

Abstract
The first order of DNA packaging is the nucleosome with the DNA wrapped around the histone octamer. This leaves the nucleosomal DNA with access restrictions, which impose a significant barrier to repair of damaged DNA. The efficiency of DNA repair has been related to nucleosome structure and chromatin status, which is modulated in part by post-translational modifications (PTMs) of histones. Numerous studies have suggested a role for acetylation of lysine at position 56 of the H3 histone (H3K56ac) in various DNA transactions, including the response to DNA damage and its association with human cancer. Biophysical studies have revealed that H3K56ac increases DNA accessibility by facilitating spontaneous and transient unwrapping motions of the DNA ends. However, how this acetylation mark modulates nucleosome structure and dynamics to promote accessibility to the damaged DNA for repair factors and other proteins is still poorly understood. Here, we utilize approximately 5–6 microseconds of atomistic molecular dynamics simulations to delineate the impact of H3K56 acetylation on the nucleosome structure and dynamics, and to elucidate how these nucleosome properties are further impacted when a bulky benzo[a]pyrene-derived DNA lesion is placed near the acetylation site. Our findings reveal that H3K56ac alone induces considerable disturbance to the histone-DNA/histone-histone interactions, and amplifies the distortions imposed by the presence of the lesion. Our work highlights the important role of H3K56 acetylation in response to DNA damage and depicts how access to DNA lesions by the repair machinery can be facilitated within the nucleosome via a key acetylation event.
Keywords: DNA- benzo[a]pyrenyl adduct, H3K56 acetylation, Posttranslational modification, Nucleosome core particle, DNA unwrapping, Nucleotide excision repair
Graphical Abstract
Introduction
The nucleosome, the basic DNA packaging unit in eukaryotes, is comprised of a nucleosome core particle (NCP) together with linker DNA; the linker interconnects individual NCPs which are further folded into the compact chromatin fiber in euchromatin.1 The NCP contains approximately 147 base pairs of the highly negatively charged DNA wrapped around the positively charged histone octamer, which consists of one (H3-H4)2 tetramer and two H2A-H2B dimers.2, 3 This leaves the occluded DNA with access restrictions even in the nucleosome, the first level of chromatin compaction,4, 5 and imposes a significant barrier to all DNA transactions. There is significant interest in understanding how accessibility to the occluded DNA is modulated to enable biological processes such as repair; one important process that fosters accessibility is post-translational modifications (PTMs) of histones,6–14 which have related the efficiency of DNA repair to nucleosome structure and chromatin status.5, 12, 15 Acetylation of lysine, one of the most studied PTMs, functions in regulating gene activity and repair of DNA damage.11, 13, 16–18 Acetylation marks, found on all core histones, are deemed to have various functions that depend on the sites of their occurrence within the nucleosome:19–24 they are implicated in DNA unwrapping near the ends, disassembly of NCPs near the dyad, and on the tails they primarily impact the chromatin structure indirectly by recruiting chromatin remodeling factors. While some molecular dynamics simulations have explored effects of certain lysine acetylations,25–31 the impact of H3K56 acetylation on a DNA lesion has never been explored.
H3K56 is evolutionarily conserved and subject to reversible acetylation,32–36 that plays a role in many DNA processes,8, 33–35, 37–47 including signaling for DNA damage checkpoints and response to DNA damage,8, 33, 37–41 and it is associated with human cancer.35, 46 Defects in H3K56ac in yeast revealed sensitivity to genotoxic agents that cause DNA strand breaks during replication.33 Lack of the H3K56 acetylation mark influences the normal level of meiotic DNA double strand breaks (DSBs) within recombination hotspots.48 H3K56ac has been shown to play a role in the oncogenic Ras-P13K signaling pathway that regulates tumor cell activity,46 and its deacetylation is involved in damaged DNA-binding protein (DDB)-mediated damage recognition, a very early event in nucleotide excision repair (NER) for UVR-induced DNA lesions.40 H3K56ac enhances the human apurinic/apyrimidinic endonuclease 1 protein activity, at an early step in DNA base excision repair (BER).47 A study in mammalian cells49 showed that the DNA damage response (DDR) is regulated by a dynamic H3K56 acetylation and deacetylation process, an important signal for completion of repair.
H3K56 is located at the first αN helix of the globular H3 core domain (Figure 1) and its side-chain extends toward the DNA major groove near the entry/exit sites of the nucleosome.2, 50 Thus, the predominantly biophysical role of the H3K56ac is to weaken the electrostatic interactions between the DNA termini and the histones.24, 51 Nucleosomes are highly dynamic;13, 52, 53 an important aspect of the intrinsic dynamics in nucleosomes is the transient and spontaneous unwrapping of the DNA termini from the histone octamer, called “DNA breathing”.13, 54 A study in vivo has suggested that the spontaneous unwrapping of DNA can facilitate rapid DNA repair.55 Notably, it has been reported that nucleosomes within homogeneous nucleosome arrays undergo DNA unwrapping similarly to the isolated nucleosomes and their DNA is similarly accessible to DNA-binding proteins,13, 56 indicating that the site exposure attributable to the presence of nucleosome neighbors within the chromatin fiber is of more modest-significance. Biophysical studies have shown that acetylation of H3K56 increases DNA accessibility by facilitating spontaneous and transient unwrapping dynamics of the DNA ends.57–61 While acetylation of H3K56 enhances the spontaneous opening dynamics of the nucleosome by about two-fold,59 the enhancement is about six-fold when H3K56 is acetylated while also bound with histone chaperone nucleosome assembly protein 1 (Nap1).61 These findings highlight the important role played by H3K56ac in changing the dynamic behavior of nucleosomes by enhancing the spontaneous and transient DNA unwrapping motion to increases site exposure that may be enhanced by the presence of a ligand moiety such as Nap1; we hypothesize that such enhancement may apply as well to a DNA lesion.
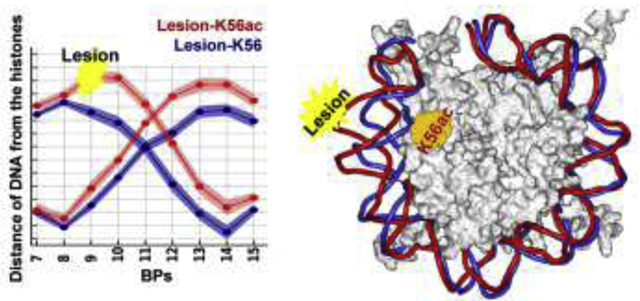
Figure 1: Overall structure of the NCP and the acetylation site of H3K56 as well as the site and structure of the DNA lesion in the duplex.
(A) The overall structure of the NCP model based on the crystal structure (PDB ID 1KX52) with truncated tails as in our prior work.62 Super helical locations (SHLs) are labeled to describe the positions of DNA wrapped around the NCP with respect to the dyad (SHL 0). The H3 αN helix is located near the DNA between SHLs 6 and 7. A DNA lesion, designated as red star, is introduced at the end of the DNA (SHL = 6.25) and placed outward toward the solvent.
(B) Zoom-in view of the region at the end of the nucleosome. The lysine 56 (K56, blue sphere) is positioned at the H3 αN-helix and faces the DNA major groove at ~ SHL 6. The inset box shows the structure of the lesion, the 10R (+)-cis-anti-B[a]P-N2-dG(cis-B[a]P-dG) adduct, (colored by atom with carbons in orange) introduced at BP9 from the end of the nucleosomal DNA. The lesion-containing DNA strand faces the solvent, and its partner dC (colored red) faces the histones. The conformation of the base-displaced/intercalated cis-B[a]P-dG adduct is based on an MD74 equilibrated NMR solution structure73 where the Watson-Crick pair at the lesion site is ruptured; the modified base G is displaced into the minor groove with linkage to the B[a]P ring system, which is intercalated into the helix with the hydroxyl-containing benzylic ring in the minor groove; the partner dC is displaced into the major groove.
The focus of our work is to elucidate how single acetylation of H3K56, at the end of the nucleosome core particle, modulates DNA accessibility both with and without a lesion, using long unconstrained and all-atom microsecond (~5–6 μs) MD simulations (Figure 1). We built a NCP model based on the crystal structure with PDB ID 1KX52 and utilized histone tail clipping as in our prior work.62 The truncation of these tails corresponds to those observed in trypsin digestion studies63 as fully detailed in reference.62 A discussion of histone tail clipping is given in Supplementary Data. While full-length tails interact with other nucleosomes or other proteins in the multi-nucleosomal context,64–67 histone tail clipping in vivo by endopeptidases has been observed in all histone proteins in several organisms and affects a wide range of biological outcomes, including development, aging, tumorigenesis, and others.68
Furthermore, we wished to establish whether a bulky lesion near position 56 of histone H3 would affect the local histone structure and dynamics differently, depending on the acetylated status of H3K56. The lesion investigated is a well-repaired adduct,69, 70 which is derived from the metabolic activation of the environmental carcinogen benzo[a]pyrene:71, 72 10R (+)-cis-anti-B[a]P-N2-dG (referred to as cis-B[a]P-dG) that adopts the base-displaced/intercalated conformation73. Our findings indicate that acetylation of K56 alone induces detectible disruptions in the histone-DNA/histone-histone interactions and further magnifies the distortions imposed by the cis-B[a]P-dG adduct, highlighting the important role of H3K56ac in response to DNA damage particularly at the ends of the DNA. Thus, our work sheds light on our understanding of how accessibility to lesions by the DNA repair machinery could be facilitated within chromatin via a key acetylation event.47, 49
Material and methods
Initial model
We wished to investigate the impact of a single acetylation of the H3 histone K56 on the local region in the NCP. We built the nucleosome core particle (NCP) model based on the crystal structure PDB ID 1KX52 with truncated histone tails according to trypsin digestion data.63 Amino acid sequences that remain in each truncated N terminal tail are given in our prior work.62 The single acetylation of K56 was modelled into only one histone H3, the one corresponding to Chain E in the PDB ID: 1KX5 NCP (H3K56ac). A NCP with unacetylated K56 was also investigated as a control (H3K56) (Figure 1). We carried out ~ 6 μs of MD simulation for each of these lesion-free NCPs.
In order to investigate whether the structural and dynamic changes induced by a lesion would be modulated when H3 K56 is acetylated, we investigated a well-repaired cis-B[a]P-dG adduct69, 70 and incorporated it at the position BP 9 from the DNA ends (SHL = 6.25), where the displaced damaged guanine oriented outward toward the solvent and its displaced partner dC inward toward the histones (Figure 1B). As in our prior study,26 we modeled the cis-B[a]P-dG adduct with its displaced partner dC based on an MD74 equilibrated NMR solution structure.73, 75 With unacetylated and acetylated K56, we generated two lesion-containing NCPs (Lesion-H3K56 and Lesion-H3K56ac) and carried out ~5μs of MD simulation for each lesion-containing NCP. Additional simulations were performed to further explore the conformations induced by this base-displaced/intercalated cis-B[a]P-dG adduct positioned at the end of the DNA in the nucleosome with ten ~500–600 ns MD simulations for each lesion-containing NCP.
