

Abstract
Along with the impressive achievements in understanding the endogenous signaling roles and mechanism(s) of action of carbon monoxide (CO), much research has demonstrated the potential of using CO as a therapeutic agent for treating various diseases. Because of CO’s toxicity at high concentrations and the observed difference in toxicity profiles of CO depending on the route of administration, this review analyzes and presents the benefits of developing orally active CO donors. Such compounds have the potential for improved safety profiles, enhancing the chance for developing CO-based therapeutics. In this review, the difference between inhalation and oral administration in terms of toxicity, CO delivery efficiency, and the potential mechanism(s) of action is analyzed. The evolution from CO gas inhalation to oral administration is also extensively analyzed by summarizing published studies up to date. The concept of “CO in a pill” can be achieved by oral administration of novel formulations of CO gas or appropriate CO donors.
Keywords: carbon monoxide, Gasotransmitter, oral delivery, Gas inhalation, CO donor, CORMs, CO prodrugs
Graphical abstract
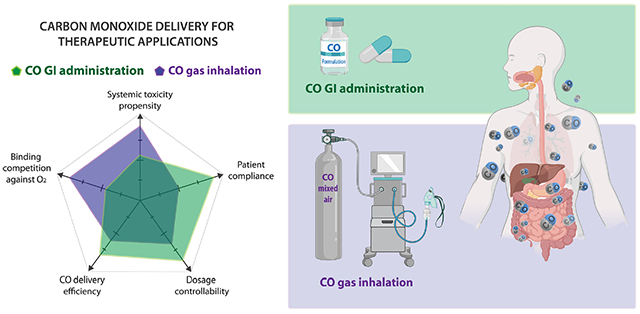
1. Introduction
Carbon monoxide (CO) is commonly known as a “silent killer” due to its lethality to humans and animals at high concentrations. However, this traditional perception of CO only as a harmful gas is changing because of the demonstrated endogenous signaling roles and potential therapeutic applications of CO [1-4]. Since Schmid demonstrated CO as the degradation product of heme by heme oxygenase in 1968 [5], a large number of researchers have made great efforts in exploring the biological mechanisms of actions and therapeutic effects of CO. Daily CO production from heme degradation as a result of red blood cell turnover has been reported to be around 400 μmol in an “average” person [6]. CO has a high affinity for heme-containing proteins (hemoproteins) at their ferrous state, with its avidity for hemoglobin (Hb) being about 240-fold higher than that of oxygen. Therefore, CO is said to mostly exist in the form of carboxyhemoglobin (COHb) and distributes to tissues through blood circulation [7] (The chemical term, “carboxyhemoglobin,” for referring to the CO-Hb complex is technically incorrect; it should be “carbonylhemoglobin.” However, to avoid confusion, we defer to convention and continue using the term carboxyhemoglobin). CO also distinguishes itself in its chemical stability from the other two gaseous signaling molecules, nitric oxide (NO) and hydrogen sulfide (H2S) [8]. It is relatively metabolically inert and is highly diffusible. CO can engage multiple targets and thus has pleiotropic effects that are essential in sustaining life [9-11]. There has been much progress in understanding CO’s biological properties and therapeutic potential in cytoprotection [12-14], anti-inflammation [15], anti-oxidation [16-18], and anti-apoptosis [19,20], all of which suggest a promising potential for developing CO as a therapeutic agent against a wide range of human diseases including inflammatory bowel disease [20,21], ischemia-reperfusion injury [20,21], bacterial infections [22], acute kidney injury [23,24], postoperative ileus [25,26], among many others.
Even though CO shows many promising therapeutic prospects, its gaseous nature has brought about several limitations for therapeutic use. A typical way of delivering CO is inhalation of gaseous CO. There have been many clinical trials approved by the US Food and Drug Administration (FDA) examining the safety and efficacy of inhaled CO. However, difficulties in controlling precise dosage, safety concerns, portability, patient acceptance and compliance, and delivery efficiency variations due to physical conditions bring upon challenges in applications, especially for managing chronic conditions [27]. Therefore, other forms of CO delivery, including CO solutions [25,28], metal-based carbonyl complexes (also referred to as CO-releasing molecules or CO-RMs) [29-31], and organic CO prodrugs [3,32-34] have been developed. Efforts have been made to deliver CO to the desired site locally or systemically [35,36]. However, along with conventional drug delivery challenges, there are other special issues related to CO delivery. Firstly, CO is a highly diffusible gas that may engage multiple biological targets depending on its binding affinity, concentration, the presence of binding competitors such as oxygen, tissue pH, and other physiological parameters. Therefore, the delivery approach used may have direct impacts on the pharmacokinetics, pharmacology, and toxicology of CO. Secondly, with its high affinity to Hb, most of the absorbed CO is believed to be in the form of COHb for distribution and elimination through exhaling. Hence, localized delivery of CO to specific tissues and the systemic effects caused by diffusion and distribution through blood circulation should be considered concomitantly. Thirdly, both safety and efficacy are key points to consider in evaluating a CO donor. The biological activity profiles and toxicity of the “byproducts” after CO release should also be weighed.
As a potential therapeutic agent, it would be desirable to develop strategies to administer CO via clinically relevant routes, including oral, intravenous, intramuscular, transdermal, and inhalation, among others, to meet the need for different applications. One may wonder how CO delivery routes would make a difference considering CO’s unique properties, including diffusivity and affinity for hemoglobin, which is ubiquitous. There are many interesting findings in the early days of toxicological studies of CO. Before we introduce the topic of oral delivery, we would like to focus our effort on discussing some historical studies, implications of different delivery routes as shown by these earlier studies, and demonstrated advantages of delivering CO via the gastrointestinal (GI) system as compared to inhalation in terms of minimizing systemic toxicity.
2. Delivery route has been shown to make a difference in CO toxicity
Generally speaking, it is well known that the route of drug delivery makes a difference in terms of both pharmacokinetics and toxicity profiles [37]. CO is no exception. However, the gaseous nature and its ability to bind to Hb make CO very unique as well, which leads to added complexity to CO delivery. Further, CO’s toxicity at high exposure levels means extra attention is needed in considering safety-related issues. There was a very well-known study in dogs that demonstrated the much more lethal nature of administering CO via inhalation than simply saturating blood hemoglobin with CO [38]. Specifically, using COHb level as the indicator of CO exposure, the study showed that 57-64% COHb was not lethal when generated by partially replacing the circulating blood with packed blood cells pre-saturated to 80% of COHb. The packed blood cells were collected from a dog after breathing 13% CO and centrifuged to remove the plasma. On the other hand, when inhalation was the route of administration, all the dogs died within 65 min after reaching a similar COHb level (54-73%) upon exposure to air with 13% CO [38]. Such results indicate that high COHb concentration per se is not the root cause for CO’s toxicity, at least not entirely. In fact, this assumption was made earlier by Haldane in 1895 as well through a series of experiments in mice showing that CO by itself was “non-poisonous” [39]. In the dog experiment discussed above, Goldbaum also showed that 2 h after the dogs were given 50, 150, and 200 ml/kg pure CO gas via intraperitoneal injection (i.p.), venous COHb level increased to 55%, 70%, and 80%, respectively. However, the dogs did not show signs of intoxication as reflected in the lack of change in the dogs’ appetite or behaviors [38]. These experiments indicate that CO administered through different routes may have distinct toxicity and activity profiles. Another good example to support such an idea was the study by McGrath and Leviseur in 1984 [40]. This study showed when pure CO gas was directly injected into the rat’s intestine between ligated pylorus and ileocecal at a dosage of 75 ml/kg (3.35 mmol/kg), the COHb level in the rats quickly increased to 35% within 5 min and reached a plateau level of 70% at the 15-min point. The elevated level was sustained until the 60-min point, which was at the end of the experiment [40]. At the 60-min point, depressed heart rate (by about 20%), blood pressure (by about 30%), and respiratory rate (by about 25% ) were observed. However, it did not lead to death. In contrast, such sustained high COHb levels were usually associated with death in rats when CO exposure was via inhalation [41-43]. Thus, it is reasonable to deduce that: (1) CO can be readily absorbed through the GI system owning to its diffusive nature, and (2) CO delivered through the GI system leads to less systemic toxicity than CO inhalation. For the second point, one may ask why the same COHb level achieved by different administration routes would make such a difference in toxicity?
There are several ways of explaining and discussing the difference in toxicity depending on the route of administration. We can dissect this question into two contributing factors. One is the cause of CO’s acute toxicity; and the other is how different administration routes make a difference in contributing to the cause of CO’s acute toxicity. Here we list five pertinent questions accordingly: (1) Does acute toxicity solely originate from inhibition of Hb’s ability to carry oxygen through the formation of COHb; (2) Are there organs that are more sensitive than others to CO in contributing to CO’s lethality; (3) Does the Hb affinity difference to CO and O2 at different sites make a difference in contributing to the toxicity and lethality of CO; (4) Is there “free CO” that is not Hb bound and is potentially more “available” to cellular targets (similar to the question we often ask of traditional non-volatile small molecule drugs as to what extent are they plasma protein (albumin) bound); and (5) Are there differences at the molecular level in CO binding to Hb depending on the route of administration?
All of these points probably need further experimental explorations in terms of validation and/or mechanistic understanding at the molecular level; and there are different levels of supporting evidence in answering these questions. In this review, we assess available data and sometimes extrapolate from the reported studies by providing our analysis. First, we examine whether an elevated level of COHb per se is the primary reason or is sufficient for lethality. The results from the aforementioned experiments by Haldane [39], Goldbaum [38], and McGrath [40] suggest that there are other factors beyond impeding O2 transport that lead to death, and the formation of COHb is not the primary root cause of the acute toxicity. In humans, there are extensive clinical data. Although an elevated COHb level can be seen in CO poisoned patients, the level of COHb does not seem to directly correlate with the severity of acute CO poisoning [44-47]. For example, patients with low COHb levels (<10%) have been found to have severe symptoms (e.g., coma) and yet patients with high COHb levels (>50%) have been found to have only mild symptoms (e.g., headaches) [46]. On the other hand, whenever intoxication or even death is shown after CO exposure, an increased COHb level seems to be a certainty. Therefore, an elevated level of COHb is a necessary but insufficient condition for CO’s acute toxicity, including lethality. There is increasing acknowledgment that CO accumulation in the tissue rather than COHb is the direct cause of acute toxicity.
As for the second point on whether some organs are more vulnerable to CO poisoning than others, a comprehensive and comparative examination of this issue would be a huge undertaking. However, available evidence does point to anoxic and subsequent oxidative neurological and cardiac injuries as a major contributing factor [44]. It should be noted that the term “anoxic” not only implies a high-COHb condition under which Hb is not able to deliver enough oxygen to the tissue but also comprises the condition in which oxygen supply is sufficient, but the utilization of oxygen in cells is deficient. CO competes with oxygen in binding to the ferrous iron of hemoproteins, which are essential to cellular respiration. Such proteins include neuroglobin [48], myoglobin [49], and cytochrome c oxidase [50], among others. Under CO poisoning situation, increased CO partial pressure in tissue allows CO to bind with these hemoproteins, thus compromising the storage and utilization of oxygen that are critical to support energy production of vital organs with high oxygen demand, such as the brain and heart. CO can be transported to these organs in two forms, as COHb and in free dissolved form. As stated in the previous discussions, at least in dogs, COHb levels of 57-64% themselves are unlikely to cause severe acute toxicity. Therefore, free CO could be responsible for the acute toxicity as seen with CO inhalation in the dog experiment.
To this point, there is the question as to whether there is available “free CO” that contributes to acute toxicity through efficient delivery of CO to vital organ tissues such as the brain and heart. This question is important in discussing the difference in toxicity among CO administration routes. In the published literature, there are mixed statements [51]. Here, we hope to analyze the results in a way that is consistent with published analytical results and observed biological phenomena by analyzing both the thermodynamic and kinetic aspects of CO binding to Hb. We first look at the affinity state of Hb at the site of the initial encounter with CO, the efficiency of CO-O2 exchange, and the associated impact on toxicity. It is well established that Hb has a high-affinity R state with a Kd of 0.3 μM for oxygen [49,52,53] and 1.67 nM for CO [52-55]. When gas exchange takes place in the lungs where Hb is mainly transformed to the R-state, CO binds to Hb with an affinity ratio over O2 of about 180 [52,53]. This affinity ratio for Hb in the R-state is lower than the typically referenced affinity constant (M, Haldane’s constant) of 246 [56]. Nevertheless, the affinity ratio of 180 still heavily favors CO binding when CO is in the ppm range. As an example, inhalation of 200 ppm of CO (1:1000 in partial pressure ratio between CO and O2) for an extended period (~1000 min) has been reported to lead to a COHb level of 30% in humans [57]. The fact that it takes at least 10 h to reach the 30% COHb plateau (presumably close to equilibrium) suggests strong competition for binding by O2 and kinetic factors/barriers for CO binding. This kinetic issue is analyzed in the next paragraph [57]. If one does a theoretical calculation of what it means to inhale 200 ppm of CO in the air, based on Henry’s law and reported solubility data [58], the theoretical concentration of CO dissolved in a cell-free serum is about 126 nM. If binding kinetics is fast, one would expect the CO to be “scavenged” by Hb to the level comparable to the Kd value of Hb for CO (0.7-1.7 nM in the R-state). On the other hand, CO binding kinetics are on the scale of minutes (discussed in the next paragraph). Thus, the true “free CO” concentration is probably somewhere between the solubility limit (126 nM) and Hb’s Kd for CO (0.7-1.7 nM). In any case, the mid- to high-nM concentration range is expected be a sufficient level of CO to engage hemoprotein targets such as myoglobin (Kd = 29 nM [59,60]) and neuroglobin (Kd = 0.2 nM [48,53]), and maybe partially engage cytochrome c oxidase (Kd = 0.3 μM [61]). The lack of lethal toxicity in the experiment of dog infusion with the CO-pre-saturated blood cells [38] also suggest that the available free CO resulting from the dissociation of COHb under the experimental conditions could be lower than that from CO inhalation when similar COHb levels are reached. Exposing a subject to such a level of “free CO” for an extended period of time is expected to offer an excellent chance for CO binding to targets critical to cellular oxygen storage and respiration, leading to toxicity. In a different scenario when pure CO is directly administered internally through GI, several factors favor CO binding to hemoglobin, which diminishes the extent of competition by binding with oxygen. It is important to note that the oxygen partial pressure in the vein (typically about 40 mmHg) is much lower than in the air (150 mmHg) and in the alveoli of the lungs (106 mmHg) [62]. As a result, the competition from oxygen is significantly diminished in the vein. Presumably, this leads to enhanced binding efficiency for CO with target hemoproteins in the epithelial tissue of GI tract and with hemoglobin in the peripheral tissue after diffusing to the capillary blood vessel. Although the increased acidity and decreased oxygen partial pressure in the tissue decrease the binding affinity for hemoglobin with CO (Kd = 1.1 μM [53]) and oxygen (Kd = 420 μM [53,63]), the ratio of the binding affinity (M) actually increases to 390 in the T-state (about 217% higher than the R-state) favoring CO binding. This is accompanied by a 75% drop in oxygen partial pressure in peripheral tissues. Since pure CO was administered into the GI tract in the described experiments, leading to a much higher local CO partial pressure than oxygen, the conditions should be favorable for efficient CO binding with hemoglobin after diffusing to the capillary vein and with hemoproteins in the tissue. The overall result is higher efficiency in engaging hemoproteins in the tissue and venous blood by CO delivered through the GI tract than via inhalation. At this moment, one may ask the question of the consequence of binding efficiency difference. Inefficient binding of CO to Hb inevitably means more “free CO” in the blood, which is readily available to engage protein targets in vital organs (e.g., brain and heart). When the CO-O2 exchange is to happen in the lungs via inhalation, the availability of “free CO” means its ready access to critical organs such as the heart, brain, liver, and kidney, which are intuitively more sensitive organs than peripheral tissues. One only needs to look at the blood flow diagrams from coronary and aortic arteries to various parts of the body to see this point. It should be noted that Goldbaum and co-authors [38] raised the same point as well; the combination of available free CO and the inhibition of cytochrome c oxidase is likely the most important mechanism of CO’s toxicity.
At this point, it is also important to bring kinetic analyses into the discussion. It is indeed true that there have been reports of low “free CO” concentrations in the blood under equilibrium conditions [38,64-67]. However, CO inhalation is a dynamic process with constant exchanges of various gaseous components. Therefore, thermodynamic analysis (equilibrium concentrations) probably represents the lower end of “free CO” concentrations. In reality, the time scale of the blood circulation in a live subject rarely allows for establishing true equilibrium. Theoretically, due to the small koff (1 × 10−2 s−1) [53] of the CO-Hb binding kinetics, the equilibration time of CO binding to Hb is on the scale of minutes [68]. Experimentally in the Goldbaum study [38], it was found that it took 5 min and 20 min to allow 26% and 91% of Hb to be converted to COHb, respectively, when blood was shaken in a 100% CO atmosphere. In contrast, an adult heart pumps about 5 L of blood per minute, which is about the equivalent of the total blood volume of an average person. Therefore, the time it takes to establish binding equilibrium is much longer than what it takes for blood to distribute to vital organs. The slow equilibrium time means that by the time COHb is measured at a high level, there is likely a substantial portion of the inhaled CO that is in the free form, capable of directly and quickly accessing critical organs such as the brain, heart, liver, and kidney through the arterial blood flow. In contrast, CO absorbed in the venous flow (GI absorption) has more time to diffuse into non-critical space and exchange with oxygen as it goes back through the lungs before reaching critical organs. This is expected to diminish the amount of “free CO” available to critical organs. Again, this is also a point raised by Goldbaum and co-authors [38]. The studies by McGrath and Leviseur [40] also support this point. Along with all these discussions, there is the important question of how blood is sampled for COHb analysis (venous vs. arterial); this is a critical issue because these two numbers are different [69] and may reflect different exchange processes as analyzed in a previous paragraph.
