

Abstract
Triple-negative breast cancer (TNBC) is an aggressive and highly lethal disease. The lack of targeted therapies and poor patient outcome have fostered efforts to discover new molecular targets to treat patients with TNBC. Here, we showed that baculoviral IAP repeat containing 6 (BIRC6) is overexpressed and positively correlated with epidermal growth factor (EGF) receptor (EGFR) in TNBC cells and tissues and that BIRC6 overexpression is associated with poor patient survival. Mechanistic studies revealed that BIRC6 stability is increased by EGF-JNK signaling, which prevents ubiquitination and degradation of BIRC6 mediated by the E3 ubiquitin ligase HECTD1. BIRC6 in turn decreases SMAC expression by inducing the ubiquitin-proteasome pathway, thereby antagonizing apoptosis and promoting the proliferation, colony formation, tumorsphere formation, and tumor growth capacity of TNBC cells. Therapeutically, the PEGylated cationic lipid nanoparticle (pCLN)-assisted delivery of BIRC6 small interfering RNA (siRNA) efficiently silences BIRC6 expression in TNBC cells, thus suppressing TNBC cell growth in vitro and in vivo, and its antitumor activity is significantly superior to that of the EGFR inhibitor gefitinib. Our findings identify an important regulatory mechanism of BIRC6 overexpression and provide a potential therapeutic option for treating TNBC.
Keywords: TNBC, BIRC6, EGFR signaling, HECTD1, lipid nanoparticles
Graphical abstract
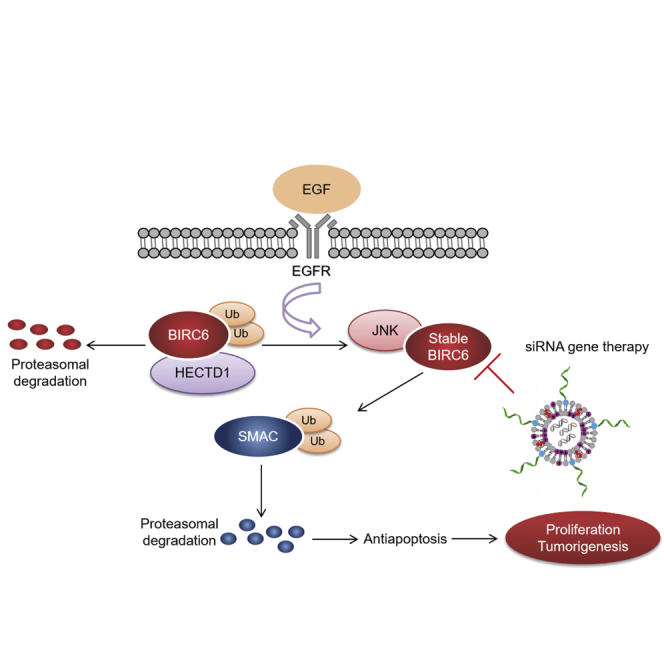
TNBC is the most aggressive subtype of breast cancer, and discovery of new targeted therapies is required. Our findings show that EGF-JNK-HECTD1 signaling can drive the overexpression of BIRC6 in TNBC. Targeting BIRC6 with lipid nanoparticle-coated siRNA represents a very promising approach for treating TNBC.
Introduction
Triple-negative breast cancer (TNBC), as defined by the absence of the estrogen receptor (ER) and the progesterone receptor (PR) and lack of human epidermal growth factor receptor 2 (HER2) overexpression or amplification, comprises 12%–17% of all breast cancers.1 Compared with other subtypes, TNBC is usually more aggressive, and patients have a higher rate of early distant recurrence and a worse prognosis.2, 3, 4, 5 PARP inhibitor and PD-L1 antibody are currently approved as therapies for BRCA-mutated and PD-L1-positive TNBC, respectively.6,7 However, targeted therapeutic treatments for TNBC are still in the early stage of development, while chemotherapy remains the standard treatment.8 Therefore, the identification of novel molecular targets that might be effectively targeted in TNBC is urgently needed.
Baculoviral IAP repeat containing 6 (BIRC6), also known as Apollon or BRUCE, is a giant (relative molecular mass of 530,000) and less-studied member of the Inhibitor of Apoptosis Proteins (IAP) family.9 BIRC6 bears a single baculoviral IAP-repeat (BIR) domain and a unique ubiquitin-conjugating (UBC) enzyme domain at the N and C termini, respectively.10 Because of the presence of UBC domain, BIRC6 functions as a chimeric E2/E3 ubiquitin ligase and facilitates proteasomal degradation of proapoptotic proteins SMAC, caspase-9, HtrA2, and p53.11, 12, 13, 14 Further investigations are needed to determine which of these proteins is the main downstream regulator of BIRC6.
Ample evidence indicated that BIRC6 is highly expressed in many cancer types including de novo acute myeloid leukemia, prostate cancer, non-small-cell lung cancer, ovarian cancer, hepatocellular carcinoma, esophageal squamous cell carcinoma, and colorectal cancer, where elevated expression of BIRC6 is significantly correlated with poor clinical outcome.14, 15, 16, 17, 18, 19, 20 However, the expression pattern and biological function of BIRC6 during TNBC tumorigenesis are not well understood. More importantly, the mechanisms underlying BIRC6 overexpression in cancer remain unclear.
Currently, there is no clinical inhibitor targeting BIRC6.21 Gene therapy using small interfering RNA (siRNA) has shown great potential to downregulate specific gene expression in cancer cells.22 However, the widespread application of unmodified siRNAs is severely limited because of their rapid degradation within serum full of nucleases and poor intracellular uptake into target tissues.23 To date, a variety of nanocarriers such as liposomes, polymeric nanoparticles, carbon nanotubes, and metallic nanoparticles have been developed to efficiently deliver siRNA.23,24 Among those formulations, lipid nanoparticles (LNPs) are the most advanced delivery systems that have enabled clinical translation of siRNA therapeutics.25 Therefore, we hypothesize that LNP-assisted delivery of BIRC6-siRNA can silence BIRC6 expression in TNBC cells with no adverse effects.
In this study, we aimed to characterize the mechanism of BIRC6 overexpression and evaluated its therapeutic potential in TNBC. We demonstrated that epidermal growth factor (EGF) receptor (EGFR) signaling was a major upstream regulator driving high expression of BIRC6. Consistent with EGFR, BIRC6 was highly expressed in TNBC cells and tissues and BIRC6 overexpression was associated with poor patient survival. Mechanistically, EGF-JNK signaling upregulated the expression of BIRC6 by blocking E3 ubiquitin ligase HECTD1-mediated ubiquitination and proteasomal degradation. BIRC6 further promoted TNBC cell survival and tumorigenesis by antagonizing SMAC-mediated apoptosis. Therapeutically, PEGylated cationic lipid nanoparticles (pCLNs) were developed to effectively deliver BIRC6-siRNA to silence BIRC6 expression in TNBC, with the aim of inhibiting TNBC tumorigenesis without adverse effects. Interestingly, BIRC6 depletion was more effective to suppress TNBC cell growth in vitro and in vivo compared to EGFR inhibition. These findings reveal the importance of targeting overexpressed BIRC6 triggered by EGF-JNK-HECTD1 signaling in TNBC.
