

Abstract
PD-L1 expression is elevated in various human cancers, including colorectal cancer. High levels of PD-L1 expressed on tumor epithelial cells are one of the potential mechanisms by which tumor cells become resistant to immune attack. However, PD-L1 regulation in tumor cells is not fully understood. Here we demonstrate that mutations in the Adenomatous Polyposis Coli (APC) gene lead to colonic epithelial cell resistance to CD8+ T cell cytotoxicity by induction of PD-L1 expression. Mechanistically this occurs as a result of the β-catenin/TCF4 complex binding to the PD-L1 promoter, leading to increased transcription. Our findings not only reveal a novel mechanism by which APC mutations induce tumor immune evasion via an immune checkpoint pathway, but also pave the way for developing β-catenin or TCF4 inhibitors as possible new options for immune checkpoint inhibition.
Keywords: Adenomatous polyposis coli (APC), PD-L1, β-catenin/TCF, tumor immune evasion, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most common malignancy and the second leading cause of cancer-related deaths in the USA. Although colonoscopy screening is an effective way to detect and prevent CRC by polyp removal, CRC still results in significant mortality in the USA, and existing therapies have limited efficacy for stage 4 disease. Understanding the molecular mechanisms responsible for colorectal adenoma initiation, growth, and progression could lead to novel strategies for CRC prevention and interception.
CRC is a heterogeneous disease, that includes at least three major forms: hereditary, sporadic, and colitis-associated CRC. Familial adenomatous polyposis (FAP) is an inherited CRC syndrome caused by germ-line mutations in one allele of the adenomatous polyposis coli (APC) tumor suppressor gene. In addition, somatic mutations of this single gatekeeper gene, APC, occurs very early as the initiating event with high frequency (85%) in sporadic colorectal adenomas and carcinomas but is not observed until much later stages of colitis-associated CRC with low frequency [1, 2]. This high mutational frequency is unique to some hereditary forms and sporadic CRC. Furthermore, animal studies provide direct evidence demonstrating that loss of Apc causes the formation of adenomas [3]. The APC protein has multiple functional domains that bind to β-catenin, Axin, and GSK3β to form a complex. This complex targets β-catenin for degradation and inhibits its entry to the nucleus. Loss of functional APC resulting from a c-terminal truncating mutations in colorectal epithelial cells leads to the disruption of complex formation, allowing β-catenin to translocate into the nucleus. Although β-catenin doesn’t bind to DNA, it acts as a transcriptional co-factor by binding to the transactivation domain of the TCF4 transcription factor [4]. The β-catenin/TCF4 complex activates transcription of a number of target genes such as c-Myc, cyclin D1, and c-Jun which regulate colorectal epithelial cell transformation and proliferation. Collectively, APC mutations initiate hereditary and sporadic colorectal adenoma formation and growth by promoting colorectal epithelial cell transformation and proliferation. However, the impact of APC mutations in transformed colorectal epithelial cells on tumor immune evasion has not been reported.
Tumor cells can evade immunosurveillance by directly impairing CD8+ T cell cytotoxic activity and proliferation through immune checkpoint receptors such as PD-1. For example, interaction of PD-1 on activated CD8+ T cells with its ligand, PD-L1 on tumor epithelial cells, suppresses CD8+ T cell cytotoxicity and proliferation. Therefore, high levels of PD-L1 expressed by tumor epithelial cells represents one of the mechanisms by which tumor cells become resistant to CD8+ T cell cytotoxicity. Indeed, PD-L1 expression is elevated in various human cancers, including CRC [5-7] and its expression is associated with poor prognosis among patients with these cancers [8-11]. However, the mechanism(s) by which PD-L1 is regulated in tumor epithelial cells is not fully understood. In this study, we investigated whether loss of APC induces transformed colonic epithelial cell resistance to CD8+ T cell cytotoxicity via induction of PD-L1 expression.
Results
Knockdown of APC induces PD-L1 expression via a β-catenin-TCF pathway
To investigate how PD-L1 is regulated, we analyzed potential transcription factor binding sites within the PD-L1 promoter. We found that the PD-L1 promoter contains three potential TCF4 binding elements (TBEs) located at TBE1 (−727 to −720), TBE2 (−592 to −585), and TBE3 (−467 to −460) relative to the transcription start site. Thus, we first examined whether knockdown of APC induces PD-L1 expression in normal colon epithelial cells. Indeed, we found that knockdown of APC using siRNA resulted in the induction of PD-L1 expression both at the protein (Fig. 1A) and mRNA levels (Fig. 1B) in HCEC-1CT cells (an immortalized normal human colonic epithelial cell line [12]). Importantly, our flow cytometry results further revealed that knockdown of APC in these cells also induced PD-L1 expression on the cell surface (Fig. 1C). In addition, knockdown of APC in these cells also led to accumulation of β-catenin (Fig. 1A), indicating that β-catenin is required for loss of APC induction of PD-L1. Importantly, knockdown of β-catenin attenuated the effect of APC loss on PD-L1 expression in HCEC-1CT cells (Fig. 1A-C), whereas overexpression of constitutively active β-catenin resulted in induction of PD-L1 expression in HCEC-1CT cells (Fig. 1D-F). These results demonstrate that β-catenin is required for APC loss-induced PD-L1 expression. Moreover, overexpression of dominant negative TCF4 (TCF4-DN) inhibited the effect of constitutively active β-catenin (β-catenin-CA) on induction of PD-L1 expression in HCEC-1CT cells (Fig. 1D-F), demonstrating that TCF4 is required for β-catenin induction of PD-L1. Similar results following APC knockdown were confirmed in 293T cells (Supplemental Fig. 1).
