

Abstract
Retinoic acids (RAs) are the most successful therapeutics for cancer differentiation therapy used in high-risk neuroblastoma (NB) maintenance therapy but are limited in effectiveness. This study identifies a strategy for improving efficacy through disruption of cancer cell identity via BET inhibitors. Mutations that block development are theorized to cause NB through retention of immature cell identities contributing to oncogenesis. NB has two interchangeable cell identities, maintained by two different core transcriptional regulatory circuitries (CRCs): a therapy-resistant mesenchymal/stem cell state and a proliferative adrenergic cell state. MYCN amplification is a common mutation of high-risk NB and recently found to block differentiation by driving high expression of the adrenergic CRC transcription factor ASCL1. We investigated whether disruption of immature CRCs can promote RA-induced differentiation since only a subset of NB patients responds to RA. We found that silencing ASCL1, a critical member of the adrenergic CRC, or global disruption of CRCs with the BET inhibitor JQ1, suppresses gene expression of multiple CRC factors, improving RA-mediated differentiation. Further, JQ1 and RA synergistically decrease proliferation and induce differentiation in NB cell lines. Our findings support preclinical studies of RA and BET inhibitors as a combination therapy in treating NB.
Keywords: neuroblastoma; ASCL1; retinoic acid; BET inhibitor JQ1; MYCN, SOX10
Graphical abstract
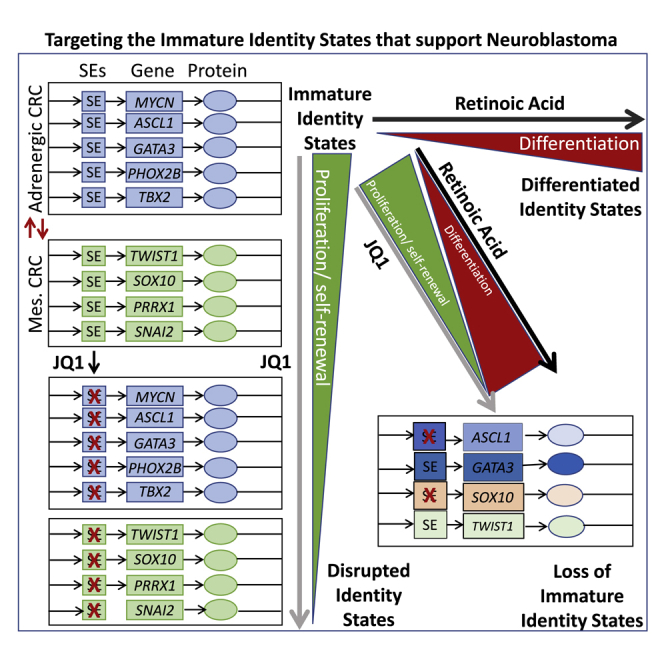
Although limited in their effectiveness, retinoic acids are the most successful therapeutics for differentiation therapy in cancer. This study identifies a strategy to improve their efficacy through use of BET bromodomain inhibitors to disrupt developmentally immature cancer cell identity states that contribute to tumor heterogeneity and oncogenic progression.
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor of infancy. It is purported to arise from a block in normal development during differentiation of neural crest cells (NCCs) into cells of the sympathetic nervous system. NB is responsible for about 15% of childhood mortality,1, 2, 3 and approximately 50%–60% of children with high-risk NB develop treatment resistance.1,4,5 Current multimodal therapies such as chemotherapy, radiation, and immune therapies prolong the survival rate in about 40% of patients.6 However, the increased risk of treatment-related toxicity, long-term complications, and the development of resistance in disease prevention and progression has been a major concern in the clinical setting.7,8 Maintenance therapy using retinoic acid (RA), a well-known differentiating agent, reduces the risk of recurrent disease after intensive multimodal treatment; however, only a subset of NB patients is responsive to RA-induced differentiation.
During development, transcription factors establish core transcriptional regulatory circuitries (CRCs) that generate and maintain cell identity by establishing and reinforcing super-enhancers upstream of other mutually regulated members. One model for NB oncogenesis is that a subset of NB mutations inappropriately reinforces early developmental CRCs, which lock NB into a developmentally immature state. For the purposes of development, that state is highly proliferative, resistant to apoptosis, migratory, and invasive. Whereas those traits are important to achieve proper development, they are dangerous if perpetuated in cancer.9 For example, MYCN gene amplification, a common mutation in high-risk NB, drives ASCL1 expression, thus preventing its reduction, which is required for cell differentiation of sympathoadrenal neuroblasts into mature neurons.10, 11, 12 ASCL1, along with transcription factors such as PHOX2B, GATA3, and TBX2, is a part of the immature sympathoadrenal CRC observed in NB.13
In addition to the proliferative sympathoadrenal identity state, NB can acquire a more immature cell-migratory, therapy-resistant mesenchymal state, which is regulated by a separate CRC. These two identity states, adrenergic and mesenchymal, likely capture NCCs early in their commitment to the sympathoadrenal lineage.14 At this stage, NCCs are transitioning from an epithelial to mesenchymal transition (EMT) program that is migratory into neuroblasts that are highly proliferative. In normal development, these cells would respond to differentiation cues like RA to become mature neurons and Schwann cells of the sympathetic nervous system.15 To differing degrees, both of these identity states co-occur as distinct cell populations within NB cell lines (NBCLs) and within tumors. NBCLs and tumors can skew toward one identity state over another, suggesting that different mutations favor different identity states.