Another issue that might affect toxicity is the total CO input in a given study. Because of the enhanced CO binding efficiency, the actual total CO intake is expected to be lower through GI administration than via inhalation to achieve the same endpoint in terms of COHb level. Take the aforementioned CO gas GI injection experiment for example, we can look at the issue of total CO intake via inhalation to sustain the same persistent COHb level of 70% for 1 h to reproduce the level achieved by GI administration. According to another study in rats with CO inhalation [43], to maintain the COHb level at about 70%, continuous exposure to 4000 ppm of CO was needed. Assuming the alveolar ventilation rate of rats of 117 ml/min in both cases as reported [70], 4000 ppm of CO inhalation for 1 h would give a total CO exposure dosage of about 94 ml/kg in a 300 g rat. It is known that when COHb exceeded 30%, the alveolar ventilation rate also increases as compensation for anoxia [43]. Therefore, an even higher total CO exposure than 94 ml/kg is expected. In contrast, the dose of 75 ml/kg CO with GI injection was sufficient to maintain the same level of COHb for 1 h. As a result, the extra CO dosage from inhalation might also contribute to the more serious toxicity observed. Given the average Hb concentration of 14 g/dL in rats [71,72], 14 ml/kg of CO is needed to bind all the Hb molecules. It should be noted that 75 ml/kg of CO is sufficient to saturate all the Hb molecules in rats. Overall, the higher level of CO intake and exposure coupled with inefficient competition for binding with hemoglobin might be the reason for the enhanced level of toxicity when CO is administered via inhalation. Or at least, this needs to be considered. It further marks the importance of studying pharmacokinetics, pharmacology, and toxicology of CO delivered through different routes, which are critical issues for developing CO into a therapeutic agent [73].
The above analysis is mostly based on the two-state (R and T) MWC allosteric binding model of hemoglobin and the Bohr effect [74]. It may need to be supplemented with an examination of molecular events in dissecting factors that could contribute to differences in toxicity profiles depending on the route of administration. The static structural model of the O2-Hb complex upon binding has been examined to the atomic level by Perutz [74], which revealed the molecular mechanism of the MWC model and Bohr effect, thus the function of hemoglobin [74]. Hb as a tetramer consists of α2β2 subunits. Due to the cooperative binding mechanism, each subunit has different binding affinities towards oxygen and CO depending on the O2/CO occupancy state of Hb and pH. The pH dependency of CO-Hb binding has been reported to be similar as O2-Hb binding [75]. The ratio in binding affinity between CO and O2 to Hb in different occupancy states is not a constant [75]. Further, under dynamic physiological conditions, the kinetic and thermodynamic changes of CO binding to Hb in the presence of oxygen in different organs are not fully understood.
When one discusses certain levels of COHb, often overlooked is the detailed composition with a range of possibilities including Hb4(O2)4, Hb4(O2)3CO, Hb4(O2)2(CO)2, Hb4(O2)1(CO)3, and Hb4(CO)4. For example, in a static in-vitro condition, it has been calculated that in blood with 25% COHb, there are 32% Hb4(O2)4, 21% Hb4(O2)2(CO)2 42% Hb4(O2)3CO, and less than 5% of other species. On the other hand, 90% COHb saturation gives rise to about 66% Hb4(CO)4, 30% Hb4(O2)1(CO)3, and less than 5% Hb4(O2)2(CO)2 [76]. According to these in-vitro experiments and calculations, the stepwise CO dissociation constants were estimated to be 0.2 μM for Hb4CO and Hb4(CO)3, and 5.6 nM for Hb4(CO)2 and Hb4(CO)4 [76]. The stepwise dissociation rate constant was measured as 0.09 s−1 for Hb4CO, 0.028 s−1 for Hb4(CO)2, 0.09 s−1 for Hb4(CO)3, and 0.036 s−1 for Hb4(CO)4. The differences in binding affinity and dissociation rate constant of each species inevitably translate into differences in their ability to bind or unload CO and oxygen in both kinetic and thermodynamic terms. It should be noted that under these static conditions, oxygen binding was not taken into calculation. It is understandable that incorporation of the oxygen binding terms will significantly convolute the picture. Nevertheless, because the occupancy status of Hb in the lungs and in the GI system is expected to be different, it is very likely that CO administered through different routes could adopt varying combinations of the O2/CO-Hb complex species, and thus the associated toxicity profiles are expected to be different. From these analyses, it is clear that much work needs to be done to gain more insights of the molecular events that take places in each case under physiological conditions.
Beyond the issue of differential toxicity between CO delivered through inhalation and GI administration, an oral route has the advantage of easy administration [17,77]. Because of its diffusivity, CO released in the GI tract can easily diffuse into the systemic circulation. The common barriers for drug absorption, such as the intestinal mucosal barrier, blood-brain barrier, and placental barrier, are not considered to be a major issue for CO. Furthermore, CO has been demonstrated to show therapeutic effects in a variety of GI disorders [78-80], including gastric injury [17,77], colitis [81], postoperative ileus [82], inflammatory bowel disease [20], among others. Oral administration can provide direct access to GI targets. With the demonstrated therapeutic effects and the goal of developing CO delivery approaches for oral administration, this review examines the various CO delivery approaches available and focuses on the formulation, pharmacology, and pharmacokinetic issues of CO administration to the GI system through oral delivery.
3. From inhalation to ingestion – the evolution of in-vivo CO delivery
CO can be delivered to the body in the gaseous form, dissolved in a homogenous solution, or in a chemically caged CO donor form. Accordingly, inhalation, injection, and oral administration can all be used for delivery. For animal experiments, intraperitoneal injection can be used as well. In this section, we briefly recap the current in-vivo delivery approaches that have been applied in animal or human studies and focus on the feasibility and applications of CO administration through oral delivery. Contrasts and comparisons between oral administration and other routes in the same type or similar studies are discussed whenever possible.
3.1. CO Inhalation
Studies of CO’s toxicology have been ongoing for more than a century. As a result, the toxicological profile of CO was well established even before the recognition of its therapeutic potential. Early human clinical trials were mostly focused on CO gas inhalation. However, newer preclinical studies also tend to use CO solutions and CO donors, such as metal-based CO-RMs and organic CO prodrugs. Completed clinical trials have addressed three basic issues of CO inhalation in humans: (1) safety and COHb levels, (2) pharmacokinetics of COHb, and (3) therapeutic potential in treating select diseases.
In studies to determine the safety margin of CO inhalation, CO was administered after mixing with air in a concentration range between 15 to 7500 ppm [23,83]. In all cases, COHb was monitored as an indicator of CO exposure level. The highest experimental COHb level in humans among these controlled studies was 24.8%, achieved by exposure to 500 ppm of CO for 114 min without indication of symptomatic outcome in the report [57]. In a few additional studies, the highest documented COHb level in human was 10%, which was said to be without significant adverse events such as fainting, headache, or discomfort. Such a COHb level was achieved by inhaling 4000 ppm of CO for less than 30 min [84], 1200 ppm of CO for less than 45 min [85], or 500 ppm of CO for 1 h [86]. It should be noted that the US National Advisory Committee for Acute Exposure Guideline Levels for Hazardous Substances set the disabling toxic AEGL-2 levels of CO to be from 27 ppm for 8 h to 420 ppm for 10 min, which generally gives rise to about 4% COHb [87]. Nevertheless, the US FDA approved 14% COHb level as the safety threshold for clinical trials in delayed graft function of kidney transplant [23]. Such a fact reflects the reality that exposure levels for therapeutic indications are not limited by threshold levels set in the context of occupational safety and daily household exposure. This is true for other therapeutics as well and is nothing unique. For example, the occupational safety level of nitric oxide (NO) is set at 25 ppm for an 8-h period [88], and yet NO is used as a therapeutic agent at a level that is orders of magnitude higher [89]. The same is true for arsenic oxide (Trisenox) [90] as well as many other therapeutic agents. Therefore, it is important to note that occupational exposure levels and therapeutic applications are two different concepts with different concentration thresholds for safety considerations.
Pharmacokinetic characteristics of CO have been well established by inhalation studies. Using COHb as the indicator of CO exposure, various models have been established to predict COHb levels after CO inhalation. Among them, the Coburn-Forster-Kane (CFK) model is the most extensively validated and is very robust [91]. The CFK model has shown excellent correlations with experimental results in healthy human volunteers [57,92-94], clinical trials [95,96], and animal models [97]. With defined physiological parameters including diffusivity in lung, blood volume, alveolar ventilation rate, rate of endogenous CO production, physical parameters such as partial pressure of oxygen and CO in the air, and vapor pressure of water at body temperature, one can predict the COHb level at a given time after CO inhalation. Further, endogenous CO production rate under various conditions can also be back-calculated by examining the involvement of endogenous CO generation in the model [98]. This feature allows for calculation of pharmacokinetics after CO administration through the GI system by mathematically assuming it is a part of endogenous CO generation.
Inhalation is an obvious method of delivering CO for pharmacological studies in humans and animals because CO gas has well-defined purity and safety profiles. In general, inhalation of 50-200 ppm of CO for no more than 2 h would give a marginal (<2%) increase of COHb level in humans [96,99]. In a phase-II clinical trial (NCT01214187) using CO to treat idiopathic pulmonary fibrosis, inhaling 200 ppm of CO gas for 2 h daily only increased the COHb level by about 1% from the basal level of about 2% [99]. This dosage did not lead to a significant change in serum matrix metalloproteinase-7 (MMP-7) concentration and respiratory scores at the endpoints of the 12-week treatment. In another clinical study to treat chronic obstructive pulmonary disease (COPD), inhalation of 100-125 ppm of CO for 2 h per day led to a median COHb level of 2.7% in COPD patients, and the COHb level for the placebo group was 0.2% [100]. A marginal trend of decrease in eosinophils and improved responsiveness to methacholine was shown, but the results were not considered statistically significant (p>0.05). Such results suggest that a higher CO exposure level may be needed to achieve statistically significant outcomes. In an initial experiment aimed at eventually treating endotoxemia in humans, inhalation of 250 ppm of CO for 2 h or 500 ppm of CO for 1 h increased COHb to 9% in the pilot trial [101]. In the main trial with 13 healthy volunteers, inhalation of 500 ppm of CO for 1 h led to an average COHb level of 7%. Then 2 ng/kg LPS was administered via bolus intravenous (i.v.) infusion, leading to acute inflammatory responses. However, CO treatment did not lead to changes in inflammatory markers in the serum, including IL-8, IL-1α, IL-1β, TNF-α, and IL-6. It should be noted that CO inhalation stopped after LPS infusion; then the COHb level began to gradually decrease to 4.5% after 4 h. Such studies suggest that a single episode of CO exposure of a short duration may not be sufficient for beneficial effects in humans, at least in the case of LPS-induced inflammation [102].
In a nonhuman primate model of lung inflammation in male cynomolgus macaques, inflammation was induced in the monkey by LPS inhalation [103]. CO was subsequently administered via inhalation of 250 or 500 ppm of CO gas for 6 h. The steady-state COHb levels were 25% from treatment with 250 ppm of CO and about 34% from 500 ppm of CO. It is interesting to see that pulmonary neutrophil count decreased by about 60% after treatment with 500 ppm of CO, while only a marginal decrease was seen with 250 ppm of CO. Levels of TNF-α in bronchoalveolar lavage fluid also showed a significant reduction (~40%) in the 500 ppm of CO study. However, the elevated levels of IL-6 and IL-8 in the lavage as a result of LPS stimulation did not respond to treatment by 500 ppm of CO. Obviously, the 34% COHb achieved in the monkey model is higher than the levels that are generally considered to be safe in humans. The results suggest the need to address the relationship between CO dosage, COHb level, and toxicity in the context of the intended therapeutic indications and species difference.
As discussed in the previous section, CO delivered through other pathways may offer a higher safety margin compared with inhalation. If a high dosage of CO is needed to achieve efficacy, inhalation may not be the best route of administration. Besides, CO needs to diffuse across the epithelial cells and matrix membrane to reach the capillary vessel, where it forms COHb and distributes to the whole body. The epithelial cell at the CO contact site may experience a higher CO concentration compared with remote tissues that rely on CO carried by COHb. As such, delivering CO directly to the target tissue through other administration routes may offer an improved chance of success.
3.2. CO solutions
Another way of delivering CO is to dissolve it in an aqueous solution. CO can dissolve in water with a solubility of 0.95 mM under 1 atm CO pressure at room temperature. Various efforts have been made to develop solubilized CO for medical use, including organ preservation solutions for transplant [104,105] and formulations for in-vivo administration. As organ preservation application is beyond the scope of this review, we mainly focus on CO solutions for internal administration in this section.
One of the pioneering studies was done by Nakao et al. with CO saturated lactated Ringer’s solution (CO-LR) [25]. CO concentration of freshly prepared CO-LR was tested to be 1.2 mM at atmospheric pressure of CO. In one study in mice, i.p. administration of CO-LR at 60 ml/kg (~72 μmol/kg) led to a quick increase in COHb level to about 7.5% within 5 min and a gradual decrease to the basal level in about 80 min. The study found that post-operative treatment with a single dose of CO-LR ameliorated postoperative ileus by inhibiting the inflammatory response in the gut wall [25]. Decreases in the levels of IL-1β, COX-2, iNOS, and ICAM-1 were also observed. Since the protective effects were abolished by pretreatment with ODQ, an sGC inhibitor, activation of sGC by CO was considered the mechanism of action. Such results indicate the sufficient solubility of CO in the LR solution for the delivery of an adequate amount of CO to mice for the intended application. As a comparison, inhalation of 250 ppm of CO led to a similar effect in preventing/treating postoperative ileus in mice [82]. Although the COHb level after CO inhalation was not reported, it is known that the steady-state COHb level by inhaling 250 ppm of CO for 3 h is about 20% [106]. Considering the alveolar ventilation rate for mice being about 0.029 L/min [107], the overall CO exposure is about 750 μmol/kg by inhaling 250 ppm of CO for 1 h. As a result, the total CO exposure dose of gas inhalation should be significantly higher than that of i.p. administration of a CO solution. Such results also further support the proposal earlier that it needs less CO intake by directly administering CO to the lesion site rather than CO inhalation. Therefore, it is important to develop other delivery forms aside from gas inhalation.
Along a similar line, Takagi et al. [108] developed a CO-saturated solution by bubbling 50% CO gas into saline to investigate the therapeutic effect of CO on gastric mucosal wound healing. Oral administration of 0.2 ml of this CO solution (500 μM CO, about 4 μmol/kg) per day for 4 days accelerated the healing of acetic acid-induced gastric ulcer in mice and significantly promoted re-epithelialization of the wound. The study did not report the COHb level. According to the dose and past literature reports, the increase of COHb is not expected to be more than 1%. Therefore, the observed activity is unlikely to be attributed to the increase of COHb level or systemic CO exposure. As a result, direct contact with the leisure tissue was probably the reason for the observed effects and can be used for CO delivery to the site of action.
Though there has been some success, the utility of a partially saturated CO solution is limited by CO’s solubility under normal pressure. For example, if a dose of 5 μmol/kg CO is needed to show efficacy in humans, a man of 70 kg would need to take 350 ml of CO saturated water, given its solubility at 1 atm of pure CO being about 1 mM. Further, such a solution would not be able to maintain its CO concentration once it is open to the atmosphere. Therefore, increasing CO’s solubility is key to improve the feasibility of using a CO solution for therapeutic purposes. To achieve this, a drinkable CO enriched formulation was described by Hillhurst Biopharmaceuticals [109]. The lead candidate HBI-002 is a saturated CO solution containing components that have been defined as “generally recognized as safe (GRAS)” by the US FDA. However, the molecular details are not described. According to a published patent, CO concentration is between 30-4400 mg/L (1.1~157 mM). The ingredients include lipid, protein, fats, and triglycerides [110]. This formulation has been studied in animal models of sickle cell disease (SCD) [110], kidney ischemia-reperfusion injury (IRI) [28], and Parkinson’s disease, among others [109]. In the studies of sickle cell disease, the formation of COHb was proposed to be a key remedy because it can decrease the level of deoxyhemoglobin, thereby reducing the sickling [111]. In evaluating HBI-002 in a mouse SCD model, a single dose via gavage at 10 mg/kg gave rise to a COHb peak between 0-10 min after administration. Then CO cleared from the circulation in approximately 3 h [110]. The peak COHb level varied depending on the mouse breed. COHb levels increased from 1.6 ± 0.2 to 5.4 ± 0.8% inNY1DD transgenic sickle mice; from 1.8 ± 0.2% to 4.7 ± 0.4% in Townes-SS transgenic sickle mice; and from 0.6 ± 0.2% to 3.0 ± 0.6% in Townes-AS control mice. The basal COHb level of non-sickle Townes-AS control mice was significantly lower than that of the sickle mice, presumably because of a markedly reduced hemolytic rate than the sickle mice. The results demonstrate that CO delivered by this solution can be readily absorbed from the gut and is bioavailable in the systemic circulation within minutes [110]. It is worth noting that similar therapeutic effects in sickle mouse models have been achieved with inhaling 250 ppm of CO gas, 1 h per day, 3 days per week for 10 weeks [112]. The steady-state COHb level was about 20% in the transgenic sickle mice and 14% in normal mice, higher than the maximum COHb levels in the HBI-002 study. The study of HBI-002 in a renal IRI model showed rescuing effects by pretreatment with HBI-002 via oral gavage at a dose of 0.2 mg/kg. Pretreatment with 250 ppm of CO for 1 h before IRI showed slightly better results than the HBI-002 solution. In the same study, an organic CO prodrug BW-CO-101 with a short half-life (2 min) (see the organic CO prodrug section for detail) dosed at 100 mg/kg (about 200 μmol/kg) showed a similar effect as gas inhalation [28]. A clinical trial (NCT03926819) for HBI-002 has already been registered with the FDA, but it seems that it is yet to start recruiting.
