

Abstract
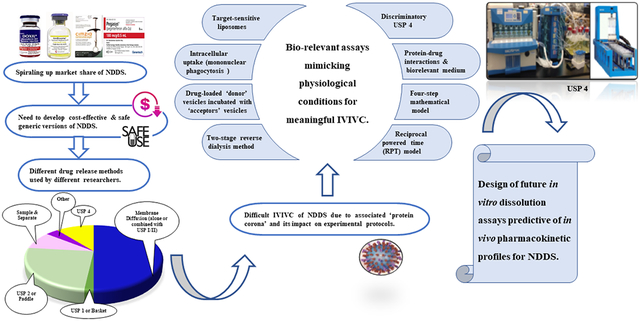
Nano drug delivery systems (NDDS) offer promising solution for the translation of future nanomedicines. As bioavailability and therapeutic outcomes can be improved by altering the drug release from these NDDS, it becomes essential to thoroughly understand their drug release kinetics. Moreover, U.S. Food and Drug Administration (FDA) requires critical evaluation of potential safety, efficacy and public health impacts of nanomaterials (FDA, 2014a). Spiraling up market share of NDDS has also stimulated the pharmaceutical industry to develop their cost-effective generic versions after the expiry of patent and associated exclusivity. However, unlike the conventional dosage forms, the in vivo disposition of NDDS is highly intricate and different from their in vitro behavior. Significant challenges exist in the establishment of in vitro-in vivo correlation (IVIVC) due to incomplete understanding of nanoparticles’ in vivo biofate and its impact on in vitro experimental protocols. A rational design of dissolution may serve as quality and quantity control tool and help develop a meaningful IVIVC for favorable economic implications. Clinically relevant drug product specifications (critical quality attributes) can be identified by establishing a link between in vitro performance and in vivo exposure. In vitro dissolution may also play a pivotal role to understand the dissolution-mediated clearance and safety of NDDS. Prevalent in vitro dissolution methods for NDDS and their limitations are discussed in this review, among which USP 4 is gaining more interest recently. Researchers are working diligently to develop biorelevant in vitro release assays to ensure optimal therapeutic performance of generic versions of these NDDS. This article focuses on these studies and presents important considerations for the future development of clinically relevant in vitro release methods.
Keywords: in vitro drug release, dissolution, nanoparticle, nano drug delivery systems, NDDS, IVIVC, USP 4, protein corona
Graphical Abstract
1 ∣. NANO DRUG DELIVERY SYSTEMS (NDDS) & CURRENT REGULATIONS
Nanotechnology is emerging as a promising approach for formulation development to improve therapeutic efficacy and safety of conventional dosage forms (Ventola, 2017). Presently, drugs in a variety of nano-sized dosage forms, such as liposomes, proteins and protein-drug conjugates, nanocrystals, polymeric nanoparticles, and other lipid-based systems, have successfully entered the market and are being used for diverse range of indications. A detailed list of commercialized NDDS is presented in Table 1. These NDDS propose innovative strategies for improving drug performances as compared to conventional formulations. NDDS can enhance drug solubility, reduce gastric degradation, provide controlled/sustained drug release or prolong circulation time to allow greater drug accumulation and better delivery efficacy (Deng et al., 2019). NDDS can circumvent physiological barriers (such as immune system, renal clearance, enzymatic and mechanical degradation) to achieve higher therapeutic drug levels, even with a lower dose. Increased permeability of vasculature and poor lymphatic drainage of tumor microenvironment allows passive targeting of NDDS (e.g., Doxil®, Baxter Healthcare Corp), well known as enhanced permeability and retention (EPR) effect. Active targeting is based on receptor-ligand binding. NDDS can be attached to a suitable ligand (e.g., antibodies, proteins) that binds to receptors present on specific cells (e.g., Trastuzumab binds to HER2 receptors) (Attia et al., 2019).
Table 1:
Marketed NDDS (Anselmo & Mitragotri, 2019; Bulbake et al., 2017; Farjadian et al., 2019; FDA, 2020a, 2020b; Wang et al., 2013; Weissig et al., 2014)
| PRODUCT | API | ROUTE | INITIAL APPROVAL |
INDICATION | COMPANY |
|---|---|---|---|---|---|
| LIPOSOME | |||||
| Doxil® | Doxorubicin Hydrochloride |
i.v. | FDA, 1995 | Ovarian, breast cancer, multiple myeloma, Kaposi’s sarcoma | Baxter Healthcare Corp |
| Curosurf® | Poractant alfa | intratracheal injection | FDA 1999 | Respiratory Distress Syndrome (RDS) | Chiesi USA Inc |
| Myocet liposomal® | Doxorubicin (Non-pegylated) | i.v. | Europe, 2000 | Breast neoplasm | Teva B. V. |
| Mepact® | Mifamurtide | i.v. | Europe, 2009 | Osteosarcoma | Takeda France SAS |
| Marqibo® | Vincristine Sulfate | i.v. | FDA, 2012 | Philadelphia chromosome -negative acute lymphoblastic leukemia | Acrotech |
| Onivyde® | Irinotecan Hydrochloride | i.v. | FDA, 1996 | Combination therapy with fluorouracil and leucovorin in metastatic adenocarcinoma of pancreas | Ipsen Inc |
| AmBisome® | Amphotericin -B | i.v. | FDA, 1997 | Systemic fungal infections | Astellas, Gilead |
| Exparel® | Bupivacaine | i.v. | FDA, 1972 | Postsurgical analgesia | Pacira Pharms Inc |
| Caelyx® | Doxorubicin Hydrochloride | i.v. | Europe, 1996 | Kaposi’s sarcoma, multiple myeloma, ovarian & breast neoplasm | Janssen Cilag International NV |
| Vyxeos™ | Cytarabine and Daunorubicin | i.v. | FDA, 2017 | Therapy-related acute myeloid leukemia (t-AML) or AML with myelodysplasia-related changes (AML-MRC) | Celator Pharms |
| DaunoXome® | Daunorubicin Citrate | i.v. | FDA 1996 (Discontinued) | Advanced HIV-associated Kaposi's sarcoma | Galen (UK) |
| DepoCyt® | Cytarabine | Intrathecal injection | FDA 1999 (Discontinued) | Lymphomatous meningitis | Pacira Pharms Inc |
| DepoDur® | Morphine sulfate | Epidural injection | FDA 2004 (Discontinued) | Pain following major surgery | Pacira Pharms Inc |
| Epaxal® | Hepatitis A virus (inactivated) | i.m. | Europe 2000 (Discontinued) | Hepatitis A vaccine | Berna Biotech Ltd |
| LIPID-BASED NANOPHARMACEUTICALS | |||||
| Abelcet® | Amphotericin B lipid complex | i.v. | FDA, 1995 | Invasive severe fungal infections | Leadiant Biosci Inc |
| Onpattro® | Patisiran (small interfering RNA) | i.v. | FDA 2018 | Polyneuropathy of hereditary transthyretin-mediated amyloidosis | Alnylam Pharms Inc |
| Pfizer-BioNTech COVID-19 Vaccine | messenger ribonucleic acid (mRNA) | i.m. | FDA 2020 | Prevention of 2019 coronavirus disease (COVID-19) for individuals 12 years of age and older | Pfizer Inc. |
| Moderna COVID-19 Vaccine | messenger ribonucleic acid (mRNA) | i.m. | FDA 2020 | Prevention of 2019 coronavirus disease (COVID-19) for individuals 18 years of age and older | ModernaTX, Inc. |
| PEGYLATED PROTEIN, POLYPEPTIDES, APTAMERS | |||||
| Cimzia® | Certolizumab pegol (Pegylated Fab’ fragment of humanized anti-tumor necrosis factor -alpha antibody) | s.c. | FDA, 2008 | Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis | UCB Inc |
| Neulasta® | Pegylated Filgrastim | s.c. | FDA, 2002 | Febrile neutropenia, hematopoietic subsyndrome of acute radiation syndrome | Amgen |
| Neupogen® | Filgrastim | s.c., i.v. | FDA 1991 | Febrile neutropenia, hematopoietic subsyndrome of acute radiation syndrome | Amgen |
| Nivestym | Filgrastim-aafi | s.c., i.v. | FDA 2018 | Febrile neutropenia, hematopoietic subsyndrome of acute radiation syndrome | Hospira Inc |
| Granix® | Tbo-filgrastim | s.c. | FDA 2012 | neutropenia in patients with non-myeloid malignancies | Sicor Biotech |
| Nyvepria™ | Pegfilgrastim-apgf | s.c. | FDA 2020 | Febrile neutropenia | Hospira Inc |
| Udenyca® | Pegfilgrastim-cbqv | s.c. | FDA 2018 | Febrile neutropenia | Coherus Biosciences Inc |
| Zarxio® | Filgrastim-sndz | s.c. | FDA 2015 | Febrile neutropenia | Sandoz Inc |
| Ziextenzo™ | Pegfilgrastim-bmez | s.c. | FDA 2019 | Febrile neutropenia | Sandoz Inc |
| Fulphila® | Pegfilgrastim-jmdb | s.c. | FDA 2018 | Febrile neutropenia | Mylan Gmbh |
| Oncaspar® | Pegaspargase | i.m., i.v. | FDA, 1994 | Acute lymphoblastic leukemia | Sigma Tau |
| Pegasys® | Pegylated Interferon alfa-2a | s.c. | FDA, 2002 | Hepatitis B and C | Hoffman-LA Roche |
| Somavert® | Pegvisomant | s.c. | FDA, 2003 | Acromegaly | Pharmacia |
| Mircera® | Methoxy polyethylene glycol-epoetin beta | i.v., s.c. | FDA, 2007 | Anemia associated with chronic kidney disease (CKD) | Hoffman-LA Roche |
| PegIntron® | Pegylated Interferon alfa-2b | s.c. | FDA, 2001 | Hepatitis C | Schering |
| Krystexxa® | Pegloticase (Pegylated uric acid specific enzyme) | i.v. | FDA, 2010 | Chronic gout | Horizon Pharma |
| Plegridy® | PEG-IFN-ß-1a | s.c. | FDA, 2014 | Multiple sclerosis (relapsing forms) | Biogen Idec Inc |
| Sylatron™ | Peginterferon alfa-2b | s.c. | FDA 2011 | melanoma with microscopic or gross nodal involvement | Schering |
| Adynovate® | Pegylated anti-hemophilic factor (Recombinant) | i.v. | FDA, 2015 | Hemophilia A (congenital factor VIII deficiency) | Baxalta US Inc. |
| Rebinyn® | Coagulation Factor IX (Recombinant), GlycoPEGylated | i.v. | FDA, 2017 | Hemophilia B | Novo Nordisk Inc. |
| Adagen® | Pegademase bovine enzyme | i.v. | FDA 1990 (Discontinued) | Severe combined immunodeficiency disease (SCID) associated with a deficiency of ADA | Leadiant Biosci Inc |
| Macugen® | Pegaptanib sodium | intravitreal injection | FDA 2004 (Discontinued) | Neovascular (wet) age-related macular degeneration | Valeant Pharms LLC |
| PROTEIN-DRUG CONJUGATES | |||||
| Abraxane® | Paclitaxel protein-bound particles (albumin-bound) | i.v. | FDA, 2005 | Metastatic breast cancer, locally advanced or metastatic non-small cell lung cancer (NSCLC), Metastatic adenocarcinoma | Abraxis Bioscience LLC |
| Ontak® | Denileukin diftitox (Recombinant fusion protein of fragment A of diphtheria toxin and interleukin-2) | i.v. | FDA, 1999 | Persistent or recurrent cutaneous T-cell lymphoma | Eisai Inc |
| Kadcyla® ado-trastuzumab emtansine) | Ado-trastuzumab emtansine (Antibody-drug conjugate) | i.v. | FDA, 2013 | HER2-positive, metastatic breast cancer (MBC), Early Breast Cancer (EBC) | Genentech |
| Taxol® | Paclitaxel | i.v. | FDA 1992 (Discontinued) | Advanced carcinoma of the ovary | HQ Spclt Pharma |
| OTHER TYPES OF POLYMER-BASED NANOPHARMACEUTICALS | |||||
| Copaxone® | Glatiramer acetate | s.c. | FDA, 1996 | Multiple sclerosis (relapsing forms) | Teva Pharms USA |
| Eligard®Kit | Leuprolide acetate incorporated in PLGH (DL-lactide / glycolide; 1/1, molar) NPs | s.c. | FDA, 2002 | Advanced prostate cancer (palliative treatment) | Tolmar Therap |
| Renagel® | Sevelamer Hydrochloride | oral | FDA, 2000 | Hyperphosphatemia (in chronic kidney disease) | Genzyme |
| Alimta® | Pemetrexed | i.v. | FDA 2004 | Advanced or metastatic, non-squamous, non-small cell lung cancer (NSCLC), Platinum chemotherapy, | Lilly |
| Genexol® | Paclitaxel micelles of poly(ethylene glycol)-poly (D,L-lactide) | i.v. | South Korea, 2001 | Metastatic breast cancer, pancreatic cancer | Samyang Biopharmaceuticals Corporation (Seoul, Korea) |
| Zinostatin stimalamer® | Neocarzinostatin (Conjugate protein or copolymer of styrene-maleic acid and an antitumor protein NCS) | Intrahepatic injection | Japan, 1994 | Primary unresectable hepatocellular carcinomas | Yamanouchi |
| Zilretta® | Triamcinolone acetonide | intra-articular injection | FDA 1957 | osteoarthritis pain of the knee | Flexion Therapeutics Inc |
| NANOCRYSTALS | |||||
| Emend® | Aprepitant | oral | FDA, 2003 | Antiemetic | Merck |
| Emend® | Fosaprepitant Dimeglumine (prodrug) | i.v. | FDA 2008 | Antiemetic | Merck and Co Inc |
| Ostim® | Calcium Hydroxyapatite | paste | EU, 2002 | Metaphyseal fractures and cysts, spinal column surgery | Osartis Gmbh & Co. KG, Germany |
| Rapamune® | Sirolimus | oral | FDA, 1999 | Immunosuppressant (mTOR inhibitor) | PF Prism CV |
| Vitoss® | ß-tricalcium phosphate (bioactive device) | i.v. | FDA, 2017 | Resorbable Synthetic Bone Voih Filler/Bone Graft Substitute | Orthovita, Inc. |
| TriCor® | Fenofibrate | oral | FDA, 1993 | Primary hypercholesterolemia or mixed dyslipidemia, hypertriglyceridemia | Abbvie Inc. |
| Vidaza® | Azacytidine (with mannitol powder for reonstitution) | i.v., s.c. | FDA, 2004 | Myelodysplastic syndromes | Celgene |
| Revlimid® | Lenalidomide | oral | FDA, 2005 | Multiple myeloma, Transfusion-dependent anemia, Mantle cell lymphoma (MCL) | Celgene |
| Megace ES® | Megestrol Acetate | oral | FDA, 1993 | Anorexia, cachexia | Endo Pharms Inc |
| Focalin®XR | Dexmethylphenidate Hydrochloride | oral | FDA, 2005 | Attention Deficit Hyperactivity Disorder | Novartis |