Fig. 1. Alteration of APC affects PD-L1 expression via a β-catenin/TCF4 pathway.
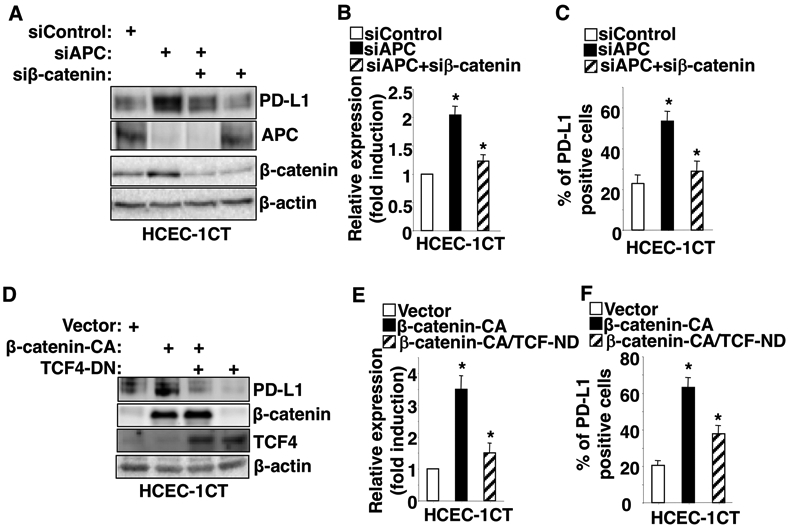
HCEC-1CT cells were transiently transfected with indicated siRNA or expression plasmid. After transfection, cells were cultured for 48 hr and subjected to Western blot, q-PCR, and flow cytometry. (A and D) PD-L1 protein levels in HCEC-1CT cells transfected with siControl, siAPC, siβ-catenin, constitutively active β-catenin (β-catenin-CA) plasmid, and/or dominant negative TCF4 (TCF4-DN) plasmid were measured by Western blot. (B and E) PD-L1 mRNA levels in HCEC-1CT cells transfected with siControl, siAPC, siβ-catenin, β-catenin-CA plasmid, and/or TCF4-DN plasmid were measured by q-PCR. (C and F) The cell surface PD-L1 protein levels in HCEC-1CT cells transfected with siControl, siAPC, siβ-catenin, β-catenin-CA plasmid, and/or TCF4-DN plasmid were measured by flow cytometry. Each Western blot image is representative of three independent experiments with similar results. Data are presented as mean ± SEM of three independent experiments. *p<0.05.
Similar to our findings in normal colonic epithelial cells, deletion of the APC gene by CRISPR/Cas9 induced PD-L1 expression and β-catenin accumulation in the nucleus of HCA-7 cells (a human CRC cell line with WT APC and WT β-catenin genes) (Fig. 2A-C). Moreover, overexpression of constitutively active β-catenin resulted in induction of PD-L1 expression in HCA-7 cells, whereas overexpression of dominant negative TCF4 inhibited the effect of activated β-catenin on induction of PD-L1 expression in these cells (Fig. 2D-F). In contrast, overexpression of WT APC (APC-OE) reduced PD-L1 expression and nuclear translocation of β-catenin in HT-29 cells (a human CRC cell line with mutant APC) (Fig. 2G-I). Collectively, these results demonstrate that loss of APC induces PD-L1 expression via a β-catenin-TCF4 pathway in human colonic epithelial cells.
Fig. 2. Knockout or overexpression of APC alters PD-L1 expression via a β-catenin/TCF4 pathway.
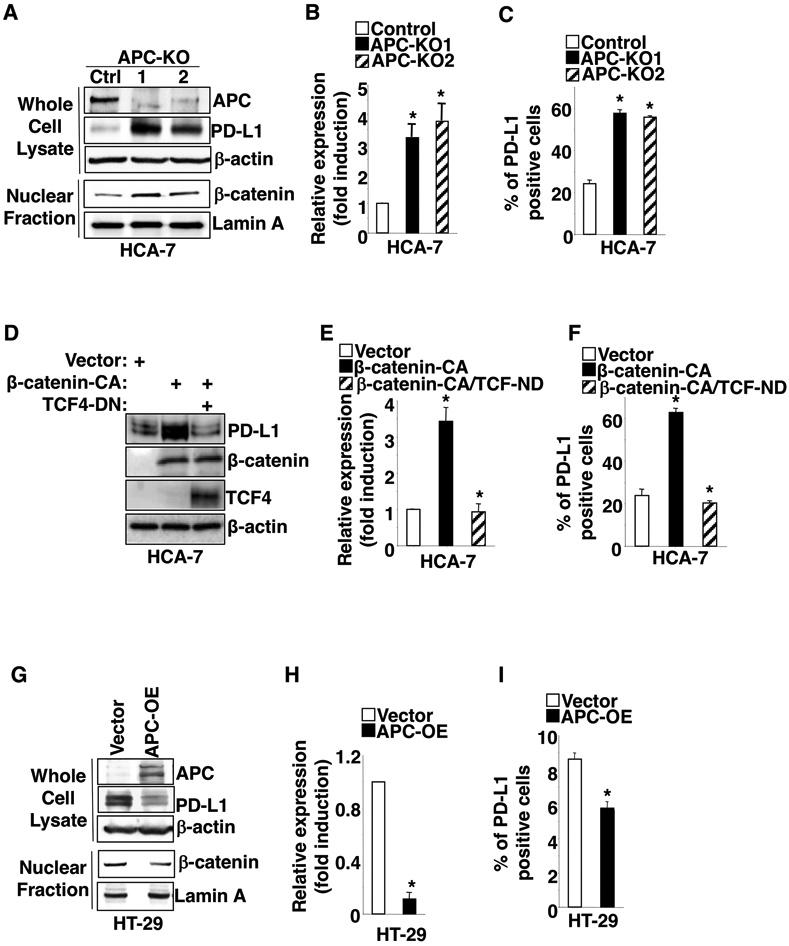
(A-C) APC gene was deleted by CRISPR/Cas9 in HCA-7 cells. The protein levels of indicated genes in whole cell lysate or nucleus were measured by Western blot (A), whereas PD-L1 levels at mRNA and cell surface were measured by q-PCR (B) and flow cytometry (C) in HCA-7/control, HCA-7/APC-KO1, and HCA-7/APC-KO1 cells. APC-KO: APC knockout. (D-F) The protein levels of indicated genes in whole cell lysate were measured by Western blot (D), whereas PD-L1 levels at mRNA and cell surface were measured by q-PCR (E) and flow cytometry (F) in