In both studies, due to a lack of information on CO concentration in HBI-002, the true dosage is not publicly known. The solubility of CO in the solution follows Henry’s law (C=kP) as the concentration of a dissolved gas (C) equals Henry’s constant (k) times the partial pressure of the gas (P). Henry’s constant is also correlated with temperature. Therefore, much the same way as soda loses its CO2 upon exposure to the air, CO solution also loses its CO when open to the air, and the rate of CO loss can be greatly influenced by physical conditions such as temperature and altitude. CO release from the solution is generally a fast process unless there are special ingredients in HBI-002 that can tune the release rate and solubility. This will be an interesting aspect to examine in order to accommodate the special needs for various release rates depending on the intended therapeutic indications.
3.3. Chemical CO donors for GI administration
The aforementioned gas and CO solutions directly deliver CO. Drawing analogy with nitroglycerin being a successful chemical donor for nitric oxide, another gaseous signaling molecule, there has been a great deal of interest in developing a “solid” form of CO that would allow for accurate dosing, tunable release rate, targeted delivery, ease of storage and transport, and convenient administration. Along this line, chemical approaches to “encage” CO in a compound have been developed. Depending on the caging chemistry, the reported CO donors are categorized into metal-based CO releasing molecules (CO-RMs) [1,29-31,113,114] and organic CO prodrugs [33,36,115-118]. Both approaches have been tested via GI administration to show therapeutic activities in animal models.
For these CO donors, several factors affect the oral bioavailability of CO, including release mechanism (trigger), release half-life, solubility, bioavailability of the donor itself, chemical reactive, and the effects of the “carrier” portion of the CO donor molecules, which can involve both chemical reactions and direct interaction with biological targets. Among all these, the release mechanism is a key factor that affects the feasibility of oral delivery. There have been reports of photo-sensitive CO-RMs. [31,119] For obvious reasons, they are not easily adapted to systemic applications and thus are not discussed in this review. In the following sections, the donors are firstly categorized by their chemical nature and then subcategorized by the release mechanism for clarity of the discussions.
3.3.1. Metal-based CO releasing molecules
The chemistry for metal coordination with CO and the subsequent release has been known for centuries. The high affinity of CO for a transition metal at the appropriate oxidation state and geometry is also the reason why the heme molecule can bind with CO. Interestingly, as early as in 1891, McKendrick and Snodgrass first studied CO delivery by inhalation of air containing nickel tetracarbonyl vapor for therapeutic application [120]. However, nickel tetracarbonyl is highly toxic and is not expected to be of therapeutic benefit. Among the modern-day CO-metal complexes, Motterlini and Mann first reported using a series of metal-carbonyl complexes as CO releasing molecules (CO-RMs) in 2001 [114]. Among them are CORM-2 and CORM-3, which are commercially available and have been widely used as CO “surrogates” in numerous in-vitro and in-vivo experiments [1]. Since an extensive discussion of CO-RMs’ chemistry and biology are beyond the scope of the current review, readers are referred to some in-depth reviews and recent publications for details [1,4,30,36], including their CO-independent effects [121-125]. In this section, we focus on the in-vivo applications of the different generations of CO-RMs that are dosed through the GI system and discuss developability issues of this approach. Figure 1 summarizes the structures of the metal-based CO-RMs to be discussed in this section based on their release mechanisms.
Figure 1.
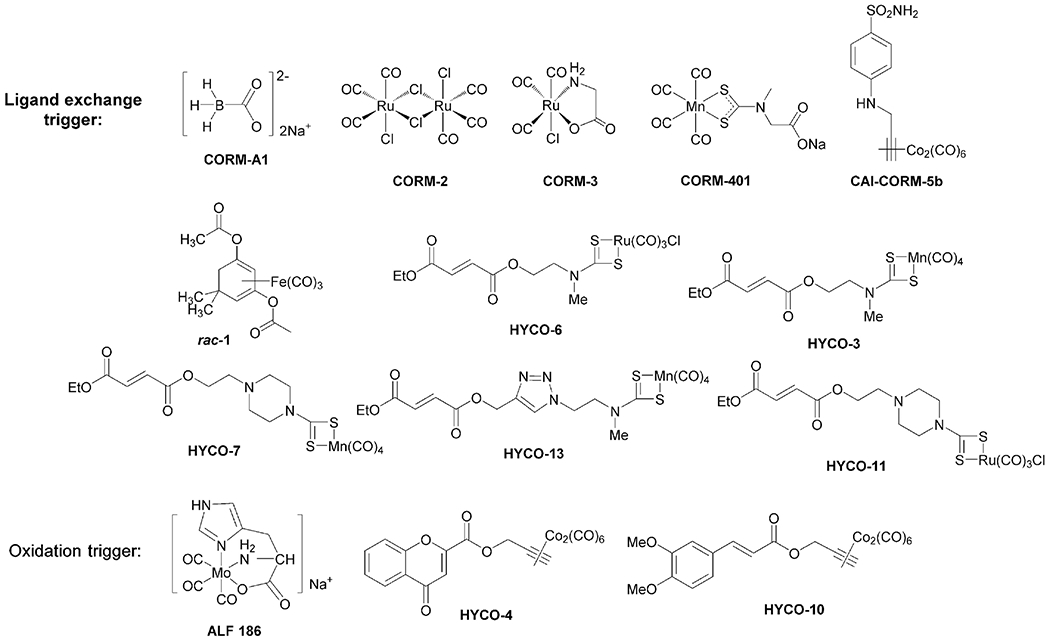
Chemical structures of select CO-RMs used in animal-model studies via oral administration.
Reaction with water as a triggering mechanism.
CORM-A1 was identified by Motterlini and coworkers as a potential CO-RM by utilizing borancarbonate decomposition chemistry to release CO in an aqueous solution. It converts myoglobin to carboxymyoglobin with a half-life of 21 min at pH 7.4 [1]. The reaction involves multiple steps with protonation likely being the rate-limiting step. If so, this would give second-order reaction kinetics and make the reaction pH dependent (and pseudo first order at a fixed pH). Indeed, the conversion can be initiated at a lower pH with a shorter half-life [126]. After releasing CO, CORM-A1 forms borane as an intermediate, which is a hydride reagent and undergoes reactions with water (or other species with an active proton or oxidizing group) to generate boric acid and hydrogen as the formal byproducts (Figure 2) [127]. The chemistry allows for release of CO in the gut without the need to absorb the compound into the systemic circulation. As a result, oral delivery of CO by CORM-A1 can be expected. Liu et al. reported oral delivery of CO via CORM-A1 to protect cerebrovascular dysfunction caused by neonatal seizures in a pig model [128]. Newborn piglets were given CORM-A1 (2 mg/kg, 20 μmol/kg) via an oral gastric feeding tube to the stomach 10 minutes before or 20 minutes after bicuculline-induced epileptic seizures. CO levels in the cortical periarachnoid cerebrospinal fluid (pCSF) increased from the basal level of 119 ± 19 pmol/mL by about 2.5 folds in 20 min and then started to decline to the basal level within about 40 min. Using intravital microscopy, dilation of the pial arterioles was observed accompanying the CO increase in pCSF. This dilation peaked at about 117% of the baseline diameter 10 min after administration and then returned to the normal level in about 20-30 min. The administration of CORM-A1 10 min before or within 20 min after seizure induction effectively reduced endothelial and astrocyte dysfunction and improved cerebral vascular outcome of neonatal seizures. COHb level in the experiments was not reported. However, oral administration of CORM-A1 at 2 mg/kg (19 μmol/kg)_did not seem to reduce blood oxygenation level as determined by monitoring arterial oxygen partial pressure, suggesting limited COHb formation. Indeed, theoretical calculations also indicate it can only increase COHb% level by 2~3 percentage points, assuming a hemoglobin concentration of 13 g/dL and all released CO is bound to hemoglobin. As a comparison, the same group reported that i.p. administration of 2 mg/kg CORM-A1 30 min before seizure induction offered similar protective effects in piglets [129] as oral dosing. As reported in another study using CO gas, inhalation of 250 ppm of CO gas for 3 h offered a similar protective effect in piglets [130]. Although COHb levels under these treatments were not reported from all these studies, a separate study showed a COHb level of about 20% in piglets after CO inhalation at this dosage, leading to disruption of mobility and thus some toxicity [131]. Therefore, oral administration CORM-A1 may be able to achieve a similar seizure protection effect with a much lower dose compared with that of CO inhalation.
Figure 2.
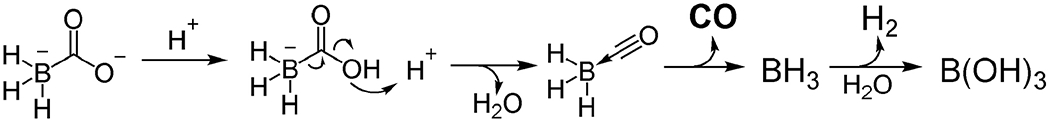
CO release chemistry of CORM-A1
CO-RMs that use ligand exchange as a triggering mechanism.
Most of the reported transition metal-based CO-RMs release CO by ligand exchange chemistry and/or photo-excited decomposition chemistry. As stated earlier, since photo-CORMs [119,132-134] are not suited for systemic application, they are not discussed in the context of oral delivery. Even though the chemistry for CO release from most metal-based CO-RMs is yet to be fully understood, ligand exchange is one of the major mechanisms that allow for CO to be “transferred” to a hemoprotein (Figure 3) [135]. Since water is not a good chelating ligand, most of the CO-RMs are stable in pure water. As a result, these CO-RMs have to react with nucleophilic ligands, such as a thiol group [136], to release CO. As such, the reactions are second order in terms of kinetics with the rate being dependent on the concentration of the CO-RM and the nature and concentration of the various participating nucleophiles. These nucleophilic ligands are available in the cell culture medium, inside the cell, in the blood, and in other body fluids.
Figure 3.
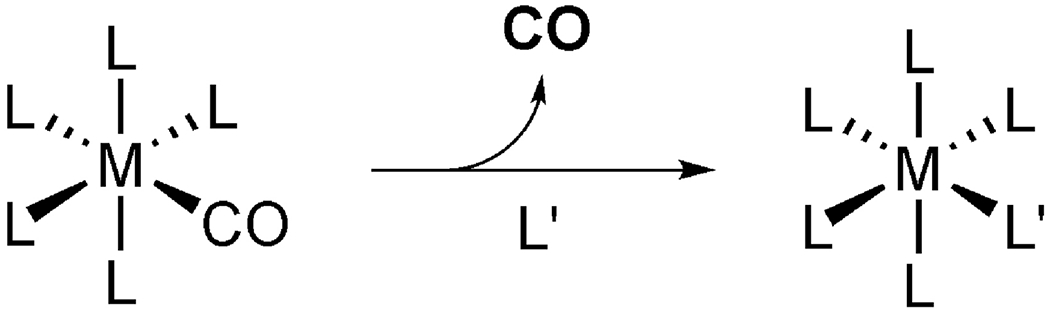
CO release by ligand exchanges.
In the CO solution section, we have described the protective effects of saline saturated with 50% CO gas against chemically induced gastric mucosal damage [108]. Before this study in 2016, Magierowski et al.[137] had reported the protective activity of orally administrated CORM-2 (Figure 1) against ethanol-induced gastric mucosa damage in rats in 2015. Pretreatment with 1–10 mg/kg CORM-2 (2-20 μmol/kg in CO dosage) decreased the area of the gastric lesion in a dose-dependent fashion and significantly (p<0.05) raised the gastric blood flow (GBF) compared with the vehicle. The dosage used is comparable to that of Takagi’s experiment with CO solution [108]. After oral administration of 5 mg/kg (10 μmol/kg) or 50 mg/kg (about 100 μmol/kg) CORM-2, CO content in the gastric mucosa increased by 83% (from 46 pmol/mg to 85 pmol/mg) and 253% (to 164 pmol/mg), respectively. Blood COHb level also increased from 0.78% (vehicle) to 1.55% (5 mg/kg CORM-2) and 2.2% (50 mg/kg CORM-2). It should be noted that the CO concentration in tissue did not differentiate CO released from CO bound hemoproteins and from absorbed CORM-2. Therefore, it is possible for absorbed CORM-2 to account for the unusually high CO concentrations in the tissue. If one converts 164 pmol/mg CO into 149 μM using a density of about 1.1 g/ml of the tissue and compares this concentration with that of the blood hemoglobin, it is an equivalent of 2% COHb. However, the concentration of hemoprotein in the tissue including cytochrome c oxidase and myoglobin (less than 100 μM in total) is not expected to be high enough to sustain such a high CO concentration in the bound form. Therefore, other factors including absorbed CORM-2 need to be considered for the high CO tissue concentrations. Nevertheless, under the low-dose condition, direct application of a CO donor to the lesion tissue was able to show efficacy without significantly increasing the COHb level. Further, a higher dosage of 100 mg/kg CORM-2 (about 200 μmol/kg) significantly increased mucosal damage as revealed by the increased area of the gastric lesion and decreased GBF. Orally administrated RuCl3 (5 mg/kg, 24 μmol/kg), which was used as the control of the “spent” donor in the study, significantly aggravated gastric damage and decreased GBF. The dosage and the inherent toxicity of the ruthenium core as seen with RuCl3 introduce complications in the assessment of such a CO donor.
Oral delivery of the manganese (Mn)-based CORM-401 [138] (Figure 1) is also feasible. It has been studied in treating high-fat diet-induced obesity and insulin resistance in mice by oral administration [139]. COHb increased from 0.3% to 2.5% or 4.5% 30 minutes after administration of CORM-401 at 15 mg/kg (45 μmol/kg) or 30 mg/kg (90 μmol/kg), respectively. Then COHb level gradually decreased to the control level within 48 h. CO contents in the adipose tissue also increased by about 2 folds with 30 mg/kg CORM-401, as determined by GC. Therapeutic effects were achieved by orally giving 30 mg/kg CORM-401 3 times a week for 14 weeks. Weight loss, improved glucose tolerance, and insulin resistance were observed in obese mice. Local and systemic insulin sensitivity in adipose tissue also increased. The mechanism was found to be CO-dependent through activation of Akt phosphorylation, uncoupling mitochondrial respiration, decreasing mitochondrial ATP production, and thus increasing ATP production through glycolysis. In comparison, i.p. administration of CORM-A1 at 5 mg/kg (45 μmol/kg) every 48 h for 30 weeks was also shown to reverse dietary-induced obesity in mice [140].
A series of cobalt (Co)- [141,142], manganese (Mn)- [143], and ruthenium (Ru)-based [144] hybrid CO complexes, namely HYCOs (Figure 1), were designed in recent years by Motterlini and co-workers by conjugating a CO complex with a nuclear factor-erythroid-2–related factor 2 (Nrf2) activator to enable a dual mode of action. Some of them have been found to be orally bioavailable for delivering CO and an Nrf2 activator simultaneously. Since release from cobalt-based HYCOs is triggered by oxidation of the cobalt, they are discussed in the next section.
The anti-inflammatory activity of HYCO-3 has been studied via oral administration [143]. HYCO-3 is a hybridized CO-RM by merging CORM-401 with dimethyl fumarate (DMF), an immunoregulatory and Nrf2 activator. Oral administration of 40 mg/kg (90 μmol/kg) HYCO-3 led to an increase of COHb level from 0.5% to a peak of 2.5% within 1 h with the ability to maintain ≥ 2% for at least 24 h. In comparison, oral dosing 40 mg/kg (120 μmol/kg) of CORM-401 increased COHb level to 5% within 1 h with a gradual decrease to 2.5% and 2% at the 6-h and 24-h points, respectively. Considering the dosage difference in molarity, CORM-401 showed better ability to increase COHb levels than HYCO-3, with the reasons to be explored. It is worth noting that challenging with 5 mg/kg LPS (i.p.) also led to a COHb level increase to about 1.3%, and the Nrf2 activator DMF alone also increased the COHb level to 2.5%. 6 h after LPS challenge and oral administration (p.o.) 40 mg/kg HYCO-3 and CORM-401, COHb levels were 4.4% and 6%, respectively. However, in Nrf2 gene knockout mice, LPS challenge did not increase COHb levels and HYCO-3 administration only increased COHb levels to 1.6%. Such results were interpreted as to show synergistic effect in elevating COHb levels by HYCO-3 and LPS. Oral administration of 40 mg/kg HYCO-3 reduced mRNA levels of pro-inflammatory markers, including TNF-α, IL-1β and IL-6 while increased the expression of the anti-inflammatory genes ARG1 and 1L-10. In comparison, DMF or CORM-401 alone or dosed together showed minimum effects in inducing anti-inflammatory genes. Overall, the results indicate that HYCO-3 may offer potent anti-inflammatory effects.
