

Abstract
Several mutations in the gene for the mitochondrial single stranded DNA binding protein (SSBP1) have recently been implicated in human disease, but initial reports are insufficient to explain the molecular mechanism of disease, including the possible role of SSBP1 heterotetramers in heterozygous patients. Here we employed molecular simulations to model the dynamics of wild type and 31 variant SSBP1 tetramer systems, including 7 variant homotetramer and 24 representative heterotetramer systems. Our simulations indicate that all variants are stable and most have stronger intermonomer interactions, reduced solvent accessible surface areas, and a net loss of positive surface charge. We then used structural alignments and phosphate binding simulations to predict DNA binding surfaces on SSBP1. Our models suggest that nearly the entire surface of SSBP1, excluding flexible loops and protruding helices, is available for DNA binding, and we observed several potential DNA binding hotspots. Changes to the protein surface in variant SSBP1 tetramers potentially alter anchor points or wrapping paths, rather than abolishing binding altogether. Overall, our findings disqualify tetramer destabilization or gross disruption of DNA binding as mechanisms of disease. Instead, they are consistent with subtle changes to DNA binding, wrapping, or release that cause rare but consequential failures of mtDNA maintenance, which, in turn, are consistent with the late onset of disease in most of the reported SSBP1 cases.
Keywords: mtSSB, single stranded DNA binding protein, mitochondrial DNA replication, molecular modeling, human disease variants
Graphical Abstract
1. Introduction
Mitochondria are the main source of ATP in eukaryotic cells, as well as hubs for numerous metabolic processes and signaling pathways. Each mitochondrion contains at least one mitochondrial genome (mtDNA), a circular molecule encoding essential components of the electron transport chain. While multiple copies of mtDNA provide a natural buffer against perturbations, compromised integrity of mtDNA gives rise to an array of multi-systemic human diseases, characterized by mtDNA depletion or large-scale deletions [1, 2]. One source of compromised mtDNA integrity is mutation of the nuclear-encoded mtDNA replication proteins, including the mitochondrial DNA polymerase (POLG), its accessory subunit (POLG2), and the mitochondrial replicative helicase (Twinkle) [2]. Only recently, with the rising accessibility of whole genome and whole exome sequencing as clinical diagnostic tools, have mutations in the gene encoding the mitochondrial single-stranded DNA binding protein (SSBP1) been identified in patients with mitochondrial disease.
In 2018, 2019, and 2020, five reports described nine point mutations in the SSBP1 gene located at 7q34 and associated with human disease in 78 patients across 16 families (Table 1) [3–7]. Two of the mutations (c.113G>A and c.320G>A) were found in multiple patients from several distinct families, while most of the others were found in a single family and often a single patient. This year, another report described a patient with inherited mutations in both SSBP1 (c.320G>A) and POLG [8].
Table 1. Summary of Human SSBP1 Disease Mutations.
Nine SSBP1 mutations have been identified in mitochondrial disease patients. M1? indicates that this mutation abolishes the primary start codon of SSBP1. The number of reported patients represents the number of individuals with relevant symptoms whose sequencing results showed the relevant mutation. For mtDNA phenotypes marked with an asterisk (*), mtDNA depletion was lower than controls, but the difference was not statistically significant. SLSMD; Single Large-Scale mtDNA Deletion.
| Genotype | Protein Variant | Number Reported | Phenotype | |||
|---|---|---|---|---|---|---|
| Mutation(s) | Zygosity | Families | Patients | Clinical Presentation | mtDNA | |
| c.3G>A m.1555A>G | +/− | M1? | 1 | 15 | Sensorineural hearing loss | Depletion Deletions |
| c.79G>A | +/− | E27K | 1 | 1 | Infantile anemia, bone marrow failure, growth failure, ptosis, ophthalmoplegia, ataxia, retinal dystrophy, sensorineural hearing loss, nephropathy, metabolic strokes, multiple endocrine deficiencies | Deletion (SLSMD) |
| c.113G>A | +/− | R38Q | 4 | 37 | Optic atrophy; sometimes foveopathy | Depletion |
| c.119G>T | +/− | G40V | 1 | 1 | Optic atrophy, retinopathy, nephropathy, sensorineural hearing loss | Depletion |
| c.184A>G | +/− | N62D | 1 | 3 | Optic atrophy, retinopathy | Depletion* |
| c.320G>A | +/− | R107Q | 5 | 18 | Optic atrophy; sometimes foveopathy, retinopathy, nephropathy, nystagmus, sensorineural hearing loss | Depletion |
| c.331G>C | +/− | E111Q | 1 | 1 | Optic atrophy | Depletion* |
| c.394A>G | −/− | I132V | 1 | 1 | Retinal dystrophy, deafness, cardiomyopathy, ataxia, nephropathy, growth retardation | Depletion |
| c.422G>A | +/− | S141N | 1 | 1 | Optic atrophy | N/R |
Nearly all of these patients suffer mainly from optic atrophy, although many also experience a variety of additional symptoms (Table 1). Neurological dysfunction relating to the eyes (retinopathy, foveopathy, ptosis, ophthalmoplegia) and ears (sensorineural hearing loss) is common among these patients. In a handful of patients, the multi-system disorders characteristic of mtDNA replication diseases are apparent; kidney disease (nephropathy), musculoskeletal issues (ataxia, growth failure, cardiomyopathy), metabolic strokes, infantile anemia, and other symptoms were reported in some patients. mtDNA depletion was reported in most of the patients, although both mtDNA deletions and depletion were reported in c.3G>A/m.1555A>G patients, only a single large-scale mtDNA deletion (SLSMD) in the c.79G>A patient, and no mtDNA phenotype for the c.422G>A patient.
All but one of the reported mutations (c.394A>G) are heterozygous in patients, and most are missense mutations, resulting in amino acid substitutions. The lone exception, c.3G>A, abolishes the start codon of SSBP1 and was reported to reduce protein translation. This mutation was found in concert with a point mutation in the mtDNA (m.1555A>G) in a patient with mtDNA depletion and multiple mtDNA deletions [3]. The majority of the other variants resulted in charge inversions in amino acid side chains in the SSBP1 polypeptide (Table 1). The residues affected by these disease mutations are well-conserved among mammals, and some are also conserved through higher eukaryotes including frogs and flies (Figure 1A).
Figure 1. Introduction to SSBP1 Protein Variants and SSBP1 Heterotetramers Tested.
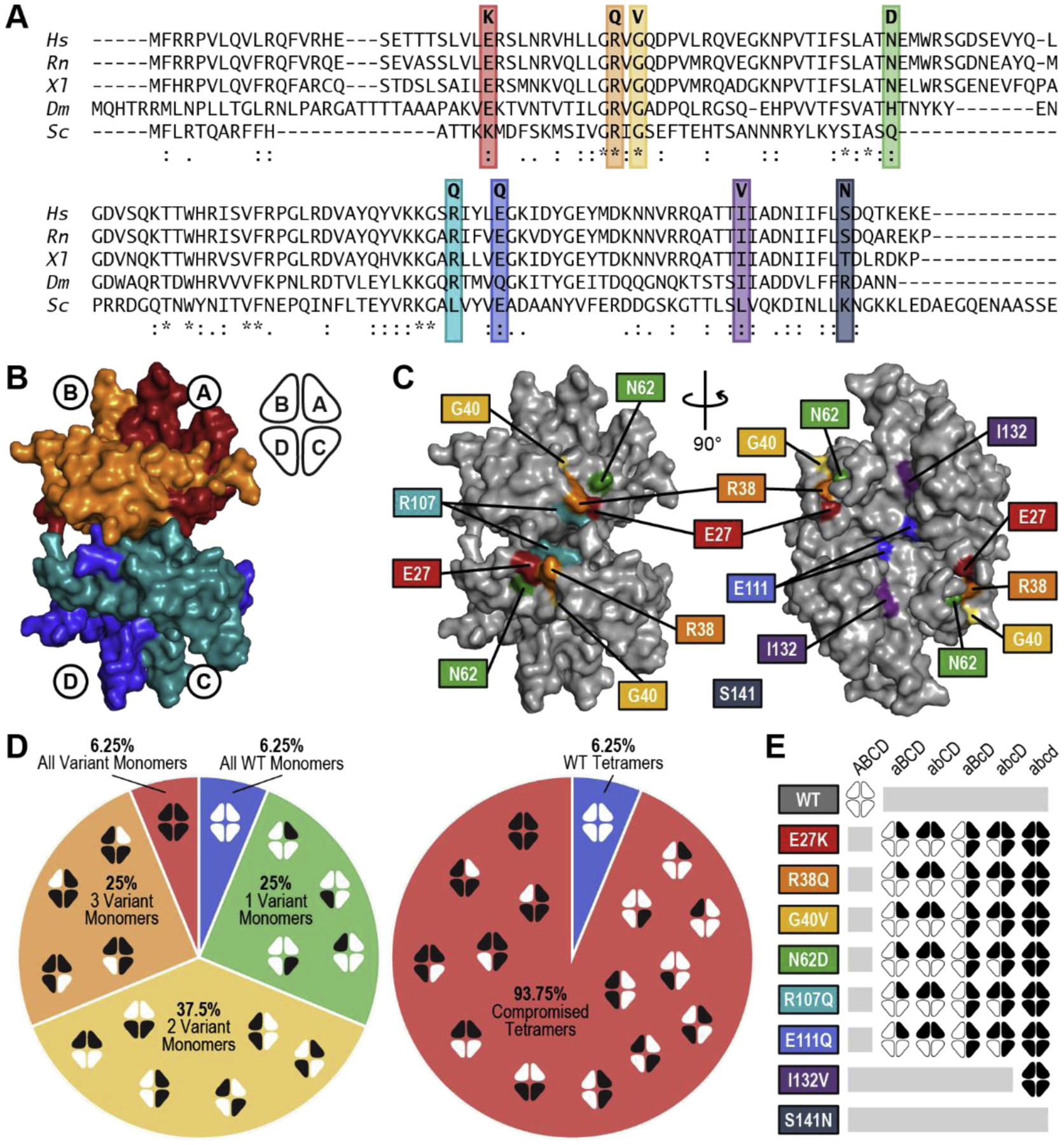
(A) Conservation of variant residues in SSBP1 from Homo sapiens (Hs) through Rattus norvegicus (Rn), Xenopus laevis (Xl), Drosophila melanogaster (Dm), and Saccharomyces cerevisiae (Sc). (*) perfect alignment; (:) strong similarity; (.) weak similarity. (B) Surface rendering of the SSBP1 tetramer (based on PBDID: 3ULL) with each monomer indicated, along with a simplified representation that will appear throughout this report. (C) Locations of the SSBP1 residues effected by disease mutations. Coordinates of S141 were not present in the reference crystal structure [9]. (D) Breakdown of all possible combinations of WT and variant monomers in SSBP1 tetramers in heterozygous patients, including ratios of all WT : single variant : double variant : triple variant : all variant tetramer systems (left) and WT : compromised tetramer systems (right). White indicates WT monomers; black indicates variant monomers. (E) Representation of 32 tetramer systems modeled in molecular dynamics simulations.
