

Abstract
Background.
Most patients with childhood-onset immune dysregulation, polyendocrinopathy and enteropathy have no genetic diagnosis for their illness. These patients may undergo empirical immunosuppressive treatment with highly variable outcomes.
Objective.
To determine the genetic basis of disease in patients referred with “IPEX-like” disease, but with no mutation in FOXP3; then to assess consequences of genetic diagnoses for clinical management.
Methods.
Genomic DNA was sequenced using a panel of 462 genes implicated in inborn errors of immunity. Candidate mutations were characterized by genomic, transcriptional, and (for some) protein analysis.
Results:
Of 123 patients with FOXP3-negative IPEX-like disease, 48 (39%) carried damaging germline mutations in one of 27 genes including AIRE, BACH2, BCL11B, CARD11, CARD14, CTLA4, IRF2BP2, ITCH, JAK1, KMT2D, LRBA, MYO5B, NFKB1, NLRC4, POLA1, POMP, RAG1, SH2D1A, SKIV2L, STAT1, STAT3, TNFAIP3, TNFRSF6/FAS, TNRSF13B/TACI, TOM1, TTC37, and XIAP. Many of these had not been previously associated with an IPEX-like diagnosis. For 42 of the 48 patients with genetic diagnoses, knowing the critical gene may have altered therapeutic management, including recommendations for targeted treatments and for or against hematopoietic cell transplantation.
Conclusion:
Many childhood disorders now bundled as “IPEX-like” disease are caused by individually rare, severe mutations in immune regulation genes. Most genetic diagnoses of these conditions yield clinically actionable findings. Barriers are lack of testing or lack of repeat testing if older technologies failed to provide a diagnosis.
Clinical Implication:
Pediatric immune dysregulation would benefit from a genetics-first approach to diagnosis: for >80% of these patients with genetic diagnoses, the genetic information offers critical guidance to clinical management.
Keywords: Immune dysregulation, molecular diagnosis, genetics, sequencing, pediatric, precision medicine, autoimmunity, inborn errors of immunity, primary immunodeficiency disorders
CAPSULE SUMMARY
Immune dysregulation, polyendocrinopathy or enteropathy in many pediatric patients results from a mutation with severe clinical effect. Identification of these causal mutations enables treatment based on genotype, supporting a genetics-first approach to diagnosis.
INTRODUCTION
Clinical presentations of immune dysregulation in children are notoriously complex1,2,3. Genetic diagnoses can help disentangle this complexity and can also guide clinical management of these patients, suggesting targeted therapies or prompting hematopoietic cell transplantation (HCT)4. Despite excellent studies revealing genetic causes of primary immunodeficiency diseases5, there are as yet no widely accepted recommendations for genetic testing of pediatric patients with immune dysregulation. Even now, a decade after next-generation sequencing became widespread, genetic testing for inborn errors of immunity often proceeds only following functional testing and in targeted panels, with the choice of gene frequently based on phenotype and serology. This approach misses genetic diagnoses of a large proportion of patients.
Among inborn errors of immunity, IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked) syndrome, caused by mutations in FOXP3, a lineage-defining gene in regulatory T cells, is a prototype of disorders affecting regulation of immune responses6,7. IPEX manifests most commonly with severe diarrhea, dermatitis, and autoimmune endocrinopathies such as type 1 diabetes or thyroid disease8. However, these clinical features are widespread in pediatric diseases of immune dysregulation, and many patients with these symptoms have no mutation in FOXP3. These patients have been termed “IPEX-like,” a distinction that is clinically significant, because patients with FOXP3 mutations usually undergo HCT9, whereas patients with IPEX-like syndrome are generally monitored and treated with immunomodulatory treatments aimed at the affected organ system. Patients with IPEX-like syndrome receive transplantation only if their disease progresses and becomes life-threatening. If HCT is ultimately undertaken, the delay in treatment can result in higher morbidity and mortality.
The scope, prevalence, and distribution of genes and mutations responsible for FOXP3-negative pediatric immune dysregulation, polyendocrinopathy or enteropathy (IPE) are not known. The goal of this project was to determine the underlying genetic causes of these conditions in pediatric patients with no mutation in FOXP3, to assess the diagnostic yield of genetic testing in these patients, and to evaluate the implications of genetic diagnosis for treatment.
METHODS
Study subjects
The study was approved by the institutional review boards of Seattle Children’s Hospital (SCH) and the University of Washington (UW). Patients were eligible for the study if referred to SCH with clinical signs suggestive of IPEX, and hence for genetic analysis of FOXP3, but with no FOXP3 mutation identified. DNA was available for 123 such patients.
Gene panel development
We designed an oligonucleotide-based sequencing panel encompassing the coding exons, 5’ and 3’ untranslated regions, and flanking intronic regions of 462 known and candidate genes for immune-mediated disease (Table EI). Genes were chosen from the classification of immune genes by the International Union of Immunological Societies (IUIS)10, and by review of the literature and consensus expert opinion of colleagues from immunology, rheumatology, and genetics. Of the 462 genes, 337 genes harbor mutations leading to inborn errors of immunity in humans; the other 125 genes are involved in immune tolerance, many with compelling murine models. Total length of the targeted genomic region was 1.5 MB. The panel enables simultaneous identification of single base pair mutations, small insertions and deletions, and exon-impacting copy number variants (CNVs) for all targeted genes. The panel was validated by blinded analysis of 33 patients with independently identified mutations. All genotypes were concordant with previous results. In addition to its research use, the panel is now in clinical use by the UW Department of Laboratory Medicine11.
Genomics
Genomic DNA was isolated from whole blood, from peripheral blood mononuclear cells (PBMCs), or from expanded PBMCs. For each sample, 500–750ng DNA was captured using the oligonucleotide panel. Molecular barcodes were added after hybridization, and 32–48 samples were multiplexed and sequenced in a single flow-cell on an Illumina HiSeq2500 instrument to obtain 100bp paired-end reads at >400x median coverage. Identification of variants was carried out as previously described12. Copy number variants (CNVs) were confirmed by TaqMan analysis and breakpoints identified by whole genome sequencing, also as described12.
Interpretation of genetic variants
Variant interpretation was based on the guidelines of the American College of Medical Genetics (ACMG)13, as applied to conditions of pediatric immune dysregulation. As described above, the challenges of these conditions are, on the one hand, phenotypes defined by an exceptionally wide range of clinical presentations and, on the other hand, candidate genes with an exceptionally wide range of biological functions, with no straightforward alignment of phenotypes and genes. For each patient, candidate variants were evaluated by several criteria and experiments: by in silico predictive tools; by published functional studies; by similarities of clinical presentations of our patient with any patients previously reported with mutations in the same gene; by transcriptional analysis in our lab when appropriate and feasible14; and by testing for de novo inheritance, if DNA from parents could be obtained. For each variant that we reported as pathogenic, likely pathogenic, or of unknown significance (VUS), we provided a narrative explanation of published evidence and of experiments carried out in our lab, as well as reporting in silico predictions and prior classification by ClinVar15. The VUS classification was used sparingly, only for mutations with plausible but uncertain links to the patient’s phenotype, for which further functional or genetic studies could add to evidence for or against causality.
RESULTS
Genetic diagnoses
Demographic and clinical features of the cohort.