HCA-7/vector, HCA-7/β-catenin-CA, and HCA-7/β-catenin-CA+TCF-DN cells. (G-I) The protein levels of indicated genes in whole cell lysate and nucleus were measured by Western blot (G), whereas PD-L1 levels at mRNA and cell surface were measured by q-PCR (H) and flow cytometry (I) in HT-29/vector and HT-29/APC-OE cells. APC-OE: APC overexpression. Each Western blot image is representative of three independent experiments with similar results. Data are presented as mean ± SEM of three independent experiments. *p<0.05.
Loss of APC induces PD-L1 transcription by inducing the binding of the β-catenin/TCF4 complex to the PD-L1 promoter
We first determined whether the β-catenin-TCF4 complex activates PD-L1 transcription. As expected, overexpression of constitutively active β-catenin resulted in induction of PD-L1 transcription, whereas overexpression of dominant negative TCF4 attenuated the effect of constitutively active β-catenin on induction of PD-L1 transcription in HCEC-1CT cells (Fig. 3A). Similar results were obtained in 293T cells (Supplemental Fig. 2A). To determine which potential TCF4 binding sites within the PD-L1 promoter are bound by the β-catenin/TCF4 complex, ChIP assays were performed. Our results showed that loss of APC induced the binding of the β-catenin/TCF4 complex to TBE3 (−467 to −460) of PD-L1 promoter in HCEC-1CT and HCA-7 cells (Fig. 3B-C), but not to TBE1 (−727 to −720) and TBE2 (−592 to −585) (data not shown). Similar results were found in 293T cells (Supplemental Fig. 2B). We further determined whether the binding of the β-catenin/TCF4 complex to TBE3 of PD-L1 promoter is required for PD-L1 transcription. Analyses of PD-L1 promoter-luciferase assays revealed that deletion of TBE3 within the PD-L1 promoter attenuated the effect of constitutively active β-catenin on induction of PD-L1 transcription in both HCEC-1CT cells and HCA-7 cells (Fig. 3D-E). In contrast, deletion of TBE1 or TBE2 did not affect activated β-catenin-induced PD-L1 transcription (Fig. 3D-E). Moreover, deletion of TBE3 within the PD-L1 promoter attenuates PD-L1 transcription induced by deletion or knockdown of APC in HCEC-1CT cells and HCA-7 cells (Fig. 3F). Similar results were also observed in 293T cells (Supplemental Fig. 2C-D). Collectively, these results demonstrate that loss of APC induces PD-L1 expression by inducing the binding of the β-catenin/TCF4 complex to the PD-L1 promoter and by initiating its transcription in both normal and tumor colorectal epithelial cells.
Fig. 3. Loss of APC induces PD-L1 transcription by binding of β-catenin/TCF4 complex to PD-L1 promoter.
