

Abstract
The small diffusible second messenger 3’,5’-cyclic adenosine monophosphate (cAMP) is found in virtually every cell in our bodies, where it mediates responses to a variety of different G protein coupled receptors (GPCRs). In the heart, cAMP plays a critical role in regulating many different aspects of cardiac myocyte function, including gene transcription, cell metabolism, and excitation-contraction coupling. Yet, not all GPCRs that stimulate cAMP production elicit the same responses. Subcellular compartmentation of cAMP is essential to explain how different receptors can utilize the same diffusible second messenger to elicit unique functional responses. However the mechanisms contributing to this behavior and its significance in producing physiological and pathological responses are incompletely understood. Mathematical modeling has played an essential role in gaining insight into these questions. This review discusses what we currently know about cAMP compartmentation in cardiac myocytes and questions that are yet to be answered.
Graphical Abstract
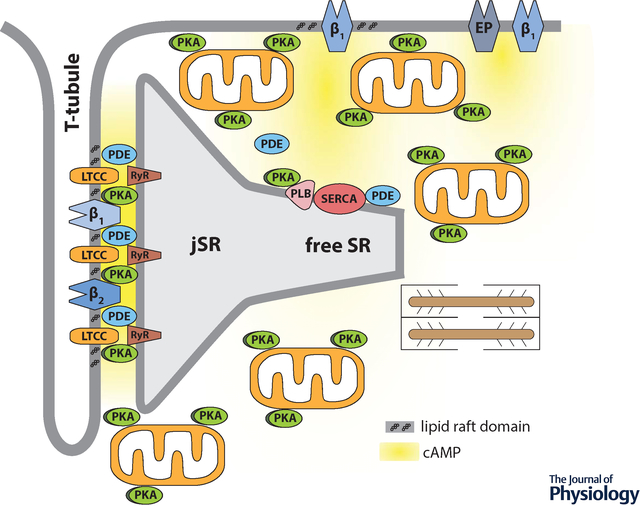
Compartmentation of receptor dependent cAMP signaling in adult cardiac ventricular myocytes depends on many factors, including localized production by different G-protein coupled receptors, localized degradation by phosphodiesterases, buffering by protein kinase A, and restricted diffusion involving tight spaces such as the dyadic cleft.
INTRODUCTION
The autonomic nervous system plays a critical role in regulating cardiac function in response to stress and exercise. Sympathetic stimulation enhances cardiac output by increasing both heart rate and contractility as part of the classic “fight or flight” response. These effects are mediated primarily through neurotransmitter stimulation of β-adrenergic receptors (βARs) and subsequent production of the small diffusible second messenger 3’,5’-cyclic adenosine monophosphate (cAMP).
In the heart, cAMP actually plays a critical role in regulating many different aspects of cardiac myocyte function, including gene transcription and cell metabolism, in addition to electrical and mechanical activity. Ironically, there are many different G-protein coupled receptors (GPCRs) capable of stimulating cAMP production in the heart, yet they do not all elicit the same responses. The classic example of this behavior was first reported more than forty years ago (Brunton et al., 1979; Hayes et al., 1979). βARs were found to stimulate cAMP production and activate protein kinase A (PKA), resulting in phosphorylation-dependent changes in contraction. On the other hand, E-type prostaglandin receptors (EPRs) were also found to increase cAMP production, but there were no concomitant functional changes. This led to the original hypothesis that cAMP signaling must be compartmentalized. In other words, this cytosolic second messenger, often thought of as being freely diffusible, must be spatially restricted in order to elicit unique cellular responses.
Compartmentalized production of cAMP is also believed to contribute to functional responses involving βARs. Cardiac myocytes express both β1 and β2AR subtypes. However, while β1ARs produce a global increase in cAMP, affecting many different processes, β2ARs produce a more localized cAMP response that selectively regulates L-type Ca2+ channel (LTCC) activity (Chen-Izu et al., 2000). The specificity of the β2AR response is attributed at least in part to the formation of a signaling complex that links β2ARs and LTCCs (Balijepalli et al., 2006). This difference in cAMP production may also contribute to the fact that β1ARs produce a strong positive inotropic response (increase in the rate and force of contraction) as well as a positive lusitropic response (increase in the rate of relaxation), while in many species, β2ARs produce a more modest increase in contraction without affecting the rate of relaxation (Xiao et al., 1994). Production of cAMP by both β1 and β2ARs regulate LTCCs associated with the plasma membrane of the transverse (t) tubules, which contributes to the increase in contractility (figure 1A). However, only β1AR production of cAMP leads to phosphorylation of phospholamban (PLN), which is found in non-junctional regions of the sarcoplasmic reticulum (SR). This later effect results in an increase in sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) activity, which not only contributes to the increase in contraction by increasing SR Ca2+ content, it also contributes to an increase in the rate of relaxation.
Figure 1.

Compartmentation of receptor-dependent cAMP signaling in adult ventricular myocytes. A, Under normal conditions, dyadic clefts are formed by tight junctions between the plasma membrane of t-tubules and the junctional sarcoplasmic reticulum (jSR). This is where β1 and β2ARs found in the plasma membrane of t-tubules are believed to be part of caveolar signaling complexes that include L-type Ca2+ channels (LTCCs), which are in close proximity to ryanodine receptors (RyRs) found in the jSR. E-type prostaglandin (EP) receptors, as well as some β1ARs, are excluded from of caveolar signaling complexes and t-tubule membranes. Phospholamban (PLN) and the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) are found outside of dyadic clefts in the free SR. Under normal conditions, β1AR production of cAMP leads to protein kinase A (PKA)-dependent phosphorylation of LTCCs, RyRs, and PLN. β2ARs lead to phosphorylation of LTCCs, but not RyRs or PLN. EP receptors do not regulate any of these effectors. Strategically placed phosphodiesterase (PDE) activity plays a critical role in cAMP compartmentation. Restricted spaces such as those created by dyadic clefts and tight mitochondrial packing as well as buffering by PKA anchored to the outer membrane of mitochondrial and other structures may also contribute to this behavior. B, Disease states, such as heart failure, are associated with changes in factors believed to contribute to compartmentalized cAMP responses. These changes include: disruption of dyadic clefts; redistribution of β2ARs from t-tubules to the peripheral sarcolemma; loss of AKAPs anchoring PKA to mitochondria; disruption of mitochondrial organization; and loss of PDE activity in some locations.
It should be noted that compartmentation of cAMP signaling is not unique to cardiac myocytes. Evidence for this type of behavior has been found in virtually every cell type in which it has been examined. Despite the ubiquitous nature of this phenomenon, our knowledge of the underlying mechanisms is incomplete. So how does a cell “decide” whether or not to produce a cAMP-dependent response following stimulation of any given receptor? One important means of ensuring the fidelity of cAMP-mediated signaling is through the formation of signaling complexes with A kinase anchoring proteins (AKAPs) (Scott et al., 2013). Disrupting AKAP interactions has been shown in numerous studies to alter cAMP responses by preventing PKA binding to and subsequent phosphorylation of specific target proteins. However, the anchoring of PKA by AKAPs alone is not sufficient to explain compartmentation. If cAMP were able to move freely throughout the cell, the same response would be expected regardless of the receptor that produced it. Therefore, there must also be mechanisms for creating discrete, localized pools of cAMP.
Over the past two decades, a number of new approaches have been developed, which have greatly impacted the study of cAMP signaling mechanisms. One of the first major advances was the generation of genetically encoded biosensors that can be used to measure cAMP activity in intact cells (Dikolayev et al., 2019). Another has been the implementation of computational modeling (Saucerman et al., 2014). This review will highlight some of the experimental evidence identifying factors contributing to cAMP compartmentation as it occurs in cardiac myocytes. We will also discuss the critical role that computational modeling has played by pointing us to new mechanisms contributing to this behavior and its significance in generating complex cellular responses. Table 1 lists those studies and indicates the aspects of cAMP compartmentation that are discussed in this review.
Table 1:
Computational models addressing mechanisms of cAMP compartmentation
| Study | Localized production | Localized degradation | PKA buffering | Restricted spaces |
|---|---|---|---|---|
| Rich et al. 2000 | X | |||
| Rich et al. 2001 | X | X | ||
| Saucerman et al. 2006 | X | X | X | X |
| Iancu et al. 2007 | X | X | X | |
| Neves et al., 2008 | X | X | ||
| Chen et al., 2008 | X | X | ||
| Oliveira et al. 2010 | X | |||
| Feinstein et al. 2012 | X | X | X | X |
| Yang et al. 2016 | X | X | X | X |
| Lohse et al. 2017 | X |
LOCALIZED PRODUCTION
Perhaps the easiest way to explain the ability of different receptors to produce unique cAMP responses is if they are found in discrete locations throughout the cell. One way in which receptors are segregated is by their inclusion or exclusion from caveolar signaling complexes (Brown, 2006; Allen et al., 2007). Caveolae are a subset of cholesterol-rich lipid raft domains of the plasma membrane that contain the multi-functional protein caveolin. There are three caveolin isoforms, with caveolin type 3 (Cav3) being the predominant version expressed in cardiac myocytes. Caveolins contain a caveolin scaffolding domain (CSD), which is believed to be involved in interactions with other signaling proteins, including certain GPCRs along with other components of the cAMP signaling cascade (Harvey & Calaghan, 2012).