Demographic and clinical characteristics of the 123 patients are shown in Table I. Most patients were male, consistent with original referral for X-linked IPEX syndrome. Patients were referred by physicians from six continents and represented a wide variety of ancestral populations, identified by self-report and by ancestral SNPs16. Clinical diagnoses were heterogeneous, including enteropathy, dermatitis, autoimmune hemolytic anemia, type I diabetes, and other autoimmune conditions.
Table I.
Patient characteristics
| N | Proportion | |
|---|---|---|
| Age at referral | ||
| 0 – 2 y | 40 | 0.33 |
| 2 – 4 y | 26 | 0.21 |
| 5 – 9 y | 18 | 0.15 |
| 10 – 14 y | 13 | 0.11 |
| 15+ y | 17 | 0.14 |
| not recorded | 9 | 0.07 |
| Total | 123 | 1.00 |
| Gender | ||
| Male | 94 | 0.76 |
| Female | 29 | 0.24 |
| Total | 123 | 1.00 |
| Ancestry | ||
| Caucasian, not Hispanic | 78 | 0.63 |
| Caucasian, Hispanic | 24 | 0.20 |
| East Asian | 9 | 0.07 |
| African American | 5 | 0.04 |
| Middle East/West Asian | 4 | 0.03 |
| Central/South Asian | 2 | 0.02 |
| Native American | 1 | 0.01 |
| Total | 123 | 1.00 |
| Major reported clinical features | ||
| Enteropathy | 69 | 0.56 |
| Dermatitis | 30 | 0.24 |
| Autoimmunity | 45 | 0.37 |
| Immunodeficiency; recurrent infections | 37 | 0.30 |
| “IPEX” only | 29 | 0.24 |
| No information recorded | 11 | 0.09 |
Genomic sequencing.
Genomic analysis of DNA samples from the 123 patients yielded median coverage across the targeted region of 406X, with 95.9% of targeted bases having >40X coverage and 99.1% of targeted bases having >8X coverage. This depth of coverage enabled identification of point mutations and small insertions and deletions in coding or regulatory regions and exon-impacting CNVs.
Frequencies and features of genetic diagnoses.
Genomic analysis yielded genetic diagnoses for 39% of patients (48 of 123), involving 27 different genes (Table II). All variants considered pathogenic, likely pathogenic, or of unknown significance but warranting further study, are indicated in Table III, with evidence bearing on causality for the child’s phenotype. Of the 28 different conditions in the 48 patients with genetic diagnoses, 19 conditions were inherited as autosomal dominant, 7 as autosomal recessive, and 2 as X-linked recessive. Damaging mutations included truncating, splice altering, and missense mutations, each private or extremely rare (Table III). Five patients carried multi-kilobase genomic deletions or amplifications in single genes (Figs E1, E2). Of the 56 different variants contributing to these 48 genetic diagnoses, 36 appeared for the first time in a patient in this series and 20 had been previously reported15 (Table III). Of the 58 reported variants, 52 were classified as pathogenic or likely pathogenic and 4 as VUS with recommendation for further functional analysis (Table III). Proportions of patients with genetic diagnoses were similar for males (37/94, or 39%) and for females (11/29, or 38%). Average age at referral for patients with a genetic diagnosis was 6.2y and for patients with no genetic diagnosis was 6.6y.
Table II.
Genetic diagnoses with phenotypic classifications of the International Union of Immunological Societies (IUIS)
| Patient | Age | Gender | Gene | AI / II* | “IPEX-like” gene** | Inh^ | Clinical features |
|---|---|---|---|---|---|---|---|
| IUIS: Diseases of immune dysregulation | |||||||
|
| |||||||
| S048 | 11 y | M | AIRE | AI | no | AR | Addison’s disease and recurrent oral thrush |
|
| |||||||
| S093 | 22 m | F | BACH2 | AI, II | no | AD | Chronic diarrhea |
|
| |||||||
| S092 | 12 y | M | CTLA4 | AI | yes | AD | Enteropathy, total parenteral nutrition dependence, alopecia totalis; HSCT |
| S175 | 9 y | M | CTLA4 | AI | yes | AD | Onset 3y. Watery diarrhea, type 1 diabetes, anti-thyroid antibodies, hemolytic anemia, thrombocytopenia, lymphadenopathy, splenomegaly, alopecia; HSCT |
| S190 | 16 y | M | CTLA4 | AI | yes | AD | Colitis, eczema, hypothyroidism, common variable immunodeficiency, cytopenias |
| S018 | 11 y | F | CTLA4, CD28, CASP8 | AI | yes | AD | Autoimmune enteropathy, eczema, interstitial pneumonitis, seizures, DD, pulmonary hypertension, vitiligo, thrombocytopenia, pan-hypogammaglobulinemia; recurrent infections including esophageal candidiasis, otitis media, and sinusitis |
|
| |||||||
| S184 | 13 y | M | FAS/TNFRSF6 | AI, II | no | AD | Enteropathy with villous atrophy from 1y, eczema, type 1 diabetes, alopecia, recurrent severe infections, short stature, seizures. Low IgA and IgG levels, and B cell lymphopenia |
|
| |||||||
| S071 | 3 y | M | ITCH | AI, II | yes | AR | Autoimmune thyroid disease, FTT, DD, chronic secretory and bloody diarrhea, alopecia, hemolytic anemia, eczema, recurrent fungal and bacterial infections. Low IgM levels; positive pANCA and anti-smooth muscle antibodies |
|
| |||||||
| S066 | 7 m | F | JAK1 | AI, II | no | AD | Dysmorphisms, enteropathy, hypogammaglobulinemia, failure to thrive, hepatitis, and decreased FOXP3 T-cells. Died from sepsis. Five siblings died in infancy. |
| S067 | 2 y | M | JAK1 | AI, II | no | AD | Enteropathy and immunodeficiency, with decreased FOXP3 expression. |
| S170 | 4 m | M | JAK1 | AI, II | no | AD | Exfoliative dermatitis, enteropathy, anemia, thrombocytopenia, neutropenia, hepatosplenomegaly. |
|
| |||||||
| S014 | 4 y | M | LRBA | AI | yes | AR | Type 1 diabetes, autoimmune enteropathy, autism, lymphoma |
| S045 | 14 y | M | LRBA | AI | yes | AR | Diarrhea from age 3 mo., malabsorption, type 1 diabetes, esophageal candidiasis, and delayed puberty. Died age 16y of intestinal adenocarcinoma |