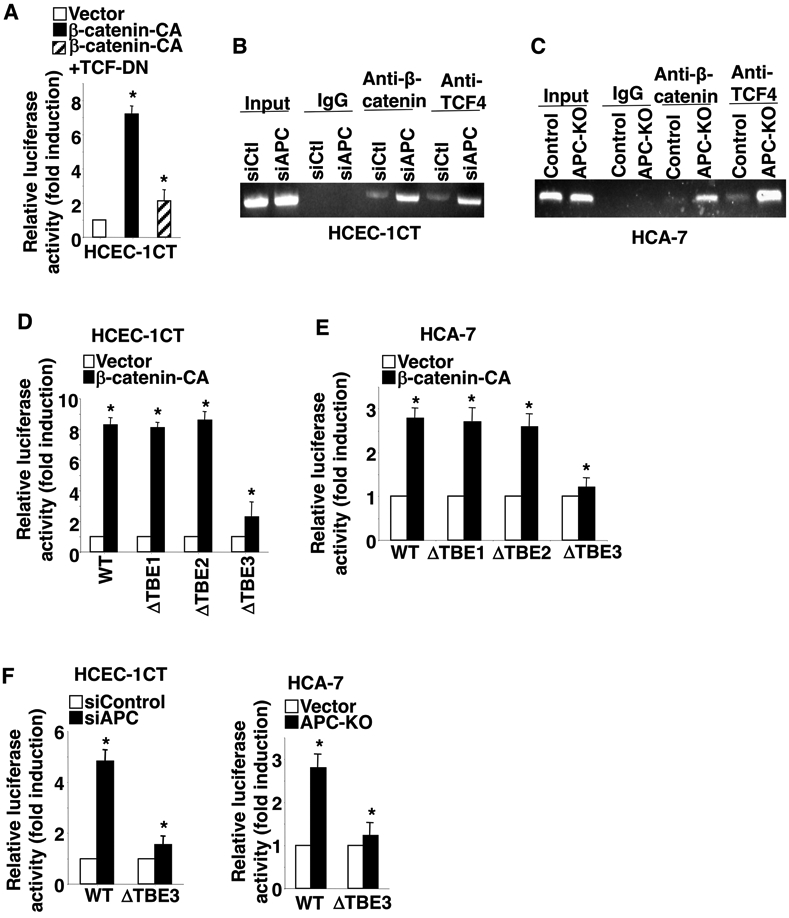
(A) HCEC-1CT cells were transiently co-transfected with PD-L1 promoter luciferase reporter and pRL-SV40 Renilla luciferase plasmids plus control vector, β-catenin-CA, and/or TCF4-DN plasmids. (B-C) A representative image of three independent ChIP assays for binding of the β-catenin/TCF4 complex to TBE3 of the PD-L1 promoter in HCEC-1CT/siControl, HCEC-1CT/siAPC, HCA-7/vector, and HCA-7/APC-OE cells. Immunoprecipitation with IgG antibody was used as a control. (D-F) Luciferase activity of the wild-type and mutant PD-L1 promoters was measured in HCEC-1CT/vector, HCEC-1CT/β-catenin-CA, HCA-7/vector, HCA-7/β-catenin-CA, HCEC-1CT/siControl, HCEC-1CT/siAPC, and HCA-7/APC-OE cells. ΔTBE1, ΔTBE2, and ΔTBE3 indicate mutant PD-L1 promoter with deletion of either TBE1, TBE2, or TBE3. Data are presented as mean ± SEM of relative luciferase activity from three independent experiments. *p<0.05.
Loss of APC induces colonic transformed epithelial cell resistance to CD8+ T cell cytotoxicity via the β-catenin-PD-L1 pathway
To evaluate relative levels of resistance of colonic transformed epithelial cells to CD8+ T cell cytotoxicity, cells were co-cultured with TALL-104 cells (a human leukemic CD8+ T cell line with high cytotoxicity against human tumor cells). As expected, HCEC-1CT cells with loss of APC or overexpression of constitutively active β-catenin are more resistant to CD8+ T cell cytotoxicity than controls (Fig. 4A). Similar results were also obtained in 293T cells (Supplemental Fig. 3A-B). Moreover, levels of cleaved PARP and cleaved caspase 3 in HCEC-1CT cells with loss of APC or overexpression of constitutively active β-catenin are lower than vector control cells after co-culture (Supplemental Fig. 3C), demonstrating that loss of APC or overexpression of constitutively active β-catenin induces resistance to CD8+ T cell cytotoxicity. In contrast, treatment of co-cultured cells with atezolizumab (an anti-PD-L1 antibody) increased sensitivity to CD8+ T cell cytotoxicity accompanied by induction of cleaved PARP and cleaved caspase 3 and completely blocked APC loss-induced cell resistance to CD8+ T cell cytotoxicity in both HCEC-1CT and HCA-7 cells (Fig. 4B-C and Supplemental Fig. 3C-D). Moreover, overexpression of PD-L1 in HCEC-1CT cells also increased resistance to CD8+ T cell cytotoxicity (Fig. 4D). Similarly, atezolizumab treatment increased sensitivity to CD8+ T cell cytotoxicity and completely inhibited active β-catenin- or PD-L1 overexpression-induced resistance to CD8+ T cell cytotoxicity in HCEC-1CT cells (Fig. 4D). In agreement with these results, knockdown of PD-L1 with shPD-L1 in HT-29 cells restored sensitivity to CD8+ T cell cytotoxicity accompanied by induction of cleaved PARP and cleaved caspase 3 (Fig. 4E and Supplemental Fig. 3E). Overall this work demonstrates that loss of APC induces colonic transformed epithelial cell resistance to CD8+ T cell cytotoxicity via the β-catenin-PD-L1 pathway.
Fig. 4. Loss of APC induces cell resistance to CD8+ T cell cytotoxicity by induction of PD-L1 via the β-catenin/TCF4 pathway.
