

Abstract
Circadian rhythms regulate a remarkable variety of physiologic functions in living organisms. Circadian disruption is associated with tumorigenesis and tumor progression through effects on cancer cell biological properties, including proliferation, DNA repair, apoptosis, metabolism, and stemness. Emerging evidence indicates that circadian clocks also play an influential role in the tumor microenvironment (TME). This review outlines the recent discoveries on how cancer cell clock components (including circadian clock and clock genes/proteins) regulate TME biology and, reciprocally, how TME clock components affect tumor growth, metastasis, and therapeutic response. An improved understanding of how clock components regulate the symbiosis between cancer cells and TME will inform the development of novel clock-oriented therapeutic strategies, including immunotherapy.
Keywords: Circadian rhythms, CLOCK, Tumor microenvironment, Crosstalk, Symbiosis
Circadian rhythm and cancer: old story but new direction
The circadian rhythm is an evolutionarily conserved phenomenon that regulates the rhythmicity of physiologic, behavioral and biochemical functions in living organisms [1, 2]. One of the most well-known circadian rhythms is the sleep-wake cycle, in which the term circadian means “around a day” [3]. Circadian rhythms are regulated by circadian clocks in mammals. Over the past few decades, the connection between circadian clocks and tumorigenesis has been well studied. Mechanistically, circadian clock disruption can promote tumor growth and progression via affecting key cancer cell biological properties [1]. However, cancer cells do not live and flourish in isolation, but are surrounded by various stromal cells and factors of the tumor microenvironment (TME). Typically, TME comprises of extracellular matrix (ECM) and a variety of cells, including innate myeloid cells [e g., tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), neutrophils, and dendritic cells], lymphocytes (e.g., T cells and NK cells), cancer-associated fibroblasts (CAFs), and endothelial cells [4]. In recent years, the importance of TME in affecting tumor progression and therapeutic efficacy is wildly recognized, and multiple TME-targeted therapies have been developed [5]. Moreover, context-dependent cancer cell-TME symbiotic interactions have been demonstrated to be critical for tumor progression and therapy resistance [6–8]. Improving the molecular understanding of circadian clock in tumorigenesis and cancer cell-TME symbiotic interactions is currently a major focus of cancer research. Therefore, we need to understand whether and how circadian clocks are essential for regulating TME biology and cancer cell-TME symbiosis. In this review, we first discuss the influence of circadian clocks on cancer cell hallmarks and TME biology. Second, we highlight and discuss how cancer cell clock components (including circadian clock and clock genes/proteins) regulate TME biology and, reciprocally, how TME clock components affect tumor growth, metastasis, and therapy resistance. Finally, we discuss immunotherapeutic potential of targeting clock component-regulated cancer cell-TME symbiosis. We believe that this emerging area of interest in cancer biology has provided and will continue to provide insights leading to novel and effective treatments for cancer patients.
Molecular circadian clock and its effect on the biology of cancer cells and TME
Circadian system is composed of central and peripheral clocks [9]. The central clock is located in the anterior hypothalamic suprachiasmatic nucleus (SCN), which can function autonomously and coordinate peripheral clocks in the body via sending signals [10]. At the molecular level, the circadian rhythms emerging from central and peripheral clocks are quite similar. The central molecular circadian clock machinery is regulated by transcription–translation feedback loops (TTFL) [1, 2, 11, 12]. Aryl hydrocarbon receptor nuclear translocator-like protein 1 [ARNTL, also named brain and muscle ARNT-like protein-1 (BMAL1)] and circadian locomotor output cycles kaput (CLOCK) constitute positive factors of the feedback loop, which can bind to the E-box motif and promote the expression of transcription repressors, including cryptochrome (e.g., CRY1 and CRY2) and period (e.g., PER1, PER2, and PER3) genes. As the repressor arm of TTFL, CRY and PER form a complex entering to nucleus and suppress the CLOCK–BMAL1 complex [13–15]. On the other hand, the CLOCK–BMAL1 complex can regulate the expression of nuclear receptors REV-ERBα/β (also known as NR1D1/2, nuclear receptor subfamily 1, group D, member 1/2) and retinoic acid receptor–related orphan receptors (RORs), which, in turn, repress and activate BMAL1, respectively, thus constituting a second feedback loop [1, 2].
Circadian clock disruption induces the pathogenesis of many types of diseases, including cancer, [16, 17]. Emerging evidence suggests that circadian clock disruption (e.g., night-shift work and ‘late-eaters’ whose eating after 9:30 PM) increases cancer risk [18–20], and is associated with increased tumor metastasis in a variety of cancer types, including breast cancer [21, 22], non-small cell lung cancer (NSCLC) [23], and colorectal cancer (CRC) [24] in humans. The importance of this line of research is also supported by mouse model studies that have shown circadian clocks as playing a prominent role in tumorigenesis and in regulating the anti-tumor efficiency of radiotherapy [25, 26] and chemotherapy [27, 28]. Mechanistically, circadian clocks can affect tumor growth and progression via regulation of multiple cancer hallmarks in cancer cells [29, 30], which include DNA damage response, apoptosis, cell cycle, and senescence [1, 18, 31, 32], proliferation [1, 2], metabolism [30, 33, 34], replicative immortality [35], as well as genome instability and mutation [34]. Cancer stem cells (CSCs) represent a subpopulation of cancer cells with self-renewal abilities that markedly contribute to tumor initiation, metastasis, and therapy resistance [6]. Increasing evidence supports that circadian clocks are essential for maintaining CSC stemness in different cancer types, including acute myeloid leukemia (AML) and glioblastoma (GBM) [36–38]. Specifically, disruption of the circadian clock machinery pharmacologically (e.g., using REV-ERB agonists SR9009 and SR9011) and genetically (e.g., using CLOCK and BMAL1 shRNAs) in AML leads to CSC differentiation [38], as well as in GBM impairs glioma stem cell (GSC) stemness and causes GSC cell-cycle arrest and apoptosis [36, 37]. These results suggest that the core components of circadian clocks regulate key cancer cell biological properties across cancer types.
Beyond its impact on cancer cells described above, circadian clock disruption also influences the TME and the interactions between cancer cells and TME. For example, in a CRC-CAF co-culture model, biological clocks of CRC cells are modulated to enhance CRC malignant phenotype via regulating cell metabolism, viability, and apoptosis, and inducing chemotherapy resistance [39]. Moreover, in vivo findings from breast cancer and melanoma mouse models demonstrate that circadian clock disruption not only significantly enhances cancer cell proliferation, dissemination, stemness, and metastasis, but also induces an immunosuppressive TME by increasing the proportion of TAMs and Tregs, inducing macrophage polarization towards an anti-inflammatory phenotype, and decreasing the infiltration and activity of CD8+ T cells [40, 41]. Similarly, computational analysis of gene expression data obtained from cancer patient samples has revealed that clock genes are associated with immune cell infiltration and cancer cell proliferation [42]. Together, these findings suggest a critical role of circadian clocks in regulating cancer cell biological properties, TME biology, and their potential symbiotic interactions. Below, we highlight specific circadian clock components in cancer cells or in cells of the TME that affect cancer cell-TME crosstalk, and discuss its therapeutic potential.