| S129 | 17 y | F | LRBA | AI | yes | AR | Hypothyroidism, inflammatory bowel disease, hepatosplenomegaly, hemolytic anemia, and hypogammaglobulinemia |
| S088 | 10 y | M | LRBA | AI | yes | AR | Early onset type 1 diabetes mellitus, autoimmune cytopenias, arthritis, interstitial lung disease, autoimmune enteropathy, uveitis, eczema, failure to thrive, recurrent warts, inflammatory brain lesions. Hypogammaglobulinemia, low T-regulatory cells |
|
| |||||||
| S123 | 11 y | M | SH2D1A | AI, II | no | AD | Alopecia totalis, neutropenia, recurrent infections, mucositis, dermatitis, colitis; HSCT |
|
| |||||||
| S041 | 6 m | M | STAT3 | AI, II | yes | AD | Chronic diarrhea, severe eczema, recurrent respiratory infections and developmental delay; HSCT |
| S068 | 8 y | M | STAT3 | AI, II | yes | AD | “IPEX” |
| S112 | 9 y | M | STAT3 | AI, II | yes | AD | Recurrent infections, type 1 diabetes, immune thrombocytopenic purpura, hypothyroidism, short stature, and mild hypogammaglobulinemia |
|
| |||||||
| S108 | 20 m | M | XIAP | AI, II | no | XR | Severe malabsorption, failure to thrive |
|
| |||||||
| IUIS: Autoinflammatory disorders | |||||||
|
| |||||||
| S030 | 4 m | M | CARD14 | II | no | AD | Severe eczema, multiple food allergies, enteropathy |
| S055 | 3 y | F | CARD14 | II | no | AD | Colitis |
|
| |||||||
| S098 | 1 m | M | NLRC4 | II | no | AD | Intractable diarrhea, distended bowel with mucosal autolysis and erosions, severe pulmonary congestion and hemorrhage, ascites, viral meningitis |
|
| |||||||
| S102 | 2 y | M | POLA1 | II | no | XR | Immune dysregulation |
| S114 | ni | M | POLA1 | II | no | XR | Unknown |
|
| |||||||
| S160 | 2 m | M | POMP | II | no | AD | Enteropathy, dermatitis, pulmonary nodules, recurrent respiratory infections, mild thrombocytopenia, and elevated IgE |
|
| |||||||
| S027 | 3 y | M | TNFAIP3 | AI, II | no | AD | Watery diarrhea, eczema, failure to thrive, autoantibodies to thyroid (without frank thyroiditis), elevated IgE. |
| S087 | 12 y | F | TNFAIP3 | AI, II | no | AD | “IPEX” |
|
| |||||||
| IUIS: Predominantly antibody deficiency | |||||||
|
| |||||||
| S125 | 4 y | F | IRF2BP2 | AI, II | no | AD | Chronic diarrhea, severe eczema, anemia, failure to thrive, fevers, short stature, recurrent infections, cataracts, hypodontia, hypotrichosis alopecia, hypogammaglobulinemia |
|
| |||||||
| S043 | 6 y | M | TACI | AI | no | AD | Eczema, enteropathy with complete villous atrophy; HSCT |
|
| |||||||
| IUIS: Combined immunodeficiency with associated features | |||||||
|
| |||||||
| S025 | 6 y | M | BCL11B | AI, II | no | AD | Failure to thrive, nutritional deficiencies, chronic emesis, pyloric stenosis, focal villous blunting of duodenum |
| S029 | 1 mo. | M | BCL11B | AI, II | no | AD | Severe dermatitis |
|
| |||||||
| S040 | 6 m | M | CARD11 | AI, II | yes | AD | Watery diarrhea with villous atrophy, eczema, type 1 diabetes, failure to thrive, alopecia areata, skin tags, club foot |
| S046 | 8 y | M | CARD11 | AI, II | yes | AD | Enteropathy, severe atopic dermatitis, failure to thrive, high IgE levels |
| S178 | 11 m | M | CARD11 | AI, II | yes | AD | Unknown |
| S179 | 1 m | F | CARD11 | AI, II | yes | AD | “IPEX” |
|
| |||||||
| S100 | 25 y | F | KMT2D | AI | no | AD | Short stature, high arched palate; autoimmune disease, enteropathy. Small intestine transplant. |
|
| |||||||
| S024 | 4 m | M | NFKB1 | AI | no | AD | “IPEX” |
|
| |||||||
| S021 | 16 y | F | RAG1 | AI | no | AR | Hypogammaglobulinemia, aplastic anemia, autoimmune enteropathy. Died at age 16y of fungal infection |
| S090 | 2 m | M | RAG1 | AI | no | AR | “IPEX” |
|
| |||||||
| S182 | 4 m | M | SKIV2L | II | no | AR | Severe enteropathy, failure to thrive, total parenteral nutrition dependence, psoriatic-type rash, watery diarrhea with villous atrophy, dilated aortic root, developmental delay. Died age 4y. |
|
| |||||||
| S097 | 2 y | M | STAT3 | AI, II | yes | AD | Chronic enteropathy and chronic liver disease. |
|
| |||||||
| S070 | 7m | M | TTC37 | II | yes | AR | Failure to thrive, small triangular face, sparse hair. Death not related to immune disease. |
| S186 | 5 y | F | TTC37 | II | yes | AR | Enteropathy with villous atrophy from age 1 y, eczema, recurrent severe upper respiratory infections and gastrointestinal infections, hemolytic anemia, thrombocytopenia, alopecia, developmental delay, trichorrhexis nodosa |
|
| |||||||
| IUIS: Defects in intrinsic and innate immunity | |||||||
|
| |||||||
| S120 | 3 y | M | STAT1 | AI, II | yes | AD | “IPEX” |
| S103 | 19 y | M | STAT1 | AI, II | yes | AD | Enteropathy, dermatitis, failure to thrive, autoimmune hemolytic anemia, recurrent infections including oropharyngeal candidiasis and herpes zoster, hypogammaglobulinemia with low T-regulatory cells |
|
| |||||||
| IUIS: Not included in IUIS categories | |||||||
|
| |||||||
| SDH | 19 y | M | MYO5B | - | no | AR | Enteropathy and failure to thrive; reduced expression of FOXP3 and of CD25. Two previously deceased siblings. |
|
| |||||||
| S015 | 2 y | M | TOM1 | II | no | AD | Congenital autoimmune enteropathy, exocrine pancreatic insufficiency, failure to thrive, hepatitis, atopic dermatitis, pityriasis alba, hypogammaglobulinemia, lymphopenia |
Role of the gene in adaptive immunity (AI), innate immunity (II), or both
Genes previously associated with diagnoses of “IPEX-like” syndrome
Inheritance of the phenotype: autosomal dominant (AD), autosomal recessive (AR), or X-linked recessive (XR). Complete genotypes are indicated in Table S2.
Table III.
