression of Na+-K+ -2Cl− cotransporter 1 is epigenetically regulated during postnatal development of hypertension. Am J Hypertens 24: 1286–1293, 2011. doi: 10.1038/ajh.2011.136. [DOI] [PubMed] [Google Scholar]
- 31.Brown RA, Chipperfield AR, Davis JP, Harper AA. Increased (Na+K+Cl) cotransport in rat arterial smooth muscle in deoxycorticosterone (DOCA)/salt-induced hypertension. J Vasc Res 36: 492–501, 1999. doi: 10.1159/000025692. [DOI] [PubMed] [Google Scholar]
- 32.Cho HM, Lee DY, Kim HY, Lee HA, Seok YM, Kim IK. Upregulation of the Na(+)-K(+)-2Cl(−) cotransporter 1 via histone modification in the aortas of angiotensin II-induced hypertensive rats. Hypertens Res 35: 819–824, 2012. doi: 10.1038/hr.2012.37. [DOI] [PubMed] [Google Scholar]
- 33.Cho HM, Lee HA, Kim HY, Lee DY, Kim IK. Recruitment of specificity protein 1 by CpG hypomethylation upregulates Na(+)-K(+)-2Cl(−) cotransporter 1 in hypertensive rats. J Hypertens 31: 1406–1413, 2013. discussion 1413 doi: 10.1097/HJH.0b013e3283610fed. [DOI] [PubMed] [Google Scholar]
- 34.Lee HA, Baek I, Seok YM, Yang E, Cho HM, Lee DY, Hong SH, Kim IK. Promoter hypomethylation upregulates Na+-K+-2Cl− cotransporter 1 in spontaneously hypertensive rats. Biochem Biophys Res Commun 396: 252–257, 2010. doi: 10.1016/j.bbrc.2010.04.074. [DOI] [PubMed] [Google Scholar]
- 35.Jiang G, Akar F, Cobbs SL, Lomashvilli K, Lakkis R, Gordon FJ, Sutliff RL, O'Neill WC. Blood pressure regulates the activity and function of the Na-K-2Cl cotransporter in vascular smooth muscle. Am J Physiol Heart Circ Physiol 286: H1552–H1557, 2004. doi: 10.1152/ajpheart.00695.2003. [DOI] [PubMed] [Google Scholar]
- 36.Kawai N, McCarron RM, Spatz M. Effect of hypoxia on Na(+)-K(+)-Cl- cotransport in cultured brain capillary endothelial cells of the rat. J Neurochem 66: 2572–2579, 1996. doi: 10.1046/j.1471-4159.1996.66062572.x. [DOI] [PubMed] [Google Scholar]
- 37.Wallace BK, Foroutan S, O'Donnell ME. Ischemia-induced stimulation of Na-K-Cl cotransport in cerebral microvascular endothelial cells involves AMP kinase. Am J Physiol Cell Physiol 301: C316–C326, 2011. doi: 10.1152/ajpcell.00517.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai Y, Tang J, Zhang JH. Role of Cl− in cerebral vascular tone and expression of Na+-K+-2Cl− co-transporter after neonatal hypoxia-ischemia. Can J Physiol Pharmacol 83: 767–773, 2005. doi: 10.1139/y05-076. [DOI] [PubMed] [Google Scholar]
- 39.Yang XL, Zeng ML, Shao L, Jiang GT, Cheng JJ, Chen TX, Han S, Yin J, Liu WH, He XH, Peng BW. NFAT5 and HIF-1alpha coordinate to regulate NKCC1 expression in hippocampal neurons after hypoxia-ischemia. Front Cell Dev Biol 7: 339, 2019. doi: 10.3389/fcell.2019.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibla JC, Khoury J, Kong T, Robinson A, Colgan SP. Transcriptional repression of Na-K-2Cl cotransporter NKCC1 by hypoxia-inducible factor-1. Am J Physiol Cell Physiol 291: C282–C289, 2006. doi: 10.1152/ajpcell.00564.2005. [DOI] [PubMed] [Google Scholar]
- 41.Soleimani M. SLC26 Cl−/HCO3− exchangers in the kidney: roles in health and disease. Kidney Int 84: 657–666, 2013. doi: 10.1038/ki.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romero MF, Chen AP, Parker MD, Boron WF. The SLC4 family of bicarbonate (HCO3−) transporters. Mol Aspects Med 34: 159–182, 2013. doi: 10.1016/j.mam.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinn DA, Honeyman TW, Joseph PM, Thompson BT, Hales CA, Scheid CR. Contribution of Na+/H+ exchange to pH regulation in pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 5: 586–591, 1991. doi: 10.1165/ajrcmb/5.6.586. [DOI] [PubMed] [Google Scholar]