Effect of cancer cell clock components on TME biology
TME is educated by cancer cells and plays an important role in tumor progression [4, 8, 43]. This section summarizes the role and underlying mechanisms of cancer cell clock components in regulation of TME biology, including angiogenesis, tumor-promoting inflammation, and immune evasion (Figure 1).
Figure 1. Clock components in cancer cells affect TME biology.
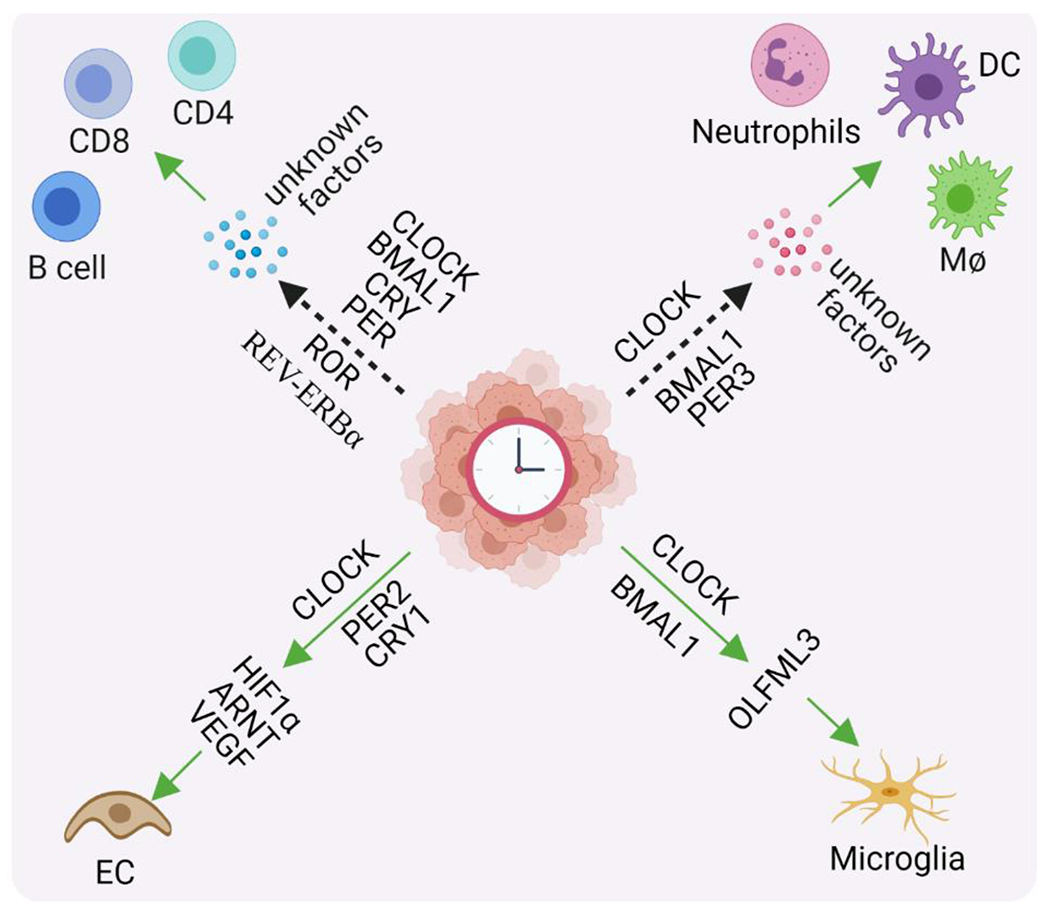
Clock components, including circadian clock and clock genes/proteins (e.g., CLOCK, ARNTL/BMAL1, PER, CRY, RORs, and REV-ERBα) in cancer cells or cancer stem cells regulate the expression and secretion of soluble factors (e.g., HIF1α, ARNT, VEGF, OLFML3, and other unidentified factors). Consequently, these secreted factors modulate TME biology, including endothelial cell (EC) biology (e.g., promoting angiogenesis and anti-angiogenic therapy resistance), infiltration of myeloid cells [e.g., macrophages (MΦ), microglia, neutrophils, and dendritic cells (DC)], as well as infiltration and activation/suppression of lymphocytes (e.g., CD8+ T cells, CD4+ T cells, and B cells). The dash arrows indicate that clock components are correlated with the infiltration of immune cells. Abbreviations: ARNT, aryl hydrocarbon receptor nuclear translocator; BMAL1, brain and muscle ARNT-like protein-1; CLOCK, circadian locomotor output cycles kaput; CRY, cryptochrome; HIF-1α, hypoxia-inducible factor 1-alpha; OLFML3, olfactomedin-like 3; PER, period; ROR, retinoic acid receptor—related orphan receptor; and VEGF, vascular endothelial growth factor.
Angiogenesis
is a TME-associated cancer hallmark, with new blood vessels forming from existing vasculatures [44–46]. Soluble factors secreted by cancer cells in the TME favor tumor angiogenesis, thus promoting tumor growth and metastasis [44, 45]. Here, we highlight recent findings that underscore the critical role of cancer cell clock components in regulating the expression of angiogenic factors, such as hypoxia-inducible factor 1 alpha (HIF-1α), aryl hydrocarbon receptor nuclear translocator (ARNT), and vascular endothelial growth factor (VEGF). Genetic studies demonstrated that the expression of these pro-angiogenic factors is decreased upon CLOCK shRNA knockdown and increased upon CLOCK overexpression in human CRC cells [47]. Furthermore, VEGF expression in xenograft tumors (including sarcoma, lung carcinoma, and melanoma) is increased under hypoxic conditions, fluctuated rhythmically in a circadian fashion (peaking during the light phase and decreasing around the early dark phase), and transcriptionally inhibited by PER2 and CRY1 [48]. Accordingly, the anti-tumor effect of anti-angiogenic therapies is more potent when drugs are administered at ZT 2 than that at ZT 14 (ZT 0 is designated as lights on and ZT 12 as lights off) [48]. On the other hand, anti-angiogenic therapy can upregulate BMAL1 expression in CRC cells, which, in turn, induces therapy resistance via upregulation of VEGF [49]. These results highlight a crucial role of cancer cell clock components in influencing tumor angiogenesis and anti-angiogenic therapy efficiency.
Inflammation
is a critical hallmark of cancer [46], supported mainly by the infiltration of innate myeloid cells [50]. Unbiased profiling of The Cancer Genome Atlas (TCGA) datasets (https://www.cancer.gov/tcga) followed by functional studies with mouse models have highlighted a strong connection between CSCs and myeloid cells by showing that cancer cell stemness inversely correlates with anti-tumor immunity signatures across cancer types [6, 51]. Specifically in GBM, high CLOCK levels in GSCs correlate with increased microglia in the TME via transcriptional regulation of chemokine olfactomedin-like 3 [36]. In addition to microglia, cancer cell clock components also contribute to the infiltration of other types of myeloid cells in mouse models. For example, in kidney renal clear cell carcinoma (KIRC) and breast cancer mouse models, the expression of clock genes (e.g., CLOCK, ARNTL and PER3) in cancer cells is fluctuated rhythmically and associated with the infiltration of macrophages, neutrophils, and dendritic cells [31, 52]. Together, these findings support the concept of cancer cell clock components that may influence tumor-promoting inflammation via effects on myeloid cell-mediated inflammation.