| Ritalin® | Methylphenidate Hydrochloride | oral | FDA, 1955 | Attention Deficit Hyperactivity Disorders (ADHD) and Narcolepsy | Novartis |
| Ritalin® LA (Extended release) | Methylphenidate Hydrochloride | oral | FDA, 1955 | Attention Deficit Hyperactivity Disorders (ADHD) and Narcolepsy | Novartis |
| Zanaflex® | Tizanidine Hydrochloride | oral | FDA, 1996 | Spasticity | Covis |
| Invega Sustenna® | Paliperidone palmitate | i.m. | FDA 2006 | Schizophrenia, schizoaffective disorder | Janssen Pharms |
| Ryanodex® | Dantrolene sodium | i.v. | FDA 1974 | Malignant hyperthermia | Eagle Pharms |
| OsSatura™ Dental | Biphasic ceramic (hydroxyapatite/tri-calcium phosphate) | irregular shaped chips (200-2000μm) | FDA 2004 | Bone Grafting Material, Synthetic | IsoTis OrthoBiologics Inc |
| OsSatura™ BCP (Biphasic Calcium Phosphate) | Biphasic ceramic (hydroxyapatite/tri-calcium phosphate) | irregular-shaped chips of different sizes | FDA 2003 | Osteoconductive bone void filler | IsoTis NV |
| NanOss™ Bone Void Filler | Calcium phosphate | pellets | FDA 2005 | Osteoconductive, resorbable, calcium phosphate implant | Angstrom Medica, Inc |
| Beta-BSM, Gamma-BSM, Equivabone | Calcium phosphate | paste | FDA 2010 | Synthetic, biocompatible bone graft substitute | ETEX Corporation |
| Aristada® | Aripiprazole lauroxil | i.m. | FDA 2015 | Schizophrenia (atypical antipsychotic) | Alkermes Inc |
| Avinza® | Morphine sulfate | oral | FDA 1941 (Discontinued) | Severe pain | King Pharms LLC |
| METAL-BASED NANOPHARMACEUTICALS | |||||
| Feraheme™ | Ferumoxytol (Superparamagnetic Iron oxide NPs coated with dextran) | i.v. | 2009 | Iron deficiency anemia | Covis |
| NanoTherm® | Aminosilane-coated superparamagnetic Iron oxide NPs | interstitial administration | Europe, 2011 | Local ablation in glioblastoma, prostate, and pancreatic cancer (intratumoral) | Magforce AG (Berlin, Germany) |
| Venofer® | Iron sucrose | i.v. | FDA 2000 | Iron deficiency anemia (IDA) in patients with chronic kidney disease (CKD) | AM Regent |
| Ferrlecit® | Sodium ferric gluconate complex in sucrose injection | i.v. | FDA 1999 | Iron deficiency anemia (IDA) | Sanofi Aventis US |
| Injectafer® | Ferric carboxymaltose | i.v. | FDA 2013 | Iron deficiency anemia (IDA) | AM Regent |
| INFeD® | Iron dextran | i.v., i.m. | FDA 1974 | Iron deficiency | Allergan |
| Dexferrum® | Iron Dextran | i.v. | FDA 2009 (Discontinued) | Iron deficiency | AM Regent |
| Feridex® | Ferumoxides | i.v. | FDA 1996 (Discontinued) | Magnetic resonance imaging (MRI) contrast media | AMAG Pharms Inc |
| Gastromark® | Ferumoxsil | oral | FDA 1996 (Discontinued) | Magnetic resonance imaging (MRI) contrast media | AMAG Pharms Inc |
| Resovist® | Iron carboxydextran colloid | i.v. | Europe, 2001 | Magnetic resonance imaging (MRI) contrast agent | Bayer Schering Pharma |
| Rienso® | Ferumoxytol | i.v. | Europe 2012 (Discontinued) | Anemia, chronic kidney failure | Takeda Pharma A/S |
| SURFACTANT-BASED NANOFORMULATIONS | |||||
| Diprivan® | Propofol (oil-in-water emulsion in soybean oil/ glycerol/egg lecithin) | i.v. | FDA, 2001 | Monitored Anesthesia Care (MAC) sedation, general anesthesia | Fresenius Kabi USA |
| Cinvanti® | Aprepitant (Emulsion) | i.v. | FDA 2003 | Antiemetic | Heron Theraps Inc |
| Visudyne® | Verteporfin | i.v. | FDA 2000 | Classic subfoveal choroidal neovascularization due to age-related macular degeneration, pathologic myopia or presumed ocular histoplasmosis | Valeant Luxembourg |
| Inflexal® V | Inactivated Hemaglutinine of Influenza virus strains A and B | i.m. | 1997 Switzerland | Influenza vaccine | Crucell (Berna Biotech) |
| Estrasorb® | Estradiol emulsion | topical | FDA 1975 (Discontinued) | Menopause (moderate to severe vasomotor symptoms) | Exeltis USA Inc |
| Janssen COVID-19 Vaccine | Recombinant, replication-incompetent adenovirus type 26 expressing the SARS-CoV-2 spike protein | i.m. | FDA 2021 | Prevention of coronavirus disease 2019 (COVID-19) for individuals 18 years of age and older | Janssen Biotech Inc., |
The Biopharmaceutics Classification System (BCS) has found widespread utility in drug product development by classifying drugs candidate based on their aqueous solubility and intestinal permeability. Low solubility and/or low permeability could lead to poor oral bioavailability. For oral administration, an NDDS (e.g., nanocrystals/nanosuspensions) with high kinetic solubility and dissolution rate improves the oral absorption due to significantly reduced particle size and increased surface area-to-volume ratio. Liposomes and other lipid-based NDDS could also enhance the drug permeability by facilitating the interactions between drug and lipophilic cell membrane (Dave et al., 2017).For parenteral administration, NDDS were able to significantly upgrade the solubility of poorly water soluble anticancer drugs without using toxic solubilizing agents e.g., polyethoxylated castor oil (e.g., paclitaxel in Abraxane® )
Nanotechnology brings both, the enthusiasm and the regulatory concern. Biocompatibility, unknown toxicity, quality control and scale-up issues are major obstacles for the growth of NDDS market. Regulatory agencies make continuous efforts to bridge the significant knowledge gap. FDA interacts with other government agencies to develop innovative, safe, and effective pharmaceutical products and technologies to avoid duplication of effort. The National Center for Toxicological Research (NCTR) develops analytical tools and procedures to assess the safety of nanomaterials. National Cancer Institute (NCI) founded the Nanotechnology Characterization Laboratory (NCL) with the cooperation of FDA and National Institute of Standards and Technology (NIST) to test the efficacy and toxicity of NPs. FDA has issued several non-binding guidances on the recommendation of FDA's Nanotechnology 2020 report. These guidance documents represent FDA's current thinking on the use of nanotechnology or nanomaterials including “ Draft Guidance for Industry – Drug Products, Including Biological Products, that Contain Nanomaterials”(FDA, 2017a); “Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology”(FDA, 2014a); “Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation”(FDA, 2018a); “Use of Nanomaterials in Food for Animals”(FDA, 2015) “Safety of Nanomaterials in Cosmetic Products”(FDA, 2014b) etc.
European Union (EU) also promotes the development of nanomedicines by publishing guidelines, ‘scientific advice’ and ‘protocol assistance’ through Scientific Advice Working Party (SAWP). Innovation task force (ITF) at the EMA provides a platform for exchange of information between innovators and regulators on the topics including nanomedicines, novel delivery methods, modelling and simulation. Innovative Medicines Initiative (IMI) is a partnership between European Commission (public partner), and European Federation of Pharmaceutical Industries and Associations (EFPIA - private partner) to promote rapid development of safe and effective innovative medicines including nanomedicines.
2 ∣. IN VITRO DISSOLUTION TESTING: SIGNIFICANCE
Throughout the drug product development in vitro dissolution testing has gained greater attention as a surrogate test for product performance to possibly avoid the expense, time, labor, and need for in vivo measurements of drug release kinetics. Kinetics of drug release is a key parameter to assess product safety and efficacy by providing critical information about dosage form behavior. Many studies have been devoted to the development of suitable dissolution method and apparatus to provide details on the release mechanism and kinetics from complex dosage forms like NDDS. It is highly important to thoroughly understand the drug release kinetics of NDDS to meet specific therapeutic objectives and to scrutinize their long-term fate in order to develop safer nanomedicines. A variety of mechanistic mathematical models have been used to describe drug release from NDDS. These models are based on the mechanism of rate limiting step of drug release, such as diffusion, swelling, erosion/ dissolution of polymer (Fredenberg et al., 2011; Zolnik & Burgess, 2008). Appropriate kinetic model (e.g., Higuchi square root law, Korsmeyer–Peppas model/power law, Hixson–Crowell model, Weibull model, reciprocal powered time (RPT) model) can be used to most accurately fit NDDS drug release data (Bruschi, 2015). Dissolution testing benefits and expedites drug development in several ways, including quality control, quality assurance, compendial testing, substantiation of label claim and evaluation of minor changes in formulation or manufacturing process.
2.1 ∣. Quality control tool for NDDS
In vitro dissolution testing is used as a valuable quality control tool for NDDS. BCS classification alone is not sufficient to predict the complex in vitro release profiles (bi- or tri-phasic) and in vivo physiologic responses of NDDS because formulation/process parameters besides the drug properties complicate the drug release (Kamaly et al., 2016). Quality of NDDS is ensured by critical quality attribute (CQA) (e.g., particle size, encapsulation efficiency, and zeta potential), which is defined as any physical, chemical or biological property that should be kept within a specified range. Like other CQA, the consistence of in vitro release profile can help establish ‘critical material attributes’ (CMA) and ‘critical process parameters’ (CPP) (Bastogne, 2017) for NDDS. CMAs include the formulation parameters (e.g., drug load, surfactant concentrations, drug-polymer ratio, drug-lipid ratio, organic to aqueous phase ratio) and CPP include the production parameters (e.g., processing temp., sonication time, stirring rate, flow, injection rate) capable to cause CQA variability. Krull S.M. et al explored the impact of various CMA (i.e., molecular weight and concentration) of film-forming polymer (HPMC; hydroxy propyl methyl cellulose) on the properties of films loaded with Griseofulvin (BCS II drug) NPs. They demonstrated that CMA of film-forming polymer can be changed to manipulate the drug release without impacting the film mechanical properties, drug content uniformity, or redispersibility of drug NPs (Krull et al., 2017).
The similarity factor (f2) and difference factor (f1), presented by equation (1) and (2) respectively (Anderson et al., 1998; FDA, 1997b) are qualitative (model independent) approaches to compare the dissolution pairs. Two profiles are considered as ‘similar’ when f2 value lies between 50 and 100 and f1 value is between 0 and 15. It is noteworthy that the f2 value being a log function, a minor difference in profiles will lead to a large drop in f2 value (Anderson et al., 1998; Kassaye & Genete, 2013; Shah et al., 1998). However, it is important to note that other CQA such as particle size, size distribution, and encapsulation efficiency etc. should be determined for NDDS (Stevens et al., 2015). Dissolution efficiency (DE), expressed by equation (3) is another quantitative approach proposed by Khan and Rhodes in 1975 to compare dissolution profiles.
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
Where, n = number of time points; Rt = mean % reference drug dissolved at time t after initiation of the study; Tt = mean % test drug dissolved at time t after initiation of the study; y = % dissolved, y100 = maximum dissolution. The area under the curve, ( ) can be estimated using model independent (trapezoid) or model dependent methods (Anderson et al., 1998) .
Yuan W. et al. established an USP 4 assay to compare the dissolution profiles (using similarity factors f2) of in-house doxorubicin liposomes and FDA approved liposome formulation, Doxil®. They concluded that such assay could serve as a quality control tool for generic liposome formulations (Yuan et al., 2017). Tang J. et al. compared Amphotericin B drug release from FDA approved liposome, AmBisome® with different generic liposomal products. For Amphonex® and Phosome®, the similarity factor values of more than 50 demonstrated that drug release from two generic formulations were similar to the innovator drug product. In this study, the USP 4 in vitro release assay was employed as a quality control tool to assist development of generic liposome formulations (Tang et al., 2019).