SSBP1 forms a stable tetramer composed of two dimers bound together across a large interface (Figure 1B) [9]. Several of the residues with human disease variants (E27, R38, G40, N62, and R107) reside on a common surface on each side of the tetramer (Figure 1C), at the monomer-monomer and dimer-dimer interface. Residue E27 of each monomer is in a position to form a salt bridge with R38 and/or R107 on the neighboring monomer in the top and bottom dimers. Additionally, these surface residues are clustered in a positively-charged patch that was identified as a potential DNA binding site in the original SSBP1 crystal structure [4, 5, 9]. Two more disease-associated residues (E111 and I132) are buried in the dimer-dimer interface, while S141 resides in a flexible region that was not observed in the crystal structure.
A few possible molecular mechanisms of disease are possible for these newly discovered mutations: destabilization of SSBP1 at the tetramer, dimer, or monomer level, perturbation of ssDNA binding or release, or interference with protein-protein interactions (though no direct protein interactions have been reported for SSBP1). The recent SSBP1 studies applied multiple approaches to investigating molecular mechanisms of disease.
Wild type SSBP1 forms a very stable, tightly bound tetramer in humans, unlike some yeast homologs which cycle between dimeric and tetrameric forms [10, 11]. Destabilization of SSBP1 tetramers could drive the molecular phenotypes observed in SSBP1 disease patients. Size exclusion chromatography of recombinant E27K and I132V SSBP1 ruled out gross destabilization or dissolution into monomers or dimers for either variant tetramer [5]. Differential scanning fluorimetry, which measures protein unfolding as temperature increases, revealed a slight downward shift in melting temperature for both E27K and I132V SSBP1 compared to WT, though neither shift would cause unfolding at biologically relevant temperatures [5]. Instead, this shift suggests subtle changes to protein or complex stability. Electrophoretic approaches for other disease variants point in the same direction. Mild denaturing SDS/PAGE and western blotting of R38Q SSBP1 patient samples showed a shift toward monomers relative to WT; crosslinking followed by SDS/PAGE and western blotting of G40V, R107Q, and I132V patient mitochondrial lysates revealed a similar shift in the ratio of tetramers to lower order species [6, 7]. While both sets of experiments were carried out under denaturing, not biological, conditions, they signal changes in tetramer structure or stability.
Because no crystal structure of SSBP1 bound to ssDNA has been determined and no specific DNA-binding regions have been identified biochemically, the roles of these disease variant residues in DNA binding could not easily be predicted. Four of the disease variants were assayed for DNA binding. Recombinant E27K SSBP1 showed a biologically insignificant reduction in the Kd of DNA binding as measured by fluorescence anisotropy, but also showed an enigmatic change in the amplitude of anisotropy that suggests some change to the binding mode [5]. SSBP1 from G40V, R107Q, and I132V patient mitochondrial lysates successfully bound biotinylated ssDNA in pull-down assays [6]. Although these results were qualitative, they exclude gross disruption of DNA binding dynamics as a disease mechanism. Any change to DNA binding affinity or mode must be subtle.
It is important to understand how these recently described SSBP1 disease mutations compromise mtDNA integrity. The initial investigations of these mutations have not resolved the starting hypotheses – that perturbations to SSBP1 stability or ssDNA binding are likely to blame – except to emphasize the subtlety of these perturbations. No single approach to mechanistic questions was used for more than three disease variants, so no unified view of a potential shared mechanism has emerged. On top of that, the fact of heterozygosity in most of the patients remains unaddressed for all but one of the mutations (c.79G>A; E27K).
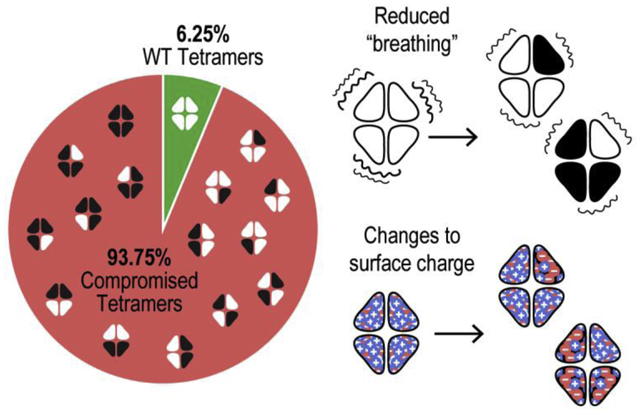
For all variants except I132V, patients would be expected to harbor mixtures of WT and variant homotetramers as well as various heterotetramers in their mitochondria (Figure 1D). We calculated the probable frequency of different tetramer systems in a mitochondrial population based on several assumptions: (1) that WT and variant monomers are equally abundant in the mitochondrial matrix and equally capable of participating in tetramer formation, (2) that monomers assemble randomly into tetramers, (3) that all tetramer systems are similarly stable, and (4) that different orientations of variant monomers qualify as distinct tetramer systems. This final assumption is rooted in our previous findings that WT and E27K SSBP1 homotetramers are not symmetrical in solution simulations [5]. Given these conditions, WT homotetramers could make up as little as 6.25% SSBP1 tetramers in patient mitochondria; variant homotetramers could make up an additional 6.25%. The remaining 87.5% of the SSBP1 pool would be a mixture of various heterotetramers.
All biochemical assays with recombinant SSBP1 disease variants to date have compared only WT homotetramers to variant homotetramers. Reported assays with patient samples cannot differentiate WT or variant homotetramers from heterotetramers. While meticulous strategies might be used to purify recombinant heterotetramers containing one, two, or three variant monomers, the orientation of those monomers would be impossible to determine. And yet, it is entirely possible that heterotetramers, making up as much as 87.5% of the SSBP1 in patient mitochondria, behave differently from WT or variant homotetramers and could therefore be the driving force behind disease.
In order to address this knowledge gap and provide a common lens through which to compare all of the reported disease variants, we have expanded our previous molecular dynamics simulations for E27K SSBP1 [5] to include all but one of the reported SSBP1 variants. In addition to the WT and variant homotetramers, we chose four representative heterotetramer systems to model for six of the SSBP1 disease variants (Figure 1E). We modeled only the homotetramer system for the disease variant I132V, as that mutation is homozygous in the patient. Because the coordinates of residue S141 were not reliably determined in the crystal structure, we were unable to model the S141N disease variant.
After carrying out a series of molecular dynamics simulations with our SSBP1 model systems, we discovered a few unifying trends across all of the variant systems, relating to system stability, intramolecular interactions, behavior of flexible regions, and changes to the tetramer surface. We also used structural alignments and phosphate binding simulations to predict DNA binding surfaces on SSBP1. Our models suggest that nearly the entire surface of SSBP1, excluding flexible loops and helices, is available for DNA binding, and we observed several binding hotspots that may be functionally important. Collectively, our results point to subtle changes in human disease variants of SSBP1 that may, for example, result in occasional failures of ssDNA wrapping or unwrapping. Such failures could contribute to mtDNA depletion and deletions, which in turn underlie patient outcomes.
2. Material and Methods
2.1. Simulating solvated structures of variant SSPB1 tetramers
We have previously reported the evaluation of solution structures for the WT and E27K variant SSBP1 tetramers using molecular dynamics simulations [5]. In that study, the initial structure of SSBP1 for simulations was taken from the homodimeric X-ray crystal structure from PDB ID 3ULL [9]. Due to poor electron densities, positions of the peptide segments of 1–25 (N-terminus), 68–76, and 141–148 (C-terminus) from chain A and 1–25, 68–80, and 142–146 from chain B were not reported in the available X-ray crystal structure. After remodeling missing loop segments 68–76 (in chain A) and 68–80 (in chain B), the reconstructed peptide segments 26–140 for monomer A and 26–141 for monomer B were used in models. The initial WT tetramer structure was created by aligning two dimer units to the tetramer in the structure of E. coli SSB (PDB ID: 1EYG). Additional variants of SSPB1, namely R38Q, G40V, N62D, R107Q, E111Q, and I132V, were introduced in the present study using the program Coot-0.8 [12]. Five representative heterogeneous tetramer systems with a single variant monomer (aBCD, where variant monomers are represented by the lower-case letters), two variant monomers (abCD and aBcD), three variant monomers (abcD), and four variant monomers (abcd) were constructed. After introducing the protons using Molprobity [13], counter ions were added, each system was solvated in a box of water, and each solvent box was selected so that box boundaries were at least 20 Å from the closest protein atom (total atoms in each system ranging from 184,000 to 184,800). Prior to equilibration, following the same molecular dynamics protocol that has been reported earlier [5], all systems were subjected to 1) 100-ps belly dynamics runs with fixed peptide, 2) minimization, 3) low temperature constant pressure dynamics with fixed protein to assure a reasonable starting density, 4) minimization, 5) step-wise slow heating molecular dynamics at constant volume, and 6) constant volume unconstrained molecular dynamics for 20 ns. All final unconstrained trajectories were calculated at 310 K under 1 atm constant pressure (for 300 ns with time step 1 fs) using the PMEMD module of Amber18 [14] to accommodate long range interactions. All protein parameters were taken from the FF14SB force field. Free energies of interactions were calculated using the MMBPSA module of Amber18. Root mean square deviations and positional fluctuations represented by B-factors were calculated using the CPPTRAJ utility program provided by Amber18. The B-factors were calculated from the last 100 ns segment of each trajectory. Average alpha-carbon deviations were calculated using an in-house program.