In 2020, the same group reported the biological activities of manganese- and ruthenium-based based HYCOs in human monocyte (THP1) and keratinocyte (HaCaT, NHEK) cell lines in vitro and anti-inflammation effects in vivo [145]. By oral administration of HYCOs at 40 mg/kg (dissolved in DMSO/sesame oil, 1:10 v:v) in mice, blood COHb levels peaked at 1 h after treatment. Among the tested compounds, HYCO-13 showed the highest increase in COHb levels, reaching about 5%, while the highest levels for HYCO-3, HYCO-6, HYCO-7 were about 2%. Moreover, the anti-inflammatory effects of HYCOs were studied in three anti-inflammation models. In an in-vivo skin wound healing model, HYCO-6 and HYCO-13 were given via gavage at 40 mg/kg (80 μmol/kg) to mice daily for 14 days. Only HYCO-6 showed accelerated wound healing. It is worth mentioning that without further explanation in the study, HYCOs did not ameliorate wound closure when applied topically, indicating inefficient bioavailability through transdermal absorption. In an in-vivo mouse model of imiquimodinduced psoriasis, HYCO-3 and HYCO-13 were given by gavage twice a day at a dose of 25 mg/kg (50 μmol/kg) for 7 days. HYCO-3 and HYCO-13 markedly attenuated erythema, epidermis thickness, and scaling in mice, and the effects were comparable to or better than the effect exerted by DMF alone. In an in-vivo mouse model of multiple sclerosis, significant improvements in clinical manifestations were achieved by oral gavage of HYCO-3 at 25 mg/kg twice a day and HYCO-13 at 25 mg/kg or 50 mg/kg once or twice a day for 40 days. Such results suggest that some HYCOs could be good candidates for oral delivery as anti-inflammation therapy.
Oxidation triggered CO-RMs.
As common reagents for reduction and carbonylation, hexacarbonyl molybdenum and octacarbonyldicobalt are widely used in organic synthesis [146,147]. The release of CO can be triggered by pyrolysis, ligand exchange, and/or oxidation of the zero-valent metal core. Among these possibilities, oxidation can take place under physiological conditions. Exchanging two CO ligands in these metal carbonyls with an amino acid or bioactive small molecule can enhance the water solubility and bioavailability of the CO-RM derivative formed in situ. At this point, it should also be mentioned that Ru-based CORM-2 and CORM-3 are also redox-active and can react with nucleophiles and certain oxidizing reagents under physiological conditions [121,123,148,149]. However, these chemistry issues are not included in this discussion. Because of the complexity of the reactions involved in CO release, the overall kinetics can be complex and can only be discussed when conditions are precisely defined. When oxidation by molecular oxygen is the mechanism of release, the reaction kinetics are expected to be dependent on the partial pressure of oxygen.
The molybdenum (Mo)-based CO-RM, fac-[Mo(0)(CO)3(histidinate)]Na (ALF186), releases CO upon oxidation and is known to have the highest CO release ratio among all CO donors [150]. In in-vitro CO release experiments, incubating ALF186 in solution without oxygen showed no release of CO. In contrast, incubation under normoxic conditions in a buffered solution at 37 °C liberated 1 equivalent of CO after 0.5 h and 2.26 equivalents (75% of the CO load) after 2 h. However, in whole blood, all three equivalents of CO were found to “instantaneously” release to hemoglobin [150]. Although the detailed reaction mechanism has not been fully explored, we reason that the strong propensity for ALF186 to reduce methemoglobin, ferromyoglobin, and ferrocytochrome c as demonstrated in the original study [150] suggests the possibility of ALF186 oxidation through extracting molecular oxygen from oxyhemoglobin, leading to the release its CO ligand. This proposed mechanism, if true, means ALF186 can “swap” the oxygen on hemoprotein with its own CO by involving a redox reaction. Through this mechanism, the exchange kinetics and equilibrium of the oxygen on hemoglobin with a CO molecule do not rely on the simple “intrinsic Kd” values. The redox chemistry involved facilitates the highly efficient formation of COHb after intravenous administration of ALF186. Experimental results also support this notion through comparison of the CO release efficiencies after i.p. and p.o. administration. Intraperitoneal injection of ALF186 at a dose of 20 mg/kg or 40 mg/kg to mice gave an increase of COHb to 15% and 25% within 10-15 min, respectively. Assuming all released CO was taken up to form COHb, the increased COHb levels corresponded to three equivalents of CO carried in the injected dose of ALF186. The delayed CO release in the peritoneal cavity is consistent with the low oxygen content (17 mmHg) at this location [151]. Thus, one would expect that COHb is formed mostly due to the absorption of intact ALF186 and then CO release in the bloodstream. The release kinetics of ALF186 through oral administration was even slower and more in line with the kinetics from the in-vitro buffer studies, which peaked at around 1 h. The maximal COHb level after gavage of 500 mg/kg ALF186 was 35-40%, which was said to correspond to roughly 50% of COHb administered by i.p. at the same dose. Absorption of ALF186 through the GI system is expected to be slow. Considering the moderate oxygen level (about 60 mmHg) in the GI, the release of CO is expected to be mainly via slow oxidation. Therefore, due to the unique oxygen-triggered release mechanism, oral administration of ALF186 is less efficient in delivering CO than that of the i.p. or i.v. route. It needs to be noted that the effect of the redox activity of ALF186 may go beyond simple CO-O2 exchange. Other observations in changes of ROS levels and decreasing oxygen partial pressure may not solely be attributed to the function of CO per se. The redox chemistry of ALF186 may convolute result interpretations and mechanistic understandings of this otherwise low-toxicity, well-tolerated, and highly efficient CORM [150]. Further, the possibility of oxygen-“scavenging” or “deprivation” effect also needs to be considered by these oxidation-sensitive CO-RMs when the concentration is sufficiently high (comparable to that of Hb).
For the cobalt-based hybrid CO-RMs, HYCO-4 and HYCO-10 [141], CO release is also found to be triggered by the oxidation of the cobalt(0) core, which could be promoted under oxidative conditions such as oxidative stress induced by inflammation [142]. The conjugated chromone (HYCO-4) and cinnamic acid (HYCO-10) moieties are known to activate Nrf2 and subsequently induce HO-1 expression. In an in-vivo study, mice were given HYCO-4 and HYCO-10 orally at a dosage of 3 mg per mouse (about 100 mg/kg, 200 μmol/kg, formulated with sesame oil). After 6 h, COHb increased from 0.5% to about 3.5% by HYCO-4 and about 1.8% by HYCO-10. It should be noted that in in-vitro studies with hemoglobin and a CO probe, HYCO-10 did not show robust CO release. Considering the relatively high dosage used, the in-vivo results are consistent with low CO delivery efficiency after oral administration. On the other hand, the dosage of HYCO-10 did show a robust increase of HO-1 expression in the lung and liver, which is consistent with the release of the Nrf2 activator part of the molecule.
3.3.2. Oral delivery formulations based on metal-based CO-RMs
As discussed in the aforementioned examples, metal-based CO-RMs have brought transition metal coordination chemistry and CO delivery to the biological system and yielded fruitful discoveries and possible drug candidates. However, questions remain in terms of the developability of CO-RMs into clinical therapies, including toxicity concerns of transition metals, especially for chronic diseases, quality control and chemical tractability issues due to the convoluted CO chemistry of metal complexes [113,148,152,153] and the uncertain stoichiometry of the released CO in some cases [148]. Beyond these, there is the question of to what extent the intrinsic chemical reactivity of the metal complexes, such as their reducing ability, thiol depletion, redox catalysis, and potential ion channel blockage, should account for the biological activities observed [23,111,123,136,148,149,152,154,155]. All these issues suggest the reactivity of these CO donors, as well as their side-products after CO release, should be taken into consideration at the molecular level when analyzing results. Some of the issues could be addressed by formulation and through material chemistry manipulations, if such efforts can prevent metal leakage.
Many efforts have been made in solving the problem of safely and effectively delivering CO-RMs through the GI system. Aim at reducing exposure to the donor molecule while enhancing target tissue exposure to free CO, elaborate drug delivery systems have been designed to deliver CO to the GI tract. It is known that CO release from CORM-2 can be triggered by sulfite species through ligand exchange [148]. Steiger et al. took advantage of this reaction and designed an oral CO release system (OCORS) for sustained and controlled CO delivery to gastric and/or intestinal sites [156]. OCORS was designed as an oral tablet microreactor (7 mm dimension) with cellulose acetate/PEG coating. The tablet core consists of citric acid-coated sodium sulfite crystals and CORM-2. When water diffuses through the semi-permeable coating membrane, the citric acid coating layer dissolves, allowing the dissolution of the sodium sulfite crystals subsequently. The liberated sulfite would react with CORM-2 to release CO. Without exposure to water, the citric acid coating of sodium sulfite crystals prevents its interaction with CORM-2 during storage. The system was shown to deliver CO for up to 10 h, with nearly linear release kinetics for a long period of time (from 30 to 240 min). Because the release is a bimolecular reaction between sulfite and the CO-RM, the linear release kinetic may mean that dissolution of the citric acid layer and/or the sulfite crystals was the rate-limiting step under the experimental conditions because of the constant nature of water concentration. Additionally, the linear nature could also simply reflect the “steady-state” concentration of available sulfite, which is the summary results of dissolution, diffusion, and reaction with CO-RM. The citric acid coating membrane not only addresses the function of controlling the pH within the tablet but also acts as an ionic strength modifier to increase CO release. Four equivalents of CO were released from such a formulation, giving almost a quantitative yield. Although the visual integrity of OCORS tablets remained unchanged throughout this study, approximately 90% of ruthenium escaped after 13 h, which still presents an issue of ruthenium exposure [157].
Two years later, the same group modified the OCORS system into a micro-scale oral carbon monoxide release system (M-OCORS) [158]. The tablet was miniaturized to a 1-mm coated tablet containing coated sodium sulfite as the trigger and uncoated CORM-2 as the CO donor. Different coating procedures and materials with varying hydrophobicity allowed for controlling water permeability and hence tunability of the CO release rate. The authors were able to tune the t1/2 (time to half-maximal release) from 0.11 h (fast release) to about 9.1 h (slow-release). Two 1-mm tablets administered to mice via oral gavage increased the COHb level from a basal level of 0.23 ± 0.03% to 0.51 ± 0.05% in 30 min (fast release formula), which declined to the basal level within 3 h. With a slow-release formula, COHb level peaked at 0.69 ± 0.14% after 3 h and then dropped to the basal level within 12 h. PK modeling of COHb levels using the Bateman equation [159] showed an acceptable correlation to the experimental data of the fast release formula. The slow-release formulation matched the murine gastrointestinal transition time and was shown to prevent TNBS-induced colitis in mice with COHb level changes of less than 1%. Such results show that local (GI) delivery of CO was able to minimize systemic CO exposure and achieve efficacy. As a comparison, a steady state of about 20% COHb was reported for inhalation CO (200-250 ppm) to show a protective effect in a colitis model [160]. However, the post-release products of M-OCORS, including CORM-2 and sodium sulfite in an aqueous solution (depleted-CORM-2) showed a significant reduction of cell survival at 0.25-1 mM. This toxicity issue of the escaped ruthenium by-products prompted the researchers to modify the OCORS by replacing the sulfite/CORM-2 system with an esterase/rac-1 system in the same study [158]. rac-1(Figure 1) is an iron-based CORM aimed at avoiding ruthenium toxicity. The new esterase-triggered OCORS (E-OCORS) is a polymer coated formulation with alternated layers of porcine liver esterase (PLE) and rac-1 which are separated by disodium hydrogen phosphate crystals as the scaffolds. It is designed to release CO upon contact with water in the GI system. Upon exposure to water, this formulation started to release CO after a lag time of about 3.5 h, and exhibited zero-order release kinetics (linear) for 13.3 h. Further experiments are yet to be done to evaluate its efficacy and toxicity. However, the release of the metal-based CO-RMs and their CO-depleted product from the tablet is still inevitable with this formulation. In 2018, the same group evaluated the M-OCORS platform to ameliorate postoperative ileus (POI) through oral delivery to the small intestine [161]. Twenty OCORS tablets with 0.5-mm diameter were administrated to mice via a custom-made gavage needle. 55% of the tablets reached the small intestine after 1 h, with the COHb level increasing to 5.2%. Although CO was reported to show a therapeutic effect on POI, orally given M-OCORS tablets did not effectively reduce surgery-induced GI muscular inflammation and transit retardation in mice.
Yin et al.[162] reported a micelle formulation to deliver CO by encapsulating CORM-2 using a biocompatible polymer, styrene-maleic acid copolymer (SMA). It addressed the water-solubility issue of CORM-2, enabled sustained CO release within ~21 h, prolonged half-life in circulation, and localized CO accumulation in inflammatory tissues. The loading ratio of CORM-2/micelle was calculated to be around 11%. The water solubility of the resulting SMA/CORM-2 micelle was determined to be >50 mg/ml in PBS. When CO release was studied in 100% fetal bovine serum, it was found that the release half-life of SMA/CORM-2 was about 8 h. In contrast, CORM-2 alone led to full release within 30 min. The overall CO release stoichiometry from SMA/CORM-2 was the same as that of CORM-2. Then CO delivery efficiency to mice was determined with GC by measuring the CO released to the headspace in the vial containing blood samples collected at different time points after administration of CO donors and NO-releasing agent NOC7, which served as a competitive CO purging agent to release CO from hemoproteins. Specifically, when SMA/CORM-2 was administered orally to mice at 20 mg/kg, the total blood CO concentration slowly increased to about 28 pmol/ml (or 28 nM) during the initial 24 h. This concentration was about 3 folds of the basal level (~10 pmol/ml). In comparison, i.v. administration at the same dose led to a peak level of 31 pmol/ml (31 nM) within the same period of 24 h. The CO concentrations were interpreted to represent COHb levels, and the results were interpreted as high systemic oral bioavailability of CO from the SMA/CORM-2 system. In a dextran sulfate sodium (DSS) induced colitis model in mice, orally administrated SMA/CORM-2 at 40 mg/kg ameliorated colitis symptoms and showed tissue-protective and anti-inflammatory activities. However, it should be noted that the CO concentration value of 30 nM only corresponds to about (3.75 × 10−4)% COHb, which is much lower (by about 1000-fold) than the reported basal CO level discussed in other studies [141,143]. Considering 0.5% COHb as a basal level, we estimate the total Hb-bound CO concentration to be about 40 μM if one assumes the concentration of mouse Hb being 8 mM [163]. The same issue could also be found in the tissue concentration, where the study showed around 5 pmol/g while most studies in the literature showed about 5 pmol/mg [164]. We have communicated with the correspondence authors of this publication, and the discrepancy seems to be the result of typographical errors in unit conversions, for which the authors indicated their intent to submit a corrigendum.
Table 1 summarizes the key features of the examples discussed above. Due to the non-negligible reactivity/toxicity of the transition metal-containing byproducts or the CORMs, some additional considerations of control experiments would improve the rigor of future studies.
Table 1.
Summary of bioactivities of metal-based CO-RMs in vivo via oral administration
| Compound | Activity | Animal model | Dosage | COHb (%) | Side product control | Ref |
|---|---|---|---|---|---|---|
| CORM-2 | gastric injury | rats | 5 mg/kg 10 mg/kg | 1.6% (5mg/kg) 2.2% (50mg/kg) | RuCl3 | [137] |
| CORM-A1 | cerebrovascular dysfunction | piglets | 2 mg/kg | NA | B(OH)3 | [128] |
| ALF-186 | acute toxicity evaluation | mice | 500 mg/kg | 35%-40% | NA | [150] |
| CORM-401 | obesity | mice | 15 mg/kg 30 mg/kg | 2.5% 4.5% | iCORM | [139] |
| HYCO-4 | inflammation | mice | 3 mg/kg | 4% | NA | [141] |
| HYCO-10 | inflammation | mice | 3 mg/kg | 2% | NA | [141] |
| HYCO-3 | inflammation | mice | 40 mg/kg | 5% | NA | [143] |
| HYCO-6 | Skin wound | mice | 40 mg/kg | 2% | NA | [145] |
| HYCO-3 HYCO-13 | Psoriasis, multiple sclerosis | mice | 25 mg/kg | 2% 5% | NA | [145] |
| CAI-CORM | rheumatoid arthritis | rats | 10-100 mg/kg | NA | NA | [165] |
| M-OCORS | colitis | mice | 2 tablets | 0.69% | abrogated - CORM-2 | [158] |
| M-OCORS | postoperative ileus | mice | 20 tablets | 5.2% | NA | [161] |
| SMA/CORM2 | Rheumatoid Arthritis | mice | 20 mg/kg | NA | NA | [162] |
3.3.3. Metal-free organic CO donors and prodrugs
To avoid metal-related issues, organic CO donors have been designed, leading to a new direction in the field [32-34,118,119,134]. In this area, extensive efforts in introducing innovative chemistry to generate CO under physiological conditions have been made in the last 6-7 years. We advocate for the term “CO prodrug” to bring pharmaceutical considerations to the forefront in designing CO-based therapeutics that go beyond simply releasing CO. In most cases, the precursor for CO is in the form of ketone, carboxylic acid, amide, ester, etc. These organic CO donors are designed to release CO under bio-relevant conditions such as light [166,167] or physiologic conditions [117]. They have tunable release rates [31,34,116,117]; are designed with single or dual-triggers to target tissue/organ [133,168,169]; and are capable of localizing in subcellular compartments such as mitochondria [4,133]. They have predictable CO release stoichiometry due to the clearly defined chemistry; showed in-vitro anti-inflammatory [34] and antibacterial effects [170]; and have been studied in animal models of colitis [116], kidney injuries [24,28], liver injuries [115,168], gastric mucosal injuries [77], and systemic inflammation [115] with promising therapeutic potentials. Readers are encouraged to consult recent reviews for in-depth coverage of this area [4,32,34,166,171]. In the following section, we mainly focus on the feasibility and developability issues of these organic CO donors for oral delivery. For the same reason, photo-triggered organic donors such as organic photoCORMs [31,166] are not included.