Immune escape
is another critical hallmark of cancer [46], a process involves in regulating the biology of lymphocytes and the expression of immune checkpoint molecules. Multi-omics analyses in KIRC and lung cancer have shown that clock genes (e.g., CLOCK, ARNTL, CRY1, CRY2, PER1, PER2, PER3, RORA, and NR1D1) not only regulate circadian rhythm and transcription factor activity of cancer cells, but also correlate with the infiltration of lymphocytes, including B cells, CD8+ T cells, and CD4+ T cells [31, 32]. Similarly, patient-derived GSCs exhibit circadian oscillation independent of tumor genetics [37], and CLOCK expression in GBM patient tumors correlates with a decreased level of activated CD8+ T cells [36]. Moreover, genome mutation analysis has revealed that clock genes are frequently mutated in cancer patients, which, in turn, can induce genome instability, associate with T cell (e.g., CD8+ and CD4+ T cell) exhaustion, and correlate with upregulation of immune inhibitory molecules, including programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [53, 54]. These results suggest an association that clock components in cancer cells correlate with the infiltration of lymphocytes and the expression of immune checkpoint molecules, thus potentially contributing to immune escape.
Collectively, these findings highlight cancer cell clock components as affecting TME-related cancer hallmarks (including angiogenesis, inflammation, and immune escape), and suggest that blockade of circadian clock-associated cancer cell-TME crosstalk may inhibit tumor progression. Nonetheless, available data provides a roadmap for further investigations of molecular mechanisms underlying the uncharacterized cancer cell clock component-associated TME characteristics, such as the presence and function of CAFs, Tregs, MDSCs, and ECM.
Impact of TME clock components on tumor progression and therapy resistance
In addition to cancer cells and CSCs, circadian clock can intrinsically regulate the behavior and function of the TME, such as those associated with immune cells [55–57], fibroblasts [58], and endothelial cells [59]. Given the importance of symbiotic cancer cell-TME interactions in tumor [36, 60–62], here we highlight the role of clock components in the TME of mouse models that affect cancer cell biological properties (Figure 2). For example, in a 4T1 breast cancer mouse model, clock genes (e.g., CLOCK, ARNTL, PER2, CRY1, and NR1D1) are rhythmically expressed in cells of the TME to induce the expression and secretion of Wnt family member 10A (WNT10A). Following the secretion, WNT10A can increase cancer cell stemness through upregulation of aldehyde dehydrogenase 3 family, member A1 (ALDH3A1) [63], an enzyme whose activity is a characteristic of CSCs and correlates with tumor malignancy [64]. As a result, time-of-day effects for the efficacy of administering ALDH inhibitor have been observed in this mouse model [63]. In MC38 colon and EO771 breast cancer mouse models, the expression of clock gene PER2 in the TME is crucial for metastatic colonization via inducing a premetastatic niche [65]. Furthermore, in a B16F10 melanoma mouse model, cisplatin treatment in the evening induces less toxicity than the same treatment administered in the morning. Notably, this difference is not apparent in PER1/2-KO mice [66]. As a result, tumor-bearing PER1/2-KO mice showed an extended survival and a more robust immune response (e.g., increased CD4+ and CD8+ T cells) in relation to tumor-bearing wild-type (WT) mice following cisplatin treatment [66]. Taken together, these findings from distinct mouse models highlight an essential role of TME clock components in regulating cancer cell stemness, metastasis, and therapy resistance, and in so doing support the potential of targeting TME clock components. As discussed earlier, TME is defined as the complex of diverse cell types. Therefore, it will be important to identify the specific cell types of the TME that contribute to clock component-regulated cancer cell biological properties and therapy resistance.
Figure 2. Impact of TME clock components on tumor growth, metastasis and stemness.
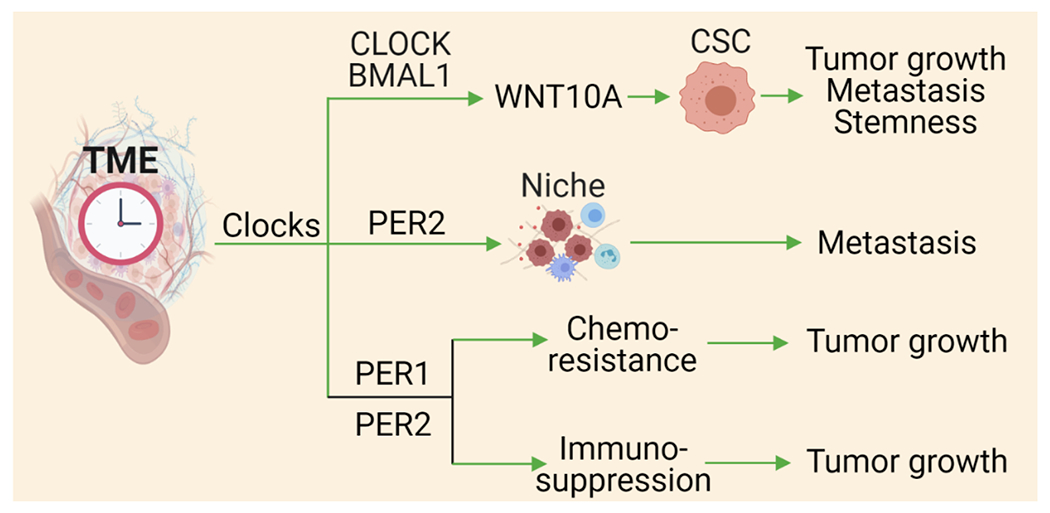
CLOCK and BMAL1 in the TME can induce the secretion of WNT10A, which, in turn, upregulates ALDH3A1 in cancer stem cells (CSCs) to promote stemness, tumor growth, and metastasis. In addition, PER1 and PER2 in the TME can induce a pre-metastatic niche to promote metastasis, and induce chemotherapy resistance and immunosuppression thus promoting tumor growth. Abbreviations: ALDH3A1, aldehyde dehydrogenase 3 family, member A1; BMAL1, brain and muscle ARNT-like protein-1; CLOCK, circadian locomotor output cycles kaput; PER, period; TME, tumor microenvironment; and WNT10A, wnt family member 10 A.