2.2. Probing DNA binding sites on the surface of SSBP1
Due to the absence of an available DNA bound structure of SSBP1, we mapped potential phosphate binding locations on the surface of SSBP1 using a dimethyl phosphate probe. The selection of dimethyl phosphate approximately represents a phosphate ion’s local environment within the DNA backbone by having two capping methyl groups representing the attached sugar carbons. The charges on the dimethyl phosphate ion were calculated using the ChelpG scheme of Gaussian-16 [15] at the B3LYP/6–31g(d) level and the regular Amber force field parameters were used for the parameters representing the internal geometry. After randomly replacing 150 water molecules in the vicinity of the SSBP1 tetramer with dimethyl phosphate ions in 4 different equilibrated solution structures, we constructed initial configurations for dynamics. Following the equilibration procedure outlined in the previous section, we performed equilibration MD simulations for 30 ns. Subsequently, we extended constant temperature (at 310 K) and constant pressure (1 atm) trajectory calculations for 500 ns as the production run for analysis. Configurations selected at each nanosecond were used to obtain information on the phosphate binding on the surface of SSBP1. The diffusion assisted dynamics brought dimethyl phosphate ions on to the surface of SSBP1 and most dimethyl phosphates did not stay bound for an extended period. If dimethyl phosphate ions stayed within 4 Å of any phosphate heavy atom on SSBP1 for 50 frames (equivalent to 50 ns), they were selected as bound. After combining the results of all four simulations (totaling 2 μs of dynamics), we binned the positions of the phosphates on the surface of SSBP1 using 1 Å3 volume elements. Any bin that contained over 9 molecules from the combined simulations was considered to be a high frequency occupation site. Note that only one phosphate can occupy a given bin at a time, but due to the dynamic nature of the binding process they associate and dissociate from the SSBP1 surface rapidly.
3. Results
3.1. Modeling and molecular dynamics simulations of SSBP1 tetramers
In order to perform molecular dynamics simulations, we needed to model WT and each variant SSBP1 tetramer system. The original crystal structure of SSBP1 (PDB ID: 3ULL), as well as a recent additional structure (PDB ID: 6RUP), had only a dimer in the asymmetric unit. These structures also had unresolved residues in each monomer, where the coordinates of a long, flexible loop could not be solved. We first modeled these loops onto the existing monomers from 3ULL, and then we used the tetrameric crystal structure of E. coli SSB (PDB ID: 1EYG) as a template to model a WT SSBP1 tetramer. In this way, Monomers C and D in our models are clones of Monomers A and B (Figure 1B). Finally, we modeled in variant residues in the appropriate monomers for each system pictured in Figure 1E. We chose to test four variant heterotetramer systems (aBCD, abCD, aBcD, and abcD) as a representative subset of all the possibilities presented in Figure 1D, along with WT and each variant homotetramer (ABCD and abcd). Throughout this report, capital monomer letters refer to WT monomers in any system, while lowercase monomer letters refer to variant monomers.
With these 32 starting models in hand, we turned to molecular dynamics simulations. After an initial ~30 ns equilibration period, each SSBP1 system was subjected to a 300 ns molecular dynamics simulation in a solvent environment at 310 K (approximately room temperature). With a GTX1080TI GPU processor, these simulations required several thousand computing hours. The data from these molecular dynamics simulations provide a great deal of information to help us predict how the modeled tetramer systems behave in solution.
3.2. All SSBP1 tetramer systems tested are stable
First, we checked whether our SSBP1 systems were stable during our simulations. The starting systems for molecular dynamics simulations are based on X-ray crystal structures of proteins, which are not necessarily representative of how those proteins behave in the solution environment of cells. Crystallized proteins are perforce rigid and have limited solvation due to the crystal environment; relatively few solvating water molecules are present around the protein in the crystal. When model systems based on crystal structures are properly solvated and permitted to breathe in simulations, root-mean-square deviations (RMSDs) of atomic positions tell us how far each protein system flexed or expanded from its starting position over the course of the simulation. RMSDs for a stable protein system in a simulation are expected to initially rise, as the model adapts to a simulated solution environment, and then plateau as a dynamic equilibrium is reached (Figure 2A, green line). On the other hand, if the modeled protein system is unstable, RMSDs will continue to rise as the protein unfolds or subunits disassemble (Figure 2A, red line).
Figure 2. Root-Mean-Square Deviations.
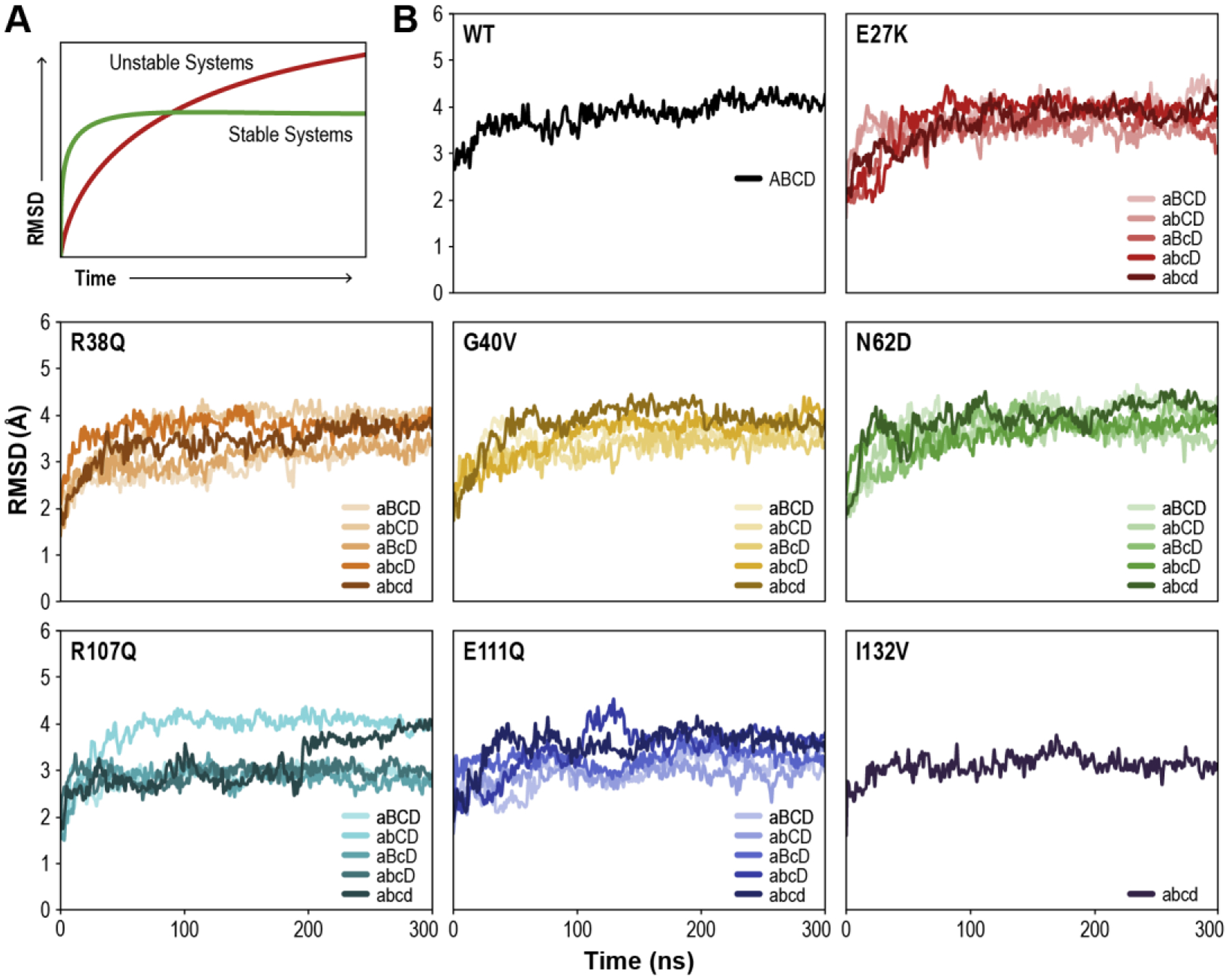
(A) Cartoon of expected root-mean-square-deviation (RMSD) values over the course of a simulation for stable (green) and unstable (red) protein systems. (B) Calculated RMSD values of all tested SSBP1 tetramer systems over the course of the simulations.
Per standard practice, we excluded the modeled loops (residues 67–79) in our SSBP1 systems from RMSD calculations. Consistent with previous in vitro studies [16–18], RMSDs confirm that the WT system was quite stable in our simulations (Figure 2B). WT system RMSDs plateaued at about 4 Å, as expected for a tetrameric system of this size assembled from a dimeric crystal structure.
All variant systems tested were also stable during our simulations (Figure 2B); none showed the characteristic steady rise in RMSDs expected for an unstable system. The variant system RMSDs plateaued in a range of values between 3 and 4 Å. This observed variation was likely due to extreme mobility of some loops and trapping of others into local minimum structures, as observed in the R107Q abcd system and some E111Q systems. Overall, our RMSD results predict that all tested systems are stable in solution.
3.3. Most SSBP1 tetramer systems tested show tighter intermonomer interactions than WT
Having established the overall stability of our simulated SSBP1 systems, we evaluated the strength of interactions among various monomers within each tetramer by comparing the calculated binding free energies of the systems. More negative binding free energy values indicate stronger intramolecular interactions, while less negative or slightly positive free energy values reflect weaker interactions. We first considered free energy values between each pair of monomers in the WT tetramer. We observed the strongest interactions between monomers within the dimers (AB and CD), followed by intradimer interactions between Monomers A and C and Monomers B and D (Figure 3A). Slightly positive free energy values for AD and BC monomer interactions indicated mild repulsion. Additionally, the total free energy of the tetramer system (ABCD) was quite negative, indicating a tightly bound tetramer and consistent with in vitro studies of SSBP1 [16–18].
Figure 3. Binding Free Energy.