Variant alleles and interpretations
| Pt | Gene | Mutation | hg19 position | Type / PPH2 / gerp | Clin Var | This study | Geno-type | Functional consequence of mutation; evidence for causality | gnomAD ** |
|---|---|---|---|---|---|---|---|---|---|
| S048 | AIRE | c.967_979del, p.D323fs | chr21:45711063 | LoF | P | P | cpd het* | Well-documented pathogenic mutation (PMID: 9837820) | 138 |
| S048 | AIRE | c.1249insC, p.L417fs | chr21:45713024 | LoF | P/LP | P | cpd het* | Well-documented pathogenic mutation (PMID: 10677297) | 5 |
|
| |||||||||
| S093 | BACH2 | c.C1586T, p.A529V | chr6:90660239 | 1.00 / 5.2 | ni | LP | het | Completely conserved site. Reduced FOXP3 expression in patient cells, consistent with BACH2 haploinsufficiency (PMID: 28530713) | 5 |
|
| |||||||||
| S025 | BCL11B | c.C779T, p.T260M | chr14:99642394 | 1.00 / 3.8 | ni | LP | het | Destroys completely conserved phosphorylation site | 9 |
|
| |||||||||
| S029 | BCL11B | c.C2421G, p.N607K | chr14:99640752 | 1.00 / 3.7 | P | P | het | Well-documented pathogenic mutation (PMID: 27959755) | 0 |
|
| |||||||||
| S040 | CARD11 | c.C1483T, p.P495S | chr7:2974122 | 0.58^ / 5.2 | ni | LP | het | Highly conserved site; enhanced TCR-induced NFKB activation (PMID:30170123) | 3 |
|
| |||||||||
| S046 | CARD11 | c.C88T, p.R30W | chr7:2987341 | 1.00 / 4.5 | P/VUS | LP | het | Completely conserved site; cosegregates with combined immunodeficiency (PMID: 28826773); leads to reduced secretion of IFN-gamma and IL-2 (PMID: 28826773) | 0 |
|
| |||||||||
| S178 | CARD11 | c.G2510A, pR837Q | chr7:2959006 | 1.00 / 4.1 | ni | LP | het | Predict loss of donor splice, transcriptional deletion of exon 18 and stop at codon 764 | 0 |
|
| |||||||||
| S179 | CARD11 | c.G1670A, p.R557H | chr7:2968316 | 1.00 / 4.9 | VUS | VUS | het | Highly conserved site; same mutation reported in another immunodeficiency patient, but no functional effect detected by saturation genome editing (PMID: 32302260) | 4 |
|
| |||||||||
| S030 | CARD14 | c.G669C, p.Q223H | chr17:78158031 | 1.00 / na | ni | LP | het | Completely conserved site; phenotype is excellent fit | 4 |
|
| |||||||||
| S055 | CARD14 | c.C605T, p.S202L | chr17:78157967 | 1.00 / 3.6 | ni | VUS | het | Highly conserved site; no functional information | 4 |
|
| |||||||||
| S092 | CTLA4 | c.G118A; p.V40M | chr2:204735317 | 1.00 / 5.3 | LP/ VUS | LP | het | Completely conserved site in homodimerization domain; reported in patients with primary immunodeficiency (PMID: 29077208, 30048690) | 0 |
|
| |||||||||
| S175 | CTLA4 | c.523dupT; p.L174fs | chr2:204736165 | LoF | ni | P | het | Truncating mutation; haploinsufficiency is pathogenic (PMID: 25329329) | 0 |
|
| |||||||||
| S190 | CTLA4 | c.C208T; p.R70W | chr2:204735407 | 1.00 / 4.3 | P | P | het | One of original CTLA4 pathogenic mutations; decreased CTLA4 T-regulatory cells (PMID: 25329329) | 0 |
|
| |||||||||
| S018 | CTLA4, CD28, CASP8 | 2.83MB deletion | chr2:201994549– 204824394 del | LoF | ni | P | het | Complete deletion of gene | 0 |
|
| |||||||||
| S125 | IRF2BP2 | c.1606insTTT, p.Q536delinsX | chr1:234743040 | LoF | ni | P | het | De novo truncating mutation at codon 536 of 587; we confirmed experimentally that the mutant message is stable | 0 |
|
| |||||||||
| S071 | ITCH | c.393dupA; p.T131fs | chr20:33001602 | LoF | ni | P | homoz | Truncating mutation. Original ITCH pathogenic mutation, cosegregating with disease (PMID: 20170897) | 0 |
|
| |||||||||
| S066 | JAK1 | c.T2414A, p.F805Y | chr1:65307274 | 1.00 / 4.6 | ni | LP | het | Private mutation at completely conserved site in kinase domain; family includes 6 children with JAK1 phenotype | 0 |
|
| |||||||||
| S067 | JAK1 | c.C1901A, p.A634D | chr1:65312418 | 1.00 / 3.9 | LP | P | het | Activating somatic mutation (PMID: 20167706); not previously observed in germline | 0 |
|
| |||||||||
| S170 | JAK1 | 25kb triplication | chr1:65337211– 65362210 | inframe tripl | ni | LP | het | Tandem triplication of exons 2–5, encoding aa 1–151 of membrane-binding FREM domain, stable mutant message experimentally confirmed (Fig S2) | 0 |
|
| |||||||||
| S100 | KMT2D | c.4132–1G>A | chr12:49441853 | inframe del | ni | P | het | Clinically dx as Kabuki. Transcriptional loss of exon 14, encoding aa 1378–1412, stable mutant message experimentally confirmed | 0 |
|
| |||||||||
| S014 | LRBA | 2347bp del | chr4:151792136– 151794482 | LoF | ni | P | cpd het* | Transcriptional loss of exons 18–19 (202 bp), stop codon 725 of 2864; maternal allele; experimentally confirmed (Fig S1) | 0 |
| S014 | LRBA | c.8349+4_8349+7del | chr4:151203595 | LoF | ni | P | cpd het* | Transcriptional loss of exon 56 (197 bp), stop codon 2755 of 2864; paternal allele; experimentally confirmed (Fig S1) | 0 |
|
| |||||||||
| S045 | LRBA | c.C6640T, p.R2214X | chr4:151392836 | LoF | P/LP | P | homoz | Known pathogenic mutation (PMID: 29867916) | 1 |
|
| |||||||||
| S129 | LRBA | c.C5047T, p.R1683X | chr4:151749456 | LoF | P | P | cpd het* | Known pathogenic mutation (PMID: 22608502) | 0 |
| S129 | LRBA | >277bp deletion | chr4:151935548–151935825 (min del) | LoF | ni | P | cpd het* | Genomic deletion of exon 2 including translation start; experimentally confirmed (Fig S1) | 0 |
|
| |||||||||
| S088 | LRBA | 79 kb deletion | chr4:151599677– 151678661 del | inframe del | ni | P | cpd het* | Deletion of exons 36–37, encoding aa 1882–1974, region highly conserved across all BEACH proteins; maternal allele; experimentally confirmed (Fig S1) | 0 |
| S088 | LRBA | c.G2736A, p.W912X | chr4:151788853 | LoF | ni | P | cpd het* | Truncating mutation; experimentally confirmed as paternal allele (Fig S1) | 0 |
|
| |||||||||
| SBDH | MYO5B | c.G946A, p.G316R | chr18:47511088 | 0.99 / 5.5 | VUS | LP | homoz | Predict loss of donor splice site and transcriptional deletion of exon 8, encoding aa 280–355 of myosin motor domain; same mutation in patient with microvillus atrophy (PMID: 21206382) | 18 |
|
| |||||||||
| S024 | NFKB1 | c.A2494C, p.N832H | chr4:103533665 | 1.00 / 5.1 | ni | LP | het | Completely conserved site in death domain, critical to programmed cell death (PMID: 10786798). Also predicted loss of splice enhancer and transcriptional deletion of exon 22 (173bp), stop at codon 829 of 969. | 0 |
|
| |||||||||
| S098 | NLRC4 | c.T1022C, p.V341A | chr2:32475911 | 0.99 / 3.3 | P | P | het | Documented gain of function, pathogenic mutation (PMID: 25217960) | 0 |
|
| |||||||||
| S114 | POLA1 | c.4243+5G>A | chrX:24948671 | LoF | ni | LP | hemiz | Splice effect experimentally confirmed, transcriptional deletion of exon 36 (97bp), stop at codon 1385 of 1463. | 2 hemiz |
|
| |||||||||
| S102 | POLA1 | c.G304T, p.D102Y | chrX:24722562 | 1.00 / 2.2 | ni | VUS | hemiz | Basepair completely conserved; predict loss of exonic splice enhancer, transcriptional deletion exon 4 encoding aa 83–109, no RNA available to validate splice effect | 0 hemiz |
|
| |||||||||
| S160 | POMP | c.342del14, p.F114fs | chr13:29246553 | LoF | P | P | het | Original pathogenic mutation for POMP, escapes NMD (PMID: 29805043) | 0 |