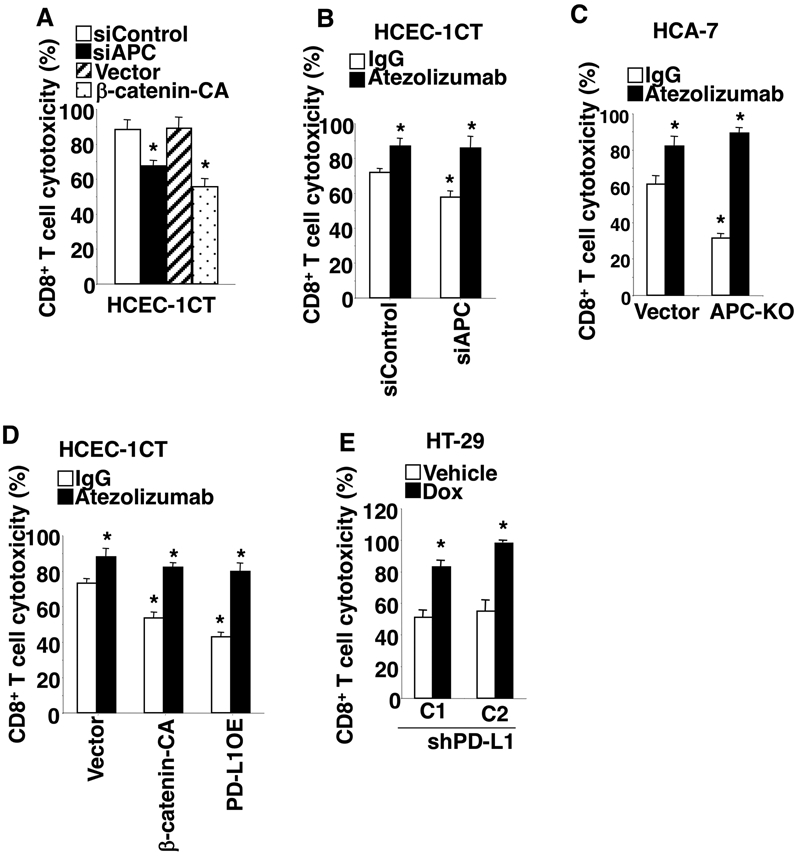
(A) Cytotoxicity of TALL-104 cells against HCEC-1CT/siControl, HCEC-1CT/siAPC, HCEC-1CT/vector, or HCEC-1CT/β-catenin-CA cells was determined as described in the Method section. (B-D) TALL-104 cells were co-cultured with either HCEC-1CT/siControl, HCEC-1CT/siAPC, HCEC-1CT/vector, HCEC-1CT/β-catenin-CA, or HCEC-1CT/PD-L1-OE, HCA-7/vector, or HCA-7/APC-KO cells and treated with IgG or atezolizumab. The TALL-104 cell cytotoxicity against indicated colon epithelial cells was determined as described above. (E) After HT-29/shPD-L1 cells were treated with vehicle or doxycycline at 1 μg/ml for 48h, these cells were co-cultured with TALL-104 cells with doxycycline. The TALL-104 cell cytotoxicity against indicated colon epithelial cells was determined as described above. Data are presented as mean ± SEM of three independent experiments. *p<0.05.
Mutation of Apc leads to induction of PD-L1 expression in transformed colonic epithelial cells accompanied by promotion of resistance to CD8+ T cell cytotoxicity in vivo
To validate our in vitro results animal experiments were performed. Deletion of Apc resulted in an increase of PD-L1-expressing colonic epithelial cells in Apcfl/fl/CDX2-Cre mice as compared to their littermate controls following tamoxifen treatment (Fig. 5A-B). Similarly, PD-L1 expression is elevated in small intestinal epithelial cells of ApcMin+/− mice as compared to WT mice (Supplemental Fig. 4). Moreover, deletion of Apc resulted in increased colonic epithelial cell resistance to CD8+ T cell cytotoxicity than controls (Fig. 5C). Importantly, treatment of Apcfl/fl/CDX2-Cre mice with an anti-PD-L1 antibody significantly inhibited colon adenoma formation and growth (Fig. 5D). Collectively, these results suggest that Apc loss of function induces colonic tumor formation and growth at least partially by inducing transformed colonic epithelial cell resistance to CD8+ T cell cytotoxicity via PD-L1.
Fig. 5. Deletion of Apc induces tumor immune evasion by induction of PD-L1 in vivo.

(A) The representative immunostaining of PD-L1 in colon tissues taken from the indicated group of mice. (B) The percentage of PD-L1-positive colon epithelial cells was determined by flow cytometry in the indicated group of mice. (C) The percentage of dead colon epithelial cells was determined by flow cytometry after co-culture with activated mouse splenic CD8+ T cells in the indicated group of mice. (D) Average numbers of polyps at different size and total that includes all sizes in mice treated with IgG or anti-PD-L1 antibody. The error bar indicates ± SEM. *p<0.05.
Discussion
For PD-1 ligands, only PD-L1 is also expressed on non-immune cells such as epithelial and endothelial cells [13, 14]. High levels of PD-L1 on tumor epithelial cells represent one of the potential mechanisms by which tumor cells become resistant to immune attack. PD-L1 expression has been shown to be regulated by tumor suppressor genes, oncogenes, pro-inflammatory cytokines, or hypoxia [15]. For example, previous in vitro studies have shown that loss of PTEN resulted in induction of PD-L1 expression in glioma and CRC cells [9, 16] and overexpression of mutant EGFR led to induction of PD-L1 in immortalized bronchial epithelial cells [17]. However, somatic mutations of PTEN occur in 2-6% of CRC [18] and EGFR mutations are somewhat rare events in CRC. Considering the importance of APC mutations in CRC, we obtained the first evidence demonstrating that loss of APC results in induction of PD-L1 in transformed colonic epithelial cells in vitro and in vivo. This finding provides one of the possible explanations responsible for PD-L1 elevation in colorectal tumor epithelial cells.
The PD-L1 promoter contains several transcription factor binding sites, including HIF1/2, MYC, NF-κB, β-catenin/TCF, and STAT1/3. For example, MYC, an oncogenic transcription factor, induced PD-L1 expression by directly binding to the PD-L1 promoter in melanoma and NSCLC cells [19]. INFγ induced PD-L1 expression via the JAK1/2-SATAT1/3 pathway in lung, ovarian, and colon cancer cell lines as well as melanoma cells [5, 20]. A recent report showed that activation of β-catenin induced PD-L1 expression in goloblastoma cells [21]. Hypoxia induced PD-L1 expression occurs via binding of HIF-1α to the PD-L1 promoter in breast and prostate cancer cell lines as well as melanoma cells [22]. Chemotherapeutic agents have been shown to induce PD-L1 expression via NF-κB in ovarian cancer cells [23]. In this study, we reveal a novel mechanism by which loss of APC results in increased PD-L1 expression by transcriptional regulation via the β-catenin/TCF4 complex binding to the PD-L1 promoter.