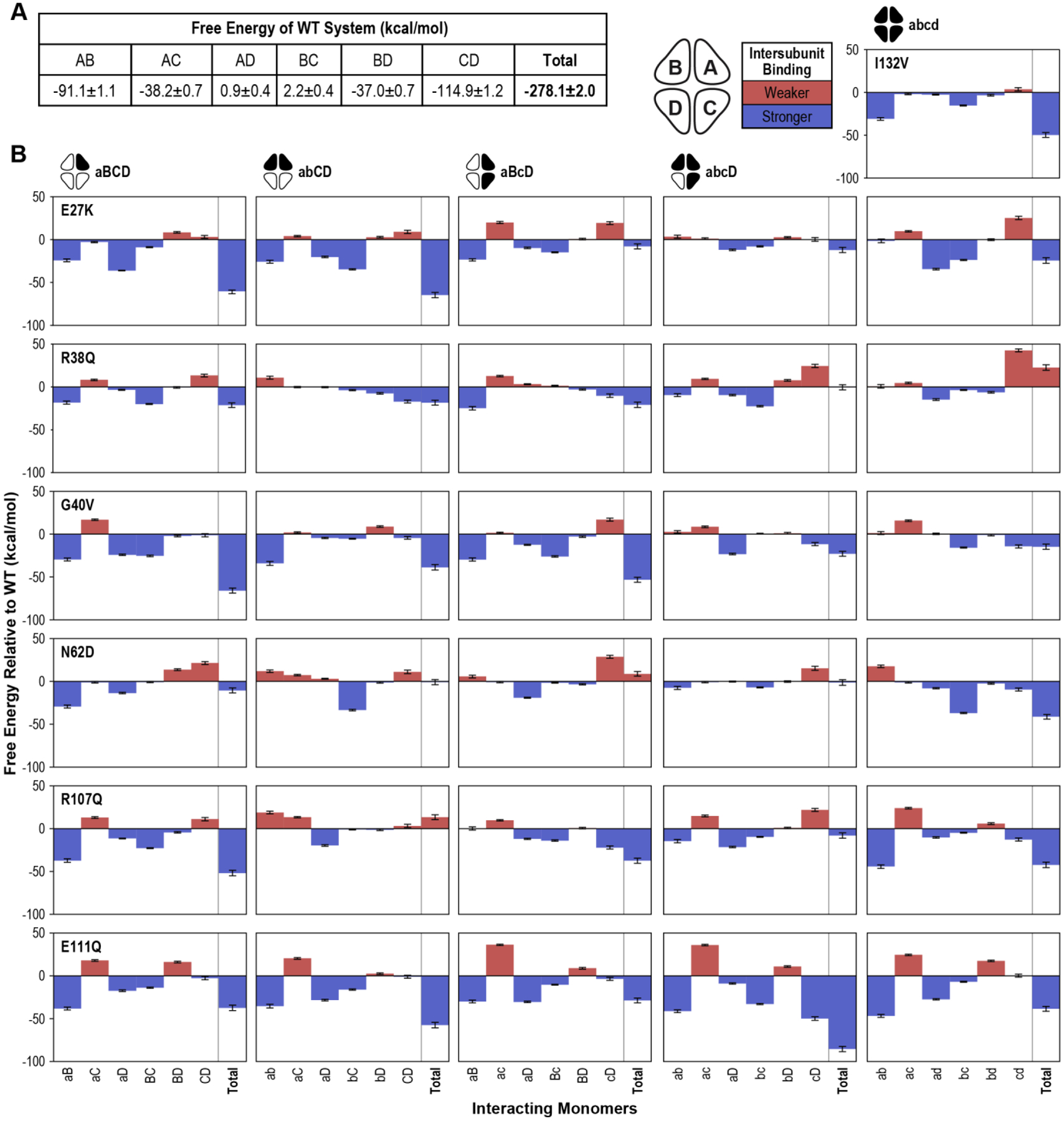
(A) Averaged interaction free energy values for the WT tetramer system were calculated for the final 100 ns of the simulation. Free energy values of each monomer pair as well as the total system are shown. (B) The average free energy values for each variant tetramer system with the corresponding WT value subtracted are plotted. Positive (red) bars indicate weaker binding than WT; negative (blue) bars indicate stronger binding. Error values and bars represent standard error of the mean.
Interestingly, the majority of variant systems had larger (more negative) total free energy values than WT; that is, most variant systems form a tighter tetramer than WT (Figure 3B). While most of the variant systems revealed the same pattern of monomer-monomer interaction strength as WT (strongest AB and CD > AC and BD > weakest AD and BC), several systems bucked this pattern, most often by tightening AD or BC interactions, occasionally at the expense of AC or BD interactions, and sometimes both (Supplemental Table 1). All variants except R38Q and R107Q had at least one system in which this intramolecular binding hierarchy was perturbed. For E111Q in particular, 4 of 5 systems were affected in these comparisons, consistent with E111Q showing significant negative/blue shifts for total free energy in all five tested systems. This is perhaps related to E111’s location at the dimer-dimer interface of SSBP1, although the I132V variant, the only other residue buried in this interface, shows a similar binding strength pattern to WT.
3.4. SSBP1 tetramers are asymmetrical in solution, and certain regions of the protein are highly flexible
We next turned our attention to data which allow us to assess dynamic characteristics of the SSBP1 systems during the simulations. Crystallographic B-factors are used to estimate thermal fluctuations of atomic positions, and similar fluctuations can be calculated from the ensemble of structures collected during molecular dynamic simulations. These B-factors reflect the mobility of regions in the system: residues in flexible loops tend to have high B-factors, while those in the structural core tend to have low B-factors. In addition, regions involved in domain-domain interactions (or intermonomer interactions, in the case of SSBP1) can also have low B-factors.
We calculated normalized B-factors for the WT and variant systems we tested. These results for the WT system revealed four flexible regions, where residues had high B-factors, in each monomer: side loops (roughly, residues 48 through 54), modeled loops (~aa 67–80), mobile helices (~aa 94–103), and top (A/B) or bottom (C/D) loops (~aa 119–130) (Figure 4A and B). The same four regions consistently stood out as flexible, with above-baseline B-factors, in most monomers of the variant systems (Supplemental Table 1), and no additional flexible features were apparent. Across the board, the regions labeled as mobile helices had higher B-factors than the baseline (rigid) regions in most monomers, but their B-factors were much smaller than those of the highly flexible loops.
Figure 4. Normalized B-Factors.
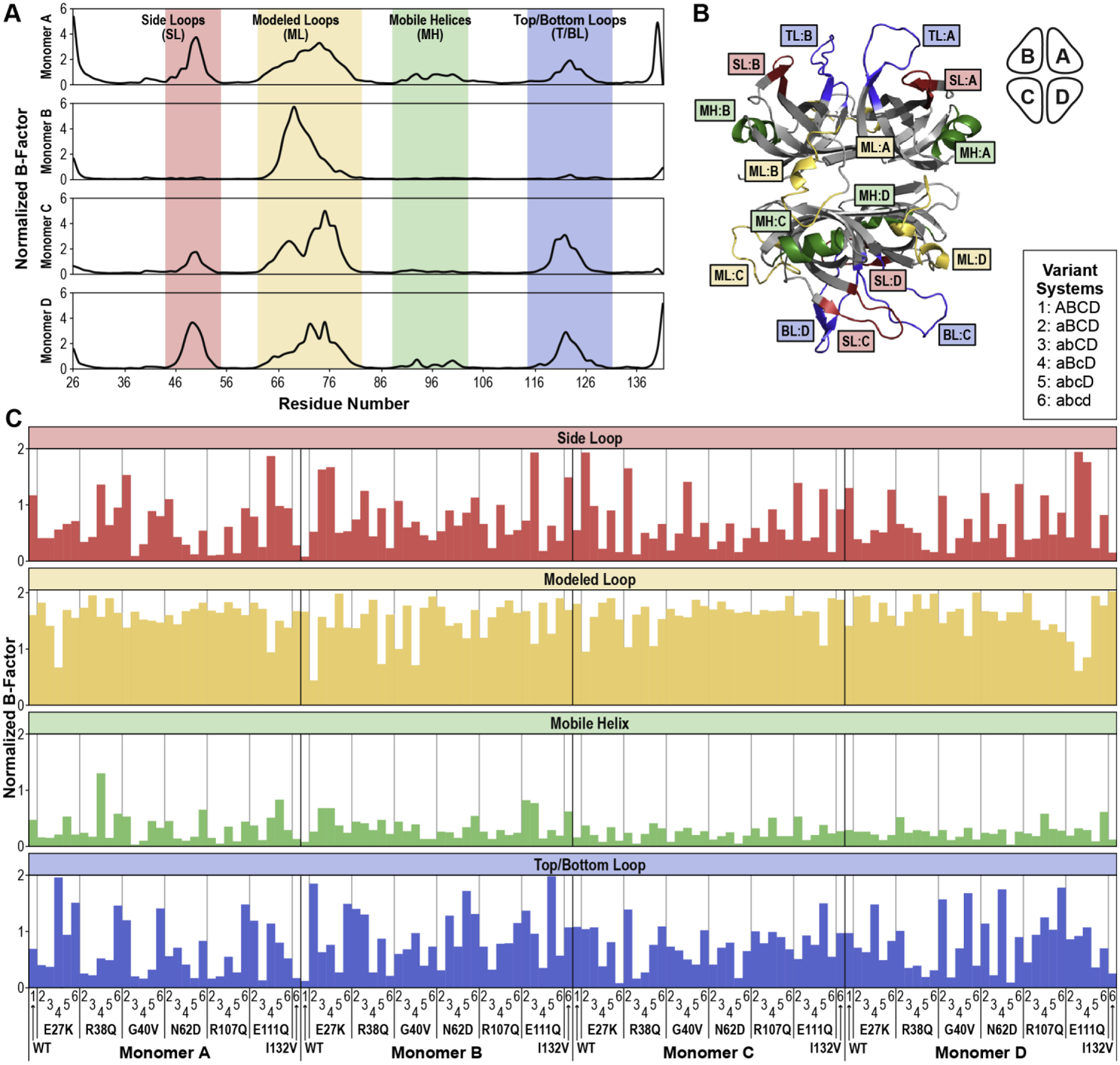
(A) Normalized B-factors for each residue of each monomer in the WT SSBP1 tetramer are plotted, with flexible regions indicated (side loops, modeled loops, mobile helices, and top/bottom loops). (B) The flexible regions identified in A are labeled on a ribbon diagram snapshot from the WT tetramer simulation. (C) Normalized B-factors averaged over all residues in a given flexible region (loop or helix) are plotted for all tested systems.