TAMs are the most prominent subpopulation of cells in the TME that display a critical role in influencing tumor growth and metastasis [67]. TAMs can be classified as polarized toward pro-inflammatory and ‘alternatively activated’ phenotypes, which exhibit immune-stimulatory (anti-tumor) and immunosuppressive (pro-tumor) effects, respectively [8, 68]. It should be noted that some TAMs may not be polarized to either state [69]. When polarized to the immune-stimulatory or immunosuppressive phenotype, BMAL1 expression in macrophages is upregulated. Depletion of BMAL1 in macrophages downregulates mitochondrial metabolism via upregulation of HIF1α and ROS accumulation, and downregulation of nuclear factor erythroid 2–related factor 2 (NRF2), thus regulating the production of pro-inflammatory cytokines [70, 71]. As a result, tumor growth and TAM alternative polarization are increased in myeloid-specific BMAL1 KO mice compared to BMAL1-WT mice [70]. Similarly, co-injection of cancer cells with BMAL1-KO macrophages promotes tumor growth, and reduces the infiltration of CD8+ T cells when compared to BMAL1-WT macrophages [70]. Thus, these emerging evidence highlight that macrophage BMAL1 is critical for suppressing tumor growth and promoting anti-tumor immune response (Figure 3).
Figure 3. Clock components in specific TME that affect tumor growth.
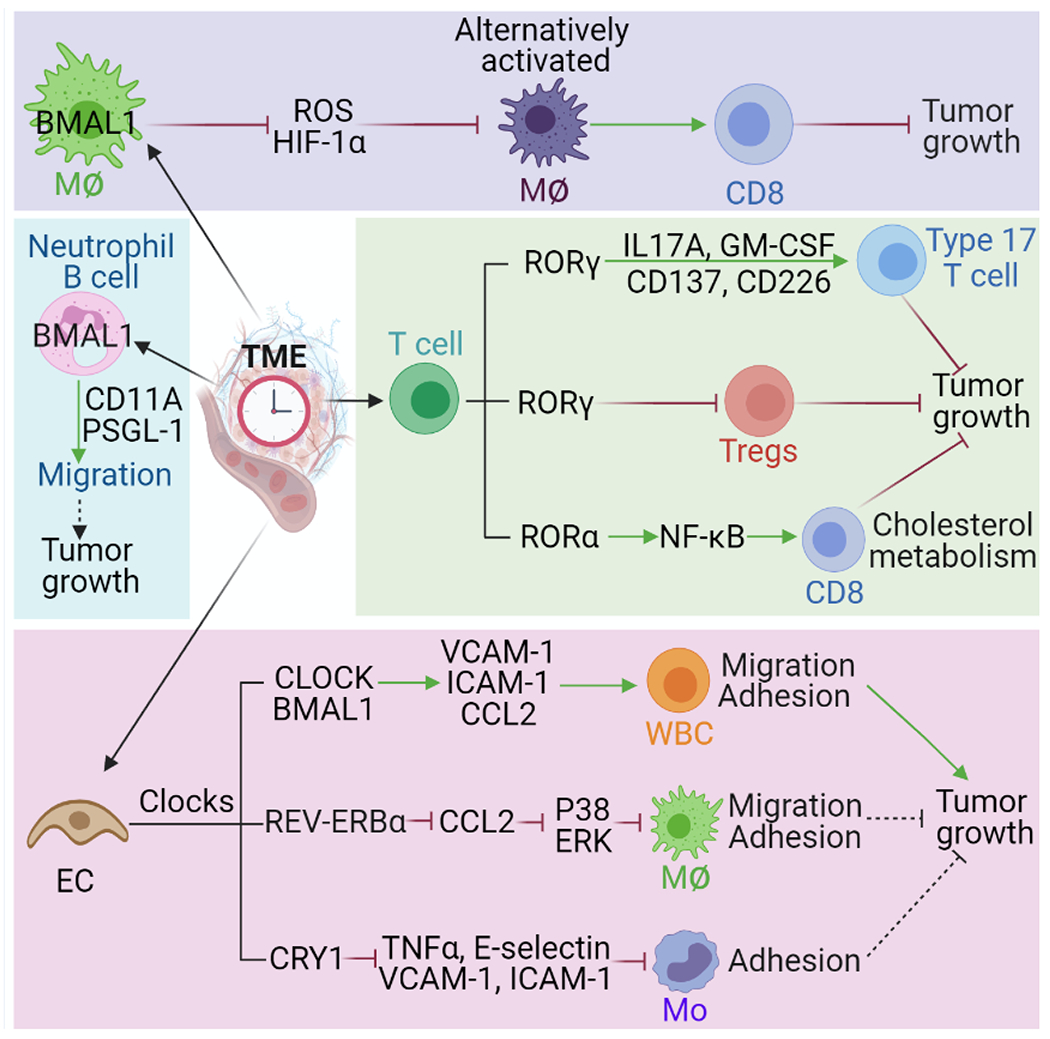
BMAL1 in macrophages (MΦ) inhibits the production of ROS and HIF1α and affects tumor growth through regulating macrophage alternative polarization and CD8+ T cell-mediated immune response. BMAL1 in B cells and neutrophils can change their migration ability via modulating the expression of pro-migratory molecules (e.g., CD11A and PSGL-1). RORγ and RORα in T cells can modulate their differentiation (e.g., CD4+ Th17 cells, CD8+ Tc cells, and Tregs) and activation via regulation of indicated factors and pathways, which, affects tumor growth and anti-tumor immune response. Endothelial cell (EC) clocks (e.g., CLOCK, BMAL1, REV-ERBα, and CRY1) can influence adhesion and migration of white blood cells (WBC, as called leukocytes), macrophages and monocytes (Mo) via regulating the expression of pro-migratory molecules (e.g., VCAM-1, ICAM-1, and E-selectin) and/or cytokines (e.g., CCL2 and TNFα), and activating ERK and P38 pathways, thus affecting tumor growth. The dash lines indicate that further studies are needed to validate this conclusion. Abbreviations: BMAL1, brain and muscle ARNT-like protein-1; CCL2, CC chemokine ligand 2; CLOCK, circadian locomotor output cycles kaput; CRY, cryptochrome; GM-CSF, granulocyte-macrophage colony-stimulating factor; HIF-1α, hypoxia-inducible factor 1-alpha; ICAM-1, intercellular adhesion molecule-1; IL-17A, interleukin 17A; NF-κB, nuclear factor kappa B; PSGL-1, P-selectin glycoprotein ligand-1; ROR, retinoic acid receptor—related orphan receptor; ROS, reactive oxygen species; TME, tumor microenvironment; TNFα, tumor necrosis factor alpha; and VCAM1, vascular cell adhesion molecule 1.
Lymphocytes display a critical role in tumor immunity, and their anti-tumor activities can be regulated by cell intrinsic clock components (Figure 3). RORγt is a transcription factor that can control the interleukin-17–producing CD4+ T helper (Th17) cell differentiation in a circadian clock-dependent manner [72]. Activation of RORγ using its synthetic agonists enhances the differentiation and effector function of Th17 cells and reduces the level of Tregs by regulating the expression of cytokines/chemokines, co-stimulatory receptors, and immunosuppressive molecules [73]. Consequently, co-culture and co-injection of RORγ agonist-treated CD8+ Tc17 cells and EG7 lymphoma cells increases apoptosis in vitro and inhibits tumor growth in vivo [73]. Moreover, translational studies in breast cancer and CRC mouse models have shown that activation of RORγ inhibits tumor growth and extends animal subject survival through influence on anti-tumor immune response [73]. It should be noted that the effect of RORγ activation on Tregs may not due to its clock function since Tregs do not have intrinsic circadian oscillators [74]. In addition to RORγ, activation of RORα can maintain the balance of cholesterol metabolism in CD8+ T cells by attenuating the nuclear factor-κB (NF-κB) pathway, which, in turn, enhances CD8+ T cell activity and function. Eventually, these activated CD8+ T cells induce CRC cancer cell apoptosis [75]. In summary, these findings suggest that the specific targeting of RORs in T cells is a potential approach for immunotherapies.