Among the strategies used to develop organic CO donors triggered by physiological conditions, there are two general types of chemistry used: (1) a cheletropic reaction to extrude CO from cyclopentadienone adopted by Larson’s group [172] and our lab [33,36,115,116,168,170,173] and (2) decarbonylation chemistry of the oxalyl moiety [24]. In the first strategy, the formation of the norbornadiene-7-one is the key and rate-limiting step to trigger CO release through cheletropic reaction (Figure 4) [23,34]. Thus, the release kinetics depend on the preceding step(s). Reactions such as intermolecular Diels-Alder reaction (second order) [168], intramolecular Diels-Alder reaction (first order) [117], and elimination reaction (pH dependent, but pseudo-first order under certain pH) [172,174] are utilized with modification to yield this intermediate. CO release can be triggered by physiological conditions such as pH, enzyme, aqueous conditions, or enrichment in mitochondria. When the triggering condition is met, CO release can happen locally at the administration site, such as the GI lumen or the peritoneal cavity, or in the vascular system after direct intravenous injection. Therefore, depending on the triggering mechanism and bioavailability of the CO prodrug itself, GI administration of organic CO prodrugs are feasible. Available conditions such as aqueous environments in the GI fluid and vascular system, esterase in the GI tract and in the blood, neutral pH in the intestinal fluid and in the blood can be harnessed to trigger the reaction to form the norbornadien-7-one intermediate followed by CO release locally in the GI tract or systemically in the bloodstream.
Figure 4.
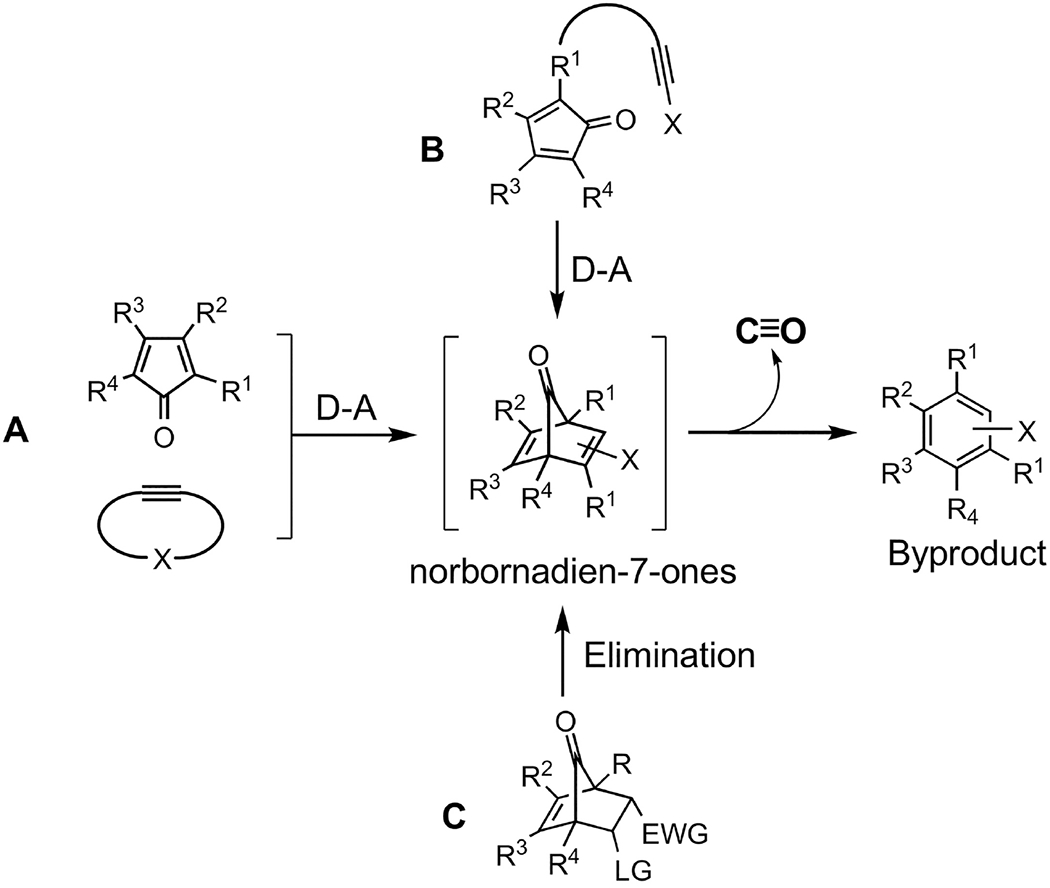
The general design of cheletropic reaction-based CO prodrugs.
In collaboration with Magierowski’s group, we investigated an organic CO prodrug CO-111 (Figure 5) for its gastroprotection activity via oral administration in rats [77]. CO-111 can release a stoichiometric amount of CO under neutral and acidic conditions in the GI tract. The half-life in the aqueous solution was determined to be about 20 min. Pretreatment with CO-111 at a dose of 0.1 mg/kg (0.25 μmol/kg) through oral gavage was able to offer significant protection against aspirin and 75% ethanol-induced mucosal injuries, while the byproduct CP-111 (0.1 and 0.5 mg/kg) as a control did not show protective or cytotoxic effect. It is important to note that at a dosage of 0.1 mg/kg, the CO saturation level of the gastric mucosa tissue increased by about 2.5 folds as determined by GC analysis. Compared with similar studies discussed in the previous sections, the effective dosage of CO-111 was much lower than CORM-2 (10 μmol/kg) [137] and the CO solution described (4 μmol/kg) [108]. The results indicate that CO-111 is very efficient in delivering CO locally to the gastric tissue. CO-111 may release CO in both the gastric fluid and the mucosal cells after being absorbed. At the molecular level, aside from attenuating pro-inflammatory biomarkers and gene expression, CO-111 also enhanced the protective PEG2 system, which was found to be involved in the physiological defensive mechanism against necrotic effects of ethanol. Overall, oral administration of CO-111 appears to be a promising and safe pharmacological approach for further studies related to gastric disorders.
Figure 5.
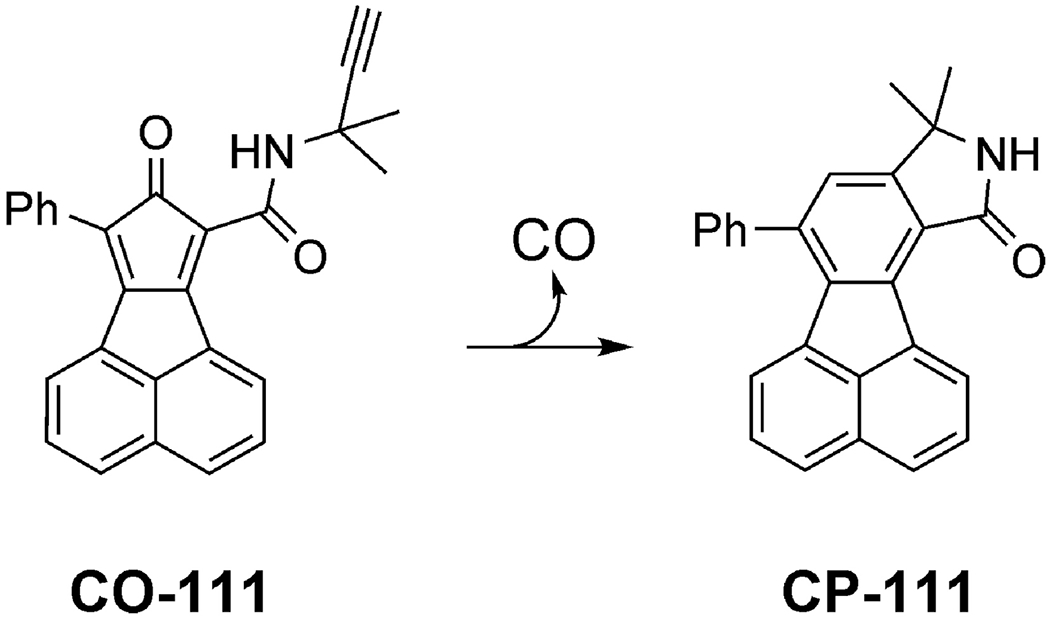
The chemical structure of CO prodrug (CO-111) used in the gastric protection studies and the corresponding CP product after CO release
Using an elimination reaction to generate norbornadiene-7-one, Larson’s group developed a CO donor, which can release CO at neutral pH (Figure 5). Rats were given oCOm-19 and oCOm-21 at a dosage of 133.3 μmol/kg and 33.3 μmol/kg in water, respectively. COHb levels of the oCOm-19 group increased from a basal level of 0.7 ± 0.28 % to 0.9% (no statistical significance) after 1 h, and then reached 1.3 ± 0.14 % after 3 h. As for oCOm-21, despite the dosage being about one-fourth of oCOm-19, COHb levels increased from 0.54 ± 0.36 to 1.52 ± 0.44 % after 1 h and 1.74 ± 0.45 % after 2 h. The author attributed the difference in the bioavailability to the less soluble nature of oCOm-19, which might lower the bioavailability of the compound itself, leading to a lower level of CO in the blood.
Recently, we reported the utility of the decarboxylation-decarbonylation chemistry to release CO from a new type of sweetener-based CO prodrugs (Figure 7)[24]. In this approach, two leaving groups chosen from either saccharin or acesulfame with pKa less than 4 were used in making oxalyl amides. Upon exposure to water in the GI lumen, hydrolysis triggers to decaroxylation-decarbonylation cascade reaction to release CO and CO2, leaving saccharin or aceulfame as the only non-gaseous byproduct (Figure 7). As FDA approved sweeteners as food additives, the safety concern of the byproducts is largely mitigated. In-vitro CO release half-lives of the saccharin-based CO-306 and the acesulfame-based CO-307 in PBS:ACN (60:40, v/v) solution were 1.3 min and 9.5 min, respectively. In-vivo CO delivery of CO-306 in mice through oral administration at a dosage of 100 mg/kg gave a peak COHb level of 5.2% within 10 min, which gradually decreased to baseline in 2 h. Lowering the dosage to 50 mg/kg led to a peak COHb level of 4.2%, which returned to the baseline in 1 h. In a mouse rhabdomyolysisinduced acute kidney injury (AKI) model, oral administration of CO-306 formulated with activated charcoal at a dosage of 50 mg/kg significantly attenuated AKI as shown by the decreased blood urea nitrogen level, tubular injury score, kidney injury molecule 1 (Kim1) biomarker transcription and expression, and increased post-AKI survival rate. The results indicate that these artificial sweetener-based CO prodrugs are capable of delivering CO through oral administration and are promising candidates for future development of CO-based therapies.
Figure 7.
Sweetener-based CO prodrugs design and CO release mechanism
4. Comparative pharmacokinetic studies of CO prodrugs after administration via different routes in mice
Pharmacokinetic (PK) studies are an essential part of drug development. In this sense, there is no difference between a small-molecule drug and a prodrug of a gaseous molecule. The discussions in the previous sections are based on reported pharmacological studies of CO donors. In most cases, COHb levels or tissue CO contents were reported as proof of CO delivery to the body and/or tissue. We felt that a dedicated pharmacokinetic study was needed to address some critical developability issues of CO prodrugs to answer the following questions: (1) can orally- delivered CO prodrugs lead to elevated levels of CO using COHb as an indicator; (2) how different delivery routes (i.v., i.p., or p.o.) compare in terms of efficiency in systemically delivering CO as measured by COHb levels; and (3) to what degree we can minimize the systemic exposure of the “carrier” molecule. Some of these questions (1 and 2) have been partially addressed through the aforementioned discussions. However, detailed PK studies of the specific organic CO donors in this section should offer a more comprehensive picture. As a pilot step to assess PK profiles in a side-by-side comparison, we selected several organic CO prodrugs to study their pharmacokinetic profiles (Figure 8) [73]. We selected these CO prodrugs because they have defined structure-release kinetic profile, quantitative release stoichiometric [117], somewhat consistent release rate regardless of minor perturbations in environmental factors such as pH, and validated therapeutic activities both in-vitro and in-vivo [28,77,115,116]. These CO prodrugs were administered through different routes (i.v., i.p. and p.o.) in mice (n≥3 for each group). The PK profiles of the prodrugs and the byproducts after CO release were evaluated (Table 2). We should note that this was the first and the only such comprehensive PK studies related to CO delivery via different routes.
Figure 8.
Chemical structures of CO prodrugs used in the PK studies described in Table 2
Table 2.
Results of PK studies of CO prodrugs
| Compound | t1/2 (h) in plasma | COHb peak (%) | Time to Peak (h) | COHb AUC (%·h) p.o. (25 mg/kg) | Oral absorption of intact CO prodrugs | Oral absorption of byproduct | CO delivery efficiency (%) |
||
|---|---|---|---|---|---|---|---|---|---|
| i.v. | i.p. | p.o. | |||||||
| CO-103 | 1.3 | 3.5 | 1 | 4.1 ± 0.3 | + | ++ | 100 | 3.7 | 11.7 |
| CO-125 | 1.6 | 3.2 | 1 | 6.1 ± 1.4 | + | + | |||
| CO-116 | 1.5 | 4.1 | 1 | 5.6 ± 1.1 | − | ++ | |||
| CO-104 | 6.0 | 2.6 | 1 | 2.3 ± 0.4 | ++ | + | |||
| CO-115 | 5.1 | 3.5 | 1 | + | |||||
Compounds ranked by their CO release rate. t1/2: release half-life of CO prodrug in SIF (50% DMSO); COHb AUC: area under the curve for blood COHb level subtracting the pre-administration baseline from time zero to the last sampling time point. Results are presented as the mean ± SD (n ≥ 3). CO delivery efficiency: ratio of COHb AUC of a given route over i.v. administration using i.v. result as 100%. Qualitative criteria: -: lower than detection limit; +: 0-2.5 μM·h; ++: >2.5 μM·h. As a comparison, i.v. dose of 25 mg/kg should give an AUC of 25 μM·h.
To answer the first question, all the test compounds are orally bioavailable for delivering a meaningful amount of CO to the body. However, the efficiency is different depending on their respective characteristics, such as release kinetics, solubility, and bioavailability of the prodrug. For instance, CO-104 with a slow-release kinetic has good oral bioavailability of the prodrug itself. However, its CO delivery efficiency is lower than CO-116 with a faster release kinetic but with much lower oral bioavailability of the prodrug itself. Such results indicate 1) CO released in the GI lumen can be readily absorbed into the bloodstream leading to elevated COHb levels, and 2) the slow releasers such as CO-104 and CO-115 have a lower overall COHb AUC (area under the curve of a drug’s concentration profile). It should be noted that the GI emptying transition time is about 5 h for mice [175]. This means that for either CO-104 or CO-115, if not absorbed into the general circulation, around half of the given dose had not fully released CO before being cleared out from the body, leading to a lower COHb AUC. Further, the COHb peak is dependent on the balance of CO generation and elimination rates, which can be fast with the t1/2 being about 5 h in humans [176] and 25 min in mice [53]. The rapid biological elimination of CO also contributes to the lower overall COHb availability for the slower releasers. Along the same line, CO donors capable of fast release are expected to provide higher efficiency as defined by AUC in the context of oral delivery.
To answer the second question, CO delivery efficiency has been studied using CO-103 as an example. The results show that i.v. injection has the highest CO delivery efficiency as measured by the COHb AUC, followed by oral administration and then intraperitoneal injection. However, when the plasma AUC of the prodrug (CO-103) itself (not CO) was examined, i.p. administration led to a higher level of bioavailability than that of oral administration. CO release from this type of CO prodrugs is promoted by an aqueous environment. Because the water content in the peritoneal cavity is low, i.p. injection is expected to mainly rely on absorption of the prodrug (CO-103) into the general circulation to trigger CO release. On the other hand, oral administration to the GI with ample water is expected to lead to quick CO release from the prodrug as compared to i.p. injection. However, the seemingly contradicting results of a lower CO bioavailability but a higher prodrug bioavailability from i.p. injection compared to the oral administration indicate convoluted kinetics for systemic CO delivery that need further studies.