Molecular dynamics simulation
Molecular topology and coordinate files for each NCP model were constructed using the tleap module of AmberTools14.76 All solvated systems were subject to energy minimization, equilibration, and ~ 5–6 μs production runs using the CUDA-enabled graphics processing units (GPUs) version of PMEMD77, 78 within the AMBER1679 package on NVIDIA Tesla board P40.80 Full details concerning initial solvated models, force fields81–84 and cis-B[a]P-dG lesion parametrization,73 and molecular dynamics protocols62, 85–87 are given in the Supplementary Methods. A discussion concerning the accuracy and time scales of current MD simulations is given in the Supplementary Discussion.
Results
We wished to elucidate how a single acetylation of H3K56 modulates the DNA accessibility at the entry-exit region of the nucleosome. We have carried out ~ 6 μs MD simulations of an intact NCP with truncated tails in the presence of the acetylated K56 (H3K56ac NCP) and the unacetylated K56 (H3K56 NCP) as a control to investigate the impact of H3K56 acetylation on the dynamics and structures of the nearby DNA and histones. Furthermore, we wished to elucidate how a well-repaired69, 70 cis-B[a]P-dG lesion may induce local structural and dynamic changes and whether such changes would be modulated by acetylation at H3K56. We placed the cis-B[a]P-dG adduct at BP 9 from the DNA ends (SHL = 6.25) with the adduct positioned outward toward the solvent (lesion-containing strand, Figure 1A) so that the sequence context is the same as it is in the lesion-free NCP. As displayed in Figure 1B, the base-displaced/intercalated cis-B[a]P-dG is oriented with the displaced base G in the minor groove directed outwardly toward the solvent (referred to as OUT position); the bulky B[a]P ring system is intercalated into the helix with the hydroxyl-containing benzylic ring in the minor groove; the partner dC opposite the adduct faces inward toward the histones (referred to as IN position). We carried out ~ 5 μs of MD simulation of the lesion-containing NCP with acetylated K56 (Lesion-H3K56ac NCP) and an unacetylated K56 (Lesion-H3K56 NCP).
Acetylation of H3K56 affects the structural stability of the H3 αN helix.
To examine the overall stability of the NCPs during the 5–6 μs MD trajectories, we calculated the RMSDs of the overall NCPs, using P atoms of DNA and the Ca atoms of octameric histones (Figure S1A); these results revealed that the whole NCP is stable after ~ 1 μs. Since we are particularly interested in the stability of the H3 αN helix where the K56 is located, we also computed the RMSDs of heavy-atoms of the H3 αN helix (Figure S1B). Our results revealed that when K56 is unacetylated, regardless of the presence of the DNA lesion, the RMSDs of H3 αN helix reached an equilibrated state after ~ 2.3 μs and ~3.6 μs for lesion-free (H3K56) and lesion-containing (Lesion-H3K56) NCPs, respectively. It was expected that it would take a longer time for the helix to exhibit stability in the presence of the lesion, which also induces dynamic changes to the local structure; an equilibrated state is still attained after 3.6 μs in the case of Lesion-H3K56. Thus, we utilized the MD trajectories after 2.3 and 3.6 μs for H3K56 and Lesion-H3K56 NCPs, respectively, for the further ensemble analyses to capture the properties of the corresponding equilibrated states. As a result, in the equilibrated states, the highly dynamic side-chains of the arginine and lysine residues (R49, R52, R53, and K56) that interact stably with nearby DNA are significantly muted (Table S1A, Figure S2, Figure S3B). These findings provide evidence that MD simulations at microsecond scale are needed to capture the dynamics and stable interactions in the presence of the K56.
In contrast, when K56 is acetylated, the RMSDs of the H3 αN helix continue to fluctuate throughout the MD simulations, indicating that a single acetylation mark (K56ac) destabilizes the interactions involving R49, R52, R53, and K56ac (Table S1B), causing these positively-charged side-chains to be more dynamic than in the unacetylated K56 (Figure S3B). Therefore, for the acetylated K56 cases (H3K56ac and Lesion-H3K56ac), we collected the MD trajectories after 1 μs to explore possible conformational distortions and dynamics induced by the presence of K56ac and DNA adduct. To highlight the structural and dynamic impact of H3K56 acetylation on the local nucleosome, we compared the results of the unstable conformations after 1 μs in acetylated nucleosomes with the results for the stable conformations in the non-acetylated cases. These results indeed show that H3K56 acetylation induces on-going dynamics to the simulation end (5–6 μs) both with and without the lesion.
In the lesion-free NCPs, a single acetylation of K56 in the H3 histone significantly weakens the histone-DNA/histone-histone interactions.
Acetylation of H3K56 weakens a stable network of interactions between the DNA, the H3 αN helix, and the α3 helix of H2A, stemming mainly from arginine residues.
K56 is positioned on the first α helix of the H3 histone (αN helix: residues 44–57) near the very end of the nucleosome between SHLs ± 6 and ± 7 (Figure 1); here the contacts between the DNA and the histones are less abundant than at any other SHL.50 It has been reported that the DNA ends are stabilized by the contacts between the DNA and the H3 αN helix,50, 54 which contain four positive charges at R49, R52, R53 and K56; thus, these contacts are dominated by the electrostatic interactions stemming from these residues. For the unacetylated K56 NCP, our simulations of the H3K56 NCP revealed that the positively-charged residues R49, R52, R53, and K56 as well as T45 have their side chains extend stably toward the backbone of the DNA (Figure 2A, Table S1A); particularly, the arginine residues (R49, R52, and R53) and the ε-amino group of K56 interact stably with the inwardly oriented backbone strand of the DNA. Furthermore, H2A R81 is conserved in almost all H2A variants.88 Notably, our simulation of H3K56 reveals that R81 of H2A is stably hydrogen bonded with Q55 of the H3 αN helix and with the carbonyl groups of G105/V107 of the H2A C terminal domain (Figure 2B, Table S2).
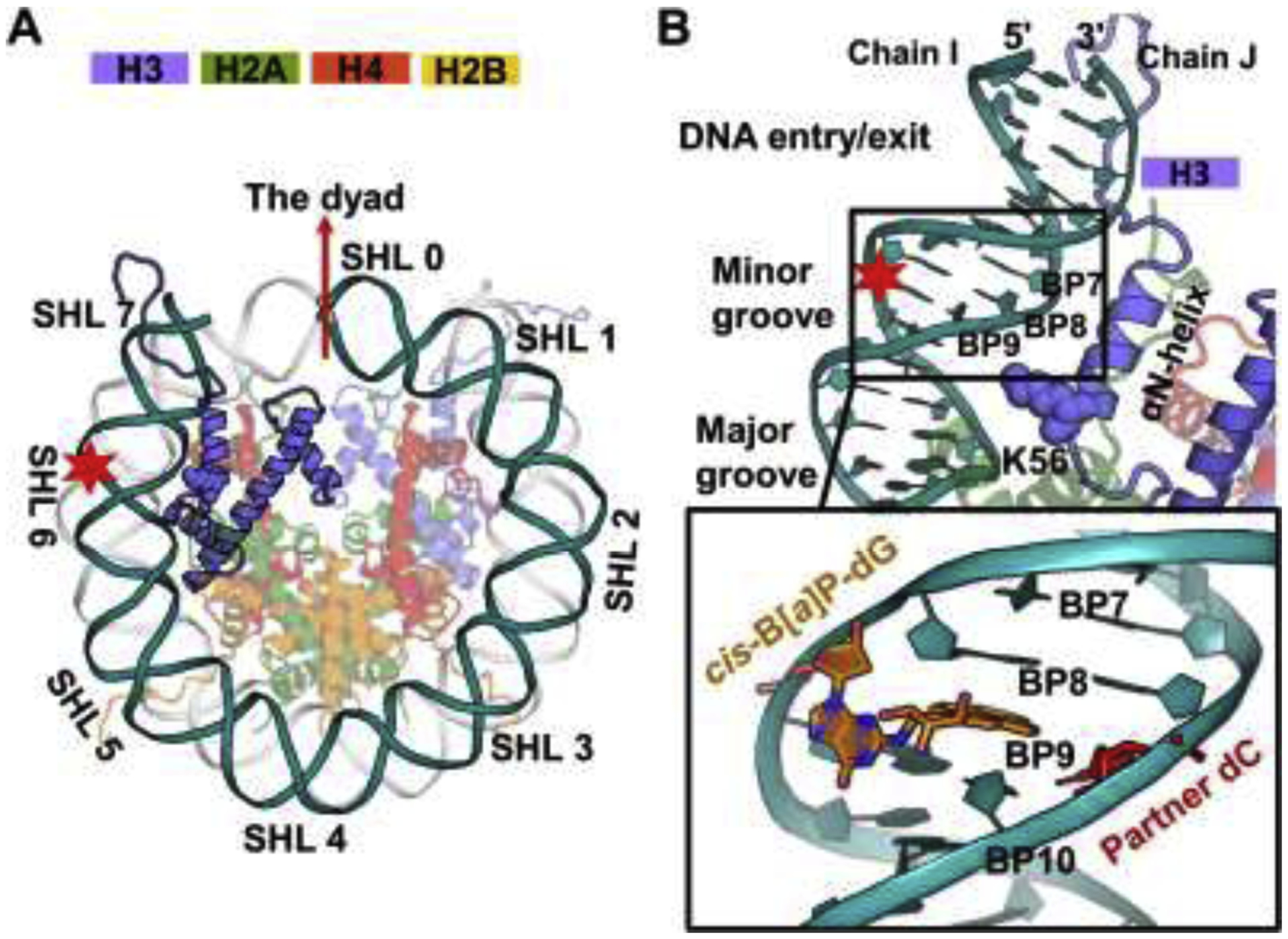
Figure 2: Acetylation of K56 of the H3 histone greatly weakens a network of interactions between DNA and the H3 αN helix as well as the R81 of H2A with the surrounding residues.
(A) Acetylation of K56 weakens the interactions between the H3 αN helix and the DNA. Best representative structure showing the interactions between the H3 αN helix and the DNA in the left panel; the corresponding hydrogen bond numbers are depicted in the right panel. Details concerning all the hydrogen bond acceptor-donor pairs are listed in Table S1.
(B) Acetylation of K56 dimishes the interactions of H2A R81 and the surrounding residues, Q55 of H3 and G105/V107 from the H2A C-loop. Best representative structure showing the interactions between H2A R81 and the surrouding residues in the left panel; the corresponding hydrogen bond numbers are shown in the right panel. Details concerning all the hydrogen bond acceptor-donor pairs are listed in Table S2.