β1-Adrenergic Receptors
β1ARs make up ~80% of the total βAR population in the hearts of most species (Steinberg, 1999). Furthermore, these receptors have been found in caveolar as well as non-caveolar fractions of the plasma membrane (Rybin et al., 2000; Ostrom et al., 2004; Balijepalli et al., 2006; Nichols et al., 2010; Agarwal et al., 2011). The effects of β1AR stimulation involve the activation of adenylyl cyclase (AC) by the stimulatory G protein Gs. Cardiac myocytes actually express multiple AC isoforms. The most abundant are AC5 and AC6, which are also found in caveolar membrane fractions (Rybin et al., 2000; Head et al., 2005; Balijepalli et al., 2006). This appears to involve direct interactions of AC with Cav3 (Rybin et al., 2000; Head et al., 2005; Balijepalli et al., 2006), although at least one study has suggested that interactions between Cav3 and AC involve an AKAP (Nichols et al., 2010).
The wide distribution of β1ARs is often associated with the production of global rather than compartmentalized cAMP responses in cardiac myocytes (Nikolaev et al., 2006). However, the effects that disrupting caveolae have on cAMP activity measured in different subcellular locations indicate that it may not be that simple. Depleting membrane cholesterol with agents such as methyl-β-cyclodextrin (MβCD) disrupts the inhibitory effect that Cav3 has on AC activity in caveolae. Agarwal et al. (2011) found that this selectively enhanced β1AR stimulation of cAMP activity detected by a FRET-based biosensor targeted to type II PKA signaling domains in adult cardiac myocytes. However, it had no effect on global changes in cAMP activity detected by a probe expressed throughout the cytosolic compartment. The increase in sensitivity of cAMP responses detected by the type II PKA probe correlated with an increase in the sensitivity of contractile and LTCC responses (Agarwal et al., 2011). These results suggest that a subpopulation of β1ARs associated with caveolae regulate target proteins involved in EC coupling, while β1ARs found elsewhere contribute to more global changes, which may be involved in regulating other responses.
β2-Adrenergic Receptors
β2ARs make up the bulk of the remaining βAR population in cardiac myocytes, and while they are capable of simulating cAMP production, the effects they produce differ from β1ARs. Some of those differences have been attributed to the ability of β2ARs to couple to the inhibitory G protein, Gi, in addition to Gs. although the Gi signaling mechanism involved is still unresolved (Xiao et al., 1995; Kuschel et al., 1999a; Xiao et al., 1999). Another difference between the sub-types of βARs in the heart is that β2ARs are believed exist exclusively in caveolar membrane domains (Ostrom et al., 2000; Rybin et al., 2000; Ostrom et al., 2001; Xiang et al., 2002; Ostrom et al., 2004; Head et al., 2005; Balijepalli et al., 2006; Head et al., 2006). Furthermore, there are distinct differences in the precise location of βAR sub-types within the 3D architecture of the cell. Adult ventricular myocytes possess an extensive t-tubule network that allows the plasma membrane to reach throughout the cell interior (see figure 1A), facilitating EC coupling. Nikolaev et al. (2010) used scanning ion conductance microscopy in combination with FRET-based biosensors to elegantly demonstrate that while β1AR stimulation produces cAMP responses associated with all areas of the plasma membrane, β2AR production of cAMP is confined to regions of the cell associated specifically with t-tubules. However, in heart failure, compartmentalized production of cAMP by β2ARs is lost, suggesting that these receptors are redistributed to the peripheral sarcolemma (see figure 1B).
βAR production of cAMP associated with t-tubules most likely occurs in dyadic clefts, which are junctional membrane complexes where LTCCs in the plasma membrane come in close proximity to ryanodine receptors (RyRs) in the junctional SR (Scriven et al., 2000) (see figure 1A). This is supported by the fact that βAR regulation of LTCCs occurs primarily in the t-tubules (Orchard & Brette, 2008), where LTCCs form signaling complexes with Cav3 that include both β1 and β2ARs (Balijepalli et al., 2006; Nichols et al., 2010).
The formation of signaling complexes that include Gi may explain how β2ARs selectively regulate LTCC function, since blocking this signaling pathway with pertussis toxin allows these receptors to produce a more global response, which includes PKA-dependent phosphorylation of PLN in the non-junctional SR, resulting in a positive lusitropic effect (Xiao et al., 1995; Kuschel et al., 1999b). It has been suggested that Gi signaling regulates PLN through a mechanism that involves changes in phosphatase activity (Kuschel et al., 1999a; Macdougall et al., 2012). However, there is evidence that β2ARs can also recruit phosphodiesterase (PDE) activity (Perry et al., 2002; Baillie et al., 2003), which breaks down cAMP, limiting its diffusion to more distant targets. Consistent with this idea, it has recently been shown that β1, but not β2AR stimulation is able to produce a cAMP response that can be detected by a FRET-based biosensor targeted to the free or non-junctional SR where PLN and SERCA are located. However, β2AR stimulation was able to produce a cAMP response detected at that more distant location after selective inhibition of PDE2 or PDE3 activity (Rudokas et al., 2021). It has also been shown that PDE4 activity prevents β2AR production of cAMP from reaching RyRs, which are presumably found just a few nanometers away, on the opposite side of the dyadic cleft. The loss of PDE4 activity can then explain how β2AR stimulation results in hyperphosphorylation of RyRs, leading to spontaneous release of Ca2+ from the SR and generation of ventricular arrhythmias in cardiac hypertrophy and heart failure (Berisha et al., 2021) (see figure 1B).
Prostaglandin Receptors
One of the first studies directly demonstrating compartmentation of cAMP produced by EPRs in adult cardiac myocytes compared responses detected by a FRET based biosensors targeted to type II PKA signaling domains and a globally expressed cytosolic biosensor in guinea pig ventricular myocytes (Warrier et al., 2007). EPR activation by PGE1 did not produce a cAMP response that could be detected by the PKA-targeted probe, which correlated with the inability of PGE1 to regulate LTCC activity. However, PGE1 was able to produce cAMP responses detected by the globally expressed biosensor. This demonstrated that EPRs are unable to stimulate cAMP production in subcellular locations where type II PKA regulation of functional responses normally occur.
Interestingly, EPR stimulation did produce a cAMP response that could be detected by the biosensor targeted to type II PKA signaling domains in adult rat ventricular myocytes. Yet, there was still no effect on myocyte contraction or LTCC activity (Agarwal et al., 2011). Furthermore, disrupting caveolae by cholesterol depletion, which enhanced β1AR responses, had no effect on the EPR mediated responses detected by the PKA targeted probe. This is consistent with the fact that the EP2 and EP4 receptor subtypes expressed in cardiac myocytes, both of which couple to Gs and stimulate cAMP production, are excluded from caveolar membrane fractions (Ostrom et al., 2001; Ostrom et al., 2004; Agarwal et al., 2011). In fact, PGE1 stimulation was found to produce changes in cAMP activity that were more readily detected by a biosensor targeted specifically to non-raft regions of the plasma (Agarwal et al., 2018). These results indicate that EPR stimulation produces a compartmentalized cAMP response associated with non-caveolar membrane domains that may be able to activate type II PKA in some species, but it is not associated with the regulation of EC coupling. The physical location of EPRs in cardiac myocytes is yet to be determined, but they are able to produce a cAMP response associated with protection from ischemia/reperfusion injury (Xiao et al., 2004; Pang et al., 2016).
Muscarinic Receptors
Cardiac myocytes also express M2 muscarinic receptors (M2Rs), which are involved in mediating parasympathetic responses. Many of these effects, especially in ventricular myocytes, involve Gi-dependent regulation of AC and changes in cAMP activity (Harvey & Belevych, 2003). The dominant effect is inhibitory, and this can be explained by direct inhibition of AC5/6 by the α subunit of Gi. However, M2Rs can also stimulate cAMP production (Warrier et al., 2005; Iancu et al., 2008). The consequence is a complex biphasic response, where upon exposure to ACh there is a rapid inhibition of cAMP that is then followed by a rebound increase upon washout of the agonist (Zakharov & Harvey, 1997; Belevych et al., 2001) (see figure 2A). In the presence of agonist, the inhibitory effect is dominant. However, upon termination of receptor activation, the inhibitory effect turns off rapidly, revealing the stimulatory response, which turns off more slowly. It has been suggested that this type of mechanism can trigger arrhythmogenic responses during transient activation of M2Rs (Song et al., 1998).
Figure 2.

M2 muscarinic receptor (M2) responses attributed to cAMP compartmentation in a cardiac ventricular myocyte. A, Time course of changes in the magnitude of the cAMP-regulated Cl− current in a guinea pig ventricular myocyte during exposure to a submaximally stimulating concentration of the βAR agonist isoproterenol (Iso), followed by addition of the M2 receptor agonist acetylcholine (ACh). Note: In the presence of ACh (+ACh), there is rapid inhibition of the current activated by Iso. Upon washout of ACh (-ACh), the inhibitory response is rapidly reversed, revealing a stimulatory effect. Adapted from Zakharov and Harvey, 1997. B, Cartoon diagram of computational model used to evaluate role of different adenylyl cyclase (AC) isoforms found in caveolar and extra-caveolar membrane domains. β1AR activation of the stimulatory G protein (Gs) increases cAMP production by AC5/6 and AC4/7 in caveolar and extra-caveolar domains, respectively. M2 muscarinic receptor activation of the inhibitory G protein (Gi) inhibits cAMP production by AC5/6 in caveolar domains, but stimulates cAMP production by AC4/7 in extra-caveolar domains. It is hypothesized that cAMP produced in the caveolar compartment is associated with most functional responses and that this can be affected by the flux (J) of cAMP from other compartments. C, Model prediction of changes in cAMP concentration in caveolar and extra-caveolar domains in response to submaximal β1 receptor stimulation by isoproterenol (Iso) followed by transient exposure to the muscarinic receptor agonist acetylcholine (ACh). Note: that exposure to ACh produces a rapid inhibition of cAMP activity in the caveolar domain, while simultaneously producing a slow increase in cAMP in the extra-caveolar domain. The model predicts that the transient changes observed in the caveolar domain upon termination of M2 receptor activation can be explained by the flux of cAMP between compartments. Adapted from Iancu et al. (2007).