2.2 ∣. Toxicity assessment for NDDS
Leveraging theranostic (therapeutic and diagnostic) advantages of engineered nanomaterials also extends unsolicited interactions with environment, biological compartments and cellular processes (Gupta & Xie, 2018). Ability of NPs to stay inside the body for long period of time is known as ‘biopersistence’. It depends not only on the clearance (physiological and mechanical) but also on biodurability of NPs (innate resistance of NPs to mechanical, chemical or biological degradations). A long residence-time reflects the greater possibility for their accumulation and subsequent higher bioavailability and potential toxicities (Gualtieri et al., 2018; OECD, 2018; Utembe et al., 2015).
Knowledge of in vitro dissolution rate and dissolution lifetime can be fundamental in understanding the dissolution-mediated clearance and safety of NDDS. Dissolution lifetime is the time required for the largest particles in the polydisperse NP population to completely dissolve (Utembe et al., 2015). The dissolution rate constant can be obtained by two methods: (i) thermodynamic approach involves calculations of Gibbs free energy (G) (for the system comprising of NPs, dissolved material and solvent) as the function of particle size (Hellmann & Tisserand, 2006; Machrafi, 2020; Tang, 2005) (ii) graphical approach uses linear plots of integrated rate laws (e.g., Zero order, First order, Higuchi square root law, Weibull model etc.).
2.3 ∣. IVIVC for NDDS
FDA defines the in vitro-in vivo correlation (IVIVC) for drug products as "a predictive mathematical model describing the relationship between an in-vitro property of a dosage form and an in-vivo response"(FDA, 1997a). It is a suitable surrogate to the single point dissolution assessment and can be used to establish correlation with in vivo pharmacokinetic parameters of various NDDS (Alshora et al., 2018; Ha et al., 2014; Kim & Baek, 2014).
IVIVC can be developed at four different levels (Cardot & Davit, 2012; FDA, 1997a; Qureshi, 2010): Level A, point to point correlation between in vitro dissolution curve and in-vivo dissolution curve (or absorption curve); Level B, correlation of mean dissolution time to mean residence time (or mean absorption time), based on the principles of statistical moment theory; Level C, single point relationship between time to have 10, 50, 90% dissolution (or rate of dissolution, dissolution-efficiency, disintegration time) and time to have 10, 50, 90% absorption (or Cmax, Tmax, Ka, AUC); Multiple level C, correlation of one or several pharmacokinetic parameters to amount of drug dissolved at several time points (minimum of 3 dissolution time points at early, middle, and late dissolution stages). Various studies have attempted to establish level A and level C correlations for a variety of NDDS (discussed in later sections).
3 ∣. IN VITRO DISSOLUTION METHODS FOR NDDS
There exists a lack of standard in vitro dissolution method for NDDS. Different drug release methods are being used by different researchers. With increasing number of marketed NDDS, a generic applicant may need to design a novel in vitro release method for their unique drug product. Dissolution rate of NPs may be determined by either in vitro (acellular or cellular) or in vivo tests as shown in Figure 1a. Current available in vitro acellular methods include three categories: dialysis membrane, sample and separate, and continuous flow. Sink conditions are usually recommended for fast and complete dissolution of dosage forms. For poorly soluble drugs, a suitable surfactant, at appropriate minimum concentration (above its critical micellar concentration (CMC)) is usually used to achieve sink conditions (USP43-NF382S-online, 2020; Weng et al., 2020). Sometimes maintenance of non-sink conditions, by using sample amounts near saturation solubility, have been reported to demonstrate slow dissolution rate and more discriminatory dissolution profiles for poorly soluble drugs in NDDS (Liu et al., 2013).
FIGURE 1.

(a) Methods for dissolution rate assessment of NPs. In vivo dissolution studies are not always feasible. Acellular in vitro tests could reflect the dissolution behavior in blood (en route to target tissues) while cellular in vitro tests could reflect dissolution inside target cells. (b) Dissolution methods to test liposomes (based on 56 publications accessed for 2010-2020). During last decade more than one thousand articles were published illustrating different in vitro dissolution studies for various NDDS. As liposomes are most widely approved NDDS an account was made for them specifically.
More than one thousand articles were published in PubMed databases during last decade about in vitro dissolution studies on various NDDS. As liposomes are the widely approved and marketed NDDS the search was run using key terms- “liposome” and “in vitro dissolution”, and the articles detailing in vitro release studies of liposomes in the past decade were retrieved (Figure 1b). The dialysis membrane methods were most widely utilized, however, the popularity of continuous flow methods using USP 4 apparatus is on the rise.
3.1 ∣. Dialysis membrane methods
In dialysis membrane methods (e.g., dialysis bag, reverse dialysis, side-by side dialysis), NPs are separated from release medium through a dialysis membrane. In the regular dialysis technique, NPs are placed inside the dialysis bag, previously soaked in dissolution medium, agitated with shaking water bath or magnetic stirrer (Figure 2a). The drug released from NPs diffuses through the dialysis membrane to the outer compartment where samples are collected at specified intervals for analysis. The molecular weight cutoff (MWCO) of the membrane is the most critical parameter that impacts drug release in the dialysis method than other key ones including agitation conditions, ratio between donor and acceptor cell volumes (Shen & Burgess, 2013). In contrast, the reverse dialysis method has an opposite setup to overcome the potential violation of sink condition (Figure 2b). (Calvo et al., 1996; Xu et al., 2012). In the side-by-side dialysis set-up, equal volume donor and receiver compartments are separated by a dialysis membrane (Figure 2c) (Chidambaram & Burgess, 1999).
FIGURE 2.

(a) Dialysis method. NPs are filled inside the dialysis bag placed into the outer medium reservoir. Samples are collected at specified intervals from outer compartment. (b) Reverse dialysis method. Opposite setup-NPs are placed in the outer compartment and the inner medium reservoir is sampled for drug release. (c) Side by side dialysis method. Equal volume donor and receiver compartments are separated by a dialysis membrane. Samples are taken from the receiver cell. (d) USP 4 (Flow Through Cell). Four different platforms comprising of dissolution platform, pump assembly, dissolution media (reservoir) and fraction collector. (e) Open Loop System. Fresh solvent from the reservoir (media selector) continuously replenishes through the flow cells to maintain infinite sink conditions. (f) Close Loop System. Small media volume is recirculated again and again from flow cell to reservoir. Sampling is done from the recirculating dissolution medium.
While using dialysis methods, the membrane size should be big enough to allow drug passage (MWCO about 100 times the drug size) (Xu et al., 2012) but avoid transfer of drug carrier (NDDS). Dialysis bags can also be used with USP apparatus 1 and 2 (Bhagav et al., 2011). Dialysis membrane are the most used techniques, however, there are some limitations of these methods:
If incorrectly sealed, leakage of media/dosage form may occur from dialysis bags.
Incomplete release data may be observed in case of non-sink conditions or high equilibration times (D’Souza, 2014).
The potential binding between drug and dialysis membrane should be checked. These methods cannot be used if a suitable membrane with adequate pore size is not available.
Inaccurate release results might be obtained if the dialysis process is rate limited by the membrane itself instead of the dissolution process (Heng et al., 2008).
3.2 ∣. Sample and separate methods
The NPs are filled into gelatin capsule shell or added directly into the release medium held in a beaker. The dispersed NPs are separated from the continuous phase at predetermined time intervals using various sample separation techniques, including ultrafiltration, ultracentrifugation, and centrifugal ultrafiltration. The recovered supernatant/filtrate is subsequently analyzed for drug release (Ham et al., 2009; Morales-Cruz et al., 2012). Alternatively, the drug content inside the separated NPs might be analyzed after restructuring them using suitable solvent (Sanna et al., 2012). The sample and separate methods offer a direct approach to monitor in vitro drug release from NDDS with the following practical challenges:
It is very difficult to efficiently separate the NPs from the release media without influencing the drug release profile. The drug release continues during the separation process, which can lead to erroneous results (Shen & Burgess, 2013).
Long and high-speed ultracentrifugation can result in destabilization of NPs (Shen & Burgess, 2013).
Clogging of filters and drug adsorption onto filter surface may happen when filtration is used as sample separation technique (Kim et al., 1997).
Variability in dissolution readings and inaccurate results may occur due to inherent wetting problems of poorly soluble drugs (Heng et al., 2008).
3.3 ∣. Continuous flow method
In vitro dissolution testing of NDDS in continuous flow can be attained with USP 4 apparatus coupled with dialysis cells (figure 2d). The test sample (NDDS) is placed in specially designed ready-to-use dialysis cells, usually made of cellulose ester (e.g., Float-A-Lyzer® G2). Different type of flow cells (available in different dimensions and molecular weight cut offs) can be used for different dosage forms. Test parameters such as volume, flow rate and temperature of dissolution media can be adjusted as per the assay requirement. USP 4 is versatile and can be used for dissolution testing of both conventional dosage forms (powders, granules, tablets, capsules, enteric-coated tablets, suppositories, and injectable suspensions) and novel drug delivery systems, covering modified-release products, implant stents, drug coated medical devices, liposomes, microsphere, and NPs etc. (Singh & Aboul-Enein, 2006; Yoshida et al., 2015). It might serve as a promising alternative to conventional dissolution test methods due to following advantages:
The composition, pH and/or flow rate of the dissolution medium can be changed over the course of test to simulate the intralumenal hydrodynamics more efficiently (Gite et al., 2016).
As dosage form is isolated from the medium reservoir, sampling and media changes can occur without disturbing the hydrodynamics inside USP 4 giving it a leverage on other conventional in vitro release methods (D'Arcy et al., 2010; Eaton et al., 2012; Singh & Aboul-Enein, 2006; Yoshida et al., 2015).
When operated in the open configuration (Figure 2e), fresh solvent from the reservoir continuously replenishes through the cell to offer infinite sink conditions for poorly soluble compounds, making the IVIVC development easier.
Closed loop configuration (Figure 2f) allows the use of small media volumes to overcome the limit of quantification issues.
No further filtration steps are needed, as any undissolved particles are kept within the cell (Heng et al., 2008).
Knowledge of operating hydrodynamics (physics of fluid flow- governed by principles of conservation of mass, momentum, and energy) can improve the robustness and discriminatory ability of a dissolution test (Shiko et al., 2011). It can guide the choice of appropriate test conditions, design of dissolution test apparatuses and even help interpret the test results. A strong degree of flow uniformity and symmetry was obtained for USP 4 when the conical part of the dissolution cell was filled with 1-mm glass beads along with the red ruby bead (Kakhi, 2009). The studies summarized in Table 2 endorse the discriminatory potential of USP 4 in testing NDDS.
Table 2.
Discriminatory USP 4 for in vitro testing of NDDS
| NDDS | In Vitro Release Methods | Study Findings | Reference |
|---|---|---|---|
| Cefuroxime Axetil NPs | •USP 4(flow rate 16 ml/min; 0.2-μm disc filter) •USP 1(100 rpm) •USP 2(100 rpm) •Dialysis bag (12-14 kDa MWCO) |
USP apparatus 4 was testified to be unequivocally the most robust dissolution method to differentiate dissolution rate ratios of Cefuroxime Axetil NPs and unprocessed drug. | (Heng et al., 2008) |
| Dexamethasone Liposomes | •USP 4(flow rate 16 ml/min; 50 kDa MWCO) •Dialysis sac (50 rpm; 50 kDa MWCO) Reverse dialysis sac (50 rpm; 50 kDa MWCO) |
USP apparatus 4 was able to discriminate between solution, suspension and also the extruded and non-extruded liposomes. | (Bhardwaj & Burgess, 2010) |
| Griseofulvin NP-laden strip-films | •USP 4(flow rate 16 ml/min; 0.2-μm disc filter) •USP 1 (50, 100, and 150 rpm) |
Particle-size discriminatory nature of USP 4 suggested its potential to provide similar results for other BCS Class II drugs. | (Sievens-Figueroa et al., 2012) |
| Vitamin E acetate oil/ water nanoemulsions | •USP 4(flow rate 16 ml/min; 50 and 100 kDa MWCO) •Reverse dialysis sac (50 and 100 kDa MWCO) |
A faster in vitro release was obtained from USP apparatus 4 as compared with dialysis sac and reverse dialysis methods. | (Morais & Burgess, 2014) |
| Atorvastatin NPs | •USP 4 (glass-bead mixing; flow rate 8,16 ml/min) •Dialysis bag in USP 1(75, 100, and 125 rpm; MWCO NA) •Dialysis bag in USP 4 (8, 16 mL/min; MWCO NA; not available) |
USP 4 dissolution method established for Atorvastatin NPs using modified sample loading (glass-bead mixing with NPs) was found to be discriminatory. | (Gite et al., 2016) |
| Doxorubicin liposome | •USP 4(flow rate 16 ml/min; 10–300 kDa MWCO) | USP 4 assay adequatly distinguished between innovator Doxorubicin liposome product Doxil® and generic formulations of different compositions, release rates, and prepared by different manufacturing techniques. | (Yuan et al., 2017) |
| Bevacizumab liposome | •USP 4(flow rate 1ml/min; MWCO NA) | USP 4 was used to test in vitro release from different composition Bevacizumab liposomes. | (Karumanchi et al., 2018) |
| Amphotericin B liposome | •USP 4(flow rate 16 ml/min; 300 kDa MWCO) | USP 4 assay was able to elicit the distinction between marketed liposome formulation of Amphotericin B AmBisome® and other in-house formulations, prepared by extrusion or homogenization processes. | (Tang et al., 2019) |
4 ∣. IN VITRO DISSOLUTION CONSIDERATIONS FOR NDDS
There are several factors related to NDDS that affect the in vivo performance of products. These factors include physicochemical characteristics of the drug substance, formulation parameters, environmental conditions, and biocompatibility. For the development of meaningful IVIVC it is important to obtain in vivo data beforehand; and a predictive in vitro dissolution test method can be designed by manipulating these factors to match in vivo profiles (Zolnik & Burgess, 2008).