|
| |||||||||
| S021 | RAG1 | c.A1336G, p.N446D | chr11:36596190 | 0.99 / 5.6 | ni | LP | cpd het* | Completely conserved in DNA-binding domain. Missense at A444V is pathogenic. | 0 |
| S021 | RAG1 | c.C2095T, p.R699W | chr11:36596949 | 1.00 / na | P | P | cpd het* | Documented pathogenic mutation with reduced T and B-cells (PMID:20956421) | 4 |
|
| |||||||||
| S090 | RAG1 | c.G2147A, p.R716Q | chr11:36597001 | 1.00 / 6.1 | LP | LP | cpd het* | Completely conserved site in RAG1 domain | 1 |
| S090 | RAG1 | c.G2924A, p.R975Q | chr11:36597778 | 1.00 / 5.6 | P | P | cpd het* | Multiple reports as damaging (PMID:20956421); impairs recombination activity (PMID: 18768869) | 12 |
|
| |||||||||
| S123 | SH2D1A | c.T160A, p.Y54N | chrX:123499633 | 1.00 / 4.8 | ni | LP | hemiz | Completely conserved site in SH2 domain; Y54C protein has reduced stability and binding affinity (PMID: 14674764) | 0 |
|
| |||||||||
| S182 | SKIV2L | c.A1907G, p.H636R | chr6:31932055 | 1.00 / 6.0 | ni | LP | cpd het* | Completely conserved site in helicase domain; cpd het with truncating mutation; good fit to phenotype | 2 |
| S182 | SKIV2L | c.C3187T, p.R1063X | chr6:31936654 | LoF | ni | LP | cpd het* | Truncating mutation | 33 |
|
| |||||||||
| S120 | STAT1 | c.T1627C, p.C543R | chr2:191845351 | 0.99 / 5.1 | ni | VUS | het | Completely conserved site in STAT-binding domain consistent with autosomal dominant immunodeficiency; no sample available for functional analysis and no DNA available from parents to test if de novo | 0 |
|
| |||||||||
| S103 | STAT1 | c.C820T, p.R274W | chr2:191859911 | 1.00 / 5.7 | ni | LP | het | R274G is known gain of function allele, increases STAT1 dependence on cytokines (PMID:21727188) | 0 |
|
| |||||||||
| S041 | STAT3 | c.G1032C, p.Q344H | chr17:40485708 | 0.99 / 4.2 | P | P | het | Documented gain of function pathogenic mutation (PMID: 25359994) | 0 |
|
| |||||||||
| S068 | STAT3 | c.C2144T, p.T715M | chr17:40469200 | 1.00 / 5.0 | P | P | het | Documented gain of function pathogenic mutation (PMID: 25359994) | 0 |
|
| |||||||||
| S097 | STAT3 | c.G1909A, p.V637M | chr17:40474492 | 1.00 / 3.7 | P | P | het | Documented gain of function pathogenic mutation (PMID:21727188) | 0 |
|
| |||||||||
| S112 | STAT3 | c.T937C, p.F313L | chr17:40485928 | 0.99 / 5.6 | ni | LP | het | Completely conserved site in STAT-alpha domain; private allele; good fit to phenotype | 0 |
|
| |||||||||
| S043 | TACI / TNFRSF13B | c.G311A, p.C104Y | chr17:16852186 | 1.00 / 5.0 | LP | LP | het | Completely conserved site; documented in families and patients with CVID (PMID:18981294, 27123465, 22884984); C104R leads to reduced immunoglobulin production (PMID: 21458042, 20889194) | 33 |
|
| |||||||||
| S027 | TNFAIP3 | c.1127delC, p.S376fs | chr6:138199709 | LoF | ni | P | het | Truncating mutation; haploinsufficiency is a causal mechanism for TNFAIP3 syndromes (PMID: 26642243) | 0 |
|
| |||||||||
| S087 | TNFAIP3 | c.G2300A, p.C767Y | chr6:138202383 | 1.00 / 5.7 | ni | LP | het | Completely conserved site at critical cysteine in zinc finger; haploinsufficiency is causal mechanism for TNFAIP3 syndromes (PMID: 26642243) | 0 |
|
| |||||||||
| S184 | TNFRSF6 / FAS | c.A260G, p.E87G | chr10:90767520 | 1.00 / 3.1 | ni | LP | het | Completely conserved site at critical turn in disulfide binding domain | 0 |
|
| |||||||||
| S015 | TOM1 | c.267+2T>C | chr22:35719172 | LoF / inframe del | ni | P | het | Splice mutation yielding two mutant transcripts: (1) intron 4 retained, leading to premature stop; (2) cryptic donor splice leading to deletion of aa 20-152 of vesicular trafficking domain. Experimentally confirmed (Fig S3) | 4 |
|
| |||||||||
| S070 | TTC37 | c.C409T, p.R137X | chr5:94876528 | LoF | P | P | homoz | Documented pathogenic mutation | 2 |
|
| |||||||||
| S186 | TTC37 | c.994+1G>A | chr5:94864704 | inframe del | ni | LP | cpd het* | Predict loss of donor splice site, transcript deletion of exon 12 and aa 298–331 | 1 |
| S186 | TTC37 | c.402+2T>G | chr5:94877007 | LoF | ni | P | cpd het* | Predict loss of donor splice site and transcriptional deletion of exon 7 and stop at codon 142 of 1564 | 0 |
|
| |||||||||
| S108 | XIAP | c.949del11, p.W317fs | chrX:123022540 | LoF | ni | P | hemiz | Truncating mutation | 0 |
All paired alleles of compound heterozygous (cpd het) genotypes were experimentally confirmed to be in trans in the patient
All gnomAD entries are heterozygotes unless otherwise indicated
Completely conserved as proline through mammals; no species has serine as reference sequence
ni: no information on ClinVar; PP2: polyphen-2 score; gerp: gerp score; LoF: loss of function
P: pathogenic; LP: likely pathogenic; VUS: unknown significance, warranting further evaluation
Biological functions of genes mutant in patients
Mutant genes in the IUIS classification system.
Genes responsible for patients’ diagnoses are involved in both adaptive and immune response (Table II), and in biological functions including signaling, cell differentiation, apoptosis, debris handling, and epithelial integrity (Figure 1). The 27 genes appeared in multiple categories of the phenotypic classification system for inborn errors of immunity of the International Union of Immunological Societies’ (IUIS) (Table II)10. Causal genes in the IUIS Immune Dysregulation category would be expected, given original clinical suspicion for IPEX. However, an equal number of genetic diagnoses were due to genes in other IUIS phenotypic categories, including Autoinflammatory Disorders, Antibody Deficiencies, Combined Immunodeficiencies, and Defects in Intrinsic and Innate Immunity. Most of the 27 genes were not previously reported in the context of “IPEX-like” disease.
Figure 1:

Genes responsible for childhood immune dysregulation, polyendocrinopathy, and enteropathy. Genes responsible for childhood IPE in 48 patients by principal biological function, based on literature review and Gene Ontogeny annotation. Genes may act primarily within the adaptive arm of the immune system (blue arc), the innate arm (maroon arc), or both.
For some genes, the mutations of these patients provide additional support for the role of the genes in IPE (Tables II, III). For example, somatic mutations in the JAK1 kinase are common in tumors, but a germline mutation in JAK1 has previously been documented in only one family17. Patients S66, S67, and S170, with severe IPE phenotypes, carry three different damaging mutations in JAK1, strongly supporting a role for JAK1 germline mutations in IPE (Figure S2). Similarly, IRF2BP2 encodes a transcriptional regulator of type I interferon and has a well-documented role in immune regulation, angiogenesis, and apoptosis18,19, but a germline mutation in IRF2BP2 has been previously documented in only one family20. Patient S125, with a similar phenotype to the previously reported patient, carries a de novo damaging mutation in IRF2BP2, supporting a role for this gene in IPE.