One underutilized strategy for treating NB is to push the cells into a more differentiated and less dangerous state through differentiation therapy. RA can drive maturation of some NBs through transition of an immature CRC to a more differentiated CRC,16 but how NB mutations interfere with RA-induced differentiation is not well understood. We investigated a therapeutic strategy for NB based on the hypothesis that blocks in differentiation are due to pathogenic CRCs, caused by multiple mechanisms including N-Myc-induced ASCL1 overexpression, and that these interfere with RA-mediated differentiation. BET bromodomain inhibitor JQ1 inhibits the epigenetic reader bromodomain-containing protein 4 (BRD4),17 which accumulates at super-enhancers to promote gene transcription. BET bromodomain inhibitors disrupt super-enhancer function and in turn CRCs. These inhibitors also show prominent anticancer activity in NB including promoting cellular differentiation in some NBCLs.18,19 We found that disrupting CRCs, either with genetic silencing of ASCL1 or with BET bromodomain inhibitors, facilitates RA differentiation, thereby identifying a promising therapeutic approach for treating NB.
Results
Basal differences of adrenergic versus mesenchymal core regulatory transcription factors in NB cells
To confirm recently reported findings,20 we examined protein expression in NBCLs that have various genomic signatures and differences in MYCN gene amplification for their identity states. We selected MYCN-amplified cell lines Kelly and SK-N-BE2 and MYCN wild-type SKNAS, GIMEN, and SY5Y cell lines to determine the basal protein expression of ASCL1, GATA3, TBX2, and PHOX2B, which are prominent adrenergic markers, and SOX10, TWIST1, PRRX1, SNAI2, and vimentin, which are prominent mesenchymal markers (Figure S1). Higher levels of ASCL1 and PHOX2B protein expression were observed in Kelly, which were also marked by low protein expression of SOX10, TWIST1, SNAI2, and vimentin, indicating that these cells favor an adrenergic cell identity. SKNAS showed a markedly lower basal protein expression of ASCL1 and PHOX2B and higher protein expression of SOX10, TWIST1, PRRX1, SNAI2, and vimentin, indicating a more mesenchymal type of identity. The other cell lines SK-N-BE2, GIMEN, and SY5Y showed an intermediate identity with a more mixed expression of adrenergic and mesenchymal markers (Figure S1). Kelly cells, an MYCN-amplified NB, had higher ASCL1 expression and the most adrenergic type of identity, thus were chosen for our ASCL1 studies.
Suppression of ASCL1 by short hairpin (sh)RNA results in the inhibition of cell proliferation and switching of cell-identity core regulatory transcription factors
Kelly cells were subjected to doxycycline (Dox)-inducible, shRNA-mediated silencing of ASCL1. After 72 h in the presence (+Dox) or absence (−Dox), quantitative PCR (qPCR) gene expression of ASCL1, normalized to ACTIN, demonstrated 57% knockdown (KD) in Sh6 and 61% knockdown in Sh8 stably transfected cells (Figures 1A and 1B). Protein expression of ASCL1 was analyzed at day 4 with Dox-treated cells, showing a significant knockdown of ASCL1 protein expression in Sh6 and Sh8 (Figures 1C and 1D). To understand the role of the proneural and neurogenic effects of ASCL1 suppression on the cells, we analyzed the expression of PHOX2B, which marks early sympathoadrenal commitments;21 GATA3, which is downstream of both ASCL1 and PHOX2B and later in sympathoadrenal commitment;22 SOX10, which is a NCC stemness marker that is lost during neuronal commitment but retained during early mesenchymal commitment;23,24 and finally, TWIST1, a marker of mesenchymal commitment.25 Knockdown of ASCL1 by Sh6 and Sh8 showed a significant increase in the gene expression of GATA3 and TWIST1 but a significant decrease of SOX10. Specifically, Sh6 ASCL1 showed a significant effect on gene expression of core transcription factors GATA3 Sh6(−Dox 1) versus knockdown (+Dox 1.23 ± 0.03), p < 0.05; SOX10 fold-change expression Sh6 (−Dox 1) versus knockdown (+Dox 0.53 ± 0.10); and TWIST1 fold-change expression in Sh6 (−Dox 1) versus knockdown (+Dox 1.22 ± 0.06), p < 0.05 (Figure 1E). Similarly, Sh8 ASCL1 knockdown showed a significant effect on gene expression of core transcription factors with an upregulation in adrenergic versus mesenchymal identity (Figure 1F). These results suggested a loss of stemness and gain of both more mesenchymal and adrenergic differentiation.
Figure 1.

Evaluation of the ASCL1 knockdown and its effects on CRC factors by Sh6 and Sh8 in Kelly
Sh6 and Sh8 induced knockdown of ASCL1 in the presence of doxycycline (Dox) at day 4, as assessed by (A and B) RT-PCR and (C and D) western blot analysis. (E and F) Gene expression of ASCL1, SOX10, and GATA3 in Sh6- and Sh8-inducible Kelly cells by RT-PCR analysis. (G and H) Protein expression of CRC transcription factors in Sh6 and Sh8 cells. (All statistical data presented as mean ± SEM with n = 3, ∗∗p < 0.01 or ∗p < 0.05.)
Due to the well-documented labile nature of TWIST1 protein,26 we wanted to determine if we observed the same changes by protein as in mRNA expression. We also expanded the number of factors that we tested for adrenergic versus mesenchymal identity core regulatory transcription factors, adding GATA3 in the adrenergic core and PRRX1 in the mesenchymal core identity. We also analyzed the rate-limiting enzyme in catecholamine biosynthesis, tyrosine hydroxylase (TH), since it is used as an expression marker that increases in accordance with further sympathoadrenal commitment.27 By protein expression, loss of ASCL1 again caused loss of the stemness marker SOX10 but a gain of multiple sympathoadrenal markers, TBX2, GATA3, and TH, at 96 h (Figures 1G and 1H). However, we did not observe obvious and consistent increases in the mesenchymal markers, TWIST1 and PRRX1, suggesting that silencing of ASCL1 promotes sympathoadrenal differentiation.