4.1 ∣. Properties of drug
Biopharmaceutics Classification System (BCS) provides a framework to define the critical quality attributes (CQA), decisive for oral bioavailability (FDA, 2018b). It can reflect the in vivo performances, essentially based on drug solubility and intestinal permeability. A drug’s occupancy within the specific BCS class may suggest optimal drug delivery technologies suitable for that drug (Figure 3a). A meaningful IVIVC is expected for BCS Class II drugs as dissolution is the rate-limiting step in absorption. For drugs in BCS class III, IVIVC is unlikely or could be possible depending on relative rates of dissolution and intestinal transit. For BCS class IV drugs, IVIVC is highly unlikely.
FIGURE 3.

Nano drug delivery systems (NDDS) for various BCS class. Class I drugs are straightforward to develop. NDDS offer promising drug delivery solutions for potential drug candidates belonging to BCS class II, III and IV by overcoming poor solubility/permeability issues. (b) DCS classification system. SLAD marks the boundary between DCS class IIa and IIb drugs. Below SLAD all the doses could be dissolved, and above SLAD only a fraction of dose is dissolved (fraction dissolved decreases with increasing dose). DCS Class IIa drugs show ‘dissolution-rate limited’ absorption. Simple particle size reduction can suitably formulate such drugs. Therefore, particle size (in contrast to dose/solubility ratio of BCS classification) can better predict the extent of absorption. DCS Class IIb drugs have ‘solubility-limited’ absorption, and they present major formulation challenges (bioavailability depends on gastric pH and intestinal precipitation can occur). These drugs remain incompletely absorbed unless suitably solubilized (e.g., lipid-based formulation or amorphous solid dispersion)
The revised BCS classification system – known as developability classification system (DCS) – provides the significance of particle size in the determination of dissolution-rate limited absorption (Butler & Dressman, 2010). As drug particle size (instead of dose/solubility ratio) is a better predictor of dissolution rate-limited absorption of DCS class IIa drugs (Figure 3b), control of particle size determines bioavailability. It is possible to achieve complete oral absorption of such crystalline drugs using a standard solid oral dosage form (due to compensatory effect of high permeability). On the other hand, DCS Class IIb compounds have solubility-limited absorption and they present major formulation challenges (as bioavailability depends on gastric pH and intestinal precipitation can occur). These drugs are incompletely absorbed unless formulated in a suitable solubilized dosage form.
Solubility/stability of drug should be tested in the target media (under relevant hydrodynamic conditions) before dissolution testing of actual dosage form to identify such issues at the earliest. However, depending on the experimental set-up, test conditions used for measuring drug solubility may not always translate during in vitro dissolution testing (e.g., agglomeration of proteins in the fine capillaries of USP 4). Alternate release methods (like dispersion releaser) should be tried under such circumstances (Janas et al., 2017).
4.2 ∣. Formulation parameters
Drug release from NDDS can be customized by manipulating the formulation parameters (e.g., lipid matrix, polymer/stabilizer concentration, solvent, drug amount) as well as the production conditions (e.g., temperature, homogenization) (Sedighi et al., 2019; Sharma et al., 2014).Shape of NPs can influence the physiological response in human cells. Needle-shaped Poly (lactic-co-glycolic acid) polyethylene glycol nanoparticles (PLGA-PEG NPs) were found to induce significant cytotoxicity compared to spherical-shaped NPs (Zhang et al., 2017). Size can influence the in vitro cytotoxicity of NPs as smaller particles tend to adsorb more biomolecules (protein-corona) (Xiong et al., 2013). Toxicity and irritancy can affect in vivo drug release due to edema and presence of inflammatory cells like macrophages and neutrophils (Zolnik & Burgess, 2008).
4.3 ∣. Environmental conditions
The spatiotemporal (depending on site and time) interplay of biological and/or environmental factors forms a spontaneous layer of surface-adsorbed biomolecules surrounding the NPs-termed as ‘protein-corona’. NPs consolidated with its protein-corona, behave as a biological entity and interact with cells and various physical, chemical and immunological barriers inside the body (Jain et al., 2017). Depending on the particle properties (e.g., size, shape, surface charge, topology, hydrophobicity, encapsulation efficiency), protein-corona can be tailored to conceive a perfect targeted drug delivery system (Lu et al., 2019; Yeo et al., 2019; Zhang et al., 2019). This protein corona can affect the biodistribution as well as the release behavior of NDDS (Behzadi et al., 2014). Therefore, it becomes very important to clearly understand the impact of bio-nano interactions on the biological consequences at both the cellular and the physiological levels.
To establish meaningful and biorelevant in vitro drug release method for NDDS, it may be desirable to determine the effect of biorelevant concentrations of various plasma components (like bile salts, phospholipids, cholesterol, albumin). Specific corona proteins should be identified and evaluated for protein-drug interactions using appropriate model (Wallenwein et al., 2019)). A synthetic (e.g., SLS, Tween 80, CTAB) or biorelevant (bile salts, phospholipids) surfactant, enzyme (e.g., pepsin) or cosolvent (e.g., polyethylene glycol 400) can be added for poorly water-soluble drugs to get clinically relevant solubility. If a protein and a surfactant, both are being incorporated in the medium, the effect of their interaction and combined impact on the solubility should be investigated. Clinically relevant media may be developed as per therapeutic indication (e.g., low albumin concentrations to reflect hypoalbuminemia). Various temperature, pH, ionic strength, stirring rate and flow rate (USP 4) conditions should be tested to obtain most robust and discriminatory dissolution test. Unique experimental set-up may be required to mimic the physiological environment depending on the specific use (e.g., two-stage drug release to mimic the drug release in circulation and drug release at target site (Xu et al., 2012); donor-acceptors vesicles to mimic drug release to phospholipid cell membrane(Shabbits et al., 2002)).
Another major challenge to in vivo delivery of NPs is their rapid clearance from circulation by reticuloendothelial system (RES), also known as mononuclear phagocyte system (MPS) — comprising the Kupffer cells of liver and the macrophages of spleen and bone marrow. Coating of NP surface with polyethylene glycol (PEG) is a commonly used approach to circumvent the immune recognition and to increase the systemic circulation time. A thorough review on “PEGylation” can be found in various previous articles (Fam et al., 2020; Suk et al., 2016).
4.4 ∣. Biocompatibility
Some excipients are cytotoxic, when used at higher concentrations. In vivo markers such as Lactate Dehydrogenase (LDH), a cytosolic enzyme, can be used to test cell membrane integrity and necrosis. Paclitaxel/Methotrexate co-loaded PLGA NPs, coated with PVA/P188, released more LDH compared to individual drug-loaded NPs or free drugs. (Madani et al., 2020). Immunohistochemical techniques such as TdT dUTP Nick End Labeling (TUNEL) assay can visualize apoptotic cells in vitro. Paclitaxel-loaded PLGA NPs were more apoptotic than the drug alone (Mo & Lim, 2005)
Many NDDS preparation techniques involve use or organic solvents. Residual solvents can be toxic and affect drug particle size, dissolution and wettability. The need to measure residual solvents was highlighted at various preparation and purification stages of liposomes and polymeric NPs for development of efficacious and safe NDDS (Dikpati et al., 2020).
International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) developed Q3C document to recommend acceptable limits for residual solvents in pharmaceuticals for patient safety. This guideline applies to all dosage forms and routes of administration. It groups the existing solvents into three classes, based on their individual health and environmental hazards (ICH, 2019). ICH has also developed maintenance procedures to include new solvents and to revise the permitted daily exposure (PDE) of solvents already listed in ICH Q3C as new toxicity data for solvents becomes available (FDA, 2017b).
FDA endorses a companion document for the ICH Q3C guidance to recommend the safe levels of residual solvents in pharmaceuticals (FDA, 2018c). Liposome drug products chemistry, manufacturing, and controls; human pharmacokinetics and bioavailability; and labeling documentation guidance requires to provide drug product specification including residual solvent(s) used in the manufacture of liposomes (FDA, 2018a). Safety of nanomaterials in cosmetic products guidance (FDA, 2014b) also necessitates the thorough understanding of manufacturing process to identify residual additives and impurities.
Dikpati A. et. al. have provided useful recommendations to minimize the residual solvents in the NDDS (Dikpati et al., 2020). First, the use of Class 3 solvents (e.g., acetone, ethyl acetate, dimethyl sulfoxide) or solvent-free/solvent-limiting preparation methods (e.g., supercritical fluid technology, microfluidics and extrusion for polymer based NDDS or high-pressure homogenization for lipid based NDDS) (Anton et al., 2012; Paliwal et al., 2014). Second, the use of dedicated or disposable measuring devices to avoid cross-contamination. Third, the rotary evaporation combined with other drying methods (e.g., lyophilization). Fourth, the multiple purification procedures (e.g., size exclusion chromatography, dialysis, ultrafiltration, centrifugation, electrophoresis)(Paliwal et al., 2014; Robertson et al., 2016).
4.5 ∣. Accelerated in vitro release testing
NDDS may be designed to sustain the drug release for several days to months. Accelerated in vitro release tests, predictive of “real-time” release, can speed up the analysis in these cases (Shen et al., 2016). However, the stress conditions (elevated temperature and extreme pH values), used to accelerate the drug release can also alter the release mechanism (Shameem et al., 1999; Zackrisson et al., 1995; Zolnik et al., 2006). It may make the process of IVIVC establishment even more exhausting (Shen & Burgess, 2012).
5 ∣. IVIVC FOR NDDS
A meaningful IVIVC can predict the in vivo performances of NDDS from their in vitro behaviors (Shen & Burgess, 2015). Compared to conventional dosage forms, the establishment of an IVIVC for NDDS products has been even more challenging due to dimension-dependent disposition of NPs and their undefined interactions with bio-interfaces (e.g., formation of protein-corona) (Jain et al., 2017; Riviere, 2013; Yuan et al., 2019) as well as the lack of compendial standard or guidance for in vitro release testing methods. It becomes important to understand the impact of physicochemical properties (particle size, size-distribution, drug loading, zeta potential etc.) and establish qualitative sameness (Q1), quantitative sameness (Q2) and in vitro drug release equivalence (Q3) along with conventional pharmacokinetic studies of these NDDS (Au, 2017; FDA, 2018a; Luke, 2017; Shah et al., 2014; Zheng et al., 2017).
Development of a successful IVIVC model requires dissolution to be the rate-limiting step in the process of drug absorption. Convolution and deconvolution are two fundamental approaches to establish an IVIVC model (Davanço et al., 2020; Gillespie, 1997; Jacob & Nair, 2018; Margolskee et al., 2016). A robust, reproducible, and reliable in vitro dissolution test method, sensitive to formulation or process variability can play a vital role in the development of meaningful IVIVC for NDDS.
5.1 ∣. Intravenous route of administration
Emerging nanoparticles tracking analysis (NTA) technologies reliably allow qualitative and quantitative identification of nanomaterials in the in vitro and in vivo samples (Kim et al., 2019; Parsons et al., 2017). In addition, design of biorelevant assays has most favorably accounted for the establishment of IVIVC for parenteral NDDS. The test conditions are optimized based on the physicochemical characteristics of the drug and the environmental conditions that the NDDS might be exposed to after intravenous administration. An account of such experiments is made to gain the experimental know-how; and to guide the design of future in vitro dissolution strategies in predicting the in vivo performances for NDDS (Table 3). From these studies it is evident that multiple factors play vital role in the bioavailability and the accumulation of drugs at the target site. Choice of in vitro release method may depend on whether the rate limiting step is diffusion, erosion, or swelling type. The biorelevant release assay should be developed with suitable understanding of what factors dictate NDDS stability, intracellular uptake and drug release profiles.