Two genes responsible for diagnoses of our patients were not previously included in the IUIS classification. TOM1 encodes a protein of endocytosis, with a missense mutation previously reported in a family with autosomal dominant immune dysregulation and impaired autophagy21. Patient S15, with a severe IPE phenotype, is heterozygous for a TOM1 splice mutation that produces a stable product lacking the vesicular trafficking domain, so likely yielding a dominant negative effect (Figure E3). Mutations in MYO5B are well documented in patients with microvillus inclusion disease22. Like patient SDH, patients with MYO5B deficiency can have extraintestinal manifestations, including hematuria, lung disease, and increased susceptibility to infection23.
Immune regulation and dysmorphology.
For four patients, immune dysregulation appeared in combination with dysmorphology. Three patients with mildly dysmorphic features harbored mutations in TTC37 or SKIV2L, both of which are responsible for tricho-hepato-enteric (THE) syndrome24,25. One patient, with a splice mutation in KMT2D, presented with features of Kabuki syndrome in addition to autoimmune dysregulation and severe enteropathy, both of which are rare but reported features of Kabuki syndrome26,27. Genetic analysis is quite frequently undertaken for patients with dysmorphic features. However, for several patients with mutations in dysmorphism genes, these features were subtle and not recognized until after the genetic diagnosis.
Genetic diagnoses and treatment
For 42 of the 48 patients with genetic diagnoses, knowing the critical gene could have guided treatment (Figure 2). Specific management implications for each patient with a genetic diagnosis are indicated in Table EII. Some patients for whom the causal gene was identified could have been treated with appropriately targeted immune modulatory drugs. Patients whose genetic diagnoses suggested life-limiting disease may have been considered for HCT early in their disease course. Conversely, patients with genetic diseases having less severe outcomes or no effective therapies may have avoided high-risk treatments.
Figure 2:
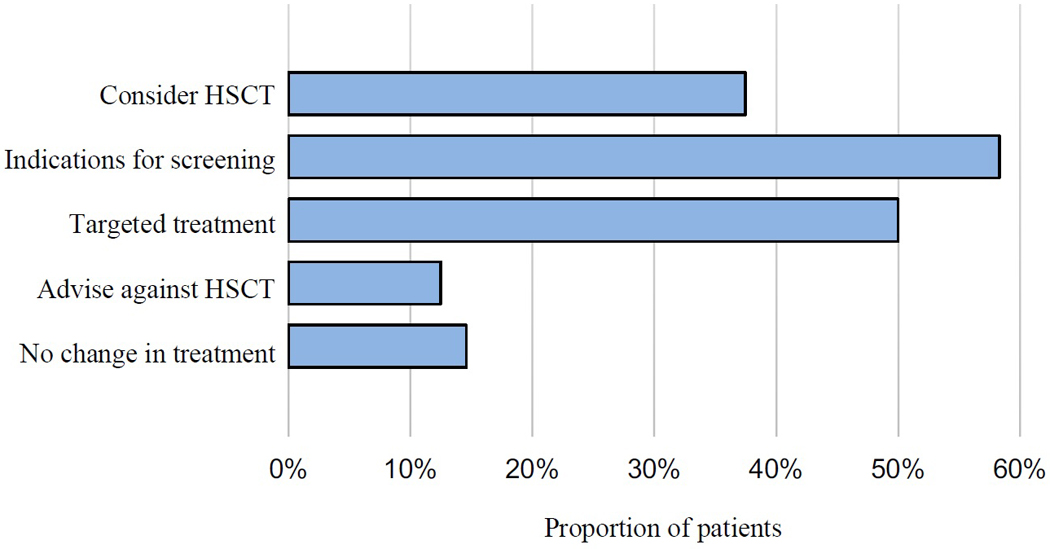
IPE disease management recommendations based on genetic diagnoses. Recommendations for therapy for each gene are presented in supplementary Table EII. Percentages add to more than 100%, because for some patients, more than one recommendation was available. HCT is hematopoietic cell transplant.
Targeted therapeutics.
For 25 patients, genetic diagnoses suggested specific targeted therapies. For example, abatacept, a CTLA-Ig fusion protein, has been reported to be an effective targeted therapy for patients with CTLA4 haploinsufficiency or LRBA deficiency28. JAK inhibitors have been shown to be effective in patients with JAK1, STAT1, or STAT3 gain-of-function mutations29. Patients with NLRC4 gain-of-function mutations have been treated successfully in preliminary studies with recombinant IL-18-binding protein30, and a clinical trial is underway evaluating this treatment in patients with XIAP mutations31. Patients with CARD14 and TNFAIP3 mutations often respond to TNF-alpha inhibitors and to IL12/IL23 inhibitors32,33.
Screening.
For 29 patients, genetic diagnoses would have altered recommendations for screening. Loss of function mutations in STAT3, FAS, and SH2D1A predispose to lymphoma, and gain of function mutations in STAT3 predispose to leukemia34. Patients with CLTA4 haploinsufficiency and LRBA deficiency also appear to be at a higher risk for cancer35,36. In our series, two of four patients with LRBA deficiency developed malignancies: patient S45 died at age 16 from gastric adenocarcinoma, and patient S14 developed lymphoma at age 3. Patients with mutations in AIRE, RAG1 and NFKB1 can develop a broad range of autoimmune phenomena that call for regular thyroid, blood, liver, renal, and pulmonary screening2,37,38. Patients with STAT3 mutations should undergo pulmonary screening39, and although there are limited cases reported, current evidence also supports pulmonary screening for patients with mutations in BACH2, ITCH and TOM121,40,41. Patients with POLA1 deficiency (figure E4) should undergo regular ophthalmologic exams, given the risk of sterile inflammation leading to cataracts, scarring and blindness42.
Hematopoietic cell transplantation (HCT).
For 18 patients, genetic diagnoses represent potential indications for HCT. The 8 patients with CTLA4 haploinsufficiency and LRBA deficiency would be strongly considered for HCT given the increasing evidence of high morbidity and mortality in these diseases. For example, patient S88 developed inflammatory brain lesions while on abatacept; because he had failed appropriate targeted therapy and had a genetic diagnosis, he underwent HCT. Patients with mutations in XIAP, RAG1, and SH2D1A are also frequently considered for early HCT43,44,45. Patient S21, who had hypogammaglobulinemia and autoimmune enteropathy beginning in early childhood, was referred for genetic testing at age 16y. She was found to have compound heterozygous RAG1 mutations, often considered an indication for HCT. She died at age 16 from a fungal infection prior to genetic diagnosis.
Genetic diagnosis can benefit patients even if gene-specific therapeutic guidelines are not yet available. For instance, severe childhood illness may lead physicians to attempt HCT in the absence of a genetic diagnosis. If genetic diagnosis reveals the cause of disease to be a gene of the hematopoietic system, bone marrow transplantation is more likely to be effective. Conversely, for the 6 patients with mutations in MYO5B, TTC37, SKIV2L, and CARD14, HCT would be unlikely to ameliorate disease, since these include epithelial defects that do not respond to HCT46. Moreover, a genetic diagnosis can be crucial to guide diagnosis in patients’ relatives. For instance, patient S92, with CTLA4 haploinsufficiency, underwent HCT due to the severity of his disease despite lacking a genetic diagnosis. He now has an infant son who can be screened for the same mutation.