This complex behavior can be explained by the ability of Gi signaling to stimulate AC4 and/or AC7, which are also present in cardiac myocytes. (Ishikawa & Homcy, 1997; Defer et al., 2000). Thus, M2Rs can inhibit cAMP production by AC5/6, while at the same time stimulating cAMP production by AC4/7 (Belevych et al., 2001; Harvey & Belevych, 2003; Warrier et al., 2005). It is hypothesized that the resulting biphasic response is due to the fact that the different AC isoforms are located in different microdomains of the plasma membrane (Iancu et al., 2007). AC5/6 are found in caveolae, while AC4/7 are excluded from those membrane domains (Ostrom & Insel, 2004; Willoughby & Cooper, 2007). The feasibility of this hypothesis was tested by developing a computational model that includes βAR and M2R signaling mechanisms affecting cAMP production in caveolar and extra-caveolar signaling domains (figure 2B). The results predict that the complex changes in cAMP activity reflect the ability of M2Rs to inhibit and stimulate cAMP production in different subcellular locations (figure 2C), with functional responses correlating most closely with changes occurring in caveolar membrane domains (Iancu et al., 2007; Iancu et al., 2008).
LOCALIZED DEGRADATION
Although localized production of cAMP is an intuitive factor to consider when explaining mechanisms contributing to compartmentation, computational analysis suggests that biochemically measured rates of AC activity are not high enough to generate significant cAMP gradients on their own (Rich et al., 2000). In addition to production of cAMP in discrete subcellular locations, the specificity of cAMP responses can only be maintained if the movement of this diffusible second messenger is somehow restricted. A number factors have been postulated to contribute to this behavior. The one that has received the most attention is localized degradation by PDE activity.
The primary mechanism for reversing the effects of cAMP production is via hydrolysis of the cyclic nucleotide by a PDE (Francis et al., 2001; Conti & Beavo, 2007), which may also serve as a means of generating cAMP gradients within a cell. PDEs are often depicted as creating functional barriers, which limit the movement of cAMP from one location to another, a concept that garners support from the fact that some PDE subtypes are known to be targeted to specific structures within the cell. In many cases this involves interactions with AKAPs (McConnachie et al., 2006).
One of the most striking examples of the role that PDE activity plays in limiting the movement of cAMP in cardiac myocytes was illustrated by the work of Jurevicius et al. (1996). Application of the βAR agonist isoproterenol to one-half of a frog ventricular myocyte caused half-maximal enhancement of the whole-cell, L-type Ca2+ current recorded using patch clamp techniques, suggesting that the only channels affected were those located in the region of the cell exposed to drug. However, when the non-selective PDE inhibitor 3-isobutyl-1-methylxanthine (IBMX) was applied together with isoproterenol, the Ca2+ current was maximally enhanced, indicating that PDE activity had been responsible for preventing cAMP at the site of production from regulating channels in more distal portions of the cell.
Subsequent development of genetically encoded biosensors has made it possible to directly monitor cAMP activity and how its movement is affected by PDE activity in live cells. One of the first studies using this approach involved a biosensor constructed using type II PKA (Zaccolo & Pozzan, 2002). Because of its interactions with AKAPs, the probe exhibits a distinct expression pattern in cardiac myocytes that corresponds with the Z line of the sarcomere. Furthermore, βAR activation produced changes in cAMP activity that remained localized to these striations. However, application of IBMX resulted in a uniform increase in cAMP, supporting the idea that movement of the second messenger produced following receptor activation had been limited by PDE activity.
There are 4 primary PDE isozymes involved in cAMP degradation in the heart – PDE1, PDE2, PDE3, and PDE4 (Osadchii, 2007). While PDE1 and PDE2 can hydrolyze both cAMP and cGMP, PDE3 preferentially hydrolyzes cAMP, and PDE4 is specific for cAMP. These PDE families also vary in how their ability to metabolize cAMP can be regulated. While PDE1 is activated in a Ca2+/calmodulin-dependent manner, PDE2 can be allosterically stimulated by cGMP, PDE3 can be competitively inhibited by cGMP, and both PDE3 and PDE4 can be activated by PKA-dependent phosphorylation (Conti & Beavo, 2007). The relative contribution of each PDE isozyme varies depending on species. Perhaps most notable is PDE4, which is in greatest abundance in the hearts of mice and rats, where it makes up as much as 60% of all cAMP hydrolytic activity (Mongillo et al., 2004; Leroy et al., 2008; Richter et al., 2011; Mika et al., 2012). However, in humans it represents only 10% of total PDE activity. Despite the striking difference in the relative amount of each PDE isoform expressed in different species, it is how each PDE isozyme is distributed throughout the cell that is likely most relevant. For example, selective inhibition of PDE4 activity in the mouse or rat results in a general increase in all phosphoproteins, whereas in human myocardium it results in phosphorylation of a specific subset of proteins (Richter et al., 2011). The use of selective pharmacologic inhibitors of the different PDE isozymes has provided important insight into the roles that each plays in contributing to cAMP production in different subcellular locations as well as the regulating specific functional responses. The exception to this is PDE1, for which the availability of selective inhibitors is limited (Vandeput et al., 2007; Miller et al., 2009).
PDE2
This PDE isozyme is associated primarily with membrane fractions of cardiac myocytes (Simmons & Hartzell, 1988; Mongillo et al., 2006), where it is believed to play a critical role in regulating sub-sarcolemmal cAMP-dependent responses. Consistent with this idea, PDE2 inhibition produced significantly greater changes in cAMP activity in subcellular locations associated with lipid raft and non-raft domains of the plasma membrane than it did in the cytosolic domain of adult ventricular myocytes. (Agarwal et al., 2018). In addition, PDE2 has been shown to play an important role in regulating the effects of βAR stimulation on LTCC activity (Hartzell & Fischmeister, 1986; Kirstein et al., 1995; Méry et al., 1995; Dittrich et al., 2001). Furthermore, FRET-based biosensors have shown that in neonatal ventricular myocytes, cGMP-signaling specifically decreases cAMP activity stimulated by βAR agonists (Stangherlin et al., 2011). Local regulation of cAMP activity by PDE2 has also been reported to selectively modify PKA-dependent regulation of LTCCs as well as RyRs, where it plays an important role in nitric oxide/cGMP dependent regulation of excitation-contraction coupling (Mohamed et al., 2011). In addition, there is evidence that PDE2 activity is involved in local control of cAMP that affects mitochondrial function (Liu et al., 2019). Additionally, PDE2 activity has been shown to play an important role in regulating cAMP-dependent responses in cardiac hypertrophy (Zoccarato et al., 2015). Those results were consistent with the finding that PDE2 expression is upregulated in cardiac hypertrophy (Mehel et al., 2013; Bastug-Ozel et al., 2019), and overexpression of PDE2 in normal cardiac myocytes can mitigate norepinephrine induced hypertrophy (Mehel et al., 2013). This suggests that targeting local control of cAMP by PDE2 may provide a therapeutic strategy. Similarly, it has been proposed that inhibition of PDE2 may be a target to selectively regulate PLN/SERCA activity and enhance relaxation in certain forms of heart failure (Rudokas et al., 2021).
PDE3
In general, PDE3 can be found in both membrane and cytosolic fractions of cardiac myocytes, although the relative distribution differs among species (Muller et al., 1992; Maurice et al., 2003; Lugnier, 2006). Consistent with these findings, selective inhibition of PDE3 activity resulted in similar changes in steady-state cAMP levels detected by FRET-based biosensors targeted to different regions of the plasma membrane as well as the bulk cytoplasmic compartment of adult rat ventricular myocytes (Agarwal et al., 2018). Interestingly, responses near the membrane were transient, suggesting that feedback regulation may play an important role in affecting sub-sarcolemmal cAMP activity. In neonatal rat ventricular myocytes, regulation of PDE3 activity by cGMP signaling pathways has been shown to modulate cAMP activity specifically associated with a soluble or type I PKA signaling domain (Stangherlin et al., 2011).
There are two PDE3 subfamilies, PDE3A and PDE3B. PDE3A is the predominant subtype found in cardiac myocytes of most species (Weishaar et al., 1987; Wechsler et al., 2002; Abi-Gerges et al., 2009). In human myocardium, alternative splicing results in at least three PDE3A isoforms. These differ in their N-terminus, which contains sites involved in feedback regulation by PKA and phosphoinositide-3 kinase γ (PI3Kγ) as well as targeting to different subcellular locations (Wechsler et al., 2002; Hambleton et al., 2005).