Table 3:
Biorelevant drug release assays for NDDS
| Sr. | Novel biorelevant method | Relevance/Purpose | Reference |
|---|---|---|---|
| 1 | ‘Two-stage reverse dialysis’ method was used to test drug release from Tenofovir liposomes. (stage 1: dialysis in pH 7.4 HEPES buffer solution and, stage 2: dialysis in 1% TX100 solution in HEPES buffer solution) |
Stage 1: represented the liposomal drug release into the blood. Stage 2: represented drug release inside targeted tissue. |
(Xu et al., 2012) |
| 2 | Drug-loaded ‘donor’ large unilamellar vesicles were incubated with ‘acceptors’ multilamellar vesicles. Drug released to the acceptor vesicles was measured (instead of drug release into the release medium). | Multilamellar vesicles mimicked the physiological lipid membrane. Drug loaded donor vesicles showed rapid drug leakage to reflect true in vivo drug retention of liposomes. |
(Shabbits et al., 2002) |
| 3 |
a) ‘Target-sensitive liposomes’ * were developed to test Streptokinase (a thrombolytic drug) drug release. More efficient drug release was found when liposomes were incubated with activated platelets (compared with resting platelets). b) Streptokinase release was also studied in PBS (pH 7.4), using sample and separate method. *having high affinity towards activated platelets- present at the site of clot formation. |
a) When target sensitive liposomes were bound to integrin receptors present on activated platelets (target) they got destabilized to release the drug more efficiently. b) Only 40% drug release in 12 h depicted that liposomes could trap the drug effectively with minimum release in systemic circulation on their way to target cells (activated platelets). |
(Vaidya et al., 2016) |
| 4 |
Dissolution/solubility of API was tested in the target media (under relevant hydrodynamic conditions) before dissolution testing from dosage form. It was suggested that stability/ solubility problems can be easily identified using the API alone. Three categories of biorelevant and clinically relevant media were used to support future development of predictive and meaningful in vivo release tests for parenteral liposomal formulations. Amphotericin B (poorly water soluble and highly protein-bound drug) was model drug. |
This study emphasized the need to identify suitably discriminating dissolution test conditions (apparatus and hydrodynamic environment) using flow-through cell dissolution test apparatus. Category 1: to test effect of albumin concentration. Category 2: to test effect of biorelevant concentrations of plasma components (bile salts, phospholipids, cholesterol, albumin). Category 3: to test effect of biorelevant or synthetic surfactants with/without albumin (simulated hypoalbuminaemic plasma medium). |
(Díaz de León-Ortega et al., 2020). |
| 5 | This study postulated that protein corona can affect biodistribution and release behavior of NDDS. Hence, it is important to evaluate the protein-drug interactions. Two different dialysis-based methods (Dispersion releaser technology in conjunction with USP 2 and A4D dialysis adapter with USP 4) were used in combination with a four-step mathematical model. Release medium comprised of PBS at a pH of 7.4 supplemented with 0.1% of ß-CD (solubilizer) and different concentrations (or absence) of fetal calf serum. Dispersion releaser technology was capable to reflect the drug transfer from liposomes to proteins. However, rapid agglomeration of proteins in the fine capillaries of USP 4 altered the hydrodynamics inside the sample cell and could not reflect the conditions defined by media composition. |
Four-step mathematical model was used to exclude the impact of membrane pore size** on drug release. Step1: Determination of concentration in donor compartment. Step2: Determination of concentration profile in acceptor compartment (assuming Fick’s law of diffusion). Step3: Determination of drug permeation coefficient (km) using non-linear regression fitting. Step4: Determination of normalized permeation of drug in donor compartment. ** In conventional diffusion methods the release kinetics is affected by both, the membrane pore size and the serum protein binding. |
(Wallenwein et al., 2019) |
| 6 |
Human pharmacokinetics of liposomal temoporfin was predicted using a hybrid
in silico
model. • Biorelevant release media, comprising PBS (pH 7.4) (0.01% Me-β-CD, 1% Pen-Strep, 10% FBS) was used and liposome stability was investigated using nanoparticle tracking analysis (NTA). • Intracellular uptake was determined into mononuclear phagocytes.• In vitro drug release was investigated with Dispersion releaser (DR) technology using two membrane sizes (MWCO of 50 kDa and 300 kDa). • Four-step model was used to normalize drug release profiles. • Three-parametric reciprocal powered time model (3RPT) was used to describe normalized drug release profiles. • Multicompartment in silico model was developed for rats and humans using Stella® Architect modeling software. Initially, in silico model was based on rat data and later adjusted to human pharmacokinetic parameters by applying allometric scaling. The simulations were verified using plasma concentration–time profiles obtained from phase I clinical trial |
• The biorelevant release medium reflected the in vivo dissolution pressure of drug (temoporfin) in blood circulation. • Uptake rate was in line with release behavior for liposomes. MWCO of 50 kDa showed effective retention of serum albumin (molecular weight 67 kDa). • Four-step model excluded the effect of membrane permeation and drug-protein transfer on the release profiles. • 3RPT model (a modified reciprocal powered time model) is based on a combination of diffusion and dissolution processes and can suitably describe the in vitro release profiles of NPs. • Deconvolution of plasma concentration-time profile into different fractions relevant for the in vivo efficacy and safety was achieved. Hybrid in silico model was able to predict liposome in vivo performance in humans. |
(Jablonka et al., 2020) |
| 7 | Ten conventional release models (zero order, first order, Hixson-Crowell, Higuchi, Power Law etc.) and three novel models, including reciprocal powered time (RPT) model were used to evaluate the release data of 32 drugs from 106 NDDS. | Novel RPT model (along with Weibull and Wagner’s log-probability models) was able to fit the drug release data most accurately from NDDS (nanosphere, nanocapsules, nanocrystal and nanoemulsion). | (Barzegar-Jalali et al., 2008) |
| 8 |
RPT model was used to describe Chlorpropamide solid dispersions. Drug release was compared with other conventional kinetic models (Power Law, First order, Weibull, Cube-root Law etc.) |
Smaller values of mean absolute percent deviation and overall mean percent deviation for RPT model reflected more accurate prediction of fraction of drug dissolved. | (Barzegar-Jalali & Dastmalchi, 2007) |
| 9 | The RPT model was successfully applied for kinetic analysis of drug release from Ibuprofen solid dispersions. | RPT model can be applied for diffusion rate limited, dissolution rate limited, and dissolution-diffusion rate limited drug release processes. (justified via unification of the Fick’s first law of diffusion and the Noyes- Whitney law of dissolution). |
(Mohammadi et al., 2010) |
| 10 |
Dispersion releaser technology effectively discriminated among different polymeric (PLGA, PLA, and Eudragit® RS PO) NDDS of flurbiprofen in the presence of biorelevant media. The effect of bovine serum albumin was studied on membrane permeability of flurbiprofen using four-step mathematical model. Two different mathematical models: reciprocal of time (RPT) and the three-parameter model. |
Acceptable curve fitting was achieved with both models. RPT (reciprocal powered time) is an easy to apply model. It explains drug release by combination of diffusion and dissolution mechanisms. Three parameter model describes the different mechanisms involved in the release behavior more appropriately. It analyzes the influence of molecular weight of polymeric drug carriers, nanoparticle diameter, and composition of release medium on drug release. |
(Janas et al., 2017) |
A silico modeling can be a suitable starting point to establish IVIVC by describing the pharmacokinetics of NDDS with preclinical animal data. The developed model can be further verified using clinical human data after applying allometric scaling. Obtained drug release profiles might need normalization (e.g., four-step model) to exclude the effect of membrane permeation and drug-protein transfer. Deconvolution of simulated pharmacokinetic profiles may enable to predict the human pharmacokinetics for novel NDDDS formulations(Jablonka et al., 2020).
5.2 ∣. Oral route of administration
Oral absorption is a complex process of several interdependent biological events. Physiologically-based pharmacokinetic (PBPK) modeling integrates these simultaneous events and simulates ADME (absorption, distribution, metabolism and elimination) to describe drug exposures in individual physiological compartments (Li et al., 2017; Utembe et al., 2020). Various biorelevant conditions have been shaped to build the IVIVC for orally administered NDDS (Table 4). Biorelevant dissolution coupled with PBPK modeling can support the oral NDDS development and minimize the animal/human testing (Kaur et al., 2018; Litou et al., 2019). However, PBPK modeling of NDDS is very challenging due to complex in vivo transport mechanisms such as mononuclear phagocyte system (MPS) uptake, enhanced permeability and retention (EPR) effect, lymphatic transport, and enzyme degradation (Li et al., 2017). Also, mechanistic modelling of NDDS flanked by organ-specific protein-coronas is still in its infancy (Utembe et al., 2020).
Table 4:
IVIVC establishment for NDDS in oral dosage forms
| Sr. No. |
Formulation | Drug | BCS Class |
In vitro dissolution |
In vivo model | IVIVC Implications | Reference |
|---|---|---|---|---|---|---|---|
| 1 | Eudragit® L100 nanodispersions (amorphous and crystalline) | Carbamazepine | 2 | Microcentrifuge dissolution or ultrafiltration dissolution with pH adjusted using 0.9 mL of intestinal fluid (FaSSIF, pH = 6.5) which was added to the simulated gastric fluid (0.01 N HCl, pH = 2) after 50 min of dissolution. Membrane-permeation dissolution using side-by-side diffusion cell with 0.01 N HCl (pH 2). After 30 min, donor compartment media was shifted to either human or canine FaSSIF (pH 6.5 or 5.2) to simulate gastrointestinal conditions. |
Mice | Level C correlation was established between in vitro drug permeated at 60 min and the in vivo AUC0-inf. Membrane-permeation in vitro test that employed canine FaSSIF as the dissolution media, 1-decanol as the acceptor solution, and porcine intestines as the permeation barrier best reflected the in vivo performance. This suggested that pH should be adjusted to correspond to subjects of in vivo studies. Microcentrifuge and ultrafiltration dissolution methods showed disconnect between in vitro tests and in vivo results due to over-estimation of free drug suggesting the need for a system to assess free drug concentrations accurately and also consider the drug diffusion across membrane. |
(Warnken et al., 2018) |
| 2 | self-nano-emulsifying drug delivery system (SNEDDS) | Etoposide (VP-16) | 4 | Dialysis membrane with simulated intestinal fluid under fast condition (FaSSIF) maintained at pH 6.5. | Sprague–Dawley rats | It was not possible to establish a IVIVC for commercial immediate release drug product due to slower absorption. For controlled release SNEDDS formulation, a good IVIVC was observed. Therefore, SNEDDS system may serve as a promising oral drug delivery system for BCS class 4 drug like VP-16. | (Khalid et al., 2018) |
| 3 | Polymeric NPs | Camptothecin | 4 | USP dissolution apparatus 2 with simulated gastrointestinal conditions of pH 1.2 for 2 hrs (stomach), pH 4.5 for 2 hrs (duodenum) followed by pH 7.4 (distal ileum and colon) for the remaining period of study stirred at 100 rpm and maintained at a temperature of 37 ± 0.5°C. | Rats | Level A IVIVC was developed between in-vitro dissolution and in-vivo absorption data. Study showed that sequential change of media to simulate gastrointestinal conditions could be a good strategy to get a meaningful IVIVC for nanoformulation development. | (Manikandan & Kannan, 2016) |
| 4 | Eudragit® L100 NPs | Atazanavir | 2 | Dialysis membrane with dissolution medium phosphate buffered saline (PBS) pH 7.4, kept at 37 ± 0.5°C and stirred at 25 rpm. | Wistar rats | For the BCS Class 2 drug establishment of good IVIVC was expected as dissolution being the rate- limiting step in absorption. Successful establishment of Level A IVIVC substantiated the judicious choice of in vitro dissolution milieu to simulate the in vivo conditions. | (Singh & Pai, 2016) |
| 5 | Liposome | Capsaicin | 2 | dialysis bag method with pH 7.4 phosphate buffer solution, pH 1.2 HCl solution and double distilled water stirred at 70 rpm and maintained at a temperature of 37 ± 1°C. | Sprague–Dawley rats | Excellent level A IVIVC was obtained between percent dissolution and percent in vivo absorption with maximum correlation coefficient in HCl solution. The study served as a working example of point-to-point correlation between in vitro dissolution and in vivo input rate of drugs for liposomal formulations. | (Zhu et al., 2015) |
| 6 | Nanocrystals | Puerarin | 4 | Basket method using purified water, HCl solution (pH 1.2), acetate buffer (pH 4.0) and phosphate buffer (pH 6.8) stirred at100 rpm and maintained at a temperature of 37 ± 0.5°C. | Beagle dogs | A good correlation between in-vitro and in-vivo drug release was found. | (Yi et al., 2015) |
| 7 | Nanosuspension | Fenofibrate | 2 | paddle dissolution apparatus using 1.0% SDS stirred at 100 rpm and maintained at a temperature of 37 ± 0.5°C. | Rats | Linear (level A) correlation was observed between in-vitro dissolution and both the in vivo absorption and the in situ intestinal absorption. This study supports that in situ closed-loop method for intestinal absorption can be used as a substitute for human studies in the optimization of nanosuspension formulations. | (Xu et al., 2014) |
| 8 | Eudragit RL 100 NPs | trans-resveratrol (t-RVT) | 2 | dialysis bag method with phosphate buffered saline (PBS) stirred at 50 rpm and maintained at a temperature of 37°C. | Rats | A point-to-point correlation (Level A IVIVC) between in-vitro and in-vivo performances may facilitate the rational development of extended-release dosage forms and serve as a tool for formulation screening, dissolution conditions optimization and a surrogate for bioequivalence testing. | (Singh & Pai, 2014) |
| 9 | Lyotropic Liquid Crystalline NPs (LCNPs) | Coenzyme Q10 (CoQ10) | 2 | Dialysis membrane, release medium comprised of 5% w/v Cremophor EL, 150 mM NaCl, and 20 mM CaCl2, maintained at 37.0 ± 1°C at 100 strokes per min. Release medium was set to SGF (pH 1.2) for 2 h, followed by SIF (pH 6.8) for 6 h, and finally to PBS (pH 7.4, containing 20% v/v plasma) for the rest of studies, in the presence/absence of lipase supplement. | Sprague-Dawley rats | Better IVIVR predictions were obtained in lipase rich dissolution media in case of digestible lipid due to rapid drug release in the presence of lipase. The established IVIVR could be applied to assist in rapid development of LCNPs from industrial perspectives. | (Swarnakar et al., 2014) |
| 10 | Self-nanoemulsifying drug delivery system (SNEDDS) | Olanzapine | 2 | USP dissolution apparatus Type I at 50 rpm at 37 ± 0.5°C with 0.1N HCl and biorelevant dissolution media to physiologically represent the in vivo biological environment. | Rabbit | Level A correlation was established. It was concluded that dissolution of SNEDDS in 0.1 N HCl can be used to predict in vivo bioavailability, instead of using biorelevant media (due to similar dissolution profiles in biorelevant dissolution media and 0.1 N HCl) | (Urmilasri, 2013) |
| 11 | Silybin (72 h-SLB) using a combination of solid dispersion, gel matrix and porous silica NPs (PSNs) | Silybin | 2 | Dialysis tubing method in paddle type dissolution apparatus using artificial gastric juice (pH 1.2), artificial intestinal juice (pH 6.8) and a series of Na2CO3 solutions (0.01 pH 11.1, 0.06 pH 11.5 and 0.08 M pH 11.6). | Beagle dogs | Both 72 h-SLB and silybin loaded PSNs indicated a level A IVIVC with 0.06 M Na2CO3. The pH value of in vitro dissolution medium was higher compared with that of the in vivo gastrointestinal environment, but in vivo absorption and in vitro dissolution exhibited an excellent linear relationship. The reason behind this phenomenon is to be explored. | (Cao et al., 2013) |
| 12 | Solid self-nanoemulsifying drug delivery systems (S-SNEDDS) | Carvedilol | 2 | USP apparatus 2 with simulated gastric fluid (SGF) containing 0.5% (w/v) SLS, stirred at 50 rpm at a temperature of 37 ± 0.5°C. | Wistar rats | Level A correlation was developed between percent drug absorbed and percent drug release. Apart from being an excellent product development tool, such IVIVC can also be exploited for obtaining biowaivers. | (Singh et al., 2013) |
| 13 | Hollow-type mesoporous silica NPs (HMSNs). | Silybin meglumine | silybin combined with meglumine shows high water solubility | dialysis tubing with paddle method using artificial gastric juice (pH 1.2), artificial intestinal juice (pH 6.8), and 0.08 M Na2CO3 stirred at 100 rpm and maintained at a temperature of 37 ± 0.5°C. | Beagle dogs | Na2CO3 solution (in contrast to artificial gastric juice and artificial intestinal juice ) showed a better correlation (level A) between in vitro dissolution and in vivo absorption. It was concluded that HMSNs together with Na2CO3 solution could achieve a promising model for sustained release of water-soluble drugs. | (Cao et al., 2012) |
| 14 | Nanostructured lipid carrier (NLC) system | Simvastatin | 2 | dialysis bag diffusion technique with pH 7.4 phosphate buffer stirred at 100 rpm and maintained at a temperature of 37 ± 0.5°C. | Mice | Level A IVIVC indicated that in-vitro release test can be used to predict the in-vivo absorption of developed formulations. This work facilitated the use of NLC systems to enhance the oral bioavailability of BCS class 2 drugs. | (Tiwari & Pathak, 2011) |
| 15 | Gelatin NPs | Indomethacin | 2 | Dialysis bag with simulated phosphate buffered saline (PBS, pH 7.4) mildly agitated and maintained at a temperature of 37°C. | Wistar albino rats | level A IVIVC was established between in-vitro dissolution and in-vivo absorption curves. This in vitro release method can be used to predict sustained release of drugs from gelatin NPs. | (Kumar et al., 2011) |
Further, the intrinsic solubility of a drug correlates well to its dissolution rate. But the release rate (and fraction of drug released) is inversely related to physical stability of NPs and lipophilicity of model drug. The stable nanoparticles (with no significant change in particle size and PDI) tend to protect the drug cargo and exhibit slow-release rates (Weng et al., 2020). Thus, drug release from NPs is not always straightforward making it difficult to develop characterization methods for oral NDDS. Researchers use different in vitro release methods to test oral NDDS (Paddle, Sample and separate and dialysis methods etc.) that gives variable results different from in vivo outcomes. Weng J et. al. proposed a novel sample and separate method combined with USP apparatus II (paddle) and well-validated centrifugal ultrafiltration technique to efficiently separate free drug from NPs to achieve better correlation between the in vitro data and the in vivo response for polymeric NPs (Weng et al., 2020). Dissolution is also a key elimination process strongly affected by pH of microenvironment (neutral extracellular fluids or low-pH intracellular fluids) (Studer et al., 2010). It requires extensive data on NDDS dissolution in these different GIT environments, focusing on dissolution rates, rate constants, orders of reaction, dissolution half-times and life-times (Utembe et al., 2015).