Notably, not all monomers in a tetramer, in WT or variant systems, showed elevated B-factors in all of the identified flexible regions (Supplemental Table 1). In the WT system, Monomer B had only one flexible region with high B-factors, its modeled loop, and the other monomers showed variation in B-factors across their flexible regions. This is consistent with our previous observations that the apparent symmetry of the SSBP1 crystal structure does not persist in solution simulations [5]. We observed comparable variability in B-factors, indicative of asymmetry, in all of the SSBP1 variant systems (Supplemental Table 1).
In order to compare these flexible regions in the variant systems more directly, we calculated the average B-factors of each flexible region in each monomer of every system (Figure 4C, Supplemental Table 2). The modeled loop, as expected, displayed the highest average B-factors in most systems, whereas the mobile helix showed high average B-factors only very rarely. Average B-factors for the side and top/bottom loops were much more variable across monomers and systems.
In most proteins linked to human disease, B-factors around variant residues are often characteristically altered compared to WT. In the case of our SSBP1 systems, the variant residues all reside in rigid regions, at least five residues away from the flexible regions that we identified, and do not directly interact with the flexible loops. We did not observe B-factor changes relative to WT near the variant residues. Rather than highlighting differences between variant and WT SSBP1 systems, these B-factor results describe common characteristics of all tested systems: four variable flexible regions, rigid structural regions, and intrinsic asymmetry in solution simulations.
For a more detailed characterization of molecular mobility of SSBP1 tetramers in our simulations, we turned to Cα deviations. Cα deviations compare the average distance between the alpha carbons of every residue pair at the end of the simulation to the corresponding distance at the beginning, usually plotted as heatmaps. These heatmaps give us a sense of how residues, or clusters of residues, are moving relative to the rest of the tetramer.
We calculated average Cα deviations for each SSBP1 system over the final 100 ns of each simulation. Examination of the WT system heatmap revealed patterns that broadly extend to the variant system heatmaps as well (Figure 5, Supplemental Figure 1). Note that these heatmaps are symmetrical across the diagonal, as the residue relationships are reciprocal.
Figure 5. Cα Deviations of the WT SSBP1 System.
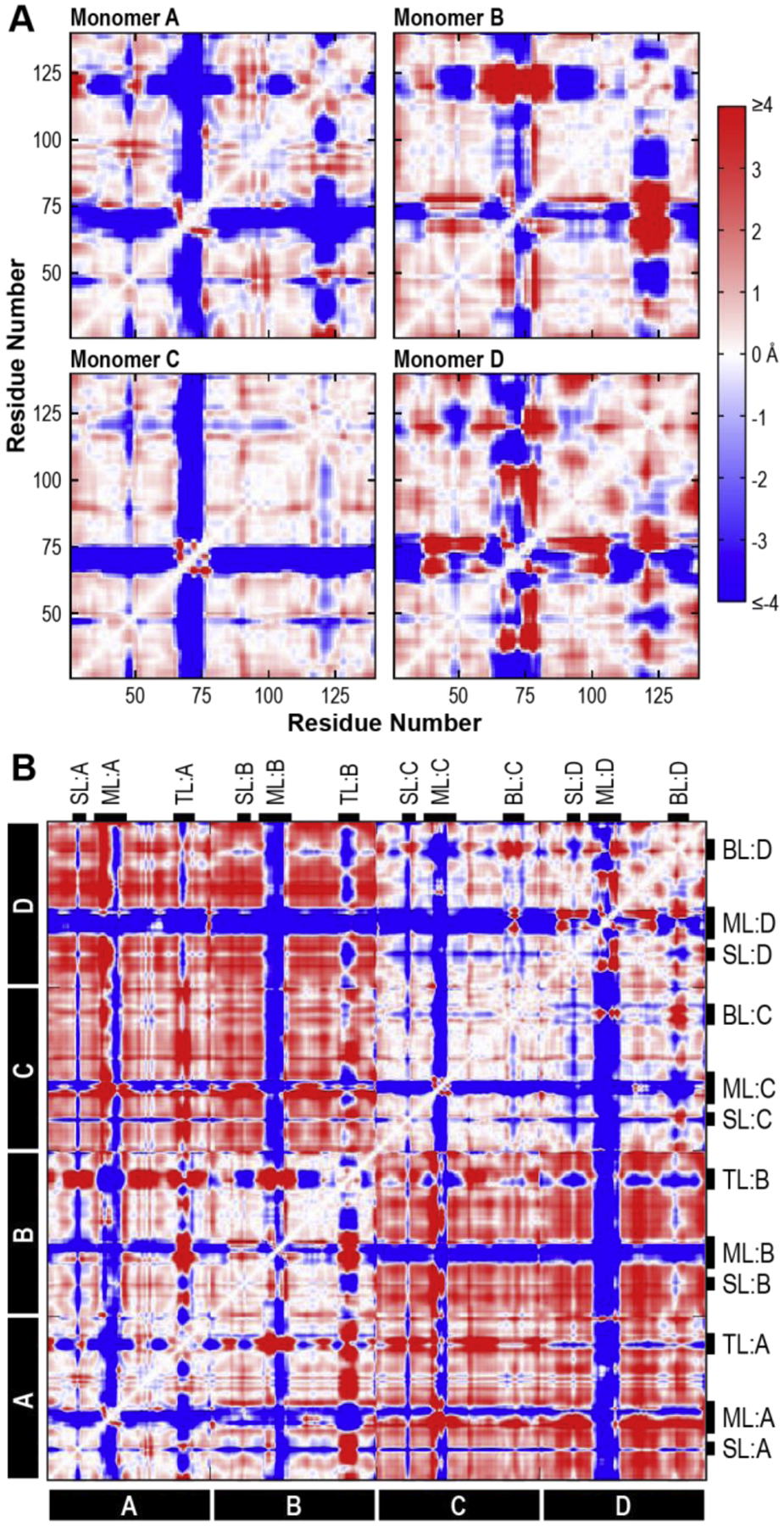
Average Cα deviations in the WT tetramer system were calculated from the final 100 ns of the WT simulation. (A) Cα deviations between residues within monomers A, B, C, and D of the WT system are plotted in heatmaps, where negative (blue) Cα deviations represent the residues moving closer over time, positive (red) Cα deviations indicate residues moving apart, and Cα deviations near zero (white) indicate no change relative to starting distances. (B) Cα deviations for every residue pair in the WT tetramer system are plotted with the same color scale as in A. Flexible features are marked with black tabs. SL, side loop; ML, modeled loop; TL, top loop; BL, bottom loop.
Focusing on individual monomers of the WT system (Figure 5A), we observed that the majority of each heatmap was white or near-white, indicating residues that deviated very little from their starting positions. These residues are most likely responsible for the structural integrity of the peptide fold, a prediction that is supported by visual examination of the SSBP1 structure: these residues participate in the β barrel cores of each monomer.
Prominent blue and red stripes represented regions of each monomer that were mobile during the simulation. These stripes coincided with the flexible loops identified by high B-factors and add detail to our understanding of their mobility during the simulation.
There were obvious and predictable similarities between the monomers in the original crystal structure and those modeled from them: the overall patterns in Monomer C are similar to those in Monomer A, and those in Monomer D are similar to those in Monomer B.
There were also clear differences between the loops of Monomers A and B (and C and D), emphasizing the lack of symmetry between monomers in solution simulations. In Monomer A, the stripe corresponding to the modeled loop (~aa 64–77) was mostly blue, indicating that the entire loop moved in one unified direction (closer to the rest of the monomer). The same stripe in Monomer B was slightly wider (~aa 62–80), suggesting that additional residues participated in loop movements, and had both positive (red) and negative (blue) segments, where different portions of the loop moved farther from or closer to the rest of the monomer. The Cα deviations of the top and bottom loops (~aa 114–129) of all four WT monomers were quite variable. This lack of a coherent pattern suggests that these loops moved independently in their local environments.
The full WT heatmap (Figure 5B) showed that the movements of Monomers A and B were synchronized (bottom left quadrant, white background), as were the movements of Monomers C and D (top right quadrant, white background). The structural cores of each dimer maintained their relative positions during the simulation. On the other hand, dimer AB moved away from the dimer CD (top left and bottom right quadrants, increased red background).
We observed variations from WT in many of the variant system heatmaps (Supplemental Figure 1). Some systems lost the clear quadrant pattern seen in WT, suggesting that the dimer-dimer expansion observed in the WT is not universal among all SSBP1 systems. However, there were no major correlations between variant or number of variant monomers and changes in Cα deviation patterns. It is important not to overinterpret the minutiae of Cα deviations, as they represent only a short snapshot of a dynamic system. Any individual loop may explore more conformations over longer time periods than we are able to model. However, these results clearly reinforce our observation that SSBP1 tetramers, WT or variant, are not symmetrical in solution simulations. Cα deviations revealed monomer-to-monomer asymmetry in both flexible loops and interdimer interactions.
3.5. Variant monomers alter the solvent accessible surfaces of SSBP1 tetramers
Because SSBP1 is a DNA-binding protein, characterization of its solvent accessible surface, where DNA interacts, is essential to understanding its function. To assess whether our modeled human disease variant systems experienced changes to their solvent accessible surfaces, we examined several metrics relating to solvent accessible area (SAA). For all variant SSBP1 systems tested, total SAA was reduced compared to WT (Figure 6, top row), meaning that the tetramer surfaces shrank in the variant systems, by as little as 0.3% (for the E27K abcD system) and as much as 12% (for the E111Q abcD system). There was no apparent pattern in the magnitude of the reduction relative to particular variants or numbers or orientations of variant monomers in a heterotetramer.
Figure 6. Solvent Accessible Area.