Another potential TME clock mechanism involves the infiltration of leukocytes, a population of immune cells essential for tumor development [76]. A growing body of evidence suggests that circadian clock-regulated pro-migratory molecules can trigger leukocyte migration. Here, we highlight recent findings that reveal the role and molecular basis of circadian clock components in specific cell types of the TME (e.g., B cells, neutrophils, and endothelial cells) in regulating the expression of pro-migratory molecules and leukocyte migration (Figure 3). For instance, specific deletion of BMAL1 in B cells, neutrophils, or endothelial cells in mice abolishes time-of-day differences in the expression of pro-migratory molecules [e.g., integrin alpha L (CD11A), P-selectin glycoprotein ligand-1 (PSGL-1), intercellular adhesion molecule 1 (ICAM-1), or vascular cell adhesion protein 1 (VCAM-1)], which, in turn, ablates the arrhythmic homing of B cells, neutrophils and leukocytes [59]. In contrast, CLOCK expression in endothelial cells can transcriptionally upregulate ICAM-1, VCAM-1, and C–C motif chemokine ligand 2 (CCL2), thus increasing the adhesion of leukocytes to endothelium [77]. In addition to the CLOCK-BMAL1 complex, other clock components also regulate the infiltration and adhesion of macrophages and monocytes. For example, activation of REV-ERBα can suppress CCL2 and its downstream signals (e.g., ERK and p38), which, in turn, inhibits macrophage adhesion and migration [78]. Moreover, overexpression of CRY1 in endothelial cells can inhibit the expression of inflammatory cytokines (e.g., IL-1β, IL-6, and TNF-α), adhesion molecules (e.g., VCAM-1, ICAM-1, and E-selectin), and activation of NF-κB pathway, all of which impairs monocyte adhesion [79]. Finally, in vivo findings from syngeneic and xenogeneic leukemia cancer models show that the clock-regulated recruitment and engraftment of leukemic cells/leukocytes increases tumor burden [59].
Beyond cell types of the TME as discussed earlier, we may also need to consider the complement system, which has a close connection with circadian clocks [80], and exhibits a pivotal role in promoting tumor growth via triggering myeloid cell infiltration and suppressing CD8+ T cell-mediated immune response [81]. Based upon the results of studies that have been described above, we propose that TME clock components play a significant role in modulating cancer cell stemness, tumor growth, metastasis, and therapeutic efficacy. Multiple examples have highlighted a critical role of clock components in myeloid cells, lymphocytes, and endothelial cells that regulate tumor growth via modulation of cancer cell apoptosis, macrophage polarization, CD8+ T cell activity, and leucocyte infiltration of tumor. These studies suggest a therapeutic potential of targeting TME clock components and their-regulated factors for disrupting TME-cancer cell symbiotic interactions.
Therapeutic potential of clock-oriented immunotherapy
TME-targeted therapeutic strategies have emerged as a promising approach for cancer treatment due to the TME-cancer cell symbiotic interactions, and the critical roles of the TME in regulating tumor progression and modulating anti-tumor efficiency of standard-of-care therapies [5, 6, 8, 82]. Among the TME, myeloid cells (e.g., TAMs and MDSCs) can suppress T cell (including CD4+ and CD8+ T cell) -mediated immune response and immunotherapy efficiency [82–85], and induce anti-angiogenic therapy resistance [86–89]. Similarly, CAFs can suppress immunotherapy efficiency through a mechanism of interacting with myeloid cells and T cells [90]. Based on these mechanistic studies, substantial attempts have been made to develop TME-targeted therapies to overcome immunotherapy resistance [91]. For example, TAM-targeted therapies (e.g., targeting TAM phagocytosis [92, 93] or reprogramming [94–97]), MDSC-targeted therapies (e.g., targeting MDSC infiltration [98–101] or activation [102–104]), CAF-targeting therapies (e.g., blocking CAF function or activation [105]), or anti-angiogenic therapies [106] show robust synergistic effects with immune checkpoint inhibitors (ICIs) in tumor mouse models. More importantly, some of these combination therapies are currently in clinical trials for treating cancer patients [67, 105–107].
Current T cell-targeted immunotherapies include those that block inhibitory immune checkpoints, and the approaches that boost adaptive immunity by genetically engineering T cells with chimeric antigen receptors (CAR) and T cell receptors [5]. Given the importance of circadian clock-regulated cancer cell-TME crosstalk in T cell biology as discussed earlier, increased understanding of this relationship could lead to strategies increasing immunotherapy efficiency. Preclinical studies in mouse models have demonstrated that administration of RORγ agonist enhances the anti-tumor activity of Th17 cells modified for expression of a CAR, and this CAR T therapy provided long-term protection against tumors [108]. In addition, clock component-regulated cancer cell-TME crosstalk has been associated with enhanced tumor infiltration of immunosuppressive myeloid cells, which, in turn, impairs infiltration and activation of CD4+ and CD8+ T cells, as well as upregulates immune checkpoint molecules [31, 32, 36, 52–54]. These findings suggest that targeting clock component-regulated TME-cancer cell crosstalk might increase the anti-tumor efficiency of ICIs. Indeed, this concept is supported by the evidence from clinical studies, where the anti-tumor efficacy of nivolumab (anti-PD1 antibody) in advanced NSCLC patients is significantly higher in the morning treatment group than that in the afternoon treatment group [109]. Moreover, a clinical trial testing RORγ agonist in combination with anti-PD-1 antibody for metastatic NSCLC patients is underway (NCT03396497). These studies suggest a clinical impact of clock components in affecting ICI therapy response. Further studies focusing on understanding clock component-regulated interactions between cancer cells and immune system will help to design and develop novel and effective clock-oriented immunotherapies.
Concluding Remarks
Although it is well established that circadian clock disruption is linked to an increased risk of cancer, details of the molecular mechanisms underlying this association are limited [29]. This review has highlighted some of the molecular circuitry that underlies clock component-regulated crosstalk between cancer cells and cells of the TME, including innate myeloid cells, lymphocytes, and endothelial cells. Currently, available results suggest a clock component-regulated cancer cell-TME symbiosis that is essential for sustained tumor growth, and that affects therapeutic response to distinct treatments, including immunotherapies.