- 44.Deshmukh G, Alper SL, Wilde DW, Brosius Iii FC. Increased expression of anion transporter AE2 mRNA in small cerebral arteries of stroke-prone spontaneously hypertensive rates (SHRSP) (abstract). Hypertension 24: 384, 1994. [Google Scholar]
- 45.Casteels R, Kitamura K, Kuriyama H, Suzuki H. The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol 271: 41–61, 1977. doi: 10.1113/jphysiol.1977.sp011989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inglefield JR, Schwartz-Bloom RD. Optical imaging of hippocampal neurons with a chloride-sensitive dye: early effects of in vitro ischemia. J Neurochem 70: 2500–2509, 1998. doi: 10.1046/j.1471-4159.1998.70062500.x. [DOI] [PubMed] [Google Scholar]
- 47.Inglefield JR, Schwartz-Bloom RD. Fluorescence imaging of changes in intracellular chloride in living brain slices. Methods 18: 197–203, 1999. doi: 10.1006/meth.1999.0772. [DOI] [PubMed] [Google Scholar]
- 48.Kreye VA, Bauer PK, Villhauer I. Evidence for furosemide-sensitive active chloride transport in vascular smooth muscle. Eur J Pharmacol 73: 91–95, 1981. doi: 10.1016/0014-2999(81)90150-3. [DOI] [PubMed] [Google Scholar]
- 49.Koncz C, Daugirdas JT. Use of MQAE for measurement of intracellular [Cl−] in cultured aortic smooth muscle cells. Am J Physiol Heart Circ Physiol 267: H2114–H2123, 1994. doi: 10.1152/ajpheart.1994.267.6.H2114. [DOI] [PubMed] [Google Scholar]
- 50.Davis JP. The effects of Na(+)-K(+)-Cl− co-transport and Cl(−)-HCO3-exchange blockade on the membrane potential and intracellular chloride levels of rat arterial smooth muscle, in vitro. Exp Physiol 77: 857–862, 1992. doi: 10.1113/expphysiol.1992.sp003652. [DOI] [PubMed] [Google Scholar]
- 51.Davis JP, Harper AA, Chipperfield AR. Stimulation of intracellular chloride accumulation by noradrenaline and hence potentiation of its depolarization of rat arterial smooth muscle in vitro. Br J Pharmacol 122: 639–642, 1997. doi: 10.1038/sj.bjp.0701431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis JP, Chien PF, Chipperfield AR, Gordon A, Harper AA. The three mechanisms of intracellular chloride accumulation in vascular smooth muscle of human umbilical and placental arteries. Pflugers Arch 441: 150–154, 2000. [Erratum in Pflugers Arch 441: 164, 2000]. doi: 10.1007/s004240000401. [DOI] [PubMed] [Google Scholar]
- 53.Chipperfield AR, Davis JP, Harper AA. An acetazolamide-sensitive inward chloride pump in vascular smooth muscle. Biochem Biophys Res Commun 194: 407–412, 1993. doi: 10.1006/bbrc.1993.1834. [DOI] [PubMed] [Google Scholar]
- 54.Davis JP, Chipperfield AR, Harper AA. Accumulation of intracellular chloride by (Na-K-Cl) co-transport in rat arterial smooth muscle is enhanced in deoxycorticosterone acetate (DOCA)/salt hypertension. J Mol Cell Cardiol 25: 233–237, 1993. doi: 10.1006/jmcc.1993.1029. [DOI] [PubMed] [Google Scholar]
- 55.Anfinogenova YJ, Baskakov MB, Kovalev IV, Kilin AA, Dulin NO, Orlov SN. Cell-volume-dependent vascular smooth muscle contraction: role of Na+, K+, 2Cl− cotransport, intracellular Cl− and L-type Ca2+ channels. Pflugers Arch 449: 42–55, 2004. doi: 10.1007/s00424-004-1316-z. [DOI] [PubMed] [Google Scholar]
- 56.Gerstheimer FP, Muhleisen M, Nehring D, Kreye VA. A chloride-bicarbonate exchanging anion carrier in vascular smooth muscle of the rabbit. Pflugers Arch 409: 60–66, 1987. doi: 10.1007/BF00584750. [DOI] [PubMed] [Google Scholar]