However, when K56 of H3 is acetylated, our MD simulation revealed that the stable network of interactions attributed to these positively charged residues is significantly disrupted (Figure 2, Table S1B, Table S2); noticeably, the acetyl group of K56ac completely loses its hydrogen bonds with the DNA and all three arginine residues of the H3 αN helix exhibit much weaker hydrogen bonds with the DNA compared to the unacetylated case (Total HB numbers ~ 7.9 in H3K56 and ~ 4.8 in H3K56ac, Figure 2A and Table S1); hydrogen bonds between R81 and its surrounding residues were also broken (HB numbers ~ 3.7 in H3K56 and ~ 0.7 in H3K56ac, Figure 2B and Table S2). Details regarding the hydrogen bond analysis are given in Supplementary Structural Analyses and Additional Results. These results indicate that K56ac directly abolishes its contact with the DNA and diminishes indirectly the interactions stemming from these arginine residues. Thus, our MD results show that these interactions, derived from these positively charged residues of the αN helix in H3 as well as R81 in H2A, are responsible for anchoring the terminal DNA.
Acetylation of H3K56 enhances the dynamics of nearby DNA and the H3αN, H2A α3- and αC-helices.
Our MD results show that the presence of K56ac disturbs a stable network of interactions between the DNA, the αN helix of H3, and the α3 helix of H2A (Figure 2, Tables S1–3); as a result, the DNA, the residues R49, R52, R53, and K56ac of H3 as well as R81 of H2A in the vicinity of the acetylation mark become more mobile than in the unacetylated case (Figure S2, Figure S3). Furthermore, we also noticed that K56ac increases the fluctuations in the αC helix of H2A, including the residues D90, E91 and E92 (Figure S2, Figure S3C), which make up part of the acidic patch. This acidic patch is comprised of eight negatively-charged glutamate/aspartate residues (E56, E61, E64, D90, E91, E92 of H2A and E102 and E110 of H2B as shown in Figure S2), forming a highly electronegative cleft region on the nucleosome surface, and acts as a binding platform89 for the H4 tail90 and many other proteins.91, 92
Acetylation of H3K56 causes the nearby DNA to be more distant from the histones.
Despite the enhanced flexibility of the DNA ends observed in our MD simulation of the acetylated K56 NCP (Figure S3A), we did not observe the very ends (first 6 BPs) of the DNA detaching from histones even in the presence of the K56ac because the real time frame for DNA unwrapping, is a slow motion in the millisecond to seconds range,52, 53, 93 that is much longer than our simulation time. However, our MD simulations did reveal that a small bulge occurred in the DNA in the vicinity of the K56ac (Figure 3A). This bulged DNA region occurred between BP 9 and 12 and particularly involves the inward oriented strand, whose distance from the H3 αN helix is increased by up to ~ 3.5 Å more than in the unacetylated K56 NCP (Figure 3B). Notably, this strand is bulged away from the acetyl group of K56ac; it moves away from the center of the NCP and the other gyre, producing more exposed DNA sites. Thus, this minimizes the unfavorable electrostatics incurred by the presence of the acetyl group of K56ac. Details regarding the distances of the DNA to the H3 histone are given in the Supplementary Structural Analyses and Additional Results.
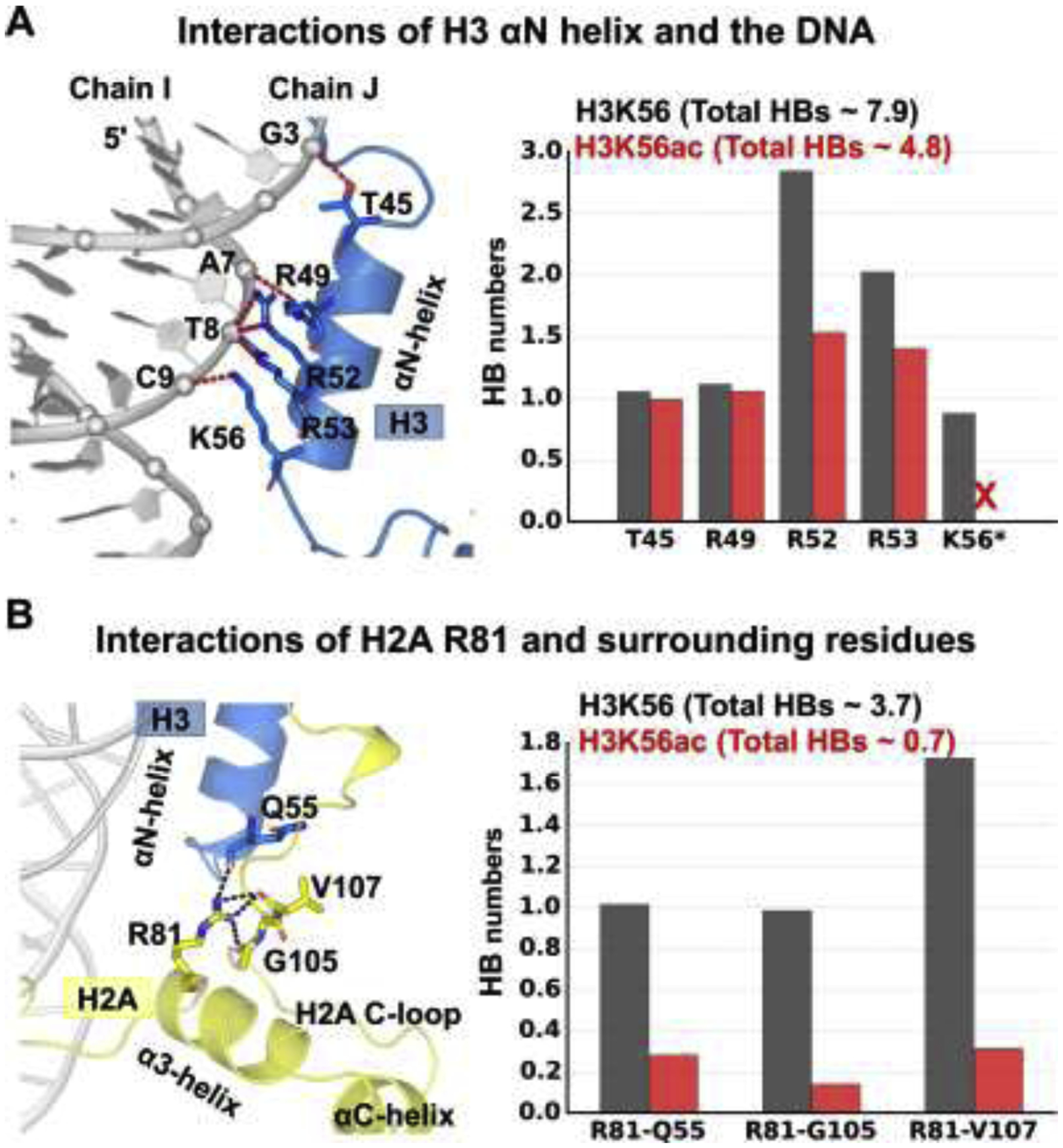
Figure 3: Acetylation of K56 causes the DNA to be more distant from the H3 αN helix than in the unacetylated K56 case.
(A) Superposition of the best representative structures of H3K56 (light-blue) and H3K56ac (red) reveals that there is a small bulge in the DNA between BP9 and BP12 in the vicinity of K56ac of the H3 histone. (B) The increased distances are mostly observed at the region BPs 9 –12 on the strand Chain I. Only half of the DNA (SHL > 0) and histones (H3, H4) near the end of the DNA are displayed for clarity. Two views illustrating how the bulged DNA is distanced away from the acetyl group of K56ac; the DNA is moved away from the NCP (View 1, view into the NCP disk axis); the DNA of gyre 1 is moved away from the other gyre (View 2, view into the two gyres with the H3 αN helix between).
(B) Ensemble averaged shortest distance between the DNA P atom and the helical axis of H3 αN. The standard deviations are displayed as shadow. Details are described in the Supplementary Structural Analyses and Additional Results. The acetylated K56 enlarges the distance of the DNA to the histones, particularly for the region between BP 9 and BP 12 on the Chain I; this region also exhibits higher dynamics than when K56 is unacetylated.
Acetylation of H3K56 induces highly dynamic side-chains and an unstable α-helical structure in the H3 αN helix.
With the presence of K56ac, the H3 αN helix becomes more flexible (Figure S4A). This is revealed in the greater fluctuations of the side-chains of residues R49, R52, R53, and K56ac (Figure S4B), the occurrence of a kink in the H3 αN helix (Figure S4C), and an unstable α-conformation of this αN helix (Figure S4D). Particularly, residues T45 and V46 near the N-terminus of H3 exhibit the most structural variations during the time interval between 3 and 5 μs when the kink occurs, with a population of less than 30% as pure α-helix and more than 60 % of the population in a turn conformation; however, in the unacetylated K56 NCP, the αN helix adopts a stable pure α-helix conformation.
In the lesion-containing NCPs, acetylated K56 of H3 magnifies the structural and dynamic distortions imposed by the lesion.
We are particularly interested in whether H3K56 acetylation may enhance access to a DNA lesion at the entry-exit region of the nucleosome and so facilitate recruitment of repair proteins. Hence, we placed a cis-B[a]P-dG lesion at BP9 within the DNA region that is influenced by the K56ac mark (Figure 3). The adduct is oriented with the displaced G directed outward toward solvent (OUT position) and its displaced partner dC faces inward toward the histones (IN position) (Figure 1B).
The lesion modestly reduces the DNA-H3 interactions, but these are further significantly weakened by the presence of K56ac.
As noted for the lesion-free NCPs, lysine acetylation at position 56 of H3 histone completely abolishes the contact of this K56ac with the DNA and diminishes greatly a network of interactions attributed to the arginine residues, R49, R52, and R53 of H3 and R81 in H2A (Figure 2, Tables S1–S2). In the lesion-containing NCPs (Figure 4A, Tables S2–3) these interactions are modestly reduced by the lesion and are further significantly weakened when K56 is acetylated. As a result, the region including the DNA vicinity, the αN helix of H3, and the α3- and αC-helices of H2A are more dynamic compared to the unacetylated K56 NCP (Figure 4B, Figure S3). Notably, the lesion-containing DNA in the vicinity of the K56ac mark is prominently distanced from the histones (Figure 4C), with the DNA noticeably kinked away from the other gyre. Thus, K56ac exposes many more sites near the damaged DNA compared to the lesion-free DNA (Figure 5); this is reflected in the greater distance of the damaged DNA from the histones and the greater number of exposed DNA base pairs (Figure 5A) as well as the enlargement of the minor groove (Figure 5B). Overall, our findings suggest that lesion-containing DNA is more responsive to the acetylated K56 than the lesion-free DNA, imposing more structural and dynamic distortions to the nucleosome, further discussed below.