Results
BIRC6 expression is positively correlated with EGFR and associated with poor prognosis in TNBC
To identify the upstream regulator of BIRC6, we analyzed the association between the expression of BIRC6 and signaling pathways by performing gene set enrichment analysis (GSEA) in the TCGA breast cancer dataset (n = 1,222) with the R package “fgsea.” Results showed that gene sets of EGFR were enriched in samples with high expression of BIRC6 (Figure 1A). Because EGFR signaling is often highly active in TNBC,26 we sought to determine whether EGFR regulated BIRC6 expression. We generated doxycycline-inducible EGFR-knockdown Hs578T and MDA-MB-468 TNBC cells with the specific short hairpin RNA (shRNA). Immunoblot analysis showed that EGFR can be efficiently depleted upon doxycycline treatment, which also led to decreased BIRC6 expression (Figure 1B). Next, we detected endogenous BIRC6 and EGFR expression in a panel of breast cancer cell lines. Compared with the normal breast epithelial cell lines or non-TNBC cell lines, BIRC6 expression tended to be higher in TNBC cell lines, which was consistent with EGFR expression (Figure 1C). We also confirmed the association between BIRC6 and EGFR in TNBC tissues by using immunohistochemistry (IHC) staining. Among 90 TNBC human tissues, both BIRC6 (66/90: 73.3%) and EGFR (34/90: 37.8%) levels were increased and showed a positive correlation (Figures 1D and 1E; p = 0.008, r = 0.2776). Therefore, BIRC6 is overexpressed and positively correlated with EGFR expression in TNBC.
Figure 1.

BIRC6 expression is positively correlated with EGFR and associated with poor survival in TNBC
(A) Gene set enrichment analysis was performed in the TCGA breast cancer dataset (n = 1,222) with R package “fgsea.” (B) Cells harboring an inducible EGFR-specific shRNA were treated with doxycycline hyclate for the indicated times, and the indicated proteins were detected by immunoblotting. (C) The expression profiles of BIRC6 and EGFR in eight TNBC and seven non-TNBC cell lines plus two breast normal cell lines were assessed by immunoblotting. The cell lines with high expression of EGFR are indicated in red. (D) Representative IHC staining for BIRC6 and EGFR in 90 tumor samples from TNBC patients. Case J07A2470 and Case J07A2313 are representative of a TNBC patient with BIRC6 and EGFR double high expression or double low expression, respectively. (E) Correlation between BIRC6 and EGFR expression in 90 tumor samples from TNBC patients. (F and G) Kaplan-Meier curves for OS (F) and RFS (G) in breast cancer patients with different BIRC6 expression. Patients were grouped into BIRC6-low or BIRC6-high based on the BIRC6 mRNA expression value.
Using 500 TCGA patient data from the OncoLnc database, we found that high BIRC6 expression was significantly associated with the poor overall survival (OS) of patients with breast invasive carcinoma (p = 0.04, Figure 1F). Similarly, another Kaplan-Meier plotter database also showed shorter relapse-free survival (RFS) in patients with high BIRC6 expression in TNBC (p = 0.0074, Figure 1G).
EGF-JNK signaling increases BIRC6 stability
Since EGFR signaling can lead to high expression of BIRC6 in TNBC, we sought to explore the mechanisms underlying EGFR activation-regulated BIRC6 expression. Immunoblot analysis showed that EGF treatment increased the protein expression of BIRC6 in Hs578T and MDA-MB-468 cells within 5–30 min (Figure 2A; Figure S1A). However, BIRC6 mRNA levels did not increase significantly in response to EGF (Figure S1B), suggesting that EGF regulates BIRC6 posttranslationally rather than transcriptionally or translationally.
Figure 2.

EGF-JNK signaling regulates BIRC6 stability
(A) After serum starvation for 48 h, cells were treated with EGF for 30 min, followed by immunoblotting as indicated. (B) After serum starvation, Hs578T cells were treated with or without SP600125 (10 μM) for 1.5 h and then treated with EGF for 30 min. Equal amounts of cell lysates were immunoblotted with the indicated antibodies. (C) Hs578T cell lysates were immunoprecipitated with an anti-BIRC6 antibody, followed by immunoblotting as indicated. (D) Hs578T cells were treated with or without EGF (100 ng/mL) and then treated with MG132 (10 μM) for another 6 h. Cell lysates were immunoprecipitated with an anti-BIRC6 antibody, followed by immunoblotting as indicated. (E) Hs578T cells were treated with or without SP600125 (10 μM) and then treated with cycloheximide (CHX, 100 μg/mL) for the indicated times. Cell lysates were immunoblotted with the indicated antibodies. (F) After serum starvation, Hs578T cells were treated with or without EGF and then treated with CHX (100 μg/mL) for the indicated times. Cell lysates were immunoblotted as indicated.
EGF treatment can activate several downstream signaling pathways, including RAS-RAF-MAPK, PI3K-AKT, PAK-JNKK-JNK, and Src kinase.27 To determine which of these pathways might be required for EGF-stimulated BIRC6 protein expression, we treated Hs578T cells with specific inhibitors of these pathways including trametinib (MEK), MK2206 (AKT), SP600125 (JNK), and saracatinib (Src). The EGF-stimulated increase in BIRC6 protein was only abrogated in the presence of the JNK inhibitor (Figure 2B; Figure S1C), suggesting that the JNK pathway is the primary signaling pathway involved in EGF-mediated BIRC6 regulation. The co-immunoprecipitation assay showed that JNK can bind to BIRC6 (Figure 2C). Furthermore, we found that EGF treatment caused JNK activation, whereas it reduced the ubiquitination level of BIRC6 (Figure 2D). We also performed pulse-chase experiments with cycloheximide (CHX) to inhibit new protein synthesis and found that SP600125 decreased BIRC6 protein stability (Figure 2E; Figure S1D), whereas EGF, which induced JNK activation, increased BIRC6 protein stability (Figure 2F; Figure S1E). Taken together, these results indicate that EGF-JNK signaling contributes to the stabilization of BIRC6.
The E3 ligase HECTD1 regulates BIRC6 stability during EGFR activation
To identify the potential E3 ubiquitin ligase responsible for BIRC6 degradation, we performed co-immunoprecipitation in myc-tagged BIRC6-transfected 293T cells by using anti-BIRC6 or anti-myc antibodies. We subsequently used mass spectrometry analysis to identify E3 ligase among BIRC6 binding partners. We found that the E3 ubiquitin ligase HECTD1 can be detected in BIRC6 and myc immunoprecipitates (Figure 3A). Also, endogenous interaction between BIRC6 and HECTD1 was found in Hs578T cells (Figure 3B).
Figure 3.

HECTD1 promotes BIRC6 ubiquitination and degradation during EGFR activation
(A) Coomassie blue staining of proteins co-precipitated with an anti-Myc or anti-BIRC6 antibody. Bottom: the number of unique peptides of HECTD1 identified by mass spectrometric analysis. (B) Hs578T cell lysates were immunoprecipitated with an HECTD1 antibody and subjected to immunoblotting analysis. (C) Hs578T cells expressing Flag-tagged HECTD1 were treated with or without MG132 for 8 h and then immunoblotted with the indicated antibodies. (D) Hs578T cells expressing Flag-tagged HECTD1 were treated with CHX (100 μg/mL) for the indicated times, followed by immunoblotting as indicated. (E) Scramble or HECTD1-knockdown Hs578T cells were treated with CHX (100 μg/mL) for the indicated times and then immunoblotted with the indicated antibodies. (F) After being transiently transfected with the indicated plasmids, 293T cells were immunoprecipitated with an anti-Myc antibody and then subjected to immunoblotting analysis. Cells were treated with 10 μM MG132 for 6 h before being lysed. (G) Scramble or HECTD1-knockdown Hs578T cells were immunoprecipitated with an anti-BIRC6 antibody and subjected to ubiquitination analysis. (H) After being serum starved overnight, scramble or HECTD1-knockdown Hs578T cells were treated with EGF for 30 min, followed by immunoblotting as indicated. (I) After being treated with or without EGF, Hs578T cells were immunoprecipitated with an anti-HECTD1 antibody and subjected to immunoblotting analysis.