Treatment of all-trans RA (ATRA) in cells with ASCL1 suppression results in upregulation of the differentiation marker TH and the inhibition of proliferation
From the observation that the knockdown of ASCL1 promoted adrenergic differentiation, we wanted to ascertain if the differentiating agent ATRA would augment differentiation when ASCL1 is silenced. We incubated the cells in +Dox or −Dox, with and without ATRA, to determine the effects of ATRA on ASCL1 expression. We observed that ATRA reduces ASCL1 gene expression to almost the same extent as caused by silencing of ASCL1 with further diminishment seen when Dox-induced silencing was combined with ATRA (Figure 2A). Next, we tested TH expression using the same combinations and observed a significant upregulation in both Sh6 and Sh8 when ASCL1 silencing was combined with ATRA (Figure 2B). By western blot, we also observe ATRA reducing ASCL1 expression and causing a further increase in TH expression when combined with ASCL1 silencing (Figure 2C). Finally, we tested the effect of suppression of ASCL1 on cell proliferation after 4 days by CyQuant assay in +Dox or −Dox, along with ATRA treatment. We observed a significant inhibition in proliferation by either ASCL1 knockdown or ATRA treatment in both Sh-ASCL1 cell lines. The treatment of ATRA in +Dox showed a significant inhibitory effect in proliferation compared to the treatments of Dox or ATRA alone in both shASCL1 Kelly cell lines (Figure 2D).
Figure 2.

Effect of ASCL1 knockdown and treatment of ATRA on proliferation and differentiation
(A and B) The gene expression of ASCL1 and TH in day-3 treatments of Dox versus Dox + ATRA in Sh6- and Sh8-inducible cells by RT-PCR assay. (C) Protein expression of ASCL1 and TH as assessed by western blots. (D) Proliferation percentage in Sh6- and Sh8-inducible cells at day-4 treatment of Dox and Dox + ATRA combination as measured by CyQuant (n = 3, ∗∗p < 0.01 or ∗p < 0.05).
Combinatorial treatment with JQ1 and ATRA alters core regulatory transcription factors in adrenergic cell types
Since the results of shRNA-mediated ASCL1 suppression on reducing proliferation and inducing differentiation in combination with ATRA were encouraging, this prompted us to test the potential of JQ1 and JQ1 + ATRA combinations in NBCLs. Whereas silencing of ASCL1 would be predicted to disrupt a pathogenic adrenergic CRC caused by MYCN amplification, we wanted to determine if broad disruption of CRCs using the BET inhibitor in combination with ATRA would recapitulate the findings observed with genetic silencing of ASCL1. Additionally, we wanted to assess if this drug combination could affect other NBCLs that are not MYCN amplified. To this end, we performed a series of experiments on Kelly and two MYCN wild-type cell lines SY5Y and SKNAS to determine the potential effect of ATRA and JQ1, either alone or in combination, on core regulatory transcription factor gene expression, proliferation, and differentiation.
A 10-fold dilution series of JQ1, ATRA, and JQ1 + ATRA was performed to determine a concentration that led to significant reduction of proliferation at 4 days of treatment but that would allow assessment for changes in gene expression on day 3 (Figure S2A). We also performed a baseline analysis of N-MYC protein expression in Kelly, SKNAS, GIMEN, and SY5Y cells to confirm differential gene expression of MYCN (Figure S2B). We then used the optimized concentration of 100 nM of JQ1 in combination with ATRA to test their effectiveness in causing differentiation and changes in core regulatory transcription factor expression. We tested the gene expression of ASCL1, GATA3, and SOX10 in the three cell lines after 72 h of treatment. The treatment of ATRA in the presence of JQ1 in Kelly and SY5Y cells showed a significant suppression of ASCL1 expression, a significant increase in GATA3 expression, and a significant decrease in the expression of SOX10 (Figures 3A and 3B), recapitulating what was observed with gene silencing of ASCL1 (Figures 1E−1H). For GATA3, which is regulated by a super-enhancer in a subset of NBCLs, we do observe a reduction of GATA3 in response to JQ1 in SY5Y cells that is offset when also treated by ATRA (Figure 3B). Whereas JQ1 is expected to cause reduced gene expression by disrupting the GATA3 super-enhancer in SY5Y, the GATA3 promoter seems directly or indirectly responsive to ATRA in SY5Y cells after JQ1 treatment. In contrast, the most mesenchymal type of cell line tested, SKNAS, did not show any significant changes in the mRNA expression of ASCL1, GATA3, or SOX10 genes (Figure 3C). However, SKNAS has the most divergent basal expression of these factors with low protein expression of ASCL1 and GATA3 in comparison to the other cell lines tested and the highest SOX10 expression, which suggests SKNAS has a different developmental block from the other cells lines (Figure S1).
Figure 3.

Effect of JQ1 and ATRA combinations on gene expression of CRC in NB cells
(A−C). Gene expression by RT-PCR of ASCL1, GATA3, and SOX10 in (A) Kelly, (B) SY5Y, and (C) SKNAS cells under the treatment of independent and combinations of JQ1 + ATRA. (All data presented as mean ± SEM with n = 3, ∗∗p < 0.01 or ∗p < 0.05.)