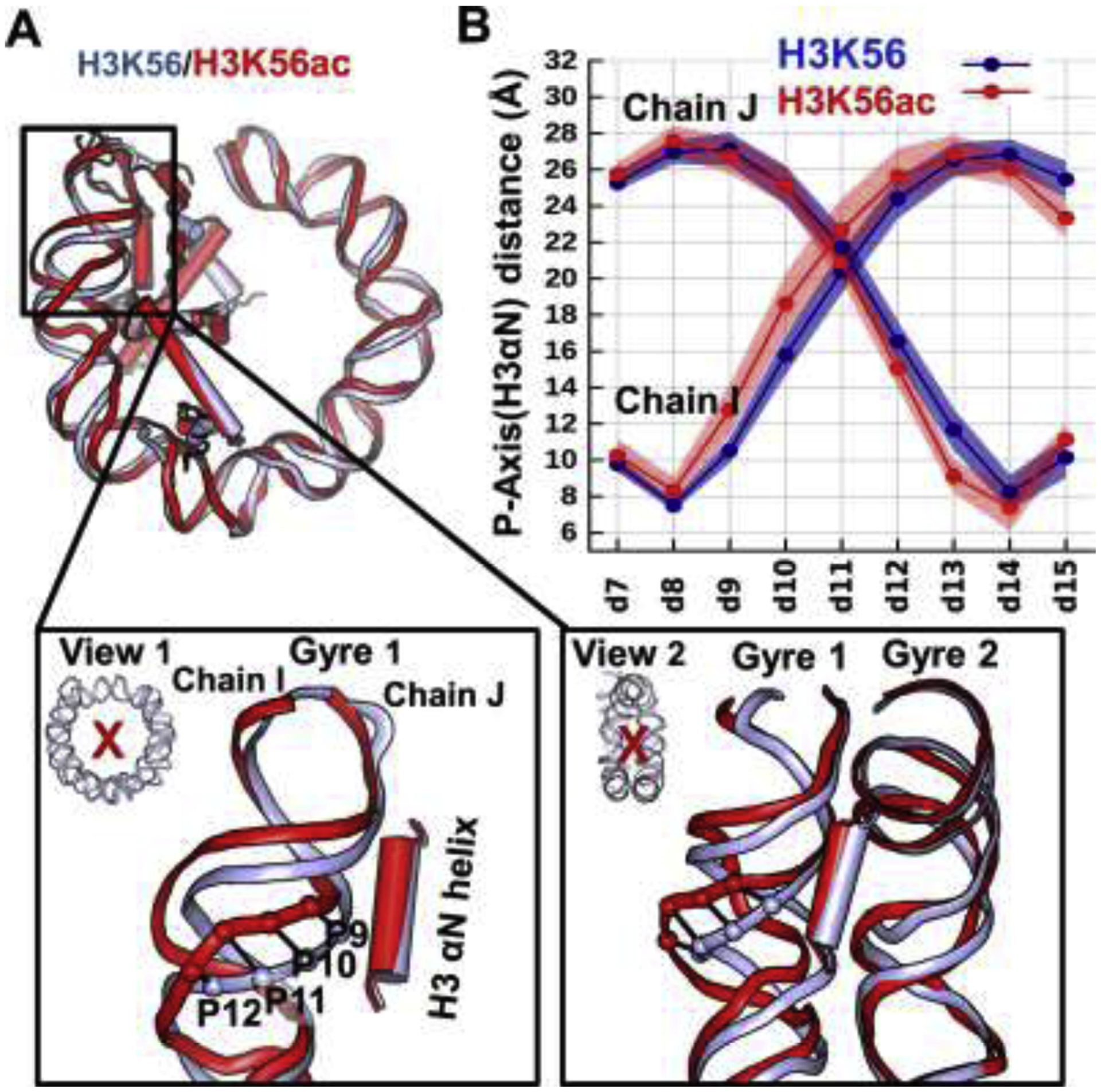
Figure 4: Impact of acetylation of H3K56 on the lesion-containing NCPs.
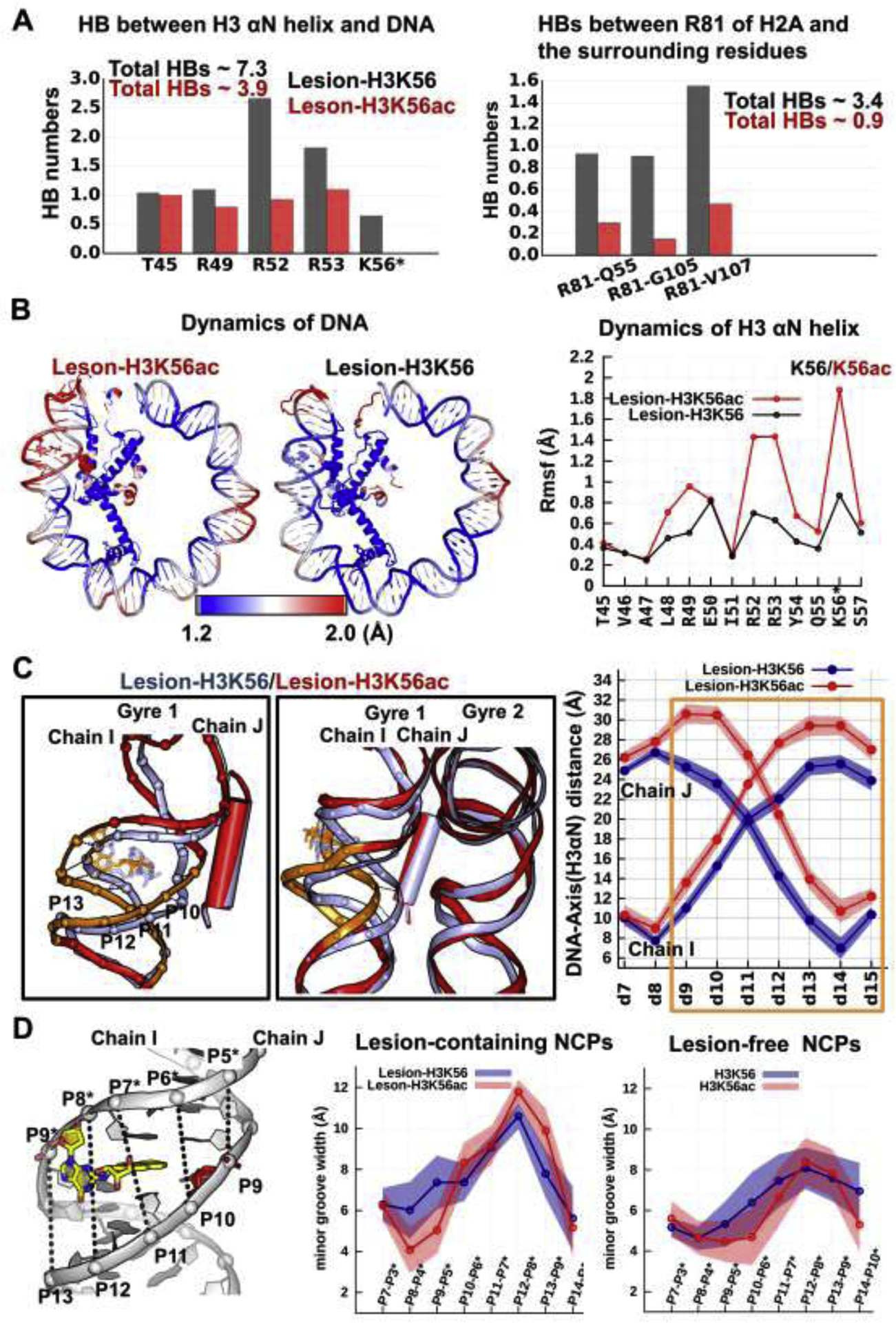
Compared to unacetylatedK56, K56ac (A) significantly weakens the DNA-histone/histone-histone interactions, (B) causes both nearby DNA and the H3 αN helix to be more dynamic, (C) causes the nearby DNA to become distanced from the histones and away from the other gyre, (D) causes the minor groove to be more enlarged.
(A) The hydrogen bonds between the H3 αN helix and the DNA (left panel) as well as the hydrogen bonds of R81 with the surrounding residues (right panel) are given for lesion-containing NCPs. Details concerning all hydrogen bond acceptor-donor pairs are listed in Tables S2–3.
(B) K56ac enhances the dynamics of the DNA near the nucleosome entry-exit region, the αN helix of H3, and the α3- and αC- helices of H2A. The RMSFs of the heavy atoms of the residues are shown by color; higher values correspond to larger fluctuations (red) and lower values to smaller fluctuations (blue) as indicated in the spectrum bar. Their corresponding RMSF values are also given in Figure S3. Only half of the DNA (SHL > 0) and histones (H3, H4) near the end of the DNA are displayed for clarity.
(C) Superposition of the best representative structures of Lesion-H3K56 (light-blue) and Lesion-H3K56ac (red) NCPs reveals that the DNA at the region highlighted as orange is markedly distanced from the H3 αN helix by the presence of K56ac, mainly by moving away from the other gyre. Two views illustrating that the lesion-containing DNA is pulled away from the center of the NCP and away from the other gyre. The ensemble averaged shortest distances between the DNA and the helical axis of H3 αN are plotted, showing that the enhanced distanced DNA segment is mainly observed at BPs 9–15, with an increased distance of up to ~7 Å.
(D) Minor groove widths were calculated by measuring the pairwise P-P distance for lesion-containing and lesion-free NCPs. When compared to the lesion-free NCPs, the lesion with its damaged dG displaced into the minor groove enlarges the minor groove width, and even more so in the presence of K56ac, particularly at P12-P8* and P13-P9* across the damaged dG. The numbering scheme: “P10” refers to the P atom at BP10 on the strand Chain I and “P10*” refers to the P atom at BP10 on the strand Chain J. A distance 5.8 Å was subtracted to account for van der Waals radius of the P atom.110
Figure 5: Impact of H3K56ac on the damaged DNA and undamaged DNA.