It is well established that APC mutations initiate hereditary and sporadic colorectal adenoma formation and growth by induction of colorectal epithelial cell transformation and proliferation. Here, we provide the first evidence that we know of showing that APC mutation-induced PD-L1 expression in colon epithelial cells resulted in increased resistance to CD8+ T cell cytotoxicity. This finding reveals a novel effect of APC loss in tumor immune evasion and expands our understanding of the role of APC in CRC.
In summary, loss of APC leads to increased PD-L1 expression via enhanced binding of the β-catenin/TCF4 complex to the PD-L1 promoter in colonic epithelial cells. Elevated PD-L1 by APC loss of function results in induction of colon epithelial resistance to CD8+ T cell cytotoxicity. In contrast, overexpression of WT APC increase the sensitivity of APC mutant colon cancer cells to CD8+ T cell cytotoxicity by reduction of PD-L1 expression. PD-L1 is required for Apc mutation-induced colon adenoma formation and growth. These findings uncover a previously unreported mechanism by which APC mutations induce adenoma formation and growth, in part, by inducing colonic epithelial resistance to CD8+ T cell cytotoxicity via PD-L1, and provide a rationale for development of β-catenin or TCF4 inhibitors as new alternative immune checkpoint inhibitors that could be useful especially in intercepting disease progression at a very early stage.
Materials and Methods
Cell culture and reagents
HCA-7, HT-29, 293T, and TALL-104 cell lines were obtained from ATCC (Manassas, VA). HCA-7 and HT-29 cells were cultured in McCoy’s 5A medium with 10% FBS. 293T cells were cultured in DMEM medium with 10% FBS. TALL-104 cells were cultured in RPMI1640 medium supplemented with recombinant IL-2 (Peprotech, Cat#200-02) and 20% FBS (Gibco, Cat#10082147). HCEC-1CT cells were cultured in DMEM medium (Corning, Cat#10-013-cv) supplemented with 25 ng/mL EGF (Invitrogen, Cat#PHG0311), 1 μg/mL hydrocortisone (Sigma, Cat#H0888), 1× insulin -transferrin-sodium selenite (Gibco, Cat# 41400045), 50 μg/mL gentamicin (Gibco, Cat#15750-060), and 2% cosmic calf serum (HyClon, Cat# SH30087.02). All cell lines have been tested by MycoProbe Mycoplasma Detection Kit (R&D) and also authenticated before the experiment according ATCC STR database.
Transient and stable transfection
siRNAs targeting APC (Cat#L-003869-00-0005) and β-catenin (Cat#L-003482-00-0005) were obtained from Horizon/Dharmacon. β-catenin-CA was a gift from Bob Weinberg (Addgene). TCF-DN was a gift from Bert Vogelstein (Addgene). APC plasmid was obtained from Horizon/Discoveries. PD-L1 plasmid was obtained from Origene. Doxycycline inducible shPD-L1 were obtained from Horizon/Discoveries. Indicated cells were transfected with indicated siRNAs, shRNAs, and/or expression plasmids by HiPerFect transfection reagents (Qiagen, Cat#301705) or Lipofectamine 3000 reagent (Cat#L3000-015 according to manufacturer’s instructions. For stable transfection (shRNAs and expression plasmids), transduced cells were selected with puromycin (ThermoFisher Scientific, Cat# A1113802) for 14 days and pooled. To generate CRISPR/Cas9 plasmids targeting APC, primers of single-guide RNAs were synthesized at Integrated DNA Technologies, annealed, ligated into LentiCRISPRv2 (Addgene, Cat#52961) and transduced to cells by lentivirus using Lenti-X packaging single shots (VSV-G, Clontech, Cat#631276). Transduced cells were selected with puromycin for 14 days and pooled. The targeted sequences are: APC-1-GGATCTGTATCAAGCCGTTC, APC-2-GATTTATTAGAGCGTCTTAA. LentiCRISPR-eGFP (Addgene) was used as the control.
Western blot analysis
Whole cell or nuclear extracts were prepared from indicated cells. Transfer membranes were blocked with 5% dry milk in TBS-T buffer for 1 h and then incubated with anti-PD-L1 (1:1000, Cat#13684), anti-β-catenin (1:1000, Cat#8480), anti-TCF4 (1:1000, Cat#2565), anti-cleaved PARP (1:1000, Cat#5625), or anti-cleaved caspase 3 (1:1000, Cat#9664) antibody for overnight at 4 °C. These antibodies were obtained from Cell Signaling Technology. Anti-APC (1:1000, Cat#MABC202) was from EMD Millipore. The blots were stripped and re-probed with β-actin (Sigma, Cat#A1978) or anti-Lamin A (Santa Cruz, Cat#SC-56137) antibody.