We next wanted to determine if the cells were differentiating; therefore, gene expression of TH and myelin basic protein (MBP), a Schwann cell marker, was analyzed in cells treated for 3 days, demonstrating significant upregulation of both markers in Kelly and SY5Y (Figures 4A and 4B) after combinatorial JQ1/ATRA treatment in comparison to independent treatments. Whereas combination treatment in SKNAS showed a significant increase in TH expression (Figure 4A), no change in MBP expression was observed (Figure 4B). The combinatorial treatment of JQ1 100 nM with ATRA 1 μΜ for 4 days showed by western blot analysis a significant suppression of SOX10 in Kelly, SY5Y, and SKNAS cells with more modest effects for TH in SY5Y and for ASCL1 in SKNAS (Figures 4C−4E). The differences in results between protein and mRNA for SOX10 in SKNAS cells in response to JQ1 with ATRA could be due to timing of the experiments with RT-PCR being done at 3 days and western blot at 4 days, or it might indicate that ATRA promotes SOX10 protein turnover when cells are also treated with JQ1. In Kelly cells, N-Myc is regulated by a super-enhancer that can be suppressed by BET inhibitors, which we confirmed; interestingly, we noted that ATRA does not further affect N-Myc expression, suggesting other factors besides N-Myc are responsible for the results of the combinatorial therapy (Figure S2C).19 Under combinatorial therapy, we do observe significant reduction of SOX10 protein expression across the cell lines, indicating a loss of mesenchymal/stem identity.
Figure 4.

Evaluation of differentiation markers under JQ1 and ATRA combinatorial treatments in NB cells
(A and B) Gene expression by RT-PCR of TH and MBP in Kelly, SY5Y, and SKNAS cells under the treatment of independent and combinations of JQ1 + ATRA. (C−E) Protein expression analyzed by western blots of TH and ASCL1 in the three cell lines under the treatment of independent and combinations of JQ1 + ATRA. (Data presented as mean ± SEM with n = 3, ∗∗p < 0.01.)
We further confirmed the effect of combination treatment on differentiation by immune fluorescent staining of TH with images captured after 4 days of treatment in Kelly and SY5Y cells (Figure S3A). The combination of JQ1 and ATRA promoted an increase in differentiation across the three genetically diverse NBCLs. In addition, our data indicate they do so by overcoming a different developmental block in SKNAS than occurs in Kelly cells based on their divergent responses to treatment with JQ1 and ATRA for the CRC factors we assayed (Figures 3A−3C) and based on their different basal expression of CRC factors and N-MYC expression pattern (Figures S1 and S2A).
Combinatorial treatment of ATRA with JQ1 is synergistic in inhibition of proliferation in NB cells
Next, we tested the effect of ATRA, JQ1, or combinations on the inhibition of cell number. A significant reduction in cell number was observed in all cell lines with a further significant decrease with combined JQ1 + ATRA therapy for Kelly (Figure 5A), SY5Y (Figure 5B), and SKNAS (Figure 5C) versus single-agent treatments. Clinically, high-risk NB patients are treated with high-dose 13-cis-RA (isotretinoin) as maintenance therapy, which converts into ATRA and 9-cis-RA, a stereoisomer of ATRA.28,29 Both 9-cis-RA and ATRA can promote differentiation. Therefore, we tested 9-cis-RA versus ATRA in combination with JQ1 to compare their effects on differentiation in the presence of JQ1 in Kelly and SY5Y cells. The combination of JQ1 with either ATRA or 9-cis-RA led to comparable increases in TH and GATA3 expression and loss of ASCL1 and SOX10 expression, indicating similar effects on cell differentiation (Figures S3B and S3C). Next, we determined the combination index (CI) for median effect concentration of JQ1 with ATRA or 9-cis-RA using the Chou and Talalay method (CompuSyn software).30 We found a CI of 0.56 against Kelly and 0.61 against SKNAS cells with dose versus fraction affected (Fa) curves for Kelly (Figure 5D) and SKNAS (Figure 5E), indicating this combination is synergistic in vitro. The findings of CI values against Fa indicate combinatorial treatment of low-dose JQ1 (100 nM) with 100 nM, 500 nM, or 1,000 nM of ATRA has CIs of 0.33, 0.40, and 0.29, respectively, indicating effective synergies, whereas high-dose JQ1 (1 μM) in combination with 100 nM or 500 nM ATRA has CIs of 1.39 and 1.03, respectively, indicating an antagonistic affect when Kelly cells are treated with high concentrations of JQ1 and lower concentrations of ATRA (Figure 5F). We had similar findings for SKNAS, where low-dose JQ1 was synergistic with ATRA, but high dose was antagonistic (Figure 5G). Additionally, we screened 9-cis-RA in combination with JQ1 analyzing the dose versus effect, with CI values in Kelly and SKNAS cells finding similar effects as we observed with ATRA (Figures S4A−S4D). However, we found 9-cis-RA to be less effective than ATRA, showing higher CI but still synergistic values at combinations of 100 nM JQ1 with either 100 nM or 500 nM of 9-cis-RA (CI values 0.82 and 0.81).
Figure 5.

Determination of dose versus response effect of JQ1 + ATRA combinatorial treatments on NB cells
(A−C) Combinatorial treatment of JQ1 and ATRA on Kelly, SKNAS, and SY5Y on percent proliferation by CyQuant assay data. (D and E) Dose versus response curves in the treatments of JQ1 with ATRA combinations on Kelly and SKNAS cells. x axis represents the concentration of drugs used; y axis represents the percent inhibition of cells as fraction affected (Fa). (F and G) The combinatorial treatment of JQ1 + ATRA in Kelly and SKNAS on the quantitative outcomes of combination index (CI) values. x axis represents percent inhibition of cells as Fa; y axis represents CI, which generates drug-effect values with a differing ratio of drug combinations. Representative data were analyzed by CompuSyn software. (Data presented as mean ± SEM with n = 3, ∗∗p < 0.01 or ∗p < 0.05; ∗significant difference between JQ1 versus JQ1 + ATRA.)