- 57.Kahle KT, Khanna AR, Alper SL, Adragna NC, Lauf PK, Sun D, Delpire E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol Med 21: 513–523, 2015. doi: 10.1016/j.molmed.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rust MB, Faulhaber J, Budack MK, Pfeffer C, Maritzen T, Didie M, Beck FX, Boettger T, Schubert R, Ehmke H, Jentsch TJ, Hubner CA. Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl cotransporter KCC3. Circ Res 98: 549–556, 2006. doi: 10.1161/01.RES.0000204449.83861.22. [DOI] [PubMed] [Google Scholar]
- 59.Akar F, Skinner E, Klein JD, Jena M, Paul RJ, O'Neill WC. Vasoconstrictors and nitrovasodilators reciprocally regulate the Na+-K+-2Cl− cotransporter in rat aorta. Am J Physiol Cell Physiol 276: C1383–C1390, 1999. doi: 10.1152/ajpcell.1999.276.6.C1383. [DOI] [PubMed] [Google Scholar]
- 60.Smith JB, Smith L. Na+/K+/Cl− cotransport in cultured vascular smooth muscle cells: stimulation by angiotensin II and calcium ionophores, inhibition by cyclic AMP and calmodulin antagonists. J Membr Biol 99: 51–63, 1987. doi: 10.1007/BF01870621. [DOI] [PubMed] [Google Scholar]
- 61.Garay R, Rosati C, Braquet M, Chabrier PE, Braquet P. Stimulation of the Na+, K+ pump and the (Na+, K+, Cl−) cotransport system by endothelin-1 in cultured vascular smooth muscle cells: involvement of cyclo-oxygenase products. J Cardiovasc Pharmacol 13, Suppl 5: S213–S215, 1989. doi: 10.1097/00005344-198900135-00062. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Breaks J, Loutzenhiser K, Loutzenhiser R. Effects of inhibition of the Na+/K+/2Cl- cotransporter on myogenic and angiotensin II responses of the rat afferent arteriole. Am J Physiol Renal Physiol 292: F999–F1006, 2007. doi: 10.1152/ajprenal.00343.2006. [DOI] [PubMed] [Google Scholar]
- 63.Koltsova SV, Kotelevtsev SV, Tremblay J, Hamet P, Orlov SN. Excitation-contraction coupling in resistance mesenteric arteries: evidence for NKCC1-mediated pathway. Biochem Biophys Res Commun 379: 1080–1083, 2009. doi: 10.1016/j.bbrc.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 64.Zeniya M, Sohara E, Kita S, Iwamoto T, Susa K, Mori T, Oi K, Chiga M, Takahashi D, Yang SS, Lin SH, Rai T, Sasaki S, Uchida S. Dietary salt intake regulates WNK3-SPAK-NKCC1 phosphorylation cascade in mouse aorta through angiotensin II. Hypertension 62: 872–878, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01543. [DOI] [PubMed] [Google Scholar]
- 65.Dai Y, Zhang J. Chloride efflux is involved in ET-1 and 5-HT-induced contraction in rabbit basilar artery. J Cardiovasc Pharmacol 44: S125–S128, 2004. doi: 10.1097/01.fjc.0000166225.96088.bd. [DOI] [PubMed] [Google Scholar]
- 66.Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, Elton TS. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol (1985) 77: 1451–1459, 1994. doi: 10.1152/jappl.1994.77.3.1451. [DOI] [PubMed] [Google Scholar]
- 67.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 328: 1732–1739, 1993. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 68.Fu XW, Nurse CA, Wong V, Cutz E. Hypoxia-induced secretion of serotonin from intact pulmonary neuroepithelial bodies in neonatal rabbit. J Physiol 539: 503–510, 2002. doi: 10.1113/jphysiol.2001.013071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kéreveur A, Callebert J, Humbert M, Hervé P, Simonneau G, Launay J-M, Drouet L, High plasma serotonin levels in primary pulmonary hypertension. Effect of long-term epoprostenol (prostacyclin) therapy. Arterioscler Thromb Vasc Biol 20: 2233–2239, 2000. doi: 10.1161/01.ATV.20.10.2233. [DOI] [PubMed] [Google Scholar]
- 70.Shimoda LA, Sham JS, Sylvester JT. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol Res 49: 549–560, 2000. [PubMed] [Google Scholar]