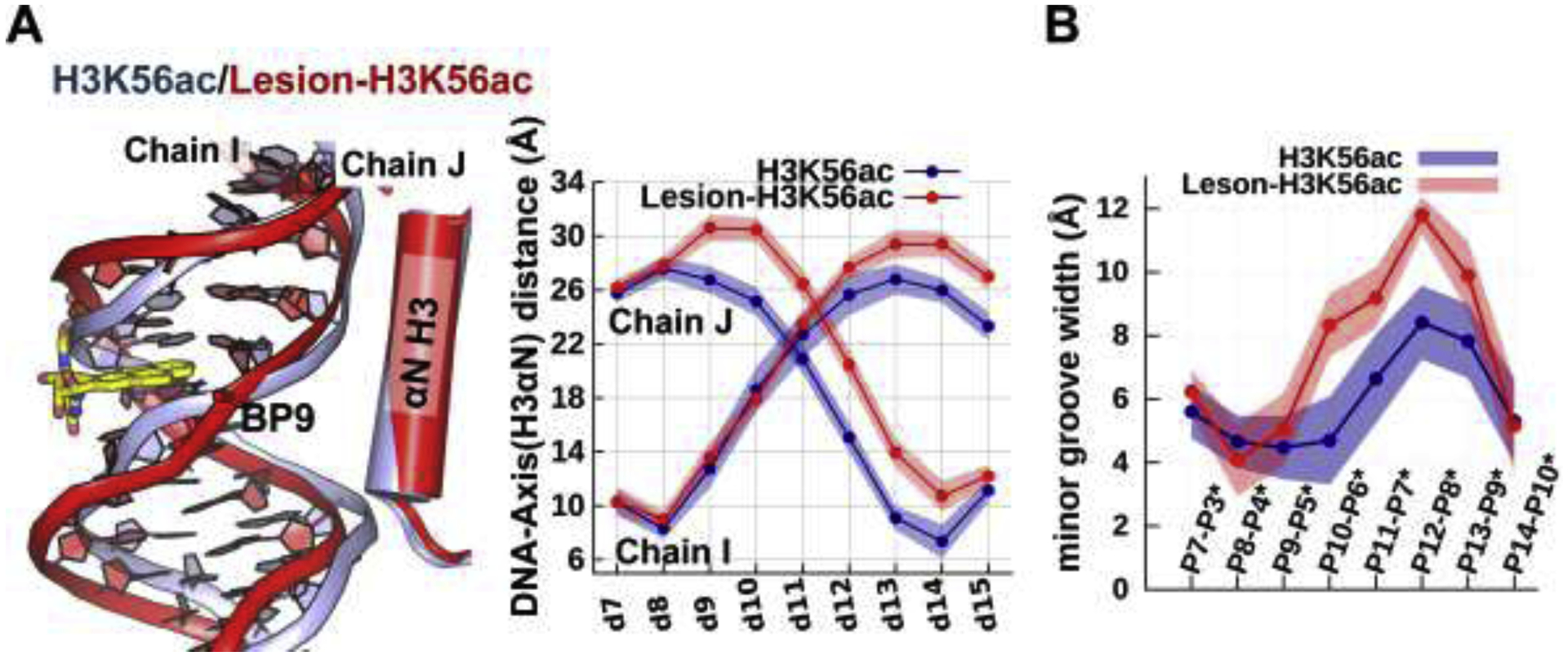
(A) Superposition of the best representative structures of H3K56ac (light-blue) and Lesion-H3K56ac (red) NCPs reveals that K56ac exposes many more sites near the damaged DNA compared to the lesion-free DNA, as shown in the greater distance of the damaged DNA from the histones than in the undamaged DNA. The ensemble averaged shortest distances between the DNA and the helical axis of H3 αN are plotted. (B) In the presence of H3K56ac, the damaged DNA has wider minor grooves than the undamaged DNA. The ensemble averaged minor grooves widths are plotted.
Structural and dynamic distortions imposed by the lesion are significantly amplified by the acetylation of H3K56.
In our MD simulation of Lesion-H3K56 NCP, the solution conformational features of the cis-B[a]P-dG adduct at the end of the nucleosome are maintained (Figure 6A, Figures S5–7); the bulky B[a]P ring system is intercalated stably between the adjacent base pairs (Figure S5, Figure S7A); both displaced base G and the hydroxyl-containing benzylic ring are in the minor groove and are exposed to the solvent. All these features that lead to structural and dynamic distortions, including the enlarged minor groove (Figure 4D), the mobility of the displaced partner dC (Figure S6, Figure S7B), the distorted duplex (Figure S7C), and a ruptured Watson-Crick pair at the lesion site (BP9, Figure S7D), characterize this lesion.26, 73, 74 We observed that the partner dC is flipped into the minor groove and fluctuates between two orientations via a dynamic network of hydrogen bonds of the partner base C with the neighboring bases, sugars and hydroxyls of the B[a]P rings (Figure 6A, Figure S6), indicative of the highly flexible partner dC and consist with its increased RMSF value (position 9 on Chain I in Figure S3A). Furthermore, this minor-groove positioned dC causes the local 3-mer duplex to be slightly over-twisted (Figure S6, Figure S7C). Overall, the structural changes imposed by the cis-B[a]P-dG adduct are well-maintained throughout the simulation of the unacetylated K56 NCP (Figure S7).
Figure 6: The dynamics of the B[a]P rings intercalated into the helix shows that the K56ac causes the B[a]P ring system to be more dynamic than in the unacetylated K56.
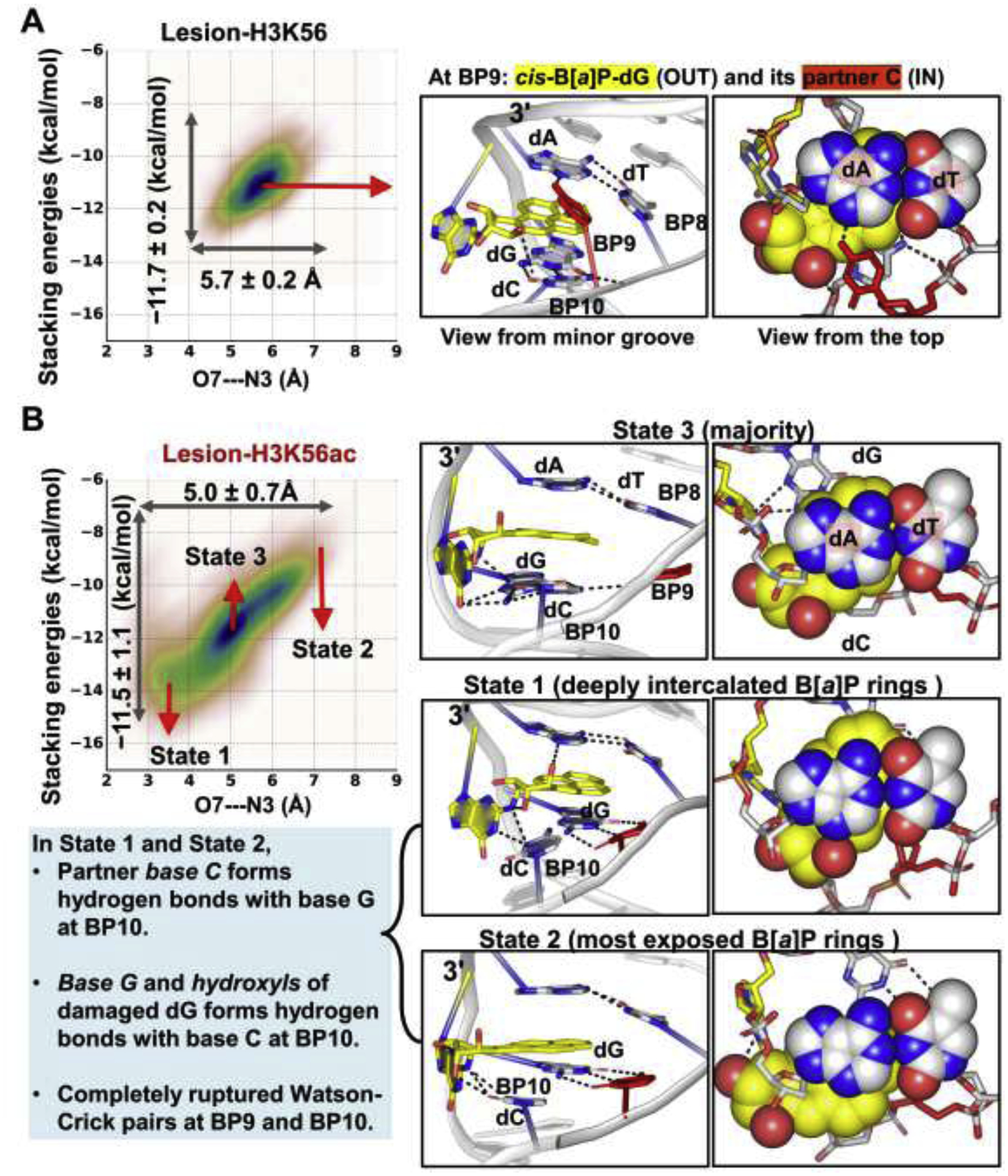
The O7—N3 distance and the stacking interactions of the B[a]P rings with the bases at BP8 are plotted against each other for the lesion-containing NCPs. Mean values and standard deviations are given; the larger range and standard deviations reflect the larger span that the B[a]P rings sample and the greater dynamics of the B[a]P rings. The larger ranges and standard deviations are revealed in the Lesion-H3K56ac NCP; when H3 K56 is acetylated, the intercalated B[a]P rings sample a larger span with enhanced dynamics compared to the unacetylated K56.
(A) In Lesion-H3K56, the B[a]P rings stack stably with the adjacent bases at BP8 (Figure S5).
(B) In Lesion-H3K56ac, the intercalated B[a]P ring system stacks dynamically with the adjacent bases between two extreme states (State 1 and State 2). In State 1, the hydroxyl group in the B[a]P rings forms a hydrogen bond with the N3 atom of the A base at BP8 (O7—N3 distance), resulting in a deeply- intercalated and least exposed B[a]P ring system. In State 2, the hydroxyls of the B[a]P rings and A base are most distanced, resulting in the least intercalated and most exposed B[a]P rings. Notably, in these two extreme states, the partner base C forms three hydrogen bonds with the base G (5′-side to the lesion) at BP10, and the damaged base G and hydroxyls of the lesion interact with base C at BP10, leading to a completely ruptured Watson-Crick base pair at BP10 and a partially ruptured WC pair at BP11 (Figure S7D); this is likely due to the neighboring sequence context. The majority conformation of the B[a]P rings is in State 3 where the Watson-Crick pair at BP10 and BP11 present as in the unacetylated K56 case. Structures indicated in the correlation plot are shown in the right panel. The 3-mer duplex GG*A with lesion (G* = cis-B[a]P-dG) at the center are rendered as sticks.