Quantitative PCR
Total RNA was isolated from indicated cells using the RNeasy Mini Kit (Qiagen, Cat#74004) and was reversely transcribed to cDNA using iScript cDNA Synthesis Kit (Bio-Rad,Cat#1708891). Real-time q-PCR was performed with Supermix (Bio-Rad, Cat#1708882) using QuantStudio 7 Flex Real-time PCR System (Life Technologies). Primers for the CD274 (PD-L1) and Actin genes were synthesized by Integrated DNA technologies. The sequences of the specific PCR primers were as follows (5' to 3'): PD-L1-forward: 5’-AAA TGG AAC CTG GCG AAA GC-3’; PD-L1-reverse: 5’-GAT GAG CCC CTC AGG CAT TT-3’; Actin-forward: 5’-ACC TTC TAC AAT GAG CTG CG-3’; Actin-reverse: 5’-CCT GGA TAG CAA CGT ACA TGG-3’. The relative expression of target gene is the averages of triplicates that are normalized against the transcription levels of Actin. Data are represented as the means ± SE of the fold induction from three independent experiments.
Luciferase reporter assays
The human PD-L1 promoter (−937 to −30) was cloned into pGL3 luciferase reporter vector (Promega). The TBE1 (−727 to −720), TBE2 (−592 to −585), or TBE3 (−467 to −460) from WT was deleted by using QuickChange II XL Site-Directed Mutagenesis Kit (Agilent, Cat#200522-5). Indicated cells were co-transfected with 0.3 μg luciferase reporter genes and 0.03 μg pRL-SV40 (Renilla luciferase) by the Lipofectamine 3000 reagent following manufacturer's protocol (Life Technologies, Inc. Rockville, MA) in 24-well plate. Luciferase activity was measured using the Dual-Luciferase Reporter Assay Kit (Promega, Cat#E1960) and a Monolight 3010 luminometer according to manufacturer’s instructions (BD Biosciences/Pharmingen, San Diego, CA). The relative luciferase activity was determined and normalized to Renilla luciferase.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using a ChIP assay kit (EMD Millipore, Cat#MAGNA0017). Briefly, indicated cells were treated with 1% formaldehyde-containing medium for 10 min at 37°C to crosslink proteins to DNA. Crosslinked chromatins were sonicated to reduce the DNA length to 200 and 1000 bp. Samples of total chromatin were taken at this point to use as a positive control (input chromatin). The cell lysates were precleared by incubation with Protein G-sepharose beads and then incubated with anti-β-catenin (1 μg, Cell Signaling, Cat#8480) or anti-TCF4 (1 μg, Cell Signaling, Cat#2565) overnight at 4°C. DNA–protein complexes were collected with Protein G-sepharose beads followed by several rounds of washing, eluted and reverse cross-linked. Following treatment with Protease K, the samples were extracted with phenol-chloroform and precipitated with ethanol. The recovered DNA was resuspended in Tris-HCl-EDTA buffer and used for the PCR amplification. The primer sequences were: forward: 5′-GAA GTC ACA GAA TCC ACG AT-3′; reverse: 5′-TCA GCA GCA GAC CCA TAT-3′.
Animal experiments
All animal experiments conformed to our animal protocols that were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. Apcfl/fl/CDX2-creERT2 mice and their control littermates (Apc+/+/CDX2-cre) were generated by conventionally crossing Apcfl/fl mice on the C57BL/6 genetic background (NCI) with CDX2-creERT2 mice on the C57BL/6J genetic background (Jackson Laboratory, Bar Harbor, Maine). Male Apcfl/fl/CDX2-cre mice aged 8 weeks and their control littermates were injected intraperitoneally with tamoxifen (40 mg/kg, Sigma, Cat#T5648-1G). In one week after tamoxifen treatment, colon tissues were collected. Excised colons from 4 mice in each group were fixed in 10% neutral-buffered formalin and embedded in paraffin. The tissue sections were subjected to IHC staining. The remaining excised colon tissues from 5 mice in each group were weighted, minced, and digested by DMEM medium supplemented with 1% FBS, 0.5 mM DTT (Sigma-Aldrich, Cat#D0632), 20μg/ml dispase II (Roche, Cat#10805400), and 75 U/ml collagenase II (Gibco, Cat#17101-015) for 3 hrs at 37 °C under slow rotation. Single-cell suspensions were subjected to flow cytometry analyses. In addition, colon epithelial cells isolated from single-cell suspensions by Flow cytometry sorting using anti-mEpCAM (CD326)-PE-Cy7 antibody (1:100, Biolegend, Cat#118216) were subjected to CD8+ T cell cytotoxicity assays. In addition, male Apcfl/fl/CDX2-cre mice aged 8 weeks were treated with tamoxifen at same dose as mentioned above and randomly divided into 2 groups treated with IgG or anti-PD-L1 antibody (100μg/per mouse) by intraperitoneal injection 3 times per week for 3 weeks. At the end of the study, excised colons from each group were used to measure polyp number and size under a dissecting microscope. For all animal experiments, the group allocation was blinded when measuring PD-L1 expression, colonic epithelial cell resistance to CD8+ T cell cytotoxicity, and tumor burden.
Flow cytometry analysis
For human cell line, single-cell suspensions in staining buffer (BD, Cat#554657) were incubated with anti-hPD-L1-APC (1:200, Biolegend, Catl#329708) for 30 min on ice. For mouse colon tissues, single-cell suspensions in staining buffer were incubated with anti-mPD-L1-PE-Cy7 (1:200, eBioscience, Cat#25-5982-82), anti-CD45-FITC (1:100, Biolegend, Cat#1031080), anti-mEpCAM (CD326)-APC-Cy7 (1:500, Biolegend, Cat#118218), anti-CD31-APC (1:200, Biolegend, Cat#102410) antibodies for 30 min on ice. After the cells were washed twice with 1 ml of staining buffer, they were analyzed on a FortessaX20 Flow Cytometer (BD Biosciences). The flow cytometric profiles were analyzed by counting 20,000 events using FLOWJO software program (FLOWJO, LLC).