Due to our observations that combinations of JQ1 and ATRA led to a reduction in SOX10 expression, which is a marker for stemness, we next performed two assays that are surrogate tests for cancer stem/progenitor cell behavior: a colony-forming assay and a tumor-sphere assay. For the colony-forming assay, we continuously treated cells with combinations of JQ1 and ATRA for 10 days with concentrations ranging from 10 nM to 1 μM for both compounds and showed a complete growth inhibition in the crystal violet assay image pattern of Kelly and SKNAS cells at a combination treatment of 10 nM JQ1 with 100 nM ATRA, which was not observed at equivalent concentrations of single agents (Figures 6A and S5). To determine the long-term treatment effects, we performed a soft-agar tumor sphere-forming assay treating with JQ1 and ATRA combinations from 10 nM and 100 nM to 1 μM concentrations for 25 days. The combination of 10 nM JQ1 with 100 nM ATRA significantly inhibited tumor-sphere formation and growth in soft agar similar to what was observed with the colony-forming assay (Figure 6B). Both the colony-forming and tumor-sphere assays indicate that BET inhibitors with RA treatment can reduced stem/progenitor behavior in NBCLs.
Figure 6.

Effect of JQ1 and ATRA combinatorial treatments on tumor-sphere formation
(A) Tumor-sphere formation in 25 days after treating cells with JQ1, ATRA, and combinations from 10 nM to 1 μM. (B) Proliferation inhibition measured by CyQuant assay of Kelly with JQ1 and ATRA combinatorial treatments for 10 days. Representative images of tumor spheres and proliferating colony of Kelly cells. Cells were imaged after adding crystal violet dye and the washing procedure.
Discussion
Dysregulated expression of CRC transcription factors in NB that control neural cell maturation is proposed to retain cells in immature oncogenic identity states. One underutilized strategy for treating NB is to push the cells into a more differentiated and less dangerous state through differentiation therapy. Although limited, RA treatments are the most successful therapeutics for differentiation therapy in cancer, and this study identifies a strategy to improve the efficacy of retinoids.31, 32, 33 NB is characterized by two identity states: an adrenergic proliferative state and a mesenchymal pro-migratory, therapy-resistant state that are maintained by different CRCs. In aggressive NB, MYCN is a frequently amplified transcription factor that can enforce high expression of ASCL1 in certain NB cell types including Kelly.34 In this study, after knockdown of ASCL1 in the MYCN-amplified Kelly cell line, we observed opposite responses of gene and protein expression in the core regulatory transcription factors GATA3 and SOX10, which regulate cell identity during development. Silencing of ASCL1 in combination with ATRA further increased TH and reduced proliferation in Kelly cells. This indicated that disrupting the developmental block caused by high ASCL1 expression in combination with ATRA is an effective strategy to drive differentiation of Kelly cells.
Other subtypes of NB that do not have a defined oncogenic driver like N-Myc and whose etiology is not as well understood also have immature CRCs.35 We investigated whether resistance to ATRA-induced differentiation could be overcome by disrupting developmentally immature CRCs irrespective of the NB subtype using the BET inhibitor JQ1. BET inhibitors suppress the super-enhancers that often increase the expression of transcription factors that regulate identity states. Treatment with JQ1 and retinoids (ATRA or 9-cis RA) resulted in the three cell lines showing (1) a significant loss of SOX10, a key neural stem cell marker that plays a prominent role in multipotency and inhibition of neuronal differentiation;36 (2) a gain of the differentiation marker TH; and (3) a significant inhibition in cell growth in multiple in vitro assays. We observed that JQ1 was sufficient to cause decreased gene expression of the mesenchymal/stem marker SOX10 in Kelly and SY5Y cells, whereas in the time frame tested, protein levels only decreased when cells were treated with both JQ1 and ATRA. Similarly, we observe that SKNAS does not show loss of SOX10 mRNA expression but does show loss of SOX10 protein when treated by the combination. This suggests ATRA promotes protein turnover of SOX10. RA has previously been shown to promote protein degradation of the insulin receptor substrate (IRS-1) and in NBCLs, repressor element-1 silencing transcription factor (REST).37, 38, 39 The reduced SOX10 expression, loss of colony formation, and loss of tumor-sphere formation in response to combinatorial treatment indicate a loss of stem/progenitor cells, the proposed cause of metastatic spread and relapse disease.40
We have demonstrated that Kelly cells are dependent on ASCL1 expression, and the changes we observe with ASCL1 silencing are also seen with JQ1/ATRA combinatorial treatment in Kelly cells. Although we have not demonstrated that SY5Y is ASCL1 dependent, this cell line has similar responses to JQ1/ATRA treatment as Kelly for the markers we have analyzed, suggesting SY5Y might have a similar developmental block as Kelly cells. In contrast, SKNAS shows multiple differing behavior with the other two cell lines in its response to JQ1/ATRA, suggesting this line has a different developmental block. It is known that SKNAS cells have a chromosomal translocation that places c-Myc under the control of the HAND2 super-enhancer, which may explain its differential response to JQ1/ATRA treatment.40 Overall, these findings suggests that the JQ1/ATRA drug combination can overcome different developmental blocks in cell differentiation that occur in NBCL and therefore could be broadly useful in treating NB with different underlying mutations.
NB patients are treated with 13-cis-RA (isotretinoin), which is metabolized into ATRA and 9-cis-RA, a stereoisomer of ATRA, and has been shown in some instances to be more effective than ATRA in differentiating NBCLs. ATRA binds the RA receptor (RAR), whereas 9-cis-RA binds both RAR and the RAR heterodimerization partner retinoid x receptor (RXR). We observed similar responses to both ATRA and 9-cis-RA in our studies; however, ATRA was more potent at lower concentrations. Interestingly, high doses of JQ1 were antagonistic to lower doses of both retinoids. We speculate that high-dose JQ1 interferes with the ability of ATRA to establish a more differentiated CRC, since BET inhibitors indiscriminately affect CRCs. This suggests that timing and dosing of BET inhibitors with retinoids will be critical in their effectiveness and should be further explored in preclinical models.