We next examined whether BIRC6 stability is regulated by HECTD1. Forced overexpression of HECTD1 reduced the endogenous BIRC6 levels, and HECTD1-mediated BIRC6 downregulation could be rescued by the proteasome inhibitor MG132 (Figure 3C), suggesting that HECTD1 induced BIRC6 protein reduction through the proteasome pathway. Conversely, HECTD1 knockdown caused accumulation of endogenous BIRC6 (Figure 3H), demonstrating that HECTD1 is a negative regulator of BIRC6. In addition, CHX chase assays showed that HECTD1 overexpression accelerated BIRC6 protein degradation (Figure 3D), whereas BIRC6 protein was more stable after HECTD1 knockdown (Figure 3E). To further investigate whether degradation of BIRC6 was mediated through ubiquitination, we cotransfected 293T cells with Myc-BIRC6, Flag-HECTD1, and HA-ubiquitin and treated the cells with MG132. A significant increase in BIRC6 ubiquitination was observed in HECTD1-transfected cells (Figure 3F). We also found that HECTD1 depletion decreased the ubiquitination of endogenous BIRC6 (Figure 3G). Therefore, HECTD1 is a E3 ligase targeting BIRC6 protein for ubiquitination and degradation.
To further determine whether HECTD1 mediated BIRC6 stability during EGFR activation, we treated HECTD1-knockdown cells with EGF. Results revealed that BIRC6 expression in HECTD1-knockdown cells did not increase significantly in response to EGF (Figure 3H). Moreover, EGF treatment attenuated the interaction between BIRC6 and HECTD1 (Figure 3I). Overall, these data reveal that HECTD1 regulates BIRC6 stability during EGFR activation.
BIRC6 promotes TNBC cell growth by inhibiting SMAC-mediated apoptosis in vitro and in vivo
To explore the biological functions of BIRC6 in TNBC, we established stable BIRC6-knockdown clones in Hs578T (Figure 4A) and MDA-MB-468 (Figure S2A) cells. Knockdown of endogenous BIRC6 in TNBC cells significantly decreased cell proliferation and colony growth (Figures 4B and 4C; Figures S2B and S2C). Moreover, BIRC6 knockdown significantly diminished the capacity of TNBC cells to form tumorspheres (Figure 4D; Figure S2D), suggesting that BIRC6 may be involved in the survival of TNBC stem cells. To investigate the function of BIRC6 in vivo, we subcutaneously injected BIRC6 stable knockdown clones of MDA-MB-468 into nude mice and monitored tumor growth. Importantly, BIRC6 knockdown completely blocked tumor growth (Figure 4E).
Figure 4.

BIRC6 promotes TNBC cell growth and tumorigenesis by blocking SMAC-mediated apoptosis
(A–D) Immunoblotting analysis (A), MTT assay showing growth (B), colony formation assay (C), and tumorsphere assay (D) were performed in Hs578T cells transduced with scramble or BIRC6 shRNA. (E) The growth curves of xenografts derived from MDA-MB-468 cells transduced with scramble or BIRC6 shRNA. (F–I) Immunoblotting analysis (F), MTT assay showing growth (G), colony formation assay (H), and tumorsphere assay (I) were performed in Hs578T cells with BIRC6 KO or BIRC6 re-expression. (J) Representative plots of cell apoptosis analysis in Hs578T cells with or without BIRC6 knockdown. (K–M) MTT assay showing growth (K), colony formation assay (L), and immunoblotting analysis (M) were performed in Hs578T cells transduced with the indicated shRNAs. (N) The growth curves of xenografts derived from Hs578T cells transduced with the indicated shRNAs. (O) Representative IHC staining for BIRC6, SMAC, and cleaved caspase-3 in tumor tissues from mice. Data are means ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001.
To further confirm that BIRC6 promotes TNBC cell growth, we engineered a BIRC6-knockout (KO) clone in Hs578T cells by using the CRISPR-Cas9 gene editing system and subsequently established BIRC6 re-expressing clone in KO cells (Figure 4F). Similar to the observations above, BIRC6 KO also significantly decreased the proliferation (Figure 4G) and colony formation (Figure 4H) and tumorsphere formation capacity (Figure 4I) of TNBC cells. Notably, re-expression of BIRC6 efficiently rescued the survival-suppressive role induced by BIRC6 KO (Figures 4G–4I). These loss-of-function studies suggest that BIRC6 promotes cell survival and tumor growth in TNBC.
Next, we sought to figure out how BIRC6 regulates cell growth in TNBC. Annexin V/propidium iodide (PI) dye binding assay revealed that BIRC6 knockdown significantly increased the proportion of apoptotic cells in TNBC cells (Figure 4J; Figure S2E). Furthermore, we also found that BIRC6 knockdown strikingly induced upregulation of apoptosis-associated proteins including cleaved PARP, cytochrome c, and cleaved caspase-3, -6, -7, and -9 (Figure S2F). Cell cycle analysis showed that BIRC6 knockdown resulted in a strong rise of a sub-G1 population, indicative of apoptotic cells, whereas no obvious changes in other phases were observed (Figure S2G). Thus, BIRC6 promotes cell growth mainly by inhibiting apoptosis.
Previous studies have demonstrated that BIRC6 can facilitate proteasomal degradation of pro-apoptotic proteins, including SMAC, caspase-9, HtrA2, and p53.11,13,14 We therefore tested whether these proteins might be involved in BIRC6 silencing-induced apoptosis. Interestingly, silencing of SMAC, but not caspase-9, p53, and HtrA2, could dramatically rescue cell proliferation (Figure 4K) and colony growth (Figure 4L) and decrease caspase cleavage (Figure 4M) in Hs578T cells with endogenous BIRC6 knockdown. Subcutaneous xenograft experiments showed that SMAC depletion largely reversed the suppression effect of BIRC6 knockdown on tumor growth (Figure 4N) by blocking apoptosis (Figure 4O; Figure S2H). Collectively, these results demonstrate that SMAC is a major downstream regulator of BIRC6 and plays a crucial role in the growth-suppressive effect of BIRC6 silencing.
BIRC6 controls SMAC protein stability in TNBC
To determine whether the EGF-JNK-BIRC6 axis regulates SMAC expression, we assessed the expression of BIRC6 and SMAC under the condition of EGF-JNK activation. BIRC6 expression was upregulated; meanwhile SMAC expression was downregulated in Hs578T and MDA-MB-468 cells after EGF treatment (Figure 5A). Moreover, knockdown of EGFR by shRNA decreased the expression of BIRC6 and increased the expression of SMAC (Figure 5B).
Figure 5.

BIRC6 decreases SMAC expression by regulating ubiquitination in TNBC
(A) After serum starvation for 48 h, cells were treated with EGF (100 ng/mL) for 30 min, followed by immunoblotting as indicated. (B) Scramble or EGFR-knockdown Hs578T and MDA-MB-468 cells were collected for immunoblotting with the indicated antibodies. (C) Hs578T cells were immunoprecipitated with an anti-BIRC6 antibody and subjected to immunoblotting analysis. (D) Immunoblotting of the indicated proteins in Hs578T cells transduced with scramble or BIRC6 shRNA. (E) Hs578T cells expressing Myc-tagged BIRC6 were treated with CHX (100 μg/mL) for the indicated times and then immunoblotted with the indicated antibodies. (F) After being transiently transfected with the indicated plasmids, 293T cells were immunoprecipitated with an anti-Flag antibody and then subjected to immunoblotting analysis. Cells were treated with 10 μM MG132 for 6 h before being lysed. (G) The expression profiles of SMAC in eight TNBC and seven non-TNBC cell lines plus two breast normal cell lines were assessed by immunoblotting. The protein expression levels were normalized to that of 184A1. (H) Correlation between BIRC6 and SMAC protein expression in eight TNBC and seven non-TNBC cell lines.
We next investigated the underlying mechanism of BIRC6 regulation of SMAC expression in TNBC. Exogenously expressed Myc-BIRC6 and Flag-SMAC were coimmunoprecipitated in 293T cells (Figure S3A). The endogenous interaction between BIRC6 and SMAC was further confirmed with Hs578T cells (Figure 5C). In addition, BIRC6 knockdown increased the steady-state expression of SMAC (Figure 5D), whereas it did not cause a significant difference in SMAC mRNA levels (Figure S3B), suggesting that BIRC6 regulates SMAC at the posttranscriptional level. CHX chase assays showed that BIRC6 overexpression led to decreased SMAC protein stability (Figure 5E). Furthermore, BIRC6 increased the ubiquitination level of co-transfected SMAC (Figure 5F). Notably, we detected the protein expression profiles of SMAC in breast cancer cell lines and found that SMAC protein levels were much higher in non-TNBC cell lines (Figure 5G) and were negatively correlated with BIRC6 protein levels (Figure 5H). Altogether, these results indicate that BIRC6 downregulates SMAC expression by promoting the ubiquitination and degradation of SMAC in TNBC.