In vitro drug release from oral dosage forms is generally tested in 0.1N hydrochloric acid (pH 1.2) followed by pH 6.8 phosphate buffer to mimic gastric and intestinal milieu (Dudhipala et al., 2018). The choice of release media may depend on the drug solubility and absorption site. Bharathi et al. studied drug release of valsartan loaded eudragit nanoparticles only at pH 6.8 as it is a weakly acidic drug and is primarily absorbed at pH greater than 5 (Bharathi et al., 2012). In contrast, the dissolution of furosemide, another weakly acidic drug, was tested at pH 1.2. Indeed, furosemide has low solubility in gastric environment, but it exhibits site specific absorption in the stomach and upper intestine. Hence improving the solubility in gastric fluid becomes crucial for improving the overall bioavailability (Shariare et al., 2019). Weng et. al. successfully evaluated in vitro drug release of three poorly water-soluble drugs from polymeric NPs. 0.1N hydrochloric acid was used for weakly basic drug (itraconazole), 0.1% (w/v ) SDS solution for non-ionic compound (cholecalciferol or VitD3) and phosphate buffered saline (PBS, pH = 7.4) for weak acidic drug (flurbiprofen) (Weng et al., 2020).
Conventional kinetic models may not be appropriate to define the complex release profiles of oral NDDS. New more suitable empirical kinetics models should be developed for oral NDDS based on the release data NPs (smaller than 200nm) are proposed to provide a “shuttle” for drug across the unstirred water layer (UWL) near the surface of intestinal epithelium. This transport mechanism results into higher unbound drug concentration for absorption. In silico nanomodified permeability model (accounting for NP solubility and size) was developed to accurately predict the in vivo absorption of poorly soluble, UWL-limited drugs from NDDS (Stewart & Grass, 2020). Other emerging computational models can be applied in near future to predict the in vivo biofate of oral NDDS more reliably. For example, Quantitative Nanostructure Activity Relationship (Nano-QSAR or QNAR) models can be employed to predict biological activity and toxicity of NDDS (Fourches et al., 2010; Kar et al., 2014; Muratov et al., 2020; Puzyn et al., 2011; Wang, 2018; Weissleder et al., 2005; Yuan et al., 2021). Quantitative structure-pharmacokinetic relationships (QSPKR) models can correlate the drug chemical structure and properties to predict various pharmacokinetic parameters such as clearance (Cl) (Dave & Morris, 2015; Zhivkova & Doytchinova, 2013), half-life(t1/2) (Durairaj et al., 2009), apparent volume of distribution (Vd (Ghafourian et al., 2004). Quantitative systems pharmacology (QSP) model illustrated that minor differences in NP attributes could result into different exposures at target site indicating that systemic bioequivalence might not accurately reflect the target site bioequivalence (Au, Lu, et al., 2019). Abbiati et. al. discussed the potential application of QSP models to determine NP delivery and in vivo residence (Au, Abbiati, et al., 2019).
6 ∣. CONCLUSION
There exists a lack of robust and biorelevant in vitro drug release method for NDDS. It leads to arbitrary selection of release methods by different researchers, among which dialysis membrane methods are most widely used. USP 4 emerges as a viable dissolution test due to various advantages including its discriminatory capabilities. However, more research is obligatory to corroborate its regulatory standing; and to confirm its suitability to test variety of drug candidates (different BCS class), formulated in different NDDS (e.g., liposomes, nanocrystals, polymeric NPs, and lipid-based systems). At present, FDA encourages discussions regarding scientific rationale, feasibility, and method validation to develop a reproducible and reliable dissolution method that should be sensitive to formulation or processes variability in generic versions of NDDS.
Further, NPs consolidated with its protein-corona, behave as a new biological entity and affect the biodistribution as well as the release behavior of NDDS. Identification of the specific corona proteins, evaluation the protein-drug interactions and their overall impact on drug solubility can be instrumental in the development of a meaningful IVIVC.
Use of biorelevant and clinically relevant test media presents a valuable approach for the development of predictive in vitro release tests for NDDS. Unique two-stage experimental set-ups to mimic the drug release at target site along with drug release in circulation may be required to simulate the physiological environment. We are hopeful that proposed considerations can guide the design of future in vitro dissolution strategies, predictive of in vivo pharmacokinetic profiles, with a net result of favorable economic implications accompanying generic versions of NDDS.
ACKNOWLEDGEMENTS
This study was funded in part by Cancer Prevention & Research Institute of Texas (CPRIT) Core Facilities Support Awards under Award Number RP180748 and the National Institute on Minority Health and Health Disparities (NIMHD) of the National Institute of Health (NIH) under Award Number 2U54MD007605. The content is solely responsibility of authors and does not necessarily represent the official views of the CPRIT and NIH.
REFERENCES
- Alshora DH, Ibrahim MA, Elzayat E, Almeanazel OT, & Alanazi F (2018). Rosuvastatin calcium nanoparticles: Improving bioavailability by formulation and stabilization codesign. PLoS One, 13(7), e0200218. 10.1371/journal.pone.0200218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NH, Bauer M, Boussac N, Khan-Malek R, Munden P, & Sardaro M (1998). An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J Pharm Biomed Anal, 17(4-5), 811–822. 10.1016/s0731-7085(98)00011-9 [DOI] [PubMed] [Google Scholar]
- Anselmo AC, & Mitragotri S (2019). Nanoparticles in the clinic: An update. Bioeng Transl Med, 4(3), e10143. 10.1002/btm2.10143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton N, Bally F, Serra CA, Ali A, Arntz Y, Mely Y, Zhao M, Marchioni E, Jakhmola A, & Vandamme TF (2012). A new microfluidic setup for precise control of the polymer nanoprecipitation process and lipophilic drug encapsulation [ 10.1039/C2SM25357G]. Soft Matter, 8(41), 10628–10635. 10.1039/C2SM25357G [DOI] [Google Scholar]
- Arafat M, Kirchhoefer C, Mikov M, Sarfraz M, & Löbenberg R (2017). Nanosized Liposomes Containing Bile Salt: A Vesicular Nanocarrier for Enhancing Oral Bioavailability of BCS Class III Drug. J Pharm Pharm Sci, 20(0), 305–318. 10.18433/J3CK88 [DOI] [PubMed] [Google Scholar]
- Attari Z, Kalvakuntla S, Reddy MS, Deshpande M, Rao CM, & Koteshwara KB (2016). Formulation and characterisation of nanosuspensions of BCS class II and IV drugs by combinative method. Journal of Experimental Nanoscience, 11(4), 276–288. 10.1080/17458080.2015.1055841 [DOI] [Google Scholar]
- Attia MF, Anton N, Wallyn J, Omran Z, & Vandamme TF (2019). An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J Pharm Pharmacol, 71(8), 1185–1198. 10.1111/jphp.13098 [DOI] [PubMed] [Google Scholar]
- Au JL-S (2017). Considerations for bioequivalence evaluation of nano-particulate/molecular medicine. Retrieved May 05 from https://www.fda.gov/media/108401/download [Google Scholar]
- Bastogne T (2017). Quality-by-design of nanopharmaceuticals - a state of the art. Nanomedicine, 13(7), 2151–2157. 10.1016/j.nano.2017.05.014 [DOI] [PubMed] [Google Scholar]
- Behzadi S, Serpooshan V, Sakhtianchi R, Müller B, Landfester K, Crespy D, & Mahmoudi M (2014). Protein corona change the drug release profile of nanocarriers: the "overlooked" factor at the nanobio interface. Colloids Surf B Biointerfaces, 123, 143–149. 10.1016/j.colsurfb.2014.09.009 [DOI] [PubMed] [Google Scholar]
- Bhagav P, Upadhyay H, & Chandran S (2011). Brimonidine tartrate-eudragit long-acting nanoparticles: formulation, optimization, in vitro and in vivo evaluation. AAPS PharmSciTech, 12(4), 1087–1101. 10.1208/s12249-011-9675-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharathi M, Chandra S, Eswari R, Raja S, Allena RT, Brito S, & Bhaskar Reddy K (2012). Preparation and in vitro & in vivo characterization of valsartan loaded eudragit nanoparticles. Der. Pharm. Sin, 3, 516–525. [Google Scholar]
- Bhardwaj U, & Burgess DJ (2010). A novel USP apparatus 4 based release testing method for dispersed systems. Int J Pharm, 388(1-2), 287–294. 10.1016/j.ijpharm.2010.01.009 [DOI] [PubMed] [Google Scholar]
- Bulbake U, Doppalapudi S, Kommineni N, & Khan W (2017). Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics, 9(2). 10.3390/pharmaceutics9020012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JM, & Dressman JB (2010). The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci, 99(12), 4940–4954. 10.1002/jps.22217 [DOI] [PubMed] [Google Scholar]
- Calvo P, Vila-Jato JL, & Alonso MJ (1996). Comparative in vitro evaluation of several colloidal systems, nanoparticles, nanocapsules, and nanoemulsions, as ocular drug carriers. J Pharm Sci, 85(5), 530–536. 10.1021/js950474+ [DOI] [PubMed] [Google Scholar]
- Cardot JM, & Davit BM (2012). In vitro-in vivo correlations: tricks and traps. AAPS J, 14(3), 491–499. 10.1208/s12248-012-9359-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidambaram N, & Burgess DJ (1999). A novel in vitro release method for submicron sized dispersed systems. AAPS PharmSci, 1(3), E11. 10.1208/ps010311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcy DM, Liu B, Bradley G, Healy AM, & Corrigan OI (2010). Hydrodynamic and species transfer simulations in the USP 4 dissolution apparatus: considerations for dissolution in a low velocity pulsing flow. Pharm Res, 27(2), 246–258. 10.1007/s11095-009-0010-4 [DOI] [PubMed] [Google Scholar]
- Davanço MG, Campos DR, & Carvalho PO (2020). In vitro - In vivo correlation in the development of oral drug formulation: A screenshot of the last two decades. Int J Pharm, 580, 119210. 10.1016/j.ijpharm.2020.119210 [DOI] [PubMed] [Google Scholar]
- Dave VS, Gupta D, Yu M, Nguyen P, & Varghese Gupta S (2017). Current and evolving approaches for improving the oral permeability of BCS Class III or analogous molecules. Drug Dev Ind Pharm, 43(2), 177–189. 10.1080/03639045.2016.1269122 [DOI] [PubMed] [Google Scholar]
- Deng Y, Zhang X, Shen H, He Q, Wu Z, Liao W, & Yuan M (2019). Application of the Nano-Drug Delivery System in Treatment of Cardiovascular Diseases. Front Bioeng Biotechnol, 7, 489. 10.3389/fbioe.2019.00489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikpati A, Mohammadi F, Greffard K, Quéant C, Arnaud P, Bastiat G, Rudkowska I, & Bertrand N (2020). Residual Solvents in Nanomedicine and Lipid-Based Drug Delivery Systems: a Case Study to Better Understand Processes. Pharm Res, 37(8), 149. 10.1007/s11095-020-02877-x [DOI] [PubMed] [Google Scholar]
- Dudhipala N, Janga KY, & Gorre T (2018). Comparative study of nisoldipine-loaded nanostructured lipid carriers and solid lipid nanoparticles for oral delivery: preparation, characterization, permeation and pharmacokinetic evaluation. Artif Cells Nanomed Biotechnol, 46(sup2), 616–625. 10.1080/21691401.2018.1465068 [DOI] [PubMed] [Google Scholar]
- D’Souza S (2014). A Review of In Vitro Drug Release Test Methods for Nano-Sized Dosage Forms. Advances in Pharmaceutics, 2014, 304757. 10.1155/2014/304757 [DOI] [Google Scholar]