In the mouse heart it has been shown that knocking out PI3Kγ increases basal cAMP levels, enhancing ventricular contraction (Crackower et al., 2002). Furthermore, it was reported that PI3Kγ binds PDE3B, but not PDE3A, leading to the conclusion that PI3Kγ activates PDE3B (Patrucco et al., 2004; Alloatti et al., 2005). Kerfant et al. (2005; 2007) demonstrated that this effect of PI3Kγ was selectively affecting cAMP in subcellular locations associated regulation of Ca2+ transients, but not LTCCs. However, Beca et al. (2013) found that knocking out expression of PDE3B had no effect on ventricular myocyte contractility, while loss of PDE3A enhanced both the SR Ca2+ content as well as the amplitude of the intracellular Ca2+ transient, without affecting LTCCs. They also demonstrated that PDE3A forms a signaling complex with both SERCA2a and PLN. These results suggest that in the adult mouse heart PDE3A is the primary subtype involved in regulating ventricular myocyte function under basal conditions, and that it does so by selectively regulating cAMP in a microdomain associated with PLN and SERCA in the non-junctional SR.
Despite the evidence that PDE3 selectively regulates cAMP in the vicinity of PLN and SERCA under basal conditions, numerous studies have demonstrated that inhibition of PDE3 activity significantly affects cAMP dependent regulation of LTCC activity in the presence of βAR stimulation in adult ventricular myocytes (Verde et al., 1999). This occurs despite reports that PDE3 activity does not play a major role in regulating the amplitude and duration of cAMP responses measured globally, as well as in type II PKA signaling domains, using FRET-based biosensors following βAR stimulation (Mongillo et al., 2004; Nikolaev et al., 2006).
PDE4
Selective inhibition of PDE4 activity under baseline conditions typically has little if any effect on cardiac function (Mika et al., 2012). This is despite the fact that changes in cAMP activity have been detected by certain FRET-based biosensors, especially those targeted to the plasma membrane or type II PKA signaling domains (Mongillo et al., 2004; Agarwal et al., 2018). These results reflect the fact the PDE4 activity in general has been shown to be more important in affecting cAMP responses in the presence of βAR activation. Notably, PDE4 activity has been demonstrated to be more important than other PDE isozymes in regulating cAMP production by β1ARs (Mongillo et al., 2004; Nikolaev et al., 2006).
There are four PDE4 family genes, but only three are commonly expressed in cardiac myocytes: PDE4A, PDE4B, and PDE4D (Mongillo et al., 2004; Mika et al., 2012). Alternative splicing yields an even larger number of PDE4 isoforms. Differences in the N-terminus often determine where each isoform is targeted. PDE4B3, PDE4D3, and PDE4D5 have been reported to represent ~90% of the total PDE4 activity in neonatal rat ventricular myocytes (Mongillo et al., 2004; Mika et al., 2014). Pharmacologic inhibitors of PDE4 activity do not differentiate between the different isoforms. However, their unique functional roles have been elucidated using transgenic mouse, siRNA knockdown, or dominant negative over-expression approaches.
Both PDE4B and PDE4D have been shown to be part of a signaling complex that includes LTCCs in cardiac myocytes and, using transgenic mice, knockout of either subtype resulted in an increase in the Ca2+ current as well as the associated Ca2+ transient and contraction under basal conditions. However, the LTCC response to βAR stimulation was only enhanced in PDE4B, but not PDE4D, knockout myocytes. Furthermore, PDE4B knockout animals exhibited an increased susceptibility to arrhythmogenesis (Leroy et al., 2011).
It was subsequently shown that genetic ablation of PDE4B selectively alters cAMP responses to β1AR stimulation measured at the plasma membrane, but not the bulk cytoplasmic compartment of neonatal mouse ventricular myocytes (Mika et al., 2014). However, there was no change in the response to either β2AR or EPR activation. Furthermore, disrupting PDE4B activity specifically affected PKA-dependent phosphorylation of LTCCs and RyRs, which are in close proximity to one another in the dyadic cleft (see figure 1). PDE4B ablation did not affect phosphorylation of PLN or troponin-I, which are believed to be more distally located.
PDE4D has also been shown to be part of a signaling complex, along with PDE3A, that includes SERCA and PLN. Moreoverr, knocking out expression of PDE4D has been shown to enhance SR Ca2+ content as well as the amplitude of the intracellular Ca2+ transient without affecting the magnitude of the L-type Ca2+ current under basal conditions (Beca et al., 2011). This correlated with changes in PKA-dependent phosphorylation of PLN but not the RyR. These results indicate that PDE4D selectively regulates local control of cAMP near the non-junctional SR in adult ventricular myocytes.
PDE4D3 has been shown to form a complex with RyRs. Deleting expression of PDE4D in the mouse heart results in development of dilated cardiomyopathy, and despite normal levels of cAMP measured globally, evidence for a local increase in cAMP activity was detected specifically at the Z line of the sarcomere using a FRET based biosensor targeted to type II PKA signaling domains (Lehnart et al., 2005). This correlated with hyperphosphorylation of the RyR, which has been shown to disrupt interactions with calstabin-2, destabilizing its activity and allowing Ca2+ to leak from the SR, increasing the incidence of arrhythmias. Interestingly, there is down regulation of PDE4D3 in failing human hearts, which may explain the increased incidence of arrhythmias and sudden cardiac death in these patients. PDE4D3 has also been found to form part of a signaling complex with the slow delayed rectifier K+ channel (Marx et al., 2002; Terrenoire et al., 2009; Li et al., 2012).
PDE4D5 has been shown to play a specific role in β2AR signaling in cardiac myocytes. Following Gs-dependent activation of AC and cAMP production, these receptors can undergo desensitization involving various mechanisms (Lefkowitz et al., 1998). One involves phosphorylation at specific sites on the receptor by G-protein coupled receptor kinases resulting in the binding of β-arrestin, which in turn recruits PDE4D5 (Perry et al., 2002; Baillie et al., 2003). Consistent with this observation, inhibition of PDE4D5 activity was shown to enhance PKA phosphorylation of the receptor, facilitating its ability to couple to other pathways, including Gi inhibition of AC as well as activation of ERK1/2 signaling (Daaka et al., 1997). PDE4D5 has also been shown to play an important role in local cAMP dependent regulation of the small heat shock protein 20 (HSP20). This chaperone protein is involved in different cardioprotective signaling mechanisms. Furthermore, this effect is mediated by cAMP/PKA-dependent phosphorylation. It has been shown that PDE4 inhibition has a greater effect on the cAMP activity detected by a FRET-based biosensor tethered to HSP20 than it does on cAMP responses detected using a cytosolic probe (Sin et al., 2011). This localized effect can be explained by the ability of PDE4D5 to interact directly with HSP20.
The concept that PDE activity is a critical factor in creating localized cAMP responses is well established. However, while PDEs may act as “functional barriers” in some circumstances, in other instances they may serve as a “sink”. This is based on predictions that basal cAMP levels in some cytoplasmic domains are quite high and that PDE activity may act to keep concentrations below the level necessary to activate effectors such as PKA under unstimulated conditions (Iancu et al., 2007; Iancu et al., 2008). However, the fact that inhibition of PDE activity can produce localized changes in cAMP activity does not mean that it alone is sufficient to explain compartmentation.
PKA BUFFERING
Computational modeling has been particularly useful in investigating the likelihood that factors other than localized production and/or localized degradation play a role in generating discrete pools of cAMP. One such element that has been implicated is buffering of cAMP movement, specifically by PKA. There are a limited number of effectors that bind cAMP. In addition to PKA, cAMP can bind the exchange protein activated by cAMP (Epac), cyclic nucleotide gated (CNG) ion channels, and Popeye (POPDC) domain containing proteins (Brand & Schindler, 2017). Of these, PKA is the most abundant and therefore the most likely to affect cAMP diffusion in cardiac myocytes. It has been estimated that the PKA buffering capacity for cAMP in cardiac myocytes is ~1.2 μM (Corbin et al., 1977; Saucerman et al., 2003). Additionally, biochemical studies have indicated that a significant fraction of total cAMP is bound to PKA, even under basal conditions (Beavo et al., 1974; Corbin et al., 1977). In the first computational model of cAMP signaling in cardiac myocytes, Saucerman et al. predicted that buffering could help stabilize cAMP at concentrations near its binding affinity for PKA (Saucerman et al., 2003). Subsequent modeling efforts suggested that buffering might also contribute to gradients between compartments by slowing cAMP diffusion (Saucerman et al., 2006). The idea that interactions with PKA may affect the movement of cAMP are not unexpected given that measurements of the cAMP concentration in cardiac myocytes are in the same range as the predicted buffering capacity of PKA (Iancu et al., 2008; Borner et al., 2011; Agarwal et al., 2018).