The pCLN/siRNA complex facilitates the cellular uptake and tumor accumulation of BIRC6-siRNA
Modifying LNPs with polyethylene glycol (PEG) is widely used to prolong their circulation following systemic delivery and is a useful strategy for maximizing the efficiency of targeting tumor tissue.28 To silence BIRC6 expression in TNBC cells, we developed a cationic 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP)-based LNP system with PEGylation (pCLNs) to encapsulate BIRC6-siRNA (Figure 6A). pCLNs showed the regular morphology and size homogeneity of LNPs in the range of ∼100 nm (Figure 6B). Negatively charged siRNA was absorbed into the positively charged lumen of pCLNs to form the pCLN/siRNA nanocomplex by incubation at room temperature. After mixing pCLNs with siRNA, the particle size changed from 100.2 ± 11.2 nm to 155.3 ± 1.6 nm, and the zeta potential changed from 40.6 ± 4.41 mV to 18.9 ± 1.31 mV, suggesting that the pCLN/siRNA complex was prepared successfully (Figure S4A). As reported, nanoparticles with particle size < 200 nm tend to accumulate in tumor sites by the enhanced permeation and retention (EPR) effect.29,30 On the other hand, the electrostatic interaction between the pCLNs and siRNA leads to condensation of the nucleic acid, which facilitates the mobility of siRNA and protects it from environmental enzymes.31 We then tested the optimal ratio of pCLNs to siRNA by gel retardation assay. The mobility of the siRNA through the gel was reduced as the amount of pCLNs increased, and the siRNA band was completely blocked in the well at a mass ratio of 60:1 (Figure S4B).
Figure 6.

pCLNs can efficiently deliver BIRC6-siRNA into TNBC cells in vitro and in vivo
(A) Schematic diagram of the synthesis process for pCLN/siRNA. (B) Size distributions and transmission electron microscope (TEM) images of pCLNs (left) and pCLN/siRNA (right). Scale bar in TEM images: 100 nm. (C) Intracellular fluorescence intensities of Cy3-siRNA were determined by flow cytometry after MDA-MB-468 cells were incubated with free Cy3-siRNA or pCLN/Cy3-siRNA for the indicated times. (D) Intracellular localization and lysosomal escape were observed by confocal laser scanning microscopy (CLSM) after MDA-MB-468 cells were incubated with pCLN/Cy3-siRNA for the indicated times. (E) In vivo imaging of MDA-MB-468 xenograft-bearing mice after tail vein injection of free Cy3-siRNA or pCLN/Cy3-siRNA for the indicated times.
We next evaluated intracellular uptake behavior of the pCLN/siRNA complex in MDA-MB-468 cells by flow cytometry and confocal microscopy. Flow cytometry analysis showed that the proportion of fluorescent cells increased with increasing incubation times of Cy3-siRNA and maintained a high level even 12 h after incubation (Figure 6C), indicating that the pCLNs were internalized. In line with this finding, confocal microscopy analysis revealed that the fluorescence intensity was significantly enhanced after 4 and 12 h of incubation (Figure 6D).
To achieve efficient cytoplasmic gene silencing, it is essential for siRNA carriers to escape from lysosomes after entering the cell. If Cy3-siRNA can escape from the lysosomes into the cytoplasm, then the fluorescence signal will not be colocalized with LysoTracker Blue and will remain red. After 4 h of incubation, the red (Cy3-siRNA) and blue (LysoTracker) fluorescence were colocalized in the MDA-MB-468 cells (Figure 6D), indicating that the Cy3-siRNA was located in the lysosomes. After 12 h of incubation, the red fluorescence was separated from the blue fluorescence, suggesting that the Cy3-siRNA had escaped from the lysosomes into the cytoplasm.
We wondered if pCLN-coated siRNA could pass through the bloodstream to reach tumor sites and is stable in circulation. Mice were injected via the tail vein with free Cy3-siRNA or pCLN/Cy3-siRNA. The biodistribution of the nanocomplexes in mice was assessed by in vivo imaging. As shown in Figure 6E, the pCLN/Cy3-siRNA-injected group demonstrated a gradual increase in fluorescence intensity in the tumor, with fluorescence maintained even at 36 h after injection, whereas no fluorescence in the tumor can be observed in the Cy3-siRNA-injected group. Furthermore, ex vivo images of major organs showed that the pCLN/siRNA complex was primarily accumulated in the tumor, whereas the Cy3-siRNA only showed negligible accumulation in the tumor (Figures S4C and S4D). These results suggest that pCLN/Cy3-siRNA can be efficiently delivered to the tumor sites after injection and is stable in circulation.
The pCLN/siBIRC6 complex is more effective than EGFR inhibitor to suppress TNBC cell growth in vitro and in vivo
Despite elevated expression of EGFR in TNBC, EGFR inhibitors showed limited clinical effect in TNBC patients.26,32 Since BIRC6 overexpression is partially attributed to EGFR activation, we sought to compare the antiproliferation and antitumor activities between BIRC6 silencing and EGFR inhibition. The pCLN/siBIRC6 complex efficiently knocked down the BIRC6 protein expression and significantly suppressed cell proliferation in high-EGFR-expressing MDA-MB-468 cells (Figure 7A). By contrast, various EGFR inhibitors did not efficiently inhibit the proliferation of MDA-MB-468 cells even at high concentrations; only afatinib, which inhibits ErbB family members other than EGFR, showed modest antiproliferation effect (Figure 7B). Annexin V/PI dye binding assay established that the pCLN/siBIRC6 complex significantly increased the level of apoptosis compared with that induced in the gefitinib-treated cells (Figures 7C and 7D).
Figure 7.

Effect of pCLN/siBIRC6 complex on TNBC cell growth in vitro and in vivo
(A) Immunoblotting of BIRC6 (left) and relative cell proliferation rates (right) in MDA-MB-468 cells treated with the indicated pCLN complexes. Data are mean ± SD relative cell proliferation rates. (B) Immunoblotting of pEGFR (top) and relative cell viability (bottom) in MDA-MB-468 cells treated with the indicated EGFR inhibitors. Data are means ± SD. (C and D) Representative plots of cell apoptosis analysis (C) and data quantification of the apoptotic cells (D) in MDA-MB-468 cells treated with gefitinib or the indicated pCLN complexes. Data are means ± SD. (E) Representative images of the tumors dissected from MDA-MB-468 xenograft-bearing mice receiving various treatments (n = 5 mice in each group). (F) Relative tumor volume in MDA-MB-468 xenograft-bearing mice receiving various treatments over time. (G) Weight of tumors dissected from mice after various treatment. (H) Representative images of IHC analysis of Ki67 and cleaved caspase-3 in MDA-MB-468 xenograft tumors. (I) Quantification analysis of mean density of Ki67 and cleaved caspase-3 staining by Image-Pro Plus. The integrated optical density (IOD) values are represented as means ± SD.
Next, we evaluated the antitumor effects of the pCLN/siBIRC6 complex and gefitinib in an orthotopic nude mouse model implanted with MDA-MB-468 cells. The pCLN/siBIRC6 treatment suppressed tumor growth more significantly than gefitinib did (Figure 7E). The tumor volumes in mice treated with pCLN/siBIRC6 were reduced by 91.58% compared with those in the gefitinib group (Figure 7F). Moreover, the tumor weight in the pCLN/siBIRC6 group was also lower than that in the gefitinib group (Figure 7G). IHC staining revealed that the pCLN/siBIRC6 treatment resulted in stronger inhibition of cell proliferation (Ki67) and induction of cell apoptosis (cleaved caspase-3) than gefitinib (Figures 7H and 7I).