- Eaton JW, Tran D, Hauck WW, & Stippler ES (2012). Development of a performance verification test for USP apparatus 4. Pharm Res, 29(2), 345–351. 10.1007/s11095-011-0559-6 [DOI] [PubMed] [Google Scholar]
- Fam SY, Chee CF, Yong CY, Ho KL, Mariatulqabtiah AR, & Tan WS (2020). Stealth Coating of Nanoparticles in Drug-Delivery Systems. Nanomaterials (Basel), 10(4). 10.3390/nano10040787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farjadian F, Ghasemi A, Gohari O, Roointan A, Karimi M, & Hamblin MR (2019). Nanopharmaceuticals and nanomedicines currently on the market: challenges and opportunities. Nanomedicine (Lond), 14(1), 93–126. 10.2217/nnm-2018-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. (1997a). Extended Release Oral Dosage Forms:Development, Evaluation, and Application of In Vitro/In Vivo Correlations. https://www.fda.gov/media/70939/download [DOI] [PubMed] [Google Scholar]
- FDA. (1997b). SUPAC-MR: Modified Release Solid Oral Dosage Forms. Retrieved April 28 from https://www.fda.gov/media/70956/download [Google Scholar]
- FDA. (2014a). Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology. Office of the Commissioner, Office of Policy, Legislation, and International Affairs, Office of Policy. Retrieved June 05 from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considering-whether-fda-regulated-product-involves-application-nanotechnology [Google Scholar]
- FDA. (2014b). Safety of Nanomaterials in Cosmetic Products. Retrieved January 03 from https://www.fda.gov/media/83957/download [Google Scholar]
- FDA. (2015). Use of Nanomaterials in Food for Animals. Retrieved January 03 from https://www.fda.gov/media/88828/download [Google Scholar]
- FDA. (2017a). Drug Products, Including Biological Products, that Contain Nanomaterials. Retrieved May 05 from https://www.fda.gov/files/drugs/published/Drug-Products--Including-Biological-Products--that-Contain-Nanomaterials---Guidance-for-Industry.pdf [Google Scholar]
- FDA. (2017b). ICH Q3C Maintenance Procedures for the Guidance for Industry Q3C Impurities: Residual Solvents. Retrieved April 14 from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ich-q3c-maintenance-procedures-guidance-industry-q3c-impurities-residual-solvents [Google Scholar]
- FDA. (2018a). Liposome Drug Products Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation. Retrieved May 05 from https://www.fda.gov/media/70837/download [Google Scholar]
- FDA. (2018b). M9 BIOPHARMACEUTICS CLASSIFICATION SYSTEM-BASED BIOWAIVERS. Retrieved April 28 from https://www.fda.gov/media/117974/download [Google Scholar]
- FDA. (2018c). Q3C — Tables and List Guidance for Industry. Retrieved April 04 from https://www.fda.gov/media/133650/download [Google Scholar]
- FDA. (2020a). Drugs@FDA: FDA-Approved Drugs. [Google Scholar]
- FDA. (2020b). Vaccines Licensed for use in the United States. [Google Scholar]
- Gillespie WR (1997). Convolution-based approaches for in vivo-in vitro correlation modeling. Adv Exp Med Biol, 423, 53–65. 10.1007/978-1-4684-6036-0_5 [DOI] [PubMed] [Google Scholar]
- Gite S, Chogale M, & Patravale V (2016). Development and validation of a discriminating dissolution method for atorvastatin delayed-release nanoparticles using a flow-through cell: A comparative study using USP apparatus 4 and 1. I. Dissolution Technologies. https://go.gale.com/ps/anonymous?p=AONE&sw=w&issn=1521298X&v=2.1&it=r&id=GALE%7CA456498201&sid=googleScholar&linkaccess=fulltext [Google Scholar]
- Gualtieri AF, Pollastri S, Bursi Gandolfi N, & Gualtieri ML (2018). In vitro acellular dissolution of mineral fibres: A comparative study. Sci Rep, 8(1), 7071. 10.1038/s41598-018-25531-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, & Xie H (2018). Nanoparticles in Daily Life: Applications, Toxicity and Regulations. J Environ Pathol Toxicol Oncol, 37(3), 209–230. 10.1615/JEnvironPatholToxicolOncol.2018026009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha ES, Choo GH, Baek IH, & Kim MS (2014). Formulation, characterization, and in vivo evaluation of celecoxib-PVP solid dispersion nanoparticles using supercritical antisolvent process. Molecules, 19(12), 20325–20339. 10.3390/molecules191220325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham AS, Cost MR, Sassi AB, Dezzutti CS, & Rohan LC (2009). Targeted delivery of PSC-RANTES for HIV-1 prevention using biodegradable nanoparticles. Pharm Res, 26(3), 502–511. 10.1007/s11095-008-9765-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann R, & Tisserand D (2006). Dissolution kinetics as a function of the Gibbs free energy of reaction: An experimental study based on albite feldspar. Geochimica et Cosmochimica Acta, 70(2), 364–383. [Google Scholar]
- Heng D, Cutler DJ, Chan HK, Yun J, & Raper JA (2008). What is a suitable dissolution method for drug nanoparticles? Pharm Res, 25(7), 1696–1701. 10.1007/s11095-008-9560-0 [DOI] [PubMed] [Google Scholar]
- ICH. (2019). ICH guideline Q3C (R6) on impurities: guideline for residual solvents. Retrieved April 14 from https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-33.pdf [Google Scholar]
- Jablonka L, Ashtikar M, Gao GF, Thurn M, Modh H, Wang JW, Preuß A, Scheglmann D, Albrecht V, Röder B, & Wacker MG (2020). Predicting human pharmacokinetics of liposomal temoporfin using a hybrid in silico model. Eur J Pharm Biopharm, 149, 121–134. 10.1016/j.ejpb.2020.02.001 [DOI] [PubMed] [Google Scholar]
- Jacob S, & Nair AB (2018). An updated overview with simple and practical approach for developing in vitro-in vivo correlation. Drug Dev Res, 79(3), 97–110. 10.1002/ddr.21427 [DOI] [PubMed] [Google Scholar]
- Jain P, Pawar RS, Pandey RS, Madan J, Pawar S, Lakshmi PK, & Sudheesh MS (2017). In-vitro in-vivo correlation (IVIVC) in nanomedicine: Is protein corona the missing link? Biotechnol Adv, 35(7), 889–904. 10.1016/j.biotechadv.2017.08.003 [DOI] [PubMed] [Google Scholar]
- Janas C, Mast MP, Kirsamer L, Angioni C, Gao F, Mäntele W, Dressman J, & Wacker MG (2017). The dispersion releaser technology is an effective method for testing drug release from nanosized drug carriers. Eur J Pharm Biopharm, 115, 73–83. 10.1016/j.ejpb.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Kakhi M (2009). Mathematical modeling of the fluid dynamics in the flow-through cell. Int J Pharm, 376(1-2), 22–40. 10.1016/j.ijpharm.2009.04.012 [DOI] [PubMed] [Google Scholar]
- Kamaly N, Yameen B, Wu J, & Farokhzad OC (2016). Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem Rev, 116(4), 2602–2663. 10.1021/acs.chemrev.5b00346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karumanchi DK, Skrypai Y, Thomas A, & Gaillard ER (2018). Rational design of liposomes for sustained release drug delivery of bevacizumab to treat ocular angiogenesis. Journal of Drug Delivery Science and Technology, 47, 275–282. [Google Scholar]
- Kassaye L, & Genete G (2013). Evaluation and comparison of in-vitro dissolution profiles for different brands of amoxicillin capsules. Afr Health Sci, 13(2), 369–375. 10.4314/ahs.v13i2.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur N, Narang A, & Bansal AK (2018). Use of biorelevant dissolution and PBPK modeling to predict oral drug absorption. Eur J Pharm Biopharm, 129, 222–246. 10.1016/j.ejpb.2018.05.024 [DOI] [PubMed] [Google Scholar]
- Kim A, Ng WB, Bernt W, & Cho NJ (2019). Validation of Size Estimation of Nanoparticle Tracking Analysis on Polydisperse Macromolecule Assembly. Sci Rep, 9(1), 2639. 10.1038/s41598-019-38915-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, & Baek IH (2014). Fabrication and evaluation of valsartan-polymer- surfactant composite nanoparticles by using the supercritical antisolvent process. Int J Nanomedicine, 9, 5167–5176. 10.2147/IJN.S71891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, Fluckiger L, Hoffman M, Lartaud-Idjouadiene I, Atkinson J, & Maincent P (1997). The antihypertensive effect of orally administered nifedipine-loaded nanoparticles in spontaneously hypertensive rats. Br J Pharmacol, 120(3), 399–404. 10.1038/sj.bjp.0700910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull SM, Ammirata J, Bawa S, Li M, Bilgili E, & Davé RN (2017). Critical Material Attributes of Strip Films Loaded With Poorly Water-Soluble Drug Nanoparticles: II. Impact of Polymer Molecular Weight. J Pharm Sci, 106(2), 619–628. 10.1016/j.xphs.2016.10.009 [DOI] [PubMed] [Google Scholar]
- Kumar S, Kaur R, Rajput R, & Singh M (2018). Bio Pharmaceutics Classification System (BCS) Class IV Drug Nanoparticles: Quantum Leap to Improve Their Therapeutic Index. Adv Pharm Bull, 8(4), 617–625. 10.15171/apb.2018.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zou P, Tyner K, & Lee S (2017). Physiologically Based Pharmacokinetic (PBPK) Modeling of Pharmaceutical Nanoparticles. AAPS J, 19(1), 26–42. 10.1208/s12248-016-0010-3 [DOI] [PubMed] [Google Scholar]
- Litou C, Patel N, Turner DB, Kostewicz E, Kuentz M, Box KJ, & Dressman J (2019). Combining biorelevant in vitro and in silico tools to simulate and better understand the in vivo performance of a nano-sized formulation of aprepitant in the fasted and fed states. Eur J Pharm Sci, 138, 105031. 10.1016/j.ejps.2019.105031 [DOI] [PubMed] [Google Scholar]
- Liu P, De Wulf O, Laru J, Heikkilä T, van Veen B, Kiesvaara J, Hirvonen J, Peltonen L, & Laaksonen T (2013). Dissolution studies of poorly soluble drug nanosuspensions in non-sink conditions. AAPS PharmSciTech, 14(2), 748–756. 10.1208/s12249-013-9960-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Xu P, Ding HM, Yu YS, Huo D, & Ma YQ (2019). Tailoring the component of protein corona via simple chemistry. Nat Commun, 10(1), 4520. 10.1038/s41467-019-12470-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke MC (2017). Equivalence of Locally-Acting Drug Products. Retrieved May 06 from https://www.fda.gov/media/105890/download [Google Scholar]
- Machrafi H (2020). On the chemical potential of nanoparticle dispersion. Physics Letters A, 384(19), 126485. [Google Scholar]
- Madani F, Esnaashari SS, Bergonzi MC, Webster TJ, Younes HM, Khosravani M, & Adabi M (2020). Paclitaxel/methotrexate co-loaded PLGA nanoparticles in glioblastoma treatment: Formulation development and in vitro antitumor activity evaluation. Life Sci, 256, 117943. 10.1016/j.lfs.2020.117943 [DOI] [PubMed] [Google Scholar]
- Margolskee A, Darwich AS, Galetin A, Rostami-Hodjegan A, & Aarons L (2016). Deconvolution and IVIVC: Exploring the Role of Rate-Limiting Conditions. AAPS J, 18(2), 321–332. 10.1208/s12248-015-9849-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, & Lim LY (2005). Paclitaxel-loaded PLGA nanoparticles: potentiation of anticancer activity by surface conjugation with wheat germ agglutinin. J Control Release, 108(2-3), 244–262. 10.1016/j.jconrel.2005.08.013 [DOI] [PubMed] [Google Scholar]
- Morais JM, & Burgess DJ (2014). In vitro release testing methods for vitamin E nanoemulsions. Int J Pharm, 475(1-2), 393–400. 10.1016/j.ijpharm.2014.08.063 [DOI] [PubMed] [Google Scholar]
- Morales-Cruz M, Flores-Fernández GM, Orellano EA, Rodriguez-Martinez JA, Ruiz M, & Griebenow K (2012). Two-step nanoprecipitation for the production of protein-loaded PLGA nanospheres. Results Pharma Sci, 2, 79–85. 10.1016/j.rinphs.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD. (2018). Assessment of Biodurability of Nanomaterials and their Surface ligands Series on the Safety of Manufactured Nanomaterials No. 86. Retrieved May 06 from http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2018)11&doclanguage=en [Google Scholar]