To answer the last question, the overall COHb AUC is not dependent on the absorption of the CO prodrug, as long as CO release happens inside the body. Therefore, it is not necessary to absorb the CO prodrug into the blood circulation, which is good for minimizing the side effect of the side product. To diminish the systemic exposure of the “carrier” byproduct molecule as well as the prodrug itself, compounds with lower bioavailability such as CO-103, -125, and -116 are preferred. The chemical property of a prodrug clearly has a significant impact on the bioavailability of the CO prodrug and the byproduct. For example, CO-125 with a morpholine moiety that forms a cation in the acidic gastric environment or even a neutral environment has a lower bioavailability. Therefore, by rational design of the compound and formulation method, systemic exposure of the CO prodrug and the side product can be further minimized without compromising the ability to deliver CO to the systemic circulation.
There are some unique technical and analytical challenges in studying the PK profiles of such prodrugs. First, although COHb level is the parameter most relevant to CO exposure, there is a need to monitor the levels of COHb, the prodrug, and the side product to gain a deep understanding of the interplay of various factors in determining the overall PK outcome. Secondly COHb determination is technically challenging, requiring very careful work. Thirdly, some vehicle may have effects on both membrane permeability and HO-1 expression level.[177] Therefore, caution is needed in comparing data from different experiments if conducted under different conditions or different vehicles were used. Fourth, the pathophysiological states of the subject may have a profound effect on CO’s tissue distribution. This is exacerbated by severe pathological states such as acidosis.
It should also be noted that the systemic COHb level may not necessarily be correlated with the therapeutic efficacy if localized CO delivery is applied. In the examples described in previous sections, such as gastroprotection by oral administration of CO-111, the elevation of local CO concentration in the tissue (gastric mucosa) was achieved by oral administration of a low dosage of CO-111 (0.1 mg/kg) [77]. Though COHb level was not assessed in the study, such a low dosage of this CO prodrug is not expected to lead to a significant elevation of systemic COHb level. As a highly diffusible gas, CO’s dissolution and diffusion across the biological membrane is dependent on the partial pressure difference according to Henry’s law and Fick’s law. Therefore, if the target tissue is accessible by the CO donor through targeted delivery or formulation, it would allow for a relatively high local CO concentration to primarily engage cellular targets in the target tissue. Lowering systemic CO exposure level may also decrease the potential toxicity of the CO donor. As such, a high systemic COHb level may not necessarily serve as a good parameter for evaluating the therapeutic effects of a targeted CO delivery approach. In such a case, tissue CO concentration should be evaluated in addition to the systemic COHb level to demonstrate the efficiency of CO delivery and understand the correlation between the pharmacological efficacy and CO delivery.
5. Concluding remarks
The therapeutic potential of CO has been well recognized through a large number of studies. To date, a good arsenal of CO donors has provided various options to deliver CO in addition to inhalation. An ideal chemical CO donor should deliver CO in a traceless fashion or use benign “carrier” components. Compared with intravenous administration, oral administration offers a pharmaceutically desirable approach for drug administration. Further, there are many inflammatory conditions of the GI system that can be treated by CO. Exogenous CO administrated by oral or parenteral routes has already been demonstrated to have therapeutic effects in animal models of gastric injury [17,77], colitis [81], diabetic gastropareses [139], postoperative ileus [25,26], inflammatory bowel disease [20,21], and sepsis [144]. Through oral administration, CO released locally in the GI system can also directly engage local targets, thus requiring a lower dose and leading to a lower level of systemic exposure to CO. Previous studies have shown that CO given through the GI tract exhibits a preferable CO-related toxicity profile than inhalation. Based on these reasons, there is a need for various options or formulations for CO “in a pill.” From a clinical application perspective, oral delivery is a preferable and convenient route for drug administration due to ease of administration and patient compliance, especially for treating chronic diseases.
Towards the idea of “CO in a pill,” much needs to be done to overcome the challenges beyond conventional drug delivery. These include designing CO donors and formulations with stoichiometric and controllable CO release; evaluating CO-dependent and independent activities, pharmacokinetics, bioavailability, and toxicity; and minimizing systemic toxicity of both CO and the carrier molecule; to name a few. All of these developmental issues call for collaborative efforts among people from different disciplines, including chemists, pharmacologists, toxicologists, and physicians.
Figure 6.
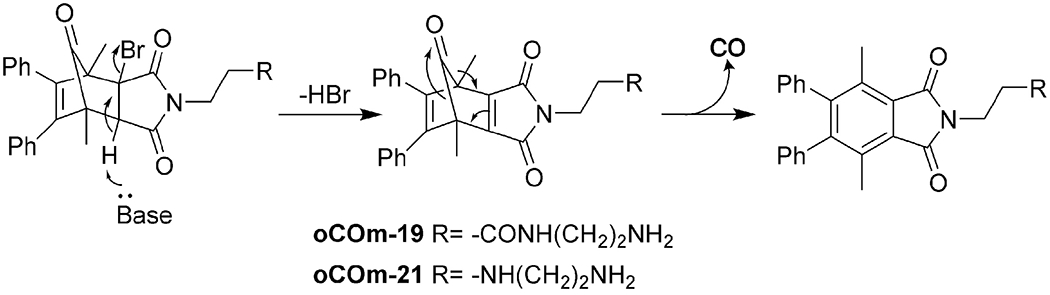
Elimination-based CO prodrug developed by Larson’s group
Acknowledgments.
We gratefully acknowledge the partial financial support by the National Institutes of Health (R01DK119202) for our CO-related work, the Georgia Research Alliance for an Eminent Scholar endowment to BW, and internal resources at Georgia State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Motterlini R, Otterbein LE, The therapeutic potential of carbon monoxide, Nat Rev Drug Discov 9 (2010) 728–743. [DOI] [PubMed] [Google Scholar]
- [2].Wu L, Wang R, Carbon monoxide: endogenous production, physiological functions, and pharmacological applications, Pharmacol Rev 57 (2005) 585–630. [DOI] [PubMed] [Google Scholar]
- [3].Yang X, Lu W, Hopper CP, Ke B, Wang B, Nature’s marvels endowed in gaseous molecules I: Carbon monoxide and its physiological and therapeutic roles, Acta Pharm Sin B (2020) In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yang X, Ke B, Lu W, Wang B, CO as a therapeutic agent: discovery and delivery forms, Chin J Nat Med 18 (2020) 284–295. [DOI] [PubMed] [Google Scholar]
- [5].Tenhunen R, Marver HS, Schmid R, The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase, Proc Natl Acad Sci U S A 61 (1968) 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Coburn RF, Williams WJ, Forster RE, Effect of Erythrocyte Destruction on Carbon Monoxide Production in Man, J Clin Invest 43 (1964) 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ryter SW, Alam J, Choi AM, Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications, Physiol Rev 86 (2006) 583–650. [DOI] [PubMed] [Google Scholar]
- [8].Papapetropoulos A, Foresti R, Ferdinandy P, Pharmacology of the ‘gasotransmitters’ NO, CO and H2S: translational opportunities, Br J Pharmacol 172 (2015) 1395–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Almeida AS, Figueiredo-Pereira C, Vieira HL, Carbon monoxide and mitochondria-modulation of cell metabolism, redox response and cell death, Front Physiol 6 (2015) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gozzelino R, Jeney V, Soares MP, Mechanisms of cell protection by heme oxygenase-1, Annu Rev Pharmacol Toxicol 50 (2010) 323–354. [DOI] [PubMed] [Google Scholar]
- [11].Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiasch E, Bach FH, Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide, Antioxid Redox Signal 4 (2002) 321–329. [DOI] [PubMed] [Google Scholar]
- [12].Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM, Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway, Nat Med 6 (2000) 422–428. [DOI] [PubMed] [Google Scholar]
- [13].Otterbein LE, Carbon monoxide: innovative anti-inflammatory properties of an age-old gas molecule, Antioxid Redox Signal 4 (2002) 309–319. [DOI] [PubMed] [Google Scholar]
- [14].Kaczorowski DJ, Zuckerbraun BS, Carbon monoxide: medicinal chemistry and biological effects, Curr Med Chem 14 (2007) 2720–2725. [DOI] [PubMed] [Google Scholar]
- [15].Brugger J, Schick MA, Brock RW, Baumann A, Muellenbach RM, Roewer N, Wunder C, Carbon monoxide has antioxidative properties in the liver involving p38 MAP kinase pathway in a murine model of systemic inflammation, Microcirculation 17 (2010) 504–513. [DOI] [PubMed] [Google Scholar]
- [16].Kim HH, Choi S, Therapeutic Aspects of Carbon Monoxide in Cardiovascular Disease, Int J Mol Sci 19 (2018) 2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Magierowska K, Brzozowski T, Magierowski M, Emerging role of carbon monoxide in regulation of cellular pathways and in the maintenance of gastric mucosal integrity, Pharmacol Res 129 (2018) 56–64. [DOI] [PubMed] [Google Scholar]
- [18].Nemecek D, Dvorakova M, Heroutova I, Chmelikova E, Sedmikova M, Anti-apoptotic properties of carbon monoxide in porcine oocyte during in vitro aging, PeerJ 5 (2017) e3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Puentes-Pardo JD, Moreno-SanJuan S, Carazo A, Leon J, Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease, Antioxidants (Basel) 9 (2020) 1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Katada K, Takagi T, Uchiyama K, Naito Y, Therapeutic roles of carbon monoxide in intestinal ischemia-reperfusion injury, J Gastroenterol Hepatol 30 Suppl 1 (2015) 46–52. [DOI] [PubMed] [Google Scholar]
- [21].Kim HJ, Joe Y, Yu JK, Chen Y, Jeong SO, Mani N, Cho GJ, Pae HO, Ryter SW, Chung HT, Carbon monoxide protects against hepatic ischemia/reperfusion injury by modulating the miR-34a/SIRT1 pathway, Biochim Biophys Acta 1852 (2015)1550–1559. [DOI] [PubMed] [Google Scholar]
- [22].Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, Soares MP, Otterbein LE, Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation, J Clin Invest 124 (2014) 4926–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang X, de Caestecker M, Otterbein LE, Wang B, Carbon monoxide: An emerging therapy for acute kidney injury, Med Res Rev 40 (2020) 1147–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De La Cruz LK, Yang X, Menshikh A, Brewer M, Lu W, Wang M, Wang S, Ji X, Cachuela A, Yang H, Gallo D, Tan C, Otterbein L, de Caestecker M, Wang B, Adapting decarbonylation chemistry for the development of prodrugs capable of in vivo delivery of carbon monoxide utilizing sweeteners as carrier molecules, Chem Sci 12 (2021) 10649–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nakao A, Toyokawa H, Tsung A, Nalesnik MA, Stolz DB, Kohmoto J, Ikeda A, Tomiyama K, Harada T, Takahashi T, Yang R, Fink MP, Morita K, Choi AM, Murase N, Ex vivo application of carbon monoxide in University of Wisconsin solution to prevent intestinal cold ischemia/reperfusion injury, Am J Transplant 6 (2006) 2243–2255. [DOI] [PubMed] [Google Scholar]
- [26].Korolkiewicz RP, Sein-Anand J, Ruczynski J, Rekowski P, Bieniaszewski L, Chodorowski Z, Petrusewicz J, Ujda M, Dabkowski J, Bitel M, Kato S, Takeuchi K, The role and interactions of nitric oxide (NO), carbon monoxide (CO), and prostanoids in the pathogenesis of postoperative ileus in rats, J Gastrointest Surg 8 (2004) 346–357. [DOI] [PubMed] [Google Scholar]
- [27].Hess DR, Inhaled Carbon Monoxide: From Toxin to Therapy, Respir Care 62 (2017) 1333. [DOI] [PubMed] [Google Scholar]
- [28].Correa-Costa M, Gallo D, Csizmadia E, Gomperts E, Lieberum JL, Hauser CJ, Ji X, Wang B, Camara NOS, Robson SC, Otterbein LE, Carbon monoxide protects the kidney through the central circadian clock and CD39, Proc Natl Acad Sci U S A 115 (2018) E2302–E2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Foresti R, Shurey C, Ansari T, Sibbons P, Mann BE, Johnson TR, Green CJ, Motterlini R, Reviewing the use of carbon monoxide-releasing molecules (CO-RMs) in biology: implications in endotoxin-mediated vascular dysfunction, Cell Mol Biol (Noisy-le-grand) 51 (2005) 409–423. [PubMed] [Google Scholar]
- [30].Romao CC, Blattler WA, Seixas JD, Bernardes GJ, Developing drug molecules for therapy with carbon monoxide, Chem Soc Rev 41 (2012) 3571–3583. [DOI] [PubMed] [Google Scholar]
- [31].Lazarus LS, Benninghoff AD, Berreau LM, Development of Triggerable, Trackable, and Targetable Carbon Monoxide Releasing Molecules, Acc Chem Res 53 (2020) 2273–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ji X, Damera K, Zheng Y, Yu B, Otterbein LE, Wang B, Toward Carbon Monoxide-Based Therapeutics: Critical Drug Delivery and Developability Issues, J Pharm Sci 105 (2016) 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ji X, Pan Z, Yu B, De La Cruz LK, Zheng Y, Ke B, Wang B, Click and release: bioorthogonal approaches to “on-demand” activation of prodrugs, Chem Soc Rev 48 (2019) 1077–1094. [DOI] [PubMed] [Google Scholar]
- [34].Ji X, Wang B, Strategies toward Organic Carbon Monoxide Prodrugs, Acc Chem Res 51 (2018) 1377–1385. [DOI] [PubMed] [Google Scholar]
- [35].Steiger C, Hermann C, Meinel L, Localized delivery of carbon monoxide, Eur J Pharm Biopharm 118 (2017) 3–12. [DOI] [PubMed] [Google Scholar]
- [36].Ji X, Wang B, Toward carbon monoxide based therapeutics: carbon monoxide in a pill, Pharm Pat Anal 6 (2017) 171–177. [DOI] [PubMed] [Google Scholar]
- [37].Wang B, Hu L, Siahaan T, Drug Delivery: Principles and Applications, in: Wang B (Ed.) Wiley Series in Drug Discovery and Development, John Wiley and Sons, Hoboken, New Jersey, 2016. [Google Scholar]
- [38].Goldbaum LR, Orellano T, Dergal E, Mechanism of the toxic action of carbon monoxide, Ann Clin Lab Sci 6 (1976) 372–376. [PubMed] [Google Scholar]
- [39].Haldane J, The Relation of the Action of Carbonic Oxide to Oxygen Tension, J Physiol 18 (1895) 201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McGrath JJ, Leviseur C, Cardiorespiratory responses to intestinal injection of carbon monoxide, Pharmacol Biochem Behav 21 Suppl 1 (1984) 103–107. [DOI] [PubMed] [Google Scholar]
- [41].Levin BC, Paabo M, Gurman JL, Harris SE, Braun E, Toxicological interactions between carbon monoxide and carbon dioxide, Toxicology 47 (1987) 135–164. [DOI] [PubMed] [Google Scholar]
- [42].Hasegawa M, Kimura K, Hieda Y, Yakabe T, Fatal and non-fatal levels of carboxyhemoglobin (COHb) in blood: An experimental study on changes in COHb level in blood due to changes in carbon monoxide level in the air, Jpn J Forensic Toxicol 20 (2002) 320–327. [Google Scholar]
- [43].Silbaugh SA, Horvath SM, Effect of acute carbon monoxide exposure on cardiopulmonary function of the awake rat, Toxicol Appl Pharmacol 66 (1982) 376–382. [DOI] [PubMed] [Google Scholar]
- [44].Rose JJ, Wang L, Xu Q, McTiernan CF, Shiva S, Tejero J, Gladwin MT, Carbon Monoxide Poisoning: Pathogenesis, Management, and Future Directions of Therapy, Am J Respir Crit Care Med 195 (2017) 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hampson NB, Hauff NM, Carboxyhemoglobin levels in carbon monoxide poisoning: do they correlate with the clinical picture?, Am J Emerg Med 26 (2008) 665–669. [DOI] [PubMed] [Google Scholar]
- [46].Myers RA, Britten JS, Are arterial blood gases of value in treatment decisions for carbon monoxide poisoning?, Crit Care Med 17 (1989) 139–142. [DOI] [PubMed] [Google Scholar]