We then investigated the in vivo toxicity of the pCLN/siBIRC6 complex. No significant changes in body weight were observed in mice treated with the pCLN/siBIRC6 complex (Figure S5A). H&E staining of the major organs, including the liver, lung, heart, spleen, and kidney, also showed no marked histological changes between the pCLN/siBIRC6 group and the other groups (Figure S5B). Taken together, these results reveal that the pCLN/siBIRC6 complex is more effective than EGFR inhibitor to suppress TNBC tumorigenesis with no adverse effects.
Discussion
TNBC is the most aggressive subtype of breast cancer, with limited therapeutic options, requiring the discovery of new targeted therapies.8 BIRC6 has been shown to be frequently overexpressed in multiple human cancers,14, 15, 16, 17, 18, 19,33, 34, 35 whereas the role of BIRC6 in TNBC and the mechanism of its overexpression in cancers remain unknown. Our study confirmed that BIRC6 expression was elevated in TNBC and led to worse survival. Targeting BIRC6 by shRNA or CRISPR-Cas9 in TNBC cell lines significantly inhibited proliferation, colony formation, and tumorsphere formation in vitro and tumor growth in vivo. Importantly, our data indicated that EGFR activation was a major upstream regulator to increase BIRC6 expression in TNBC. EGFR signaling leads to the activation of several downstream cascades; we demonstrated that EGF-mediated increase in BIRC6 protein expression primarily involved activation of the JNK signaling pathway. A limitation of this study is that we have not clarified how JNK regulates BIRC6 protein stability. One could speculate that EGF-JNK-induced stabilization of BIRC6 involves phosphorylation, which deserves to be further explored experimentally. Moreover, IHC staining with a TNBC tissue microarray showed a nearly 2-fold difference in the sample size between BIRC6-positive and EGFR-positive patients (Figure 1E, 66 versus 34), suggesting that other upstream factors regulating BIRC6 expression need to be identified in addition to EGFR signaling.
A previous study reported that the RING finger-containing ubiquitin ligase Nrdp1 promoted ubiquitination and proteasomal degradation of BIRC6.36 By BIRC6 co-immunoprecipitation followed by mass spectrometry, we identified HECTD1 as a novel E3 ubiquitin ligase of BIRC6. Our finding indicated that HECTD1 regulated BIRC6 protein stability by ubiquitination during EGFR activation. EGF stimulation attenuated the interaction between BIRC6 and HECTD1, thus reducing the ubiquitin level of BIRC6. Notably, the E3 ligase Nrdp1 was not involved in regulating the protein stability of BIRC6 during EGFR activation.
The non-adherent tumorsphere assay has been widely used to enrich for cancer stem cells.37 To resist apoptotic stimuli, cancer stem cells usually express high levels of anti-apoptotic proteins.38 Previous studies revealed that BIRC6 was highly expressed in colonospheres and mammospheres through proteomic and microarray analysis, respectively.39,40 EGF is a routinely added growth factor in tumorsphere culture, which may help cancer stem cells to survive by resisting anoikis.41 Sippel et al. found that downregulation of BIRC6 reduced neural progenitor cell (NPC) number and removal of EGF from the culture medium of neurospheres decreased the levels of BIRC6, suggesting that the level of BIRC6 is important in controlling cell proliferation in NPCs.42 In this study, we showed that EGF treatment upregulated BIRC6 expression by increasing protein stability, and BIRC6 depletion dramatically reduced tumorsphere formation and increased the ratio of apoptosis in TNBC. Together, we can speculate that BIRC6 may promote cancer stem cell survival by inhibiting apoptosis upon EGF stimulation.
BIRC6 is a giant IAP protein with an N-terminal single BIR domain and a C-terminal UBC domain.43 Via the UBC domain, BIRC6 is reported to ubiquitinate and facilitate proteasomal degradation of proapoptotic proteins, including SMAC, caspase-9, p53, and HtrA2.11, 12, 13, 14 Our data showed that the suppression effect of BIRC6 knockdown on cell survival was rescued only by SMAC depletion and not by silencing of caspase-9, p53, and HtrA2. Furthermore, BIRC6 knockdown increased SMAC protein stability, thus inhibiting caspase cascade reaction and ultimate apoptosis. Therefore, UBC domain-mediated SMAC ubiquitination was crucial for BIRC6 to inhibit apoptosis and promote cell survival in TNBC.
Gene therapy using siRNA has emerged as a promising therapeutic method to treat various diseases by silencing specific gene expression.44 LNPs are the leading delivery system for enabling the therapeutic potential of siRNA.45 In this study, pCLNs based on a LNP system were prepared to efficiently deliver BIRC6-siRNA into TNBC cells in vitro and in vivo. The siRNA markedly downregulated BIRC6 expression in TNBC, thus inhibiting cell growth and tumorigenesis, with no adverse effects. In line with previous findings, our results demonstrated that EGFR was abnormally overexpressed in TNBC, whereas inhibitors against EGFR did not significantly suppress TNBC cell growth. By contrast, BIRC6 silencing caused by pCLN/siBIRC6 complex was more effective to inhibit TNBC cell proliferation and tumor growth, suggesting that BIRC6 may be a more potential target to treat TNBC than EGFR. In summary, our study provides the evidence that BIRC6 is a new therapeutic target in TNBC and pCLN-assisted delivery of BIRC6-siRNA holds promise as a therapeutic approach for TNBC.
Materials and methods
Cell culture and compounds
All the cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were confirmed by short tandem repeat (STR) identification. Two breast epithelial cell lines, MCF10A and 184A1, were cultured in Dulbecco’s modified Eagle’s medium (DMEM)-F12 medium, supplemented with 5% fetal bovine serum (FBS), 10 μg/mL insulin, 20 ng/mL EGF, 100 ng/mL cholera toxin, 0.5 μM calcium chloride, and 500 ng/mL hydrocortisone. All other cell lines were cultured in DMEM-F12 medium containing 10% FBS in a 37°C incubator supplemented with 5% CO2. Antibiotics (penicillin and streptomycin) were added to the medium according to the manufacturer’s instructions.
Compounds including SP600125, trametinib, MK2206, saracatinib, CHX, MG132, erlotinib, gefitinib, afatinib, dacomitinib, rociletinib, and osimertinib were purchased from Selleck Chemicals. Doxycycline hyclate was obtained from Santa Cruz Biotechnology.
Tissue microarray and immunohistochemical staining
The human TNBC tissue microarray (HBreD090Bc01) was purchased from Shanghai Outdo Biotech. The tissue microarray was incubated with primary antibodies against BIRC6 (1:1,000, Abcam, ab19609) or EGFR (1:750, Santa Cruz, sc-373746), detected with biotin-conjugated secondary antibody and streptavidin-horseradish peroxidase (HRP), and visualized by 3,3′-diaminobenzidine (DAB). Images were scanned by Aperio ScanScope XT (Leica Microsystems, Germany).
For xenografts, the tumor mass was isolated from mice and fixed with 4% paraformaldehyde. IHC was performed on 5-μm-thick paraffin sections. The sections were deparaffinized in xylene, rehydrated with graded ethanol, and then processed for antigen retrieval by microwave heating for 20 min in 1 mM EDTA (pH 8.0). To block the endogenous peroxidase, the sections were incubated with 3% hydrogen peroxide for 10–15 min. After pre-incubation in 5% normal goat serum to prevent nonspecific binding, the samples were incubated with primary antibodies specific to BIRC6, SMAC, cleaved caspase-3, and Ki67. Subsequently, the sections were incubated with biotin-conjugated secondary antibody and streptavidin-HRP. Immunoreactive signal was detected by DAB as the location of antigens. After counterstaining with hematoxylin, slides were mounted for imaging.