- Paliwal R, Babu RJ, & Palakurthi S (2014). Nanomedicine scale-up technologies: feasibilities and challenges. AAPS PharmSciTech, 15(6), 1527–1534. 10.1208/s12249-014-0177-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MEM, McParland D, Szklanna PB, Guang MHZ, O’Connell K, O’Connor HD, McGuigan C, Ní Áinle F, McCann A, & Maguire PB (2017). A Protocol for Improved Precision and Increased Confidence in Nanoparticle Tracking Analysis Concentration Measurements between 50 and 120 nm in Biological Fluids. Front Cardiovasc Med, 4, 68. 10.3389/fcvm.2017.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi SA (2010). In Vitro-In Vivo Correlation (IVIVC) and Determining Drug Concentrations in Blood from Dissolution Testing – A Simple and Practical Approach. The Open Drug Delivery Journal, 4, 10. 10.2174/1874126601004010038 [DOI] [Google Scholar]
- Riviere JE (2013). Of mice, men and nanoparticle biocoronas: are in vitro to in vivo correlations and interspecies extrapolations realistic? Nanomedicine (Lond), 8(9), 1357–1359. 10.2217/nnm.13.129 [DOI] [PubMed] [Google Scholar]
- Robertson JD, Rizzello L, Avila-Olias M, Gaitzsch J, Contini C, Magoń MS, Renshaw SA, & Battaglia G (2016). Purification of Nanoparticles by Size and Shape. Sci Rep, 6, 27494. 10.1038/srep27494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna V, Roggio AM, Siliani S, Piccinini M, Marceddu S, Mariani A, & Sechi M (2012). Development of novel cationic chitosan-and anionic alginate-coated poly(D,L-lactide-co-glycolide) nanoparticles for controlled release and light protection of resveratrol. Int J Nanomedicine, 7, 5501–5516. 10.2147/IJN.S36684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedighi M, Sieber S, Rahimi F, Shahbazi MA, Rezayan AH, Huwyler J, & Witzigmann D (2019). Rapid optimization of liposome characteristics using a combined microfluidics and design-of-experiment approach. Drug Deliv Transl Res, 9(1), 404–413. 10.1007/s13346-018-0587-4 [DOI] [PubMed] [Google Scholar]
- Shabbits JA, Chiu GN, & Mayer LD (2002). Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems. J Control Release, 84(3), 161–170. 10.1016/s0168-3659(02)00294-8 [DOI] [PubMed] [Google Scholar]
- Shah VP, Cecil TL, Srinivasan SV, & Williams RL (2014). Progressively reducing regulatory burden. AAPS J, 16(4), 621–624. 10.1208/s12248-014-9601-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah VP, Tsong Y, Sathe P, & Liu JP (1998). In vitro dissolution profile comparison--statistics and analysis of the similarity factor, f2. Pharm Res, 15(6), 889–896. 10.1023/a:1011976615750 [DOI] [PubMed] [Google Scholar]
- Shameem M, Lee H, & DeLuca PP (1999). A short term (accelerated release) approach to evaluate peptide release from PLGA depot-formulations. AAPS PharmSci, 1(3), E7. 10.1208/ps010307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shariare MH, Altamimi MA, Marzan AL, Tabassum R, Jahan B, Reza HM, Rahman M, Ahsan GU, & Kazi M (2019). dissolution and bioavailability study of furosemide nanosuspension prepared using design of experiment (DoE). Saudi Pharm J, 27(1), 96–105. 10.1016/j.jsps.2018.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Maheshwari D, Philip G, Rana R, Bhatia S, Singh M, Gabrani R, Sharma SK, Ali J, Sharma RK, & Dang S (2014). Formulation and optimization of polymeric nanoparticles for intranasal delivery of lorazepam using Box-Behnken design: in vitro and in vivo evaluation. Biomed Res Int, 2014, 156010. 10.1155/2014/156010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, & Burgess DJ (2012). Accelerated in vitro release testing of implantable PLGA microsphere/PVA hydrogel composite coatings. Int J Pharm, 422(1-2), 341–348. 10.1016/j.ijpharm.2011.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, & Burgess DJ (2013). Dissolution Testing Strategies for Nanoparticulate Drug Delivery Systems: Recent Developments and Challenges. Drug Deliv Transl Res, 3(5), 409–415. 10.1007/s13346-013-0129-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, & Burgess DJ (2015). In vitro-in vivo correlation for complex non-oral drug products: Where do we stand? J Control Release, 219, 644–651. 10.1016/j.jconrel.2015.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Lee K, Choi S, Qu W, Wang Y, & Burgess DJ (2016). A reproducible accelerated in vitro release testing method for PLGA microspheres. Int J Pharm, 498(1-2), 274–282. 10.1016/j.ijpharm.2015.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiko G, Gladden LF, Sederman AJ, Connolly PC, & Butler JM (2011). MRI studies of the hydrodynamics in a USP 4 dissolution testing cell. J Pharm Sci, 100(3), 976–991. 10.1002/jps.22343 [DOI] [PubMed] [Google Scholar]
- Sievens-Figueroa L, Pandya N, Bhakay A, Keyvan G, Michniak-Kohn B, Bilgili E, & Davé RN (2012). Using USP I and USP IV for discriminating dissolution rates of nano- and microparticle-loaded pharmaceutical strip-films. AAPS PharmSciTech, 13(4), 1473–1482. 10.1208/s12249-012-9875-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, & Aboul-Enein HY (2006). Advantages of USP Apparatus IV (flow-through cell apparatus) in dissolution studies. Journal of the Iranian Chemical Society, 3(3), 220–222. 10.1007/BF03247211 [DOI] [Google Scholar]
- Stevens RE, Gray V, Dorantes A, Gold L, & Pham L (2015). Scientific and regulatory standards for assessing product performance using the similarity factor, f2. AAPS J, 17(2), 301–306. 10.1208/s12248-015-9723-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AM, & Grass ME (2020). Practical Approach to Modeling the Impact of Amorphous Drug Nanoparticles on the Oral Absorption of Poorly Soluble Drugs. Mol Pharm, 17(1), 180–189. 10.1021/acs.molpharmaceut.9b00889 [DOI] [PubMed] [Google Scholar]
- Studer AM, Limbach LK, Van Duc L, Krumeich F, Athanassiou EK, Gerber LC, Moch H, & Stark WJ (2010). Nanoparticle cytotoxicity depends on intracellular solubility: comparison of stabilized copper metal and degradable copper oxide nanoparticles. Toxicol Lett, 197(3), 169–174. 10.1016/j.toxlet.2010.05.012 [DOI] [PubMed] [Google Scholar]
- Suk JS, Xu Q, Kim N, Hanes J, & Ensign LM (2016). PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev, 99(Pt A), 28–51. 10.1016/j.addr.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Srinivasan S, Yuan W, Ming R, Liu Y, Dai Z, Noble CO, Hayes ME, Zheng N, Jiang W, Szoka FC, & Schwendeman A (2019). Development of a flow-through USP 4 apparatus drug release assay for the evaluation of amphotericin B liposome. Eur J Pharm Biopharm, 134, 107–116. 10.1016/j.ejpb.2018.11.010 [DOI] [PubMed] [Google Scholar]
- Tang R (2005). Progress in the studies of interfacial energy and kinetics of crystal growth/dissolution. Progress in Chemistry, 17, 369–376. [Google Scholar]
- USP43-NF382S-online. (2020). 〈 1092 〉 THE DISSOLUTION PROCEDURE: DEVELOPMENT AND VALIDATION. text-autospace:none">https://online.uspnf.com/uspnf/document/1_GUID-CE0902BA-77AC-422D-8BF0-A221B5DE6012_5_en-US [Google Scholar]
- Utembe W, Clewell H, Sanabria N, Doganis P, & Gulumian M (2020). Current Approaches and Techniques in Physiologically Based Pharmacokinetic (PBPK) Modelling of Nanomaterials. Nanomaterials (Basel), 10(7). 10.3390/nano10071267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utembe W, Potgieter K, Stefaniak AB, & Gulumian M (2015). Dissolution and biodurability: Important parameters needed for risk assessment of nanomaterials. Part Fibre Toxicol, 12, 11. 10.1186/s12989-015-0088-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventola CL (2017). Progress in Nanomedicine: Approved and Investigational Nanodrugs. P T, 42(12), 742–755. [PMC free article] [PubMed] [Google Scholar]
- Wallenwein CM, Nova MV, Janas C, Jablonka L, Gao GF, Thurn M, Albrecht V, Wiehe A, & Wacker MG (2019). A dialysis-based in vitro drug release assay to study dynamics of the drug-protein transfer of temoporfin liposomes. Eur J Pharm Biopharm, 143, 44–50. 10.1016/j.ejpb.2019.08.010 [DOI] [PubMed] [Google Scholar]
- Wang R, Billone PS, & Mullett WM (2013). Nanomedicine in Action: An Overview of Cancer Nanomedicine on the Market and in Clinical Trials. Journal of Nanomaterials, 2013, 629681. 10.1155/2013/629681 [DOI] [Google Scholar]
- Weissig V, Pettinger TK, & Murdock N (2014). Nanopharmaceuticals (part 1): products on the market. Int J Nanomedicine, 9, 4357–4373. 10.2147/IJN.S46900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Tong HHY, & Chow SF (2020). In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method. Pharmaceutics, 12(8). 10.3390/pharmaceutics12080732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong S, George S, Yu H, Damoiseaux R, France B, Ng KW, & Loo JS (2013). Size influences the cytotoxicity of poly (lactic-co-glycolic acid) (PLGA) and titanium dioxide (TiO(2)) nanoparticles. Arch Toxicol, 87(6), 1075–1086. 10.1007/s00204-012-0938-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Khan MA, & Burgess DJ (2012). A two-stage reverse dialysis in vitro dissolution testing method for passive targeted liposomes. Int J Pharm, 426(1-2), 211–218. 10.1016/j.ijpharm.2012.01.030 [DOI] [PubMed] [Google Scholar]
- Yeo ELL, Cheah JUJ, Thong PSP, Soo KC, & Kah JCY (2019). Gold Nanorods Coated with Apolipoprotein E Protein Corona for Drug Delivery. ACS Applied Nano Materials, 2(10), 6220–6229. 10.1021/acsanm.9b01196 [DOI] [Google Scholar]
- Yoshida H, Kuwana A, Shibata H, Izutsu K, & Goda Y (2015). Particle Image Velocimetry Evaluation of Fluid Flow Profiles in USP 4 Flow-Through Dissolution Cells. Pharm Res, 32(9), 2950–2959. 10.1007/s11095-015-1676-4 [DOI] [PubMed] [Google Scholar]
- Yuan D, He H, Wu Y, Fan J, & Cao Y (2019). Physiologically Based Pharmacokinetic Modeling of Nanoparticles. J Pharm Sci, 108(1), 58–72. 10.1016/j.xphs.2018.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Kuai R, Dai Z, Yuan Y, Zheng N, Jiang W, Noble C, Hayes M, Szoka FC, & Schwendeman A (2017). Development of a Flow-Through USP-4 Apparatus Drug Release Assay to Evaluate Doxorubicin Liposomes. AAPS J, 19(1), 150–160. 10.1208/s12248-016-9958-2 [DOI] [PubMed] [Google Scholar]
- Zackrisson G, Östling G, Skagerberg B, & Anfält T (1995). ACcelerated Dissolution Rate Analysis (ACDRA) for controlled release drugs. Application to Roxiam®. Journal of Pharmaceutical and Biomedical Analysis, 13(4), 377–383. [DOI] [PubMed] [Google Scholar]
- Zhang B, Sai Lung P, Zhao S, Chu Z, Chrzanowski W, & Li Q (2017). Shape dependent cytotoxicity of PLGA-PEG nanoparticles on human cells. Sci Rep, 7(1), 7315. 10.1038/s41598-017-07588-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Guan J, Jiang Z, Yang Y, Liu J, Hua W, Mao Y, Li C, Lu W, Qian J, & Zhan C (2019). Brain-targeted drug delivery by manipulating protein corona functions. Nature Communications, 10(1), 3561. 10.1038/s41467-019-11593-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Sun DD, Zou P, & Jiang W (2017). Scientific and Regulatory Considerations for Generic Complex Drug Products Containing Nanomaterials. AAPS J, 19(3), 619–631. 10.1208/s12248-017-0044-1 [DOI] [PubMed] [Google Scholar]
- Zolnik BS, & Burgess DJ (2008). In Vitro–In Vivo Correlation on Parenteral Dosage Forms. . Springer. [Google Scholar]
- Zolnik BS, Leary PE, & Burgess DJ (2006). Elevated temperature accelerated release testing of PLGA microspheres. J Control Release, 112(3), 293–300. 10.1016/j.jconrel.2006.02.015 [DOI] [PubMed] [Google Scholar]