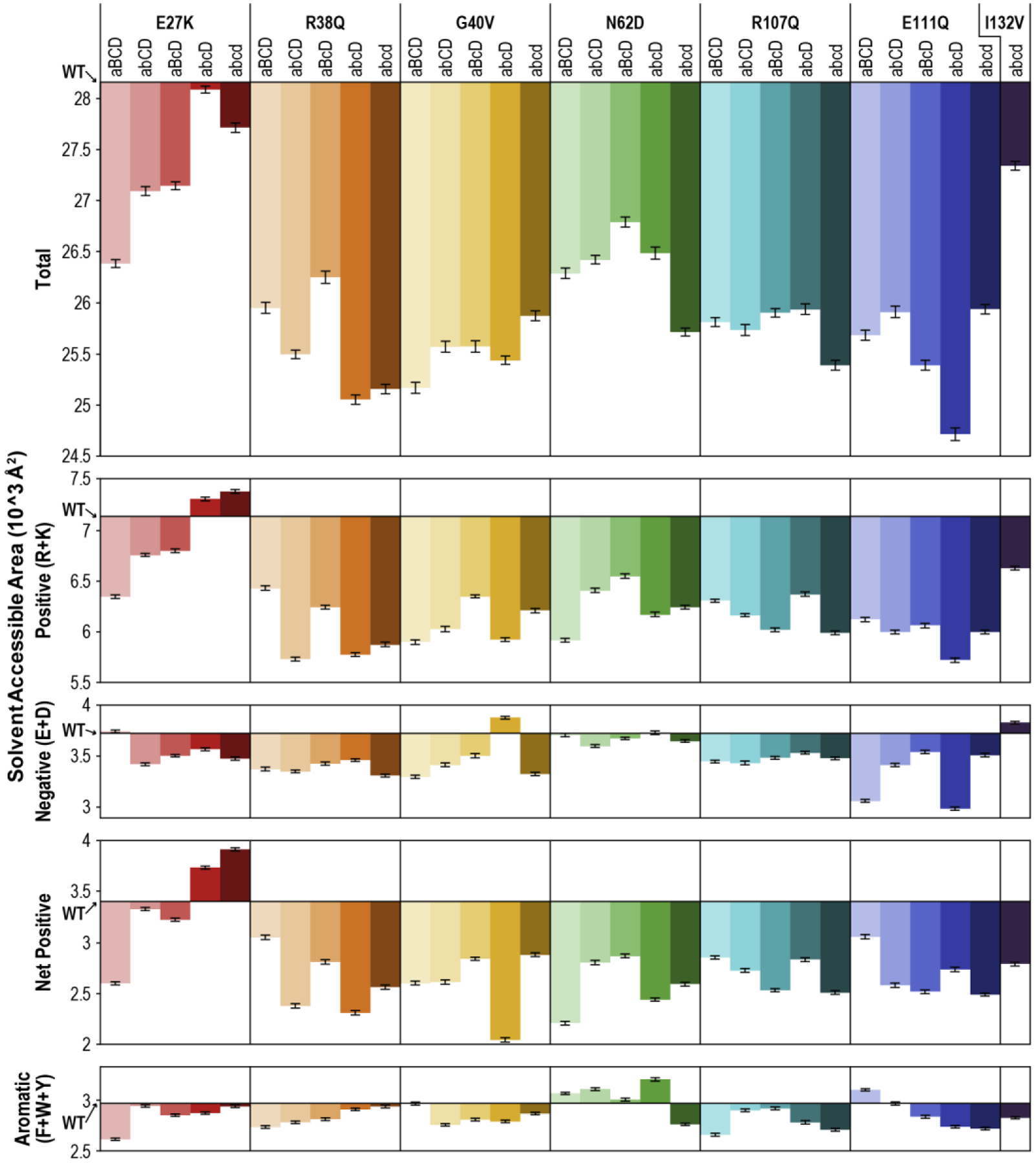
Total solvent accessible area (SAA), positive (basic) SAA, negative (acidic) SAA, net positive (net basic) SAA, and aromatic SAA were averaged over the final 100 ns of the simulation for each SSBP1 variant system. X-axes are set at the respective WT value for each plot. Positive SAA was calculated as the sum of SAA contributed by arginine (R) and lysine (K) residues, negative SAA as that contributed by aspartate (D) and glutamate (E) residues, and net positive SAA as the difference between positive and negative SAA. Aromatic SAA was calculated as the sum of SAA contributed by phenylalanine (F), tryptophan (W), and tyrosine (Y). Error bars represent standard error of the mean, propagated through subtracting WT from each variant value. WT SAA values (in 10^3 Å2) are as follows: Total, 28.16; Positive, 7.13; Negative, 3.72; Net Positive, 3.40; Aromatic, 2.97.
SSBP1 does not bind DNA in a sequence specific manner, so binding likely relies on interactions between the protein surface and the negatively-charged phosphate backbone of the DNA. The presence of basic residues will lend the protein surface positive charge and promote DNA binding, while the presence of acidic residues on the surface may repel or divert DNA from certain areas.
Therefore, we calculated positive SAA (contributed by arginine or lysine residues) and negative SAA (contributed by aspartate and glutamate residues) of each system tested (Figure 6, second and third rows). The WT system had a very charged surface overall: a little over 25% of the total SAA in the WT system is positively charged, while an additional 13% is negatively charged. In nearly all of the variant systems, positive SAA was reduced. The two exceptions were in the only variant, E27K, which results in a side chain charge inversion from negative to positive, and even then, only in the two systems with at least three variant monomers (abcD and abcd), which netted an extra 6 and 8 positive surface charges, respectively. Most of the systems also showed a small reduction in negative SAA. On the whole, the net positive SAA of all but two variant systems (again, E27K abcD and abcd) was smaller than WT (Figure 6, fourth row). This has potential implications for the DNA-binding function of these SSBP1 variant tetramers.
Lastly, we observed that SSBP1 has a surprising number of surface aromatic residues which, because they are hydrophobic, are more often found in protein cores. Surface aromatic residues have been implicated in DNA binding by other DNA binding proteins, either as guides for DNA wrapping or stabilizers for DNA bases [19–23]. We therefore compared the aromatic SAA (contributed by phenylalanine, tryptophan, and tyrosine residues) of our SSBP1 variant systems to that of WT SSBP1. However, we found only minor changes to aromatic SAA in the variant systems (Figure 6, bottom panel), so this seems unlikely to cause aberrant ssDNA binding.
3.6. Homologous SSB structures and phosphate probing predict potential ssDNA binding surfaces on SSBP1
Given the global stability of the variant SSBP1 systems in our simulations, and the role that local changes in mobility and surface properties may play in DNA binding, we sought to improve existing models of WT SSBP1 DNA binding. In the absence of any direct experimental information or DNA-bound SSBP1 structures, we first examined what is known about SSB DNA binding from the few existing crystallographic studies of various DNA-bound SSB homologs. The majority of homologs that have been successfully crystallized with DNA are bacterial; the lone exception is SSB from Plasmodium falciparum, one of the five unicellular parasites that cause malaria in humans.
There are well-documented differences between human mitochondrial SSBP1 (HsmtSSBP1) and bacterial SSBs. Long C-terminal tails are common among bacterial SSBs but missing from HsmtSSBP1 (and from bacterial SSB crystal structures). In bacteria, these tails facilitate cooperative ssDNA binding by SSB as well as protein-protein interactions [24]. HsmtSSBP1 does not appear to bind ssDNA cooperatively [18, 25, 26] and little is known about its direct interactions with other proteins. Bacterial SSBs also switch between static and sliding ssDNA binding modes, which have not been observed in HsmtSSBP1 studies [24, 27–29]. Sequence homology is low among HsmtSSBP1 and these bacterial and P. falciparum SSBs (Supplemental Figure 2).
Despite these differences, the overall architecture of the apo HsmtSSBP1 structure (shown alone in Figure 7A with the human disease-relevant residues marked) aligned remarkably well with the other SSBs (Figure 7B). Heterogeneity observed in the aligned structures mostly derived from flexible loops, though the origins of these loops coincided. The modeled loop in HsmtSSBP1 is longer than its counterparts in the other SSBs, and it also contains three isolated negatively-charged residues, whereas matching loops in the other SSBs have fewer negatively-charged residues as well as positive charges. It may be that this loop has some function in mammalian mitochondrial SSBs that is absent in bacterial SSBs.
Figure 7. DNA-Bound Structure Alignments and Dimethyl Phosphate Probing.
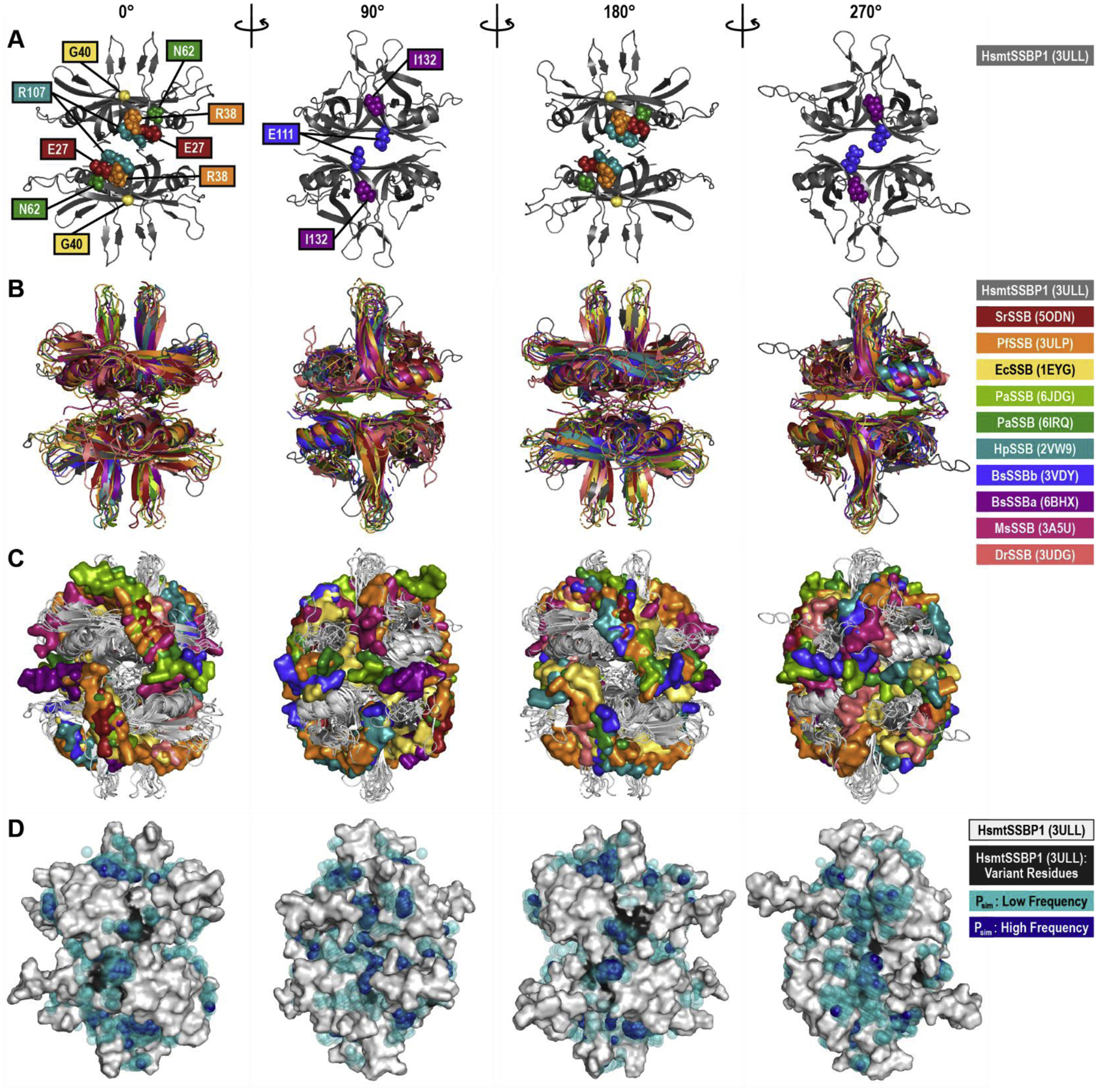
(A) The WT human mitochondrial (Hsmt) SSBP1 structure, based on PDB ID 3ULL and with missing loops modeled, is shown as a cartoon in four orientations, with residues linked to human disease indicated by spheres. Note that only the front-facing disease residues are shown in each orientation. This HsmtSSBP1 model was aligned with 10 other SSB proteins for which DNA-bound structures have been solved. This structural alignment is presented as cartoons of the proteins alone (B) and with the proteins rendered in white and each structure’s DNA rendered as surfaces (C). PDB IDs are listed in parentheses. (D) Predicted phosphate binding sites from molecular dynamics simulations are mapped onto a white surface rendering of HsmtSSBP1. Transparent teal spheres indicate low frequency dimethyl phosphate positions; dark blue opaque spheres indicate high frequency dimethyl phosphate positions. Black areas on the protein mark disease residue locations. Hs, Homo sapiens; Sr, Salinibacter ruber; Pf, Plasmodium falciparum; Ec, Escherichia coli; Pa, Pseudomonas aeruginosa; Hp, Helicobacter pylori; Bs, Bacillus subtilis; Ms, Mycolicibacterium smegmatis; Dr, Deinococcus radiodurans.