Plasmids
The coding sequence of human SMAC was cloned from the cDNA of 293T cells and inserted into a pCMV10-3xFlag vector. The pcDNA3-myc-BIRC6 plasmid was kindly provided by Prof. Xin-Yuan Liu (Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai, China). Flag-tagged HECTD1 was purchased from OriGene (RC221760). Transfection was performed with Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The shRNA oligonucleotides were cloned into the pLKO.1 or Tet-pLKO-puro vector using AgeI and EcoRI sites. The shRNA oligonucleotide sequences are listed in Table S1. All the constructs were confirmed by DNA sequencing.
Lentivirus production and infection
Lentiviral shRNA constructs, packaging plasmids pMDLg/pRRE, pRSV-Rev, and pMD2.G, were transfected together into 293T cells to produce viruses. Culture medium was collected and filtered with a 0.4-μm filter 48 and 72 h after transfection. The viral medium was concentrated with PEG8000, re-suspended with BSA, and saved for infection. Target cells were infected with viral particles for 24 h in the presence of polybrene (10 μg/ml). Positive cells were selected with puromycin for 5–7 days.
CRISPR-Cas9-mediated gene deletion
BIRC6-deficient cells were established with the CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 system.46 The plasmid expressing Cas9, single-guide RNA (sgRNA), and puromycin was transfected into Hs578T cells. After transfection and transient selection with puromycin for 3 days, the single-cell suspension was seeded into 96-well plates without puromycin at a density of 0.5 cells per 100 μL. KO clones were selected by immunoblot analysis for the lack of BIRC6 protein and further confirmed by sequencing. The guide sequence used was 5′-gggaccatcaaagtcatcga-3′ for BIRC6.
BIRC6 KO clones were transfected with the plasmid expressing the coding sequence of human BIRC6 and neomycin. After transfection and selection with G418, BIRC6 KO clones re-expressing BIRC6 were isolated by single-cell dilution cloning and identified by immunoblot analysis for expression of BIRC6 protein.
Immunoblotting
Cells were lysed in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS), containing 1 mM Na3VO4, 5 mM NaF, and cOmplete Protease Inhibitor Cocktail (Roche, 11873580001). Lysed cells were scraped and sonicated for 15 s. Equal amounts of protein samples were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) or nitrocellulose membranes. Membranes were blocked with 5% milk and then incubated with the indicated primary antibodies overnight at 4°C. HRP-conjugated secondary antibodies (Jackson ImmunoResearch) were further incubated for 1 h at room temperature. Chemiluminescence signals were detected by the Tanon 6100 Gel Image System with ECL substrate (Pierce, 32106 or Bio-Rad, 1705061).
The following primary antibodies were used: antibodies against BIRC6 (Abcam, ab19609), EGFR (Santa Cruz, sc-373746), JNK (Santa Cruz, sc-7345), pJNK (Cell Signaling, 4668), pERK1/2 (Cell Signaling, 4370), pAKT (Cell Signaling, 4060), pSrc (Cell Signaling, 6943), Ubiquitin (Cell Signaling, 3936), HECTD1 (Santa Cruz, sc-134976), PARP (Cell Signaling, 9542), cytochrome c (Santa Cruz, sc-13156), cleaved caspase-3 (Cell Signaling, 9664), cleaved caspase-6 (Cell Signaling, 9761), cleaved caspase-7 (Cell Signaling, 8438), cleaved caspase-9 (Cell Signaling, 7237), SMAC (Santa Cruz, sc-393118), Myc (Abcam, ab9132), HA (Santa Cruz, sc-7392), Flag (Sigma, F3165), pEGFR (Cell Signaling, 3777), Vinculin (Santa Cruz, sc-25336), and β-tubulin (Abcam, ab135209).
Co-immunoprecipitation
Cells were lysed in cell lysis buffer (20 mM Tris-HCl pH 7.5, 0.15 M NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate) containing 5 mM NaF and cOmplete Protease Inhibitor Cocktail. Lysates were pre-cleared with protein A/G agarose beads (Santa Cruz, SC-2003) and the appropriate immunoglobulin G (IgG) for 30 min and were then immunoprecipitated with the indicated antibodies overnight at 4°C. The beads were added in each sample for another 3 h the next day, followed by washing 5 times with cell lysis buffer. Finally, the beads were boiled in loading buffer for 5 min, and samples were further subjected to immunoblotting.
Immunoprecipitation and mass spectrometric analysis of BIRC6-associated proteins
After transient transfection with myc-tagged BIRC6 for 48 h, 293T cells were lysed with cell lysis buffer and were further immunoprecipitated with anti-Myc or anti-BIRC6 antibody. The precipitants were subjected to SDS-PAGE, and total proteins were visualized by Coomassie blue staining and excised for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
RNA isolation and real-time PCR
Total RNA was isolated with TRIzol reagent and was then reverse transcribed into cDNA with a PrimeScript RT Reagent Kit (TakaRa). Quantitative PCR was performed with a SYBR Green PCR Kit (QIAGEN, 208054) according to the manufacturer’s instructions. Relative mRNA levels of target genes were normalized to ACTB mRNA. Primers used were for BIRC6 forward 5′-cagcagctcttatcagcatgt-3′ and reverse 5′-aactgtggcccacttagcaac-3′; for SMAC forward 5′-cgcgcagcgtaacttcattc-3′ and reverse 5′-ccaaagccaatcgtcacagtttt-3′; and for ACTB forward 5′-tccatcatgaagtgtgacgt-3′ and reverse 5′-gagcaatgatcttgatcttcat-3′.
Cell proliferation and viability assay
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess the cell growth rate and viability. Cells were seeded on 96-well plates in 100 μL of DMEM-F12 supplemented with 10% FBS. For the cell proliferation experiment, 20 μL of MTT (Amresco, 5 mg/mL) was added to the cells every day. For cell viability analysis, when cells were treated with the indicated inhibitors or LNP formulations for 72 h, 20 μL of MTT was added to each well. After incubation for 4 h, 50 μL of triplex solution (10% SDS, 5% isobutanol, and 12 mM HCl) was added to each well and cells were incubated for 12–20 h. The absorbance was then measured with a spectrophotometer at a wavelength of 570 nm. Cell proliferation rate was normalized to day 0 in each group. Cell viability rate was normalized to DMSO-treated control.
Colony formation assay
Cells were plated in 6-well plates at a density of 2,000 (Hs578T) or 6,000 (MDA-MB-468) cells per well and incubated at 37°C for 10–14 days to ensure that colonies reached >50 cells per plate. Colonies were fixed and stained with a mixture of 6.0% glutaraldehyde and 0.5% crystal violet for at least 30 min. The colony number was determined by the Colony Counter developed by Tanon.
Mammosphere culture
Single suspension cells were plated into ultralow-attachment 6-well plates (Corning, 3457) at a density of 10,000 (Hs578T) or 15,000 (MDA-MB-468) cells per well. Cells were cultured in a serum-free DMEM-F12 medium supplemented with 1% penicillin-streptomycin, 2% B27 (Gibco, 17504-044), 20 ng/mL EGF (PeproTech, AF-100-15), 20 ng/mL basic fibroblast growth factor (bFGF) (PeproTech, 100-18B), and 4 μg/mL heparin. To prevent cell aggregation, 0.5% methylcellulose (Sigma) was added to the medium. The mammospheres larger than 50 μm in diameter were counted 7–10 days after culturing.
Flow cytometric analysis
For cell cycle analysis, single suspension cells were fixed with 70% ice-cold ethanol for 12–24 h at 4°C and then stained with a Cell Cycle and Apoptosis Analysis Kit (Beyotime Biotechnology, C1052). Flow cytometry was then used to detect the DNA content in PI-stained samples.
An Annexin V-FITC (fluorescein isothiocyanate) Apoptosis Detection Kit (Beyotime Biotechnology, C1063) was used to assess the apoptotic rate according to the manufacturer’s protocol. All the data were analyzed by ModFit or FlowJo software.
In vivo tumor xenograft model
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Shenzhen University. All the mice were purchased from the Shanghai Slac Laboratory Animal Co. Ltd.