DISCUSSION
Enabling precision medicine for children with immune dysregulation, polyendocrinopathy or enteropathy requires embracing genetic heterogeneity. That is, despite all patients in this project being referred for the same clinical concern (IPEX), many different genes were responsible for their illnesses, including some genes known to cause IPEX-like disease and others not previously associated with this syndrome. The diversity of IUIS phenotypes among these patients highlights the difficulty of inferring genotype from phenotype and the value of comprehensive genomic analysis early in the course of disease.
This genetic heterogeneity reflects the clinical reality that the features of childhood immune disorders are widely encountered and overlapping. The search for a genetic cause cannot realistically be based only on clinical phenotype, because immune dysregulation conditions have ill-defined phenotypic boundaries and can be due to any of multiple different genes. A genetics-first paradigm avoids unhelpful classifications that may lead to errors in diagnosis or therapy. To assist this practice, we suggest that when a causal link between gene and phenotype is established, the gene responsible for an immune disorder be included in its name (e.g. LRBA immunodeficiency). Diagnoses impact treatment options and define cohorts for scientific studies. By including the responsible gene in the name of each disease, clinicians can avoid ambiguity and immediately access treatments most promising for the patient.
Genetic testing platforms vary widely in capacity and limitations47. Given rapidly improving technology, it is important both to seek early genetic diagnosis and to re-evaluate if original results are negative. Sequencing to high coverage and including critical intronic and regulatory regions among the targets greatly increases detection sensitivity for copy number variants (CNVs), which for our patients represented 10% of all mutations. CNVs were particularly frequent in LRBA and represent a growing class of defects for which genetic diagnosis dictates targeted therapy. Custom-designed gene panels detect this class of mutations, but most exome sequencing does not. In practice, most commercial clinical sequencing is now exome sequencing, with “gene panels” in fact simply exome sequence data with only a subset of genes reported to the physician. Approximately 10% of the mutations of our patients would have been missed by exome sequencing.
If genetic disease remains a consideration despite a negative genetic test, it is worth considering more complete and current genomic analysis. False negatives on genetic tests can result from coverage that is inadequate to detect structural variants, from incorrect variant interpretation, from limitations in knowledge of gene function and hence of pathogenic variants, or from lab error. Errors of variant interpretation can be subtle and include failure to detect effects on transcription caused by mutations that do not alter amino acid sequence, failure to detect mutations at sites other than exon-intron boundaries that alter splicing, failure to distinguish variation in true genes from variation in pseudogenes, and so on. The pace of gene discovery, knowledge about individual genes, knowledge of classes of cryptic mutations, and technological advance all support additional genetic testing as platforms improve. Immunologists will likely find it useful to consult with academic centers to interpret increasingly complex genetic information. Conversely, clinically well-characterized patients will very greatly contribute to functional analysis of new mutations.
In our experience, negative results from tests of single genes or narrow panels have almost no value, and furthermore may impair the ability of clinicians to do follow-up testing due to insurance limitations. The solution is for the first genetic test to include as many genes and as many classes of mutations as possible, either from a large gene panel or from whole exome sequencing.
This project had several limitations, largely as the result of some patients being originally referred several years ago. First, for some patients, clinical information was limited and referring physicians were no longer available. Second, only probands were available for initial analysis, although a few families were re-contacted through the referring providers. Analysis of a child and both parents is critical to identifying de novo mutations, an important part of genetic diagnosis48. Analysis of the complete patient-parent-parent trio is particularly important in the evaluation of mutations in genes such as CTLA4. Severe mutations in CTLA4 can lead to severe phenotypes, as in our patients (Tables II and III), but in some families, mutations in CTLA4 have been identified in relatives with no, or late-onset, clinical signs49,50. It is possible that CTLA4 mutations with severe, early onset phenotypes are de novo in affected children, but this speculation can only be tested with DNA from parents. Both these limitations reduced the number of genetic diagnoses in which we had confidence.
A third limitation was that our gene-panel sequencing strategy detected all mutations in coding sequence and all gene-impacting copy number variants but did not detect mutations in distant non-coding genomic regions. Such distant non-coding mutations would be revealed by whole genome sequencing. The cost of whole genome sequencing is rapidly decreasing, but the cost of interpreting non-coding variants in whole genome sequence is very high. Insurance reimbursement is the practical limitation to applying whole genome sequencing to genetic diagnosis to these or other complex conditions.
Despite these limitations, the yield of reportable mutations among the children in this series was quite high (39%). Analysis of an active clinical population, with patient-parent-parent trios available for testing, would yield more solved cases with correspondingly greater benefit for management.
In conclusion, genetic diagnoses of children with immune dysregulation, polyendocrinopathy and enteropathy, but no mutation in FOXP3, revealed a wide array of clinical immune diseases and disruption of a wide array of biological pathways. More than 80% of genetic diagnoses were clinically actionable. We suggest that these results support a “genetics first” approach for patients with severe, early-onset immune dysregulation. We propose that all patients with childhood-onset immune dysregulation undergo comprehensive genomic analysis, rather than single-gene testing or no genetic testing at all. Treatments targeting immune pathways are rapidly advancing. To harness the promise of these treatments, it is critical to fully exploit genetic testing.