In all of these DNA-bound structures, the length of bound ssDNA varied from 2 to 28 nucleotides. In Figure 7C, these ssDNA fragments are shown on the surface of the tetramer orientations given in Figure 7B. As can be seen from these panels, only certain areas on the tetramer surface were readily accessible to DNA, with the surfaces of mobile loops and the mobile helix excluded from the DNA binding. We also observed DNA “hotspots” on the 0° and 180° faces of these alignments, where DNA from more than five structures overlaps in the upper right and lower left of each orientation. The collective DNA footprint in these aligned structures suggests that the entire surface of these SSB tetramers, excluding the mobile loops and helices, was available for DNA binding, an observation that may reasonably be extended to HsmtSSBP1.
Interestingly, a number of aromatic residues aligned sequentially very well between HsmtSSBP1 and these other SSBs (Supplemental Figure 2, orange hexagons), including a few residues (HsmtSSBP1 W65, W84, and F90) that have been experimentally linked to ssDNA binding [25, 30]. Indeed, the prevalence of conserved surface aromatic residues suggests a universal role in SSB-ssDNA binding that likely complements electrostatic interactions.
Because none of these bacterial SSB-DNA structures captures DNA wrapping with a biologically relevant length of DNA, questions about actual wrapping paths and the process of DNA wrapping remain. A recent molecular dynamics study described these transitions in the E. coli SSB system, where the authors used trinucleotides to probe DNA binding surfaces and identified both transient and anchoring interactions [31].
We employed a similar molecular dynamics approach to probe the ssDNA binding surfaces of WT human mitochondrial SSBP1. We ran molecular dynamics simulations in which free dimethyl phosphate ions stood in for the negatively-charged phosphates of the DNA backbone. At the start of each simulation, we randomly placed 150 dimethyl phosphates in the vicinity of the solvated SSBP1 tetramer. These dimethyl phosphates were then permitted to diffuse within the system for 500 ns. We repeated the simulation four times, with varied initial phosphate placements.
To analyze the positions occupied by dimethyl phosphates during the simulations, we first eliminated any dimethyl phosphates that spent fewer than 50 ns of each simulation bound to SSBP1, about 50% of all dimethyl phosphates. We then aligned and averaged the results of all four simulations to map common surfaces occupied by dimethyl phosphates, merging overlapping dimethyl phosphates into 1 Å2 bins (Figure 7D). Transparent teal spheres represent bins occupied by up to 9 dimethyl phosphates for greater than 50 ns. Solid deep blue spheres represent bins occupied by greater than 10 dimethyl phosphates for more than 50 ns, highlighting high occupancy areas on the surface of SSBP1.
Despite strong ionic interactions between dimethyl phosphates and the surface of SSBP1, these interactions were very dynamic: individual dimethyl phosphate ions tended to diffuse rapidly in and out of contact with the protein surface over the course of our simulations. In any given nanosecond snapshot, only 20–30 dimethyl phosphates were bound to the SSBP1 surface. While dimethyl phosphates are clearly smaller and more agile molecules than continuous ssDNA, this might reflect a real behavior, where backbone phosphates probe SSBP1’s surface before finding an anchoring position to begin wrapping. Indeed, we observed a handful of dimethyl phosphate hotspots in our alignments, which dimethyl phosphate ions occupied with high frequency. Four of these hotspots coincided with DNA hotspots on orientations 0° and 180° in Figure 7C, which were also proximal to several of the human disease-linked residues in SSBP1 (E27, R38, G40, N62, and R107). In addition to basic residues, there are several conserved aromatic residues near these hotspots (SSBP1 W65, W84, F90, and Y119) which could participate in ssDNA binding. These hotspot positions on the surface of SSBP1 seem to be important, perhaps for ssDNA probing or for anchoring to promote wrapping.
4. Discussion
Molecular dynamics simulations give us tools to understand systems that can’t be probed experimentally, like the heterotetramer SSBP1 systems described here. Of course, such approaches are not free from limitations. We make necessary assumptions and simplifications in the course of these simulations, and the results are always based on models that mimic actual systems as closely as possible. We made several assumptions here about how the SSBP1 disease variants might behave in vivo. Our predictions about expected ratios of homotetramers and heterotetramers in patient mitochondria may be wrong if variant and WT SSBP1 monomers are not equally abundant and able to randomly sort into tetramers. We cannot rule out co-translational, co-translocational tetramer assembly, which would yield only homotetramers. While our results support our assumption that heterotetramers and homotetramers are equally stable, if this is untrue in vivo, various tetramer combinations may not be evenly distributed. And we are limited in the length of the simulations that it is feasible to run and analyze, so we must interpret snapshots of molecular time to model long term SSBP1 dynamics. Because of this, rather than treating our models as firm proof SSBP1 molecular dynamics, we view this study as a framework for predicting real world protein behavior. We can use the results of these simulations to eliminate unlikely explanations for biological phenomena and to support likely explanations, and we hope that these explanations will guide future experimental approaches to similar biological questions.
One hypothesis for the role of SSBP1 variants in human disease is that variant monomers disrupt tetramer stability. Our RMSD data indicate global stability of all tested systems, ruling out gross destabilization. Our binding free energy results support a dimer of dimers structure of SSBP1 tetramers, but they also show that in most cases, variant tetramers are more tightly bound than WT, which undermines a hypothesis of variant tetramer disassembly into dimers or monomers. On the other hand, the observation that variant tetramers “breathe” less than WT may have implications for ssDNA binding or release, if the inherent wiggle room in the WT tetramer is important for these transitions. Through B-factor and Cα deviation data, we observed comparable rigid core regions and flexible loops in both WT and variant SSBP1 systems, belying an explanation in which the presence of variant residues alters the terrain of the protein core or the behavior of mobile loops and impacts stability or function. Our analysis of the solvent accessible areas of the variant SSBP1 systems showed that all of the tetramers shrank at least slightly, consistent with the tighter binding free energies and reduced “breathing”. We also observed that most systems experienced a reduction in net positive surface area, which may be consistent with subtle changes in ssDNA binding. Our analyses of SSBP1 homologs’ DNA binding patterns and phosphate probing simulations suggest that DNA can bind nearly anywhere on the SSBP1 surface except the mobile loops and helices, with potential hotspots for ssDNA binding near the location of several disease variant residues. These results predict local perturbations in specific DNA interactions, rather than global disruption of DNA binding.
Overall, the results of our simulations, considered with findings in previous reports, seem to favor a mechanism of disease in which subtle changes in DNA binding by SSBP1 leave ssDNA vulnerable to damage. Many unknowns remain, but several possible factors merit consideration. First, the surface of human mitochondrial SSBP1 boasts an unusually high number of basic residues, even compared to the other SSBs presented at the end of this study. Possibly these extra positive charges are required to compensate for the basic pH of the mitochondrial matrix in which SSBP1 must function, and the loss of some of net positive charge in the variant tetramers may compromise ssDNA binding more than we predict. Given reports of cooperation between basic and aromatic surface residues in ssDNA binding by other SSBs [19–23], it is conceivable that the surface reorganizations and overall tightening of most variant SSBP1 tetramers disrupts a finely tuned network of basic and aromatic residues that normally coordinate ssDNA binding, wrapping, or release.
We know very little about how SSBP1 transitions from individual transient interactions with ssDNA to the tightly wrapped state. The proximity of five of the human disease variant residues to DNA and phosphate binding hotspots found in our analyses suggests a tempting hypothesis: that the presence of these variant residues interferes with initial anchoring of ssDNA at these hotspots, slowing the transition to a stable, wrapped state. We also have little information about the process of ssDNA release from SSBP1, so we can only speculate about the role that molecular dynamics changes in variant SSBP1 tetramers might play in altering this process.
Lastly, we cannot rule out the possibility that any of the variant residues might disrupt crucial protein-protein interactions between SSBP1 and other proteins involved in mtDNA maintenance or replication. Direct protein interactions with SSBP1 are largely uncharacterized but not improbable. Therefore, it remains possible that disruption of such interactions drives SSBP1 disease. However, due to the positions of the variant residues and the lack of major structural rearrangements, a role for these residues in protein-protein interactions is fairly unlikely.