When H3 K56 is acetylated, we observed that the structural changes imposed by the lesion are more distorting and dynamic compared to the unacetylated K56 case (Figures 4 and 6, Figures S5–7). Notably, we observed aberrant and dynamic changes particularly during 0.5 to 3 μs of the MD simulation of the Lesion-H3K56ac NCP. During this period of time, the intercalated B[a]P ring system stacks dynamically with the adjacent base pair with its rings spanning two main subspaces of structures (State 1 and State 2 in Figure 6B and Figure S5); in State 1, the O7 hydroxyl group in the B[a]P rings form a hydrogen bond with the N3 atom of the A base at BP8, resulting in a deeply-intercalated and least exposed B[a]P ring system; in State 2, the hydroxyls of the B[a]P rings and A base are most distanced, resulting in a least intercalated and most exposed B[a]P ring system. Details concerning the intercalated B[a]P rings are given in the Supplementary Structural Analyses and Additional Results. Furthermore, the partner dC at BP9 that is flipped into the major groove forms three hydrogen bonds with the base G at BP10 (Figure 6B, Movie S1), a sequence context effect. As a result, the Watson-Crick pair at BP10 is completely ruptured and the Watson-Crick pair at BP11 also becomes partially ruptured (Figure S7D), causing the local 3-mer duplex to be greatly under-twisted by ~30.0° together with the enhanced dynamics (Figure S7C). After 3μs, this aberrant conformation is transitioned to a less-distorted state where the Watson-Cricks pairs at BP10 and 11 are restored; nevertheless, these lesion-induced structural distortions still exhibit modestly enhanced dynamics compared to the unacetylated K56 case. Details concerning the flipping dynamics of the partner dC and the under-twisted duplex are given in the Supplementary Structural Analyses and Additional Results. Overall, our observations indicate that K56ac greatly amplifies the local dynamic distortions of the duplex imposed by the lesion.
IN-facing partner dC is conformationally flexible and becomes more so in the presence of the acetylated K56 of H3.
To further scrutinize whether the conformational orientation and flexibility of the partner dC that faces the histones are dependent on the acetylated status of K56, we also carried out additional 10 independent ~ 600 ns MD simulations for each Lesion-containing NCPs. Figure S8 shows that, at SHL 6.25, IN-facing partner dC spans a wide range of orientations in the duplex, from residing in the major groove to the minor groove, regardless of the acetylated status of the K56 of H3. The greater dynamics in the presence of K56ac of the partner dC is also revealed in the higher standard deviation values.
Discussion
Lysine acetylation at position 56 of histone H3 disrupts histone-DNA/histone-histone interactions involving mainly arginine residues.
During many important cellular processes, nucleosomes must permit DNA-binding proteins to gain access to the DNA. Spontaneous DNA unwrapping motions that expose the binding sites would allow free but transient/rapid access to protein.93 This unwrapping dynamics of DNA ends is facilitated by the acetylation of K56 on histone H3,57–61 which weakens the electrostatic interactions between DNA termini and the histones.24, 51 The spontaneous DNA unwrapping occurs once in the range of milliseconds to seconds,52, 53, 93 which is still far beyond the present microsecond (~6μs) time scale of out all-atom MD simulations. While coarse-grained MD studies94, 95 have obtained insights on the molecular mechanism of DNA unwrapping and rewrapping as they can investigate the appropriate time spans, they cannot provide functionally relevant motions and interactions at atomic resolution such as the hydrogen bonding interactions that is our interest, but do provide insights that the longer time-frames permit. Watanabe et al.96 have obtained a crystal structure of a nucleosome containing H3K56Q as a surrogate for H3K56ac. They found that the H3K56Q mutation that mimics the acetylation does not affect the DNA conformation, while it had been experimentally observed60 that H3K56 acetylation increased DNA breathing. However, this was not captured in the crystal structure, which was attributed to crystal packing that stabilized the DNA in a ‘closed’ conformation. In our MD simulations we did observe distinct structural and dynamic perturbations upon acetylation, which highlights the value of the solution MD simulations.
Our MD study provides a detailed analysis of the histone-DNA interactions near the DNA ends with atomic level detail and microsecond (5–6μs) time scale to elucidate the structural and dynamic impacts of acetylation of K56 on the lesion-containing DNA. Our 5–6 μs simulations did not reveal directly the detachment of the ends (first 6 BPs) of the DNA from the histones, even in the presence of the H3K56ac. However, we did observe that the acetylated H3K56 causes the H3 αN helix to be unstable, concomitant with enlargement of the distance between the nearby DNA and the histones. As a result, a bulge occurs in the DNA at BPs 9–12, in the vicinity of the K56ac. A cryo-EM structure of the NCP with unwrapped DNA reported by Bilokapic et al.54 also showed that the DNA bulges at one entry-exit site but the DNA end remains attached to the octamer. Furthermore, similarly bulged DNA has been shown bound to the Snf2 chromatin remodeler.97 We speculate that the unstable αN helix and the bulge may initiate the slow microsecond to millisecond scale changes in histone-DNA interactions that are responsible for DNA unwrapping dynamics in this time range.52, 53, 93
We observed a stable network of interactions of the H3 αN helix with the DNA as well as with the H2A (Figure 2, Tables S1–3); R49, R52, R53, and K56 of the H3 αN helix maintain hydrogen bonds with the phosphate backbone of the DNA, and the R81 of the H2A α3 domain interacts stably with Q55 of the H3 αN helix and with the G105/V107 of the H2A C terminal domain. However, when K56 is acetylated, the acetylation directly abolishes its contact with the DNA and also diminishes indirectly the interactions stemming from the arginine residues, which are the key residues in holding the terminal DNA.21, 95, 98–101 This possibly explains why a single acetylation mark at position 56 of H3 imposes the strong outcome that facilitates the unwrapping opening motions of DNA.57–61
It has been suggested that the arginine residues in the H3 αN helix contribute to the tight wrapping of nucleosomal DNA.95, 98, 100, 101 A coarse-graine MD study95 showed that within the wrapped DNA in the NCP, residues R49, R52, R53 and K56 interact stably with the ends of the DNA. An alanine mutation experiment101 showed that the point mutation R49A causes the greatest decrease in the DNA end-to-end FRET ratio, followed by R52A and R53A, indicating that the DNA wrapping is severely altered by these arginine residues. Mutants R49K and R53K of H3 increase the local flexibility and lead to a preference for an open DNA entry/exit arrangement, indicating that an arginine residue is more likely to form hydrogen bonds via its guanidinium group as compared to lysine with its amine group.100 Furthermore, the R81of H2A that interacts with H3 Q55 contributes to the stabilization of the electrostatic contacts between DNA and the H3 αN helix, which is extremely crucial for holding the DNA entry-exit site; Langowski et al.21, 99 have demonstrated that point mutation R81A or R81E of H2A abolished most contacts between H2A R81 and the surrounding residues (H3 Q55/K56, H2A G105/V107), which is also seen in our K56ac case (Figure 2B, Table S2). Thus, these studies combined with our results suggest that K56ac can impose similar effects as the site-specific mutations R49K or R81A/R81E by similarly altering the electrostatic environment of the H3 αN helix or the H2A α3 domain, further accounting for the strong impact of acetylation. Altogether, K56ac significantly weakens the histone-DNA/histone-histone interactions stemming from the key residues that aid in unwrapping the DNA from the nucleosome, increasing DNA accessibility to the transcription machinery or repair proteins.
Histone acetylation amplifies the structural and dynamic distortions imposed by the lesion, suggesting increased accessibility to the lesion for repair.
The structural changes that are induced by the cis-B[a]P-dG lesion (Figures 4 and 6, Figures S5–7): minor groove enlargement, ruptured Watson-Crick pairing at the lesion site, and the displaced partner dC and the distorted helical conformation at the lesion site, provide a signal for lesion recognition by damage-sensing proteins.26, 73, 74 Our MD results revealed that the reduced DNA-H3 interactions imposed by the lesion are significantly further weakened by the presence of acetylated K56, causing the lesion-containing duplex to be much less restrained and more exposed; both OUT-facing damaged dG and its IN-facing partner dC become more dynamic than in the unacetylated case, suggesting that the signals sent by the lesion can be amplified by the acetylation of K56 of H3. Thus, it may assist the GG-NER system in recognizing the damage and facilitating repair of the lesion by initiating recruitment of repair proteins.102, 103 Delaney et al.57 reported an increased activity of hOGG1 at approximately 20 base-pairs away from the end of the DNA in response to acetylation of the H2B tail, emphasizing the effect of histone acetylation on the activity of the BER enzyme.
Although, Smerdon et al.104 have reported that H3K56ac or H3K14ac do not significantly contribute to removal of uracils, regardless their translational and rotational settings within the NCPs, by uracil DNA glycosylase; this may well be lesion-dependent. Many factors contribute to the initiation or the efficiency of repair, including the surrounding histones, addition of chromatin remodeling complexes, the specific lesion sites, and the local DNA sequence composition as well as the type of lesion.70, 105 Lesions with different chemical structures accommodated in the nucleosome have different structural and dynamic impacts on the local distortions and dynamic destablizations,70, 106 which may be either minimally or markedly affected by histone lysine acetylation.
Lesion-induced structural distortions and destabilizations in a nucleosome vary depending on the SHLs, which may contribute to the varying accessibility of the lesion and contribute to subsequent differences in GG-NER efficiencies.
The local structural and dynamic distortions of the duplex imposed by the DNA adduct, rather than the nature of the lesion itself, provide the basis for the recognition by damage-sensing proteins in the GG-NER mechanism.70 GG-NER studies with nucleosomes containing HeLa histones from the Geacintov laboratory107 reported higher GG-NER excision efficiency for an IN-facing cis-B[a]P-dG lesion near the dyad and a lower GG-NER excision efficiency for the OUT-facing case; this was mainly attributed to the displaced and dynamic partner dC opposite the lesion in the nucleosome based on MD simulations by Cai et al.106 The MD results revealed that the damaged dG near the dyad is inherently restrained due to its linkage to the intercalated B[a]P rings regardless of their IN-facing (SHL = −0.25) or OUT-facing (SHL = −0.75) position. However, its partner dC exhibits different flipped out positions with varied dynamics for the IN vs. OUT cases; the partner dC that faces histones is constrained via interactions with histones. As a result, IN-facing dC is less dynamic and less extruded into the major groove than OUT-facing dC. These results suggest that interactions with the histones near the dyad play a role in determining this bulky lesion’s accessibility and thus play a role in its repair efficiency in the dyad vicinity. However, when this adduct is placed at the OUT position near the DNA exist/entry region as in the present case (SHL = 6.25, Figure 1), its IN-facing partner dC still exhibits conformational flexibility (position 9 on Chain I in Figure S3A). This is indicated by the partner dC sampling a wide conformational range in the H3K56 NCP (Figures S6, S8). These findings support better accessibility of the lesion and subsequent repair close to the nucleosomal DNA exit/entry sites compared to the restricted sites at the dyad. A similar trend has been observed with DNA base lesions subject to the BER pathway. Delaney et al. have reported the activity of hOGG1 near the nucleosome exit-entry region;57 however, this activity was completely inhibited at the dyad axis, regardless of the rotational orientation of the lesion at the dyad,108 highlighting the impact of the local histone environment and the corresponding histone-DNA interactions.109
Conclusion
In this present study, using all atom molecular dynamics simulations of 5–6 microseconds, we have demonstrated how a single acetylation mark at position 56 of the H3 histone exposes a DNA damaged site and enhances its mobility. Our analyses of the interactions in the vicinity of the K56ac mark at the nucleosome entry-exit region suggest that K56ac modulates key interactions of the histone residues, notably involving arginine residues, that aid in unwrapping the DNA from the histones by altering the electrostatic environment. Notably, our findings suggest that lesion-containing DNA is more responsive to K56ac than the lesion-free DNA; K56ac exposes sites more on the damaged DNA and imposes more structural and dynamic distortions to the damaged DNA than on the undamaged case, which should facilitate access to the lesion for repair.