- [47].Ilano AL, Raffin TA, Management of carbon monoxide poisoning, Chest 97 (1990) 165–169. [DOI] [PubMed] [Google Scholar]
- [48].Dewilde S, Kiger L, Burmester T, Hankeln T, Baudin-Creuza V, Aerts T, Marden MC, Caubergs R, Moens L, Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family, J Biol Chem 276 (2001) 38949–38955. [DOI] [PubMed] [Google Scholar]
- [49].Brunori M, Bonaventura J, Bonaventura C, Antonini E, Wyman J, Carbon monoxide binding by hemoglobin and myoglobin under photodissociating conditions, Proc Natl Acad Sci U S A 69 (1972) 868–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wald G, Allen DW, The equilibrium between cytochrome oxidase and carbon monoxide, J Gen Physiol 40 (1957) 593–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oliverio S, Varlet V, Total Blood Carbon Monoxide: Alternative to Carboxyhemoglobin as Biological Marker for Carbon Monoxide Poisoning Determination, J Anal Toxicol 43 (2019) 79–87. [DOI] [PubMed] [Google Scholar]
- [52].Cooper CE, Nitric oxide and iron proteins, Biochim Biophys Acta Bioenerg 1411 (1999) 290–309. [DOI] [PubMed] [Google Scholar]
- [53].Azarov I, Wang L, Rose JJ, Xu Q, Huang XN, Belanger A, Wang Y, Guo L, Liu C, Ucer KB, McTiernan CF, O’Donnell CP, Shiva S, Tejero J, Kim-Shapiro DB, Gladwin MT, Five-coordinate H64Q neuroglobin as a ligand-trap antidote for carbon monoxide poisoning, Sci Transl Med 8 (2016) 368ra173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vandegriff KD, Le Tellier YC, Winslow RM, Rohlfs RJ, Olson JS, Determination of the rate and equilibrium constants for oxygen and carbon monoxide binding to R-state human hemoglobin cross-linked between the alpha subunits at lysine 99, J Biol Chem 266 (1991) 17049–17059. [PubMed] [Google Scholar]
- [55].Unzai S, Eich R, Shibayama N, Olson JS, Morimoto H, Rate constants for O2 and CO binding to the alpha and beta subunits within the R and T states of human hemoglobin, J Biol Chem 273 (1998) 23150–23159. [DOI] [PubMed] [Google Scholar]
- [56].Douglas CG, Haldane JS, Haldane JB, The laws of combination of haemoglobin with carbon monoxide and oxygen, J Physiol 44 (1912) 275–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Peterson JE, Stewart RD, Absorption and elimination of carbon monoxide by inactive young men, Arch Environ Health 21 (1970) 165–171. [DOI] [PubMed] [Google Scholar]
- [58].O’Brien HR, Parker WL, Solubility of carbon monoxide in serum and plasma, J Biol Chem 50 (1922) 289–300. [Google Scholar]
- [59].Moffet DA, Case MA, House JC, Vogel K, Williams RD, Spiro TG, McLendon GL, Hecht MH, Carbon monoxide binding by de novo heme proteins derived from designed combinatorial libraries, J Am Chem Soc 123 (2001) 2109–2115. [DOI] [PubMed] [Google Scholar]
- [60].Gibson QH, Olson JS, McKinnie RE, Rohlfs RJ, A kinetic description of ligand binding to sperm whale myoglobin, J Biol Chem 261 (1986) 10228–10239. [PubMed] [Google Scholar]
- [61].Yoshikawa S, Choc MG, O’Toole MC, Caughey WS, An infrared study of CO binding to heart cytochrome c oxidase and hemoglobin A. Implications re O2 reactions, J Biol Chem 252 (1977) 5498–5508. [PubMed] [Google Scholar]
- [62].Ortiz-Prado E, Dunn JF, Vasconez J, Castillo D, Viscor G, Partial pressure of oxygen in the human body: a general review, Am J Blood Res 9 (2019) 1–14. [PMC free article] [PubMed] [Google Scholar]
- [63].Sharma VS, Geibel JF, Ranney HM, “Tension” on heme by the proximal base and ligand reactivity: conclusions drawn from model compounds for the reaction of hemoglobin, Proc Natl Acad Sci U S A 75 (1978) 3747–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Goldbaum LR, Ramirez RG, Absalon KB, What is the mechanism of carbon monoxide toxicity?, Aviat Space Environ Med 46 (1975) 1289–1291. [PubMed] [Google Scholar]
- [65].Ernst A, Zibrak JD, Carbon monoxide poisoning, N Engl J Med 339 (1998) 1603–1608. [DOI] [PubMed] [Google Scholar]
- [66].Turner M, Hamilton-Farrell MR, Clark RJ, Carbon monoxide poisoning: an update, J Accid Emerg Med 16 (1999) 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Oliverio S, Varlet V, Carbon monoxide analysis method in human blood by Airtight Gas Syringe - Gas Chromatography - Mass Spectrometry (AGS-GC-MS): Relevance for postmortem poisoning diagnosis, J Chromatogr B: Anal Technol Biomed Life Sci 1090 (2018) 81–89. [DOI] [PubMed] [Google Scholar]
- [68].Jarmoskaite I, AlSadhan I, Vaidyanathan PP, Herschlag D, How to measure and evaluate binding affinities, Elife 9 (2020) e57264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Naples R, Laskowski D, McCarthy K, Mattox E, Comhair SA, Erzurum SC, Carboxyhemoglobin and methemoglobin in asthma, Lung 193 (2015) 183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP, Physiological parameter values for physiologically based pharmacokinetic models, Toxicol Ind Health 13 (1997) 407–484. [DOI] [PubMed] [Google Scholar]
- [71].Teixeira MA, Chaguri LDCAG, Carissimi AS, Souza NLD, Mori CMC, Gomes VMW, Poli Neto A, Nonoyama K, Merusse JLB, Hematological and biochemical profiles of rats (Rattus norvegicus) kept under microenvironmental ventilation system, Braz J Vet Res Anim Sci 37 (2000) 00–00. [Google Scholar]
- [72].Lee HB, Blaufox MD, Blood volume in the rat, J Nucl Med 26 (1985) 72–76. [PubMed] [Google Scholar]
- [73].Wang M, Yang X, Pan Z, Wang Y, De La Cruz LK, Wang B, Tan C, Towards “CO in a pill”: Pharmacokinetic studies of carbon monoxide prodrugs in mice, J Control Release 327 (2020) 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Eaton WA, Henry ER, Hofrichter J, Mozzarelli A, Is cooperative oxygen binding by hemoglobin really understood?, Nat Struct Biol 6 (1999) 351–358. [DOI] [PubMed] [Google Scholar]
- [75].Joels N, Pugh LG, The carbon monoxide dissociation curve of human blood, J Physiol 142 (1958) 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sharma VS, Schmidt MR, Ranney HM, Dissociation of CO from carboxyhemoglobin, J Biol Chem 251 (1976) 4267–4272. [PubMed] [Google Scholar]
- [77].Bakalarz D, Surmiak M, Yang X, Wójcik D, Korbut E, Śliwowski Z, Ginter G, Buszewicz G, Brzozowski T, Cieszkowski J, Głowacka U, Magierowska K, Pan Z, Wang B, Magierowski M, Organic carbon monoxide prodrug, BW-CO-111, in protection against chemically-induced gastric mucosal damage, Acta Pharm Sin B (2020) 456–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jasnos K, Magierowski M, Kwiecien S, Brzozowski T, [Carbon monoxide in human physiology--its role in the gastrointestinal tract], Postepy Hig Med Dosw (Online) 68 (2014) 101–109. [DOI] [PubMed] [Google Scholar]
- [79].Gibbons SJ, Farrugia G, The role of carbon monoxide in the gastrointestinal tract, J Physiol 556 (2004) 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Farrugia G, Lei S, Lin X, Miller SM, Nath KA, Ferris CD, Levitt M, Szurszewski JH, A major role for carbon monoxide as an endogenous hyperpolarizing factor in the gastrointestinal tract, Proc Natl Acad Sci U S A 100 (2003) 8567–8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hegazi RAF, Rao KN, Mayle A, Sepulveda AR, Otterbein LE, Plevy SE, Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1–dependent pathway, J Exp Med 202 (2005) 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Moore BA, Otterbein LE, TÜrler A, Choi AMK, Bauer AJ, Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine, Gastroenterology 124 (2003) 377–391. [DOI] [PubMed] [Google Scholar]
- [83].Barn P, Giles L, Héroux M-E, Kosatsky T, A review of the experimental evidence on the toxicokinetics of carbon monoxide: the potential role of pathophysiology among susceptible groups, Environ Health 17 (2018) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ren X, Dorrington KL, Robbins PA, Respiratory control in humans after 8 h of lowered arterial Po 2, hemodilution, or carboxyhemoglobinemia, J Appl Physiol 90 (2001) 1189–1195. [DOI] [PubMed] [Google Scholar]
- [85].Vesely AE, Somogyi RB, Sasano H, Sasano N, Fisher JA, Duffin J, The effects of carbon monoxide on respiratory chemoreflexes in humans, Environmental Research 94 (2004) 227–233. [DOI] [PubMed] [Google Scholar]
- [86].Resch H, Zawinka C, Weigert G.n., Schmetterer L, Garhöfer G, Inhaled Carbon Monoxide Increases Retinal and Choroidal Blood Flow in Healthy Humans, Invest Ophthalmol Vis Sci 46 (2005) 4275–4280. [DOI] [PubMed] [Google Scholar]
- [87].N.R.C.U.C.o. AEGL, National Research Council (US) Committee on Acute Exposure Guideline Levels. Acute Exposure Guideline Levels for Selected Airborne : Volume 8., Washington (DC): National Academies Press; 2010. [PubMed] [Google Scholar]
- [88].OSHA, Nitric Oxide, in, Technical Assistance Branch, Division of Occupational Health and Safety, Office of Research Services, 2019. [Google Scholar]
- [89].Sherlock LG, Wright CJ, Kinsella JP, Delaney C, Inhaled nitric oxide use in neonates: Balancing what is evidence-based and what is physiologically sound, Nitric Oxide 95 (2020) 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhu HH, Hu J, Lo-Coco F, Jin J, The simpler, the better: oral arsenic for acute promyelocytic leukemia, Blood 134 (2019) 597–605. [DOI] [PubMed] [Google Scholar]
- [91].Coburn RF, Forster RE, Kane PB, Considerations of the physiological variables that determine the blood carboxyhemoglobin concentration in man, J Clin Invest 44 (1965) 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Andiman WA, Jacobson RI, Tucker G, Leukocyte-associated viremia with adenovirus type 2 in an infant with lower-respiratory-tract disease, N Engl J Med 297 (1977) 100–101. [DOI] [PubMed] [Google Scholar]
- [93].Peterson JE, Stewart RD, Predicting the carboxyhemoglobin levels resulting from carbon monoxide exposures, J Appl Physiol 39 (1975) 633–638. [DOI] [PubMed] [Google Scholar]
- [94].Woehlck Harvey J., Mei D, Dunning Marshall B., Ruiz F, Mathematical Modeling of Carbon Monoxide Exposures from Anesthetic Breakdown: Effect of Subject Size, Hematocrit, Fraction of Inspired Oxygen, and Quantity of Carbon Monoxide, Anesthesiology 94 (2001) 457–460. [DOI] [PubMed] [Google Scholar]
- [95].Kuo Y-W, Harris RS, Hess DR, Dieffenbach PB, Choi AMK, Fredenburgh LE, Winkler T, Endogenous Carbon Monoxide Production and Diffusing Capacity of the Lung for Carbon Monoxide in Sepsis-Induced Acute Respiratory Distress Syndrome, Critical Care Explorations 2 (2020) e0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fredenburgh LE, Perrella MA, Barragan-Bradford D, Hess DR, Peters E, Welty-Wolf KE, Kraft BD, Harris RS, Maurer R, Nakahira K, Oromendia C, Davies JD, Higuera A, Schiffer KT, Englert JA, Dieffenbach PB, Berlin DA, Lagambina S, Bouthot M, Sullivan AI, Nuccio PF, Kone MT, Malik MJ, Porras MAP, Finkelsztein E, Winkler T, Hurwitz S, Serhan CN, Piantadosi CA, Baron RM, Thompson BT, Choi AM, A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS, JCI Insight 3 (2018) e124039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kimmel EC, Carpenter RL, Reboulet JE, Still KR, A physiological model for predicting carboxyhemoglobin formation from exposure to carbon monoxide in rats, J Appl Physiol 86 (1999) 1977–1983. [DOI] [PubMed] [Google Scholar]
- [98].Kuo YW, Harris RS, Hess DR, Dieffenbach PB, Choi AMK, Fredenburgh LE, Winkler T, Endogenous Carbon Monoxide Production and Diffusing Capacity of the Lung for Carbon Monoxide in Sepsis-Induced Acute Respiratory Distress Syndrome, Crit Care Explor 2 (2020) e0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rosas IO, Goldberg HJ, Collard HR, El-Chemaly S, Flaherty K, Hunninghake GM, Lasky JA, Lederer DJ, Machado R, Martinez FJ, Maurer R, Teller D, Noth I, Peters E, Raghu G, Garcia JGN, Choi AMK, A Phase II Clinical Trial of Low-Dose Inhaled Carbon Monoxide in Idiopathic Pulmonary Fibrosis, Chest 153 (2018) 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Bathoorn E, Slebos DJ, Postma DS, Koeter GH, van Oosterhout AJ, van derToorn M, Boezen HM, Kerstjens HA, Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study, Eur Respir J 30 (2007) 1131–1137. [DOI] [PubMed] [Google Scholar]
- [101].Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B, Effects of carbon monoxide inhalation during experimental endotoxemia in humans, Am J Respir Crit Care Med 171 (2005) 354–360. [DOI] [PubMed] [Google Scholar]
- [102].Nakahira K, Choi AM, Carbon monoxide in the treatment of sepsis, Am J Physiol Lung Cell Mol Physiol 309 (2015) L1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Mitchell LA, Channell MM, Royer CM, Ryter SW, Choi AM, McDonald JD, Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation, Am J Physiol Lung Cell Mol Physiol 299 (2010) L891–897. [DOI] [PubMed] [Google Scholar]
- [104].Kohmoto J, Nakao A, Sugimoto R, Wang Y, Zhan J, Ueda H, McCurry KR, Carbon monoxide-saturated preservation solution protects lung grafts from ischemia-reperfusion injury, J Thorac Cardiovasc Surg 136 (2008) 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Nakao A, Huang CS, Stolz DB, Wang Y, Franks JM, Tochigi N, Billiar TR, Toyoda Y, Tzeng E, McCurry KR, Ex vivo carbon monoxide delivery inhibits intimal hyperplasia in arterialized vein grafts, Cardiovasc Res 89 (2011) 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zeynalov E, Dore S, Low doses of carbon monoxide protect against experimental focal brain ischemia, Neurotox Res 15 (2009) 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Vijayaraghavan R, Schaper M, Thompson R, Stock MF, Alarie Y, Characteristic modifications of the breathing pattern of mice to evaluate the effects of airborne chemicals on the respiratory tract, Arch Toxicol 67 (1993) 478–490. [DOI] [PubMed] [Google Scholar]
- [108].Takagi T, Naito Y, Uchiyama K, Mizuhima K, Suzuki T, Horie R, Hirata I, Tsuboi H, Yoshikawa T, Carbon monoxide promotes gastric wound healing in mice via the protein kinase C pathway, Free Radic Res 50 (2016) 1098–1105. [DOI] [PubMed] [Google Scholar]
- [109].in. [Google Scholar]
- [110].Belcher JD, Gomperts E, Nguyen J, Chen C, Abdulla F, Kiser ZM, Gallo D, Levy H, Otterbein LE, Vercellotti GM, Oral carbon monoxide therapy in murine sickle cell disease: Beneficial effects on vaso-occlusion, inflammation and anemia, PLoS One 13 (2018) e0205194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sears DA, Udden MM, Thomas LJ, Carboxyhemoglobin levels in patients with sickle-cell anemia: relationship to hemolytic and vasoocclusive severity, Am J Med Sci 322 (2001) 345–348. [DOI] [PubMed] [Google Scholar]
- [112].Beckman JD, Belcher JD, Vineyard JV, Chen C, Nguyen J, Nwaneri MO, O’Sullivan MG, Gulbahce E, Hebbel RP, Vercellotti GM, Inhaled carbon monoxide reduces leukocytosis in a murine model of sickle cell disease, Am J Physiol Heart Circ 297 (2009) H1243–H1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Mann BE, CO-Releasing Molecules: A Personal View, Organometallics 31 (2012) 5728–5735. [Google Scholar]
- [114].Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ, Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities, Circ Res 90 (2002) E17–24. [DOI] [PubMed] [Google Scholar]
- [115].Ji X, Pan Z, Li C, Kang T, De La Cruz LKC, Yang L, Yuan Z, Ke B, Wang B, Esterase-Sensitive and pH-Controlled Carbon Monoxide Prodrugs for Treating Systemic Inflammation, J Med Chem 62 (2019) 3163–3168. [DOI] [PubMed] [Google Scholar]
- [116].Ji X, Zhou C, Ji K, Aghoghovbia RE, Pan Z, Chittavong V, Ke B, Wang B, Click and Release: A Chemical Strategy toward Developing Gasotransmitter Prodrugs by Using an Intramolecular Diels-Alder Reaction, Angew Chem Int Ed Engl 55 (2016) 15846–15851. [DOI] [PubMed] [Google Scholar]