The regulation of clock component-oriented cancer cell-TME crosstalk is complicated. As discussed earlier, BMAL1 depletion in GSCs inhibits tumor growth by reducing the infiltration of immunosuppressive microglia in GBM: a result supporting an oncogenic effect of BMAL1 expression [36]. However, BMAL1 expression in macrophages exhibits a tumor suppressive effect, as indicated by BMAL1 deficiency promoting an immunosuppressive phenotype that stimulates tumor growth in melanoma [70]. Such results highlight that clock component-regulated cancer cell-TME interactions are context dependent. Due to this complexity, cancer cells and cells of the TME within different cancer types can express distinct immune checkpoint molecules, which might provide various ICI treatment options. Further studies using genetic mouse models and advanced technologies, such as cytometry by time of flight (CyTOF) and single-cell RNA sequencing (scRNA-seq) [110, 111], will increase our understanding of context-dependent signaling axis (including immune checkpoint profile) underlying clock component-regulated cancer cell-TME interactions. Such knowledge will enable a more informed use of ICI therapies that, in turn, will lead to improved outcomes for cancer patients receiving ICI therapy.
In summary, recent studies have provided information highlighting the influence of clock component-regulated cancer cell-TME interactions on tumor growth, metastasis, and therapy resistance. However, multiple open questions remain regarding molecular mechanisms underlying this symbiosis and approaches for disrupting these symbiotic interactions (see Outstanding Questions). We anticipate that future studies will increase understanding of these interactions, and in so doing will reveal novel approaches for cancer treatments.
Outstanding Questions.
Which clock components and related cancer cell-TME interactions are the drivers that regulate tumorigenesis, metastasis and therapy resistance? What are the specific differences and similarities regarding cancer cell-TME symbiosis among different clock components under specific cancer types?
What signaling pathways are critical for regulation of clock component-regulated cancer cell-TME interactions?
Can scRNA-seq and CyTOF identify new clock component-regulated cancer cell-TME interactions and new immune checkpoint molecules during this interaction?
What’s the molecular mechanism underlying interactions between cancer cell clock components and adaptive immune response?
Whether and how clock component-regulated cancer cell-TME symbiosis affects the anti-tumor activity of immunotherapies and conventional therapies? Whether targeting this symbiosis has synergistic effects with immunotherapies or conventional therapies? If yes, what is the best combination strategy?
Highlights.
Circadian clocks contribute to tumor growth and metastasis by regulating the biology of cancer cells and tumor microenvironment (TME), as well as their symbiotic interactions.
Cancer cell clock components affect TME biology via regulation of angiogenesis, tumor-promoting inflammation, and immune escape.
TME clock components affect tumor progression via regulating cancer cell biological properties directly and affecting pro-tumor TME indirectly.
Characterizing clock component-regulated cancer cell-TME symbiosis might reveal unique therapeutic strategies, and targeting this symbiosis might increase the efficiency of immunotherapy.
Acknowledgments
This work was supported in part by NIH R00 CA240896 (to P.C.), NIH P50CA221747 (to P.C., Brain cancer SPORE CEP Award), Cancer Research Foundation Young Investigator Award (to P.C.), Lynn Sage Scholar Award (to P.C.), and Northwestern University start-up funds (to P.C.).
Glossary
- Cancer hallmarks
a group of key biological capabilities that acquired during tumor development.
- Cancer-associated fibroblasts (CAFs)
a component of the tumor microenvironment with diverse functions, including matrix remodeling, immunosuppression and extensive reciprocal interactions with cancer cells and other cells.
- CAR T therapy
a kind of therapy where T cells are genetically engineered to produce an artificial receptor for immunotherapy.
- Circadian rhythm
an evolutionarily conserved physiological process that regulates the sleep-wake during a 24-hour cycle.
- Cytometry by time of flight (CyTOF)
a valuable approach in the high-dimensional analysis of single cells, especially immune cells, in tumor tissues.
- Extracellular matrix (ECM)
non-cellular component in mammalian organs and tissues that not only provides structural support but also exerts critical biological functions.
- Glioma stem cells (GSCs)
a small population of cells in gliomas with self-renewal ability that can induce tumorigenesis and therapeutic resistance.
- Hypoxia-inducible factor 1-alpha
the master transcriptional regulator of cellular response under hypoxic conditions.
- Inflammation
a process by which the immune system protects the body from harmful agents. Tumor-promoting inflammation is supported mainly by innate immune cells and plays an important role in tumor growth and metastasis.
- Immune checkpoint inhibitors (ICIs)
a class of agents that trigger immune response by targeting immune checkpoint molecules.
- Immune evasion
a strategy used by tumors to evade host’s immune response, thus maximizing their probability for tumor growth and metastasis.
- Lymphocytes
one of the main types of immune cells in the body, which include T cells, NK cells and B cells.
- Macrophage polarization
macrophage activation into distinct phenotypes characterized by pro-inflammatory and anti-inflammatory gene expression profiles.
- Metastasis
the spreading of tumor from primary sites to other organs, or from one part to another part in the same organ.
- Microglia
specialized macrophages in the brain that originate from embryonic yolk sac progenitors.
- Myeloid cells
originates from hematopoietic stem cells in the bone marrow and includes macrophages, neutrophils, monocytes, dendritic cells, myeloid derived suppressor cells, and mast cells.
- Myeloid derived suppressor cells (MDSCs)
a subset of myeloid cells in tumor tissues and in periphery of tumor-bearing hosts exhibiting immunosuppressive function.
- Single-cell RNA sequencing (scRNA-seq)
a technology that can provide the expression profiles of an individual cell, thus offering a better understanding of the phenotype, state and function of an individual cell in the context of its microenvironment.
- Stemness
a molecular process underlying the core stem cell properties of self-renewal.
- Symbiosis
a type of relationship between two species or cell types in which at least one component benefits.
- Th17 cells
a subpopulation of CD4+ T cells, which are characterized by producing high levels of interleukin-17.
- The Cancer Genome Atlas (TCGA)
a landmark cancer genomics program that characterizes more than 20,000 primary cancer and matched normal samples across 33 cancer types.
- Tregs
a population of specialized T cells that suppress an anti-tumor immune response
- Tumor-associated macrophage (TAMs)
macrophages presented in tumor tissues that display a pro-tumor and immunosuppressive function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
No interests are declared.