Supplementary Material
Highlights.
Acetylation of H3K56 weakens DNA-histone interactions, especially with nearby arginine residues.
H3K56ac induces the nearby DNA to bulge away from the H3 histone and it distorts the H3 αN helix.
H3K56ac causes more DNA exposure when it is damaged by a benzo[a]pyrene-derived lesion than when unmodified.
H3K56ac amplifies the structural and dynamic distortions imposed by the DNA lesion.
Acknowledgments
We gratefully acknowledge resources provided by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (NSF) Grant MCB060037 (to S.B.), and the NYU IT High Performance Computing Resources and Services. This work was supported by the National Institutes of Environmental Health Sciences Grants R01-ES025987 (to S.B.), R01-ES024050 (to N.E.G.), and R01-CA168469 (to N.E.G.).
Abbreviations
- PTM
post-translational modification
- GG-NER
global genomic nucleotide excision repair
- hOGG1
human oxoguanine glycosylase 1
- Nap1
nucleosome assembly protein 1
- BER
base excision repair
- H3K56ac
histone H3 lysine 56 acetylation
- DSB
double strand break
- DDB
damaged DNA binding protein
- DDR
DNA damage response
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare no competing financial interest.
Appendix A. Supplementary Data
The Supplementary Data is available free of charge alongside the electronic version of the article.
References
- [1].Li G, and Reinberg D (2011) Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev 21, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Davey CA, Sargent DF, Luger K, Maeder AW, and Richmond TJ (2002) Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- [3].Luger K, Mader AW, Richmond RK, Sargent DF, and Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- [4].Hara R, Mo J, and Sancar A (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell Biol 20, 9173–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rodriguez Y, and Smerdon MJ (2013) The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. Journal of Biological Chemistry 288, 13863–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smerdon MJ (1991) DNA repair and the role of chromatin structure. Curr Opin Cell Biol 3, 422–428. [DOI] [PubMed] [Google Scholar]
- [7].Downs JA, Nussenzweig MC, and Nussenzweig A (2007) Chromatin dynamics and the preservation of genetic information. Nature 447, 951–958. [DOI] [PubMed] [Google Scholar]
- [8].Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, and Tyler JK (2008) Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang H, Maertens AM, Hyland EM, Dai J, Norris A, Boeke JD, and Bader JS (2009) HistoneHits: a database for histone mutations and their phenotypes. Genome Res 19, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Luger K, Dechassa ML, and Tremethick DJ (2012) New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mao P, and Wyrick JJ (2016) Emerging roles for histone modifications in DNA excision repair. Fems Yeast Research 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Adar S, Hu J, Lieb JD, and Sancar A (2016) Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc Natl Acad Sci U S A 113, E2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou K, Gaullier G, and Luger K (2019) Nucleosome structure and dynamics are coming of age. Nature structural & molecular biology 26, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zsidó BZ, and Hetényi C (2020) Molecular structure, binding affinity, and biological activity in the epigenome. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mao P, and Wyrick JJ (2019) Organization of DNA damage, excision repair, and mutagenesis in chromatin: A genomic perspective. DNA repair 81, 102645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- [17].Suganuma T, and Workman JL (2018) Chromatin and Metabolism. Annu Rev Biochem 87, 27–49. [DOI] [PubMed] [Google Scholar]
- [18].Roth SY, Denu JM, and Allis CD (2001) Histone acetyltransferases. Annu Rev Biochem 70, 81–120. [DOI] [PubMed] [Google Scholar]
- [19].Fenley AT, Adams DA, and Onufriev AV (2010) Charge state of the globular histone core controls stability of the nucleosome. Biophysical journal 99, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fenley AT, Anandakrishnan R, Kidane YH, and Onufriev AV (2018) Modulation of nucleosomal DNA accessibility via charge-altering post-translational modifications in histone core. Epigenetics Chromatin 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Biswas M, Voltz K, Smith JC, and Langowski J (2011) Role of histone tails in structural stability of the nucleosome. PLoS Comput Biol 7, e1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tessarz P, and Kouzarides T (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15, 703–708. [DOI] [PubMed] [Google Scholar]
- [23].Tropberger P, and Schneider R (2013) Scratching the (lateral) surface of chromatin regulation by histone modifications. Nature structural & molecular biology 20, 657–661. [DOI] [PubMed] [Google Scholar]
- [24].Buning R, and van Noort J (2010) Single- pair FRET experiments on nucleosome conformational dynamics. Biochimie [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- [25].Fu I, Cai Y, Zhang Y, Geacintov NE, and Broyde S (2016) Entrapment of a histone tail by a DNA lesion in a nucleosome suggests the lesion impacts epigenetic marking: a molecular dynamics study. Biochemistry 55, 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cai Y, Fu I, Geacintov NE, Zhang Y, and Broyde S (2018) Synergistic effects of H3 and H4 nucleosome tails on structure and dynamics of a lesion-containing DNA: Binding of a displaced lesion partner base to the H3 tail for GG-NER recognition. DNA repair 65, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Korolev N, Yu H, Lyubartsev AP, and Nordenskiold L (2014) Molecular dynamics simulations demonstrate the regulation of DNA-DNA attraction by H4 histone tail acetylations and mutations. Biopolymers 101, 1051–1064. [DOI] [PubMed] [Google Scholar]
- [28].Rajagopalan M, Balasubramanian S, Ioshikhes I, and Ramaswamy A (2017) Structural dynamics of nucleosome mediated by acetylations at H3K56 and H3K115,122. Eur Biophys J 46, 471–484. [DOI] [PubMed] [Google Scholar]
- [29].Zhang R, Erler J, and Langowski J (2017) Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophysical journal 112, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ikebe J, Sakuraba S, and Kono H (2016) H3 histone tail conformation within the nucleosome and the impact of K14 acetylation studied using enhanced sampling simulation. PLoS Comput. Biol 12, e1004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huertas J, and Cojocaru V (2021) Breaths, Twists, and Turns of Atomistic Nucleosomes. Journal of molecular biology 433, 166744. [DOI] [PubMed] [Google Scholar]
- [32].Che J, Smith S, Kim YJ, Shim EY, Myung K, and Lee SE (2015) Hyper-Acetylation of Histone H3K56 Limits Break-Induced Replication by Inhibiting Extensive Repair Synthesis. PLoS Genet 11, e1004990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Masumoto H, Hawke D, Kobayashi R, and Verreault A (2005) A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436, 294–298. [DOI] [PubMed] [Google Scholar]
- [34].Xu F, Zhang K, and Grunstein M (2005) Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121, 375–385. [DOI] [PubMed] [Google Scholar]
- [35].Das C, Lucia MS, Hansen KC, and Tyler JK (2009) CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Maas NL, Miller KM, DeFazio LG, and Toczyski DP (2006) Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Molecular cell 23, 109–119. [DOI] [PubMed] [Google Scholar]
- [37].Ozdemir A, Masumoto H, Fitzjohn P, Verreault A, and Logie C (2006) Histone H3 lysine 56 acetylation: a new twist in the chromosome cycle. Cell Cycle 5, 2602–2608. [DOI] [PubMed] [Google Scholar]
- [38].Downs JA (2008) Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA repair 7, 2020–2024. [DOI] [PubMed] [Google Scholar]
- [39].Tjeertes JV, Miller KM, and Jackson SP (2009) Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. Embo J 28, 1878–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhu Q, Battu A, Ray A, Wani G, Qian J, He J, Wang QE, and Wani AA (2015) Damaged DNA-binding protein down-regulates epigenetic mark H3K56Ac through histone deacetylase 1 and 2. Mutat Res 776, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wurtele H, Kaiser GS, Bacal J, St-Hilaire E, Lee EH, Tsao S, Dorn J, Maddox P, Lisby M, Pasero P, and Verreault A (2012) Histone H3 lysine 56 acetylation and the response to DNA replication fork damage. Mol Cell Biol 32, 154–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hyland EM, Cosgrove MS, Molina H, Wang D, Pandey A, Cottee RJ, and Boeke JD (2005) Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol Cell Biol 25, 10060–10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gospodinov A, and Herceg Z (2013) Shaping chromatin for repair. Mutat Res 752, 45–60. [DOI] [PubMed] [Google Scholar]
- [44].Unnikrishnan A, Gafken PR, and Tsukiyama T (2010) Dynamic changes in histone acetylation regulate origins of DNA replication. Nature structural & molecular biology 17, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Xie W, Song C, Young NL, Sperling AS, Xu F, Sridharan R, Conway AE, Garcia BA, Plath K, Clark AT, and Grunstein M (2009) Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Molecular cell 33, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu Y, Wang DL, Chen S, Zhao L, and Sun FL (2012) Oncogene Ras/phosphatidylinositol 3-kinase signaling targets histone H3 acetylation at lysine 56. The Journal of biological chemistry 287, 41469–41480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rodriguez Y, Horton JK, and Wilson SH (2019) Histone H3 Lysine 56 Acetylation Enhances AP Endonuclease 1-Mediated Repair of AP Sites in Nucleosome Core Particles. Biochemistry 58, 3646–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Karányi Z, Hornyák L, and Székvölgyi L (2020) Histone H3 Lysine 56 Acetylation Is Required for Formation of Normal Levels of Meiotic DNA Breaks in S. cerevisiae. Front Cell Dev Biol 7, 364–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Battu A, Ray A, and Wani AA (2011) ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 39, 7931–7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Luger K, and Richmond TJ (1998) DNA binding within the nucleosome core. Curr Opin Struct Biol 8, 33–40. [DOI] [PubMed] [Google Scholar]
- [51].Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, and Poirier MG (2011) Histone fold modifications control nucleosome unwrapping and disassembly. Proceedings of the National Academy of Sciences 108, 12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kono H, and Ishida H (2020) Nucleosome unwrapping and unstacking. Current Opinion in Structural Biology 64, 119–125. [DOI] [PubMed] [Google Scholar]
- [53].Fierz B, and Poirier MG (2019) Biophysics of chromatin dynamics. Annual Review of Biophysics 48, 321–345. [DOI] [PubMed] [Google Scholar]