Based on size alone, the predicted diffusion coefficient for cAMP in an aqueous solution devoid of any other factors that might affect its movement (free diffusion) is ~300 μm2/s (Neves et al., 2008; Agarwal et al., 2016). Early attempts to measure cAMP diffusion coefficients in various cell types came up with values ranging anywhere from approximately one-half to more than twice the rate of free diffusion (Bacskai et al., 1993; Chen et al., 1999; Nikolaev et al., 2004; Nikolaev et al., 2006; Saucerman et al., 2006). Many of these initial estimates involved the use of various FRET based biosensors to monitor the spread of cAMP within a cell following receptor activation. More recently Agarwal et al. (2016) used the technique of raster image correlation spectroscopy (RICS) to measure the diffusion coefficient of fluorescently labeled cAMP in intact, adult ventricular myocytes. This technique applies the principle of fluorescence correlation spectroscopy to images from a laser scanning confocal microscope on a pixel-by-pixel basis (Rossow et al., 2010). In these experiments, the fluorescent cAMP molecule used was 8-[Pharos-450]-cAMP (φ450-cAMP) (Moll et al., 2008). Attaching the Pharos dye to cAMP did not affect its affinity for PKA binding, but it did render it resistant to PDE hydrolysis (Moll et al., 2008). Although the dye alone has a molecular weight nearly identical to cAMP, estimates using Stokes-Einstein theory indicate that this would not significantly affect diffusion. This made φ450-cAMP an ideal choice to study cAMP diffusion independent of PDE activity. Using this approach, it was found that under basal conditions φ450-cAMP has a diffusion coefficient of 10 μm2/s, dramatically slower than previous estimates.
A major factor contributing to the slow rate of cAMP diffusion was revealed in the images of cells loaded with φ450-cAMP. Rather than being uniformly distributed throughout the cell, cAMP co-localized with mitochondria due to interactions with PKA anchored to the outer mitochondrial membrane (Agarwal et al., 2016). This was confirmed by demonstrating that the pattern could be disrupted by blocking interactions between the regulatory subunit of PKA and AKAPs. This maneuver also increased the diffusion coefficient of cAMP. Consistent with the idea that PKA buffering plays an important role in slowing cAMP diffusion, Nikolaev et al. (2010) demonstrated that disrupting AKAP anchoring allowed cAMP generated by β2ARs, which is normally not detectable away from the site of production in adult cardiac myocytes, to propagate throughout the entire cell. Mitochondria make up approximately 30% of the intracellular volume of a cardiac myocyte (Schaper et al., 1985; Barth et al., 1992). Therefore, anchoring a buffer to the outer membrane of these organelles would be an effective means of limiting the movement of cAMP as it attempts to diffuse throughout the cell.
Interestingly, the diffusion coefficients of the free pharos dye alone or fluorescein alone, both of, which have molecular weights similar to that of cAMP, were estimated to be ~60 μm2/s (Agarwal et al., 2016). This suggests that even in the absence of PKA binding, the diffusion coefficient of cAMP should still be significantly slower than the rate of free diffusion. This can be attributed to non-binding interactions with other solutes and macromolecules in the cytoplasm, which is referred to as molecular crowding. It is also consistent with the observation that cytoplasmic diffusion of small molecules is typically 3 to 8 times slower than their rate of free diffusion (Dix & Verkman, 2008). Furthermore, the diffusion coefficient of these molecules was found to be virtually the same in adult cardiac myocytes and morphologically simpler HEK293 cells, suggesting that factors slowing the movement of cAMP are similar across cell types. It should be noted, however, that Bock et al. have argued that the slow rate of cAMP diffusion in HEK293 cells may be due to mechanisms other than PKA buffering (Bock et al., 2020).
These results are largely consistent with modeling predictions, supporting the idea that slow diffusion of cAMP is an important factor contributing to cAMP compartmentation, but experimental evidence providing more direct proof of this hypothesis is needed. If substantiated, these initial studies raise some intriguing questions. Yet designing experiments that might provide that kind of proof requires additional information about the buffering effect itself. For example, why does cAMP seem to associate specifically with mitochondria? Is it because PKA is more heavily concentrated in this location? Or could it be that there is something different about the PKA found there? Could it be that there is more type I PKA targeted to that location? Type II PKA is most often thought of as being anchored by AKAPs. However, it is now known that there are type I specific AKAPs as well as dual specific AKAPs that interact with both type I and type II PKA. Furthermore, type I PKA has been reported to have a higher affinity for cAMP, which might then explain the tighter association with mitochondria. Another interesting possibility is that cAMP is interacting with free regulatory subunits of PKA, not the holoenzyme. Normally, we think of PKA as a heterotetrameric complex, with equal numbers of catalytic and regulatory subunits. However, it has been demonstrated that number of regulatory subunits far exceeds that of the catalytic subunits in most cell types. In cardiac myocytes the ratio is 6 to 1 (Walker-Gray et al., 2017). Furthermore, the cAMP affinity of free regulatory subunits is in the nanomolar range, while that of the holoenzyme is in the micromolar range (Dao et al., 2006). This would suggest the intriguing possibility that free regulatory subunits may exist for the purpose of buffering the movement of cAMP. It is also interesting to speculate about the role that changes in cAMP buffering and compartmentation play in the development of heart failure and injury due to myocardial infarction that are associated with the loss of mitochondrial AKAPs (Marin, 2020) (see figure 1B). These are all ideas that merit further investigation.
RESTRICTED SPACES
Some computational models have suggested that PDE activity alone is sufficient to generate cAMP gradients, even if it is assumed that cAMP can move at rates equal to free diffusion. For example, Oliveira et al. used a stochastic modeling approach to demonstrate the high levels of PDE activity are theoretically sufficient to explain cAMP compartmentation in HEK 293 cells (Oliveira et al., 2010). However, the rates of cAMP synthesis and degradation used in those simulations were significantly higher than experimental values measured in most cells (Saucerman et al., 2014). Other modeling studies have predicted that PDE activity alone is not sufficient to create cAMP gradients associated with compartmentation (Chen et al., 2008; Lohse et al., 2017). Neves et al. (2008) predicted that cAMP gradients can occur in the presence of more realistic levels of PDE activity when the cytosolic space is restricted to the geometry of a neuron, where the surface-to-volume ratio in the dendrites is quite high. Similarly, Feinstein et al. (2012) predicted that while the surface-to-volume ratio alone was not sufficient to explain cAMP compartmentation in endothelial cells, it did affect the potential role of other variables affecting cAMP diffusion. Early computational modeling also pointed to the idea that restriction of cAMP diffusion due to physical barriers between the plasma membrane and the cytosol could contribute to cAMP compartmentation in simple cells or cell free systems, although the exact nature of those barriers was not defined (Rich et al., 2000; Rich et al., 2001). Modeling by Iancu et al. (2007; 2008) also predicted that there must be some factor other than PDE activity that limits the flux of cAMP between compartments in cardiac myocytes.
Yang et al. (2016) addressed the question of whether or not PDE activity alone can act as a functional barrier by developing a 3D stochastic model of cAMP diffusion that represented the cytosolic space of an adult ventricular myocyte. The amount of PDE activity calculated to exist within a single myocyte was assumed to be distributed uniformly along a plane 100 nm from the inner surface of the t-tubule membrane, with no other physical barriers to diffusion. In these simulations, realistic estimates of PDE activity alone were not sufficient to prevent uniform distribution of cAMP throughout that space, even if diffusion was slowed to mimic the effects of molecular crowding or PKA buffering (figure 3A). Gradients could be generated by the model, but only when PDE activity was increased to unrealistically high levels. Yet, under those conditions, the total cAMP concentration was predicted to drop below levels necessary to activate known effectors such as PKA.
Figure 3.

A, Stochastic 3D simulation of cAMP diffusion. Left, four snapshots in time of the distribution of individual cAMP molecules (green spheres), generated by adenylyl cyclase (AC) activity (green box) in a caveolar membrane domain on the left, with cytosolic space to the right. Phosphodiesterase (PDE) molecules were placed in a plane (red bar) 100 nm from the plasma membrane to simulate a functional barrier. Right, average concentration of cAMP at various distances from the site of production when the number of PDE molecules and diffusion coefficient were varied. Normal PDE activity (415 μM), high PDE activity (41.5 mM). Free diffusion (200 μm2/s), molecular crowding (60 μm2/s), PKA buffering (10 μm2/s) B, 3D Continuum model of cAMP diffusion in an anatomically restricted space. Left, Cross section (1040 × 765 × 415 nm) through a 3D reconstruction of the dyadic space between the sarcoplasmic reticulum (SR) and t-tubule in an adult ventricular myocyte generated by cryo-transmission electron microscopy z-stacks. Location of AC activity is represented in red and PDE activity is represented in green. Right, Effect of diffusion coefficient on cAMP concentration in dyadic cleft in presence of normal PDE activity. Adapted from Yang et al. (2016).
The authors then evaluated the possibility that a more realistic representation of the space where cAMP signaling occurs might have an effect on the predicted outcome. Cryo-transmission electron microscopy images of adult mouse ventricular myocytes were used to generate a 3D reconstruction of the space surrounding the dyadic cleft. These junctional membrane complexes are already known to be essential for creating localized Ca2+ signaling domains (Bers, 2001; Winslow & Greenstein, 2011). Evidence that subpopulations of βARs are components of signaling complexes with LTCCs found at those sites suggests that cAMP signaling is likely to be confined within that space as well (Scriven et al., 2000; Balijepalli et al., 2006; Nichols et al., 2010). This anatomically restricted space is also surrounded by mitochondria that might further restrict cAMP diffusion. The 3D model of cAMP signaling was then implemented within this framework (Yang et al., 2016) (figure 3B). Adenylyl cyclase production of cAMP was initiated at the plasma membrane in the center of the dyadic cleft, and realistic levels of PDE activity were simulated as a barrier surrounding this space. T-tubules, SR, and mitochondria were treated as impenetrable barriers limiting the movement of cAMP. No significant gradients were predicted when cAMP was assumed to move at rates mimicking free diffusion or even at the slower rates attributed to molecular crowding. However, significant gradients were predicted when the diffusion rate was reduced to levels taking into account the potential effect of PKA buffering. These results demonstrated a plausible explanation for how production of cAMP by receptors within the dyadic cleft could lead to an increase in cAMP that is limited to that restricted space. The same mechanisms are also likely to limit the ability of cAMP produced by receptors located outside the dyadic cleft from entering. These results also suggest that the disruption of dyadic clefts that occurs in diseases states such as heart failure might be expected to affect compartmentalized cAMP responses (Zhang et al., 2013) (see figure 1B). However, these are predictions that still need experimental confirmation.