References
- 1.Sancar A and Van Gelder RN (2021) Clocks, cancer, and chronochemotherapy. Science 371 (6524). [DOI] [PubMed] [Google Scholar]
- 2.Shafi AA and Knudsen KE (2019) Cancer and the Circadian Clock. Cancer Res 79 (15), 3806–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mauvoisin D et al. (2015) Proteomics and circadian rhythms: it’s all about signaling! Proteomics 15 (2-3), 310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quail DF and Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19 (11), 1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bejarano L et al. (2021) Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov 11 (4), 933–959. [DOI] [PubMed] [Google Scholar]
- 6.Chen P et al. (2021) Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep 34 (1), 108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wellenstein MD and de Visser KE (2018) Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48 (3), 399–416. [DOI] [PubMed] [Google Scholar]
- 8.Xuan W et al. (2021) Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms. Trends Immunol 42 (4), 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turek FW (2016) Circadian clocks: Not your grandfather’s clock. Science 354 (6315), 992–993. [DOI] [PubMed] [Google Scholar]
- 10.Mohawk JA et al. (2012) Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 35, 445–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Partch CL et al. (2014) Molecular architecture of the mammalian circadian clock. Trends Cell Biol 24 (2), 90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patke A et al. (2020) Molecular mechanisms and physiological importance of circadian rhythms. Nature Reviews Molecular Cell Biology 21 (2), 67–84. [DOI] [PubMed] [Google Scholar]
- 13.Kume K et al. (1999) mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98 (2), 193–205. [DOI] [PubMed] [Google Scholar]
- 14.Griffin EA Jr. et al. (1999) Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science 286 (5440), 768–71. [DOI] [PubMed] [Google Scholar]
- 15.Sangoram AM et al. (1998) Mammalian circadian autoregulatory loop: a timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron 21 (5), 1101–13. [DOI] [PubMed] [Google Scholar]
- 16.Xie YL et al. (2019) New Insights Into the Circadian Rhythm and Its Related Diseases. Frontiers in Physiology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leng Y et al. (2019) Association between circadian rhythms and neurodegenerative diseases. Lancet Neurology 18 (3), 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koritala BSC et al. (2021) Night shift schedule causes circadian dysregulation of DNA repair genes and elevated DNA damage in humans. J Pineal Res, e12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dun AS et al. (2020) Association Between Night-Shift Work and Cancer Risk: Updated Systematic Review and Meta-Analysis. Frontiers in Oncology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srour B et al. (2018) Circadian nutritional behaviours and cancer risk: New insights from the NutriNet-sante prospective cohort study: Disclaimers. Int J Cancer 143 (10), 2369–2379. [DOI] [PubMed] [Google Scholar]
- 21.Numata M et al. (2021) Metastasis of Breast Cancer Promoted by Circadian Rhythm Disruption due to Light/Dark Shift and its Prevention by Dietary Quercetin in Mice. J Circadian Rhythms 19, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha NH et al. (2016) The Circadian Rhythm Gene Arntl2 Is a Metastasis Susceptibility Gene for Estrogen Receptor-Negative Breast Cancer. Plos Genetics 12 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J et al. (2020) Downregulation of the circadian rhythm regulator HLF promotes multiple-organ distant metastases in non-small cell lung cancer through PPAR/NF-kappab signaling. Cancer Lett 482, 56–71. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y et al. (2017) Upregulation of circadian gene ‘hClock’ contribution to metastasis of colorectal cancer. Int J Oncol 50 (6), 2191–2199. [DOI] [PubMed] [Google Scholar]
- 25.Zhanfeng N et al. (2015) Circadian genes Per1 and Per2 increase radiosensitivity of glioma in vivo. Oncotarget 6 (12), 9951–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen H et al. (2020) Hypoxia, metabolism, and the circadian clock: new links to overcome radiation resistance in high-grade gliomas. J Exp Clin Cancer Res 39 (1), 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner PM et al. (2021) Temporal regulation of tumor growth in nocturnal mammals: In vivo studies and chemotherapeutical potential. FASEB J 35 (2), e21231. [DOI] [PubMed] [Google Scholar]
- 28.Katamune C et al. (2019) Mutation of the gene encoding the circadian clock component PERIOD2 in oncogenic cells confers chemoresistance by up-regulating the Aldh3a1 gene. J Biol Chem 294 (2), 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruan W et al. (2021) Circadian rhythm as a therapeutic target. Nature Reviews Drug Discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sulli G et al. (2019) Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends in Cancer 5 (8), 475–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou LC et al. (2020) Circadian clock is associated with tumor microenvironment in kidney renal clear cell carcinoma. Aging-Us 12 (14), 14620–14632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y et al. (2019) Circadian clock associates with tumor microenvironment in thoracic cancers. Aging (Albany NY) 11 (24), 11814–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinouchi K and Sassone-Corsi P (2020) Metabolic rivalry: circadian homeostasis and tumorigenesis. Nat Rev Cancer 20 (11), 645–661. [DOI] [PubMed] [Google Scholar]
- 34.Pavlova NN and Thompson CB (2016) The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23 (1), 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen WD et al. (2014) The circadian rhythm controls telomeres and telomerase activity. Biochem Biophys Res Commun 451 (3), 408–14. [DOI] [PubMed] [Google Scholar]
- 36.Chen P et al. (2020) Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov 10 (3), 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong Z et al. (2019) Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov 9 (11), 1556–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puram RV et al. (2016) Core Circadian Clock Genes Regulate Leukemia Stem Cells in AML. Cell 165 (2), 303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuhr L et al. (2019) The Interplay between Colon Cancer Cells and Tumour-Associated Stromal Cells Impacts the Biological Clock and Enhances Malignant Phenotypes. Cancers (Basel) 11 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hadadi E et al. (2020) Chronic circadian disruption modulates breast cancer stemness and immune microenvironment to drive metastasis in mice. Nature Communications 11 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aiello I et al. (2020) Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation. Science Advances 6 (42). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z et al. (2021) Circadian clock genes promote glioma progression by affecting tumour immune infiltration and tumour cell proliferation. Cell Prolif, e12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaymak I et al. (2021) Immunometabolic Interplay in the Tumor Microenvironment. Cancer Cell 39 (1), 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Z et al. (2015) Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: focus on the cancer hallmark of tumor angiogenesis. Carcinogenesis 36 Suppl 1, S184–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang X et al. (2020) The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res 39 (1), 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 47.Wang YP et al. (2017) Upregulation of circadian gene ‘hClock’ contribution to metastasis of colorectal cancer. International Journal of Oncology 50 (6), 2191–2199. [DOI] [PubMed] [Google Scholar]
- 48.Koyanagi S et al. (2003) A molecular mechanism regulating circadian expression of vascular endothelial growth factor in tumor cells. Cancer Res 63 (21), 7277–83. [PubMed] [Google Scholar]
- 49.Burgermeister E et al. (2019) Aryl hydrocarbon receptor nuclear translocator-like (ARNTL/BMAL1) is associated with bevacizumab resistance in colorectal cancer via regulation of vascular endothelial growth factor A. EBioMedicine 45, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalapour S and Karin M (2019) Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 51 (1), 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miranda A et al. (2019) Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A 116 (18), 9020–9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos CA et al. (2020) A Non-canonical Function of BMAL1 Metabolically Limits Obesity-Promoted Triple-Negative Breast Cancer. iScience 23 (2), 100839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Y et al. (2019) Pan-Cancer Analysis Reveals Disrupted Circadian Clock Associates With T Cell Exhaustion. Front Immunol 10, 2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou J et al. (2020) The aberrant expression of rhythm genes affects the genome instability and regulates the cancer immunity in pan-cancer. Cancer Med 9 (5), 1818–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheiermann C et al. (2018) Clocking in to immunity. Nat Rev Immunol 18 (7), 423–437. [DOI] [PubMed] [Google Scholar]