Supplementary Material
Acknowledgments
Funding: NIH R35CA197458 (M.C.K) and King Lab Gift Fund
ABBREVIATIONS
- IPEX
Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked
- CNV
copy number variant
- IPE
Immune dysregulation, polyendocrinopathy, enteropathy
- IUIS
International Union of Immunological Societies
- UW
University of Washington
- SCH
Seattle Children’s Hospital
- HCT
hematopoietic cell transplantation
- PBMC
peripheral blood mononuclear cell
- ACMG
American College of Medical Genetics
- THE
trichohepatoenteric
Footnotes
Disclosures: Tom Walsh PhD is a consultant for Color Genomics. Troy Torgerson MD PhD is currently employed by the Allen Institute for Immunology, Seattle. The rest of the authors declare that they have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Leiding JW, Forbes LR. Mechanism-based precision therapy for the treatment of primary immunodeficiency and primary immunodysregulatory diseases. J Allergy Clin Immunol Pract. 2019;7(3):761–773. DOI: 10.1016/j.jaip.2018.12.017 [DOI] [PubMed] [Google Scholar]
- 2.Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract. 2019;7(6):1970–1985. DOI: 10.1016/j.jaip.2019.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223–230. doi: 10.1016/j.jaci.2015.09.025. [DOI] [PubMed] [Google Scholar]
- 4.Delmonte OM, Notarangelo LD. Targeted therapy with biologicals and small molecules in primary immunodeficiencies. Med Princ Pract. 2020;29(2):101–112. doi: 10.1159/000503997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139:232–245. doi: 10.1016/j.jaci.2016.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 7.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 8.Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Primary Immune Deficiency Treatment Consortium (PIDTC) and the Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT). Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol 2018;141:1036–1049. doi: 10.1016/j.jaci.2017.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burroughs LM, Torgerson TR, Storb R, Carpenter PA, Rawlings DJ, Sanders J, et al. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol 2010;126:1000–5. doi: 10.1016/j.jaci.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019. Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. doi: 10.1007/s10875-019-00737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.University of Washington Department of Laboratory Medicine and Pathology: https://testguide.labmed.uw.edu/public/view/IMD
- 12.Abu Rayyan A, Kamal L, Casadei S, Brownstein Z, Zahdeh F, Shahin H, et al. Genomic analysis of inherited hearing loss in the Palestinian population. Proc Natl Acad Sci USA 2020;117(33):20070–6. doi: 10.1073/pnas.2009628117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casadei S, Gulsuner S, Shirts BH, Mandell JB, Kortbawi HM, Norquist BS, et al. Characterization of splice-altering mutations in inherited predisposition to cancer. Proc Natl Acad Sci USA. 2019;116(52):26798–807. doi: 10.1073/pnas.1915608116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ClinVar: http://www.ncbi.nlm.nih.gov/clinvar/
- 16.Gulsuner S, Stein DJ, Susser ES, Sibeko G, Pretorius A, Walsh T, et al. Genetics of schizophrenia in the South African Xhosa. Science. 2020;367(6477):569–73. doi: 10.1126/science.aay8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol. 2017;139(6):2016–2020. doi: 10.1016/j.jaci.2016.12.957. [DOI] [PubMed] [Google Scholar]
- 18.Chen HH, Keyhanian K, Zhou X, Vilmundarson RO, Almontashiri NA, Cruz SA, et al. IRF2BP2 reduces macrophage inflammation and susceptibility to atherosclerosis. Circ Res 2015;117:671–683. doi: 10.1161/CIRCRESAHA.114.305777. [DOI] [PubMed] [Google Scholar]
- 19.Ramalho-Oliveira R, Oliveira-Vieira B, Viola JPB. IRF2BP2: A new player in the regulation of cell homeostasis. J Leukoc Biol 106:717–723. doi: 10.1002/JLB.MR1218-507R. [DOI] [PubMed] [Google Scholar]
- 20.Keller MD, Pandey R, Li D, Glessner J, Tian L, Henrickson SE, et al. Mutation in IRF2BP2 is responsible for a familial form of common variable immunodeficiency disorder. J Allergy Clin Immunol. 2016;138(2):544–550. doi: 10.1016/j.jaci.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keskitalo S, Haapaniemi EM, Glumoff V, Liu X, Lehtinen V, Fogarty C, et al. Dominant TOM1 mutation associated with combined immunodeficiency and autoimmune disease. NPJ Genom Med. 2019;4:14. doi: 10.1038/s41525-019-0088-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz-Erian P, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40(10):1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 23.Siahanidou T, Koutsounaki E, Skiathitou AV, Stefanaki K, Marinos E, Panajiotou I, et al. Extraintestinal manifestations in an infant with microvillus inclusion disease: complications or features of the disease?. Eur J Pediatr. 2013;172(9):1271–1275. doi: 10.1007/s00431-013-1948-0. [DOI] [PubMed] [Google Scholar]
- 24.Fabre A, Martinez-Vinson C, Roquelaure B, Missirian C, André N, Breton A, et al. Novel mutations in TTC37 associated with tricho-hepato-enteric syndrome. Hum Mutat. 2011;32(3):277–281. doi: 10.1002/humu.21420. [DOI] [PubMed] [Google Scholar]
- 25.Fabre A, Charroux B, Martinez-Vinson C, Roquelaure B, Odul E, Sayar E, et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet. 2012;90(4):689–692. doi: 10.1016/j.ajhg.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindsley AW, Saal HM, Burrow TA, Hopkin RJ, Shchelochkov O, Khandelwal P, et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol. 2016;137(1):179–187. doi: 10.1016/j.jaci.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang YR, Xu NX, Wang J, Wang XM. Kabuki syndrome: review of the clinical features, diagnosis and epigenetic mechanisms. World J Pediatr. 2019;15(6):528–535. doi: 10.1007/s12519-019-00309-4. [DOI] [PubMed] [Google Scholar]
- 28.Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932–1946. doi: 10.1016/j.jaci.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delmonte OM, Notarangelo LD. Targeted therapy with biologicals and small molecules in primary immunodeficiencies. Med Princ Pract. 2020;29(2):101–112. doi: 10.1159/000503997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017;139(5):1698–1701. doi: 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT03113760
- 32.Craiglow BG, Boyden LM, Hu R, Virtanen M, Su J, Rodriguez G, et al. CARD14-associated papulosquamous eruption: A spectrum including features of psoriasis and pityriasis rubra pilaris. J Am Acad Dermatol. 2018;79(3):487–494. doi: 10.1016/j.jaad.2018.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders. J Allergy Clin Immunol. 2018;141(4):1485–1488. doi: 10.1016/j.jaci.2017.10.039. [DOI] [PubMed] [Google Scholar]
- 34.Chandrasekaran P, Zimmerman O, Paulson M, Sampaio EP, Freeman AF, Sowerwine KJ, et al. Distinct mutations at the same positions of STAT3 cause either loss or gain of function. J Allergy Clin Immunol. 2016;138(4):1222–1224.e2. doi: 10.1016/j.jaci.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habibi S, Zaki-Dizaji M, Rafiemanesh H, Lo B, Jamee M, Gámez-Díaz L, et al. Clinical, immunologic, and molecular spectrum of patients with lps-responsive beige-like anchor protein deficiency: a systematic review. J Allergy Clin Immunol Pract. 2019;7(7):2379–2386. doi: 10.1016/j.jaip.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932–1946. doi: 10.1016/j.jaci.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev. 2019;287(1):103–120. doi: 10.1111/imr.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol. 2018;142(4):1285–1296. doi: 10.1016/j.jaci.2018.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich PS, et al. Clinical Aspects of STAT3 Gain-of-Function Germline Mutations: A Systematic Review. J Allergy Clin Immunol Pract. 2019;7(6):1958–1969.e9. Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich PS, [DOI] [PubMed] [Google Scholar]
- 40.Afzali B, Grönholm J, Vandrovcova J, O’Brien C, Sun HW, Vanderleyden I, et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat Immunol. 2017;18(7):813–823. doi: 10.1038/ni.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet 2010;86:447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Starokadomskyy P, Wilton KM, Krzewski K, Lopez A, Sifuentes-Dominguez L, Overlee B, et al. NK cell defects in X-linked pigmentary reticulate disorder. JCI Insight. 2019;4(21):e125688. doi: 10.1172/jci.insight.125688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ono S, Okano T, Hoshino A, Yanagimachi M, Hamamoto K, Nakazawa Y, et al. Hematopoietic stem cell transplantation for XIAP deficiency in Japan. J Clin Immunol. 2017;37(1):85–91. doi: 10.1007/s10875-016-0348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135(16):1332–1343. doi: 10.1182/blood.2019000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract. 2019;7(6):1970–1985.e4. doi: 10.1016/j.jaip.2019.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fabre A, Bourgeois P, Coste ME, Roman C, Barlogis V, Badens C. Management of syndromic diarrhea/tricho-hepato-enteric syndrome: A review of the literature. Intractable Rare Dis Res. 2017;6(3):152–157. doi: 10.5582/irdr.2017.01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol. 2018;38(3):320–329. doi: 10.1007/s10875-018-0489-8. [DOI] [PubMed] [Google Scholar]
- 48.Baxter SK, King M-C. A time to sequence. JAMA Pediatr 2017;171(12):e173435. doi: 10.1001/jamapediatrics.2017.3435. [DOI] [PubMed] [Google Scholar]
- 49.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932–1946. doi: 10.1016/j.jaci.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.