To evaluate the effect of BIRC6 knockdown on tumor growth, 5 × 106 MDA-MB-468 cells were injected subcutaneously into the right flank of female nude mice at 4–5 weeks of age. To examine the effect of SMAC depletion on the BIRC6 knockdown-mediated tumor-suppressive role, 4 × 106 Hs578T cells were mixed with Matrigel (1:1) and were injected subcutaneously into the right flank of female non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice at 6–8 weeks of age. Tumor was measured at the indicated time points, and tumor volume was calculated by the formula 0.5 × length × width2. After mice were sacrificed, tumor samples were collected and analyzed by IHC analysis.
To detect the antitumor effect of pCLN/siBIRC6 complexes and gefitinib, 5 × 106 MDA-MB-468 cells were injected into the mammary fat pads of female nude mice at 4–5 weeks of age. When the tumor volume reached ∼100 mm3, the mice were randomly assigned to four treatment groups. Gefitinib was administered at 100 mg/kg daily by oral gavage. pCLN/siBIRC6 (20 μg of siBIRC6 per injection) or an equal dose of pCLN/siRNA negative control (siNC) was intravenously injected. The injections were repeated once a week for 6 weeks. Tumor volume and body weight of mice were observed. Heart, liver, spleen, lung, kidney, and tumor tissues were harvested for histological analysis.
pCLN/siRNA preparation and characterization
pCLNs were synthesized via the solvent diffusion method as described previously.47 In brief, 0.75 mg of PEG-distearoyl phosphatidylethanolamine (DSPE), 3 mg of DOTAP, 10.5 mg of dipalmitoylphosphatidylcholine (DPPC), and 0.75 mg of cholesterol were dissolved in ethanol (1.5 mL) and maintained at 60°C. Subsequently, this dissolved solution was rapidly dispersed in F68 solution (15 mL, 0.1% w/v) at 60°C under the condition of 400 rpm stirring. After 5 min, the newly prepared solution was placed at room temperature to obtain the pCLNs.
pCLNs were mixed with BIRC6-siRNA (sequence: 5′-GUUUCAAAGCAGGAUGAUG tt-3′) at room temperature for 30 min to form the pCLN/siBIRC6 complex. The size distribution and zeta potential of the pCLN/siRNA complex were determined with a dynamic light scattering (DLS) spectrometer (3000HS; Malvern Instruments, Malvern, UK), and the morphology of the complex was observed by transmission electron microscopy (TEM).
Agarose gel electrophoresis
The efficiency of siRNA binding was assessed via agarose gel electrophoresis. pCLNs were mixed with siRNA in different pCLN:-to-siRNA mass ratios (10:1, 20:1, 40:1, 60:1, 100:1) at room temperature for 30 min. The suspension of pCLN/siRNA complex was analyzed on a 1% agarose gel with GelRed in tris-acetate-EDTA buffer. Electrophoresis was performed at 100 V for 30 min, and the bands were visualized on an ultraviolet transilluminator.
Cellular uptake and lysosomal escape of siRNA
Cy3-labeled siRNA was used in cellular uptake and lysosomal escape studies of pCLN/siRNA. MDA-MB-468 cells were seeded in 24-well plates or 35-mm glass-bottom culture dishes and incubated overnight, after which they were transfected with pCLN/Cy3-siRNA for 1 h, 4 h, and 12 h or free Cy3-siRNA for 1 h at a final siRNA concentration of 100 nM. To assess cellular uptake of siRNA, the cells were collected by trypsin treatment and subjected to fluorescence analysis by flow cytometry. To study the escape of siRNA from lysosomes, the lysosomes was stained with LysoTracker Blue at 37°C for 30 min. Co-localization of LysoTracker Blue and Cy3-siRNA was observed by confocal laser scanning microscopy (CLSM).
In vivo imaging of siRNA
To assess the biodistribution of pCLN/Cy3-siRNA in tumor of mice, MDA-MB-468 cells were implanted into the mammary fat pads of female nude mice to generate an orthotopic model of TNBC. When the tumor volume reached ∼100 mm3, pCLN/Cy3-siRNA or free Cy3-siRNA was injected intravenously at a siRNA dose of 20 μg. At the appointed time after injection, mice were imaged with a Maestro in vivo Imaging System (CRI).
Statistical analysis
The data are presented as means ± SD. The statistical significance between different groups was analyzed by Student’s t test, except that log rank was used for survival analysis. Statistical significances are denoted as not significant (n.s.; p > 0.05), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Acknowledgments
We are grateful to Dr. Jingyu Lang (Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, China) for helpful discussion and suggestions. We thank Dr. Xinyuan Liu for kindly providing materials (Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai, China). This work was supported by grants from the National Key Research and Development Program of China (2017YFA0105900), the National Natural Science Foundation of China (82003791, 82003307), Guangdong Basic and Applied Basic Research Foundation (2020A1515110911, 2019A1515110669), Shenzhen Key Laboratory Program (ZDSYS20190902092857146), and the China Postdoctoral Science Foundation (2020M672899, 2021T140671, 2021M693200).
Author contributions
Y.P.L., S.W., and Y.T. conceived the projects. Y.P.L. and Y.T. designed, performed, and interpreted experiments. Y.P.L. conducted the immunoprecipitation, immunoblot, mass spectrometry analysis, and cell biology experiments. Y.T. performed most of the experiments related to lipid nanoparticles, including preparation, in vivo imaging, and effectiveness evaluation. L.W. helped with lipid nanoparticle preparation and characterization. Z.X. and K.W. contributed to bioinformatics analysis. C.W., H.S., Y.Q.L., Q.L., and L.Z. helped to provide critical advice and reagents for the study. The manuscript was written by Y.P.L. and edited by S.W.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.09.011.