- 56.Adrover JM et al. (2019) A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 50 (2), 390–402 e10. [DOI] [PubMed] [Google Scholar]
- 57.Curtis AM et al. (2014) Circadian clock proteins and immunity. Immunity 40 (2), 178–86. [DOI] [PubMed] [Google Scholar]
- 58.Nagoshi E et al. (2004) Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 119 (5), 693–705. [DOI] [PubMed] [Google Scholar]
- 59.He WY et al. (2018) Circadian Expression of Migratory Factors Establishes Lineage-Specific Signatures that Guide the Homing of Leukocyte Subsets to Tissues. Immunity 49 (6), 1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen P et al. (2019) Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 35 (6), 868–884 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen P et al. (2017) Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A 114 (3), 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao D et al. (2020) Chromatin Regulator CHD1 Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Deficient Prostate Cancer. Cancer Discov 10 (9), 1374–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsunaga N et al. (2018) Optimized Dosing Schedule Based on Circadian Dynamics of Mouse Breast Cancer Stem Cells Improves the Antitumor Effects of Aldehyde Dehydrogenase Inhibitor. Cancer Res 78 (13), 3698–3708. [DOI] [PubMed] [Google Scholar]
- 64.Chefetz I et al. (2019) A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep 26 (11), 3061–3075 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaashua L et al. (2020) Stromal Expression of the Core Clock Gene Period 2 Is Essential for Tumor Initiation and Metastatic Colonization. Front Cell Dev Biol 8, 587697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dakup PP et al. (2018) The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget 9 (18), 14524–14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pathria P et al. (2019) Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol 40 (4), 310–327. [DOI] [PubMed] [Google Scholar]
- 68.Mehla K and Singh PK (2019) Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 5 (12), 822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gabrusiewicz K et al. (2016) Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. Jci Insight 1 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alexander RK et al. (2020) Bmal1 integrates mitochondrial metabolism and macrophage activation. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Early JO et al. (2018) Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc Natl Acad Sci U S A 115 (36), E8460–E8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu XF et al. (2013) T(H)17 Cell Differentiation Is Regulated by the Circadian Clock. Science 342 (6159), 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu X et al. (2016) Synthetic RORgamma agonists regulate multiple pathways to enhance antitumor immunity. Oncoimmunology 5 (12), e1254854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hand LE et al. (2020) Regulatory T cells confer a circadian signature on inflammatory arthritis. Nat Commun 11 (1), 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee IK et al. (2020) RORalpha Regulates Cholesterol Metabolism of CD8(+) T Cells for Anticancer Immunity. Cancers (Basel) 12 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Newman AM and Alizadeh AA (2016) High-throughput genomic profiling of tumor-infiltrating leukocytes. Curr Opin Immunol 41, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao Y et al. (2014) Clock upregulates intercellular adhesion molecule-1 expression and promotes mononuclear cells adhesion to endothelial cells. Biochem Biophys Res Commun 443 (2), 586–91. [DOI] [PubMed] [Google Scholar]
- 78.Sato S et al. (2014) A Circadian Clock Gene, Rev-erb alpha, Modulates the Inflammatory Function of Macrophages through the Negative Regulation of Ccl2 Expression. Journal of Immunology 192 (1), 407–417. [DOI] [PubMed] [Google Scholar]
- 79.Qin B and Deng Y (2015) Overexpression of circadian clock protein cryptochrome (CRY) 1 alleviates sleep deprivation-induced vascular inflammation in a mouse model. Immunol Lett 163 (1), 76–83. [DOI] [PubMed] [Google Scholar]
- 80.Shivshankar P et al. (2020) Circadian Clock and Complement Immune System-Complementary Control of Physiology and Pathology? Front Cell Infect Microbiol 10, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Markiewski MM et al. (2008) Modulation of the antitumor immune response by complement. Nat Immunol 9 (11), 1225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engblom C et al. (2016) The role of myeloid cells in cancer therapies. Nat Rev Cancer 16 (7), 447–62. [DOI] [PubMed] [Google Scholar]
- 83.Duhan V and Smyth MJ (2021) Innate myeloid cells in the tumor microenvironment. Curr Opin Immunol 69, 18–28. [DOI] [PubMed] [Google Scholar]
- 84.Wculek SK et al. (2020) Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 20 (1), 7–24. [DOI] [PubMed] [Google Scholar]
- 85.DeNardo DG and Ruffell B (2019) Macrophages as regulators of tumour immunity and immunotherapy. Nature Reviews Immunology 19 (6), 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ruffell B and Coussens LM (2015) Macrophages and therapeutic resistance in cancer. Cancer Cell 27 (4), 462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Palma M and Lewis CE (2013) Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 23 (3), 277–286. [DOI] [PubMed] [Google Scholar]
- 88.Rivera LB et al. (2015) Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep 11 (4), 577–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Keklikoglou I et al. (2018) Periostin Limits Tumor Response to VEGFA Inhibition. Cell Rep 22 (10), 2530–2540. [DOI] [PubMed] [Google Scholar]
- 90.Barrett R and Pure E (2020) Cancer-associated fibroblasts: key determinants of tumor immunity and immunotherapy. Curr Opin Immunol 64, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meric-Bernstam F et al. (2021) Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 397 (10278), 1010–1022. [DOI] [PubMed] [Google Scholar]
- 92.Lian S et al. (2019) Dual blockage of both PD-L1 and CD47 enhances immunotherapy against circulating tumor cells. Scientific Reports 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu X et al. (2018) Dual Targeting of Innate and Adaptive Checkpoints on Tumor Cells Limits Immune Evasion. Cell Rep 24 (8), 2101–2111. [DOI] [PubMed] [Google Scholar]
- 94.Baer C et al. (2016) Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol 18 (7), 790–802. [DOI] [PubMed] [Google Scholar]
- 95.Guerriero JL et al. (2017) Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543 (7645), 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kaneda MM et al. (2016) PI3K gamma is a molecular switch that controls immune suppression. Nature 539 (7629), 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhu Y et al. (2014) CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74 (18), 5057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao D et al. (2020) Chromatin Regulator, CHD1, Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Deficient Prostate Cancer. Cancer Discov 10(9):1374–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liao W et al. (2019) KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 35 (4), 559–572 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Highfill SL et al. (2014) Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med 6 (237), 237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Flores-Toro JA et al. (2020) CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U S A 117 (2), 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu X et al. (2017) Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 543 (7647), 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu M et al. (2020) Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut 69 (2), 365–379. [DOI] [PubMed] [Google Scholar]
- 104.Davis RJ et al. (2017) Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3K delta/gamma. Cancer Research 77 (10), 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu T et al. (2019) Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol 12 (1), 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rahma OE and Hodi FS (2019) The Intersection between Tumor Angiogenesis and Immune Suppression. Clinical Cancer Research 25 (18), 5449–5457. [DOI] [PubMed] [Google Scholar]
- 107.Hou A et al. (2020) Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front Immunol 11, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu X et al. (2018) In Vitro Priming of Adoptively Transferred T Cells with a RORgamma Agonist Confers Durable Memory and Stemness In Vivo. Cancer Res 78 (14), 3888–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karaboué A et al. (2020) Circadian variation in nivolumab efficacy in patients with advanced non-small cell lung cancer. Journal of Clinical Oncology 38 (15), e21585. [Google Scholar]
- 110.Goswami S et al. (2020) Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nature Medicine 26 (1), 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharma P and Allison JP (2020) Dissecting the mechanisms of immune checkpoint therapy. Nature Reviews Immunology 20 (2), 75–76. [DOI] [PubMed] [Google Scholar]