We present the results of this study as a common scaffold to understand these recently identified SSBP1 disease variants and any others that may emerge over time. Overall, our results are consistent with the biology of these diseases. The effects of these disease variants on SSBP1 function are likely subtle, only rarely compromising mtDNA replication to the extent that they lead to mtDNA depletion or deletions, rather than causing catastrophic failure of mtDNA maintenance. Whereas such a catastrophic failure might cause embryonic lethality, subtle DNA-binding changes and rare DNA-damaging events are consistent with SSBP1 mutations that most often result in isolated, adult-onset optic atrophy. It is our hope that this study may be used to generate testable hypotheses around the molecular mechanisms of SSBP1 diseases and to drive further advances in our understanding of SSBP1 function.
Supplementary Material
Highlights.
Recently reported human disease mutations in SSBP1 lack mechanistic explanations.
SSBP1 variant tetramers are stable in molecular dynamics simulation experiments.
Simulations suggest that SSBP1 variants breathe less and have altered surfaces.
Structural alignments and phosphate probing predict DNA binding surfaces on SSBP1.
SSBP1 variants may cause subtle defects in binding, wrapping, or release of ssDNA.
Acknowledgements
We thank Drs. Matthew J. Longley, Geoffrey A. Mueller, and Eric D. Sullivan for helpful comments on this manuscript.
Funding
This work was supported by Projects Z01 ES065078 to W.C.C. and Z01 ES043010 to L.P. from the Division of Intramural Research of the NIH, NIEHS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Young MJ, Copeland WC, Human mitochondrial DNA replication machinery and disease, Curr Opin Genet Dev, 38 (2016) 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Copeland WC, Inherited mitochondrial diseases of DNA replication, Annual review of medicine, 59 (2008) 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kullar PJ, Gomez-Duran A, Gammage PA, Garone C, Minczuk M, Golder Z, Wilson J, Montoya J, Hakli S, Karppa M, Horvath R, Majamaa K, Chinnery PF, Heterozygous SSBP1 start loss mutation co-segregates with hearing loss and the m.1555A>G mtDNA variant in a large multigenerational family, Brain, 141 (2018) 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jurkute N, Leu C, Pogoda HM, Arno G, Robson AG, Nurnberg G, Altmuller J, Thiele H, Motameny S, Toliat MR, Powell K, Hohne W, Michaelides M, Webster AR, Moore AT, Hammerschmidt M, Nurnberg P, Yu-Wai-Man P, Votruba M, SSBP1 mutations in dominant optic atrophy with variable retinal degeneration, Ann Neurol, 86 (2019) 368–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gustafson MA, McCormick EM, Perera L, Longley MJ, Bai R, Kong J, Dulik M, Shen L, Goldstein AC, McCormack SE, Laskin BL, Leroy BP, Ortiz-Gonzalez XR, Ellington MG, Copeland WC, Falk MJ, Mitochondrial single-stranded DNA binding protein novel de novo SSBP1 mutation in a child with single large-scale mtDNA deletion (SLSMD) clinically manifesting as Pearson, Kearns-Sayre, and Leigh syndromes, PLoS One, 14 (2019) e0221829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Del Dotto V, Ullah F, Di Meo I, Magini P, Gusic M, Maresca A, Caporali L, Palombo F, Tagliavini F, Baugh EH, Macao B, Szilagyi Z, Peron C, Gustafson MA, Khan K, La Morgia C, Barboni P, Carbonelli M, Valentino ML, Liguori R, Shashi V, Sullivan J, Nagaraj S, El-Dairi M, Iannaccone A, Cutcutache I, Bertini E, Carrozzo R, Emma F, Diomedi-Camassei F, Zanna C, Armstrong M, Page M, Stong N, Boesch S, Kopajtich R, Wortmann S, Sperl W, Davis EE, Copeland WC, Seri M, Falkenberg M, Prokisch H, Katsanis N, Tiranti V, Pippucci T, Carelli V, SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder, J Clin Invest, 130 (2020) 108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Piro-Mégy C, Sarzi E, Tarres-Sole A, Pequignot M, Hensen F, Quiles M, Manes G, Chakraborty A, Senechal A, Bocquet B, Cazevieille C, Roubertie A, Muller A, Charif M, Goudenege D, Lenaers G, Wilhelm H, Kellner U, Weisschuh N, Wissinger B, Zanlonghi X, Hamel C, Spelbrink JN, Sola M, Delettre C, Dominant mutations in mtDNA maintenance gene SSBP1 cause optic atrophy and foveopathy, J Clin Invest, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee Y, Kim T, Lee M, So S, Karagozlu MZ, Seo GH, Choi IH, Lee PCW, Kim CJ, Kang E, Lee BH, De Novo Development of mtDNA Deletion Due to Decreased POLG and SSBP1 Expression in Humans, Genes (Basel), 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang C, Curth U, Urbanke C, Kang C, Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 A resolution, Nat Struct Biol, 4 (1997) 153–157. [DOI] [PubMed] [Google Scholar]
- [10].Singh SP, Kukshal V, Galletto R, A stable tetramer is not the only oligomeric state that mitochondrial single-stranded DNA binding proteins can adopt, J Biol Chem, 294 (2019) 4137–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Singh SP, Kukshal V, De Bona P, Antony E, Galletto R, The mitochondrial single-stranded DNA binding protein from S. cerevisiae, Rim1, does not form stable homotetramers and binds DNA as a dimer of dimers, Nucleic Acids Res, 46 (2018) 7193–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallogr D Biol Crystallogr, 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [13].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr D Biol Crystallogr, 66 (2010) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham I, E. T, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, Gohlke H, Goetz AW, Greene D, Harris R, Homeyer N, Huang Y, Izadi S, Kovalenko A, Kurtzman T, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein DJ, Merz KM, Miao Y, Monard G, Nguyen C, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling CL, Smith J, SalomonFerrer R, Swails J, Walker RC, Wang J, Wei H, Wolf RM, Wu X, Xiao L, York DM, Kollman PA, AMBER, University of California, San Francisco, (2018). [Google Scholar]
- [15].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ Gaussian 16 Rev. C.01, in, Wallingford, CT, 2016. [Google Scholar]
- [16].Longley MJ, Smith LA, Copeland WC, Preparation of human mitochondrial single-stranded DNA-binding protein, Methods Mol Biol, 554 (2009) 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Qian Y, Johnson KA, The human mitochondrial single-stranded DNA-binding protein displays distinct kinetics and thermodynamics of DNA binding and exchange, J Biol Chem, 292 (2017) 13068–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kaur P, Longley MJ, Pan H, Wang H, Copeland WC, Single-molecule DREEM imaging reveals DNA wrapping around human mitochondrial single-stranded DNA binding protein, Nucleic Acids Res, 46 (2018) 11287–11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raghunathan S, Kozlov AG, Lohman TM, Waksman G, Structure of the DNA binding domain of E. coli SSB bound to ssDNA, Nat Struct Biol, 7 (2000) 648–652. [DOI] [PubMed] [Google Scholar]
- [20].Chan KW, Lee YJ, Wang CH, Huang H, Sun YJ, Single-stranded DNA-binding protein complex from Helicobacter pylori suggests an ssDNA-binding surface, J Mol Biol, 388 (2009) 508–519. [DOI] [PubMed] [Google Scholar]
- [21].Khamis MI, Casas-Finet JR, Maki AH, Stacking interactions of tryptophan residues and nucleotide bases in complexes formed between Escherichia coli single-stranded DNA binding protein and heavy atom-modified poly(uridylic) acid. A study by optically detected magnetic resonance spectroscopy, J Biol Chem, 262 (1987) 1725–1733. [PubMed] [Google Scholar]
- [22].Touma C, Kariawasam R, Gimenez AX, Bernardo RE, Ashton NW, Adams MN, Paquet N, Croll TI, O’Byrne KJ, Richard DJ, Cubeddu L, Gamsjaeger R, A structural analysis of DNA binding by hSSB1 (NABP2/OBFC2B) in solution, Nucleic Acids Res, 44 (2016) 7963–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Croft LV, Bolderson E, Adams MN, El-Kamand S, Kariawasam R, Cubeddu L, Gamsjaeger R, Richard DJ, Human single-stranded DNA binding protein 1 (hSSB1, OBFC2B), a critical component of the DNA damage response, Semin Cell Dev Biol, 86 (2019) 121–128. [DOI] [PubMed] [Google Scholar]
- [24].Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL, SSB as an organizer/mobilizer of genome maintenance complexes, Crit Rev Biochem Mol Biol, 43 (2008) 289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Curth U, Urbanke C, Greipel J, Gerberding H, Tiranti V, Zeviani M, Single-stranded-DNA-binding proteins from human mitochondria and Escherichia coli have analogous physicochemical properties, Eur J Biochem, 221 (1994) 435–443. [DOI] [PubMed] [Google Scholar]
- [26].Mikhailov VS, Bogenhagen DF, Effects of Xenopus laevis mitochondrial single-stranded DNA-binding protein on primer-template binding and 3’->5’ exonuclease activity of DNA polymerase gamma, Journal of Biological Chemistry, 271 (1996) 18939–18946. [DOI] [PubMed] [Google Scholar]
- [27].Lohman TM, Overman LB, Datta S, Salt-dependent changes in the DNA binding co-operativity of Escherichia coli single strand binding protein, J Mol Biol, 187 (1986) 603–615. [DOI] [PubMed] [Google Scholar]
- [28].Roy R, Kozlov AG, Lohman TM, Ha T, SSB protein diffusion on single-stranded DNA stimulates RecA filament formation, Nature, 461 (2009) 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dubiel K, Myers AR, Kozlov AG, Yang O, Zhang J, Ha T, Lohman TM, Keck JL, Structural Mechanisms of Cooperative DNA Binding by Bacterial Single-Stranded DNA-Binding Proteins, J Mol Biol, 431 (2019) 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Merrill BM, Williams KR, Chase JW, Konigsberg WH, Photochemical cross-linking of the Escherichia coli single-stranded DNA-binding protein to oligodeoxynucleotides. Identification of phenylalanine 60 as the site of cross-linking, J Biol Chem, 259 (1984) 10850–10856. [PubMed] [Google Scholar]
- [31].Maffeo C, Aksimentiev A, Molecular mechanism of DNA association with single-stranded DNA binding protein, Nucleic Acids Res, 45 (2017) 12125–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.