Immunohistochemical staining
Paraffin-embedded tissue sections (5 μm thick; n=5 per animal) were stained with anti-PD-L1 antibody (1:50, Cell Signaling, Cat#64988T) in 4°C for overnight. After wash, tissue sections were incubated with biotinylated goal anti-rabbit IgG (1:200, Vector, Cat#PK-6101). The immunohistochemical staining was completed by using a DAB substrate Kit, Peroxidase (Vector, Cat#SK-4100) according to the manufacturer’s instructions.
CD8+ T cell cytotoxicity assays
For human colon epithelial cells, TALL-104 cells were co-cultured with 1 x 104 indicated epithelial cells in 96-well round-bottomed plate at ratios (E:T=1:1) for 18 hr. In some experiments, co-cultured cells were treated with either IgG or 10 μg/ml atezolizumab (Selleck Chemicals, Cat#A2004). The cytotoxicity was measured using a CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Cat#G1781) according to manufacturer’s instructions. For mouse colon epithelial cells, excised spleen was smashed on 70 μm cell strainer. CD8+ T cells were isolated from spleen by using a mouse CD8+ T Cell Isolation Kit (Miltenyi Biotec, Cat#130-104-075) according to the manufacturer’s instructions. Isolated CD8+ T cells with a purity of 98% were activated by anti-CD3/CD28 mAb-coated beads (Dynabeads Mouse T-Activator CD3/CD28, Gibco, Cat#11452D) according to the manufacturer’s instructions. Activated CD8+ T cells were co-cultured with 0.5 x 106 isolated colonic epithelial cells in RPMI1640 with 10% FBS at ratios (E:T= 2:1) for 16 hr. After incubation, cells were incubated with anti-mCD3-APC (1:100, BD Biosciences, Cat#100312) and anti-EpCAM (CD326)-PE-Cy7 (1:100, Biolegend, Cat#118216) antibodies in staining buffer (Biolegend) for 30 min on ice. After the cells were washed twice with 1 ml of staining buffer, the cells were stained by propidium iodide (PI) using TACS™ Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s instructions (R&D System, cat#4830-01-K). The percentage of dead epithelial cells was determined as EpCAM+CD3−PI+ cells divided by EpCAM+CD3− cells by flow cytometry.
Statistical analysis
Each in vitro experiment was done at least 3 times and each in vivo experiment was conducted at least twice. Data are presented as mean ± SEM. Comparisons among multiple groups were performed by factorial analysis of variance, followed by Bonferroni test. Comparisons between two groups were performed with Student’s t-test or Mann-Whitney U test where appropriate. Fischer’s exact test was used for categorical variables. p<0.05 was considered significant. No samples from in vitro and in vivo experiments were excluded from the analysis.
Supplementary Material
Acknowledgements:
We thank the National Colorectal Cancer Research Alliance (NCCRA) for its generous support (R.N.D.). This work was supported in part by Flow Cytometry & Cell Sorting Unit, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313).
Footnotes
Conflict of interest disclosure statement: All authors have no any conflict interests
References:
- 1.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359: 235–237. [DOI] [PubMed] [Google Scholar]
- 2.Tarmin L, Yin J, Harpaz N, Kozam M, Noordzij J, Antonio LB et al. Adenomatous polyposis coli gene mutations in ulcerative colitis-associated dysplasias and cancers versus sporadic colon neoplasms. Cancer research (Research Support, U.S. Gov't, P.H.S.) 1995; 55: 2035–2038. [PubMed] [Google Scholar]
- 3.McIntyre RE, Buczacki SJ, Arends MJ, Adams DJ. Mouse models of colorectal cancer as preclinical models. Bioessays 2015; 37: 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999; 99: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 6.Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res 2003; 63: 6501–6505. [PubMed] [Google Scholar]
- 7.Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y et al. MiR-20b, −21, and −130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Human immunology 2014; 75: 348–353. [DOI] [PubMed] [Google Scholar]
- 8.Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res 2005; 11: 2947–2953. [DOI] [PubMed] [Google Scholar]
- 9.Song M, Chen D, Lu B, Wang C, Zhang J, Huang L et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PloS one 2013; 8: e65821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson RH, Webster WS, Cheville JC, Lohse CM, Dong H, Leibovich BC et al. B7-H1 glycoprotein blockade: a novel strategy to enhance immunotherapy in patients with renal cell carcinoma. Urology 2005; 66: 10–14. [DOI] [PubMed] [Google Scholar]
- 11.Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A 2007; 104: 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roig AI, Eskiocak U, Hight SK, Kim SB, Delgado O, Souza RF et al. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 2010; 138: 1012–1021 e1011-1015. [DOI] [PubMed] [Google Scholar]
- 13.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annual review of immunology 2008; 26: 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192: 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell 2019; 76: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007; 13: 84–88. [DOI] [PubMed] [Google Scholar]
- 17.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery 2013; 3: 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salvatore L, Calegari MA, Loupakis F, Fassan M, Di Stefano B, Bensi M et al. PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target. Cancers (Basel) 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016; 352: 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 2017; 19: 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du L, Lee JH, Jiang H, Wang C, Wang S, Zheng Z et al. beta-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J Exp Med 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 2014; 74: 665–674. [DOI] [PubMed] [Google Scholar]
- 23.Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res 2015; 75: 5034–5045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.