Materials and methods
Cell lines and culture methods
Human NBCLs Kelly, SY5Y, and SKNAS from the European collection of cell cultures and American Type Culture Collection (ATCC; Manassas, VA, USA) and Gimen cells were purchased from Cell Line Service (Germany). These NB cells were cultured as previously described.41,42 Briefly, Kelly cell-culture media include RPMI with 10% fetal bovine serum (FBS); SY5Y cells cultured in MEMα with Ham’s F12, NEAA, and 10% FBS; and SKNAS cells in DMEM, 1% NEAA, and 10% FBS. Gimen cells were grown on DMEM plus 10% FBS.
Dox-inducible shRNA system
Two different mission shRNAs from the TRC1 library (Sigma-Aldrich; TRCN0000013550 and TRCN0000235656, referred to in the manuscript as sh6 and sh8, respectively) targeting ASCL1 and one non-targeting shRNA control (NTC) were used to generate Kelly NBCLs with ASCL1 knockdown. ASCL1-specific shRNAs were cloned into the Tet-pLKO-puro plasmid (Addgene). Lentiviral particles were produced in 293T cells, and Kelly cells were infected and incubated with viral particles for 24 h with 4 μg/mL Polybrene. Selection with puromycin (Invitrogen) at 500 ng/mL was performed 48 h after infection and maintained during all culture experiments. ASCL1 knockdown efficacy was assessed by immunoblot 72 h after the addition of Dox (1 μg/mL).
Drug treatments
RA from Sigma (cat. #R2625), BET inhibitor JQ1 from Cayman Chemical (cat. #11232), puromycin (cat. #11138-10; Gibco), Dox (cat. #D9891; Sigma), and DMSO (cat. #BP231-100; Fisher Scientific) were prepared in 10 mM or 10 mg/mL stocks depending on requirements, filter sterilized, and stored in −80°C until further use.
Cell proliferation assays
In a 96-well tissue-culture plate, 1 × 104 cells were plated in each well. Plates were incubated for 4 days in the presence of DMSO, RA, and JQ1 drug treatments and then submitted to CyQuant Cell Direct Proliferation Assay (Thermo Fisher Scientific), according to the manufacturer’s instructions, and read with a Synergy HT Multi-mode microplate reader (BioTek Instruments).
Soft agar and crystal violet staining assay
Kelly and SKNAS cells 5 × 104 were incubated in a soft agar mixture of 0.3% upper layer- and 0.6% agar-quoted 12-well culture plates. Cells were incubated for 21 days in DMSO, ATRA, JQ1, and combinations, changing the media every 3 days. Images of tumor sphere were observed and captured by Microphot FX (Nikon) by treating the plates with 0.01% crystal violet and subsequent gentle washing with PBS. A 10-day incubation of Kelly and SKNAS in drug treatments was done in normal plates, and plates were stained with crystal violet to observe the effect of drug treatments on proliferation.
Immunofluorescence and confocal microscopy
Kelly and SY5Y cells were cultured on 8-well chamber slides under the treatments of DMSO, RA, JQ1, and JQ1 + RA condition for 4−7 days. After completion of the time course, cells were immune stained with TH primary antibody and Alexa Fluor 488 secondary antibody (Invitrogen) and for the imaging under confocal microscopy (Zeiss 5000) at the University of Tennessee Health Science Center (UTHSC) neuro-imaging facility.
Gene-expression analysis
RNA was isolated using the RNeasy Mini Kit (QIAGEN USA), following the manufacturer’s protocol. 1 μg of total RNA from each sample was converted to cDNA using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific). The resulting cDNA sample served as a template for real-time qPCR using TaqMan probes and accompanying TaqMan Master Mix (Applied Biosystems, Foster City, CA, USA). PCR amplification was carried out using the Quantstudio3 (Applied Biosystems) system with cycle conditions of the initial cycle: 50°C for 2 min and initial denaturation at 95°C for 15 s. This was followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension of 60°C for 1 min. The expression levels of target gene transcripts were determined using the 2−DDCt method and normalized to PPIB. TaqMan probes used in the study (Applied Biosystems) include PPIB (cat. #Hs00168719), TH (cat. #Hs00165941), ASCL1 (cat. #Hs04187546), GATA3 (cat. #Hs Hs00231122), SOX10 (cat. #Hs00366918), TWIST1 (cat. #Hs04989912), MBP (cat. #Hs00921945), and GFAP (cat. #Hs00157674).
Protein blot analysis
After incubation of the cells with designated treatment conditions, the lysates were prepared, and protein expression was measured by western blot analysis using anti-ASCL1 (cat. #ABE1025; Sigma/Millipore), anti-TH (cat. #2792; CST [Cell Signaling Technology], Danvers, MA, USA), anti-GATA3 (cat. #5852; CST), anti-PHOX2B (cat. #AB227719; Abcam), anti-SOX9 (cat. #82630; CST), anti-SOX10 antibody (cat. #89356; CST), anti-vimentin (cat. #5741; CST), anti-SNAI2 (cat. #9585; CST), anti-TWIST1 (cat. #4670; CST), anti-ACTIN (cat. #3700), anti-PRRX1 (cat. #B2380; LS Bio), and secondary goat anti-rabbit Alexa Fluor 488 antibody (BD Biosciences); secondary antibodies with IR Dyes 700 (mouse) and 800 (rabbit) were purchased from LI-COR Biosciences. Western blot images were captured on nitrocellulose (Whatman) by the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Quantification of the protein bands on blots was performed by Image Studio Lite version 5.2 (LI-COR Odyssey).
Statistics
All experiments were replicated three times. Differences were tested for statistical significance using ANOVA groups or as unpaired t test for two groups (GraphPad Prism) with data presented as the mean ± standard error of the mean (SEM), with a p value of <0.05 considered significant from three independent experiments.