- [117].Pan Z, Chittavong V, Li W, Zhang J, Ji K, Zhu M, Ji X, Wang B, Organic CO Prodrugs: Structure—CO-Release Rate Relationship Studies, Chem Eur J 23 (2017) 9838–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Thiang Brian Kueh J, Seifert-Simpson JM, Thwaite SH, Rodgers GD, Harrison JC, Sammut IA, Larsen DS, Studies towards Non-toxic, Water Soluble, Vasoactive Norbornene Organic Carbon Monoxide Releasing Molecules, Asian J Org Chem 9 (2020) 2127–2135. [Google Scholar]
- [119].Popova M, Lazarus LS, Benninghoff AD, Berreau LM, CO Sense and Release Flavonols: Progress toward the Development of an Analyte Replacement PhotoCORM for Use in Living Cells, ACS Omega 5 (2020) 10021–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].McKendrick JG, Snodgrass W, On the Physiological Action of Carbon Monoxide of Nickel, Br Med J 1 (1891) 1215–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yuan Z, Yang X, Ye Y, Tripathi R, Wang B, Chemical Reactivities of Two Widely Used Ruthenium-Based COReleasing Molecules with a Range of Biologically Important Reagents and Molecules, Anal Chem 93 (2021) 5317–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Southam HM, Williamson MP, Chapman JA, Lyon RL, Trevitt CR, Henderson PJF, Poole RK, ‘Carbon-Monoxide-Releasing Molecule-2 (CORM-2)’ Is a Misnomer: Ruthenium Toxicity, Not CO Release, Accounts for Its Antimicrobial Effects, Antioxidants 10 (2021) 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Yuan Z, Yang X, Wang B, Redox and Catalase-like Activities of Four Widely Used Carbon Monoxide Releasing Molecules (CO-RMs), Chem Sci (2021) Manuscript accepted pending minor revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Nielsen VG, The anticoagulant effect of Apis mellifera phospholipase A(2) is inhibited by CORM-2 via a carbon monoxide-independent mechanism, J Thromb. Thrombolysis 49 (2020) 100–107. [DOI] [PubMed] [Google Scholar]
- [125].Gessner G, Sahoo N, Swain SM, Hirth G, Schönherr R, Mede R, Westerhausen M, Brewitz HH, Heimer P, Imhof D, Hoshi T, Heinemann SH, CO-independent modification of K(+) channels by tricarbonyldichlororuthenium(II) dimer (CORM-2), Eur. J. Pharmacol 815 (2017) 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, Green CJ, CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule, FASEB J 19 (2005) 284–286. [DOI] [PubMed] [Google Scholar]
- [127].Klein M, Neugebauer U, Schmitt M, Popp J, Elucidation of the CO-Release Kinetics of CORM-A1 by Means of Vibrational Spectroscopy, Chemphyschem 17 (2016) 985–993. [DOI] [PubMed] [Google Scholar]
- [128].Liu J, Fedinec AL, Leffler CW, Parfenova H, Enteral supplements of a carbon monoxide donor CORM-A1 protect against cerebrovascular dysfunction caused by neonatal seizures, J Cereb Blood Flow Metab 35 (2015) 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Parfenova H, Leffler CW, Basuroy S, Liu J, Fedinec AL, Antioxidant roles of heme oxygenase, carbon monoxide, and bilirubin in cerebral circulation during seizures, J Cereb Blood Flow Metab 32 (2012) 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Mahan VL, Zurakowski D, Otterbein LE, Pigula FA, Inhaled carbon monoxide provides cerebral cytoprotection in pigs, PLoS One 7 (2012) e41982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Morris GL, Curtis SE, Simon J, Perinatal piglets under sublethal concentrations of atmospheric carbon monoxide, J Anim Sci 61 (1985) 1070–1079. [DOI] [PubMed] [Google Scholar]
- [132].Klinger-Strobel M, Glaser S, Makarewicz O, Wyrwa R, Weisser J, Pletz MW, Schiller A, Bactericidal Effect of a Photoresponsive Carbon Monoxide-Releasing Nonwoven against Staphylococcus aureus Biofilms, Antimicrob Agents Chemother 60 (2016) 4037–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Lazarus LS, Esquer HJ, Anderson SN, Berreau LM, Benninghoff AD, Mitochondrial-Localized Versus Cytosolic Intracellular CO-Releasing Organic PhotoCORMs: Evaluation of CO Effects Using Bioenergetics, ACS Chemical Biology 13 (2018)2220–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Peng P, Wang C, Shi Z, Johns VK, Ma L, Oyer J, Copik A, Igarashi R, Liao Y, Visible-light activatable organic CO-releasing molecules (PhotoCORMs) that simultaneously generate fluorophores, Org Biomol Chem 11 (2013) 6671–6674. [DOI] [PubMed] [Google Scholar]
- [135].Schatzschneider U, Novel lead structures and activation mechanisms for CO-releasing molecules (CORMs), Br J Pharmacol 172 (2015) 1638–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Southam HM, Smith TW, Lyon RL, Liao C, Trevitt CR, Middlemiss LA, Cox FL, Chapman JA, El-Khamisy SF, Hippier M, Williamson MP, Henderson PJF, Poole RK, A thiol-reactive Ru(II) ion, not CO release, underlies the potent antimicrobial and cytotoxic properties of CO-releasing molecule-3, Redox Biol 18 (2018) 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Magierowska K, Magierowski M, Hubalewska-Mazgaj M, Adamski J, Surmiak M, Sliwowski Z, Kwiecien S, Brzozowski T, Carbon Monoxide (CO) Released from Tricarbonyldichlororuthenium (II) Dimer (CORM-2) in Gastroprotection against Experimental Ethanol-Induced Gastric Damage, PLoS One 10 (2015) e0140493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Crook SH, Mann BE, Meijer AJ, Adams H, Sawle P, Scapens D, Motterlini R, [Mn(CO)4{S2CNMe(CH2CO2H)}], a new water-soluble CO-releasing molecule, Dalton Trans 40 (2011) 4230–4235. [DOI] [PubMed] [Google Scholar]
- [139].Braud L, Pini M, Muchova L, Manin S, Kitagishi H, Sawaki D, Czibik G, Ternacle J, Derumeaux G, Foresti R, Motterlini R, Carbon monoxide-induced metabolic switch in adipocytes improves insulin resistance in obese mice, JCI Insight 3 (2018) e123485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hosick PA, AlAmodi AA, Hankins MW, Stec DE, Chronic treatment with a carbon monoxide releasing molecule reverses dietary induced obesity in mice, Adipocyte 5 (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Nikam A, Ollivier A, Rivard M, Wilson JL, Mebarki K, Martens T, Dubois-Rande JL, Motterlini R, Foresti R, Diverse Nrf2 Activators Coordinated to Cobalt Carbonyls Induce Heme Oxygenase-1 and Release Carbon Monoxide in Vitro and in Vivo, J Med Chem 59 (2016) 756–762. [DOI] [PubMed] [Google Scholar]
- [142].Wilson JL, Fayad Kobeissi S, Oudir S, Haas B, Michel B, Dubois Rande JL, Ollivier A, Martens T, Rivard M, Motterlini R, Foresti R, Design and synthesis of new hybrid molecules that activate the transcription factor Nrf2 and simultaneously release carbon monoxide, Chemistry 20 (2014) 14698–14704. [DOI] [PubMed] [Google Scholar]
- [143].Motterlini R, Nikam A, Manin S, Ollivier A, Wilson JL, Djouadi S, Muchova L, Martens T, Rivard M, Foresti R, HYCO-3, a dual CO-releaser/Nrf2 activator, reduces tissue inflammation in mice challenged with lipopolysaccharide, Redox Biol 20 (2019) 334–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ollivier A, Foresti R, El Ali Z, Martens T, Kitagishi H, Motterlini R, Rivard M, Design and Biological Evaluation of Manganese- and Ruthenium-Based Hybrid CO-RMs (HYCOs), ChemMedChem 14 (2019) 1684–1691. [DOI] [PubMed] [Google Scholar]
- [145].El Ali Z, Ollivier A, Manin S, Rivard M, Motterlini R, Foresti R, Therapeutic effects of CO-releaser/Nrf2 activator hybrids (HYCOs) in the treatment of skin wound, psoriasis and multiple sclerosis, Redox Biol 34 (2020) 101521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Khurana JM, Chauhan S, Agrawal A, Molybdenum in organic synthesis. A review Org Prep Proced Int 36 (2004) 201–276. [Google Scholar]
- [147].Fischer S, Dicobalt octacarbonyl, Synlett 9 (2002) 1558–1559. [Google Scholar]
- [148].McLean S, Mann BE, Poole RK, Sulfite species enhance carbon monoxide release from CO-releasing molecules: implications for the deoxymyoglobin assay of activity, Anal Biochem 427 (2012) 36–40. [DOI] [PubMed] [Google Scholar]
- [149].Yuan Z, Yang X, De La Cruz LK, Wang B, Nitro reduction-based fluorescent probes for carbon monoxide require reactivity involving a ruthenium carbonyl moiety, Chem Commun (Camb) 56 (2020) 2190–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Seixas JD, Mukhopadhyay A, Santos-Silva T, Otterbein LE, Gallo DJ, Rodrigues SS, Guerreiro BH, Goncalves AM, Penacho N, Marques AR, Coelho AC, Reis PM, Romao MJ, Romao CC, Characterization of a versatile organometallic pro-drug (CORM) for experimental CO based therapeutics, Dalton Trans 42 (2013) 5985–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lutz J, Nöth U, Morrissey SP, Adolf H, Deichmann R, Haase A, Measurement of Oxygen Tensions in the Abdominal Cavity and in the Skeletal Muscle Using 19F-MRI of Neat PFC Droplets, in: Harrison DK, Delpy DT (Eds.) Oxygen Transport to Tissue XIX, Springer US, Boston, MA, 1997, pp. 569–572. [DOI] [PubMed] [Google Scholar]
- [152].Santos-Silva T, Mukhopadhyay A, Seixas JD, Bernardes GJ, Romao CC, Romao MJ, CORM-3 reactivity toward proteins: the crystal structure of a Ru(II) dicarbonyl-lysozyme complex, J Am Chem Soc 133 (2011) 1192–1195. [DOI] [PubMed] [Google Scholar]
- [153].Chaves-Ferreira M, Albuquerque IS, Matak-Vinkovic D, Coelho AC, Carvalho SM, Saraiva LM, Romao CC, Bernardes GJ, Spontaneous CO release from Ru(II)(CO)2-protein complexes in aqueous solution, cells, and mice, Angew Chem Int Ed Engl 54 (2015) 1172–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Dray A, Forbes CA, Burgess GM, Ruthenium red blocks the capsaicin-induced increase in intracellular calcium and activation of membrane currents in sensory neurones as well as the activation of peripheral nociceptors in vitro, Neurosci Lett 110 (1990) 52–59. [DOI] [PubMed] [Google Scholar]
- [155].Duesterberg CK, Mylon SE, Waite TD, pH Effects on Iron-Catalyzed Oxidation using Fenton’s Reagent, Environ Sci Technol. 42 (2008) 8522–8527. [DOI] [PubMed] [Google Scholar]
- [156].Steiger C, Luhmann T, Meinel L, Oral drug delivery of therapeutic gases - carbon monoxide release for gastrointestinal diseases, J Control Release 189 (2014) 46–53. [DOI] [PubMed] [Google Scholar]
- [157].Winburn IC, Gunatunga K, McKernan RD, Walker RJ, Sammut IA, Harrison JC, Cell damage following carbon monoxide releasing molecule exposure: implications for therapeutic applications, Basic Clin Pharmacol Toxicol 111 (2012) 31–41. [DOI] [PubMed] [Google Scholar]
- [158].Steiger C, Uchiyama K, Takagi T, Mizushima K, Higashimura Y, Gutmann M, Hermann C, Botov S, Schmalz HG, Naito Y, Meinel L, Prevention of colitis by controlled oral drug delivery of carbon monoxide, J Control Release 239 (2016) 128–136. [DOI] [PubMed] [Google Scholar]
- [159].Teorell T, Kinetics of distribution of substances administered to the body, I : The extravascular modes of administration, Archives Internationales de pharmacodynamie et de therapie 57 (1937) 205–225. [Google Scholar]
- [160].Takagi T, Naito Y, Mizushima K, Akagiri S, Suzuki T, Hirata I, Omatsu T, Handa O, Kokura S, Ichikawa H, Yoshikawa T, Inhalation of carbon monoxide ameliorates TNBS-induced colitis in mice through the inhibition of TNF-alpha expression, Dig Dis Sci 55 (2010) 2797–2804. [DOI] [PubMed] [Google Scholar]
- [161].Van Dingenen J, Steiger C, Zehe M, Meinel L, Lefebvre RA, Investigation of orally delivered carbon monoxide for postoperative ileus, Eur J Pharm Biopharm 130 (2018) 306–313. [DOI] [PubMed] [Google Scholar]
- [162].Yin H, Fang J, Liao L, Nakamura H, Maeda H, Styrene-maleic acid copolymer-encapsulated CORM2, a water-soluble carbon monoxide (CO) donor with a constant CO-releasing property, exhibits therapeutic potential for inflammatory bowel disease, J Control Release 187 (2014) 14–21. [DOI] [PubMed] [Google Scholar]
- [163].Raabe BM, Artwohl JE, Purcell JE, Lovaglio J, Fortman JD, Effects of weekly blood collection in C57BL/6 mice, J Am Assoc Lab Anim Sci 50 (2011) 680–685. [PMC free article] [PubMed] [Google Scholar]
- [164].Vreman HJ, Wong RJ, Kadotani T, Stevenson DK, Determination of carbon monoxide (CO) in rodent tissue: effect of heme administration and environmental CO exposure, Anal Biochem 341 (2005) 280–289. [DOI] [PubMed] [Google Scholar]
- [165].Berrino E, Milazzo L, Micheli L, Vullo D, Angeli A, Bozdag M, Nocentini A, Menicatti M, Bartolucci G, di Cesare Mannelli L, Ghelardini C, Supuran CT, Carta F, Synthesis and Evaluation of Carbonic Anhydrase Inhibitors with Carbon Monoxide Releasing Properties for the Management of Rheumatoid Arthritis, J Med Chem 62 (2019) 7233–7249. [DOI] [PubMed] [Google Scholar]
- [166].Soboleva T, Berreau LM, 3-Hydroxyflavones and 3-Hydroxy-4-oxoquinolines as Carbon Monoxide-Releasing Molecules, Molecules 24 (2019) 1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Antony LA, Slanina T, Sebej P, Solomek T, Klan P, Fluorescein analogue xanthene-9-carboxylic acid: a transition-metal-free CO releasing molecule activated by green light, Org Lett 15 (2013) 4552–4555. [DOI] [PubMed] [Google Scholar]
- [168].Zheng Y, Ji X, Yu B, Ji K, Gallo D, Csizmadia E, Zhu M, Choudhury MR, De La Cruz LKC, Chittavong V, Pan Z, Yuan Z, Otterbein LE, Wang B, Enrichment-triggered prodrug activation demonstrated through mitochondria-targeted delivery of doxorubicin and carbon monoxide, Nat Chem 10 (2018) 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Popova M, Lazarus LS, Ayad S, Benninghoff AD, Berreau LM, Visible-Light-Activated Quinolone Carbon-Monoxide-Releasing Molecule: Prodrug and Albumin-Assisted Delivery Enables Anticancer and Potent Anti-Inflammatory Effects, J Am Chem Soc 140 (2018) 9721–9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].De La Cruz LKC, Benoit SL, Pan Z, Yu B, Maier RJ, Ji X, Wang B, Click, Release, and Fluoresce: A Chemical Strategy for a Cascade Prodrug System for Codelivery of Carbon Monoxide, a Drug Payload, and a Fluorescent Reporter, Org Lett 20 (2018) 897–900. [DOI] [PubMed] [Google Scholar]
- [171].Ling K, Men F, Wang WC, Zhou YQ, Zhang HW, Ye DW, Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs), J Med Chem 61 (2018) 2611–2635. [DOI] [PubMed] [Google Scholar]
- [172].Kueh JTB, Stanley NJ, Hewitt RJ, Woods LM, Larsen L, Harrison JC, Rennison D, Brimble MA, Sammut IA, Larsen DS, Norborn-2-en-7-ones as physiologically-triggered carbon monoxide-releasing prodrugs, Chem Sci 8 (2017) 5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Wang D, Viennois E, Ji K, Damera K, Draganov A, Zheng Y, Dai C, Merlin D, Wang B, A click-and-release approach to CO prodrugs, Chem Commun (Camb) 50 (2014) 15890–15893. [DOI] [PubMed] [Google Scholar]
- [174].Ji X, De La Cruz LKC, Pan Z, Chittavong V, Wang B, pH-Sensitive metal-free carbon monoxide prodrugs with tunable and predictable release rates, Chem Commun (Camb) 53 (2017) 9628–9631. [DOI] [PubMed] [Google Scholar]
- [175].Schwarz R, Kaspar A, Seelig J, Künnecke B, Gastrointestinal transit times in mice and humans measured with 27Al and 19F nuclear magnetic resonance, Magn Reson Med 48 (2002) 255–261. [DOI] [PubMed] [Google Scholar]
- [176].Culnan DM, Craft-Coffman B, Bitz GH, Capek KD, Tu Y, Lineaweaver WC, Kuhlmann-Capek MJ, Carbon Monoxide and Cyanide Poisoning in the Burned Pregnant Patient: An Indication for Hyperbaric Oxygen Therapy, Ann Plast Surg 80 (2018) S106–S112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Noh EM, Cho DH, Lee YR, Jeong YJ, Kim JH, Chae HS, Park J, Jung WS, Park SJ, Kim JS, Dimethylsulfoxide (DMSO) induces downregulation of heme oxygenase-1 (HO-1) in HL-60 cells: involvement of HO-1 in HL-60 cell differentiation, BMB Rep 44 (2011) 753–757. [DOI] [PubMed] [Google Scholar]