Supplemental information
References
- 1.Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Dent R., Trudeau M., Pritchard K.I., Hanna W.M., Kahn H.K., Sawka C.A., Lickley L.A., Rawlinson E., Sun P., Narod S.A. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 3.Dignam J.J., Dukic V., Anderson S.J., Mamounas E.P., Wickerham D.L., Wolmark N. Hazard of recurrence and adjuvant treatment effects over time in lymph node-negative breast cancer. Breast Cancer Res. Treat. 2009;116:595–602. doi: 10.1007/s10549-008-0200-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venkitaraman R. Triple-negative/basal-like breast cancer: clinical, pathologic and molecular features. Expert Rev. Anticancer Ther. 2010;10:199–207. doi: 10.1586/era.09.189. [DOI] [PubMed] [Google Scholar]
- 5.Collignon J., Lousberg L., Schroeder H., Jerusalem G. Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer (Dove Med. Press) 2016;8:93–107. doi: 10.2147/BCTT.S69488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robson M., Im S.A., Senkus E., Xu B., Domchek S.M., Masuda N., Delaloge S., Li W., Tung N., Armstrong A. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017;377:523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 7.Schmid P., Adams S., Rugo H.S., Schneeweiss A., Barrios C.H., Iwata H., Diéras V., Hegg R., Im S.A., Shaw Wright G., IMpassion130 Trial Investigators Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 8.Bianchini G., Balko J.M., Mayer I.A., Sanders M.E., Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srinivasula S.M., Ashwell J.D. IAPs: what’s in a name? Mol. Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z., Naito M., Hori S., Mashima T., Yamori T., Tsuruo T. A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem. Biophys. Res. Commun. 1999;264:847–854. doi: 10.1006/bbrc.1999.1585. [DOI] [PubMed] [Google Scholar]
- 11.Hao Y., Sekine K., Kawabata A., Nakamura H., Ishioka T., Ohata H., Katayama R., Hashimoto C., Zhang X., Noda T. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 2004;6:849–860. doi: 10.1038/ncb1159. [DOI] [PubMed] [Google Scholar]
- 12.Qiu X.B., Goldberg A.L. The membrane-associated inhibitor of apoptosis protein, BRUCE/Apollon, antagonizes both the precursor and mature forms of Smac and caspase-9. J. Biol. Chem. 2005;280:174–182. doi: 10.1074/jbc.M411430200. [DOI] [PubMed] [Google Scholar]
- 13.Sekine K., Hao Y., Suzuki Y., Takahashi R., Tsuruo T., Naito M. HtrA2 cleaves Apollon and induces cell death by IAP-binding motif in Apollon-deficient cells. Biochem. Biophys. Res. Commun. 2005;330:279–285. doi: 10.1016/j.bbrc.2005.02.165. [DOI] [PubMed] [Google Scholar]
- 14.Tang W., Xue R., Weng S., Wu J., Fang Y., Wang Y., Ji L., Hu T., Liu T., Huang X. BIRC6 promotes hepatocellular carcinogenesis: interaction of BIRC6 with p53 facilitating p53 degradation. Int. J. Cancer. 2015;136:E475–E487. doi: 10.1002/ijc.29194. [DOI] [PubMed] [Google Scholar]
- 15.Sung K.W., Choi J., Hwang Y.K., Lee S.J., Kim H.J., Lee S.H., Yoo K.H., Jung H.L., Koo H.H. Overexpression of Apollon, an antiapoptotic protein, is associated with poor prognosis in childhood de novo acute myeloid leukemia. Clin. Cancer Res. 2007;13:5109–5114. doi: 10.1158/1078-0432.CCR-07-0693. [DOI] [PubMed] [Google Scholar]
- 16.Dong X., Lin D., Low C., Vucic E.A., English J.C., Yee J., Murray N., Lam W.L., Ling V., Lam S. Elevated expression of BIRC6 protein in non-small-cell lung cancers is associated with cancer recurrence and chemoresistance. J. Thorac. Oncol. 2013;8:161–170. doi: 10.1097/JTO.0b013e31827d5237. [DOI] [PubMed] [Google Scholar]
- 17.Wang L., Chen Y.J., Hou J., Wang Y.Y., Tang W.Q., Shen X.Z., Tu R.Q. Expression and clinical significance of BIRC6 in human epithelial ovarian cancer. Tumour Biol. 2014;35:4891–4896. doi: 10.1007/s13277-014-1641-6. [DOI] [PubMed] [Google Scholar]
- 18.Zhang S., Tang W., Weng S., Liu X., Rao B., Gu J., Chen S., Wang Q., Shen X., Xue R., Dong L. Apollon modulates chemosensitivity in human esophageal squamous cell carcinoma. Oncotarget. 2014;5:7183–7197. doi: 10.18632/oncotarget.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu T., Weng S., Tang W., Xue R., Chen S., Cai G., Cai Y., Shen X., Zhang S., Dong L. Overexpression of BIRC6 Is a Predictor of Prognosis for Colorectal Cancer. PLoS ONE. 2015;10:e0125281. doi: 10.1371/journal.pone.0125281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luk S.U., Xue H., Cheng H., Lin D., Gout P.W., Fazli L., Collins C.C., Gleave M.E., Wang Y. The BIRC6 gene as a novel target for therapy of prostate cancer: dual targeting of inhibitors of apoptosis. Oncotarget. 2014;5:6896–6908. doi: 10.18632/oncotarget.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulda S., Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 22.Davidson B.L., McCray P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 24.Gharpure K.M., Wu S.Y., Li C., Lopez-Berestein G., Sood A.K. Nanotechnology: Future of Oncotherapy. Clin. Cancer Res. 2015;21:3121–3130. doi: 10.1158/1078-0432.CCR-14-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni J.A., Cullis P.R., van der Meel R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018;28:146–157. doi: 10.1089/nat.2018.0721. [DOI] [PubMed] [Google Scholar]
- 26.Costa R., Shah A.N., Santa-Maria C.A., Cruz M.R., Mahalingam D., Carneiro B.A., Chae Y.K., Cristofanilli M., Gradishar W.J., Giles F.J. Targeting Epidermal Growth Factor Receptor in triple negative breast cancer: New discoveries and practical insights for drug development. Cancer Treat. Rev. 2017;53:111–119. doi: 10.1016/j.ctrv.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 28.Allen T.M., Hansen C., Martin F., Redemann C., Yau-Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim. Biophys. Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 29.Maeda H., Sawa T., Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J. Control. Release. 2001;74:47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 30.Maeda H., Bharate G.Y., Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Del Pozo-Rodríguez A., Solinís M.Á., Rodríguez-Gascón A. Applications of lipid nanoparticles in gene therapy. Eur. J. Pharm. Biopharm. 2016;109:184–193. doi: 10.1016/j.ejpb.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Nakai K., Hung M.C., Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016;6:1609–1623. [PMC free article] [PubMed] [Google Scholar]
- 33.Lamers F., Schild L., Koster J., Speleman F., Øra I., Westerhout E.M., van Sluis P., Versteeg R., Caron H.N., Molenaar J.J. Identification of BIRC6 as a novel intervention target for neuroblastoma therapy. BMC Cancer. 2012;12:285. doi: 10.1186/1471-2407-12-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tassi E., Zanon M., Vegetti C., Molla A., Bersani I., Perotti V., Pennati M., Zaffaroni N., Milella M., Ferrone S., Carlo-Stella C. Role of Apollon in human melanoma resistance to antitumor agents that activate the intrinsic or the extrinsic apoptosis pathways. Clin. Cancer Res. 2012;18:3316–3327. doi: 10.1158/1078-0432.CCR-11-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Low C.G., Luk I.S., Lin D., Fazli L., Yang K., Xu Y., Gleave M., Gout P.W., Wang Y. BIRC6 protein, an inhibitor of apoptosis: role in survival of human prostate cancer cells. PLoS ONE. 2013;8:e55837. doi: 10.1371/journal.pone.0055837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu X.B., Markant S.L., Yuan J., Goldberg A.L. Nrdp1-mediated degradation of the gigantic IAP, BRUCE, is a novel pathway for triggering apoptosis. EMBO J. 2004;23:800–810. doi: 10.1038/sj.emboj.7600075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visvader J.E., Lindeman G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 38.Signore M., Ricci-Vitiani L., De Maria R. Targeting apoptosis pathways in cancer stem cells. Cancer Lett. 2013;332:374–382. doi: 10.1016/j.canlet.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Kok M., Koornstra R.H., Margarido T.C., Fles R., Armstrong N.J., Linn S.C., Van’t Veer L.J., Weigelt B. Mammosphere-derived gene set predicts outcome in patients with ER-positive breast cancer. J. Pathol. 2009;218:316–326. doi: 10.1002/path.2544. [DOI] [PubMed] [Google Scholar]
- 40.Van Houdt W.J., Emmink B.L., Pham T.V., Piersma S.R., Verheem A., Vries R.G., Fratantoni S.A., Pronk A., Clevers H., Borel Rinkes I.H.M. Comparative proteomics of colon cancer stem cells and differentiated tumor cells identifies BIRC6 as a potential therapeutic target. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.011353. M111.011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sippel M., Rajala R., Korhonen L., Bornhauser B., Sokka A.L., Naito M., Lindholm D. Dexamethasone regulates expression of BRUCE/Apollon and the proliferation of neural progenitor cells. FEBS Lett. 2009;583:2213–2217. doi: 10.1016/j.febslet.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Hauser H.P., Bardroff M., Pyrowolakis G., Jentsch S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 1998;141:1415–1422. doi: 10.1083/jcb.141.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F. RNA-Targeted Therapeutics. Cell Metab. 2018;27:714–739. doi: 10.1016/j.cmet.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Setten R.L., Rossi J.J., Han S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019;18:421–446. doi: 10.1038/s41573-019-0017-4. [DOI] [PubMed] [Google Scholar]
- 46.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu F.Q., Yuan H., Zhang H.H., Fang M. Preparation of solid lipid nanoparticles with clobetasol propionate by a novel solvent diffusion method in aqueous system and physicochemical characterization. Int. J. Pharm. 2002;239:121–128. doi: 10.1016/s0378-5173(02)00081-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.