Acknowledgments
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: US National Institutes of Health (R01CA216394 to K.W.F.) and US Department of Defense (W81XWH-18-1-0477 to K.W.F.).
Author contributions
Conception and design, K.W.F. and S.A.; development of methodology, K.W.F., S.A., and N.A.; acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.), S.A.; analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis), K.W.F. and S.A.; writing, review, and/or revision of the manuscript, K.W.F. and S.A.; administrative and technical support (reporting or organizing data and constructing databases), S.A., N.A., and M.M.; study supervision, K.W.F. and S.A.
Declaration of interest
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.09.004.
Supplemental information
References
- 1.Maris J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung N.K., Dyer M.A. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J.R., Eggert A., Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol. Oncol. Clin. North Am. 2010;24:65–86. doi: 10.1016/j.hoc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 4.Peaston A.E., Gardaneh M., Franco A.V., Hocker J.E., Murphy K.M., Farnsworth M.L., Catchpoole D.R., Haber M., Norris M.D., Lock R.B., Marshall G.M. MRP1 gene expression level regulates the death and differentiation response of neuroblastoma cells. Br. J. Cancer. 2001;85:1564–1571. doi: 10.1054/bjoc.2001.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Applebaum M.A., Henderson T.O., Lee S.M., Pinto N., Volchenboum S.L., Cohn S.L. Second malignancies in patients with neuroblastoma: the effects of risk-based therapy. Pediatr. Blood Cancer. 2015;62:128–133. doi: 10.1002/pbc.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K., Diskin S.J., Zhang H., Attiyeh E.F., Winter C., Hou C., Schnepp R.W., Diamond M., Bosse K., Mayes P.A. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011;469:216–220. doi: 10.1038/nature09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laverdière C., Cheung N.K., Kushner B.H., Kramer K., Modak S., LaQuaglia M.P., Wolden S., Ness K.K., Gurney J.G., Sklar C.A. Long-term complications in survivors of advanced stage neuroblastoma. Pediatr. Blood Cancer. 2005;45:324–332. doi: 10.1002/pbc.20331. [DOI] [PubMed] [Google Scholar]
- 8.Pinto N.R., Applebaum M.A., Volchenboum S.L., Matthay K.K., London W.B., Ambros P.F., Nakagawara A., Berthold F., Schleiermacher G., Park J.R. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015;33:3008–3017. doi: 10.1200/JCO.2014.59.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomolonis J.A., Agarwal S., Shohet J.M. Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res. 2018;372:245–262. doi: 10.1007/s00441-017-2747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H.S., Redmond T.M., Kubish G.M., Gupta S., Thompson R.C., Turner D.L., Uhler M.D. Transcriptional regulatory events initiated by Ascl1 and Neurog2 during neuronal differentiation of P19 embryonic carcinoma cells. J. Mol. Neurosci. 2015;55:684–705. doi: 10.1007/s12031-014-0408-2. [DOI] [PubMed] [Google Scholar]
- 11.Decaesteker B., De Preter K., Speleman F. DREAM target reactivation by core transcriptional regulators supports neuroblastoma growth. Mol. Cell. Oncol. 2019;6:1565470. doi: 10.1080/23723556.2019.1565470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almutairi B., Charlet J., Dallosso A.R., Szemes M., Etchevers H.C., Malik K.T.A., Brown K.W. Epigenetic deregulation of GATA3 in neuroblastoma is associated with increased GATA3 protein expression and with poor outcomes. Sci. Rep. 2019;9:18934. doi: 10.1038/s41598-019-55382-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boeva V., Louis-Brennetot C., Peltier A., Durand S., Pierre-Eugène C., Raynal V., Etchevers H.C., Thomas S., Lermine A., Daudigeos-Dubus E. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017;49:1408–1413. doi: 10.1038/ng.3921. [DOI] [PubMed] [Google Scholar]
- 14.Soldatov R., Kaucka M., Kastriti M.E., Petersen J., Chontorotzea T., Englmaier L., Akkuratova N., Yang Y., Häring M., Dyachuk V. Spatiotemporal structure of cell fate decisions in murine neural crest. Science. 2019;364:eaas9536. doi: 10.1126/science.aas9536. [DOI] [PubMed] [Google Scholar]
- 15.Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007;8:755–765. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]
- 16.Zimmerman M.W., Durbin A.D., He S. Retinoic acid rewires the adrenergic core regulatory circuitry of neuroblastoma but can be subverted by enhancer hijacking of MYC or MYCN. bioRxiv. 2020 2020.2007.2023.218834. [Google Scholar]
- 17.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S., Rellinger E.J., Kim K.W., Craig B.T., Romain C.V., Qiao J., Chung D.H. Bromodomain and extraterminal inhibition blocks tumor progression and promotes differentiation in neuroblastoma. Surgery. 2015;158:819–826. doi: 10.1016/j.surg.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puissant A., Frumm S.M., Alexe G., Bassil C.F., Qi J., Chanthery Y.H., Nekritz E.A., Zeid R., Gustafson W.C., Greninger P. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Groningen T., Koster J., Valentijn L.J., Zwijnenburg D.A., Akogul N., Hasselt N.E., Broekmans M., Haneveld F., Nowakowska N.E., Bras J. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017;49:1261–1266. doi: 10.1038/ng.3899. [DOI] [PubMed] [Google Scholar]