- [54].Bilokapic S, Strauss M, and Halic M (2018) Histone octamer rearranges to adapt to DNA unwrapping. Nature structural & molecular biology 25, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bucceri A, Kapitza K, and Thoma F (2006) Rapid accessibility of nucleosomal DNA in yeast on a second time scale. The EMBO journal 25, 3123–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Poirier MG, Bussiek M, Langowski J, and Widom J (2008) Spontaneous access to DNA target sites in folded chromatin fibers. Journal of molecular biology 379, 772–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bilotti K, Kennedy EE, Li C, and Delaney S (2017) Human OGG1 activity in nucleosomes is facilitated by transient unwrapping of DNA and is influenced by the local histone environment. DNA Rep. 59, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wei S, Falk SJ, Black BE, and Lee TH (2015) A novel hybrid single molecule approach reveals spontaneous DNA motion in the nucleosome. Nucleic Acids Res 43, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kim J, Lee J, and Lee TH (2015) Lysine Acetylation Facilitates Spontaneous DNA Dynamics in the Nucleosome. J Phys Chem B 119, 15001–15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, and Chin JW (2009) A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Molecular cell 36, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lee J, and Lee TH (2019) How Protein Binding Sensitizes the Nucleosome to Histone H3K56 Acetylation. ACS Chem Biol 14, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fu I, Smith DJ, and Broyde S (2019) Rotational and translational positions determine the structural and dynamic impact of a single ribonucleotide incorporated in the nucleosome. DNA repair 73, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brower-Toland B, Wacker DA, Fulbright RM, Lis JT, Kraus WL, and Wang MD (2005) Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J Mol Biol. 346, 135–146. [DOI] [PubMed] [Google Scholar]
- [64].Garcia-Ramirez M, Dong F, and Ausio J (1992) Role of the histone “tails” in the folding of oligonucleosomes depleted of histone H1. The Journal of biological chemistry 267, 19587–19595. [PubMed] [Google Scholar]
- [65].Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, and Peterson CL (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- [66].Bannister AJ, and Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kulaeva OI, Zheng G, Polikanov YS, Colasanti AV, Clauvelin N, Mukhopadhyay S, Sengupta AM, Studitsky VM, and Olson WK (2012) Internucleosomal interactions mediated by histone tails allow distant communication in chromatin. The Journal of biological chemistry 287, 20248–20257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Azad GK, Swagatika S, Kumawat M, Kumawat R, and Tomar RS (2018) Modifying Chromatin by Histone Tail Clipping. Journal of molecular biology 430, 3051–3067. [DOI] [PubMed] [Google Scholar]
- [69].Hess MT, Gunz D, Luneva N, Geacintov NE, and Naegeli H (1997) Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol Cell Biol 17, 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Geacintov NE, and Broyde S (2017) Repair-resistant DNA lesions. Chemical research in toxicology 30, 1517–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Conney AH (1982) Induction of Microsomal Enzymes by Foreign Chemicals and Carcinogenesis by Polycyclic Aromatic Hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Research 42, 4875–4917. [PubMed] [Google Scholar]
- [72].Phillips DH (1983) Fifty years of benzo(a)pyrene. Nature 303, 468–472. [DOI] [PubMed] [Google Scholar]
- [73].Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, Harvey R, Geacintov NE, Broyde S, and Patel DJ (1993) Solution Conformation of the (+)-cis-anti-[BP]dG Adduct in a DNA Duplex: Intercalation of the Covalently Attached Benzo[a]pyrenyl Ring into the Helix and Displacement of the Modified Deoxyguanosine. Biochemistry 32, 4145–4155. [DOI] [PubMed] [Google Scholar]
- [74].Reeves DA, Mu H, Kropachev K, Cai Y, Ding S, Kolbanovskiy A, Kolbanovskiy M, Chen Y, Krzeminski J, Amin S, Patel DJ, Broyde S, and Geacintov NE (2011) Resistance of bulky DNA lesions to nucleotide excision repair can result from extensive aromatic lesion-base stacking interactions. Nucleic Acids Res 39, 8752–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mocquet V, Kropachev K, Kolbanovskiy M, Kolbanovskiy A, Tapias A, Cai Y, Broyde S, Geacintov NE, and Egly JM (2007) The human DNA repair factor XPC-HR23B distinguishes stereoisomeric benzo[a]pyrenyl-DNA lesions. Embo J 26, 2923–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE 3rd, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C , Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, and Kollman PA (2014), AMBER 14, University of California, San Francisco. [Google Scholar]
- [77].Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, and Walker RC (2012) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. Journal of chemical theory and computation 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent particle mesh Ewald. J. Chem. Theory Comput 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- [79].Case DA, Betz RM, Cerutti DS, Cheatham I, T. E, , Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, and Kollman PA (2016) AMBER 2016, University of California, San Francisco. [Google Scholar]
- [80].Le Grand S, Gotz AW, and Walker RC (2013) SPFP: speed without compromise-a mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Physics Commun 184, 374–380. [Google Scholar]
- [81].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the cccuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ivani I, Dans PD, Noy A, Perez A, Faustino I, Hospital A, Walther J, Andrio P, Goni R, Balaceanu A, Portella G, Battistini F, Gelpi JL, Gonzalez C, Vendruscolo M, Laughton CA, Harris SA, Case DA, and Orozco M (2016) Parmbsc1: a refined force field for DNA simulations. Nature Methods 13, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Papamokos GV, Tziatzos G, Papageorgiou DG, Georgatos SD, Politou AS, and Kaxiras E (2012) Structural role of RKS motifs in chromatin interactions: a molecular dynamics study of HP1 bound to a variably modified histone tail. Biophys. J 102, 1926–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jorgensen WL, Chandreskhar J, Madura JD, Imprey RW, and Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- [85].Darden T, York D, and Pedersen L (1993) Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092. [Google Scholar]
- [86].Cheatham TE, Miller JL, Fox T, Darden TA, and Kollman PA (1995) Molecular-Dynamics simulations on solvated biomolecular systems - the particle mesh Ewald eethod leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc 117, 4193–4194. [Google Scholar]
- [87].Ryckaert JP, Ciccotti G, and C. BHJ (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 23, 327–341. [Google Scholar]
- [88].Bönisch C, and Hake SB (2012) Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res 40, 10719–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, and Hansen JC (2013) The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface 10, 20121022–20121022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Fan JY, Rangasamy D, Luger K, and Tremethick DJ (2004) H2A.Z Alters the Nucleosome Surface to Promote HP1α-Mediated Chromatin Fiber Folding. Molecular cell 16, 655–661. [DOI] [PubMed] [Google Scholar]
- [91].Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, and Kaye KM (2006) The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–861. [DOI] [PubMed] [Google Scholar]
- [92].Roussel L, Erard M, Cayrol C, and Girard J-P (2008) Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep 9, 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Li G, Levitus M, Bustamante C, and Widom J (2005) Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol 12, 46–53. [DOI] [PubMed] [Google Scholar]
- [94].Kenzaki H, and Takada S (2015) Partial Unwrapping and Histone Tail Dynamics in Nucleosome Revealed by Coarse-Grained Molecular Simulations. PLoS Comput Biol 11, e1004443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kono H, Sakuraba S, and Ishida H (2018) Free energy profiles for unwrapping the outer superhelical turn of nucleosomal DNA. PLoS computational biology 14, e1006024–e1006024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Watanabe S, Resch M, Lilyestrom W, Clark N, Hansen JC, Peterson C, and Luger K (2010) Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochimica et biophysica acta 1799, 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu X, Li M, Xia X, Li X, and Chen Z (2017) Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 544, 440–445. [DOI] [PubMed] [Google Scholar]
- [98].Panchenko T, Sorensen TC, Woodcock CL, Kan ZY, Wood S, Resch MG, Luger K, Englander SW, Hansen JC, and Black BE (2011) Replacement of histone H3 with CENP-A directs global nucleosome array condensation and loosening of nucleosome superhelical termini. Proceedings of the National Academy of Sciences of the United States of America 108, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lehmann K, Zhang R, Schwarz N, Gansen A, Mücke N, Langowski J, and Toth K (2017) Effects of charge-modifying mutations in histone H2A α3-domain on nucleosome stability assessed by single-pair FRET and MD simulations. Scientific Reports 7, 13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kono H, Shirayama K, Arimura Y, Tachiwana H, and Kurumizaka H (2015) Two arginine residues suppress the flexibility of nucleosomal DNA in the canonical nucleosome core. PloS one 10, e0120635–e0120635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ferreira H, Somers J, Webster R, Flaus A, and Owen-Hughes T (2007) Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol Cell Biol 27, 4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ramanathan B, and Smerdon MJ (1989) Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J. Biol. Chem 264, 11026–11034. [PubMed] [Google Scholar]
- [103].Duan MR, and Smerdon MJ (2010) UV Damage in DNA Promotes Nucleosome Unwrapping. Journal of Biological Chemistry 285, 26295–26303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rodriguez Y, Hinz JM, Laughery MF, Wyrick JJ, and Smerdon MJ (2016) Site-specific Acetylation of Histone H3 Decreases Polymerase beta Activity on Nucleosome Core Particles in Vitro. The Journal of biological chemistry 291, 11434–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wood RD (1999) DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 81, 39–44. [DOI] [PubMed] [Google Scholar]
- [106].Cai Y, Geacintov NE, and Broyde S (2020) Variable impact of conformationally distinct DNA lesions on nucleosome structure and dynamics: Implications for nucleotide excision repair. DNA repair 87, 102768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Shafirovich V, Kolbanovskiy M, Kropachev K, Liu Z, Cai Y, Terzidis MA, Masi A, Chatgilialoglu C, Amin S, Dadali A, Broyde S, and Geacintov NE (2019) Nucleotide Excision Repair and Impact of Site-Specific 5′,8-Cyclopurine and Bulky DNA Lesions on the Physical Properties of Nucleosomes. Biochemistry 58, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Olmon ED, and Delaney S (2017) Differential Ability of Five DNA Glycosylases to Recognize and Repair Damage on Nucleosomal DNA. ACS Chem Biol 12, 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Caffrey PJ, and Delaney S (2021) Nucleosome Core Particles Lacking H2B or H3 Tails Are Altered Structurally and Have Differential Base Excision Repair Fingerprints. Biochemistry 60, 210–218. [DOI] [PubMed] [Google Scholar]
- [110].Fratini AV, Kopka ML, Drew HR, and Dickerson RE (1982) Reversible bending and helix geometry in a B-DNA dodecamer: CGCGAATTBrCGCG. J. Biol. Chem 257, 14686–14707. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.