The dyadic cleft is just one example of spatial restriction contributing to cAMP compartmentation in the heart. Richards et al. (2016) addressed the question of cAMP mobility in adult ventricular myocytes expressing a cytosolic FRET-based biosensor together with a microfluidics system, which made it possible to apply an agonist to one-half of a cell while monitoring changes in cAMP activity in both halves simultaneously. The diffusion coefficient was then estimated by fitting the time course of the responses, taking into account predicted rates of synthesis and degradation. Using this method the authors suggested that the cAMP diffusion coefficient in adult cardiac ventricular myocytes is 35 μm2/s. Like the results obtained by Agarwal et al. (2016), this is significantly slower than the expected rate of free diffusion. However, Richards et al. concluded that this was not due to PKA buffering, since the calculated diffusion coefficient was not affected by the presence of a cAMP analog that prevented PKA binding. Switching to a fluorescence recovery after photobleaching (FRAP) approach, the authors determined that the diffusion coefficient of fluorescein, a molecule the same size as cAMP, is faster in neonatal cardiac myocytes than is in adult ventricular myocytes. Because neonatal cells or reported to have a lower density of mitochondria, it was concluded that the slow rate of diffusion in adult cardiac myocytes must be a function of cellular tortuosity due to the presence of mitochondria, although other structural differences were not considered. If the density of mitochondria does limit the movement cAMP in adult myocytes, the loss of mitochondrial organization that occurs in disease states such as heart failure (Miragoli et al., 2016) might be expected to alter compartmentalized responses (see figure 1B).
Predictions based on the studies like those described above support the idea that physically restricted spaces play an essential role in explaining the mechanisms of cAMP compartmentation. Proof of this concept awaits further experimental evidence.
CONCLUSIONS AND FUTURE DIRECTIONS
Our understanding of compartmentalized cAMP signaling in cardiac myocytes and the mechanisms that contribute to this behavior have advanced significantly over last 20 years, largely due the development of new experimental and computational approaches. The evidence is now quite clear that cAMP is not uniformly distributed throughout the cell and that a number of factors contribute to this behavior. Experimental approaches thus far have largely focused on the roles of localized production and localized degradation of cAMP. However, predictions advanced by computational modeling have pointed to other factors affecting cAMP diffusion such as PKA buffering and restricted physical spaces. Early experiments support the idea that these mechanisms contribute to this complex behavior as well, but additional experimental evidence is needed to confirm these predictions. Other factors that should be considered, but were not discussed here include the role of liquid phase separation of the cytosolic space (Zhang et al., 2020) and localized cAMP extrusion by membrane transporters.
The functional role of cAMP compartmentation is perhaps most clearly illustrated by comparing the responses between βARs and EPRs or between βAR subtypes. However, various experimental approaches have highlighted the functional significance that changes in local cAMP activity play in different disease states. Yet there are likely to be even more examples of functional responses that can be attributed to cAMP compartmentation. Again, this is where the power of computational modeling can be leveraged to provide additional insight. One example highlighted here is the prediction that this process may contribute to the complex behavior associated M2 receptor activation. But there are likely other examples yet to be discovered. Such approaches can provide insight that may be used to design appropriate experiments to test the predictions that arise from the emergent behavior identified by such models.
There has been substantial improvement in understanding mechanisms related to restricted signaling by cAMP. These advances have arisen from multiple new techniques and technologies in the experimental and computational space. As protein structural approaches continue to mature and allow the exploration of protein complexes through visualization, and atomistic scale modeling provides insights into the dynamics of signaling, there is the promise of a complete understanding of signaling via the ubiquitous second messenger cAMP.
ACKNOWLEDGMENTS
The authors wish to thank Michael Rudokas with help in generating figure 1 and Shailesh Agarwal, Rinzhin Sherpa, Karni Moshal, and Chase Fiore for reading the final manuscript.
FUNDING
This work was supported by grants R01 HL145778, R01 GM107904, OT2 OD023848, OT2 OD026580, and the Center for Biological Research Excellence P20 GM130459 from the National Institutes of Health.
Biography

Robert Harvey is a Professor of Pharmacology at the University of Nevada, Reno School of Medicine. His research interests include autonomic regulation of cardiac myocyte function with the goal of understanding how changes in sympathetic and parasympathetic tone contribute to the generation of arrhythmogenic responses associated with sudden cardiac death.

Colleen Clancy is a Professor of Physiology and Membrane Biology and the Vice Chancellor for Academic Personnel at the University of California Davis School of Medicine. Her research interests include computational approaches to reveal mechanisms of normal and pathological excitability in the heart and nervous system. Key areas of research focus include computational approaches to safety pharmacology, autonomic control of the cardiovascular system, multiscale modeling and simulation, machine learning and interdisciplinary approaches to problem solving.
Footnotes
COMPETING INTERESTS
The authors have no conflicts of interests to declare.
REFERENCES
- Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel JL, Lugnier C, Conti M, Fischmeister R & Vandecasteele G (2009) Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res 105, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Clancy CE & Harvey RD (2016) Mechanisms restricting diffusion of intracellular cAMP. Sci Rep 6, 10.1038/srep19577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Gratwohl J, Cozad M, Yang PC, Clancy CE & Harvey RD (2018) Compartmentalized cAMP Signaling Associated With Lipid Raft and Non-raft Membrane Domains in Adult Ventricular Myocytes. Front Pharmacol 9, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Macdougall DA, Tyser R, Pugh SD, Calaghan SC & Harvey RD (2011) Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J Mol Cell Cardiol 50, 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Halverson-Tamboli RA & Rasenick MM (2007) Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8, 128–140. [DOI] [PubMed] [Google Scholar]
- Alloatti G, Marcantoni A, Levi R, Gallo MP, Del SL, Patrucco E, Barberis L, Malan D, Azzolino O, Wymann M, Hirsch E & Montrucchio G (2005) Phosphoinositide 3-kinase gamma controls autonomic regulation of the mouse heart through Gi-independent downregulation of cAMP level. FEBS Lett 579, 133–140. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER & Tsien RY (1993) Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science 260, 222–226. [DOI] [PubMed] [Google Scholar]
- Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ & Houslay MD (2003) beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA 100, 940–945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Balijepalli RC, Foell JD, Hall DD, Hell JW & Kamp TJ (2006) Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci USA 103, 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth E, Stämmler G, Speiser B & Schaper J (1992) Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 24, 669–681. [DOI] [PubMed] [Google Scholar]
- Bastug-Ozel Z, Wright PT, Kraft AE, Pavlovic D, Howie J, Froese A, Fuller W, Gorelik J, Shattock MJ & Nikolaev VO (2019) Heart failure leads to altered beta2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain. Cardiovasc Res 115, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavo JA, Bechtel PJ & Krebs EG (1974) Activation of protein kinase by physiological concentrations of cyclic AMP. Proc Natl Acad Sci USA 71, 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beca S, Ahmad F, Shen W, Liu J, Makary S, Polidovitch N, Sun J, Hockman S, Chung YW, Movsesian M, Murphy E, Manganiello V & Backx PH (2013) Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res 112, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beca S, Helli PB, Simpson JA, Zhao D, Farman GP, Jones PP, Tian X, Wilson LS, Ahmad F, Chen SR, Movsesian MA, Manganiello V, Maurice DH, Conti M & Backx PH (2011) Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res 109, 1024–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Sims C & Harvey RD (2001) ACh-induced rebound stimulation of L-type Ca(2+) current in guinea-pig ventricular myocytes, mediated by Gbetagamma-dependent activation of adenylyl cyclase. J Physiol (Lond) 536, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berisha F, Götz K, Wegener JW, Brandenburg S, Subramanian H, Molina CE, Rueffer A, Petersen J, Bernhardt A, Girdauskas E, Jungen C, Pape U, Kraft AE, Warnke S, Lindner D, Westermann D, Blankenberg S, Meyer C, Hasenfuß G, Lehnart SE & Nikolaev VO (2021) cAMP Imaging at Ryanodine Receptors Reveals β2-Adrenoceptor Driven Arrhythmias. Circ Res. [DOI] [PubMed] [Google Scholar]
- Bers DM (2001) Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer, Dordrecht. [Google Scholar]
- Bock A, Annibale P, Konrad C, Hannawacker A, Anton SE, Maiellaro I, Zabel U, Sivaramakrishnan S, Falcke M & Lohse MJ (2020) Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 182, 1519–1530.e1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ & Nikolaev VO (2011) FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc 6, 427–438. [DOI] [PubMed] [Google Scholar]
- Brand T & Schindler R (2017) New kids on the block: The Popeye domain containing (POPDC) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cellular signalling 40, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA (2006) Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21, 430–439. [DOI] [PubMed] [Google Scholar]
- Brunton LL, Hayes JS & Mayer SE (1979) Hormonally specific phosphorylation of cardiac troponin I and activation of glycogen phosphorylase. Nature 280, 78–80. [DOI] [PubMed] [Google Scholar]