- 21.Amiel J., Laudier B., Attié-Bitach T., Trang H., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 22.Moriguchi T., Takako N., Hamada M., Maeda A., Fujioka Y., Kuroha T., Huber R.E., Hasegawa S.L., Rao A., Yamamoto M. Gata3 participates in a complex transcriptional feedback network to regulate sympathoadrenal differentiation. Development. 2006;133:3871–3881. doi: 10.1242/dev.02553. [DOI] [PubMed] [Google Scholar]
- 23.Aquino J.B., Hjerling-Leffler J., Koltzenburg M., Edlund T., Villar M.J., Ernfors P. In vitro and in vivo differentiation of boundary cap neural crest stem cells into mature Schwann cells. Exp. Neurol. 2006;198:438–449. doi: 10.1016/j.expneurol.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Aldskogius H., Berens C., Kanaykina N., Liakhovitskaia A., Medvinsky A., Sandelin M., Schreiner S., Wegner M., Hjerling-Leffler J., Kozlova E.N. Regulation of boundary cap neural crest stem cell differentiation after transplantation. Stem Cells. 2009;27:1592–1603. doi: 10.1002/stem.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isenmann S., Arthur A., Zannettino A.C., Turner J.L., Shi S., Glackin C.A., Gronthos S. TWIST family of basic helix-loop-helix transcription factors mediate human mesenchymal stem cell growth and commitment. Stem Cells. 2009;27:2457–2468. doi: 10.1002/stem.181. [DOI] [PubMed] [Google Scholar]
- 26.Díaz V.M., Viñas-Castells R., García de Herreros A. Regulation of the protein stability of EMT transcription factors. Cell Adhes. Migr. 2014;8:418–428. doi: 10.4161/19336918.2014.969998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwase K., Nagasaka A., Nagatsu I., Kiuchi K., Nagatsu T., Funahashi H., Tsujimura T., Inagaki A., Nakai A., Kishikawa T. Tyrosine hydroxylase indicates cell differentiation of catecholamine biosynthesis in neuroendocrine tumors. J. Endocrinol. Invest. 1994;17:235–239. doi: 10.1007/BF03348964. [DOI] [PubMed] [Google Scholar]
- 28.Veal G.J., Cole M., Errington J., Pearson A.D., Foot A.B., Whyman G., Boddy A.V., UKCCSG Pharmacology Working Group Pharmacokinetics and metabolism of 13-cis-retinoic acid (isotretinoin) in children with high-risk neuroblastoma - a study of the United Kingdom Children’s Cancer Study Group. Br. J. Cancer. 2007;96:424–431. doi: 10.1038/sj.bjc.6603554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthay K.K., Villablanca J.G., Seeger R.C., Stram D.O., Harris R.E., Ramsay N.K., Swift P., Shimada H., Black C.T., Brodeur G.M., Children’s Cancer Group Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. N. Engl. J. Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 30.Chou T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 31.Matthay K.K., Reynolds C.P., Seeger R.C., Shimada H., Adkins E.S., Haas-Kogan D., Gerbing R.B., London W.B., Villablanca J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J. Clin. Oncol. 2009;27:1007–1013. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohler J.A., Imeson J., Ellershaw C., Lie S.O. A randomized trial of 13-Cis retinoic acid in children with advanced neuroblastoma after high-dose therapy. Br. J. Cancer. 2000;83:1124–1127. doi: 10.1054/bjoc.2000.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peinemann F., van Dalen E.C., Tushabe D.A., Berthold F. Retinoic acid post consolidation therapy for high-risk neuroblastoma patients treated with autologous hematopoietic stem cell transplantation. Cochrane Database Syst. Rev. 2015;1:CD010685. doi: 10.1002/14651858.CD010685.pub2. [DOI] [PubMed] [Google Scholar]
- 34.Westermann F., Muth D., Benner A., Bauer T., Henrich K.O., Oberthuer A., Brors B., Beissbarth T., Vandesompele J., Pattyn F. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008;9:R150. doi: 10.1186/gb-2008-9-10-r150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L., Tan T.K., Durbin A.D., Zimmerman M.W., Abraham B.J., Tan S.H., Ngoc P.C.T., Weichert-Leahey N., Akahane K., Lawton L.N. ASCL1 is a MYCN- and LMO1-dependent member of the adrenergic neuroblastoma core regulatory circuitry. Nat. Commun. 2019;10:5622. doi: 10.1038/s41467-019-13515-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J., Lo L., Dormand E., Anderson D.J. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 37.del Rincón S.V., Guo Q., Morelli C., Shiu H.Y., Surmacz E., Miller W.H. Retinoic acid mediates degradation of IRS-1 by the ubiquitin-proteasome pathway, via a PKC-dependant mechanism. Oncogene. 2004;23:9269–9279. doi: 10.1038/sj.onc.1208104. [DOI] [PubMed] [Google Scholar]
- 38.Singh A., Rokes C., Gireud M., Fletcher S., Baumgartner J., Fuller G., Stewart J., Zage P., Gopalakrishnan V. Retinoic acid induces REST degradation and neuronal differentiation by modulating the expression of SCF(β-TRCP) in neuroblastoma cells. Cancer. 2011;117:5189–5202. doi: 10.1002/cncr.26145. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y., Zhang D., Tang Z., Zhang Y., Gao H., Ni N., Shen B., Sun H., Gu P. REST, regulated by RA through miR-29a and the proteasome pathway, plays a crucial role in RPC proliferation and differentiation. Cell Death Dis. 2018;9:444. doi: 10.1038/s41419-018-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visvader J.E., Lindeman G.J. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Edsjö A., Nilsson H., Vandesompele J., Karlsson J., Pattyn F., Culp L.A., Speleman F., Påhlman S. Neuroblastoma cells with overexpressed MYCN retain their capacity to undergo neuronal differentiation. Lab. Invest. 2004;84:406–417. doi: 10.1038/labinvest.3700061. [DOI] [PubMed] [Google Scholar]
- 42.Biedler J.L., Helson L., Spengler B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973;33:2643–2652. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.