- Chen-Izu Y, Xiao RP, Izu LT, Cheng H, Kuschel M, Spurgeon H & Lakatta EG (2000) G(i)-dependent localization of beta(2)-adrenergic receptor signaling to L-type Ca(2+) channels. Biophys J 79, 2547–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Nakamura T & Koutalos Y (1999) Cyclic AMP diffusion coefficient in frog olfactory cilia. Biophys J 76, 2861–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Levine H & Rappel WJ (2008) A mathematical analysis of second messenger compartmentalization. Phys Biol 5, 046006. [DOI] [PubMed] [Google Scholar]
- Conti M & Beavo J (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76, 481–511. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Sugden PH, Lincoln TM & Keely SL (1977) Compartmentalization of adenosine 3’:5’-monophosphate and adenosine 3’:5’-monophosphate-dependent protein kinase in heart tissue. J Biol Chem 252, 3854–3861. [PubMed] [Google Scholar]
- Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH & Penninger JM (2002) Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110, 737–749. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM & Lefkowitz RJ (1997) Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91. [DOI] [PubMed] [Google Scholar]
- Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A & Doskeland SO (2006) Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J Biol Chem 281, 21500–21511. [DOI] [PubMed] [Google Scholar]
- Defer N, Best-Belpomme M & Hanoune J (2000) Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am J Physiol 279, F400–F416. [DOI] [PubMed] [Google Scholar]
- Dikolayev V, Tuganbekov T & Nikolaev VO (2019) Visualizing Cyclic Adenosine Monophosphate in Cardiac Microdomains Involved in Ion Homeostasis. Front Physiol 10, 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich M, Jurevicius J, Georget M, Rochais F, Fleischmann B, Hescheler J & Fischmeister R (2001) Local response of L-type Ca(2+) current to nitric oxide in frog ventricular myocytes. J Physiol (Lond) 534, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix JA & Verkman AS (2008) Crowding effects on diffusion in solutions and cells. Annu Rev Biophys 37, 247–263. [DOI] [PubMed] [Google Scholar]
- Feinstein WP, Zhu B, Leavesley SJ, Sayner SL & Rich TC (2012) Assessment of cellular mechanisms contributing to cAMP compartmentalization in pulmonary microvascular endothelial cells. Am J Physiol Cell Physiol 302, C839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Turko IV & Corbin JD (2001) Cyclic nucleotide phosphodiesterases: relating structure and function. Prog Nucleic Acid Res Mol Biol 65, 1–52. [DOI] [PubMed] [Google Scholar]
- Hambleton R, Krall J, Tikishvili E, Honeggar M, Ahmad F, Manganiello VC & Movsesian MA (2005) Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to camp hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem 280, 39168–39174. [DOI] [PubMed] [Google Scholar]
- Hartzell HC & Fischmeister R (1986) Opposite effects of cyclic GMP and cAMP on Ca2+ current in single heart cells. Nature 323, 273–275. [DOI] [PubMed] [Google Scholar]
- Harvey RD & Belevych AE (2003) Muscarinic regulation of cardiac ion channels. Br J Pharmacol 139, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RD & Calaghan SC (2012) Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J Mol Cell Cardiol 52, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JS, Brunton LL, Brown JH, Reese JB & Mayer SE (1979) Hormonally specific expression of cardiac protein kinase activity. Proc Natl Acad Sci USA 76, 1570–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG & Insel PA (2005) G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem 280, 31036–31044. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG & Insel PA (2006) Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem 281, 26391–26399. [DOI] [PubMed] [Google Scholar]
- Iancu RV, Jones SW & Harvey RD (2007) Compartmentation of cAMP signaling in cardiac myocytes: a computational study. Biophys J 92, 3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu RV, Ramamurthy G, Warrier S, Nikolaev VO, Lohse MJ, Jones SW & Harvey RD (2008) Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am J Physiol Cell Physiol 295, C414–C422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y & Homcy CJ (1997) The adenylyl cyclases as integrators of transmembrane signal transduction. Circ Res 80, 297–304. [DOI] [PubMed] [Google Scholar]
- Jurevicius J & Fischmeister R (1996) cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc Natl Acad Sci USA 93, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfant BG, Gidrewicz D, Sun H, Oudit GY, Penninger JM & Backx PH (2005) Cardiac sarcoplasmic reticulum calcium release and load are enhanced by subcellular cAMP elevations in PI3Kgamma-deficient mice. Circ Res 96, 1079–1086. [DOI] [PubMed] [Google Scholar]
- Kerfant BG, Zhao D, Lorenzen-Schmidt I, Wilson LS, Cai S, Chen SR, Maurice DH & Backx PH (2007) PI3Kgamma is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res 101, 400–408. [DOI] [PubMed] [Google Scholar]
- Kirstein M, Rivet-Bastide M, Hatem S, Bénardeau A, Mercadier J-J & Fischmeister R (1995) Nitric oxide regulates the calcium current in isolated human atrial myocytes. J Clin Invest 95, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschel M, Zhou YY, Cheng H, Zhang SJ, Chen Y, Lakatta EG & Xiao RP (1999a) G(i) protein-mediated functional compartmentalization of cardiac beta(2)-adrenergic signaling. J Biol Chem 274, 22048–22052. [DOI] [PubMed] [Google Scholar]
- Kuschel M, Zhou YY, Spurgeon HA, Bartel S, Karczewski P, Zhang SJ, Krause EG, Lakatta EG & Xiao RP (1999b) Beta2-adrenergic cAMP signaling is uncoupled from phosphorylation of cytoplasmic proteins in canine heart. Circulation 99, 2458–2465. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Pitcher J, Krueger K & Daaka Y (1998) Mechanisms of beta-adrenergic receptor desensitization and resensitization. Adv Pharmacol 42:416–20, 416–420. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M & Marks AR (2005) Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R & Vandecasteele G (2008) Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res 102, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Leroy J, Richter W, Mika D, Castro LR, Abi-Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Mateo P, Westenbroek R, Catterall WA, Charpentier F, Conti M, Fischmeister R & Vandecasteele G (2011) Phosphodiesterase 4B in the cardiac L-type Ca(2)(+) channel complex regulates Ca(2)(+) current and protects against ventricular arrhythmias in mice. J Clin Invest 121, 2651–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen L, Kass RS & Dessauer CW (2012) The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol Chem 287, 29815–29824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Wang Z, Nicolas V, Lindner M, Mika D, Vandecasteele G, Fischmeister R & Brenner C (2019) PDE2 regulates membrane potential, respiration and permeability transition of rodent subsarcolemmal cardiac mitochondria. Mitochondrion 47, 64–75. [DOI] [PubMed] [Google Scholar]
- Lohse C, Bock A, Maiellaro I, Hannawacker A, Schad LR, Lohse MJ & Bauer WR (2017) Experimental and mathematical analysis of cAMP nanodomains. PLoS One 12, e0174856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugnier C (2006) Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther 109, 366–398. [DOI] [PubMed] [Google Scholar]
- Macdougall DA, Agarwal SR, Stopford EA, Chu H, Collins JA, Longster AL, Colyer J, Harvey RD & Calaghan S (2012) Caveolae compartmentalise beta2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J Mol Cell Cardiol 52, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin W (2020) A-kinase anchoring protein 1 (AKAP1) and its role in some cardiovascular diseases. J Mol Cell Cardiol 138, 99–109. [DOI] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR & Kass RS (2002) Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295, 496–499. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS & Jimmo SL (2003) Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol 64, 533–546. [DOI] [PubMed] [Google Scholar]
- McConnachie G, Langeberg LK & Scott JD (2006) AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med 12, 317–323. [DOI] [PubMed] [Google Scholar]
- Mehel H, Emons J, Vettel C, Wittkopper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechene P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R & El-Armouche A (2013) Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 62, 1596–1606. [DOI] [PubMed] [Google Scholar]
- Méry P-F, Pavoine C, Pecker F & Fischmeister R (1995) Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP- stimulated phosphodiesterase in isolated cardiac myocytes. Mol Pharmacol 48, 121–130. [PubMed] [Google Scholar]
- Mika D, Leroy J, Vandecasteele G & Fischmeister R (2012) PDEs create local domains of cAMP signaling. J Mol Cell Cardiol 52, 323–329. [DOI] [PubMed] [Google Scholar]
- Mika D, Richter W, Westenbroek RE, Catterall WA & Conti M (2014) PDE4B mediates local feedback regulation of beta(1)-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J Cell Sci 127, 1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X, Xu H, Florio V, Rybalkin SD, Beavo JA, Chen YF, Li JD, Blaxall BC, Abe J & Yan C (2009) Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 105, 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miragoli M, Sanchez-Alonso JL, Bhargava A, Wright PT, Sikkel M, Schobesberger S, Diakonov I, Novak P, Castaldi A, Cattaneo P, Lyon AR, Lab MJ & Gorelik J (2016) Microtubule-Dependent Mitochondria Alignment Regulates Calcium Release in Response to Nanomechanical Stimulus in Heart Myocytes. Cell reports 14, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed TMA, Oceandy D, Zi M, Prehar S, Alatwi N, Wang Y, Shaheen Ma, Abou-Leisa R, Schelcher C, Hegab Z, Baudoin F, Emerson M, Mamas M, Di Benedetto G, Zaccolo M, Lei M, Cartwright EJ & Neyses L (2011) Plasma membrane calcium pump (PMCA4)/neuronal nitric oxide synthase complex regulates cardiac contractility through modulation of a compartmentalized cyclic nucleotide microdomain. J Biol Chem 286, 41520–41529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll D, Prinz A, Brendel CM, Berrera M, Guske K, Zaccolo M, Genieser HG & Herberg FW (2008) Biochemical characterization and cellular imaging of a novel, membrane permeable fluorescent cAMP analog. In BMC biochemistry, pp. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD & Zaccolo M (2004) Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res 95, 67–75. [DOI] [PubMed] [Google Scholar]
- Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD & Zaccolo M (2006) Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res 98, 226–234. [DOI] [PubMed] [Google Scholar]
- Muller B, Stoclet JC & Lugnier C (1992) Cytosolic and membrane-bound cyclic nucleotide phosphodiesterases from guinea pig cardiac ventricles. Eur J Pharmacol 225, 263–272. [DOI] [PubMed] [Google Scholar]
- Neves SR, Tsokas P, Sarkar A, Grace EA, Rangamani P, Taubenfeld SM, Alberini CM, Schaff JC, Blitzer RD, Moraru II & Iyengar R (2008) Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell 133, 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF & McKnight GS (2010) Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res 107, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A & Lohse MJ (2004) Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem 279, 37215–37218. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ & Engelhardt S (2006) Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta-1 adrenergic but locally confined beta-2 adrenergic receptor-mediated signaling. Circ Res 99, 1084–1091. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE & Gorelik J (2010) Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327, 1653–1657. [DOI] [PubMed] [Google Scholar]
- Oliveira RF, Terrin A, Di Benedetto G, Cannon RC, Koh W, Kim M, Zaccolo M & Blackwell KT (2010) The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PLoS One 5, e11725–e11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard C & Brette F (2008) t-Tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. CardiovascRes 77, 237–244. [DOI] [PubMed] [Google Scholar]
- Osadchii OE (2007) Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease. Cardiovasc Drugs Ther 21, 171–194. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Bundey RA & Insel PA (2004) Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes. J Biol Chem 279, 19846–19853. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW & Insel PA (2001) Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J Biol Chem 276, 42063–42069. [DOI] [PubMed] [Google Scholar]
- Ostrom RS & Insel PA (2004) The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol 143, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom RS, Violin JD, Coleman S & Insel PA (2000) Selective enhancement of beta-adrenergic receptor signaling by overexpression of adenylyl cyclase type 6: colocalization of receptor and adenylyl cyclase in caveolae of cardiac myocytes. Mol Pharmacol 57, 1075–1079. [PubMed] [Google Scholar]
- Pang L, Cai Y, Tang EH, Irwin MG, Ma H & Xia Z (2016) Prostaglandin E Receptor Subtype 4 Signaling in the Heart: Role in Ischemia/Reperfusion Injury and Cardiac Hypertrophy. Journal of diabetes research 2016, 1324347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G & Hirsch E (2004) PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118, 375–387. [DOI] [PubMed] [Google Scholar]
- Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD & Lefkowitz RJ (2002) Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298, 834–836. [DOI] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM & Karpen JW (2000) Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol 116, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Tse TE, Rohan JG, Schaack J & Karpen JW (2001) In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J Gen Physiol 118, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards M, Lomas O, Jalink K, Ford KL, Vaughan-Jones RD, Lefkimmiatis K & Swietach P (2016) Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc Res 110, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA & Conti M (2011) Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol 106, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow MJ, Sasaki JM, Digman Ma & Gratton E (2010) Raster image correlation spectroscopy in live cells. Nat Protoc 5, 1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudokas MW, Post JP, Sataray-Rodriguez A, Sherpa RT, Moshal KS, Agarwal SR & Harvey RD (2021) Compartmentation of β(2) -adrenoceptor stimulated cAMP responses by phosphodiesterase types 2 and 3 in cardiac ventricular myocytes. Br J Pharmacol 178, 1574–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybin VO, Xu X, Lisanti MP & Steinberg SF (2000) Differential targeting of beta -adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem 275, 41447–41457. [DOI] [PubMed] [Google Scholar]
- Saucerman JJ, Brunton LL, Michailova AP & McCulloch AD (2003) Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem 278, 47997–48003. [DOI] [PubMed] [Google Scholar]
- Saucerman JJ, Greenwald EC & Polanowska-Grabowska R (2014) Mechanisms of cyclic AMP compartmentation revealed by computational models. J Gen Physiol 143, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY & McCulloch AD (2006) Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci USA 103, 12923–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper J, Meiser E & Stämmler G (1985) Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res 56, 377–391. [DOI] [PubMed] [Google Scholar]
- Scott JD, Dessauer CW & Tasken K (2013) Creating Order from Chaos: Cellular Regulation by Kinase Anchoring. Annu Rev Pharmacol Toxicol 53, 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriven DR, Dan P & Moore ED (2000) Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J 79, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons MA & Hartzell HC (1988) Role of phosphodiesterase in regulation of calcium current in isolated cardiac myocytes. Mol Pharmacol 33, 664–671. [PubMed] [Google Scholar]
- Sin YY, Edwards HV, Li X, Day JP, Christian F, Dunlop AJ, Adams DR, Zaccolo M, Houslay MD & Baillie GS (2011) Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the beta-agonist induced hypertrophic response in cardiac myocytes. J Mol Cell Cardiol 50, 872–883. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC & Belardinelli L (1998) Potentiating effect of acetylcholine on stimulation by isoproterenol of L-type Ca2+ current and arrhythmogenic triggered activity in guinea pig ventricular myocytes. JCardiovascElectrophysiol 9, 718–726. [DOI] [PubMed] [Google Scholar]
- Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G & Zaccolo M (2011) cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res 108, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SF (1999) The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes. Circ Res 85, 1101–1111. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Houslay MD, Baillie GS & Kass RS (2009) The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem 284, 9140–9146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V & Movsesian MA (2007) Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. The Journal of biological chemistry 282, 32749–32757. [DOI] [PubMed] [Google Scholar]
- Verde I, Vandecasteele G, Lezoualc’h F & Fischmeister R (1999) Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol 127, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker-Gray R, Stengel F & Gold MG (2017) Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proceedings of the National Academy of Sciences of the United States of America 114, 10414–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier S, Belevych AE, Ruse M, Eckert RL, Zaccolo M, Pozzan T & Harvey RD (2005) Beta-adrenergic and muscarinic receptor induced changes in cAMP activity in adult cardiac myocytes detected using a FRET based biosensor. Am J Physiol Cell Physiol 289, C455–C461. [DOI] [PubMed] [Google Scholar]
- Warrier S, Ramamurthy G, Eckert RL, Nikolaev VO, Lohse MJ & Harvey RD (2007) cAMP microdomains and L-type Ca2+ channel regulation in guinea-pig ventricular myocytes. J Physiol (Lond) 580, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC & Movsesian MA (2002) Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem 277, 38072–38078. [DOI] [PubMed] [Google Scholar]
- Weishaar RE, Kobylarz-Singer DC & Kaplan HR (1987) Subclasses of cyclic AMP phosphodiesterase in cardiac muscle. J Mol Cell Cardiol 19, 1025–1036. [DOI] [PubMed] [Google Scholar]
- Willoughby D & Cooper DM (2007) Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev 87, 965–1010. [DOI] [PubMed] [Google Scholar]
- Winslow RL & Greenstein JL (2011) Cardiac myocytes and local signaling in nanodomains. Progress in biophysics and molecular biology, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Rybin VO, Steinberg SF & Kobilka B (2002) Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem 277, 34280–34286. [DOI] [PubMed] [Google Scholar]
- Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi T, Narumiya S & Ushikubi F (2004) Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation 109, 2462–2468. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ & Lakatta EG (1999) Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res 84, 43–52. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Hohl C, Altschuld R, Jones L, Livingston B, Ziman B, Tantini B & Lakatta EG (1994) Beta 2-adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J Biol Chem 269, 19151–19156. [PubMed] [Google Scholar]
- Xiao RP, Ji X & Lakatta EG (1995) Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol 47, 322–329. [PubMed] [Google Scholar]
- Yang PC, Boras BW, Jeng MT, Docken SS, Lewis TJ, McCulloch AD, Harvey RD & Clancy CE (2016) A computational modeling and simulation approach to investigate mechanisms of subcellular cAMP compartmentation. PLoS Comput Biol 12, e1005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M & Pozzan T (2002) Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295, 1711–1715. [DOI] [PubMed] [Google Scholar]
- Zakharov SI & Harvey RD (1997) Rebound stimulation of the cAMP-regulated Cl- current by acetylcholine in guinea-pig ventricular myocytes. J Physiol (Lond) 499, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HB, Li RC, Xu M, Xu SM, Lai YS, Wu HD, Xie XJ, Gao W, Ye H, Zhang YY, Meng X & Wang SQ (2013) Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res 98, 269–276. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Lu TW, Stolerman LM, Tenner B, Yang JR, Zhang JF, Falcke M, Rangamani P, Taylor SS, Mehta S & Zhang J (2020) Phase Separation of a PKA Regulatory Subunit Controls cAMP Compartmentation and Oncogenic Signaling. Cell 182, 1531–1544.e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I & Zaccolo M (2015) Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ Res 117, 707–719. [DOI] [PubMed] [Google Scholar]